

Abstract
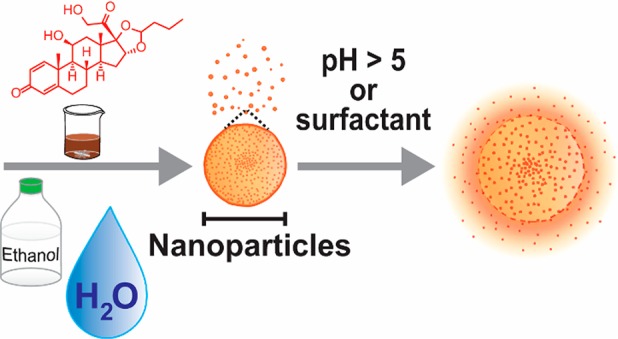
The mechanism of lignin nanoprecipitation and subsequent self-assembly was elucidated by studying generation of lignin nanoparticles (LNPs) from aqueous ethanol. LNP formation was found to follow a kinetically controlled nucleation–growth mechanism in which large lignin molecules formed the initial critical nuclei. Using this information, we demonstrate entrapment of budesonide in LNPs and subsequent pH-triggered and surfactant-responsive release of this synthetic anti-inflammatory corticosteroid. Overall, our results not only provide a promising intestinal delivery system for budesonide but also deliver fundamental mechanistic understanding for the entrapment of actives in LNPs with controlled size and release properties.
Keywords: Assembly, Association, Colloids, Mechanism, Nanotechnology
Short abstract
A fundamental understanding of the self-assembly mechanism of lignin nanoparticles enables size-controlled entrapment of budesonide for surfactant- and pH-triggered intestinal release.
Introduction
Lignins are natural polyphenols emerging in large amounts from pulp and paper and modern second-generation biorefinery processes. The sustainability of future biorefineries largely depends on the possible exploitation of the potentialities of lignin as the most abundant renewable aromatic polymer.1 To date lignin is still mostly burnt to support energy needs of the processes. The lack of significant lignin valorization is due to the low added value applications developed so far. A sustainable lignin biorefinery would credibly rely upon differentiated processes leading to high value added chemicals and materials.2−4 From this perspective, the valorization and exploitation of intrinsic structural features of lignin is of pivotal importance.
Lignins are biocompatible biopolymers exhibiting significant antioxidant activity5,6 and pH-dependent aqueous solubility.7 They also present intrinsic amphiphilicity due to the presence of phenolic and aliphatic hydroxyl groups, carboxylic groups, and aromatic rings, respectively, which are present in various quantities along polymeric and oligomeric backbones, depending on the chosen type of technical lignin.8−16 Such structural features strongly favor aggregation phenomena, exploitable in the formation of nanostructures upon supramolecular self-assembly.17−20 Among various promising applications, lignin particles in nanoscale have been used as biocompatible photoluminescence emitters21 and fluorescent sensors to detect formaldehyde vapor.22
Prior studies have fabricated versatile lignin-based active carriers. Nano- and microscale lignin capsules have been formed from vegetable oil-in-water emulsions for the entrapment of coumarin 6 as a model compound,23−25 while lignin nanocapsules with an aqueous core were synthesized for the entrapment of sulforhodamine.26 Complex micelles from cationized pine alkali lignin and sodium dodecylbenzenesulfonate were used to entrap ibuprofen.27 Lignin nanoparticles (LNPs), also referred to as colloidal lignin particles (CLPs),28,29 are compact submicron spheres that have been used to entrap anticancer medicines,30,31 the food flavor 2-propylpyridine,32 and the natural phenolic compound resveratrol.33 Despite the scattered occurrence of such reports, to date little is known about the mechanism of lignin nanoprecipitation and nanoparticle self-assembly. There is a lack of general understanding of the driving forces triggering particle formation and a potential subsequent release of entrapped actives. This is especially relevant when targeting medical applications that have to integrate a controlled release behavior in the target tissue with a reliable supply of that active. Furthermore, a mechanistic understanding of the active upload in LNP and the downstream release is pivotal when attempting to couple specific activity of the entrapped active with the intrinsic antioxidant (and therefore anti-inflammatory) activity of lignin. Besides, previous studies used odiferous Kraft lignin and nonbiocompatible solvents for the synthesis of LNPs and performed chemical modifications without subsequent trace analysis in the formed particles.
The use of lignin with better organoleptic and chemical properties and the development of a process that involves only biocompatible solvents are major issues that need attention to validate lignin-based materials for biomedical applications. Sufficient organoleptic quality is important for all delivery routes and especially crucial with respect to oral dosing. Orally administered pharmaceuticals face varying acidity from pH 1–2 in the stomach to pH 7–8 in the small intestine, and pH 5–7 in the large intestine.34 Coating and entrapment protect actives from the gastric acid, avoid irritation of the stomach, and facilitate site-specific release.35 The use of natural polymers for enteric coating and as matrix-forming substances should be preferred over fossil oil-based acrylic polymers that currently dominate this application.36−43 So far, mainly various polysaccharides have been used,36,39,40,44 but more recently research has seen an increasing general interest in the use of lignin in biomaterial applications.29,45−47
Interestingly, none of the aforementioned lignin-based drug carriers have attempted to target drug release in a system as complex as the human gastrointestinal (GI) tract. One example of actives used in this area is budesonide that is a synthetic corticosteroid for the treatment of inflammatory bowel diseases (IBS) such as Chron’s disease.48 Maximizing local activity of this sparingly water-soluble drug in the colonic mucosa is a key challenge for the controlled release.49 Current commercial budesonide formulations are capsules that among other excipients contain gelatin and/or Eudragit acrylic polymers. Budesonide microsphere dispersions have been prepared for inhalation products,50 whereas nanoparticles formed from mesoporous silica, poly(lactic-co-glycolic) acid, and acrylic resins have been prepared for oral dosing.38,43,51 However, information is lacking regarding the possible use of LNPs in this application.
This work was thus ultimately aiming at understanding the mechanism of formation of LNPs in a biocompatible system and exploiting this insight for the development of efficient entrapment of budesonide into LNPs suitable for a pH-triggered release protocol. The first step of the study consisted of careful selection of the lignin source in order to use a starting biopolymer containing significant amounts of carboxylic acid groups, which dissociate in the pH region of intestinal fluid and thus expectedly facilitate the release of actives. More specifically, wheat straw soda lignin was selected as the carrier biopolymer, and aqueous ethanol was used as a biocompatible solvent system for the preparation of LNPs. Among the commercially available lignins, wheat straw soda lignin was considered most suitable with respect to the above-mentioned criteria. It contains 3.9 wt % of carbohydrates (mainly arabinoglucuronoxylan, analyzed after 4% sulfuric acid hydrolysis with HPLC) which were expectedly separated as insoluble residues from the 70% ethanol-soluble fraction. Carboxylic groups should provide control over dissolution of budesonide from the LNPs. The carboxylic acid content of the soda lignin (1.18 mmol/g) is more than twice the COOH content of an industrial softwood Kraft lignin.28 Besides proving to be an efficient method for entrapping budesonide for extended pH-dependent release, the formation of LNPs was understood as a result of a kinetically controlled nucleation–growth self-assembly mechanism.
Results
Controlling Lignin Nanoparticle Preparation in Aqueous Ethanol
Preparation of LNPs commonly employs aqueous organic solvents such as tetrahydrofuran, dimethyl sulfoxide, and acetone. For biomedical applications, utilization of biocompatible solvents is a prerequisite, as any solvent residues may impart safety of the product. We selected ethanol as the solvent and water as the nonsolvent for wheat straw soda lignin and determined the important process conditions under which spherical LNPs are formed. The solubility screening of soda lignin provided initial hints of the regions in which lignin undergoes self-assembly. The highest solubility of lignin (87%) was obtained in 7:3 ethanol–water v/v solvent mixture that was selected for this work (Figure S-1 in the Supporting Information).
