

Abstract
Amorphous titanium dioxide (a-TiO2) combined with an electrocatalyst has shown to be a promising coating for stabilizing traditional semiconductor materials used in artificial photosynthesis for efficient photoelectrochemical solar-to-fuel energy conversion. In this study we report a detailed analysis of two methods of modifying an undoped thin film of atomic layer deposited (ALD) a-TiO2 without an electrocatalyst to affect its performance in water splitting reaction as a protective photoelectrode coating. The methods are high-temperature annealing in ultrahigh vacuum and atomic hydrogen exposure. A key feature in both methods is that they preserve the amorphous structure of the film. Special attention is paid to the changes in the molecular and electronic structure of a-TiO2 induced by these treatments. On the basis of the photoelectrochemical results, the a-TiO2 is susceptible to photocorrosion but significant improvement in stability is achieved after heat treatment in vacuum at temperatures above 500 °C. On the other hand, the hydrogen treatment does not increase the stability despite the ostensibly similar reduction of a-TiO2. The surface analysis allows us to interpret the improved stability to the thermally induced formation of O– species within a-TiO2 that are essentially electronic defects in the anionic framework.
Introduction
The ever-growing demand for energy and increasing concern for sustainable development are forcing humankind to seek for new technologies for harnessing energy from renewable sources. One promising alternative is the production of hydrogen by a solar energy-driven process called artificial photosynthesis. In this process, the energy of the incoming photons is converted into chemical energy of the molecular bonds by dissociating water molecules into hydrogen and oxygen.1 The dissociation reaction requires semiconducting electrodes that are stable in electrochemical conditions, can efficiently absorb solar energy, can support the redox reaction of water molecules on their surfaces, and are cost-efficient. Several traditional semiconductor materials such as silicon, gallium arsenide, and gallium phosphide have electronic structures with suitable band potentials to enable the water oxidation half reaction. However, these materials are unstable under oxidizing conditions. Thus, the oxidation of the electrode material itself competes with the water oxidation leading to either photocorrosion or passivation of the electrode surface.1−3 For this reason the electrode surface must be coated with a suitable protective layer. One promising material for this application is titanium dioxide (TiO2), which has attracted tremendous research interest during the last decades.
Traditionally the TiO2 research has concentrated on its crystalline anatase, rutile, and brookite polymorphs, but in recent years several studies have revealed that the amorphous counterpart might actually be more advantageous due to its electric “leakiness”.2,4−6 In other words, the electronically defective nature of the amorphous TiO2 makes it more conductive. Good conductivity is important in avoiding tunneling barriers that reduce the photocurrent or increase the required overpotential.7 In insulating, tunneling based protection layers these losses would constitute a significant disadvantage because the film has to be at least several nanometers thick to avoid pinhole defects.8,9 In the chemical sense, the increased conductivity can be attributed to the reduction of Ti4+ cations to the Ti3+ state. This can be achieved, for example, by n-type doping, where tetravalent Ti is substituted by pentavalent cation such as Nb or Sn or by substituting O with F.10 Another option is to modify the bond structure by creating O vacancies or Ti3+ interstitials.10 The advantage of the latter method is that no new elements have to be introduced to the structure.
In the present study we report a detailed analysis of two approaches for modifying ALD grown a-TiO2 ultrathin films to address the question of intrinsic stability of a-TiO2 in photoelectrochemical (PEC) energy conversion: thermal treatment and atomic hydrogen treatment. The PEC properties and the electrochemical stability of the post-treated a-TiO2 ultrathin films are investigated with standard electrochemical measurements in conjunction with simulated solar irradiation and correlated with the molecular and electronic structure obtained by photoelectron spectroscopy analysis. In both thermal and atomic hydrogen treatment, a 28.7 nm thick a-TiO2 film deposited on a Si single crystal is used as a substrate. The first modification method is based on high-temperature annealing (up to 900 °C) in ultrahigh vacuum (UHV). Interestingly, X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD) results reveal that the amorphous structure is preserved despite the high annealing temperature. In the thermal method the improved ”leakiness” is shown to be induced by charge transfer within the ionic framework of a-TiO2. Moreover, the elemental composition of the a-TiO2 film is not altered by the treatment. On the other hand, in atomic H treatment, the H atoms react with O leading to the desorption of water molecules and creation of O vacancies. Ultraviolet photoelectron spectroscopy (UPS) is used to show that in both methods the valence band structure of a-TiO2 is severely modified by occupied Ti 3d derived states above the valence band maximum (VBM) of a stoichiometric TiO2.10,11 Furthermore, our PEC results reveal that a-TiO2 is susceptible to severe photocorrosion under photoelectrochemical water splitting conditions. Achieving the PEC stability for a bare a-TiO2 coating without an electrocatalyst requires the thermally induced formation of oxide defects in the ionic framework. These findings regarding the intrinsic stability of thermally treated a-TiO2 are likely to promote the use of a-TiO2 thin films in photoelectrochemical and other functional applications. Moreover, the a-TiO2 combined with an active electrocatalyst exhibits advantageous approach stabilizing traditional semiconductor materials used for water splitting reaction.
Materials and Methods
The P-doped (resistivity 1–20 Ω cm) n-type Si(100) wafers were purchased from Wafer World, Inc. (Florida). The 400 μm thick, 3-in. diameter prepolished wafers had been cut in (100) orientation with ±1° accuracy. For the experiments 10 × 10 mm2 squares were cleaved. The Si substrates were first cleaned by sonicating them for 45 min in 99.5% EtOH followed by a combination of annealing and atomic hydrogen treatments in UHV. The details of the UHV cleaning procedure are described in ref (12). In short, the samples were first annealed to 1000 °C to remove native oxide. After this they were exposed to atomic hydrogen at 800 °C (10 min) and 400 °C (10 min) at pH = 1.0 × 10–7 mbar, which removed the segregated Cu and Ni impurities, respectively. In all stages, the sample temperature was monitored with a pyrometer (Land Cyclops 160B) using an emissivity value of ε = 0.60. The pyrometer reading was calibrated against type K thermocouple measurements in a separate system. After annealing, the surface cleanness and structure were verified by X-ray photoelectron spectroscopy and low energy electron diffraction (LEED) (Figure S1 in the Supporting Information and ref (12)). After the UHV cleaning, the samples were cooled down in vacuum and transferred to the ALD system through the atmosphere. The exposure to air was kept less than 5 min.
ALD deposition of a-TiO2 was carried out using a Picosun Sunale ALD R200 Advanced reactor. Tetrakis(dimethylamido)titanium(IV) (Ti(N(CH3)2)4, TDMAT, 99%, Strem Chemicals Inc., France), deionized water, and N2 (99.9999%, Oy AGA Ab, Finland) were used as the Ti precursor, O precursor, and carrier/purge/venting gas, respectively. a-TiO2 films were prepared with two different thicknesses: 28.7 nm for PEC and XPS/UPS measurements and 200 nm for XRD measurements because the thinner film did not give sufficient XRD signal intensity. The number of ALD pulses for the films were 804 and 5600, respectively. The film thickness was verified by ellipsometry (Rudolph Auto EL III Ellipsometer, Rudolph Research Analytical).
During the ALD, the Si substrate temperature was kept at 200 °C. The vapor pressure of the TDMAT was increased to 3.6 mbar by heating the precursor bubbler to 76 °C, and the precursor gas delivery line was heated to 85 °C to prevent condensation. The water bubbler was sustained at 18 °C by a Peltier element for stability control. The substrate temperature was stabilized for 30 min before starting the deposition. The 200 °C ALD growth temperature was selected because it results in amorphous growth whereas higher ALD temperatures produce strongly crystallized anatase TiO2.13,14 On the other hand, much lower temperatures would result in incomplete precursor dissociation leading to higher remnant impurity concentrations, especially nitrogen from TDMAT. Low temperatures also produce more stoichiometric TiO2 which, based on our preliminary experiments, cannot be modified by the post-treatments as effectively as the films grown at 200 °C (Figure S2).
