

A novel mTOR complex assembled by the ETS transcription factor ETV7 contributes to rapamycin resistance in cancer.
Abstract
The mechanistic target of rapamycin (mTOR) serine/threonine kinase, a critical regulator of cell proliferation, is frequently deregulated in human cancer. Although rapamycin inhibits the two canonical mTOR complexes, mTORC1 and mTORC2, it often shows minimal benefit as an anticancer drug. This is caused by rapamycin resistance of many different tumors, and we show that a third mTOR complex, mTORC3, contributes to this resistance. The ETS (E26 transformation–specific) transcription factor ETV7 interacts with mTOR in the cytoplasm and assembles mTORC3, which is independent of ETV7’s transcriptional activity. This complex exhibits bimodal mTORC1/2 activity but is devoid of crucial mTORC1/2 components. Many human cancers activate mTORC3 at considerable frequency, and tumor cell lines that lose mTORC3 expression become rapamycin-sensitive. We show mTORC3’s tumorigenicity in a rhabdomyosarcoma mouse model in which transgenic ETV7 expression accelerates tumor onset and promotes tumor penetrance. Discovery of mTORC3 represents an mTOR paradigm shift and identifies a novel target for anticancer drug development.
INTRODUCTION
The mechanistic target of rapamycin (mTOR) is a phosphatidylinositol 3-kinase (PI3K)–related kinase that regulates cell growth through control of ribosome biogenesis, translation of mRNAs, metabolism, cytoskeleton organization, and autophagy [reviewed in (1)]. It is a master effector of many cell signaling pathways and is frequently deregulated in human cancer. Two distinct mTOR-containing complexes have been described, mTORC1 and mTORC2, each showing nonoverlapping kinase activity toward unique protein targets. mTORC1 contains mLST8, Raptor, FKBP-38, Deptor, and PRAS40, while mTORC2 contains mLST8, SIN1, Rictor, Deptor, and Protor/PRR5 or PPR5L. Among many targets, mTORC1 phosphorylates the protein synthesis regulators P70S6K and 4E-BP1 and 4E-BP2 (1) and is associated with lysosomes (1), while mTORC2 phosphorylates AKT (1), protein kinase C family members (PKCα, δ, ε, γ, and ζ) (1), and the serum- and glucocorticoid-induced protein kinase–1 (SGK-1) (1). In addition, mTORC2 also plays a role in translation elongation via interaction with the ribosomal proteins L23 and L26 in translating ribosomes (2, 3). Both complexes are large dimers of mTOR associated with mLST8, Deptor, Raptor or Rictor, and the other complex-specific proteins (4, 5). The macrolide antibiotic rapamycin in complex with FKBP-12 inhibits mTORC1 signaling at nanomolar concentration by preventing substrates from entering the mTOR catalytic cleft (6), whereas long-term treatment (≥24 hours) also inhibits mTORC2 (1). Despite the common activation of mTOR signaling in cancer, clinical application is hampered by rapamycin resistance of many tumors. The development of second-generation mTOR adenosine 5′-triphosphate (ATP)–competitive kinase inhibitors uncovered a rapamycin-resistant mTOR activity (7–9), which has been ascribed to incomplete inhibition of mTORC1 by rapamycin (8, 9). Inhibition of mTORC1 signaling is measured by the reduction in 4E-BP1Thr37/46 phosphorylation levels, which is much more profound with second-generation mTOR inhibitors such as AZD-8055 (7), KU-0063794 (8), or Torin (9) than with rapamycin or its chemical analogs (rapalogs). 4E-BP1 phosphorylation correlates with tumor progression and adverse prognosis in breast, ovarian, and prostate cancer (10). The phosphorylation of 4E-BP1 was shown to control mTORC1-mediated cell proliferation (11). In addition, phosphorylation of 4E-BP1 on Thr37/46 was sufficient to prevent association with eIF4E, leading to cell cycle progression (12). Rapamycin resistance of acute myeloid leukemia (AML) cells also correlated with mTOR-dependent 4E-BP1 phosphorylation. This was maintained after Raptor knockdown (13), potentially pointing to the existence of an mTORC1-independent activity.
ETV7 is a member of the PNT (pointed) domain–containing ETS (E26 transformation–specific) family of transcription factors (14) present in all vertebrates with the exception of a subset of rodents, including the mouse (www.ensembl.org). The ETS domain binds DNA, whereas the PNT domain mediates protein-protein interactions and confers the ability to form homodimers/oligomers and heterodimers with ETV6 (14). ETV7 overexpression is associated with tumorigenic transformation (15, 16). Here, we report the discovery of a rapamycin-resistant mTOR complex, mTORC3, which is assembled upon expression of ETV7 in many human cancers and increases tumor incidence and penetrance in a mouse model of embryonal rhabdomyosarcoma (ERMS).
RESULTS
ETV7 expression is up-regulated in many types of cancer
Given that ETV7 overexpression is associated with tumorigenesis (15–17), we analyzed available St. Jude tumor expression arrays, which showed up-regulated ETV7 expression in 70% of acute lymphoblastic leukemia and AML samples (18, 19) and 48% of pediatric solid tumor xenografts (fig. S1, A to C) (20). Among the latter, expression was lower in Ewing sarcoma and Wilms tumor xenografts compared to other tumor types (fig. S1D). Medulloblastoma expression arrays (21) showed ETV7 up-regulation in 85% of cases (fig. S1E), while a proteomics study identified ETV7 as 1 of the 10 most up-regulated proteins in human hepatocellular carcinoma (22). Analysis of expression array data in Oncomine (www.oncomine.org) showed ETV7 to be among the top 10% up-regulated genes in many cancers (table S1A), thus correlating endogenous ETV7 up-regulation with tumorigenesis.
ETV7 expression alters mTOR signaling
Forced ETV7 expression in mouse precursor B cells (pre-B cells) increases proliferation twofold and inhibits apoptosis (15). Analysis of proliferation pathways identified increased mTOR signaling in ETV7-expressing wild-type (WT) and Arf−/− mouse pre-B cells. Western blots of whole-cell lysates (Fig. 1A) showed increased phosphorylation of direct mTORC1 and mTORC2 targets, including p-P70S6KThr389, p–4E-BP1Thr37/46, p–4E-BP1Ser65, p–4E-BP1Thr70, p-AKTSer473, and p-NDRG1Thr346 [a readout of mTORC2-activated SGK-1 (1)], while total mTOR, P70S6K, AKT, NDRGI, 4E-BP1/2, and eIF4E levels remained constant. Figure 1A also showed that ETV7 expression did not increase the level of phosphoinositide-dependent protein kinase 1 (PDK1), p-PDK1Ser241, or p-AKTThr308 (readouts of PI3K signaling), suggesting that there was no increased receptor signaling. Affymetrix expression analysis showed that increased mTOR activity in Arf−/− pre-B cells was not due to differential transcription of upstream regulatory genes such as PTEN or TSC1/2, while there was a modest up-regulation of a negative regulator of mTOR signaling, that is, the catalytic subunit of AMPK (table S1B). There was also little change in expression of known mTORC1/2 components or associated proteins (table S1B), nor was there gross up-regulation of receptor or nonreceptor tyrosine kinases, growth factors, cytokines, or their receptors (table S1, C and D). Although expression of protein tyrosine kinase 2 (PTK2) was up-regulated threefold, activated p-PTKTyr397, a known activator of PI3K signaling (23), was only slightly higher in Arf−/− ETV7 than in Arf−/− vector pre-B cells and was considerably lower in WT pre-B vector cells (fig. S2A) and therefore unlikely to trigger increased PI3K signaling. In agreement with these results, a phospho-tyrosine (p-tyr) Western blot of whole-cell lysates of vector or ETV7 Arf −/− pre-B cells did not show an obvious difference in overall p-tyr levels (fig. S2B). Together, this suggested that ETV7 did not transcriptionally up-regulate genes that hyperactivate mTORC1/2 signaling pathways. Nonetheless, gene set enrichment analysis using the Hallmark and canonical pathway databases indicated, among others, up-regulation of MYC targets and mTORC1 signaling (table S1E).