The initial lignin concentration must be carefully selected to obtain colloidal LNPs. Aggregation of particles beyond 3 g L–1 lignin concentrations was observed from SEM images (Figure S-2) and DLS analysis that indicated a linearly increasing average particle diameter within the concentration range 1–20 g L–1, while surface charge of the particles showed a parabolic trend (Figure 1a). Adding water by means of a syringe pump into 3 g L–1 lignin solution in 70% ethanol changed the appearance of the dispersions from transparent to opaque, when the resulting volume fraction of ethanol decreased from 62% to 13% (Figure 1b). The dilution rate exhibited another important effect on particle size, when other factors were maintained constant (initial lignin concentration 3 g L–1, final ethanol concentration 13%). There was a distinct pivot point at a dilution rate of 0.15 v v–1 min–1, above which the particle diameter increased sharply (Figure 1c). In accordance with the SEM image analysis (mean particle diameter 271 ± 110 nm, N = 160), DLS-based particle size of 312 nm was obtained at the dilution rate of 0.06 v v–1 min–1.
Figure 1.

Development of colloidal lignin particles from aqueous ethanol solution. (a) Effect of initial lignin concentration on particle size and surface charge (ζ-potential) of lignin dispersions formed by adding distilled water at a dilution rate of 0.06 v v–1 min–1. (b) Appearance of dispersions diluted at a rate of 0.06 v v–1 min–1 (0.03 mL min–1) from 3 g L–1 initial lignin concentrations in 70% ethanol solution to gradually decreasing final ethanol and lignin concentrations. (c) Effect of dilution rate on particle size of LNPs. Initial 3 g L–1 lignin solution in 7:3 ethanol–water (v/v) was diluted with distilled water using a syringe pump. (d) SEM images show appearance of the dispersions produced at dilution rates of 0.03 (d1–2), 1.16 (d3–4), and >300 v v–1 min–1 (d5–6, pipetting water into lignin solution).
Properties of LNPs
Stability of LNPs is important regarding not only the scale-up and processability of the active formulation, but also due to the contrastive acidity across the human GI tract. Surface charge, conveniently measured as ζ-potential, predominantly influences colloidal stability of nano- or microparticles. LNPs exhibited decreasing ζ-potentials as the pH increased (Figure 2a). At neutral pH 7, LNPs, more precisely the numerous carboxylic acid groups on their surfaces, exhibited a ζ-potential of −43 mV, similar to the values of LNPs prepared from Kraft lignin in aqueous THF.28 Colloidal stability was also assessed by adjusting dispersions at pH 2 through pH 10 with dilute hydrochloric acid or sodium hydroxide and measuring particle sizes after 4 h standing of the dispersions at room temperature. Particle sizes of LNPs decreased only slightly when pH increased to 10, but aggregation occurred at pH 2.1, which is close to the isoelectric point (Figure 2b).
Figure 2.
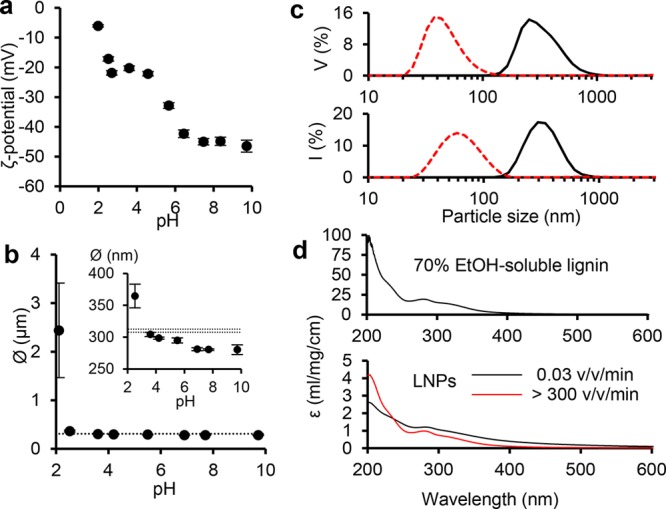
Surface charge and stability of LNPs. (a) ζ-potentials of LNPs as a function of pH. (b) Particle diameters of LNPs at the native pH 4.9 (dashed horizontal lines) and after 4 h holding at room temperature of dispersions adjusted to pH 2–4 (dilute HCl) and pH 5–10 (dilute NaOH). (c) Volume (V) and intensity (I) distributions of particle sizes of LNPs produced at a dilution rate 0.06 v v–1 min–1 (black solid line) and by rapid pouring of water to lignin solution (dilution rate >300 v v–1 min–1, red dashed line). (d) UV–vis mass extinction coefficients of soluble lignin, and LNPs produced at slow and high dilution rates.
Intrigued by the possibility to control the particle size of LNPs, we prepared another sample by rapidly pouring water into magnetically stirred lignin solution. As a result, a clear transparent dispersion with mean particle size of 67 ± 1 nm was obtained (Figure 1c). UV–vis spectra of LNPs produced at contrasting dilution rates of 0.03 v v–1 min–1 and approximately 300 v v–1 min–1 expectedly showed higher visible light absorption by the larger particles, whereas the small particle exhibited rather similar spectrum compared to the starting lignin solution in 70% ethanol. The significantly lower mass extinction coefficients of LNPs compared to the values of soluble lignin evidenced, however, that the majority of lignin precipitated regardless of the dilution rate. The correlation of particle size and dilution rate was confirmed by SEM imaging (Figure 1d).
Mechanism of LNP Self-Assembly and Nanoprecipitation
The observed effects of ethanol concentration and dilution rate on the formation of LNPs indicate kinetically controlled self-assembly, which is postulated to involve electronic interactions between aromatic moieties. If this reasoning is correct, then large lignin molecules ought to precipitate at initial stages of water addition, as their solubility decreases. To assess this, we determined molecular weight distributions of soluble and insoluble lignin fractions generated at different ethanol concentrations. At 62% ethanol concentration, no solid fraction sedimented from the dispersion upon centrifugation at 12000 rpm because of the small size of the colloidal particles. An important molecular weight-dependent fractionation was observed at lower ethanol concentrations. Compared to the molecular weight distribution of the starting lignin soluble in 70% ethanol (dashed line, Figure 3), the insoluble LNPs consisted of systematically larger lignin fragments. There was a marked shift toward the polymeric region at 45% ethanol concentration, while precipitation of gradually smaller molecules occurred thereafter. The soluble fractions exhibited an opposite trend, with increasing enrichment of low molecular weight fragments as the ethanol concentration decreased.
Figure 3.
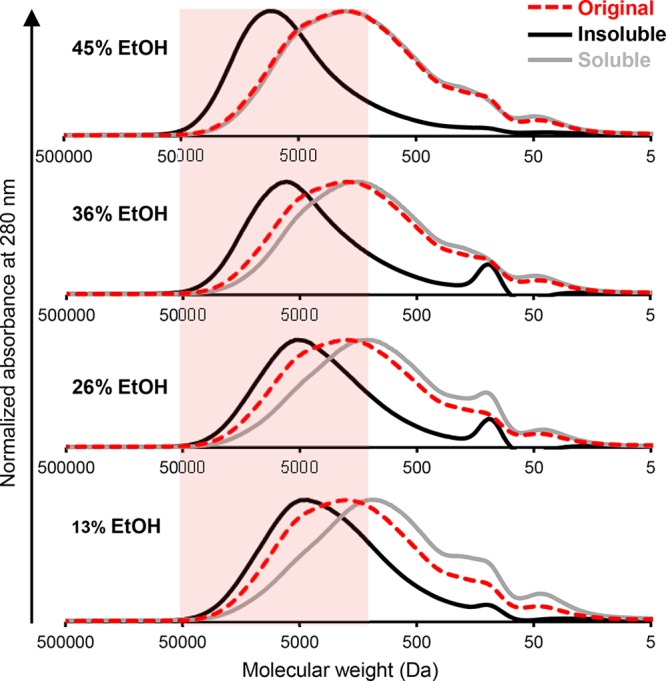
Fractionation of lignin during LNP formation into insoluble (black line) and soluble (gray line) fractions from the starting solution in 70% ethanol (red dashed line). GPC chromatograms show normalized absorbance at 280 nm.