After the ALD deposition, the samples were cooled down in nitrogen gas before transferring them back to UHV for post-treatments and photoelectron spectroscopy (PES) measurements. The exposure to ambient atmosphere during the transfer was approximately 5 min.
The post-treatments were performed in the preparation chamber of the NanoESCA spectromicroscopy system (Omicron NanoTechnology GmbH)15 and they were divided into two sets: (1) thermal treatment and (2) atomic hydrogen treatment. Thermal treatments were conducted cumulatively in 100 °C steps from 400 to 900 °C in UHV keeping the chamber pressure in the 10–9 mbar range during the annealings. The upper limit for the annealing series was selected based on our previous studies where a-TiO2 became completely reduced above 950 °C.12 The heating setup consisted of a resistive PBN-heating element mounted to a manipulator close to the backside of the sample and the sample held in a Mo sample plate. The temperature was increased to the target value in approximately 3 min and then kept constant for 10 min after which the sample was let to cool down to <100 °C. The temperature was monitored with a pyrometer. After each annealing step, the sample was transferred to the analysis chamber under UHV conditions for PES measurements or alternatively removed from the UHV system for PEC studies.
The atomic hydrogen treatments were performed in the same preparation chamber as the thermal treatment series. The sample temperature was kept at 300 °C during the atomic hydrogen exposure. The hydrogen treatment series was made cumulatively so that the total exposure times of 1, 5, 10, and 50 min were achieved. The hydrogen flux was produced by using EFM H Atomic Hydrogen Source (Omicron NanoTechnology GmbH). The source is based on leaking hydrogen gas through a heated tungsten capillary which causes thermal dissociation of hydrogen molecules with a dissociation efficiency close to 100%.16 The following parameters were used: e-beam acceleration voltage = 1000 V, e-beam emission current = 40 mA, filament current = 2.00 A. Hydrogen gas pressure in the preparation chamber was adjusted to 1.0 × 10–6 mbar, and the sample was set in line with the capillary tube. After each hydrogen exposure step the sample was transferred to the analysis chamber for PES.
The PES measurements were conducted in the analysis chamber of the NanoESCA system with a base pressure below 1 × 10–10 mbar. Focused monochromatized Al Kα radiation (hν = 1486.5 eV) was utilized for core level XPS whereas valence band UPS spectra were measured with a focused nonmonochromatized He Iα radiation (hν = 21.22 eV) using HIS 13 VUV Source (Focus GmbH). The spectra were collected with a photoemission electron microscope (PEEM) paired with a single hemispherical energy analyzer. The energy resolution of the analyzer was set to 400 meV (pass energy 100 eV, slit 1 μm) and 100 meV (pass energy 50 eV, slit 0.5 μm) for XPS and UPS, respectively. The analysis area was set to 230 μm in diameter for XPS and 95 μm for UPS, corresponding to the maximum spot sizes of the radiation sources. Large analysis areas ensured that the results represent the average surface composition. Previous small area XPS results have shown that both the ALD grown and post-treated surfaces are homogeneous.
The chemical states of the elements were determined from the core level XP spectra by least-squares fitting of asymmetric Gaussian–Lorentzian line shapes after subtracting a Shirley type background (Table S1). The determination of the electron band structure near the Fermi edge was made by analyzing the UPS valence band spectra. Because of large inelastic secondary electron background a Tougaard background was subtracted and the remaining intensity was fitted with Finite Lorentzian Asymmetric peaks (Table S2). The analysis was made in CasaXPS software version 2.3.17PR1.117 using the Scofield photoionization cross sections as relative sensitivity factors. The binding energy (Eb) scale was calibrated by setting the O 1s main peak (O in a-TiO2) to 530.20 eV.
The influence depth of the hydrogen treatment was estimated by comparing the measured Ti 2p photoelectron spectroscopy results with a simulation. For this comparison the Ti 2p attenuation length was calculated by using the TPP-2M18 formula in the QUASES-Tougaard software package.19 For the 1028 eV kinetic energy (corresponding to Ti 2p3/2), the formula gave an attenuation length of λ = 2.2 nm.
Thermal treatment of ALD grown a-TiO2 is known to induce crystallinity in some cases.13,20,21 For this reason, structural characterization of the 200 nm thick samples both in as-deposited (after ALD growth) and thermally treated conditions was carried out by using XRD (Panalytical Empyrean multipurpose diffractometer) with Cu Kα radiation (λ = 1.5405 Å) and 45 kV and 40 mA cathode voltage and current, respectively. The samples were scanned in 2θ between 20 and 40°. The XRD data was postprocessed by subtracting the background and removing the Cu Kα2 X-ray satellite peaks.
Photoelectrochemical performance was studied in a homemade PEC cell (PTFE body, volume 3.3 cm3), using a three-electrode system controlled by Autolab PGSTAT12 potentiostat (Metrohm AG). The studied samples were inserted between a rubber O-ring and a stainless steel plate. The steel plate on the back side provided the electrical contact and the O ring ensured a well-defined 0.28 cm2 planar projected electrode surface area. An Ag/AgCl electrode (Leak-Free LF-2, Warner Instruments, LLC) and Pt wire (surface area 0.82 cm2) were used as reference and counter electrodes, respectively, in an aqueous solution of 1 M NaOH (pH = 13.6). The potential values were converted to the reversible hydrogen electrode (RHE) scale by the equation VRHE = VAg/AgCl + 0.197 V + pH × 0.059 V. A simulated solar spectrum was produced with a HAL-C100 solar simulator (Asahi Spectra Co., Ltd., JIS Class A at 400–1100 nm with an AM1.5G filter), and the intensity was adjusted to 1.00 Sun using a 1 sun checker (model CS-30, Asahi Spectra Co., Ltd.). The photon flux was directed to the sample front surface through a 5 mm thick quartz glass window and 18 mm thick electrolyte layer. The PEC testing was started by studying the current–voltage characteristics in dark and under simulated sunlight (Figure S3). After this linear sweep voltammetry, the sample was subjected to an electrochemical stability test by biasing the sample to water redox potential of +1.23 V vs RHE under simulated solar light. Both light and dark currents were measured by chopping the light off and on every 5 min. The data collection was continued for 60 min corresponding to a total of six light/dark cycles. The as-deposited and 500 °C UHV annealed samples were also subjected for a longer stability test under continuous illumination.
Results and Discussion
Photoelectrochemical Activity and Stability
The PEC behavior of the thermally and hydrogen treated a-TiO2 samples was tested by studying their PEC responses at +1.23 V vs RHE. Figure 1 illustrates the photocurrent density as a function of time for four samples. The highest current was actually reached with the untreated as-deposited sample. However, after about 10 min illumination, the photocurrent started to decrease sharply. This indicates that the current is not due to the oxygen evolution reaction but instead caused by the dissolution of the a-TiO2 layer and therefore an indication of a-TiO2 photocorrosion. The result was confirmed with a long exposure in 1 M NaOH which dissolved the 30 nm film completely in 5 h as shown by the current density drop and the photograph in the inset of the Figure 1. Similar results were obtained for the hydrogen treated and the 400 °C thermally treated surfaces. On the other hand, all samples annealed at 500 °C or higher showed stable photocurrents for at least 20 h and close to zero dark current. It was also found that as the thermal treatment temperature approached 900 °C, the photocurrent declined. This is likely due to the insulating Si oxide layer that is formed at the Si–a-TiO2 interface, as will be discussed later.
Figure 1.