Fig. 1. ETV7 induces rapamycin resistance in mouse WT and Arf−/− pre-B cells.
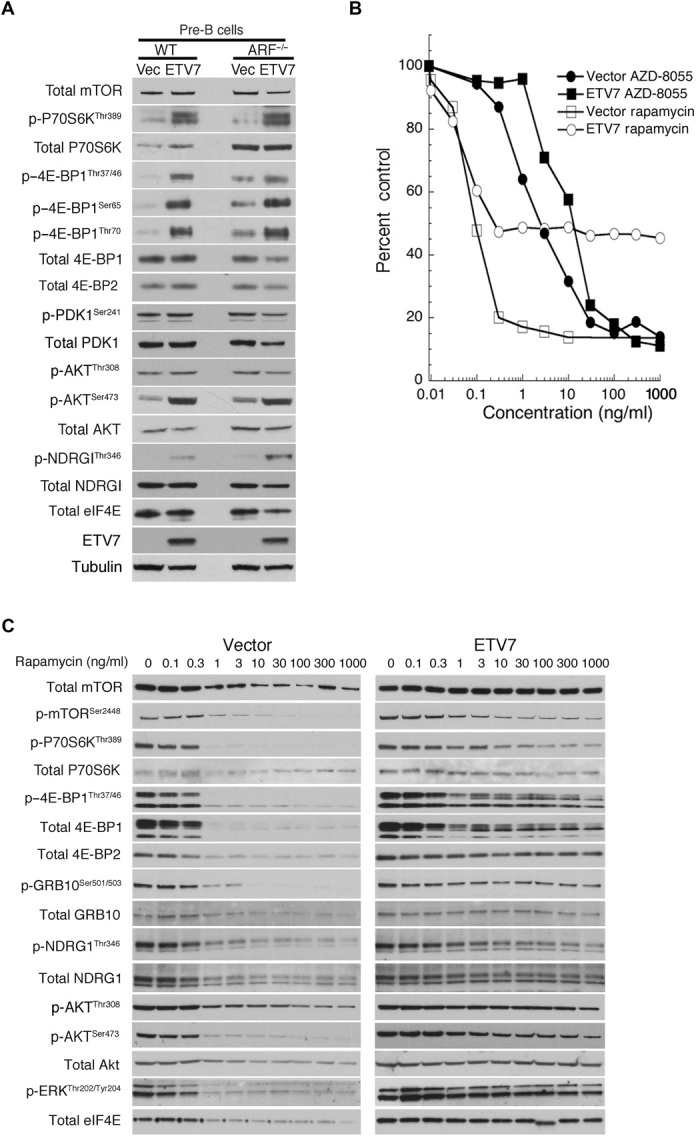
(A) Cell lysates from WT and Arf−/−mouse pre-B cells (ARF−/−) expressing GFP (Vec) or ETV7 (ETV7) immunoblotted for mTOR, p-P70S6KThr389, total P70S6K, p–4E-BP1Thr37/46, p–4E-BP1Ser65, p–4E-BP1Thr70, total 4E-BP1, total 4E-BP2, p-PDK1Ser241, total PDK1, p-AKTThr308, p-AKTSer473, total AKT, p-NDRG1Thr346, total NDRG1, total eIF4E, ETV7, and tubulin as a loading control. (B) Proliferating Arf−/− mouse pre-B cells transduced with murine stem cell virus (MSCV)–internal ribosomal entry site (IRES)–green fluorescent protein (GFP) (vector) or MSCV-ETV7-IRES-GFP (ETV7) were treated with increasing amounts (0.1, 0.3, 1.3, 10, 30, 100, 300, and 1000 ng/ml) of rapamycin or AZD-8055 for three population doublings. Cell densities (percent control) were plotted as the percentage of cells treated with vehicle. Data are means ± SEM from three independent experiments. (C) Cell lysates of proliferating Arf−/− mouse pre-B cells transduced with MSCV-IRES-GFP (vector) or MSCV-ETV7-IRES-GFP (ETV7) were immunoblotted after treatment of the cells with increasing amounts (0.1, 0.3, 1.3, 10, 30, 100, 300, and 1000 ng/ml) of rapamycin for three population doublings. The blots were probed for total mTOR, mTORSer2448, p-P70S6KThr389, total P70S6K, p–4E-BP1Thr37/46, total 4E-BP1, total 4E-BP2, p-GRB10Ser501/503, total GRB-10, p-NDRG1Thr346, total NDRG1, p-AKTThr308, p-AKTSer473, total AKT, p-ERKThr202/Tyr204, and total eIF4E.
ETV7-expressing mouse Arf−/− pre-B cells showed altered sensitivity to treatment with increasing amounts of rapamycin for three population doublings (72 hours). Amounts of drug ≥1 ng/ml completely halted proliferation of Arf−/− vector pre-B cells, while its ETV7-expressing counterpart continued to proliferate at half pace at these concentrations (Fig. 1B), which equaled the proliferation rate of untreated vector-expressing pre-B cells. Western blots (Fig. 1C) of cell lysates of the rapamycin-treated Arf−/− vector pre-B cells showed loss of mTORC1/2 signaling. This was deduced from loss of the following mTORC1/2 phosphorylation markers: p–4E-BP1/2Thr36/47, p-GRB10Ser501/503, p-P70S6KThr389 (see also fig. S10), and its target p-mTORSer2448 (24), p-AKTSer473, and p-NDRG1Thr346. p-AKTThr308, the target of PDK1, was somewhat diminished, likely due to overall reduced protein synthesis, as indicated by the decreased total amounts of mTOR, p70S6K, AKT, NDRG1, and eIF4E. p-ERK1/2Thr202/Tyr204 was also lost, which activates the MNK1/2 kinases (25, 26) that phosphorylate p-eIF4ESer209, which, in turn, is essential for translation of eIF4E-sensitive, that is, 5′ oligopyrimidine–containing, mRNAs (25, 27). The 4E-BP–eIF4E axis in lymphoid cells is uniquely sensitive to rapamycin inhibition (28) owing to the relatively elevated expression of 4E-BP2, whose phosphorylation is sensitive to rapamycin (29). However, after 72 hours of rapamycin treatment of Arf−/− vector cells, 4E-BP1 had mostly disappeared and 4E-BP2 was much reduced, which, together with reduced eIF4E, indicated loss of mTORC1 signaling (Fig. 1C) (30). In contrast, these mTORC1/2 markers remained on in ETV7 Arf−/− cells, even at high rapamycin concentrations, albeit at somewhat decreased levels (Fig. 1C). Maintenance of p-GRB10Ser501/503 phosphorylation and total GRB10 suggested maintenance of mTORC1-like phosphorylation activity (1). Lack of GRB10 accumulation (31) and loss (vector cells) or maintenance (ETV7 cells) of ERK1/2 signaling indicated no up-regulation of the mTOR-PI3K-AKT feedback loop, which activates AKT and other AGC kinases (32). The clear difference in mTOR-dependent phosphorylation between vector and ETV7 pre-B cells at rapamycin concentrations ≥1 ng/ml underscores the profound effect ETV7 has on rapamycin-resistant mTOR signaling.
