

Abstract
The spliceosome is a dynamic macromolecular machine that undergoes a series of conformational rearrangements as it transitions between the several states required for accurate splicing. The transition from the B to Bact is a key part of spliceosome assembly and is defined by the departure of several proteins, including essential U5 component Dib1. Recent structural studies suggest that Dib1 has a role in preventing premature spliceosome activation, as it is positioned adjacent to the U6 snRNA ACAGAGA and the U5 loop I, but its mechanism is unknown. Our data indicate that Dib1 is a robust protein that tolerates incorporation of many mutations, even at positions thought to be key for its folding stability. However, we have identified two temperature-sensitive mutants that stall in vitro splicing prior to the first catalytic step and block assembly at the B complex. Additionally, Dib1 readily exchanges in splicing extracts despite being a central component of the U5 snRNP, suggesting that the binding site of Dib1 is flexible. Structural analyses show that the overall conformation of Dib1 and the mutants are not affected by temperature, so the temperature sensitive defects most likely result from altered interactions between Dib1 and other spliceosomal components. Together, these data lead to a new understanding of Dib1’s role in the B to Bact transition and provide a model for how dynamic protein-RNA interactions contribute to the correct assembly of a complex molecular machine.
Keywords: pre-mRNA splicing, spliceosome, Burn-McKeown, U5 snRNP, protein reconstitution
Graphical abstract
Introduction
Pre-messenger RNA (pre-mRNA) splicing removes non-coding intronic sequences from pre-messenger RNA via two trans-esterification steps. This process is facilitated by the spliceosome, a large macromolecular protein-RNA complex. The spliceosome is composed of five small nuclear RNA-protein complexes (snRNP) that, along with over 100 additional protein factors, assembles anew on each pre-mRNA (reviewed in [1,2]). Assembly of the spliceosome is highly regulated at each step. After U1 and U2 snRNPs bind to the pre-mRNA, the U4/U6-U5 triple snRNP joins forming the B complex (Figure 1). After B complex formation, Brr2, a DEIH- box helicase, is primed to unwind the U4/U6 snRNA duplex, allowing U4 snRNA to depart. This departure is critical for progression to a catalytically active spliceosome and is accompanied by a series of snRNA-snRNA rearrangements (reviewed in [3,4]). Once U4 snRNA is no longer bound to the U6 snRNA, the U6 ACAGAGA stem unwinds to interact with the 5’ splice site. Additionally, the U5 loop I interacts with nucleotides adjacent to the 5’ splice site, helping to orient the catalytic core of the spliceosome. These two newly formed RNA-RNA interactions, 5’ splice site with U6 ACAGAGA stem and U5 loop I with nucleotides adjacent to the 5’ splice site are hallmarks of the Bact complex and must be regulated in order to prevent aberrant splicing which could cause or contribute to human disease.
Figure 1.
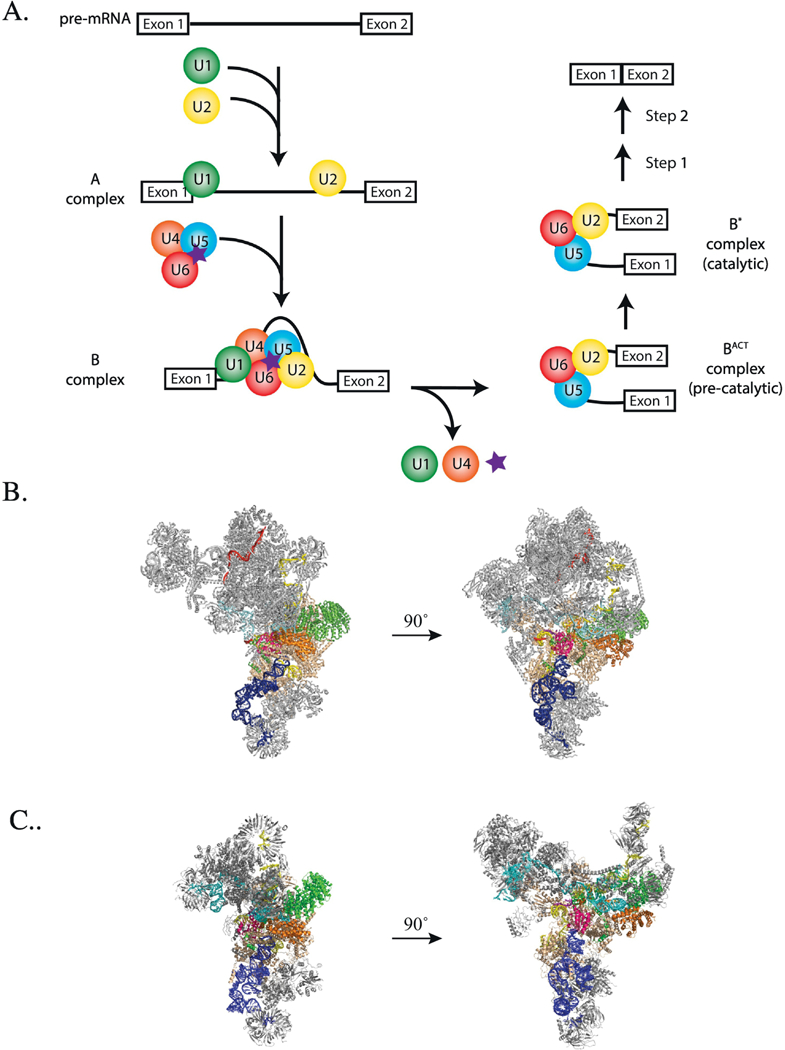
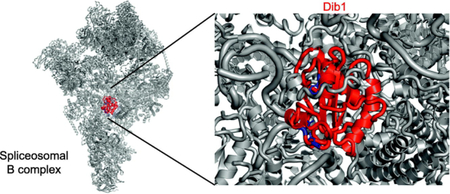
A. Diagram of spliceosome assembly and activation highlighting Dib1 presence and departure (purple star). B and C. Location of Dibl in the yeast B complex (panel B, pdb 5nrl, [8]) and yeast U4/U6-U5 triple snRNP (panel C, pdb 5GAN, [7]) Colored are Dib1 (magenta), U4 snRNA (cyan), U5 snRNA (blue), U6 snRNA (yellow) with ACAGAGA (purple), Prp8 (beige), Prp6 (green), Prp31 (orange), mapped pre-mRNA (red).
In addition to the shifts in RNA-RNA interactions, there are several other compositional changes that occur between the B and Bact complexes [5]. Upon departure of U4 snRNP, a host of proteins depart as well, including several U5 associated proteins, Snu66, Prp6 and Dib1, and B complex proteins, Snu23, Prp38 and Spp381. These departures are accompanied by the arrival of the nineteen complex proteins (NTC) and the NTC-related proteins joining the splicing complex. However, despite their clear importance in ensuring that the spliceosome does not departures and additions are not well characterized. This study examines the role of U5 snRNP protein, Dib1, in spliceosome assembly.