The accumulated insight on the LNP formation allowed us to deduce a mechanism for the self-assembly of lignin in ethanol–water solvent mixture (Figure 4a). As could be anticipated from the distinct visual appearance of LNPs (Figure 1b), clear differences were observed in the particle size at different ethanol concentrations (Figure 4b–c). Upon addition of water into lignin solution, the particle formation initiates by molecular weight-dependent precipitation of lignin, from high to low Mw. Small, approximately 40–70 nm, nuclei prevailed at ethanol concentrations from 62% to 36% (Figure 4b). At ethanol concentrations of 45%, their mean diameter was 65 ± 16 nm (SEM image analysis, N = 15). As shown by the GPC results (Figure 3), these nuclei formed from large molecules and further grew by aggregation of these intermediate particles. At 36% ethanol concentration, SEM images revealed crumpled particles with incompletely fused small particles at their surfaces (Figure 4c). Such a tendency of LNPs to agglomerate was prevalent especially at higher lignin concentrations that did not result in stable colloidal dispersions (Figure S-2). Once the ethanol concentration decreased to 26%, adsorption of smaller molecules rendered the particles gradually smoother and the small nuclei were no longer observed. While the majority of the LNPs were spherical (Figure 1d), irregular droplet-shaped particles occasionally formed due to the incomplete fusing of the particles. Finally, at 13% ethanol concentration, there were few small particles (<50 nm), with the majority of particles in the size range of 250–350 nm (Figures 2b and S-2). Other analyses, such as the determination of surface area and porosity have not been obtained at this stage. Variations in surface characteristics or pore size distributions as a function of the production protocols will be studied in due course as part of ongoing research.
Figure 4.

Lignin nanoparticle formation in ethanol–water system. (a) Schematic mechanism of slow self-assembly and rapid nanoprecipitation. Collision of the soft nuclei of 10–50 nm and their fusing with larger particles lead to the growth of crumpled nanoparticle spheres. Spherical colloidal lignin particles form upon adsorption of low molecular weight and more polar lignin fragments on the preformed particles when the ethanol concentration falls below 30%. (b) SEM images of LNP dispersions (dilution rate 0.06 v v–1 min–1) at varying ethanol concentrations. (c) SEM image of crinkly particles in the LNP dispersion at 36% ethanol concentration. Scale bars = 500 nm.
Budesonide Entrapment in LNPs
Entrapment of budesonide in LNPs was studied under the optimized self-assembly conditions and compared to the nanoprecipitation approach. The first obvious task was to determine the weight proportion of budesonide that can be entrapped in LNPs. Budesonide crystallized from a 7:3 v/v ethanol–water solution when water was added by means of a syringe pump in the absence of lignin (Figure S-3). Some crystals and aggregated LNPs were observed at 50% budesonide weight fraction (relative to total weight of budesonide-LNPs), while at 25% only a few aggregates were present. The 10% budesonide concentration was used as a loading ratio hereafter.
Entrapment efficiencies (EE) of 34–37% were determined from the particles washed by repeated centrifugal filtration (30 kDa membranes) in water dispersions (Figure 5). Importantly, similar EE values were obtained regardless of the contrastive LNP particle diameters of 312 and 67 nm resulting from the regular self-assembly and the nanoprecipitation approach, respectively. These values mean that the oral dosing of approximately 250 mg of budesonide–LNPs suffices to meet the required daily dosing of 9 mg, encouraging further development of LNP-based controlled release products.
Figure 5.
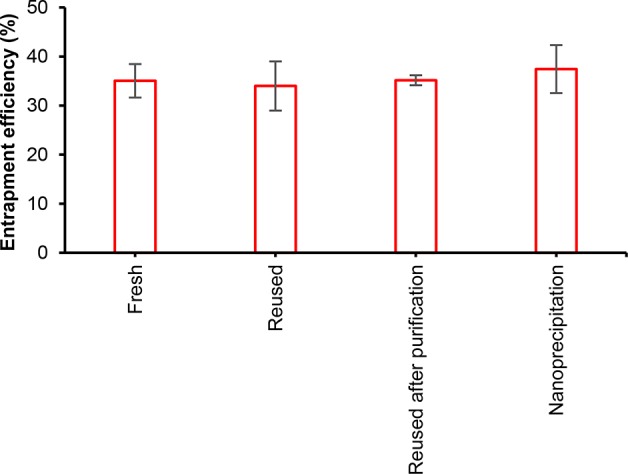
Entrapment efficiencies (EE) of budesonide in LNPs. Comparison of EE when using fresh budesonide solution (mean ± SD, N = 4), reused budesonide from permeate obtained in the purification step as such (mean ± absolute deviation, N = 2), or after prior treatment with an ion-exchange resin that adsorbed lignin (mean ± absolute deviation, N = 2). Nanoprecipitation consisted of rapid pouring of water into the solution mixture of fresh budesonide and lignin (mean ± SD, N = 4). SD, standard deviation.
Comparison of the essentially similar EE values achieved using contrastive self-assembly and nanoprecipitation methods suggest that budesonide entrapment was a matrix-type process, since the increased surface area of the smaller LNPs did not influence the EE. The permeate fractions of two repeated entrapment experiments contained 67 ± 1% of the initial amount of budesonide, signifying a mass balance closure of 97%. Regarding the anticipated repeated reuse of budesonide from the permeate fraction, it would be eventually required to reduce the associated soluble lignin concentration. As a possible solution, we demonstrated that it is possible to reduce the lignin concentration by 65% by adsorption on Amberlite IRA-900 Cl ion-exchange resin (Figure S-4). The resin-purified budesonide fraction exhibited a similar EE compared to the use of fresh budesonide (Figure 5). The overall mass recovery yield of purified LNPs (58 ± 1% N = 2) was not affected by the incorporation of budesonide (51 ± 7% N = 5). It is noted that mass recovery yields based on lignin quantification by UV−vis spectrophotometry (78 ± 7%, N = 3) were higher than that obtained by weighing the lyophilized materials. For the active release study, the larger LNPs from the self-assembly process were selected due to the ease of the liquid–solid separation for HPLC analysis.
Triggered Release of Budesonide under Simulated Physiological Conditions
Budesonide release was determined at 37 °C under acidity conditions simulating pH of the stomach, small intestine, and colon. The active release involved disintegration of LNPs, and hence dissolution of lignin, which challenged HPLC analysis of budesonide. The method developed to overcome overlapping budesonide and lignin peaks consisted in using an alkaline mobile phase, which led to a rapid elution of lignin due to its charged groups and higher solubility in the aqueous eluent compared to that of budesonide (Figure S-5a). Another important point that needed attention was the aqueous solubility of budesonide. To ensure that the active release rate from LNPs was not solubility-limited, the theoretical maximum concentration was set at 3.6 mg L–1, which was within the range of linear dissolution kinetics regardless of different pH values (Figure S-5b).
The release rate of budesonide from the LNPs increased with increasing pH, approaching the dissolution rate of the pure drug at pH 7.4 under saturation conditions (Figure 6a). The released proportion plateaued in 3 h at 83% at pH 7.4 and remained at this level until 25 h. In contrast, particles incubated at pH 2 and pH 5.5 exhibited much lower 25 h extents of release of 57% and 74%, respectively. The particles aggregated at pH 2 (Figure S-6, Figure 2b), which is beneficial since it reduces the available surface area and reduced the diffusion rate of budesonide from the particles under acidic conditions. The release half time (t1/2) were 10 h, 90 min, and 5 min at pH 2, 5.5, and 7.4. The release kinetics are favorable if compared to the acidity across the GI tract. The short residence time of less than 2 h in the stomach expectedly retains the majority of budesonide until the small intestine in which alkaline pancreatic fluid triggers the release by increasing pH and additionally by providing surface-active lecithin.
Figure 6.