To determine whether ETV7 affected the composition of mTOR complexes in ETV-7 pre-B cells, we performed mTOR/ETV7 coimmunoprecipitation (co-IP) experiments, followed by immunoblotting using anti-ETV7 and anti-mTOR antibodies on lysates of vector- or ETV7-expressing Arf−/− pre-B cells. On the basis of the co-IP of ETV7 and mTOR, ETV7 interacts with mTOR (Fig. 2A), but auxiliary mTORC1/2 components, including Raptor, Rictor, SIN1, and MLST-8, did not coimmunoprecipitate with anti-ETV7 antibodies, but did so with anti-mTOR antibodies under the same experimental conditions. Identical co-IPs using the endogenous ETV7-expressing human cell lines Karpas-299, K562, and OS-17 produced the same results (Fig. 2B) with again, the absence of mTORC1/2-associated proteins in the anti-ETV7 IPs. Figure 2B also shows co-IP of p–4E-BP1Thr37/46 with anti-ETV7 and anti-mTOR antibodies in these cell lines, in the absence of Raptor in the ETV7 IP. Thus, p–4E-BP1Thr37/46 interaction with ETV7-mTOR seems Raptor-independent, contrary to its Raptor-mediated recruitment to mTORC1 (33). This suggests that ETV7 is associated with a form of mTOR distinct from mTORC1/2. In support of this possibility, we performed IPs of K562 lysates using anti-mTOR (mTORC1+2+ETV7/mTOR), anti-ETV7 (ETV7/mTOR), anti-Raptor (mTORC1), or anti-Rictor (mTORC2) antibodies and probed immunoblots of these IPs with the same antibodies and anti-mSIN1 and anti-mLST8 antibodies (Fig. 2C). This demonstrated that Raptor and Rictor IPs did not contain ETV7, consistent with ETV7 IPs not containing Raptor or Rictor (Fig. 2, A to C). Because ETV7 is associated with mTOR and p–4E-BP1 (Fig. 2B) and enhances mTOR kinase activity (Fig. 1A), we tentatively dubbed this novel form of mTOR, mTORC3. To determine its subcellular localization, we performed anti-mTOR and anti-ETV7 co-IPs of nuclear, cytoplasmic, and membrane fractions of Karpas-299 cells. ETV7 was present in the nucleus and cytoplasm, whereas mTOR was present in all three fractions (Fig. 2D), but only cytoplasmic ETV7 coimmunoprecipitated mTOR and vice versa, thereby localizing mTORC3 to the cytoplasm. ETV7 IPs also demonstrated the presence of mTORC3 in lysates of medulloblastoma (BT28) and glioblastoma (BT39) tumor xenografts (20), as shown by co-IP of mTOR and p–4E-BP1Thr37/46 (fig. S3A). Immunohistochemistry with anti-ETV7 antibody on sections of these xenografts showed ETV7 staining predominantly in the cytoplasm of tumor cells (fig. S3B).
Fig. 2. ETV7 expression induces mTORC3 assembly.
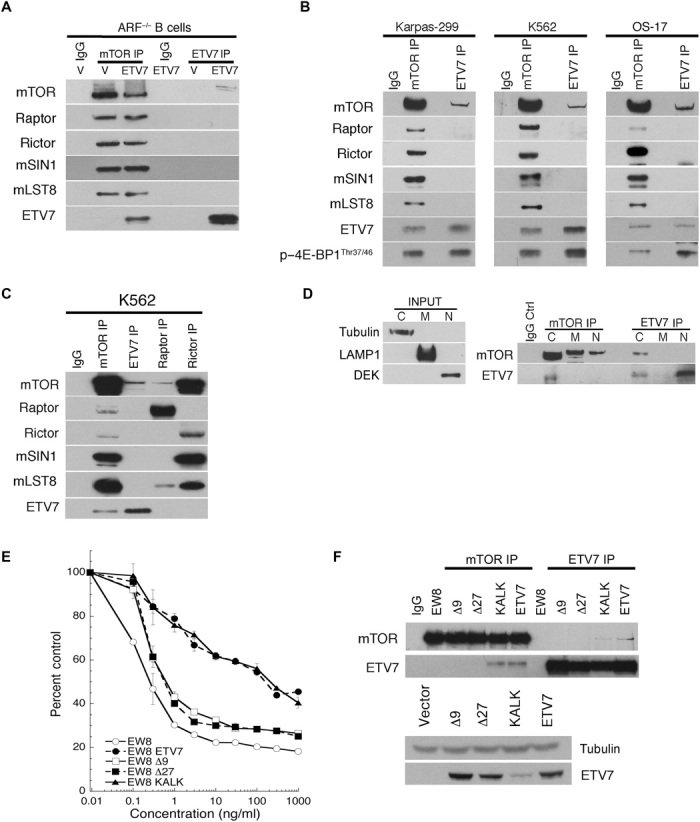
(A) IPs [mTOR IP or ETV7 IP, nonrelevant immunoglobulin G (IgG) IP] of Arf−/− pre-B cells expressing vector (V) or ETV7 (ETV7), immunoblotted for mTOR, Raptor, Rictor, mSIN1, mLST8, and ETV7. (B) IPs of lysates of Karpas-299 cells, K562 cells, or OS-17 cells immunoblotted for mTOR, Raptor, Rictor, mSIN1, mLST8, ETV7, and p–4E-BP1Thr37/46. (C) IgG IP (IgG), mTOR IP, ETV7 IP, Raptor IP, or Rictor IP of K562 cell lysate immunoblotted for mTOR, ETV7, Raptor, Rictor, mSIN1, and mLST8. (D) Left: Immunoblot of fractions probed for tubulin (cytoplasm), LAMP1 (membrane), and DEK (nucleus). Right: IPs (mTOR IP and ETV7 IP) of cytoplasmic (C), membrane (M), and nuclear (N) fractions of Karpas-299 cells immunoblotted for mTOR and ETV7. (E) EW8 cells or EW8 cells transduced with lentiviral vectors encoding ETV7-Δ9, ETV7-Δ27, ETV7-KALK, or ETV7 were treated for three cell doublings (>72 hours) with an increasing concentration of rapamycin (0.1, 0.3, 1.3, 10, 30, 100, 300, and 1000 ng/ml). Cell densities (percent control) were plotted as the percentage of cells treated with vehicle. Data are means ± SEM from three independent experiments. Thirty percent control or less means no proliferation. (F) Top: Lysates of cells in (G) were immunoprecipitated with ETV7 or mTOR antibodies, followed by immunoblotting for ETV7 or mTOR. Bottom: Western blot of lysates of the same cells showing ETV7 levels in EW8 cells expressing vector, ETV7-Δ9, ETV7-Δ27, ETV7-KALK, or ETV7.
To validate the specificity of the ETV7/mTOR interaction, we created two ETV7 PNT domain mutants by deleting sequences, which are involved in dimerization and/or multimerization in the ETV6 PNT domain (34). Because the PNT domains of ETV6 and ETV7 are highly conserved (fig. S4) (14), we reasoned that these mutations might affect ETV7 PNT domain protein-protein interactions. A third ETV7 mutant we tested does not bind DNA due to mutation of two critical arginines (R281 and R284) to lysines in the ETS domain (fig. S4), which directly interact with DNA (35). The three mutants, ΔE81MN83 (Δ9), ΔM82NGRALCIL90 (Δ27), and K281ALK284 (KALK), and WT ETV7 were each introduced into human EW8 Ewing sarcoma cells, which lack endogenous ETV7 and are rapamycin-sensitive. These four transduced cell lines and WT EW8 were each treated with increasing amounts of rapamycin for three population doublings (≥72 hours). EW8 cells and those expressing ETV7-Δ9 or ETV7-Δ27 were rapamycin-sensitive, while cells expressing WT ETV7 or ETV7-KALK were rapamycin-resistant and, like ETV7-expressing mouse pre-B cells, proliferated at half-speed at high rapamycin concentrations (Fig. 2E). Two-way IP Western blots of cell lysates using anti-ETV7 and anti-mTOR antibodies showed that in contrast with cells expressing ETV7-KALK or WT ETV7, those expressing the Δ9 or Δ27 PNT domain mutants failed to assemble mTORC3 (no mTOR/ETV7 co-IP; Fig. 2F, top). A Western blot of the cell lysates showed that the PNT domain mutants and ETV7 were expressed at a similar level, while the level of the KALK mutant was approximately threefold lower (Fig. 2F, bottom). Together, this supports the specificity of the ETV7/mTOR co-IPs and implicates the ETV7 PNT domain in mTORC3 assembly. The KALK mutation suggests that mTORC3 assembly by ETV7 is independent of its transcriptional activity. Affymetrix array analysis of EW8 cells expressing ETV7 or ETV7-KALK further supported the KALK mutation’s loss of transcriptional activity given that compared with ETV7 cells, 65% of the genes in KALK cells were up-regulated (table S1F), which is in agreement with the observation that ETV7 mostly acts as a transcriptional repressor (17).