Recent cryo-electron microscopy studies have suggested potential roles for Dib1 in regulating spliceosome assembly and preventing premature spliceosome activation. In crosslinking and structural studies of isolated S. cerevisiae U4/U6-U5 triple snRNP and B complex, Dib1 is adjacent to the U5 loop 1, occluding the U6 ACAGAGA sequence required to position the pre-mRNA for splicing (Figure 1 )[6–8]. This is consistent with the early finding that an uncharacterized 16-kDa protein crosslinked to the U5 loop [9]. In order for the activated spliceosome hallmark interactions between the pre-mRNA, U6 ACAGAGA, and U5 loop to occur, Dib1 must depart [5,10]. The timing and interactions that trigger this departure are unknown.
S. cerevisiae Dib1 (Dim1 in S. pombe or TXNL4A/U5–15k in humans) is 16.8 kDa protein that is well conserved with 66% identity from yeast to humans. Human Dib1 (U5–15k) possesses a thioredoxin-like fold with three a- helices surrounding a core of four β- sheets and a flexible tail extension [11] (Figure 2). In S. cerevisiae, S. pombe and C. elegans, Dib1 is essential [11–15]. In humans, alleles of TXNL4A gene are linked to Burn-McKeown Syndrome, a rare condition that results in craniofacial abnormalities, including cleft palettes and neural deafness [13]. Patients afflicted with Burn-McKeown carry one mutated allele that likely results in a non-functional protein, compounded by a promoter deletion on the second allele that lowers the level of mRNA transcription [13]. Parents with one defective allele have no physiological defects.
Figure 2.
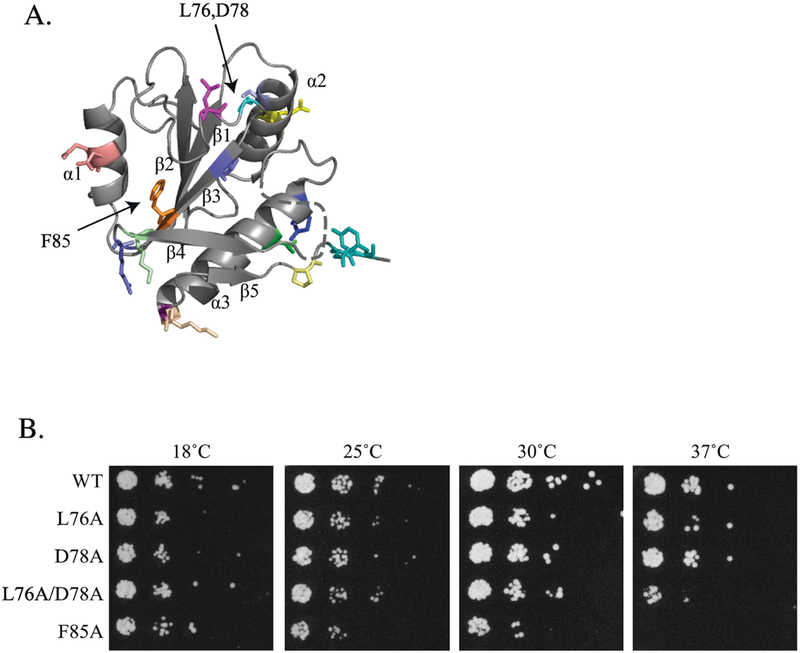
A. Crystal structure of U5–15Kd, the human homolog of Dib1 (pdb lqgv, [11]). Mutants constructed in this study are colored as indicated in Table 1. B. Growth phenotypes of dib1 mutants. Serial dilutions of S. cerevisae harboring dib1 mutants as the sole copy of dib1 were spotted onto YPD and grown for five days at 18°C, three days at 25°C and two days at 30 and 37°C.
Using a rational design approach, we have identified two novel mutants of Dib1 that have temperature-sensitive growth phenotypes with varying severity upon heat treatment. We present the effects of these Dib1 mutants on in vitro splicing and spliceosome assembly, showing that they stall splicing prior to the first step of splicing. We further show that Dib1 has the ability to exchange within the splicing machinery. Structural analyses of the Dib1 mutants suggest that the effects of the mutations may act via altered interactions with other components of the spliceosome, since the structures of these mutants are not further altered at increased temperatures. Together our data provide mechanistic insight into how Dib1 prevents premature spliceosome activation in S. cerevisiae.
Results
Identification of novel mutants in dib1 that stall growth
To identify regions that may be important for Dib1’s biological function, we designed a series of mutants in which residues were modified to alanine. Previous structural analysis of U5– 15kD or hDim1, the human homolog of Dib1, identified regions that were proposed as potential sites of interactions with other proteins or RNA [11]. These included hydrophobic clefts and grooves as well as acidic and basic regions. Specifically, the hydrophobic pocket created by a cleft between the β3 and β4 strands was identified as a potential protein binding site [11] and was targeted in this study. Additionally, flexible loop regions are potential interaction sites, so we identified several loops of interest based on the crystal structure of the human homolog, U5–15k [11]. Residues of interest were then mapped onto B complex and U4/U6-U5 triple snRNP structures to identify potential disruption to interactions with neighboring protein (shown in Figure 2 and listed in Table 1).
Table 1.
Growth Phenotypes and Location of dib1 mutants. Mutants are mapped in Figure 2 and colored as listed.