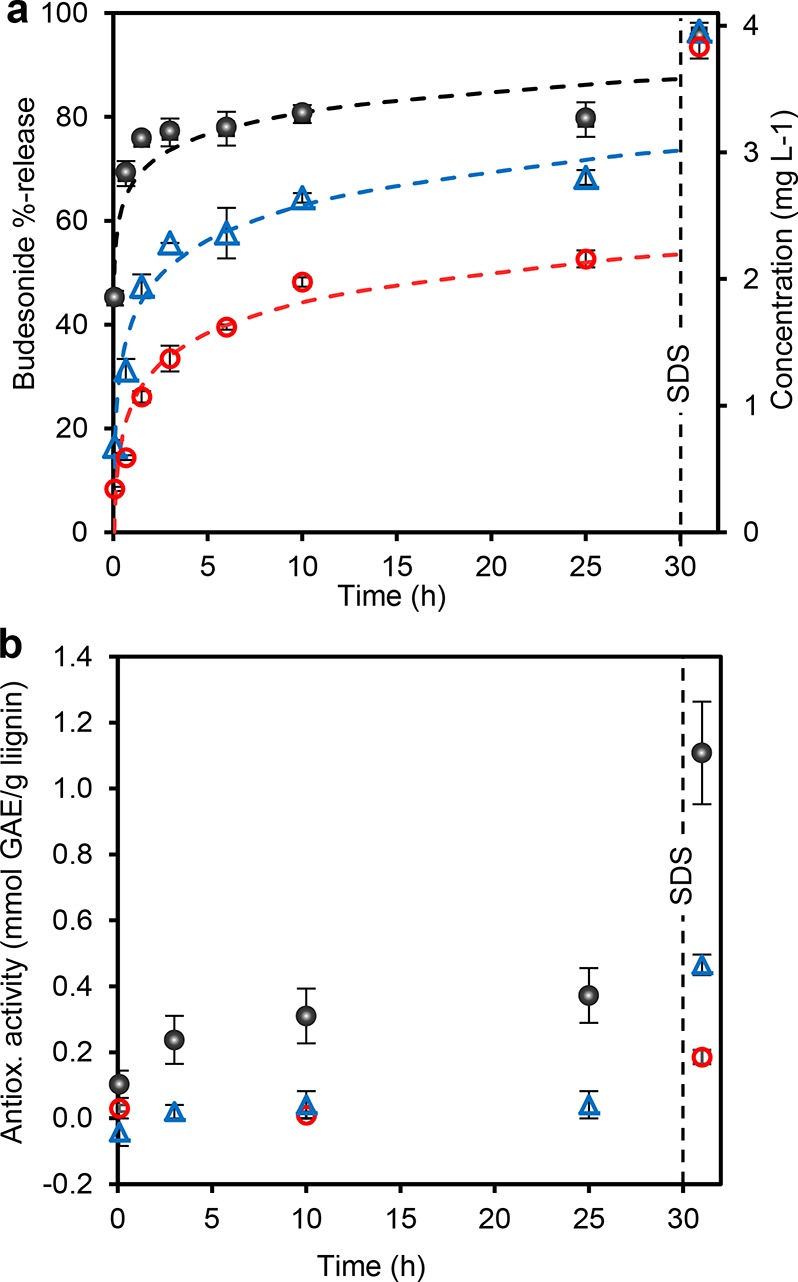
Active release from lignin nanoparticles. (a) Release kinetics of entrapped budesonide from LNPs at pH 2 (0.05 M HCl–KCl), 5.5 (0.05 M sodium phosphate buffer), and 7.4 (0.05 M sodium phosphate buffer). Mean values ± average deviations of two replicate experiments are shown. (b) Released antioxidant activity (as gallic acid equivalents, GAE). The vertical dashed lines indicates the point of SDS supplementation (20 g L–1 in the dispersion). Dashed curves are logarithmic fits extrapolated to 30 h. Error bars indicate absolute deviations from the mean.
The effect of surface-active substances on the release of the active cargo was tested by supplementing SDS in the dispersions at the time point of 30 h at which the active was incompletely released. Regardless of the pH, budesonide was released within 1 h from the SDS supplementation that disintegrated the particles (Figure S-5c). At pH 7.4, the released percentage of the active after the SDS trigger was 10%, while at pH 2 it was 40% (Figure 6a).
Dissolution of lignin occurred in parallel with the release of budesonide. The antioxidant activity (AA) profiles measured from the supernatants revealed kinetically increasing AA at pH 7.4, whereas at pH 5.5 there was only a slight increasing trend. Unaltered AA levels at pH 2 resulted from the low solubility of lignin under acidic conditions (Figure 6b). Again, SDS served as a trigger, but in this case, the final AA levels correlated with the pH. The value of 1.11 mmol GAE g–1 lignin at pH 7.4 is five times as high as the value of LNPs (0.24 ± 0.00, N = 3) formed when precipitating lignin in the ABTS•+ radical cation solution. This difference is due to the dissolution of the majority of LNPs in the presence of SDS (Figure S-5c). Without SDS, the mean diameter of the particles after 25 h stirring at pH 7.4 was 264 ± 70 nm (SEM image analysis, N = 683) that is only slightly lower than the particle size of LNPs without budesonide (312 nm by DLS). Therefore, the majority of budesonide was released regardless of the incomplete disintegration of the particles. In addition to particles that appeared intact, SEM images showed disintegrated LNP debris (Figure S-5c). Such material may have arisen from the dissolution of surface adsorbed charged lignin fragments that increased porosity of LNPs and liberated budesonide that was observed from dried release dispersions as recrystallized flakes and small NPs with a diameter of 50 ± 7 nm (N = 29).
Discussion
There are two important contributions from this work. The first finding relates to the mechanism of lignin nanoparticle formation. We studied the effects of lignin concentration, dilution rate, and nonsolvent concentration on LNP properties and molecular weight fractionation. Consequently, we propose a novel mechanism of lignin self-assembly in aqueous ethanol solution involving a nucleation–growth process that has occupied colloid scientists for a long time.52
LNP formation via a nucleation–growth route has been considered earlier,19 but our results show that the nucleation initiates by precipitation of the large lignin fragments as precursors for the critical nuclei. The growth proceeds via collision-driven aggregation and fusing of the particles into gradually larger crinkly intermediate particles. The formation completes by two parallel events: orientation of the hydrophilic segments toward the particle surfaces and adsorption of small polar lignin fragments on the existing particles. Our GPC and SEM analyses provide indirect support for the final adsorption step. This clarifies the mystique that has prevailed around LNP formation in aqueous organic media.
There are fragments of prior data that support our conclusions, but a lucid synthesis of the LNP formation mechanism has been lacking. We showed that molecular weight governs the initiation of lignin precipitation when ethanol concentration decreases. A similar trend of molecular weight fractionation was previously observed from the precipitation of aqueous ethanol solutions of Kraft lignin by adding water,53 but that work did not study particle formation. The inverse nonlinear correlation between the dilution rate and particle diameter when adding water into a solution of lignin in organic solvents has been shown before,54,55 but the important correlation to molecular weight of lignin has been missing. Xiong et al. proposed that LNPs form by layer-by-layer (LbL) mechanism from THF upon adding water as nonsolvent.56 It is possible that adsorption-based particle growth occurred in parallel with particle–particle combination in our case of aqueous ethanol. However, SEM data suggest that adsorption plays a more significant role in the finalization stage of the LNP formation. We decided to use SEM imaging to observe directly particle morphologies at various ethanol concentrations, because DLS can give ambiguous data from colloidally unstable systems such as incompletely formed LNPs.
SEM images of LNPs (Figure 4b and c) exhibit strikingly similar patterns of particle ripening at decreasing ethanol concentration despite the different lignin sources and coprecipitation of electrostatic complexes of cationic Kraft lignin and sodium dodecyl benzenesulfonate.27 The formation of complex micelles was ascribed to the self-assembly via electronic interactions between aromatic moieties, which drives the LNP formation in THF using water as a nonsolvent,19,56 and are reasoned to be central also in the present work. Li et al.27 indicated that the complex micelles grow by aggregation of small particles, and suggested that the process completes by the collapse of the loosely associate hydrophobic segments to the micelle core. While minimization of surface energy is essential for the LNP formation as well, our results revealed the important role of molecular weight and collision-driven growth of the nuclei. Future work should assess whether this mechanism can be generalized to LNP formation in other solvent–nonsolvent systems.