mTORC3 has mTOR-specific kinase activity in vitro
We next determined whether mTORC3 immunoprecipitated from Karpas-299 cell lysates had in vitro mTOR kinase activity and whether this activity was sensitive to mTOR inhibitors. As in Fig. 2C, the different mTORCs in Karpas-299 cells were immunoprecipitated from whole-cell lysates with anti-mTOR, anti-ETV7, anti-Raptor, or anti-Rictor antibody, while control IPs were carried out with nonrelevant IgG. Immunoblots were performed with the immunoprecipitated material (Fig. 3, top). Parallel IPs were used for in vitro kinase assays by incubating each with ATP and recombinant P70S6KThr389-peptide–glutathione S-transferase (GST) fusion protein, a substrate of both mTORC1 and mTORC2 (36). Each kinase assay was performed after preincubation of the immunoprecipitated material with rapamycin/FKBP-12 or the second-generation mTOR inhibitors OSI-27 or AZD-8055, or without inhibitor, respectively. P70S6K peptide phosphorylation was visualized by immunoblotting with anti–p-P70S6KThr389 antibody (Fig. 3, bottom). With the exception of the control IgG IP, all four IPs contained mTOR-specific kinase activity. Preincubation of the immunoprecipitated material with rapamycin/FKBP-12 diminished the kinase activity of TORC1+2+3, virtually eliminated mTORC1 kinase activity, reduced mTORC2 kinase activity, but did not affect mTORC3 kinase activity. In contrast, preincubation with OSI-27 or AZD-8055 at an mTOR-specific concentration inhibited the kinase activity of all mTORCs. Therefore, the in vitro mTORC3 kinase activity is rapamycin-insensitive but OSI-27– or AZD-8055–sensitive, which concurs with the observation that ETV7 expression renders mouse pre-B cells and human EW8 cells rapamycin-resistant via an induced rapamycin-insensitive mTOR kinase activity (Figs. 1B and 2E).
Fig. 3. mTORC3 has mTOR-specific kinase activity in vitro.
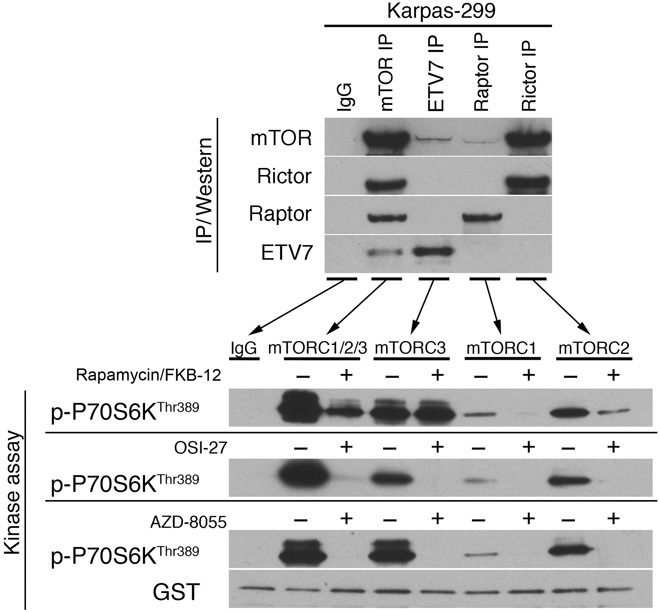
Top: IgG IP (IgG), mTOR IP, ETV7 IP, Raptor IP, or Rictor IP of Karpas-299 cell lysate immunoblotted for mTOR, ETV7, Raptor, or Rictor. Bottom: Immunoblot of IgG IP (IgG), mTOR IP (mTORC1/2/3), ETV7 IP (mTORC3), Raptor IP (mTORC1), or Rictor IP (mTORC2) of Karpas-299 lysate incubated with recombinant GST-tagged P70S6K peptide after preincubation with vehicle (−) or rapamycin/FKBP-12 (+) (top blot), vehicle (−) or OSI-27 (+) (middle blot), and vehicle (−) or AZD-8055 (+) (bottom blot) was probed for p-P70S6KThr389. GST was used as a loading control.
mTOR-ETV7 binding in vitro and mTORC3 size estimation
We next coincubated purified ETV7 and mTOR protein to determine whether they associated in vitro after validating that these preparations were free of mTORC1/2 auxiliary proteins using mass spectrometry (MS) analysis (table S1, G and H). To check specificity, we controlled binding of ETV7 and mTOR to purified RUVBL2, a protein not present in co-IPs, with anti-ETV7 or anti-mTOR antibody from Karpas-299 lysates (fig. S5A). After coincubation for 8 or 24 hours, a similar fraction of ETV7 and mTOR protein coimmunoprecipitated (Fig. 4A), indicating binding. Anti-ETV7 and anti-mTOR antibodies did not coimmunoprecipitate RUVBL2, which supports the specificity of the ETV7/mTOR interaction. Since the fraction of associated protein did not increase beyond 8 hours of coincubation, it suggested that only a portion could associate under our experimental conditions. This limited binding agrees with observations in U937 cells in vivo in which increased exogenous ETV7 expression failed to assemble more mTORC3 (fig. S5B), indicating that mTORC3 assembly might be tightly regulated.
Fig. 4. In vitro association of mTOR and ETV7 and gel filtration of mTORC1, mTORC2, and mTORC3 in Karpas-299 cell lysates.
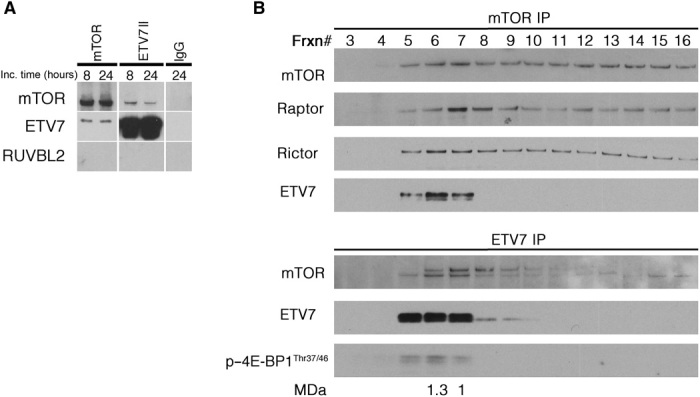
(A) IPs (mTOR and ETV7) of coincubated (8 or 24 hours) purified mTOR, ETV7, and RUVBL2 proteins immunoblotted for mTOR, ETV7, and RUVBL2. (B) IPs [mTOR IP (top) and ETV7 IP (bottom)] of Superose 6 fast protein liquid chromatography (FPLC) gel filtration fractions of Karpas-299 lysates immunoblotted for mTOR, Raptor, Rictor, and ETV7 (mTOR IP) and mTOR, ETV7, and p–4E-BP1Thr37/46 (ETV7 IP). Frxn# = column fraction number. The Raptor peak fraction is used as the molecular mass marker for mTORC1 (1 MDa) and the Rictor peak fraction is used as the molecular mass marker for mTORC2 (1.3 MDa). The peak fraction of mTORC3 (ETV7) coincides with that of mTORC2 (Rictor).
To compare the size of mTORC3 with that of mTORC1 and mTORC2, which were estimated to be approximately 1 and 1.3 MDa, respectively (4, 5), we separated Karpas-299 cell lysates on a Superose 6 gel filtration column. Column fractions were immunoprecipitated with anti-mTOR or anti-ETV7 antibody, and the mTOR IPs were immunoblotted for mTOR, ETV7, Raptor, and Rictor, while ETV7 IPs were immunoblotted for mTOR, ETV7, and p–4E-BP1Thr37/46. The IP/Western blots showed that the peak fraction of Rictor coincided with that of ETV7 (fraction 6), while the Raptor peak eluted one fraction later (fraction 7) (Fig. 4B), suggesting that mTORC3 has a size comparable to that of mTORC2.