| Mutant | Phenotype at 37°C* | Location in hDim1(Dib1) | Residue in Humans | Color |
|---|---|---|---|---|
| D16A/Q17A | wt | Al | D15/Q16 | Salmon |
| C39A | wt | a2 | C38 | Light Blue |
| E44A | wt | a2 | E43 | Yellow |
| L76A | ts (mild) | Loop between β2/β3 | L75 | Magenta |
| L76A/D78A | ts | Loop between β2/β3 | L75/D77 | Magenta/Cyan |
| D78A | wt | Loop between β2/β3 | D77 | Cyan |
| V82L/H87A | wt | β3 and Loop between β3/β4 | V81/R86 | Slate |
| V82L/H87R | wt | β3 and Loop between β3/β4 | V81/R86 | Slate |
| F85A | ts | β3 | F84 | Orange |
| K89A | wt | β4 | K88 | Pale Green |
| K102A | wt | Loop between β4/α3 | K101 | ** |
| Q111A | wt | α4 | Q110 | Blue |
| D115A | wt | α4 | D114 | Green |
| K126A | wt | α4 | K125 | Wheat |
| N127G | wt | Tail | G126 | Purple |
| N127D | wt | Tail | G126 | Purple |
| P134A | wt | Tail | P133 | Pale Yellow |
| D136A/Y137A | wt | Tail | D135/Y136 | Teal |
wt: wild-type growth phenotype, ts: slower growth or death at 37°C
Unmodelled flexible loop
While the function of thioredoxin and its orthologs is dependent on a Cys-X-X-Cys motif, Dib1 lacks this motif, replacing it with a conserved Asp-X-X-Cys (aa 36–39 in S. cerevisiae) [14]. Studies of hDim1 (U5–15kD) in the presence and absence of reducing agent suggested that although a disulfide bond can form between C38 and C79, the stability of the protein is lowered in the presence of the disulfide [16]. This finding, in combination with the reducing environment of the cell and the lack of conservation of C79, which is a methionine in S. cerevisiae, suggested that hDim1 does not function like a thioredoxin, despite its similar fold. Previous work showed a limited role for the Asp-X-X-Cys motif in S. pombe [16]. However, the function of this motif in S. cerevisiae has not been explored, and a potential role may be for C39 to participate in a disulfide bond necessary for protein stability. Therefore we sought to create mutants to disrupt potential disulfide bonds to determine whether this interaction is necessary for protein function.
Dib1 mutants were constructed using PCR-based site directed mutagenesis of a HIS3- marked yeast expression plasmid in which the sequence 1000 nt upstream and downstream of the DIB1 coding region was incorporated. This ensured the inclusion of native endogenous regulatory elements necessary for dib1 transcription. Plasmids encoding dib1 mutants were then transformed into a S. cerevisiae yeast strain lacking a chromosomal copy of dib1 but harboring DIB1 on an URA3-marked yeast expression plasmid (which grew identically to the genomically encoded DIB1 base strain), because of the essential nature of dibl. Presence of dibl mutants was achieved via plasmid swapping with counter-selection on 5-FOA.
Most of the modifications constructed in dib1 were tolerated by the cell and showed little effect on cell growth (Supplemental Figure 1, Table 1). However, two mutants showed significant effects on S. cerevisiae growth and were found to be temperature sensitive (ts) (Figure 2, Table 1). First, the dib1-F85A mutant showed similar growth as a wild-type yeast at 18, 25, and 30°C, but was severely sick at 37°C and lethal at 38°C (Figure 2B, Supplemental Figure 2). F85 is located in a hydrophobic pocket that has been proposed as a potential protein binding region based on the high propensity of solvent-exposed hydrophobic side chains that are in a shallow cleft region [11]. The dib1-F85A mutant changes the large bulky aromatic phenylalanine into a small methyl group, thereby disrupting potential pi-pi stacking interactions. In the second mutant, residues L76 and D78 are located in a flexible loop of Dib1 between the β2 and β3 strands. In this case, the dib1- L76A/D78A mutant resulted in similar cell growth similar as wild type at lower temperatures but was impaired at 37°C and lethal at 38°C (Figure 2B, Supplemental Figure 2). Cell growth of dib1-L76A alone was mildly impaired at 38°C (Supplemental Figure 2), while dib1-D78A showed no effect on cell growth at any temperature (Figure 2B). This suggests that the double mutant has a synthetic phenotype.
Purified Dib1 mutants have altered secondary structure
Mutations in Dib1 could cause significant perturbations to the secondary and tertiary structure of the protein, leading to the observed cell growth defects. Therefore, it was important to compare the structure of mutant Dib1 to wild-type Dib1. Circular dichroism (CD) spectroscopy has been used to characterize the structural fold by monitoring changes in absorbance of circularly polarized light by secondary structure elements. Using this technique, we sought to compare wild-type and mutant proteins at both permissive and non-permissive temperatures.
Wild-type Dib1 protein was purified from a Ni-terminal 6x His-tag bacterial expression system. The expression plasmid was constructed in a pET expression plasmid (Novagen) that has a thrombin cleavage site between the N-terminal 6x His-tag and the DIB1 ORF. Thrombin was used to remove the 6xHis-tag from the purified protein after isolation from the Ni-NTA column. This was followed by a Q-sepharose ion exchange chromatography step. Because S. pombe Dib1 has been shown to auto-cleave its own C-terminal tail after lengthy storage at 4°C or room temperature [17]. purified Dib1 was stored in the −80°C shortly after purification, and the quality of the protein was assessed by SDS-PAGE to ensure Dib1 remained full-length. CD spectra of the wild-type protein at both 30°C and 37°C were identical indicating that the wild-type Dib1 is not affected by temperature (Figure 3).
Figure 3.
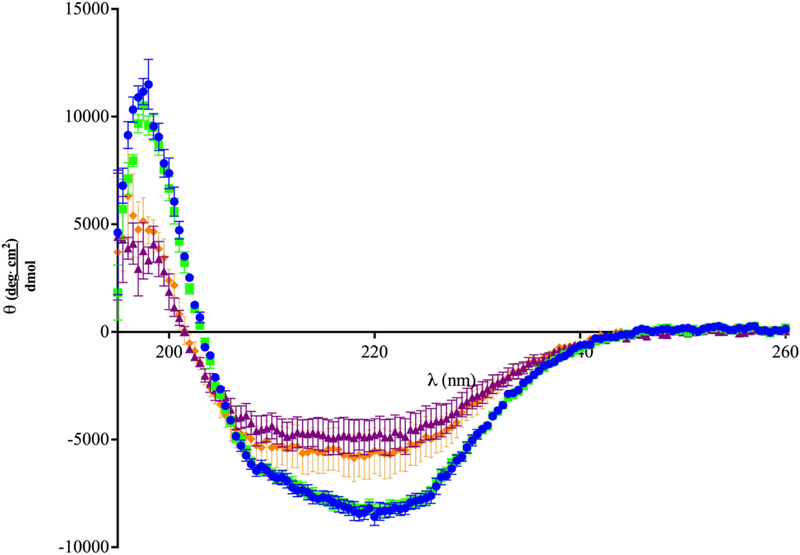
Structural Analysis of Recombinant Wild-type Dib1 and Dib1 L76A/D78A. CD spectra of wild-type (blue circles) and L76A/D78A (orange diamonds) Dib1 protein were taken from 195nm to 260nm at 30°C. Spectra for wild-type (green squares) and L76A/D78A (purple triangles) Dib1 were taken at 37°C. Data points are the average of three trials and error bars indicate one standard deviation.