The second significant contribution from this work is the developed one-pot process for the facile entrapment of budesonide in LNPs, purification of the particles, and reuse of the active. The release of the active cargo was demonstrated by pH and surfactant triggers. The importance of matching the lignin source to the target applications is frequently undervalued. We used sulfur-free wheat straw soda lignin without chemical modifications. Instead of using cytotoxic methanol in LNP preparation,33 the demonstrated use of ethanol is another advantage of the current work. Purification of the nanoscale particles from the solvent and the remaining soluble actives is equally important, yet often neglected. We demonstrated that centrifugation filtration is a convenient method to remove practically all of the ethanol and budesonide for reuse. This is important because, in addition to the processing costs, it is important to develop a closed cycle for the entrapment of expensive pharmaceutical actives.
The LNP concentration that remains colloidal when preparing particles from aqueous ethanol solution is slightly higher compared to that of water-to-ethanol precipitation of Kraft lignin55 but lower compared to the concentrations of dispersions obtained from aqueous THF solutions using water as a nonsolvent.28,54 However, neither the low LNP concentration nor the moderate loading percentage represents bottlenecks for this technology, since the daily dosing of budesonide is only 9 mg. This illustrates the importance of finding matching application for the LNPs.
The release rate of budesonide from LNPs increased with increasing pH. This is explained by the presence of ionizable carboxylic acid (pKa 2–5) and phenolic hydroxyl (pKa 7–11) groups57 in soda lignin. At pH 2, only 27% of budesonide was released in 2 h. This is slightly more compared to the release of 20% budesonide from NPs formed from Eudragit acrylic resins43 and significantly less compared to the burst release of 75% of loaded budesonide from PLGA nanoparticles at pH 1.2 in 2 h.38 These works employed an emulsion solvent evaporation method for nanoparticle preparation and coating, which used ethyl acetate, acetone, and ethanol as solvents, and is more complicated than our one-pot coprecipitation approach. However, Eudragit S100-coating of budesonide-loaded PLGA NPs reduced the extent of release to 18% after 2 h in pH 1.2. Coating might enable tailoring of the sustained release properties of budesonide-loaded LNPs. The compression coating method using Albizia procera gum could be a starting point.44
The burst release of budesonide was faster at pH 7.4 compared to the release from mixed Eudragit polymer NPs, but almost similar compared to the release from NPs consisting of pH-responsive polymers only.43 The excellent stability of the budesonide–LNPs at pH 7.4 for up to 48 h (Figure S-5c) suggests that ionization of the carboxylic groups is not sufficient to disintegrate the aggregated lignin molecules. Compared to the dissolution rates observed under acidic conditions, swelling of LNPs and desorption of their outermost lignin layers may have facilitated release of budesonide. Consequently, 83% of budesonide was released after 10 h stirring in the release medium. Previously, entrapment of budesonide in cellulose acetate butyrate led to complete release within 7 h at pH 7.5.37 Embedding the microparticles in Eudragit S polymer matrix reduced the dissolution rate, but in this case only 70% was released after 24 h. LNPs disintegrated more extensively in the presence of SDS that expectedly weakened intermolecular association forces. All of these data indirectly support our findings on the LNP formation.
Among lignin materials, the dissolution of ibuprofen from cationic lignin-sodium dodecylbenzenesulfonate micelles27 was slower compared to our results with budesonide, which can be due to the electrostatic interactions between the quaternary ammonium groups and carboxylate groups of ibuprofen. Complete release of benzazulene and sorafenib, two poorly water-soluble hydrophobic drugs, was observed from LNPs at pH 7.4 after 2 and 6 h, respectively.23 These authors reported elevated solubility rates in the presence of lignin, which was detected in the present work with budesonide (Figure S-7). It appears, therefore, that facilitated codissolution of actives may be a more general intrinsic property of amphiphilic lignins.
Conclusions
We elucidated the formation mechanism to enable preparation of lignin nanoparticles with controlled properties for the entrapment and release of actives. The practical benefit of this new knowledge is the control over size and shape of the particles, as well as insight into the entrapment of actives in the course of molecular weight-dependent precipitation of lignin fragments. Budesonide was entrapped in LNPs and its pH- and surfactant-dependent release was demonstrated. Under physiological conditions, secretion of alkaline pancreatic juice that increases pH in the small intestine between pH 7–8 is a likely trigger for the release of the active. Overall, our results demonstrate that LNPs provide a feasible platform for the entrapment and controlled the release of actives, especially those with low required dosing such as numerous steroids drugs. From the point of view of lignin valorization, biomedical applications will involve limited amounts. However, by increasing knowledge on the possible controlled release systems, it may be possible to transfer some of the developed technologies to high-volume applications in other fields of sustainable chemistry, e.g. in agriculture and food.
Experimental Section
Materials
Wheat straw soda lignin was obtained from GreenValue SA (Switzerland). Size-exclusion chromatography indicated that this soda lignin has a weight-average molecular weight of 2820 g mol–1,58 while 31P nuclear magnetic resonance (NMR) spectroscopy analysis59,60 showed the following functionalities (in mmol g–1): COOH 1.18, phenolic OH 2.55, aliphatic OH 1.92.61 Pharmaceutical grade budesonide was purchased from Sigma-Aldrich. Anhydrous ethanol and all reagent grade chemicals were used as such, if not stated otherwise.
Colloidal Lignin Nanoparticle Preparation
All experiments were made using wheat straw soda lignin dissolved in ethanol–water 7:3 v/v solvent mixture that was found to maximize the solubility of this lignin (Figure S-1). Development of LNP formation procedure consisted of diluting a magnetically stirred (500 rpm) lignin solution in ethanol–water 7:3 v/v solvent mixture by adding distilled water dropwise using a syringe pump (Model BSP 99, Braintree Scientific, Inc.). The initial lignin concentration, dilution rate, and final volume fraction of ethanol were varied. Alternatively, a “nanoprecipitation” was done by pouring water rapidly into a magnetically stirred lignin solution.
Budesonide Entrapment in LNPs
A typical procedure for active entrapment in LNPs consisted of adding 35 mL of distilled water as nonsolvent at a dilution rate of 0.06 v v–1 min–1 to a 10 mL solution of lignin (3 g L–1) and budesonide (0.33 g L–1) in ethanol–water 7:3 v/v. Stirring rate was maintained at 500 rpm at room temperature. Another approach employed the nanoprecipitation with distilled water as nonsolvent of the solution mixture of lignin and budesonide in ethanol–water 7:3 v/v. The final ethanol concentration was 16% in both of the procedures. The colloidal dispersions were purified and concentrated by repeated 5–10 min centrifugal utltrafiltration using 30 kDa Amicon Ultra-15 membranes (Millipore). Entrapment efficiency was determined by extracting 3 mg of freeze-dried budesonide-LNPs with 1 mL of ethanol under sonication during 1 h, followed by 5 h rotation mixing at room temperature to ensure complete release of the active.
Ion-Exchange Resin Purification of Budesonide for Reuse
The permeate fraction from the washing step of budesonide-LNPs was evaporated to a small volume under reduced pressure and dissolved in ethanol–water 7:3 v/v. Ten milliliters of this solution was rotated with 5 g of Amberlite IRA-900 Cl ion-exchange resin during 4 h at room temperature. The resin was separated from the liquid phase by filtration and washed with ethanol–water 7:3 v/v and distilled water. The liquid fractions were combined, evaporated to a small volume under reduced pressure, and dissolved in ethanol–water 7:3 v/v. After analysis of budesonide and lignin concentrations, this purified fraction was used in the entrapment of budesonide in LNPs.
Particle Analysis
A Gemini Supra 35 Leo Scanning Electron Microscope (SEM) was used to record images of the particle dispersions. Samples deposited on aluminum surfaces were sputtered with gold (Emitech K550X) and imaged at an acceleration voltage of 5 kV using a secondary electron detector. ImageJ software was used to analyze particle diameter from the SEM images. Dynamic light scattering (DLS) analysis of particle sizes and surface charges of LNPs and budesonide–LNPs utilized a Malvern Zetasizer Nano-ZS90 instrument. A dip cell probe was used in ζ-potential measurements. Mean values of at least three measurements of Z-average particle diameters and ζ-potentials are reported.