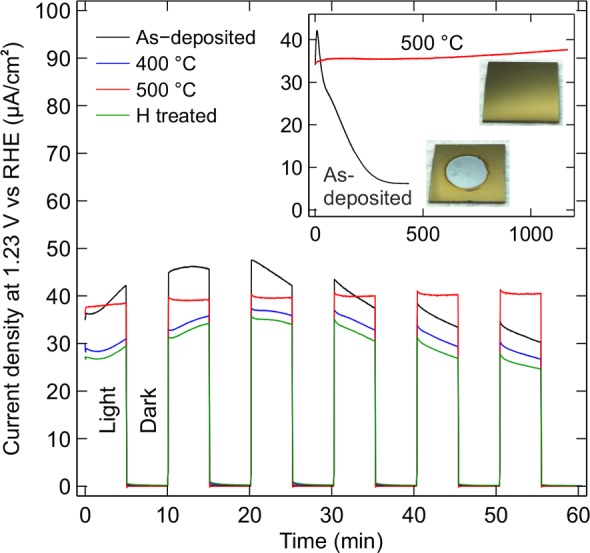
Chronoamperometric testing of four a-TiO2/Si electrodes in 1 M NaOH (aq.) under the illumination intensity of 1.00 Sun. Black: as-deposited film without post-treatment, blue: annealed at 400 °C, red: annealed at 500 °C, green: hydrogen treated for 10 min at 300 °C. The inset shows the photocurrent density of the as-deposited and 500 °C annealed samples during a long stability test under continuous irradiation. The photographs illustrate the surface at the end of the stability test. Photoanodes were kept at constant +1.23 V vs reversible hydrogen electrode (RHE).
The instability of the as-deposited, hydrogen treated, and below 500 °C annealed samples was somewhat surprising, because there are reports of apparently similar ALD grown amorphous TiO2 films used as PEC electrodes.4,22 In many cases, the TiO2 film has been even much thinner than 30 nm.7 It should also be noted that in most studies, Ni, IrO2, or some other electrocatalyst material has been evaporated or sputtered on top of the TiO2 layer.4,7,8,22,23 On the basis of our results, the protective properties of the a-TiO2 film may not originate from the native film itself, but they may be attributed to the combination of TiO2 and the electrocatalyst. However, with a thermal treatment the as-deposited a-TiO2 film can also be made stable for PEC applications.
Molecular Structure of Amorphous TiO2
To achieve molecular level understanding of the reasons why thermally and hydrogen treated a-TiO2 films exhibit such different photoelectrochemical behaviors, we analyzed the samples by XPS. Special attention was paid to the formation of the different Ti 2p oxidation states in both treatment series. The measured O 1s and Ti 2p XP spectra are shown in Figure 2.
Figure 2.

Ti 2p and O 1s XP spectra for (a) thermal treatment and (b) hydrogen treatment series. The Ti 2p spectra are fitted for both 3/2 and 1/2 doublets with three synthetic oxide components in each spin–orbit coupled state.
Ti was identified in three different oxidation states, which correspond to Ti4+ (stoichiometric a-TiO2) at 459.00 ± 0.20 eV, Ti3+ at 457.20 ± 0.20 eV, and Ti2+ at 455.95 ± 0.20 eV. All subsequent analysis is based only on the 3/2 spin–orbit coupled state but both 1/2 and 3/2 states had to be fitted because the splitting is only about 5.7 eV thus causing overlap between Ti3/24+ and Ti1/2 components. The 1/2 states were fitted with an area ratio of 0.43 ± 0.03 compared to the corresponding 3/2 state. It is noteworthy that no metallic Ti was observed at any stage of the post-treatments. In O 1s spectra, four different chemical states were identified. In all cases the main peak (set to 530.20 eV in calibration) corresponded to O2– anions in a-TiO2. In the thermal treatment series another state was identified at 531.33 eV, which is denoted as O–. In the literature, this binding energy region is usually associated with hydroxyl groups,24,25 carbonate-like species or other carbon related impurities,26 or oxygen-deficient titanium oxide (such as TiOx, where 1.35 < x < 1.65).27 The first two explanations would be plausible on the as-deposited surface, because it may have hydroxyl groups or carbon impurities that have adsorbed from air or remained in the structure as residues from the incompletely reacted ALD precursors. However, the peak at 531.33 eV is visible only after 500 °C and higher thermal treatments when the hydroxyl groups and carbon impurities have desorbed from the surface (Figure S4). Above all, our results clearly show that the O 1s peak at 531.33 eV is not associated with the decrease of oxygen content, because the O/Ti ratio remains constant throughout the thermal treatment series (see Figure 3). Thus, the buildup of O– peak is assumed to result from localized charge transfer between anionic and cationic frameworks during thermally induced reduction, as will be discussed later. Second minor component (Eb = 532.25 ± 0.10 eV) in O 1s spectra appeared during high temperature annealings at 800 and 900 °C. This peak, denoted as OSi, is associated with Si oxide impurities. At these temperatures small amount (<1.8 at %) of oxidized Si was observed in Si 2p spectra (Figure S5). Importantly, both O– and OSi were observed only in the thermal treatment series. On the other hand, the spectra measured from the hydrogen exposed samples started to show a slowly growing component at 531.50 eV as the hydrogen exposure time was increased. This component was associated with a process where a chemisorbed hydrogen atom does not desorb from the surface but forms Ti–O–H compounds. The concentration of the OH– component is in all cases less than 6% of all O atoms, which indicates that most of the reactions between a-TiO2 and H lead to the desorption of O as water molecules.
Figure 3.
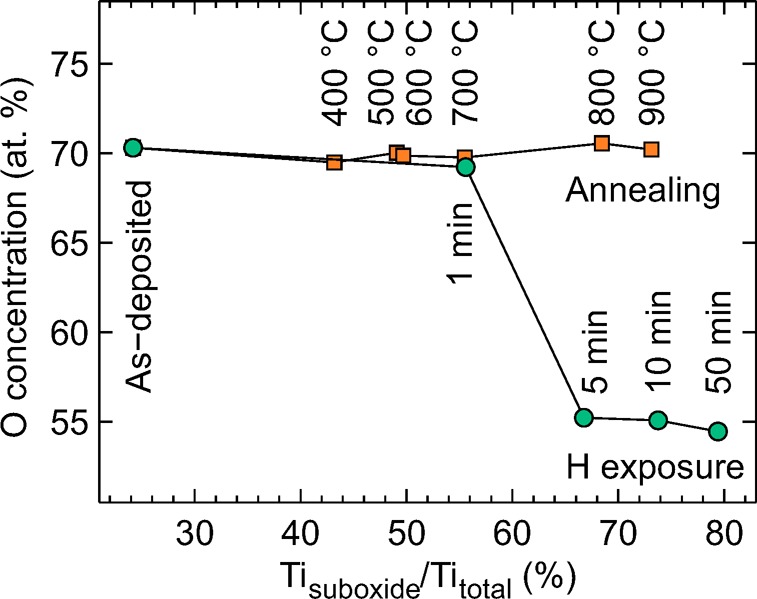
O concentration as a function of the relative Ti3+/2+ concentration. For the annealing series, the O content remains constant despite the strong reduction observed in Ti 2p transition. In the hydrogen treatment series, the O concentration approached the 50% limit which implies a TiO stoichiometry.
The bottom spectrum in Figure 2 noted as as-deposited represents the situation directly after ALD growth without any post-treatment. In this case the O 1s spectrum can be fitted with one asymmetric Gaussian–Lorentzian component. In addition to O and Ti, the as-deposited surface had approximately 1.5 at % of carbon and <0.3 at % of nitrogen impurities (Figure S4). These are however completely removed after the first thermal or hydrogen treatment and do not contribute to our results. Otherwise the surfaces were clean with the exception of the previously mentioned Si oxide after 800 and 900 °C annealing steps. The origin of the Si was traced back to the TDMAT precursor material by continuing the annealing at 900 °C for a total of 30 min. After this prolonged treatment, the Si had disappeared from the surface. This implies that there is a limited source of segregating Si trapped inside the ALD film instead of Si from the substrate diffusing through the film. From the photoelectrochemical point of view this small Si impurity concentration may actually be beneficial because Si doping has been shown to improve photocatalytic activity of TiO2 nanotubes.28
It is noteworthy that the ALD grown surface is not stoichiometric TiO2 but has approximately 26% of Ti in Ti3+ oxidation state, which can be accounted for interstitial Ti ions or O vacancies.10,29 As the postprocessing advances to higher temperatures or longer hydrogen exposure times the amount of Ti suboxides (Ti3+ and Ti2+) starts to increase. This increases the number of excess electrons in the structure that occupy the electronic states near the Fermi edge, which in turn improves the conductivity.10 For PEC applications this is an important aspect as the photocurrent must be transferred through the a-TiO2 film. Despite the somewhat similar reduction behavior of Ti in both the thermal and hydrogen treatment series, the chemical compositions of the surfaces are strongly distinct when comparing the different reduction methods. This is illustrated in Figure 3 which shows the relative O concentration on the surface. The calculation is based on the total area of the components shown in Figure 2.