Purification of Dib1 L76A/D78A was performed using a 6x His-tag bacterial expression system similar to wild-type Dib1. Dib1 L76A/D78A was isolated under similar conditions to the wild-type protein. CD analysis of Dib1 L76A/D78A showed an altered spectrum compared to the wild-type protein (Figure 3). In particular, the trough located around 220 nm appears to be lessened and the high peak around 200 nm is decreased. CD analysis for each protein and condition was repeated at least three times with proteins isolated from independent protein purifications. These findings suggest changes to the secondary structure of the Dib1 L76A/DDib1protein, specifically the α-helical nature of the protein, although the severity cannot be determined. Although subtly different, no statistically significant changes in the spectra of Dib1 L76A/D78A resulted from performing the CD analysis at 37°C (Figure 3). This similarity in protein folding at 30°C and 37°C suggests the growth defects are not exclusively the result of structural changes of Dib1, but perhaps also loss of interactions with other spliceosomal components.
Several attempts were made to purify Dib1 F85A protein. While the protein appeared to express well in induction assays, purification attempts were all unsuccessful. Different E.coli expression cell lines, induction temperatures and purification conditions were attempted. This finding suggests that Dib1 F85A is significantly destabilized. Based on the growth phenotypes (above) and the in vitro splicing assays (below) for dib1-F85A, the protein may be stably folded under cellular conditions used for in vitro splicing assays rather than our in vitro conditions. Thus, we observe the ability of the dib1-F85A extract to splice at permissive temperatures.
In vitro splicing is negatively affected by Dib1 mutants
To determine the effects of these two novel Dib1 mutants on splicing, in vitro splicing assays were performed [18]. Whole cell extracts were collected from yeast cells with mutant dib1 as its sole copy and grown at permissive temperatures. Splicing of fluorescently labeled ACT1 pre-mRNA in whole cell extracts was monitored on denaturing polyacrylamide gels. Splicing defects were observed for both the dib1-F85A and dib1-L76A D78A mutants, but the phenotype was dependent on the temperature of the assay (Figure 4). For dib1-F85A extracts, splicing at 25°C had no observable differences from a wild type splicing reaction. However, when whole cell extracts harboring the dib1-F85A were heated to 37°C prior to the reaction, no splicing or splicing intermediates could be observed, suggesting a defect prior to the first step in splicing (Figure 4). This finding aligns with the mass spectrometry findings that Dib1 is present in the B complex, but absent in the Bact complex [5], and therefore must have a role prior to the first step.
Figure 4.
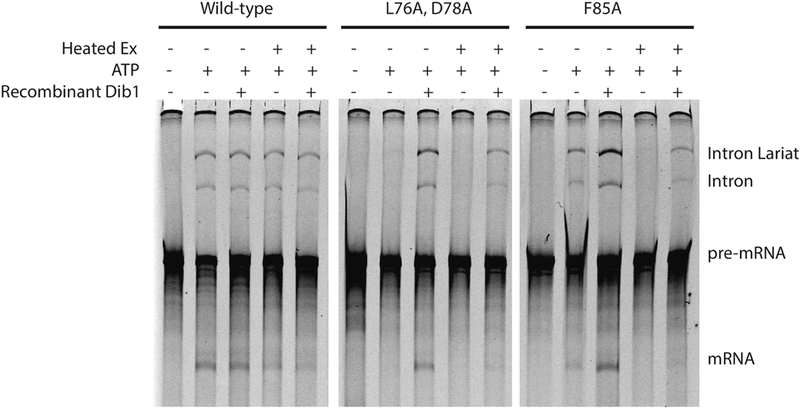
Dib1 mutants disrupts splicing but is rescued by exogenous Dib1. Conditions in the assay are indicated. Heated Ex indicates splicing extract was heated at 37°C prior to the splicing reaction. For samples with ATP, ATP was added to a final concentration of 2.5 mM. For samples reconstituted with Dib1, recombinant Dib1 was added to extract and incubated for 20 minutes at 25°C prior to addition of the pre-mRNA.
In contrast to the temperature dependent phenotype of dib1-F85A, the dib1-L76A D78A extract exhibited splicing difference from wild-type even at permissive temperatures. Weak to no splicing was observed for the dib1-L76A D78A extract at 30°C. This negative effect was compounded in the heat-treated extracts with no observable splicing. Heat treatment at 37°C reproducibly had minor to no effect on splicing of the wild type extract, and splicing was observed to be ATP dependent as expected. Because the structure of Dib1-L76A D78A is minimally affected at 37°C, these results suggest that the cell growth and splicing defects observed result from disruption of other interactions in the spliceosome.
Exogenous Dib1 protein can rescue dib1 splicing defects
To attribute the splicing defects observed in the in vitro splicing assays to Dib1 defects, we sought to restore splicing activity with the addition of a functional Dib1 protein. Dib1 protein was added to the whole cell extracts and allowed to incubate in the extract for 10 to 15 minutes at 25°C. These newly reconstituted extracts were then used in in vitro splicing assays. Surprisingly, although Dib1 is a core U5 snRNP component, addition of exogenous wild type Dib1 was able to significantly rescue splicing defects in both the dib1-F85A and dib1- L76A/D78A heat treated extracts (Figure 4). Addition of Dib1 also improved splicing activity in in vitro splicing reactions of the dibl1F85A extracts at permissive temperatures, suggesting that dib1-F85A does have some minor effect on splicing activity at permissive temperatures (Figure 4). These data suggest that though Dib1 arrives at the pre-mRNA as part of the U5 snRNP, it is able to dissociate and re-associate independently either in the context of the U4/U6-U5 triple snRNP or in the B complex.
Spliceosome assembly of dib 1 mutants is stalled at B complex
Because splicing was stalled prior to the first trans-esterification step of splicing and Dib1 is not present in the Bact complex [5], we sought to determine the effects of the dib1 mutants on spliceosome assembly. Native gel analysis was performed on splicing reactions at permissive and non-permissive temperatures similar to Cheng and Abelson [19] with the exception that fluorescently-labeled ACT1 pre-mRNA was used. Addition of exogenous wild type Dib1 to mutant dib1 splicing extracts was also examined by native gel electrophoresis to monitor restoration of spliceosome assembly. To determine the identity of the splicing complexes, control reactions were performed in which extracts were ATP depleted with glucose [20], incubated with EDTA to stall at Mg2+ dependent steps [19], or depleted of U6 snRNA using endogenous RNase H activity [21]. Multiple splicing assembly gels were run for at least three independently isolated splicing extracts and showed consistent phenotypes.