Effects of ETV7 loss on mTOR signaling
To address the contribution of endogenous mTORC3 signaling to overall mTOR kinase activity during proliferation, we performed knockdown of ETV7 in proliferating Karpas-299 cells. This induced massive cell death 6 days later, which prevented us from obtaining reliable mTOR signaling information. To illustrate this cell death, we recorded time-lapse movies of ETV7 knockdown in the attached growing human DAOY medulloblastoma cell line (movies S1 and S2). To circumvent knockdown, we isolated a single-cell clone (K-E7−) from a stationary culture of Karpas-299 that grew noticeably slower. An ETV7 IP/Western blot showed that K-E7− did not express ETV7 (Fig. 5A, inset), while reintroduction of ETV7 with a lentiviral vector (K-E7−–ETV7) restored its proliferation rate to that of parental Karpas-299 cells (Fig. 5A). To avoid drawing conclusions from a single clone, we generated additional ETV7-negative Karpas-299 clones by using clustered regularly interspaced short palindromic repeats/CRISPR-associated 9 (CRISPR-Cas9) targeting of the ATG encoding methionine 82 (M82) in the ETV7 PNT domain. These clones proliferated at the same rate as K-E7− and did not express ETV7 as assayed by ETV7 IP/Western blotting, one of which, K-E7Δ120/Δ154+60 (fig. S4), is shown in Fig. 5A (inset). Treatment of Karpas-299, K-E7−, K-E7−–ETV7, and K-E7Δ120/Δ154+60 cells with increasing amounts of rapamycin for three population doublings (72 hours) halved the proliferation rate of Karpas-299 and K-E7–ETV7 cells at concentrations ≥10 ng/ml but was cytostatic for K-E7− and K-E7Δ120/Δ154+60 cells at these concentrations (Fig. 5B). In contrast, treatment of Karpas-299, K-E7−–ETV7 cells (Fig. 5C), or ETV7-expressing mouse pre-B and EW8 cells (Fig. 1B and fig. S6A) with increasing amounts of AZD-8055 completely halted proliferation of all these cell lines at concentrations ≥100 ng/ml.
Fig. 5. mTORC3 kinase activity is insensitive to rapamycin, but sensitive to AZD-8055.
(A) Growth curves of Karpas-299, K-E7−, K-E7−–ETV7, and K-EΔ120/Δ159+60 cells each inoculated at a density of 6 × 104 cells/ml and grown in liquid spinner cultures for 72 hours. Data are means ± SEM from three independent experiments. The inset shows an ETV7 IP/Western blot of lysates of 2 × 107 Karpas-299, K-E7−, K-E7−–ETV7, and K-EΔ120/Δ159+60 cells. (B) Graph representing cell densities (percent control) of Karpas-299, K-E7−, K-E7−–ETV7, and K-EΔ120/Δ159+60 cells cultured for three population doublings with increasing amounts of rapamycin (0.1, 0.3, 1.3, 10, 30, 100, 300, and 1000 ng/ml), plotted as the percentage of cells treated with vehicle. Data are means ± SEM from three independent experiments. Thirty percent control or less indicates no proliferation. (C) Graph representing cell densities (percent control) of Karpas-299 and K-E7− cells cultured for three population doublings with increasing amounts of AZD-8055 (0.1, 0.3, 1.3, 10, 30, 100, 300, and 1000 ng/ml), plotted as the percentage of cells treated with vehicle. Data are means ± SEM from three independent experiments. Thirty percent control or less indicates no proliferation. (D) Immunoblots of lysates from equal numbers of Karpas-299, K-E7−, and K-E7−–ETV7 cells treated with increasing amounts of rapamycin (0.1, 0.3, 1.3, 10, 30, 100, 300, and 1000 ng/ml) probed for p-mTORSer2448 p-AKTSer473, p-AKTThr308, p-ERK1/2Thr202/Tyr204, p-p70S6KThr389, p–4E-BP1Thr37/46, total 4E-BP1, p-GRB10Ser501/503, total GRB-10, LC3BI/II, and SQSTM1/p62. (E) mTOR IP (mTOR IP) of Karpas-299 cells treated with increasing amounts of rapamycin (0.1, 0.3, 1.3, 10, 30, 100, 300, 1000 ng/ml) immunoblotted for mTOR, Rictor, Raptor, and ETV7. (F) Lysates of the AZD-8055–treated Karpas-299 and K-E7− cells immunoblotted for p-mTORSer2448, p-AKTSer473, p-AKTThr308, p-ERK1/2Thr202/Tyr204, p-P70S6KThr389, p–4E-BP1Thr37/46, total 4E-BP1, KI-67, LC3BI/II, and SQSTM1/p62.
Analysis of the difference in mTOR signaling in Karpas-299, K-E7−, K-E7−–ETV7, and K-E7Δ120/Δ154+60 cells with increasing amounts of rapamycin (Fig. 5D and fig. S6B) showed persistence of mTORC1/2-like signaling in Karpas-299 and K-E7−–ETV7 cells at high rapamycin concentrations (partial maintenance of p–4E-BP1Thr37/46, p-GRB10Ser471/473, and p-P70S6KThr389, and increased p-AKTSer473). In comparison, mTORC1 signaling in K-E7− and K-E7Δ120/Δ154+60 cells was strongly reduced at rapamycin concentrations ≥3 ng/ml [loss of p-P70S6KThr389 (see also fig. S10) and mTORSer2448, strongly reduced p–4E-BP1Thr37/46 and loss of p-GRB10Ser471/473], with mTORC2 signaling (p-AKTSer473) tapering off more gradually. It has been reported that 4E-BP1Thr37/46 phosphorylation can be mediated by glycogen synthase kinase 3-β (GSK3-β) in rapamycin-resistant breast and bladder cancer cell lines, thereby maintaining protein synthesis (37). In Karpas-299 and K-E7− cells, the p–GSK3-βSer9 signal diminished with increasing amounts of rapamycin, normally a sign of GSK3-β activation (38), but here loss was caused by loss of total GSK3-β expression (fig. S6C). In support of this conclusion, densitometric comparison of the relative intensities of bands representing p–GSK3-βSer9 and total GSK3-β at each rapamycin concentration showed that they remained approximately constant (fig. S6D). Therefore, maintenance of p–4E-BP137/46 was likely mediated by mTORC3 rather than by GSK3-β.
p-AKTThr308 increased slightly in Karpas-299 and K-E7−–ETV7 but remained level in K-E7− and K-EΔ120/Δ154+60 cells, while there was no increase in total GRB10 or in ERK1/2 signaling in all four cell lines, suggesting lack of up-regulation of the P70S6K-IRS1-AKT feedback loop (32). Still, the increase in p-AKTSer473 with rising rapamycin concentration in Karpas-299 and KE-7−–ETV7 cells would normally indicate activation of the feedback loop. Notably, this increase in p-AKTSer473 did not occur in rapamycin-treated ETV7-expressing Arf−/− pre-B cells (Fig. 1C). We speculated that this was caused by the constitutively active nucleophosmin–anaplastic lymphoma kinase (NPM-ALK) tyrosine kinase in Karpas-299 and KE-7−–ETV7, which is known to increase p-AKTSer473 (39). Partial inhibition of NPM-ALK with the ALK inhibitor alectinib in combination with increasing amounts of rapamycin greatly reduced this induction of p-AKTSer473 (fig. S7, A and B). In addition, the relative amount of FKBP-12 over FKBP-51 in Karpas-299 is less favorable to inhibit mTORC2 activity with rapamycin than in 293T cells (fig. S7C), in which this relative amount was shown to be insufficient (40). This suggests that their levels are similarly insufficient in Karpas-299 cells. Therefore, there is likely combined mTORC2/3 phosphorylation of AKTSer473 in Karpas-299 in the presence of rapamycin, a possibility supported by the tapering off of p-AKTSer473 in K-E7− and K-E7Δ120/Δ154+60 cells with increasing rapamycin levels. As a possible alternative cause of the p-AKTSer473 effect, we also analyzed the fate of two other kinases reported to phosphorylate p-AKTSer473, IκB kinase ε (IKKε), and TANK-binding kinase 1 (TBK1) (41), which showed that they became partially (phospho-IKKεSer172) and almost completely (phospho-TBK1Ser172) inactivated (fig. S6C), disproving this possibility.