For stoichiometric TiO2, the theoretical O concentration is 67 at %. In our case, the value should be slightly lower because 26% of Ti is in the Ti3+ oxidation state already in the as-deposited film. The discrepancy between the theoretical value and the observed 70 at % value originates mainly from a small error in the relative sensitivity factor of Ti 2p, which does not account for the TiO2 satellite features outside the analyzed energy region. The most pronounced feature in Figure 3 is, however, the difference between thermally and H treated samples. In the H treatment series, the O content decreases with increasing H exposure time. This is as expected because, as shown in Figure 2b, the Ti becomes strongly reduced but almost all of the remaining O stays in the original O2– oxidation state. This implies that O reacting with H atoms is removed from the material, most probably as water, and only a small concentration of Ti–OH compounds is formed. The removal of a neutral O atom from the structure leaves two excess electrons on the empty states of Ti cations. The electrons can be either localized or distributed among the 5-fold coordinated Ti3+ ions surrounding the O vacancy. According to the simulations conducted by Di Valentin et al.,10 the energetically most favorable situations are when the electrons are localized to the Ti3+ ion next to the vacancy and the next nearest neighboring Ti cation or alternatively one electron is localized to 5-fold coordinated Ti3+ and the other electron is completely delocalized.
Contrary to the hydrogen treatment, the thermal treatment leaves the O concentration practically constant at 70 at % in all studied annealing temperatures. This result is unexpected because Ti is strongly reduced and one could assume that this leads to a decreasing O content. The explanations for this anomalous behavior are scarce in the literature because most of the Ti oxide research has traditionally concentrated on crystalline anatase or rutile forms instead of the amorphous phase. In crystalline TiO2, the thermal treatment usually leads to a reduction of only a couple of percent of Ti cations.30 In that case, the question of what the O/Ti ratio represents is not valid nor easy to study. For example, Ti3+ cations are known to readily diffuse toward the bulk,11,31 which may alter the results with small concentrations.
One possible explanation for the high O content can be trapped molecular O2. This is however unlikely, because in crystalline TiO2 molecular O2 is known to desorb at about 110 °C,32 and the desorption energy should not differ too much for amorphous TiO2. Thus, the desorption is expected to occur at much lower temperature than what is used in the thermal treatment series. More plausible explanation is localized charge transfer from anions to cations. For example, Pfeifer et al. have studied charge transfer in amorphous IrO2, which has a structure very similar to amorphous TiO2. They have introduced an IrO2 reduction model, where the creation of an Ir vacancy results in two Ir3+ ions and six localized reactive O– species.33−35 Interestingly, we also observe the O– formation in a-TiO2, even without the Ti vacancy formation. If sufficient amount of thermal energy is introduced to a Ti–O bond, the electron transfer from anionic to cationic framework can result in stable 5-fold-coordinated Ti3+ and 2-fold-coordinated O– states in the matrix. This model is supported by the observed binding energy changes in the Ti 2p transition. In the as-deposited film, the Ti4+ peak is located at 458.80 eV but starts to shift linearly toward higher binding energies (up to 459.20 eV) as the temperature of the thermal treatment is increased. This can be explained by the increased number of O– species, which attract electrons from the neighboring, still 6-fold-coordinated Ti4+ species, thus increasing their effective nuclear charge.
More detailed analysis of the Ti reduction can be obtained by analyzing the relative concentrations of different Ti oxidation states in Figure 2. Figure 4 illustrates the evolution of these states as the total Ti suboxide (Ti3+ and Ti2+) concentration increases. In the thermally treated samples, the reduction starts with only Ti3+ being formed at temperatures below 400 °C. At higher temperatures, however, the formation rate of both Ti3+ and Ti2+ suboxide species is somewhat similar and follows the total suboxide concentration linearly. In contrast, at the hydrogen treated surfaces the initial reduction is strongly focused on the formation of the Ti3+ states, and the amount of Ti2+ species starts to increase significantly only after the total suboxide concentration has reached about 70%.
Figure 4.
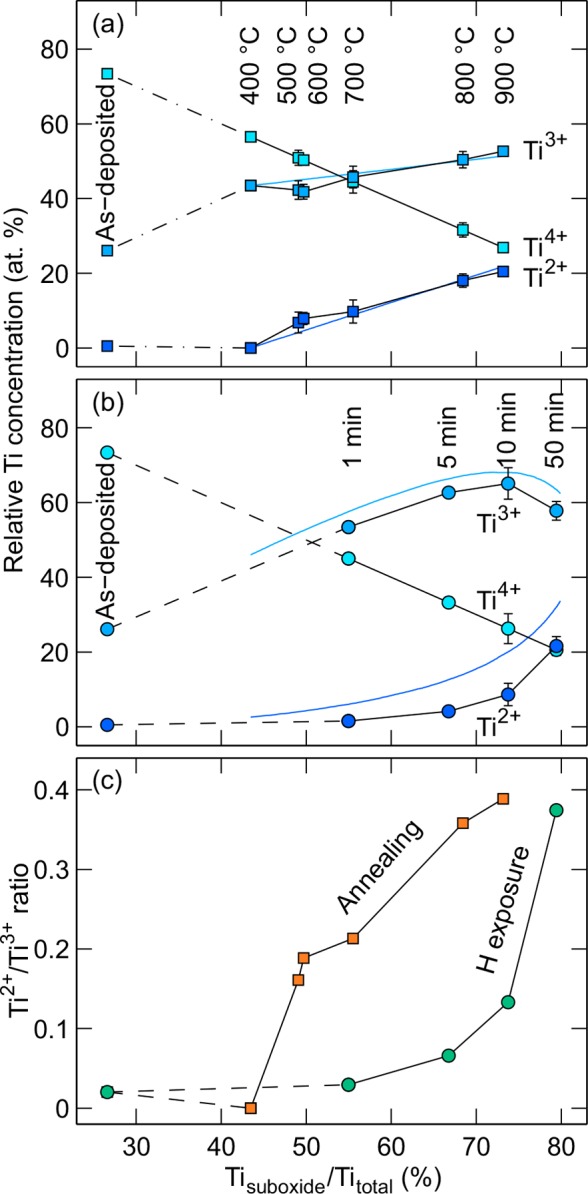
Relative concentration of Ti oxidation states in (a) thermal and (b) hydrogen treatment series and (c) the Ti2+/Ti3+ ratio as a function of the total suboxide concentration. All experimental values (squares and circles) are determined from the Ti 2p transition shown in Figure 2a. The error bars represent the range of variation from repeated measurements. The solid blue lines represent the Monte Carlo simulation results.
To better understand the difference between these reduction mechanisms, we conducted Monte Carlo simulations with two different reduction mechanisms (see the Supporting Information for details). The reason for these simulations was to qualitatively validate the reduction mechanisms that were deduced based on the XPS data of the Ti oxidation states.
For thermally treated samples, the best agreement between the simulation (solid blue lines in Figure 4a) and experimental data was obtained with a mechanism where the original Ti4+ state can be reduced directly to Ti3+ or Ti2+. The reduction from Ti4+ to Ti2+ state is a possible alternative for the reduction from Ti4+ to Ti3+ oxidation state because the process does not involve external atoms, such as hydrogen, to be introduced into the matrix. On the other hand, for hydrogen treatment a double reduction from Ti4+ to Ti2+ state is not likely because a reduction event can be considered as a process where one hydrogen atom at a time encounters the titanium oxide surface and removes one O atom. This process can take place if the Ti atom is either a part of the original TiO2 structure or it can already be in a partially reduced Ti2O3 form. Thus, the removal of one O atom as OH compound leads to Ti2O3 or TiO configuration, respectively. The simulation results (blue lines in Figure 4b) support this type of “one step reduction” mechanism. The exclusive reduction options either from Ti4+ to Ti3+ or from Ti3+ to Ti2+ lead to a behavior, where the formation of Ti3+ cations is pronounced at the beginning. However, as the amount of Ti4+ cations decreases and Ti3+ cations increases, the probability for Ti3+ to Ti2+ reduction increases and the relative Ti2+ concentration starts to increase sharply as shown by the Ti2+/Ti3+ ratio in the Figure 4c.