For both dib1-L76A/D78A and dib1-F85A extracts, formation beyond A complex is impeded (Figure 5). Because of the nature of these complex gels, it is difficult to confidently determine whether spliceosome assembly at non-permissive temperatures appears significantly different from assembly at permissive temperatures. However, addition of exogenous Dib1 does restore spliceosome assembly and results in formation of B and post-B complexes. (Figure 5). We observed a reproducible difference between dib1-L76A/D78A extracts with and without exogenous Dib1, which aligned with the in vitro splicing assays. Addition of exogenous Dib1 resulted in greater accumulation of post-A complexes at both temperatures, suggesting a moderate restoration of splicing. In the case of dib1-F85A extracts, we consistently observed minor to no difference in post-A complex formation with or without exogenous Dib1 at permissive temperature. However, at non-permissive temperatures, for dib1-F85A extracts, addition of exogenous Dib1 reproducibly increased the presence of post-A complexes. Surprisingly, addition of exogenous Dib1 to wild type extract appears to progress complex formation increasing the abundance of B and post-B complexes. This finding was observed for several independently isolated wild-type extracts.
Figure 5.
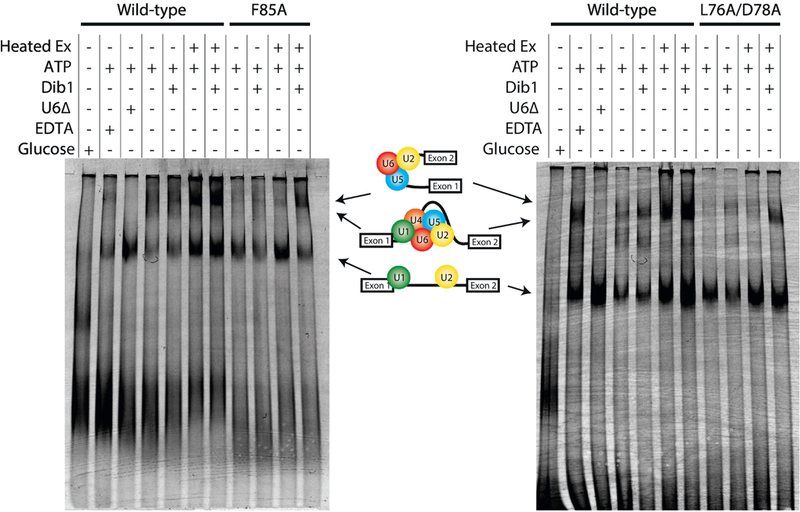
Wild-type Dib1 promotes formation of the splicing machinery beyond B complex. Spliceosome assembly of dibl-F85A (A) and dibl-L76A D78A (B). Splicing extracts were subjected to a variety of conditions prior to incorporation into splicing reactions. The presence or absence of a treatment is indicated by - or +. Extract was heat treated at 37°C prior to the reaction indicated by Heat Ex. Three control reactions are shown: addition of oligo D1 depletes U6 snRNA by RNase H activity (indicated by U6A), addition of EDTA depletes magnesium stalling splicing at B* formation (indicated by EDTA), addition of glucose was used to deplete ATP (indicated by glucose). Splicing reactions were run on a 4% native polyacrylamide gel.
Discussion
We have created several targeted point mutations in S. cerevisicie Dib1 and characterized their effects on yeast cell growth, in vitro splicing, and spliceosome assembly. Target residues were chosen by examining the crystal structure of U5–15kD [11] to identify residues, which, if mutated, may potentially affect interactions with neighboring proteins and RNA or may disrupt protein structure. The majority of the dib1 mutants examined were aphenotypic. The fact that extensively changing these residues caused overall little effect seems surprising. However, the small, compact structure of Dib1 may be able to tolerate these changes without noticeable effect. Also, examining the location of Dib1 in the U4/U6-U5 triple snRNP and B complex structures suggest single amino acid changes might be tolerated and that modification to multiple amino acids simultaneously may be necessary to affect splicing.
Two dib1 mutations resulted in significant growth defects in yeast. In the F85A mutant, changing a bulky phenylalanine to alanine in a core β-sheet resulted in a temperature-sensitive growth defect. While this phenylalanine residue has the ability to stack with neighboring aromatic residues, it is also part of a hydrophobic groove in which a small segment of Prp6, residues 22 to 25, appear to sit. Therefore, F85A could have compounding effects by weakening the structural stability of Dib1 by perturbing stacking interactions and disrupting an important interface with Prp6 (Figure 6). Direct Prp6-Dib1 interactions have previously been reported [22]. Either of these biological effects would be significantly enhanced at 37°C due to increased thermal fluctuations. In the case of F85A, the ts growth phenotype is directly mirrored in the in vitro splicing assays with defects in splicing only observed upon heat treatment of the whole cell extract.
Figure 6.
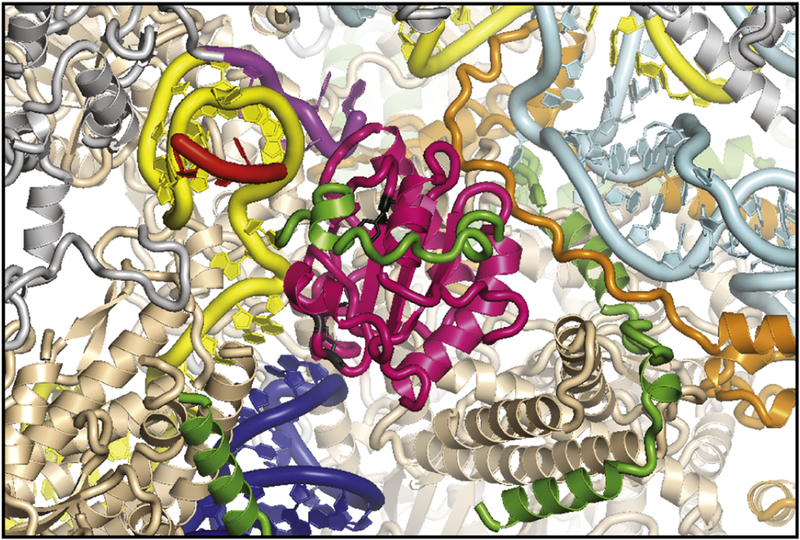
Prp6 and Prp31 surround Dib1 in the B complex (pdb 5nrl, [8]). Prp6 (green) is not completely resolved, but a strand of Prp6 covers Dib1 (magenta) locking it into place. Prp31 (orange) is immediately adjacent to Dib1. U6 ACAGAGA (purple) is adjacent to Dib1. Dib1 L76/D78 and Dib1 F85A are shown in black. Other spliceosomal components proteins are highlighted U4 snRNA (cyan), U5 snRNA (blue), U6 snRNA (yellow) with ACAGAGA (purple), Prp8 (beige), Prp6 (green), Prp31 (orange), mapped pre-mRNA (red).