Budesonide Release Study
The release of entrapped budesonide from LNPs was studied in aqueous 0.05 M buffer solutions to simulate acidity in the stomach (pH 2, HCl–KCl buffer), small intestine (pH 7.4, sodium phosphate buffer), and colon (pH 5.5, sodium phosphate buffer). One milliliter of purified budesonide–LNPs was stirred with 9 mL of the buffer solution at 150 rpm in a thermostatic water bath at 37 °C. After 30 h, an aliquot was mixed with sodium dodecyl sulfate (SDS, concentration of 20 g L–1 in the dispersion) and incubated separately at 37 °C. Homogeneous aliquots were taken at various time intervals and centrifuged for 5 min at 12000 rpm to separate particles from the liquid phase that was passed through a 0.45 μm syringe filter. Soluble budesonide was analyzed by HPLC using an YMC-Pack Polymer C18 column (250 mm × 4.6 mm, 6 μm particle size) eluted with a mixture of (A) 1% formic acid in methanol and (B) 0.05 M aq sodium borate buffer (pH 11) in a volume ratio of 75:25. The run time at 0.3 mL min–1 flow rate was 60 min. A photodiode array detector signal at 254 nm was used to detect budesonide at room temperature. The HPLC calibration curves are given in the SI (Figure S-7).
ABTS Antioxidant Assay
Undiluted liquid phases separated from the budesonide release dispersions were subjected to the antioxidant assay62 with a few modifications. The aqueous aliquot, calibration standard, or distilled water blank (10 μL) was mixed with 1 mL with the ABTS•+ radical cation solution at 30 °C. The absorbance at 730 nm was measured exactly one min after mixing of the components using a spectrophotometer with a thermostatic cell container equilibrated at 30 °C. Reduction in the absorbance was calculated relative to the blank value of 0.708. Two replicates from the budesonide release study were measured as single determinations, while triplicate measurements were made for LNPs formed in situ from aqueous ethanol solution of soda lignin. Aqueous gallic acid in the concentration range of 0.15–1.67 mM was used for calibration (Figure S-8). Mean values were calculated and expressed as gallic acid equivalents (GAE) relative to the dry weight of lignin present in the original dispersion, i.e. GAE/lignin (mmol g–1).
Lignin Characterization
Gel permeation chromatography (GPC) analysis of lignin employed a PLgel 5 μm MiniMIX-C column (Agilent, 250 mm × 4.6 mm) eluted at 70 °C with DMSO containing 0.1% lithium chloride. The run time at 0.25 mL min–1 flow rate was 20 min. Molecular weights were calculated from a linear calibration (R2 = 0.999) constructed with poly(styrenesulfonic acid) polymers (4.3–2600 kDa) and tannins (170–941 Da).
Acknowledgments
This work was supported by the Academy of Finland (grant number 296547). The authors thank Paola Gianní for helpful discussions regarding the antioxidant assay.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssuschemeng.8b01652.
Effect of ethanol concentration on solubility of wheat straw soda lignin, effect of initial lignin concentration on aggregation of LNPs, optimization of the loading ratio of budesonide for entrapment in LNPs, UV–vis absorbance spectra, HPLC chromatograms and dissolution kinetics of budesonide released from LNPs, digital photographs of budesonide–LNP dispersions, dissolution of budesonide from the unpurified budesonide–LNPs, HPLC calibration curves for budesonide, calibration curves used in the ABTS antioxidant assay (PDF).
Author Present Address
§ Center for Advanced Materials, Strategic Research Division, Forestry and Forest Products Research Institute, Tsukuba 305-8687, Japan.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ragauskas A. J.; Beckham G. T.; Biddy M. J.; Chandra R.; Chen F.; Davis M. F.; Davison B. H.; Dixon R. a.; Gilna P.; Keller M.; et al. Lignin Valorization: Improving Lignin Processing in the Biorefinery. Science 2014, 344 (6185), 1246843. 10.1126/science.1246843. [DOI] [PubMed] [Google Scholar]
- Lignin: Properties and Materials; Glasser W. G., Sarkanen S., Eds.; American Chemical Society, 1989; Vol. 397. [Google Scholar]
- Chemical Modification, Properties, and Usage of Lignin; Hu T. Q., Ed.; Kluwer Academic/Plenum: New York, 2002. [Google Scholar]
- Lignin: Historical, Biological, and Materials Perspectives; Glasser W. G., Northey R. A., Schultz T. P., Eds.; American Chemical Society: Washington, D.C., 1999. [Google Scholar]
- Vinardell M. P.; Ugartondo V.; Mitjans M. Potential Applications of Antioxidant Lignins from Different Sources. Ind. Crops Prod. 2008, 27 (2), 220–223. 10.1016/j.indcrop.2007.07.011. [DOI] [Google Scholar]
- Catignani G. L.; Carter M. E. Antioxidant Properties of Lignin. J. Food Sci. 1982, 47 (5), 1745–1745. 10.1111/j.1365-2621.1982.tb05029.x. [DOI] [Google Scholar]
- Sipponen M. H.; Rojas O. J.; Pihlajaniemi V.; Lintinen K.; Österberg M. Calcium Chelation of Lignin from Pulping Spent Liquor for Water-Resistant Slow-Release Urea Fertilizer Systems. ACS Sustainable Chem. Eng. 2017, 5 (1), 1054–1061. 10.1021/acssuschemeng.6b02348. [DOI] [Google Scholar]
- Huang S. Y.; Chang H. L.; Goto M. Preparation of Surfactant-Coated Lipase for the Esterification of Geraniol and Acetic Acid in Organic Solvents. Enzyme Microb. Technol. 1998, 22 (7), 552–557. 10.1016/S0141-0229(97)00257-3. [DOI] [Google Scholar]
- Raton B.; New L.; Group F.. Lignin and Lignans Advances in Chemistry; Heitner C., Dimmel D. R., Schmidt J. A., Eds.; CRC Press: New York, 2010. [Google Scholar]
- Buranov A. U.; Mazza G. Lignin in Straw of Herbaceous Crops. Ind. Crops Prod. 2008, 28 (3), 237–259. 10.1016/j.indcrop.2008.03.008. [DOI] [Google Scholar]
- Crestini C.; Lange H.; Sette M.; Argyropoulos D. S. On the Structure of Softwood Kraft Lignin. Green Chem. 2017, 19, 4104–4121. 10.1039/C7GC01812F. [DOI] [Google Scholar]
- Crestini C.; Melone F.; Sette M.; Saladino R. Milled Wood Lignin: A Linear Oligomer. Biomacromolecules 2011, 12 (11), 3928–3935. 10.1021/bm200948r. [DOI] [PubMed] [Google Scholar]
- Crestini C.; Argyropoulos D. S. Structural Analysis of Wheat Straw Lignin by Quantitative 31P and 2D NMR Spectroscopy. The Occurrence of Ester Bonds and α-O-4 Substructures. J. Agric. Food Chem. 1997, 45 (4), 1212–1219. 10.1021/jf960568k. [DOI] [Google Scholar]