In addition to the different reduction mechanisms in thermal and hydrogen treatment simulations, the latter required a modification in the x-axis scaling. The simulated curves had to be scaled down by 17% to match with the measured data points. This can be interpreted so that 17% of the Ti atoms inside the XPS information depth are deeper than the influence depth of the hydrogen exposure. Thus, these atoms can never be reduced which leads to a sharp decrease in the Ti3+ concentration earlier than what the unscaled simulation predicts. On the basis of the Beer–Lambert law and the attenuation length of the Ti 2p photoelectrons (λ = 2.2 nm), the hydrogen exposure influence depth is calculated to be 4.0 nm.
The above-discussed reduction mechanisms are summarized in eqs 1 and 2 for thermal treatment and hydrogen treatment, respectively. The ↑ sign in eq 2 depicts the desorption of water molecules from the surface.
 |
1 |
| 2 |
In thermal treatment at above 400 °C, the reduction process may lead directly to either Ti3+ or Ti2+ formation whereas in hydrogen treated samples the Ti3+ is the preferred reduction product as long as there is a significant amount of stoichiometric TiO2 available.
In addition to the oxidation states, the thermal and hydrogen treatments may affect the molecular ordering of the a-TiO2 films. Qualitative information about these structural changes can be obtained by analyzing the XPS peak shapes. Accurate curve fitting of the Ti 2p spectra in Figure 2 required that the full width at half-maximum (fwhm) of the peaks was allowed to vary. Blue squares and circles in Figure 5 illustrate the variation of the Ti4+ component fwhm. Other oxidation states gave qualitatively similar results. In the hydrogen exposure series, the fwhm growth is only 24% whereas in thermally treated samples the peak width increases 66% as the suboxide concentration is increased from the original 26 to 73% (900 °C and 10 min treatments). The increase in peak width is a sign of increasing structural disorder.34 This can be interpreted as a relaxation of internal stress among the chains of the octahedral TiO2 units. Thus, it can be concluded that the thermal breaking of Ti–O bonds and the formation of Ti3+ and O– species introduce structural changes in the amorphous material. Figure 5 also shows the relative concentration of O– (in thermal treatment) and OH– (in hydrogen treatment) states compared to the total O content. For hydrogen treated samples, the OH– increase is relatively weak, because most of the O ions participating in the structural changes desorb from the material as water molecules. For the thermally treated samples, the increase in O– states correlates excellently with the Ti 2p fwhm changes thus consolidating the model that the peak widening is caused by changes in molecular bonding and subsequent deformation of the amorphous structure. Interestingly, the onset temperature for the O– species formation coincides with the temperature where photoelectrochemical stability was obtained. Thus, we may conclude that the O– species stabilizes the a-TiO2 from photocorrosion.
Figure 5.
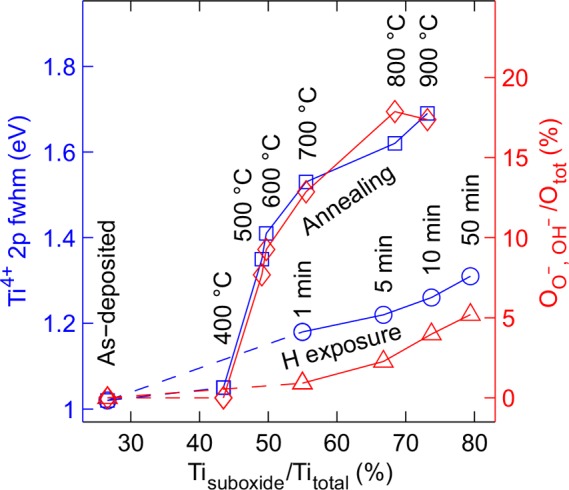
Structural disorder of thermally and hydrogen treated surfaces. For Ti, the disorder increase is interpreted from the fwhm widening of the Ti4+ 2p state (blue squares and circles). For O, a qualitatively similar behavior can be seen in the increase of the area of the shoulder peaks (peaks O– and OH– in Figure 2) (red diamonds and triangles).
Phase Stability of the Amorphous TiO2
Thermal treatment of a-TiO2 at high temperature is known to induce partial crystallization, especially if the annealing is made in atmospheric pressure.2,13 Our analysis of the Ti 2p spectral features clearly indicated that the annealing in UHV increases the structural disorder. This was further supported by XRD measurements of similarly heat treated 200 nm thick a-TiO2 films as shown in Figure 6 along with the spectra measured from rutile and anatase reference samples.
Figure 6.
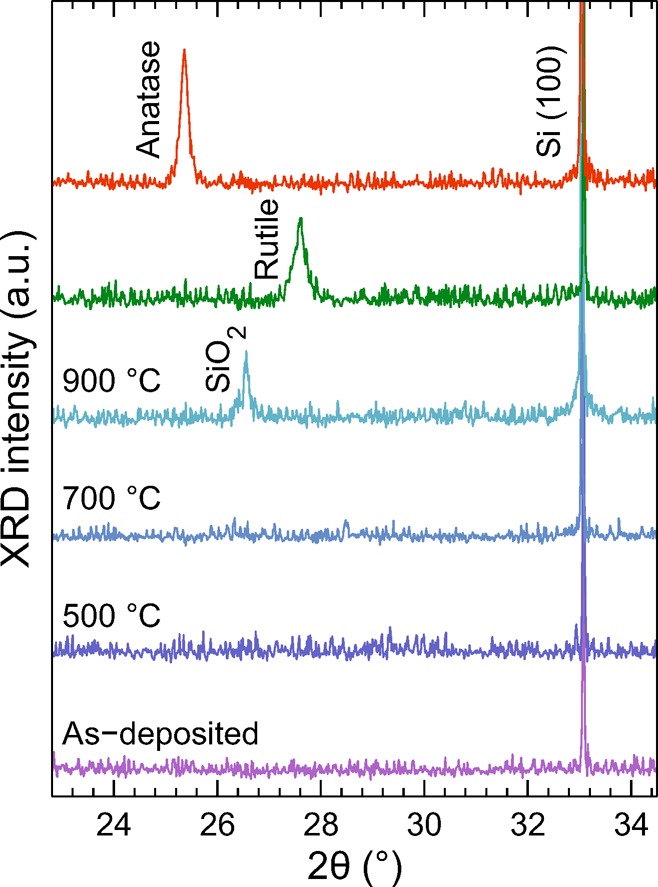
XRD patterns from the thermally treated a-TiO2 films in addition to the spectra measured from the rutile and anatase reference samples. The films do not show any crystalline TiO2 features at temperatures from 200 °C (as-deposited) to 900 °C. The SiO2 peak in the 900 °C diffractogram is due to thin SiO2 film at the Si–a-TiO2 interface. The anatase and rutile references were obtained from TiO2 films grown at 100 and 200 °C followed by annealing in air at 700 °C for 45 min, respectively.
The XRD results were consistent with the XPS results showing no indication of crystalline phases of TiO2 in the thermally treated films. This is most advantageous in terms of using the films for PEC applications, because amorphous films are known to protect the underlying semiconductor better than crystalline films and also exhibit higher photocurrent conductivity.2 The only emerging diffraction feature as a result of annealing was the SiO2 related peak which appeared during the 900 °C thermal treatment. TiO2 is known to donate O to Si at high temperatures, which produces a thin SiO2 film at the Si–TiO2 interface.36 Importantly, the insulating SiO2 film at the interface limits the current transfer required for efficient photoelectrochemical water splitting. This explains the observed PEC response where higher annealing temperatures produced smaller photocurrent densities.