The Dib1 L76A/D78A mutant displayed a subtler cell growth phenotype with slight growth at 37°C. Leucine-76 and aspartate-78 are part of a flexible loop between the β2 and β3 strands. In the B complex, the nearest mapped neighboring protein is Prp8 around residues −700–735; however, the region between residues 735 and 751 is not resolved in the B complex and appears to have a trajectory adjacent to Dib1’s a flexible loop between the β2 and β3 strands, which contains L76/D78 [8]. Additionally, a large segment of Prp6 is unresolved and appears to be in this area. Modifications of a branched leucine and a negatively charged aspartate have the potential to change interactions with nearby Prp8 or Prp6. Dib1 L76A/D78A has negative effects on in vitro splicing and spliceosome assembly at both permissive and non-permissive temperatures, even though significant defects to growth arise only at higher temperatures. As with the F85 A mutant, the loss of function may result from a combination of indirect effects of overall protein stability losses and direct effects of weakened interaction with neighboring proteins. Indeed, the defects observed for in vitro splicing are more significant after heat inactivation of the whole cell extract. This would be consistent with the CD analysis that suggests that L76A/D78A has a structure altered from wild-type, but is not completely unfolded.
The central location of Dib1 in the U4/U6-U5 triple snRNP opens some interesting questions about Dib1’s role in the spliceosome. Dib1 must depart in order for the splicing cycle to proceed, but the interactions that lead to this departure are uncharacterized. In the B complex, Dib1 appears to serve as a placeholder, simply blocking formation of RNA-RNA interactions and prohibiting the joining of NTC components or the necessary movements to reach the Bact complex. With the identification of novel mutants, two potential Dib1 regions which may stabilize the U4/U6-U5 triple snRNP or the B complex have been identified. While our data cannot distinguish between defects in the U4/U6-U5 triple snRNP assembly or B complex formation, these two mutants would, in theory, perturb both complexes by disrupting interactions that stabilize the complexes. Interestingly, Dib1 may also directly target NTC components as U5–15k, the human homolog of Dib1, was shown to directly interact with PQBP1 [15]. Transition from B to Bact may require Dib1’s involvement in the recruitment of NTC and then Dib1’s departure allows U6 snRNA to interact with the 5’ss. Therefore, Dib1 may essentially be a lynchpin in spliceosome assembly.
A key finding in this study is that exogenous wild-type Dib1 can be added to heat inactivated cell extracts and restore splicing. The mechanism by which exogenous Dib1 is able to restore splicing is still unknown. Cryo-EM structures of both U4/U6-U5 triple snRNP and B complex have a resolved portion of Prp6 that appears to block Dib1 from readily exchanging in either structure [7,8]. However, fluctations of Prp6 may allow exchange to occur within formed U4/U6/U5 triple snRNPs or in the B complex. Alternatively, exogenous Dib1 may be incorporated into newly forming U4/U6-U5 triple snRNPs after they are recycled in the extract. Indeed, addition of ATP is known to result in U4/U6-U5 triple snRNP disassembly[23], so addition of ATP to proceed with splicing may allow formation of new U4/U6-U5 triple snRNPs with exogenous Dib1. Dib1 adds to only a handful of splicing proteins that can be reconstituted in extract, for example, Prp2, Prpl8 and Prp5 have been shown to be able to be reconstituted in extract [24–26].
The early steps in spliceosome assembly and activation involve a myriad of highly ordered and regulated changes in spliceosome composition. Amongst these changes is the departure of U5 snRNP protein Dib1, which occurs during the transition from B to Bact. The role of this small, essential protein in splicing has not been previously known. Here, we have identified two Dib1 mutants that stall splicing prior to the first step and disrupt spliceosome assembly. In addition, we have determined that mutant Dib1 is readily exchangeable with wild type Dib1 in the splicing machinery. Together these findings support a role of Dib1 in the transition from B to Bact, promoting productive splicing. Future studies will focus on how the interactions of Prp6 and Prp31 with Dib1 and their role in regulating the departure of Dib1 from the spliceosome, thereby aiding the transition of B to Bact.
Materials and Methods
Strains and Plasmids
Yeast expression plasmids were created by inserting the yeast DIB1 gene cassette into pSE360 and pSE362 at the XbaI and Sac I restriction sites [27]. The Dib1 gene cassette was created via PCR using S. cerevisiae genomic DNA as the template and the primers oCM118 and oCM134, which have XbaI and SacI sites incorporated and anneal −1kb and +1kb from the DIB1 ORF.
To create a dib1 A haploid, a heterozygous diploid deletion, MAT a MAT a, his3Δ1/his3Δ1 leu2Δ0/leu2Δ lys2˚0/+ met15˚0/+ura3Δ0/ura3Δ0 ypr082c::KanMX4 (ATCC, [28]) was transformed with pSE360-DIBl and sporulated. Tetrads were dissected using a Nikon H550S dissection microscope. Isolated colonies were tested for growth on G418 and on SD-Ura plates. For colonies that grew on the SD -Ura and G418 plates, PCR of the DIB1 and KANMX genes was used for strain confirmation. The dib1 Δ haploid used in this study has the following genotype: MAT a, his3Al leu 2 AO met15° ura3Δ0 ypr082c::KanMX4.
A bacterial expression vector was created by ligating the DIB1 coding sequence isolated from S. cerevisae genomic DNA into pET15b (Novagen). The DIB1 coding sequence was amplified via PCR, using the primers oCM120 and oCM165, which contained Ndel and BglII restriction sites on the forward and reverse primers, respectively. Restriction enzymes BglII and NdeI were used to digest the plasmid and DIB1 coding sequence, and the DIB1 coding sequence was then ligated using T4 DNA ligase.
Dib1 mutants were generated on the pSE362-DIB1 and pET15b-DIB1 plasmids via site- directed PCR mutagenesis using Bio-rad iProof high-fidelity DNA polymerase. Oligonucleotides used to generate each mutant are listed in Supplemental Table 1. Newly constructed plasmids were sequenced by Quintara Biosciences (Berkeley, C A).
Mutant dib1 plasmids created were transformed into the S. cerevisiae dib1Δ strain. Transformants were plated on SD -HIS plates. Once colonies were observed on the SD -HIS plates, the colonies were restreaked onto SD -HIS plates and allowed to grow for two days at 30 °C. The yeast cells were then transferred to 5-fluoroorotic acid (5-FOA) plates and allowed to grow at 30 °C. The yeast cells that grew on 5-FOA were then transferred to both SD -URA and SD -HIS plates and allowed to grow at 30 °C to verify loss of the pSE360-DIB1 plasmid and maintenance of the pSE362-dib1 plasmid.
Splicing extracts
Whole cell yeast extracts were purified as described previously [18] with the following changes: Cells were grown to 1.6–1.8 OD600in 2 L of media containing yeast extract, peptone, and glucose. Extracts were dialyzed in buffer contained 20 mM HEPES pH 7.9, 0.2 mM EDTA, 0.5 mM DTT, 50 mM KC1, 20% (v/v) glycerol, 0.2 mM PMSF, 0.2 mM benzamidine.