- Zeng J.; Tong Z.; Wang L.; Zhu J. Y.; Ingram L. Isolation and Structural Characterization of Sugarcane Bagasse Lignin after Dilute Phosphoric Acid plus Steam Explosion Pretreatment and Its Effect on Cellulose Hydrolysis. Bioresour. Technol. 2014, 154, 274–281. 10.1016/j.biortech.2013.12.072. [DOI] [PubMed] [Google Scholar]
- Lange H.; Schiffels P.; Sette M.; Sevastyanova O.; Crestini C. Fractional Precipitation of Wheat Straw Organosolv Lignin – Macroscopic Properties and Structural Insights. ACS Sustainable Chem. Eng. 2016, 4, 5136. 10.1021/acssuschemeng.6b01475. [DOI] [Google Scholar]
- Zikeli F.; Ters T.; Fackler K.; Srebotnik E.; Li J. Successive and Quantitative Fractionation and Extensive Structural Characterization of Lignin from Wheat Straw. Ind. Crops Prod. 2014, 61, 249–257. 10.1016/j.indcrop.2014.07.013. [DOI] [Google Scholar]
- Ago M.; Tardy B. L.; Wang L.; Guo J.; Khakalo A.; Rojas O. J. Supramolecular Assemblies of Lignin into Nano- and Microparticles. MRS Bull. 2017, 42 (5), 371–378. 10.1557/mrs.2017.88. [DOI] [Google Scholar]
- Deng Y.; Feng X.; Zhou M.; Qian Y.; Yu H.; Qiu X. Investigation of Aggregation and Assembly of Alkali Lignin Using Iodine as a Probe. Biomacromolecules 2011, 12 (4), 1116–1125. 10.1021/bm101449b. [DOI] [PubMed] [Google Scholar]
- Lievonen M.; Valle-Delgado J. J.; Mattinen M.-L.; Hult E.-L.; Lintinen K.; Kostiainen M. a.; Paananen A.; Szilvay G. R.; Setälä H.; Österberg M. Simple Process for Lignin Nanoparticle Preparation. Green Chem. 2016, 18, 1416–1422. 10.1039/C5GC01436K. [DOI] [Google Scholar]
- Norgren M.; Edlund H.; Wågberg L.; Lindström B.; Annergren G. Aggregation of Kraft Lignin Derivatives under Conditions Relevant to the Process, Part I: Phase Behaviour. Colloids Surf., A 2001, 194 (1–3), 85–96. 10.1016/S0927-7757(01)00753-1. [DOI] [Google Scholar]
- Niu N.; Ma Z.; He F.; Li S.; Li J.; Liu S.; Yang P. Preparation of Carbon Dots for Cellular Imaging by the Molecular Aggregation of Cellulolytic Enzyme Lignin. Langmuir 2017, 33 (23), 5786–5795. 10.1021/acs.langmuir.7b00617. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Xiong Z.; Yang D.; Pang Y.; Wang D.; Qiu X. Preparation of Slow Release Nanopesticide Microspheres from Benzoyl Lignin. Holzforschung 2018, 10.1515/hf-2017-0155. [DOI] [Google Scholar]
- Bartzoka E. D.; Lange H.; Thiel K.; Crestini C. Coordination Complexes and One-Step Assembly of Lignin for Versatile Nanocapsule Engineering. ACS Sustainable Chem. Eng. 2016, 4 (10), 5194–5203. 10.1021/acssuschemeng.6b00904. [DOI] [Google Scholar]
- Chen N.; Dempere L. A.; Tong Z. Synthesis of pH-Responsive Lignin-Based Nanocapsules for Controlled Release of Hydrophobic Molecules. ACS Sustainable Chem. Eng. 2016, 4 (10), 5204–5211. 10.1021/acssuschemeng.6b01209. [DOI] [Google Scholar]
- Tortora M.; Cavalieri F.; Mosesso P.; Ciaffardini F.; Melone F.; Crestini C. Ultrasound Driven Assembly of Lignin into Microcapsules for Storage and Delivery of Hydrophobic Molecules. Biomacromolecules 2014, 15 (5), 1634–1643. 10.1021/bm500015j. [DOI] [PubMed] [Google Scholar]
- Yiamsawas D.; Baier G.; Thines E.; Landfester K.; Wurm F. R. Biodegradable Lignin Nanocontainers. RSC Adv. 2014, 4 (23), 11661. 10.1039/C3RA47971D. [DOI] [Google Scholar]
- Li Y.; Qiu X.; Qian Y.; Xiong W.; Yang D. pH-Responsive Lignin-Based Complex Micelles: Preparation, Characterization and Application in Oral Drug Delivery. Chem. Eng. J. 2017, 327, 1176–1183. 10.1016/j.cej.2017.07.022. [DOI] [Google Scholar]
- Sipponen M. H.; Smyth M.; Leskinen T.; Johansson L.-S.; Österberg M. All-Lignin Approach to Prepare Cationic Colloidal Lignin Particles: Stabilization of Durable Pickering Emulsions. Green Chem. 2017, 19, 5831–5840. 10.1039/C7GC02900D. [DOI] [Google Scholar]
- Leskinen T.; Witos J.; Valle Delgado J. J.; Lintinen K. S.; Kostiainen M. A.; Wiedmer S. K.; Österberg M.; Mattinen M.-L. Adsorption of Proteins on Colloidal Lignin Particles for Advanced Biomaterials. Biomacromolecules 2017, 18, 2767–2776. 10.1021/acs.biomac.7b00676. [DOI] [PubMed] [Google Scholar]
- Figueiredo P.; Lintinen K.; Kiriazis A.; Hynninen V.; Liu Z.; Bauleth-Ramos T.; Rahikkala A.; Correia A.; Kohout T.; Sarmento B.; et al. In Vitro Evaluation of Biodegradable Lignin-Based Nanoparticles for Drug Delivery and Enhanced Antiproliferation Effect in Cancer Cells. Biomaterials 2017, 121, 97–108. 10.1016/j.biomaterials.2016.12.034. [DOI] [PubMed] [Google Scholar]
- Figueiredo P.; Ferro C.; Kemell M.; Liu Z.; Kiriazis A.; Lintinen K.; Florindo H. F.; Yli-Kauhaluoma J.; Hirvonen J.; Kostiainen M. A.; et al. Functionalization of Carboxylated Lignin Nanoparticles for Targeted and pH-Responsive Delivery of Anticancer Drugs. Nanomedicine 2017, 12 (21), 2581. 10.2217/nnm-2017-0219. [DOI] [PubMed] [Google Scholar]
- Yiamsawas D.; Beckers S.; Lu H.; Landfester K.; Wurm F. R. Morphology-Controlled Synthesis of Lignin Nanocarriers for Drug Delivery and Carbon-Materials. ACS Biomater. Sci. Eng. 2017, 3 (10), 2375–2383. 10.1021/acsbiomaterials.7b00278. [DOI] [PubMed] [Google Scholar]
- Dai L.; Liu R.; Hu L.; Zou Z.; Si C. Lignin Nanoparticle as a Novel Green Carrier for the Efficient Delivery of Resveratrol. ACS Sustainable Chem. Eng. 2017, 5 (9), 8241–8249. 10.1021/acssuschemeng.7b01903. [DOI] [Google Scholar]
- Cook M. T.; Tzortzis G.; Charalampopoulos D.; Khutoryanskiy V. V. Microencapsulation of Probiotics for Gastrointestinal Delivery. J. Controlled Release 2012, 162 (1), 56–67. 10.1016/j.jconrel.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Leonard F.; Ali H.; Collnot E. M.; Crielaard B. J.; Lammers T.; Storm G.; Lehr C. M. Screening of Budesonide Nanoformulations for Treatment of Inflammatory Bowel Disease in an Inflamed 3D Cell-Culture Model. ALTEX 2012, 29 (3), 275–285. 10.14573/altex.2012.3.275. [DOI] [PubMed] [Google Scholar]
- Kaur G.; Rana V.; Jain S.; Tiwary A. K. Colon Delivery of Budesonide: Evaluation of Chitosan–Chondroitin Sulfate Interpolymer Complex. AAPS PharmSciTech 2010, 11 (1), 36–45. 10.1208/s12249-009-9353-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M.; Antúnez J. a.; Taboada C.; Seijo B.; Torres D. Colon-Specific Delivery of Budesonide from Microencapsulated Cellulosic Cores: Evaluation of the Efficacy against Colonic Inflammation in Rats. J. Pharm. Pharmacol. 2001, 53 (9), 1207–1215. 10.1211/0022357011776658. [DOI] [PubMed] [Google Scholar]