Another sign of persistent mTORC1-like signaling in Karpas-299 and K-E7−–ETV7 cells at high rapamycin concentration was demonstrated by the absence of autophagy, a cellular response blocked by mTORC1 signaling (1). Both cell lines showed steady relative intensities of LC3BI/II protein bands at all rapamycin concentrations, contrary to the observed conversion of nonlipidated LC3BI to lipidated autophagosome-associated LC3BII in lysates of K-E7− and, to a lesser extent, in lysates of K-E7Δ120/Δ154+60 at rapamycin concentrations ≥100 ng/ml (Fig. 5D and fig. S6B). In addition, immunoblotting for p62/SQSTM1 in lysates of all four cell lines showed that p62/SQSTM1 levels remained constant in Karpas-299 and K-E7−–ETV7 with increasing rapamycin concentration but diminished in K-E7− and K-E7Δ120/Δ154+60 cells, further corroborating the induction of autophagy in the latter two cell lines (Fig. 5D and fig. S6B). These observations were complemented in Karpas-299 cells with immunofluorescence showing dispersed staining of exogenous LCBI/II-mCherry fusion protein at high rapamycin concentration, whereas eliminating mTOR signaling with AZD-8055 (Fig. 3) produced punctate LCBI/II-mCherry immunofluorescent staining, implying active autophagy (fig. S6E).
The effect of rapamycin treatment on the three mTORCs in lysates of rapamycin-treated Karpas-299 cells further showed that mTORC1 and mTORC2 became destabilized at 3 ng/ml, as indicated by the loss of Raptor and Rictor co-IP in our IP buffer containing phosphate (Fig. 5E) (42). In contrast, ETV7 remained associated with mTOR at all rapamycin concentrations, with a slight increase in the amount of coprecipitated ETV7 at concentrations ≥3 ng/ml, where mTORC1 and mTORC2 were destabilized (Fig. 5E).
As predicted by Fig. 5C, lysates of Karpas-299 and K-E7− cells treated with increasing amounts of AZD-8055 showed identical loss of p-mTORSer2448, p–4E-BP1Thr37/46, p-P70S6KThr389, and p-AKTSer473 phosphorylation at concentrations between 10 and 100 ng/ml. At 100 ng/ml, all cells had transitioned to the G0 phase of the cell cycle, as shown by loss of the Ki-67 antigen (Fig. 5F). P-AKTThr308, the target of membrane-receptor signaling via PI3K and PDK, remained unaltered, underlining the idea that inhibition was mTOR kinase–specific. Starting at 100 ng/ml, AZD-8055–induced loss of mTOR signaling activated autophagy, as shown by the change in relative intensities of the LC3BI/II bands (Fig. 5F) in Karpas-299 and K-E7− cells and by loss of p62/SQSTM1. Hence, phosphorylation of mTOR targets was maintained by mTORC3 at rapamycin concentrations and incubation times (72 hours) that inhibit mTORC1/2 signaling, while mTORC3 signaling was extinguished by AZD-8055 at concentrations specific for mTOR kinase. mTORC3 signaling was absent in the Karpas-299 derivatives K-E7− and K-E7Δ120/Δ154+60, which had lost ETV7 expression.
mTORC3 kinase activity in vivo is insensitive to Raptor or Rictor knockdown
To verify that mTORC3 signaling is independent of Raptor or Rictor, we analyzed mTOR signaling in EW8 cells engineered to express ETV7-IRES-GFP or GFP alone after 120 hours of Raptor-short hairpin RNA (shRNA), Rictor-shRNA, or scrambled-shRNA knockdown. Western blots of cell lysates showed substantial loss of mTORC1 signaling in GFP/Raptor-shRNA (reduced p-P70S6KThr389 and p–4E-BP1Thr37/46) or mTORC2 signaling in GFP/Rictor-shRNA cells (reduced p-AKTSer473 and p-NDRG1Thr346), but not in ETV7/Raptor-shRNA– and ETV7/Rictor-shRNA–expressing cells (maintenance of p-P70S6KThr389, p–4E-BP1Thr37/46, p-AKTSer473, and p-NDRG1Thr346; fig. S8A). Hence, exogenous ETV7 expression maintains mTORC1/2-like signaling in the absence of mTORC1 or mTORC2. In addition, ETV7 or mTOR IPs from Raptor or Rictor knockdown cell lysates followed by Western blotting for mTOR, Rictor, Raptor, mSin1, mLST8, and ETV7 showed that knockdown did not eliminate the presence of mTORC3 (fig. S8B). Western blot analysis of cell lysates of an identical knockdown experiment in Karpas-299 cells showed sustained mTOR signaling despite efficient Raptor or Rictor knockdown. Phosphorylation of p-P70S6KThr389, p–4E-BP1Thr37/46, p-AKTSer473, and p-NDRG1Thr346 was maintained throughout the duration of the experiment (fig. S8C). Finally, Rictorfl/fl fibroblasts were transduced with ETV7 or empty lentiviral vectors and selected for blasticidin resistance. These cells and untransduced Rictorfl/fl cells were then infected with adenovirus-GFP-iCre and cell lysates prepared 90 hours later. Knockout of Rictor resulted in maintenance of mTORC2-like signaling in ETV7-expressing cells (p-AKTSer473 and p-NDRG1Thr364 remain) but loss of mTORC2 signaling in untransduced and vector cells (fig.S8D).
ETV7/mTORC3 expression enhances tumor formation in a mouse model of ERMS
Given that mice lack the Etv7 gene, we generated a transgenic mouse carrying a single copy of a human ETV7-containing bacterial artificial chromosome (BAC) transgene (details of the generation and ETV7 expression patterns to be published elsewhere). ETV7TG+/− mice were phenotypically normal, and maintenance for 24 months showed that they were not tumor-prone. To assess the tumorigenic potential of the transgene, we crossed the ETV7TG+/− mice onto the Ptch1+/− background, which predisposes to medulloblastoma and ERMS development (43). Ptch1+/− and Ptch1+/−/ETV7TG+/− mice were monitored for tumor formation for 500 days. Mice were palpated for tumors when presenting with distended abdomen or masses on the front or hind limbs. Kaplan-Meier survival curves (Fig. 6A) represent death by tumor, or tumor mass ≤20% of body mass. Tumors arose most frequently in the hind limbs (n = 9) but also occurred in the thoracic cavity (n = 6), associated with the spine (n = 5) or the intestinal tract (n = 4). Hematoxylin and eosin (H&E) staining of tumor sections revealed a well-differentiated skeletal muscle morphology (arrowheads in Fig. 6B), with multifocal positivity for the skeletal muscle–specific marker myogenin (Fig. 6B). These tumors also expressed general muscle markers, including muscle-specific actin (MSA), smooth-muscle actin (SMA), and desmin (fig. S9A), which led to the diagnosis of ERMS. In addition, we checked expression levels of the mTORC1 and mTORC3 target p–4E-BP1Thr37/46, but all ERMS tumor specimens showed similar cytoplasmic staining for p–4E-BP1Thr37/46 (Fig. 6B). Staining of sections from Ptch1+/−/ETV7TG+/− ERMS tumors revealed a cytoplasmic signal of transgene-expressed ETV7, which was blocked by preincubation with ETV7 peptide. Ptch1+/− ERMS tumors did not stain with ETV7 antibody (Fig. 6B). To confirm the presence of mTORC3, we isolated cell lines from primary ERMS tumors. Ptch1+/−/ETV7TG+/− tumors readily produced cell lines in culture, while Ptch1+/− ERMS tumors did not. Unlike primary tumors (Fig. 6B), Ptch1+/−/ETV7TG+/− ERMS cell lines had no striation (fig. S9B), did not express myogenin, but did express MSA, SMA, and desmin (fig. S9B), as did the original tumors (fig. S9A). Given the heterogeneous myogenin expression in the primary tumors (Fig. 6B), this might reflect selection in culture of a less differentiated proliferating cell population, as evidenced by expression of the early skeletal muscle markers Myf5 and MyoD (fig. S9C). To confirm the presence of functional mTORC3, we next treated the Ptch1+/−/ETV7TG+/− cell lines with rapamycin. Ptch1+/−/ETV7TG+/− cell lines continued to proliferate when treated with rapamycin (Fig. 6C, left), much like human tumor cell lines expressing endogenous ETV7 (Karpas-299, Fig. 5B) or human and murine cells expressing exogenous ETV7 (Figs. 1B, 2E, and 5A and fig. S6B). To show mTORC3’s contribution to rapamycin resistance in human ERMS cell lines, we treated RD cells and a separately isolated cell line, JR-1, from the same patient [identical short tandem repeats; (44)] with rapamycin, which revealed that RD was resistant, whereas JR-1 stopped proliferating at concentrations ≥1 ng/ml (Fig. 6C, right). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) confirmed that RD expresses ETV7 RNA, while JR does not (fig. S9D). Rapamycin resistance correlated with the presence of mTORC3: mTOR IP of lysates of both Ptch1+/−/ETV7TG+/− cell lines and RD cells coprecipitated ETV7, and ETV7 IP brought down mTOR (Fig. 6D). Together, this suggested that Ptch1+/−/ETV7TG+/− tumors and cell lines contain mTORC3 and that rapamycin resistance in ERMS cell lines is at least in part caused by mTORC3. To support this conclusion, we used the ETV7TG+/− mouse to obtain an ETV7TGΔ9+/− mouse by CRISPR-Cas9 targeting of the M82 codon in the PNT domain. The resulting Δ9 mutation is identical to that in the EW8-ETV7Δ9 cell line, which cannot assemble mTORC3 (Fig. 2, E and F). After crossing ETV7TGΔ9+/− mice onto the Ptch+/− background, the Ptch+/−/ETV7TGΔ9+/− mice were monitored for the formation of rhabdomyosarcomas for 365 days, which showed that the incidence in the double mutants had reverted to that observed in Ptch+/− mice (Fig. 6A). This supports the notion that mTORC3 activity caused the increased rhabdomyosarcoma incidence in Ptch1+/−/ETV7TG+/− mice. The Ptch+/−/ETV7TGΔ9+/− rhabdomyosarcomas occurred in the skeletal muscle (n = 1), along the vas deferens (n = 1) and the spine (n = 1). To support the relevance of these data for human ERMS, we queried primary ERMS RNA-sequence data generated at St. Jude for the presence of ETV7 exon junction reads and found that seven of seven ERMS samples contained mature ETV7 mRNA at variable frequencies (Fig. 6E).