Serial dilution growth assays
Overnight yeast cultures were diluted to an OD600 0.1 and allowed to grow for two hours. Once in log-phase growth, the yeast cultures were diluted down to an OD600 = 0.1 again. These cultures were then further serially diluted using a dilution factor of 1 to 6 for each dilution. Cultures were pinned on to YEPD plates and grown at 18, 25, 30 and 37°C.
In vitro splicing with Dib1 recovery
Splicing extracts were mixed with MgCl2 and ATP to a final concentration of 1.5 mM and 0.2 mM respectively. The extracts were then either heated at 37°C for 18 min or kept on ice during the duration. Then MgCl2, PEG 8000, KPO4, and either purified recombinant WT Dib1 protein or the final dialysis buffer from the Dib1 purification was added to the extracts. The mixtures were incubated at 25°C for 10–15 minutes. Cy5-labeled ACT1 pre-mRNA, prepared with 5% Cy5-UTP, and ATP was then added to the reactions, and the splicing reactions were incubated at 25°C for 15 min. Final reactions were in a volume of 10 μL, with concentrations of 2.5mM MgCl2, 3% PEG 8000, 61 mM KPO4 pH 7.0, 1.5–3 pM Dib1, 2 mM ATP, 10mM NaC1, 0.5% glycerol, and 40% splicing extract. Reactions were quenched by adding 5 μL of stop solution containing 2.5 mg/mL proteinase K, 1% SDS, 50 mM Tris pH 8.0, and 50 mM EDTA and heating at 55°C for 10 min. Results were imaged on a Typhoon FLA 9500 imager after running the quenched reactions on a 6% 7 M urea denaturing polyacrylamide (19:1) gel for 80 min at 20 W.
Spliceosome assembly with Dibl recovery
Splicing reactions for native gel analysis were assembled similar to in vitro splicing reactions with the following modifications: the -ATP reactions had their extracts incubated in 4 mM glucose for 30 min at 25°C after the initial addition of MgCl2. The U6 depletion reactions were incubated in 6 μM D1 oligonucleotide [21] for 30 min at 30 °C after the initial addition of MgCl2 and ATP. The +EDTA reactions had EDTA added to the reactions to a final concentration of 5 mM during the initial addition of MgCl2 and ATP. The extracts were heated for 17–21 min at 37°C. Splicing reactions were incubated for 30–40’ at 25°C. Reactions were quenched by adding in 5 μL of a solution of 60 mM KPO4, 3 mM MgCl2, 3.3% PEG 8000, 8% glycerol, and 4 mg/mL heparin. Splicing reactions were run on a 4% native polyacrylamide (80:1) gel for 6 hrs and at 200 V. The gel was pre-run for 3 hrs at 200 V, and a peristaltic pump was used to cycle buffer. The gels were imaged on a Typhoon FLA 9500 imager.
Protein Purification
Dib1 was purified from Rosetta (Novagen) expression cells transformed with pET15b- Dib1. Two liters of cells were grown in LB media at 37°C to an OD600 of 0.3, induced with 1 mM IPTG and allowed to grow overnight at 18°C. Cells were spun down at 5 krpm for 10 min and resuspended in 20mL of 50 mM phosphate buffer, 10 mM imidazole, 300 mM NaCl. Cells were sonicated with amplitude of 50 Hz for 2 min (30 sec on, 30sec off) and subsequently spun at 18 krpm for 45 min. The supernatant was collected and re-spun at 18 krpm for 30 min. The cell lysate was loaded onto a 3.5 mL column volume of Ni-NTA resin (Qiagen) and allowed to flow through.
The resin was then washed with buffer containing 50 mM phosphate buffer, 300 mM NaCl, and increasing concentrations of imidazole (10, 25, 50, 100, 250, and 500 mM) at pH 7. A total of six 15 mL fractions were collected, and samples were run on a SDS-PAGE gel to determine which fraction contained protein. Typically the protein eluted at 250 mM imidazole. The samples were then dialyzed twice against 1.5 L of pre-thrombin buffer containing 50 mM Tris pH 8.0, 100 mM NaCl, 1 mM DTT, 5% glycerol. To remove the His-tag, the sample was incubated with thrombin at room temperature for 3 hrs. The sample was then dialyzed twice against 1.5 L of dialysis buffer containing 20 mM Tris pH 8.0, 10 mM NaCl, 1 mM DTT, 5% glycerol. After dialysis the sample was loaded onto a Q-sepharose anion-exchange column, which was then washed with buffer containing 20 mM Tris and increasing concentrations of NaCl (10 mM, 20 mM, 50 mM, 100 mM, 150 mM, 200 mM) at pH 8. A total of six 10 mL fractions were collected. The fractions from the 150 mM and 200 mM washes were collected in 1mL fractions and then run on a gel to determine which fractions contained the Dib1 protein. For storage, the protein sample was dialyzed twice against 1.5 L of storage buffer containing 10 mM phosphate buffer pH 8.0, 100 mM NaCl, 5% glycerol. The sample was then snap-frozen with liquid nitrogen.
Circular Dichroism Studies
Recombinant Dib1 proteins (1 mL) were dialyzed twice against 1L of CD buffer containing 10 mM phosphate buffer pH 8.0 and 100 mM NaCl to remove any glycerol from the storage buffer. CD samples were prepared at a final protein concentration of 10 μM, and full spectra were taken on a JASCO J-815 CD spectrometer, using a JASCO PTC-423S/15 accessory to regulate the cell temperature. Full spectra were taken from 195 nm to 260 nm at both 30°C and 37°C with data pitch of 0.5 nm, a continuous scanning mode, 20 nm/min speed, 2 sec response, 10 nm bandwidth with three accumulations. Raw data was then graphed and analyzed using GraphPad Prism 7.
Structural Analysis
Figures were constructed using PyMol (Schrodinger LLC) and Adobe Illustrator.
Supplementary Material
Highlights.
Spliceosomal protein Dib1 is centrally located in the putative active site of the spliceosome.
Novel Dib1 mutants stall spliceosome assembly or activation.
Exogeneous Dib1 protein can rescue dib1 mutants in whole cell yeast extracts.
Structural analyses suggest Dib1 mutants functionally affect interactions with other spliceosomal proteins.
Acknowledgements
We thank Megan Mayerle for discussions and comments on the manuscript and Jon Staley for technical help with the assembly assays. This work is supported by grants to CM from National Institute Of General Medical Sciences of the National Institutes of Health under Award Number R15GM120720, the Robert A Welch Foundation Grant W-1905 and the John L. Santikos Charitable Foundation Fund of the San Antonio Area Foundation.