Modification of the Valence Band Structure
Figure 7a,b shows the UP spectra of the thermal and hydrogen treated a-TiO2/Si samples. The valence band (VB) (Eb ≈ 3.3–12 eV) is fitted with three states corresponding to TiO2 orbitals.37,38 The highest energy band is associated with the nonbonding O 2p orbitals (π-type). Below that are the π- and σ-type molecular orbitals of TiO2. In the spectrum of the as-deposited sample there is an additional peak at 10.2 eV (denoted as γ) which is produced by the impurities such as OH or CO groups on the surface.11,39 This state disappears immediately during the first thermal or hydrogen treatment, which together with the core level XP spectra verifies the purity of the studied films. Additionally, a new peak with the same binding energy appears after 800 and 900 °C thermal treatments. This is caused by Si oxide formed when the residual Si impurity from the film segregates to the surface.40 The UPS spectral feature observed at the 2–3 eV range is a combination of the valence band structure and He Iβ satellite line. The latter accounts for approximately 30% of the total intensity at this energy range.
Figure 7.

Ultraviolet photoemission valence band (VB) spectra after (a) thermal and (b) hydrogen treatments of a-TiO2. The main components originating from the a-TiO2 film are the σ and π molecular orbitals together with the O 2p nonbonding orbitals. In addition to these components, the reduced a-TiO2 has a significant Ti 3d component as shown in blue in the magnified views. In the case of the as-deposited surface the γ component can be associated with the σ states of hydroxyl groups adsorbed from the atmosphere.11 For 800 and 900 °C annealed samples, the same component position corresponds to the electronic states in SiO2 which is formed by the surface segregated Si.40 Part c illustrates the area of the Ti3+/2+ oxidation state related Ti 3d peak. For hydrogen treatment series the area of Ti 3d suboxide states increases linearly whereas in thermal treatment the area starts to saturate after 700 °C annealing.
For pure stoichiometric TiO2, the band gap is about 3.0–3.4 eV depending on the crystal structure38,41,42 and the conduction band minimum is located close to the Fermi energy. This is in good agreement with our as-deposited sample where the valence band maximum (VBM) is located at a binding energy of 3.3 eV as determined by extrapolating the linear region of the lower energy side of the VB to the zero intensity baseline. During the treatments, the VBM shows minor gradual shift to 3.5 or 3.6 eV for thermally and hydrogen treated samples, respectively. In hydrogen treatment series the density of the nonbonding O 2p states decreases with increasing exposure time which is in concordance with the aforementioned removal of O atoms from the structure. On the other hand, in thermally treated samples the density of the O 2pn/b states remains rather constant from 400 to 700 °C. After this, there is a sudden decrease in the density which can be associated with some of the O atoms reacting with the surface segregated Si atoms resulting in the formation of the γ state.
The Ti 3d state just below the Fermi edge is playing a major role in determining the photoactivity of the a-TiO2/Si surface. These in-gap states provide a pathway for electron transfer between the bulk electrode material and the electrolyte9,41 and also effectively narrow the band gap thus improving the absorption of longer wavelengths.43 The in-gap states divide the band gap in two smaller sub gaps thus providing an intermediate stepping stone for photoexcitation at lower energies.44 The in-gap states of TiO2 have often been ascribed to dopants or impurities (e.g., C, S, N, W, Mn),10,38,43−45 Ti3+/2+ interstitials,10,11 or bridging O vacancies.10,37 In our case, the impurity contribution can be excluded because the as-deposited surface has the highest C and N concentration accumulated from the air exposure and ALD precursor residues but still shows no in-gap density of states. Instead, when the surface is exposed to elevated temperatures or hydrogen atoms, the area of the Ti3+/2+ 3d component starts to increase rapidly. For hydrogen treated samples, the density of the in-gap state shows rather linear increase as a function of the total hydrogen exposure induced suboxide concentration, as illustrated in Figure 7c. The position of the peak remains constant at Eb = 0.54 eV, which suggests that the peak is related to only one type of chemical state. The observed binding energy is in good agreement with the theoretically calculated value for Ti3+/2+ 3d states caused by O vacancies,10 thus supporting the model that the peak is produced solely by the removal of O atoms.
In the thermal treatment series, the growth of the Ti3+/2+ 3d component is more subtle, especially above 60% suboxide concentration, and the peak position shifts toward the Fermi edge with the increasing treatment temperature. The shift from Eb = 0.72 to 0.50 eV can be interpreted such that the peak is a convolution of two different chemical states: the Ti3+ cation where Ti has only one localized excess electron or Ti2+ with two localized excess electrons as discussed previously in the reduction mechanism section. Thus, the increasing Ti2+/Ti3+ ratio pushes the total in-gap density of states to smaller binding energy.
Conclusions
The presented results show two methods for modifying the molecular structure and electronic states of the ALD grown a-TiO2 thin film on a semiconductor electrode surface. Both the thermal treatment in UHV and the atomic hydrogen treatment lead to a partially restructured amorphous phase, where the appearance of the in-gap Ti3+/2+ 3d electronic states narrows the effective band gap. Importantly, both methods preserve the amorphous phase of the a-TiO2 film. This is advantageous for photoelectrochemical applications as several recent studies have shown that the underlying semiconductor electrodes can be protected more efficiently with a conformal a-TiO2 film rather than with its crystalline counterparts.
The studied post-treatment methods lead to considerably distinct chemical compositions as summarized in Figure 8. In the thermal treatment series, the relative concentration of O and Ti atoms remains constant but some electrons are transferred from anionic to cationic network. This leads to the formation of localized Ti3+ and Ti2+ species and O– species. Importantly, the annealing procedure in UHV does not introduce any crystallization, but on the contrary decreases the structural order by relieving the strain in the amorphous lattice. By comparison, the atomic hydrogen treatment removes O atoms creating O vacancies surrounded by Ti cations in Ti3+ or Ti2+ valence state.
Figure 8.
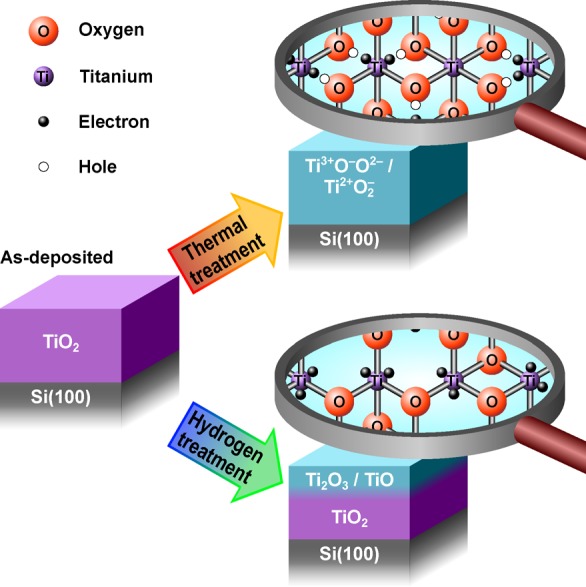
Schematic representation of the chemical and electronic changes in the a-TiO2 ultrathin films as a result of the thermal and hydrogen post-treatments. The thermal treatment breaks bonds between Ti and O atoms leaving the elemental composition intact. The electron transfer from O to Ti results in excess electrons that occupy the Ti 3d state, which improves charge transfer properties. Additionally, the thermal treatment enhances the photoelectrochemical stability of the film via formation of O– species that are essentially electronic defects in the anionic framework. In contrast, atomic hydrogen treatment creates an electronically “leaky” film via formation of O vacancies resulting in a photoelectrochemically unstable film.
From the photoelectrochemical point of view, the as-deposited a-TiO2 is susceptible to severe photocorrosion under photoelectrochemical water oxidation conditions. The stability of a-TiO2 can be obtained after heat treatment at >500 °C in vacuum. This was attributed to the formation of O– species that are essentially electronic defects in the anionic framework. At elevated temperatures above 500 °C, the a-TiO2 thin film starts to react with the underlying Si substrate which produces an insulating SiO2 interface layer thus lowering the photocurrent. On the other hand, the hydrogen treated samples were found to be unstable in alkaline electrochemical conditions. On the basis of the photocurrent measurements, the photocorrosion of the hydrogen treated samples was comparable to that of the as-deposited samples.