Fig. 6. ETV7 expression cooperates with Ptch1 heterozygosity in a mouse model of ERMS.
(A) Ptch1+/− mice (red) show 21% (n = 33) versus Ptch1+/−/ETV7TG mice (blue) 65% ERMS incidence (n = 42) at 500 days (P < 0.001). Ptch1+/−/ETV7TGΔ9 mice (green) show 14% ERMS incidence (n = 22) at 365 days. Non–ERMS-related events were excluded. All mice were sacrificed when tumors reached a size equal to 20% of body weight, when tumors obstructed normal movement, or when mice were moribund. (B) Representative example of formalin-fixed paraffin-embedded sections of a Ptch1+/− (top) and Ptch1+/−/ETV7TG (bottom) primary tumor stained with H&E, myogenin antibody, p–4E-BP1Thr37/46 antibody, ETV7 antibody, and peptide blocked ETV7 antibody. Scale bars, 20 μm. (C) Graphs representing cell counts of two Ptch1+/−/ETV7TG cell lines (left) and human ERMS cell lines RD and JR-1 (right), cultured for three population doublings with increasing amounts of rapamycin (0.1, 0.3, 1.3, 10, 30, 100, 300, and 1000 ng/ml) charted as the percentage of cells treated with vehicle. Data are means ± SEM from three independent experiments. (D) Immunoblot of IgG IP, mTOR IP, and ETV7 IP of two Ptch1+/−/ETV7TG cell lines and human ERMS cell line RD lysate using anti-mTOR and anti-ETV7 antibodies. (E) Table listing the number of ETV7 exon junction reads in RNA sequence analysis of seven of seven primary human ERMS RNA samples.
DISCUSSION
We describe a rapamycin-resistant cytoplasmic mTOR complex, mTORC3, which assembles upon expression of the transcription factor ETV7 that interacts with mTOR. The presence of ETV7 and therefore mTORC3 stimulates cell proliferation and, when expressed exogenously, promotes tumorigenic transformation in mice [(15, 16); this work]. In addition, ETV7 expression is up-regulated at variable frequencies in many human tumor types.
Our conclusion that mTORC3 represents a rapamycin-resistant mTOR complex distinct from mTORC1/2 is based on the following observations: (i) Anti-ETV7–immunoprecipitated material contains mTOR and p–4E-BP1Thr37/46, but not Raptor, Rictor, mLST8, or SIN1. (ii) Anti-Raptor– or anti-Rictor–immunoprecipitated material contains mTOR and mTORC1/2 auxiliary proteins, but not ETV7. (iii) mTORC3 has rapamycin-resistant but second-generation mTOR inhibitor–sensitive mTOR-specific kinase activity in vitro and in vivo. (iv) Exogenous ETV7 expression in ETV7-negative cell lines induces mTORC3 assembly, rapamycin resistance, and increased cell proliferation. (v) Loss of ETV7 in a rapamycin-resistant mTORC3-expressing cell line induces rapamycin sensitivity and reduced proliferation. (vi) Raptor or Rictor knockdown or Rictor knockout does not affect mTORC3’s assembly or kinase activity. (vii) The ETV7 PNT domain participates in mTORC3 assembly. (viii) ETV7’s transcriptional activity is dispensable for mTORC3 assembly and activity.
Inclusion of expression of two ETV7 PNT domain mutants in EW8 cells verified that the interaction between ETV7 and mTOR is not an artifact of the methodology. Although we do not know the impact of these deletions on the three-dimensional (3D) structure of the PNT domain, we can conclude that this domain contributes to mTOR binding, but we have not determined whether its presence is also sufficient for binding. Inclusion of the ETV7KALK DNA binding mutant suggests that ETV7’s mTORC3 assembly activity is independent of its transcriptional activity. Affymetrix expression analysis of EW8 cells expressing ETV7 or ETV7KALK showed that 65% of the genes in the latter were up-regulated, confirming the notion that ETV7 mainly functions as a transcriptional repressor. Together, the ETV7 mutants showed that there is a strict correlation between mTOR/ETV7 co-IP and induced rapamycin resistance.
Our in vitro kinase assays showed that mTORC3 has intrinsic rapamycin-resistant mTOR kinase activity. Yang and coworkers (6) reported that mTORC2 is sensitive to micromolar amounts of free rapamycin that binds to the Ser2035 rapamycin contact point in the FKBP-12–rapamycin binding (FRB) domain of mTOR, a condition we used for our inhibitor kinase assays (3 μM FKBP-12 + 10 μM rapamycin). This would explain the rapamycin-induced partial inhibition of the mTORC2 kinase activity in Fig. 3. Because this condition did not inhibit mTORC3 activity, we speculate that Ser2035 in mTORC3 is less accessible to rapamycin than it is in mTORC2, but further experimentation is needed to substantiate this possibility. Inhibition of in vitro mTORC3 kinase activity by the ATP-competitive inhibitors AZD-8055 and OSI-27 at mTOR kinase specific concentrations confirms that phosphorylation of the GST-p70S6KThr389 fusion protein is the result of mTOR kinase activity and not of another kinase, given that the specificity of inhibition is 1000-fold [AZD-8055; (7)] and 100-fold [OSI-27; (45)] more potent for mTOR than for PI3Kα, PI3Kβ, PI3Kγ, or the PI3K-related protein kinase family members ataxia telangiectasia mutated and DNA-dependent protein kinase or 260 (AZD-8055) and 26 (OSI-027) other serine/threonine and tyrosine kinases.
mTORC3 phosphorylates target proteins such as 4E-BP1, P70S6K, and AKT without Raptor or Rictor in the complex. Raptor is the scaffold in mTORC1 that recruits substrates via binding to their mTOR-signaling (TOS) motif (33). We speculate that future identification of mTORC3-associated proteins may provide an answer to this question. Raptor and Rictor knockdown and Rictor knockout experiments further confirm that mTORC3 is distinct from mTORC1/2 as its activity is independent of Raptor or Rictor for the duration of these experiments. Inactivation of mTORC3 via ETV7 knockdown proved to be technically unattainable. None of the commercially available ETV7 shRNAs induced complete knockdown and proliferating cells started dying after 5 to 6 days of knockdown, as illustrated with DAOY cells (movies S1 and S2). The reason that we could obtain ETV7-null Karpas-299 clones using CRISPR-Cas9 (Fig. 5A and fig. S6B) was we kept the cultures sufficiently dense (≥105/ml). We believe that cell-cell contact provided the survival signal, as addition of Karpas-299–conditioned medium to thin (<105 cells/ml) knockout cultures failed to salvage the cells.