Footnotes
Declarations of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Will CL, Luhrmann R, Spliceosome structure and function, Cold Spring Harbor Perspectives in Biology. 3 (2011) a003707–a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wahl MC, Will CL, Luhrmann R, The spliceosome: design principles of a dynamic RNP machine, Cell. 136 (2009) 701–718. [DOI] [PubMed] [Google Scholar]
- [3].Turner IA, Norman CM, Churcher MJ, Newman AJ, Roles of the U5 snRNP in spliceosome dynamics and catalysis, Biochem Soc Trans. 32 (2004) 928–931. [DOI] [PubMed] [Google Scholar]
- [4].Staley JP, Guthrie C, Mechanical devices of the spliceosome: motors, clocks, springs, and things, Cell. 92 (1998) 315–326. [DOI] [PubMed] [Google Scholar]
- [5].Fabrizio P, Dannenberg J, Dube P, Kastner B, Stark H, Urlaub H, et al. , The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome, Mol Cell. 36 (2009) 593–608. [DOI] [PubMed] [Google Scholar]
- [6].Wan R, Yan C, Bai R, Wang L, Huang M, Wong CCL, et al. , The 3.8 A structure of the U4/U6.U5 tri-snRNP: Insights into spliceosome assembly and catalysis, Science. 351(2016)466–475. [DOI] [PubMed] [Google Scholar]
- [7].Nguyen THD, Galej WP, Bai X-C, Oubridge C, Newman AJ, Scheres SHW, et al. , Cryo-EM structure of the yeast U4/U6.U5 tri-snRNP at 3.7 A resolution, Nature. 530 (2016)298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Plaschka C, Lin P-C, Nagai K, Structure of a pre-catalytic spliceosome, Nature. 546 (2017)617–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dix I, Russell CS, O’Keefe RT, Newman AJ, Beggs JD, Protein-RNA interactions in the U5 snRNP of Saccharomyces cerevisiae, RNA. 4 (1998) 1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Makarov EM, Makarova OV, Urlaub H, Gentzel M, Will CL, Wilm M, et al. , Small nuclear ribonucleoprotein remodeling during catalytic activation of the spliceosome, Science. 298 (2002) 2205–2208. [DOI] [PubMed] [Google Scholar]
- [11].Reuter K, Nottrott S, Fabrizio P, Luhrmann R, Ficner R, Identification, characterization and crystal structure analysis of the human spliceosomal U5 snRNP - specific 15 kD protein, J Mol Biol. 294 (1999) 515–525. [DOI] [PubMed] [Google Scholar]
- [12].Stevens SW, Barta I, Ge HY, Moore RE, Young MK, Lee TD, et al. , Biochemical and genetic analyses of the U5, U6, and U4/U6 x U5 small nuclear ribonucleoproteins from Saccharomyces cerevisiae, RNA. 7 (2001) 1543–1553. [PMC free article] [PubMed] [Google Scholar]
- [13].Wieczorek D, Newman WG, Wieland T, Berulava T, Kaffe M, Falkenstein D, et al. , Compound Heterozygosity of Low-Frequency Promoter Deletions and Rare Loss-of- Function Mutationsin TXNL4A Causes Bum-McKeown Syndrome, The American Journal of Human Genetics. 95 (2014) 698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang YZ, Gould KL, Dunbrack RL JR, Cheng H, Roder H, Golemis EA, The evolutionarily conserved Dim1 protein defines a novel branch of the thioredoxin fold superfamily, Physiol Genomics. 1 (1999) 109–118. [DOI] [PubMed] [Google Scholar]
- [15].Zhang Y, Lindblom T, Chang A, Sudol M, Sluder AE, Golemis EA, Evidence that dim1 associates with proteins involved in pre-mRNA splicing, and delineation of residues essential for dim1 interactions with hnRNP F and Npw38/PQBP-1, Gene. 257 (2000)33–43. [DOI] [PubMed] [Google Scholar]
- [16].Zhang Y-Z, Cheng H, Gould KL, Golemis EA, Roder H, Structure, stability, and function of hDim1 investigated by NMR, circular dichroism, and mutational analysis, Biochemistry. 42 (2003) 9609–9618. [DOI] [PubMed] [Google Scholar]
- [17].Jin T, Guo F, Wang Y, Zhang Y-Z, Identification of human diml as a peptidase with autocleavage activity, Chem Biol Drug Des. 68 (2006) 266–272. [DOI] [PubMed] [Google Scholar]
- [18].Lin RJ, Newman AJ, Cheng SC, Abelson J, Yeast mRNA splicing in vitro, J Biol Chem. 260(1985) 14780–14792. [PubMed] [Google Scholar]
- [19].Cheng SC, Abelson J, Spliceosome assembly in yeast, Genes Dev. 1 (1987) 1014–1027. [DOI] [PubMed] [Google Scholar]
- [20].Liao XC, Colot HV, Wang Y, Rosbash M, Requirements for U2 snRNP addition to yeast pre-mRNA, Nucleic Acids Res. 20 (1992) 4237–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fabrizio P, McPheeters DS, Abelson J, In vitro assembly of yeast U6 snRNP: a functional assay, Genes Dev. 3 (1989) 2137–2150. [DOI] [PubMed] [Google Scholar]
- [22].Liu S, Rauhut R, Vomlocher H-P, Luhrmann R, The network of protein-protein interactions within the human U4/U6.U5 tri-snRNP, RNA. 12 (2006) 1418–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Raghunathan PL, Guthrie C, RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2, Curr Biol. 8 (1998) 847–855. [DOI] [PubMed] [Google Scholar]
- [24].Kim SH, Smith J, Claude A, Lin RJ, The purified yeast pre-mRNA splicing factor PRP2 is an RNA-dependent NTPase, Embo J. 11 (1992) 2319–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].O’Day CL, Dalbadie-McFarland G, Abelson J, The Saccharomyces cerevisiae Prp5 protein has RNA-dependent ATPase activity with specificity for U2 small nuclear RNA, J Biol Chem. 271 (1996) 33261–33267. [DOI] [PubMed] [Google Scholar]
- [26].Horowitz DS, Abelson J, Stages in the second reaction of pre-mRNA splicing: the final step is ATP independent, Genes Dev. 7 (1993) 320–329. [DOI] [PubMed] [Google Scholar]
- [27].Elledge SJ, Davis RW, A family of versatile centromeric vectors designed for use in the sectoring-shuffle mutagenesis assay in Saccharomyces cerevisiae, Gene. 70 (1988) 303–312. [DOI] [PubMed] [Google Scholar]
- [28].Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, et al. , Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis, Science. 285 (1999) 901–906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.