In addition to optimizing the surface chemical and electronic structure, large scale manufacturing of cost-efficient water splitting devices would require optimization of the morphological structure, such as utilization of porous nanomaterials or nanowires.46,47 This would increase the reactive surface area thus increasing the achievable photocurrent. However, the planar model systems used in this study provide valuable information about the possible a-TiO2 post-treatment routes. Combining the knowledge obtained here with the nanostructured substrates and highly active electrocatalyst materials2,4,8 can lead to a significant improvement in photoelectrodes utilizing a-TiO2 ultrathin film coatings.
Acknowledgments
This work was supported by the Academy of Finland (Decision Numbers 141481, 286713, and 309920). M.H. was supported by the Tampere University of Technology Graduate School. H.A.-L. was supported by the Jenny and Antti Wihuri Foundation.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemmater.7b02938.
Additional XPS spectra, photoelectrochemical current–voltage curves, XPS/UPS fitting parameters, and details of the Monte Carlo simulations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Walter M. G.; Warren E. L.; McKone J. R.; Boettcher S. W.; Mi Q.; Santori E. A.; Lewis N. S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473. 10.1021/cr1002326. [DOI] [PubMed] [Google Scholar]
- Hu S.; Shaner M. R.; Beardslee J. A.; Lichterman M.; Brunschwig B. S.; Lewis N. S. Amorphous TiO2 Coatings Stabilize Si, GaAs, and GaP Photoanodes for Efficient Water Oxidation. Science 2014, 344, 1005–1009. 10.1126/science.1251428. [DOI] [PubMed] [Google Scholar]
- Chen S.; Wang L.-W. Thermodynamic Oxidation and Reduction Potentials of Photocatalytic Semiconductors in Aqueous Solution. Chem. Mater. 2012, 24, 3659–3666. 10.1021/cm302533s. [DOI] [Google Scholar]
- Hu S.; Richter M. H.; Lichterman M. F.; Beardslee J.; Mayer T.; Brunschwig B. S.; Lewis N. S. Electrical, Photoelectrochemical, and Photoelectron Spectroscopic Investigation of the Interfacial Transport and Energetics of Amorphous TiO2/Si Heterojunctions. J. Phys. Chem. C 2016, 120, 3117–3129. 10.1021/acs.jpcc.5b09121. [DOI] [Google Scholar]
- Pham H. H.; Wang L.-W. Oxygen Vacancy and Hole Conduction in Amorphous TiO2. Phys. Chem. Chem. Phys. 2015, 17, 541–550. 10.1039/C4CP04209C. [DOI] [PubMed] [Google Scholar]
- Scheuermann A. G.; Lawrence J. P.; Kemp K. W.; Ito T.; Walsh A.; Chidsey C. E. D.; Hurley P. K.; McIntyre P. C. Design Principles for Maximizing Photovoltage in Metal-oxide-protected Water-Splitting Photoanodes. Nat. Mater. 2016, 15, 99–105. 10.1038/nmat4451. [DOI] [PubMed] [Google Scholar]
- Chen Y. W.; Prange J. D.; Dühnen S.; Park Y.; Gunji M.; Chidsey C. E. D.; McIntyre P. C. Atomic Layer-Deposited Tunnel Oxide Stabilizes Silicon Photoanodes for Water Oxidation. Nat. Mater. 2011, 10, 539–544. 10.1038/nmat3047. [DOI] [PubMed] [Google Scholar]
- Scheuermann A. G.; Prange J. D.; Gunji M.; Chidsey C. E. D.; McIntyre P. C. Effects of Catalyst Material and Atomic Layer Deposited TiO2 Oxide Thickness on the Water Oxidation Performance of Metal-Insulator-Silicon Anodes. Energy Environ. Sci. 2013, 6, 2487–2496. 10.1039/c3ee41178h. [DOI] [Google Scholar]
- Sivula K. Defects Give New Life to an Old Material: Electronically Leaky Titania as a Photoanode Protection Layer. ChemCatChem 2014, 6, 2796–2797. 10.1002/cctc.201402532. [DOI] [Google Scholar]
- Di Valentin C.; Pacchioni G.; Selloni A. Reduced and n-Type Doped TiO2: Nature of Ti3+ Species. J. Phys. Chem. C 2009, 113, 20543–20552. 10.1021/jp9061797. [DOI] [Google Scholar]
- Wendt S.; Sprunger P. T.; Lira E.; Madsen G. K. H.; Li Z.; Hansen J. Ø.; Matthiesen J.; Blekinge-Rasmussen A.; Lægsgaard E.; Hammer B.; Besenbacher F. The Role of Interstitial Sites in the Ti3d Defect State in the Band Gap of Titania. Science 2008, 320, 1755–1759. 10.1126/science.1159846. [DOI] [PubMed] [Google Scholar]
- Hannula M.; Lahtonen K.; Ali-Löytty H.; Zakharov A.; Isotalo T.; Saari J.; Valden M. Fabrication of Topographically Microstructured Titanium Silicide Interface for Advanced Photonic Applications. Scr. Mater. 2016, 119, 76–81. 10.1016/j.scriptamat.2016.03.016. [DOI] [Google Scholar]
- Jin C.; Liu B.; Lei Z.; Sun J. Structure and Photoluminescence of the TiO2 Films Grown by Atomic Layer Deposition Using Tetrakis-Dimethylamino Titanium and Ozone. Nanoscale Res. Lett. 2015, 10, 95. 10.1186/s11671-015-0790-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarik J.; Aidla A.; Kiisler A.-A.; Uustare T.; Sammelselg V. Effect of Crystal Structure on Optical Properties of TiO2 Films Grown by Atomic Layer Deposition. Thin Solid Films 1997, 305, 270–273. 10.1016/S0040-6090(97)00135-1. [DOI] [Google Scholar]
- Omicron Nanotechnology. NanoESCA, 2017; http://www.scientaomicron.com.
- Omicron Nanotechnology. Atomic Hydrogen Source EFM-H, 2017; http://www.scientaomicron.direct/16/thin-film-deposition-products/efm-h.
- CasaXPS: Processing Software for XPS, AES, SIMS and More, 2017; http://www.casaxps.com.
- Tanuma S.; Powell C. J.; Penn D. R. Calculations of Electron Inelastic Mean Free Paths. V. Data for 14 Organic Compounds Over the 50–2000 eV Range. Surf. Interface Anal. 1994, 21, 165–176. 10.1002/sia.740210302. [DOI] [Google Scholar]
- Tougaard S.QUASES: Software for Quantitative XPS/AES of Surface Nano-Structure by Analysis of the Peak Shape and Background, version 2.2; University of Southern Denmark: Odense, Denmark, 2002; http://www.quases.com.