- Ali H.; Weigmann B.; Neurath M. F.; Collnot E. M.; Windbergs M.; Lehr C. M. Budesonide Loaded Nanoparticles with pH-Sensitive Coating for Improved Mucosal Targeting in Mouse Models of Inflammatory Bowel Diseases. J. Controlled Release 2014, 183 (1), 167–177. 10.1016/j.jconrel.2014.03.039. [DOI] [PubMed] [Google Scholar]
- Varshosaz J.; Emami J.; Tavakoli N.; Minaiyan M.; Rahmani N.; Dorkoosh F.; Mahzouni P. Pectin Film Coated Pellets for Colon-Targeted Delivery of Budesonide: In-vitro/In-Vivo Evaluation in Induced Ulcerative Colitis in Rat. Iran. J. Pharm. Res. 2012, 11 (3), 733–745. [PMC free article] [PubMed] [Google Scholar]
- Crcarevska M. S.; Dodov M. G.; Goracinova K. Chitosan Coated Ca-Alginate Microparticles Loaded with Budesonide for Delivery to the Inflamed Colonic Mucosa. Eur. J. Pharm. Biopharm. 2008, 68 (3), 565–578. 10.1016/j.ejpb.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Raval M. K.; Ramani R. V.; Sheth N. R. Formulation and Evaluation of Sustained Release Enteric-Coated Pellets of Budesonide for Intestinal Delivery. Int. J. Pharm. Investig. 2013, 3 (4), 203–211. 10.4103/2230-973X.121294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt H.; Naik B.; Dharamsi A. Solubility Enhancement of Budesonide and Statistical Optimization of Coating Variables for Targeted Drug Delivery. J. Pharm. 2014, 2014, 1–13. 10.1155/2014/262194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J.-W.; Choi M.; Cao J.; Lee Y.; Ikram M.; Yoon S.; Lee J.; Moon H. R.; Kim M. S.; Jung Y.; Naeem M.; et al. Colon-Targeted Delivery of Budesonide Using Dual pH- and Time-Dependent Polymeric Nanoparticles for Colitis Therapy. Drug Des., Dev. Ther. 2015, 9, 3789–3799. 10.2147/DDDT.S88672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachuau L.; Mazumder B. Evaluation of Albizia Procera Gum as Compression Coating Material for Colonic Delivery of Budesonide. Int. J. Biol. Macromol. 2013, 61, 333–339. 10.1016/j.ijbiomac.2013.07.017. [DOI] [PubMed] [Google Scholar]
- Vinardell M. P.; Mitjans M. Lignins and Their Derivatives with Beneficial Effects on Human Health. Int. J. Mol. Sci. 2017, 10.3390/ijms18061219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai D.; Ren W.; Tian L.; Chee P. L.; Liu Y.; Ramakrishna S.; Loh X. J. Engineering Poly(lactide)-Lignin Nanofibers with Antioxidant Activity for Biomedical Application. ACS Sustainable Chem. Eng. 2016, 4 (10), 5268–5276. 10.1021/acssuschemeng.6b00478. [DOI] [Google Scholar]
- Mattinen M. L.; Valle-Delgado J. J.; Leskinen T.; Anttila T.; Riviere G.; Sipponen M.; Paananen A.; Lintinen K.; Kostiainen M.; Österberg M. Enzymatically and Chemically Oxidized Lignin Nanoparticles for Biomaterial Applications. Enzyme Microb. Technol. 2018, 111, 48–56. 10.1016/j.enzmictec.2018.01.005. [DOI] [PubMed] [Google Scholar]
- Thomsen O. Ø.; Cortot A.; Jewell D.; Wright J. P.; Winter T.; Veloso F. T.; Vatn M.; Persson T.; Pettersson E. A Comparison of Budesonide and Mesalamine for Active Chron’s Disease. N. Engl. J. Med. 1998, 339 (6), 370–374. 10.1056/NEJM199808063390603. [DOI] [PubMed] [Google Scholar]
- Travis S. P. L.; Danese S.; Kupcinskas L.; Alexeeva O.; D’Haens G.; Gibson P. R.; Moro L.; Jones R.; Ballard E. D.; Masure J.; et al. Once-Daily Budesonide MMX in Active, Mild-to-Moderate Ulcerative Colitis: Results from the Randomised CORE II Study. Gut 2014, 63 (3), 433–441. 10.1136/gutjnl-2012-304258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruch F.; Matijević E. Preparation of Micrometer Size Budesonide Particles by Precipitation. J. Colloid Interface Sci. 2000, 229 (1), 207–211. 10.1006/jcis.2000.7012. [DOI] [PubMed] [Google Scholar]
- Yoncheva K.; Popova M.; Szegedi A.; Mihaly J.; Tzankov B.; Lambov N.; Konstantinov S.; Tzankova V.; Pessina F.; Valoti M. Functionalized Mesoporous Silica Nanoparticles for Oral Delivery of Budesonide. J. Solid State Chem. 2014, 211, 154–161. 10.1016/j.jssc.2013.12.020. [DOI] [Google Scholar]
- Horn D.; Rieger J. Organic Nanoparticles in the Aqueous Phase - Theory, Experiment, and Use. Angew. Chem., Int. Ed. 2001, 40 (23), 4330–4361. . [DOI] [PubMed] [Google Scholar]
- Jääskeläinen A. S.; Liitiä T.; Mikkelson A.; Tamminen T. Aqueous Organic Solvent Fractionation as Means to Improve Lignin Homogeneity and Purity. Ind. Crops Prod. 2017, 103, 51–58. 10.1016/j.indcrop.2017.03.039. [DOI] [Google Scholar]
- Leskinen T.; Smyth M.; Xiao Y.; Lintinen K.; Mattinen M.; Kostiainen M. A.; Oinas P.; Osterberg M. Scaling Up Production of Colloidal Lignin Particles. Nord. Pulp Pap. Res. J. 2017, 32 (4), 586–596. 10.3183/NPPRJ-2017-32-04-p586-596. [DOI] [Google Scholar]
- Li H.; Deng Y.; Liu B.; Ren Y.; Liang J.; Qian Y.; Qiu X.; Li C.; Zheng D. Preparation of Nanocapsules via the Self-Assembly of Kraft Lignin: A Totally Green Process with Renewable Resources. ACS Sustainable Chem. Eng. 2016, 4 (4), 1946–1953. 10.1021/acssuschemeng.5b01066. [DOI] [Google Scholar]
- Xiong F.; Han Y.; Wang S.; Li G.; Qin T.; Chen Y.; Chu F. Preparation and Formation Mechanism of Size-Controlled Lignin Nanospheres by Self-Assembly. Ind. Crops Prod. 2017, 100, 146–152. 10.1016/j.indcrop.2017.02.025. [DOI] [Google Scholar]
- Ragnar M.; Lindgren C. T.; Nilvebrant N.-O. pKa-Values of Guaiacyl and Syringyl Phenols Related to Lignin. J. Wood Chem. Technol. 2000, 20 (3), 277–305. 10.1080/02773810009349637. [DOI] [Google Scholar]
- Sipponen M. H.Effect of Lignin Structure on Enzymatic Hydrolysis of Plant Residues. Ph.D. dissertation, Aalto University, 2015. [Google Scholar]
- Granata A.; Argyropoulos D. S. 2-Chloro-4,4,5,5-Tetramethyl-1,3,2-Dioxaphospholane, a Reagent for the Accurate Determination of the Uncondensed and Condensed Phenolic Moieties in Lignins. J. Agric. Food Chem. 1995, 43 (6), 1538–1544. 10.1021/jf00054a023. [DOI] [Google Scholar]
- Jiang Z. H.; Argyropoulos D. S.; Granata A. Correlation Analysis of 31P-NMR Chemical Shifts with Substituent Effect of Phenols. Magn. Reson. Chem. 1995, 33, 375. 10.1002/mrc.1260330509. [DOI] [Google Scholar]
- Sipponen M. H.; Pihlajaniemi V.; Littunen K.; Pastinen O.; Laakso S. Determination of Surface-Accessible Acidic Hydroxyls and Surface Area of Lignin by Cationic Dye Adsorption. Bioresour. Technol. 2014, 169, 80–87. 10.1016/j.biortech.2014.06.073. [DOI] [PubMed] [Google Scholar]
- Re R.; Pellegrini N.; Proteggente A.; Pannala A.; Yang M.; Rice-Evans C. Antioxidant Activity Applying an Improved Abts Radical. Free Radical Biol. Med. 1999, 26 (98), 1231–1237. 10.1016/S0891-5849(98)00315-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.