- Cai J.; Wang Y.; Zhu Y.; Wu M.; Zhang H.; Li X.; Jiang Z.; Meng M. In Situ Formation of Disorder-Engineered TiO2(B)-Anatase Heterophase Junction for Enhanced Photocatalytic Hydrogen Evolution. ACS Appl. Mater. Interfaces 2015, 7, 24987–24992. 10.1021/acsami.5b07318. [DOI] [PubMed] [Google Scholar]
- Iancu A. T.; Logar M.; Park J.; Prinz F. B. Atomic Layer Deposition of Undoped TiO2 Exhibiting p-Type Conductivity. ACS Appl. Mater. Interfaces 2015, 7, 5134–5140. 10.1021/am5072223. [DOI] [PubMed] [Google Scholar]
- Lichterman M. F.; Hu S.; Richter M. H.; Crumlin E. J.; Axnanda S.; Favaro M.; Drisdell W.; Hussain Z.; Mayer T.; Brunschwig B. S.; Lewis N. S.; Liu Z.; Lewerenz H.-J. Direct Observation of the Energetics at a Semiconductor/Liquid Junction by Operando X-ray Photoelectron Spectroscopy. Energy Environ. Sci. 2015, 8, 2409–2416. 10.1039/C5EE01014D. [DOI] [Google Scholar]
- McDowell M. T.; Lichterman M. F.; Carim A. I.; Liu R.; Hu S.; Brunschwig B. S.; Lewis N. S. The Influence of Structure and Processing on the Behavior of TiO2 Protective Layers for Stabilization of n-Si/TiO2/Ni Photoanodes for Water Oxidation. ACS Appl. Mater. Interfaces 2015, 7, 15189–15199. 10.1021/acsami.5b00379. [DOI] [PubMed] [Google Scholar]
- Fan C.; Chen C.; Wang J.; Fu X.; Ren Z.; Qian G.; Wang Z. Black Hydroxylated Titanium Dioxide Prepared via Ultrasonication with Enhanced Photocatalytic Activity. Sci. Rep. 2015, 5, 11712. 10.1038/srep11712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Feng C.; Zhang M.; Yang J.; Zhang Z. Enhanced visible light photocatalytic activity of N-doped TiO2 in relation to single-electron-trapped oxygen vacancy and doped-nitrogen. Appl. Catal., B 2010, 100, 84–90. 10.1016/j.apcatb.2010.07.015. [DOI] [Google Scholar]
- Zhang L.; Tse M. S.; Tan O. K.; Wang Y. X.; Han M. Facile fabrication and characterization of multi-type carbon-doped TiO2 for visible light-activated photocatalytic mineralization of gaseous toluene. J. Mater. Chem. A 2013, 1, 4497–4507. 10.1039/c3ta01366a. [DOI] [Google Scholar]
- Kuznetsov M.; Zhuravlev J.; Zhilyaev V.; Gubanov V. XPS study of the nitrides, oxides and oxynitrides of titanium. J. Electron Spectrosc. Relat. Phenom. 1992, 58, 1–9. 10.1016/0368-2048(92)80001-O. [DOI] [Google Scholar]
- Zhang Y.; Li X.; Chen D.; Ma N.; Hua X.; Wang H. Si Doping Effects on the Photocatalytic Activity of TiO2 Nanotubes Film Prepared by an Anodization Process. Scr. Mater. 2009, 60, 543–546. 10.1016/j.scriptamat.2008.12.004. [DOI] [Google Scholar]
- Henderson M. A. A Surface Perspective on Self-Diffusion in Rutile TiO2. Surf. Sci. 1999, 419, 174–187. 10.1016/S0039-6028(98)00778-X. [DOI] [Google Scholar]
- Diebold U. The Surface Science of Titanium Dioxide. Surf. Sci. Rep. 2003, 48, 53–229. 10.1016/S0167-5729(02)00100-0. [DOI] [Google Scholar]
- Park K. T.; Pan M.; Meunier V.; Plummer E. W. Reoxidation of TiO2(110) via Ti Interstitials and Line Defects. Phys. Rev. B: Condens. Matter Mater. Phys. 2007, 75, 245415. 10.1103/PhysRevB.75.245415. [DOI] [Google Scholar]
- Perkins C. L.; Henderson M. A. Photodesorption and Trapping of Molecular Oxygen at the TiO2(110)-Water Ice Interface. J. Phys. Chem. B 2001, 105, 3856–3863. 10.1021/jp0031799. [DOI] [Google Scholar]
- Pfeifer V.; et al. The Electronic Structure of Iridium Oxide Electrodes Active in Water Splitting. Phys. Chem. Chem. Phys. 2016, 18, 2292–2296. 10.1039/C5CP06997A. [DOI] [PubMed] [Google Scholar]
- Pfeifer V.; et al. The Electronic Structure of Iridium and Its Oxides. Surf. Interface Anal. 2016, 48, 261–273. 10.1002/sia.5895. [DOI] [Google Scholar]
- Pfeifer V.; Jones T. E.; Wrabetz S.; Massué C.; Velasco Vélez J. J.; Arrigo R.; Scherzer M.; Piccinin S.; Hävecker M.; Knop-Gericke A.; Schlögl R. Reactive Oxygen Species in Iridium-Based OER Catalysts. Chem. Sci. 2016, 7, 6791–6795. 10.1039/C6SC01860B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuan A.; Yoon M.; Medvedev V.; Ono Y.; Ma Y.; Rogers J. Jr. Interface Control in the Chemical Vapor Deposition of Titanium Dioxide on Silicon(100). Thin Solid Films 2000, 377–378, 766–771. 10.1016/S0040-6090(00)01453-X. [DOI] [Google Scholar]
- Fleming L.; Fulton C. C.; Lucovsky G.; Rowe J. E.; Ulrich M. D.; Lüning J. Local Bonding Analysis of the Valence and Conduction Band Features of TiO2. J. Appl. Phys. 2007, 102, 033707. 10.1063/1.2764004. [DOI] [Google Scholar]
- Pandiyan R.; Delegan N.; Dirany A.; Drogui P.; el Khakani M. A. Probing the Electronic Surface Properties and Bandgap Narrowing of in situ N, W, and (W,N) Doped Magnetron-Sputtered TiO2 Films Intended for Electro-Photocatalytic Applications. J. Phys. Chem. C 2016, 120, 631–638. 10.1021/acs.jpcc.5b08057. [DOI] [Google Scholar]
- Sanjinés R.; Tang H.; Berger H.; Gozzo F.; Margaritondo G.; Lévy F. Electronic Structure of Anatase TiO2 Oxide. J. Appl. Phys. 1994, 75, 2945–2951. 10.1063/1.356190. [DOI] [Google Scholar]
- Fulton C. C.; Lucovsky G.; Nemanich R. J. Electronic States at the Interface of Ti-Si Oxide on Si(100). J. Vac. Sci. Technol., B: Microelectron. Process. Phenom. 2002, 20, 1726–1731. 10.1116/1.1493785. [DOI] [Google Scholar]
- Yaghoubi H.; Li Z.; Chen Y.; Ngo H. T.; Bhethanabotla V. R.; Joseph B.; Ma S.; Schlaf R.; Takshi A. Toward a Visible Light-Driven Photocatalyst: The Effect of Midgap-States-Induced Energy Gap of Undoped TiO2 Nanoparticles. ACS Catal. 2015, 5, 327–335. 10.1021/cs501539q. [DOI] [Google Scholar]
- Perego M.; Seguini G.; Scarel G.; Fanciulli M.; Wallrapp F. Energy Band Alignment at TiO2/Si Interface with Various Interlayers. J. Appl. Phys. 2008, 103, 043509. 10.1063/1.2885109. [DOI] [Google Scholar]
- Chen X.; Burda C. The Electronic Origin of the Visible-Light Absorption Properties of C-, N- and S-Doped TiO2 Nanomaterials. J. Am. Chem. Soc. 2008, 130, 5018–5019. 10.1021/ja711023z. [DOI] [PubMed] [Google Scholar]
- Xia X.; Lu L.; Walton A.; Ward M.; Han X.; Brydson R.; Luo J.; Shao G. Origin of Significant Visible-Light Absorption Properties of Mn-Doped TiO2 Thin Films. Acta Mater. 2012, 60, 1974–1985. 10.1016/j.actamat.2012.01.006. [DOI] [Google Scholar]
- Asahi R.; Morikawa T.; Ohwaki T.; Aoki K.; Taga Y. Visible-Light Photocatalysis in Nitrogen-Doped Titanium Oxides. Science 2001, 293, 269–271. 10.1126/science.1061051. [DOI] [PubMed] [Google Scholar]
- Wang G.; Wang H.; Ling Y.; Tang Y.; Yang X.; Fitzmorris R. C.; Wang C.; Zhang J. Z.; Li Y. Hydrogen-Treated TiO2 Nanowire Arrays for Photoelectrochemical Water Splitting. Nano Lett. 2011, 11, 3026–3033. 10.1021/nl201766h. [DOI] [PubMed] [Google Scholar]
- Kibria M. G.; Zhao S.; Chowdhury F. A.; Wang Q.; Nguyen H. P. T.; Trudeau M. L.; Guo H.; Mi Z. Tuning the Surface Fermi Level on p-Type Gallium Nitride Nanowires for Efficient Overall Water Splitting. Nat. Commun. 2014, 5, 3825. 10.1038/ncomms4825. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.