Regulation of mTORC3 appears to be complex and is likely executed at the level of ETV7 gene transcription or ETV7 half-life, while ETV7 nuclear/cytoplasmic distribution and kinase activity of the complex are other regulatory options. We base this on the observations that endogenous or forced ETV7 expression is sufficient to generate mTORC3 activity. Most (if not all) cytoplasmic ETV7 is associated with mTOR, as shown by the nearly equal ETV7 signals on the Western blot of the mTOR- and ETV7-coimmunoprecipitated material from Karpas-299 cytoplasmic fractions (Fig. 2D), while nuclear mTOR and ETV7 do not interact. Increasing overexpression of ETV7 in U937 cells failed to boost mTORC3 assembly (fig. S5B). Thus, assembly and activity of mTORC3 seem stringently controlled, conceivably via posttranslational modification. In contrast, increased expression of Raptor and Rictor directly increases mTORC1 and mTORC2 complex formation and activity (42, 46, 47).
The in vitro association of purified ETV7 and mTOR suggests direct binding (Fig. 4A). Although MS analysis of these preparations showed absence of mTORC1/2-associated proteins, we cannot exclude the idea that the contaminating proteins in these preparations (tables S1, G and H) could serve as binding intermediates.
The presence of mTORC3 dominantly confers resistance to rapamycin, whether induced exogenously (mouse Arf−/− pre-B cells, EW8, K-E7− cells, and Ptch+/−/ETV7TG+/− ERMS cells) or present endogenously (Karpas-299 and RD), while loss of ETV7/mTORC3 reverts to rapamycin sensitivity (K-E7− and K-E7Δ120/154+60 cells). Primary mouse lymphocytes are much more sensitive to rapamycin than mouse fibroblasts or most tumor cell lines (8, 9) because their 4E-BP–eIF4E axis is more dependent on 4E-BP2, whose phosphorylation by mTORC1 is rapamycin-sensitive (29). Nonetheless, p–4E-BP1/2Thr37/46 is partially maintained by ETV7/mTORC3 expression in these cells at high rapamycin concentration, without up-regulation of pathways that would increase PI3K-AKT-mTOR signaling.
The prominent increase in mTOR signaling seen in primary mouse pre-B cells (Fig. 1A) is not detected in Karpas-299 cells as compared with K-E7− or K-E7Δ120/154+60 cells (Fig. 5C and fig. S6B). The reason for this is currently unknown, but this may be due to overall increased signaling in these tumor cell lines by NPM-ALK. Although mTOR kinase inactivation by rapamycin is less pronounced in transformed Karpas-299 cells (Fig. 5D), ETV7’s effect on rapamycin resistance is the same as that in pre-B cells. Unlike derivative clones that lost ETV7 expression (K-E7− and K-E7Δ120/Δ154+60), all mTOR signaling remains active, which allows Karpas-299 to proliferate at half-speed at high rapamycin concentrations. Alternative pathways for rapamycin-induced AKT phosphorylation involving, among others, src-family kinases have been proposed (48). Given that the relative levels of FKBP-12 and FKBP-51 are likely insufficient to block mTORC2 in the presence of rapamycin (40) and that NPM-ALK in Karpas-299 activates pp60-c-SRC (49), the combination of activated pp60-cSRC and mTORC3 might be responsible for p-AKTSer473 phosphorylation (fig. S7B). Because the PI3K-AKT-mTOR feedback loop is not activated by rapamycin treatment in Karpas-299, we speculate that an as yet unidentified feedback loop affecting mTORC3 signaling is responsible for the increase in p-AKTSer473 in the presence of rapamycin (≥1 ng/ml). Notably, p-AKTSer473 up-regulation does not occur in mouse Arf−/− ETV7 pre-B cells or K-E7− and K-E7Δ120/Δ154+60 cells. On the basis of the 1.5-fold down-regulation of CD19 in ETV7 pre-B cells (Affymetrix array mouse Arf−/− vector pre-B cells versus Arf−/− ETV7 pre-B cells), a signaling costimulator and activator of SRC kinases (50), these cells are unlikely to contain hyperactivated pp60-c-SRC, while K-E7− and K-E7Δ120/Δ154+60 cells do not contain mTORC3.
The description of mTORC3 here and the recent identification of an mTOR/GIT1/β-PIX complex lacking Raptor or Rictor in mouse astrocytes (51) suggest that the scope of mTOR complexes in different cell types is more diverse than generally accepted.
Our data suggest that proteins in mouse necessary to assemble mTORC3 are present, as exogenous ETV7 induced active mTORC3. This is further strengthened by the observation that rhabdomyosarcomas from Ptch+/−/ETV7 BAC transgenic mice yielded mTORC3-expressing cell lines.
ETV7 is expressed in many tumor types. This and the strict correlation between ETV7 expression and mTORC3 assembly in cell lines and xenografts suggest that tumors expressing ETV7 contain mTORC3.
We believe that mTORC3 signaling contributes to the poor efficacy of rapamycin and rapalogs as antitumor agents. This underlines the importance of clinical testing of available second-generation mTOR inhibitors, which do inactivate mTORC3. The potential drawback of these inhibitors would be toxicity to normal tissues, as indicated by induction of cell death in vector-transduced Arf−/− pre-B cells when treated with AZD-8055 (Fig. 2B). Results from initial clinical trials suggest that these inhibitors indeed displayed dose-limiting toxicity when used in human treatment protocols (52–54), while tolerated dosing in patients with advanced solid tumors or multiple myeloma reduced mTOR signaling but had modest antitumor effects (52). In addition, AZD-8055 treatment has been shown to increase autophagy in tumor cells in vivo, leading to an increase in chemotherapy resistance, further closing the therapeutic window using these types of inhibitors (55). Specific inhibition of mTORC3 while maintaining mTORC1 signaling should prevent the activation of autophagy, thereby avoiding chemotherapy resistance in the absence of normal tissue toxicity. Since cycling ETV7-expressing tumor cells are dependent on its presence, inhibitors specifically targeting mTORC3 in combination with survival pathway inhibitors might present a novel therapeutic window, given that ETV7 and therefore mTORC3 are activated in many cancers but are notably absent in most normal tissue counterparts. Efficacy of these treatment protocols was demonstrated by the combination of second-generation mTOR inhibitors with the block of the antiapoptotic BCL2 family members by ABT-737 or its analog ABT-263 (56). Second-generation mTOR inhibitors potently sensitized cancer cell lines to apoptosis by BCL2 inhibition, which was not achieved by rapamycin treatment. This suggested that rapamycin-resistant mTOR signaling, which we believe can be caused by mTORC3, was able to protect tumor cell lines from apoptosis (56). The latter is supported by our observation that an ETV7-expressing ERMS, Ewing sarcoma, and Karpas-299 cell lines are rapamycin-resistant, whereas ETV7-negative ERMS, Karpas-299 K-E7− and K-E7Δ120/Δ154+60 cell lines, and cell lines expressing mutant ETV7 unable to assemble mTORC3 are rapamycin-sensitive.
MATERIALS AND METHODS
Experimental design
The main goal of this study was to describe the existence of a novel rapamycin-resistant mTOR complex in human tumors and tumor cell lines. All biochemical experiments described in this manuscript were repeated at least three times, including IP/Western blotting, in vitro kinase assays, mTOR inhibitor assays, gel filtration assays, knockdown experiments, and Western blotting. In an earlier study, we had characterized ETV7 as a potential oncogene in mouse pre-B cells. Therefore, we first analyzed publicly available expression array databases of human tumors and found frequently increased ETV7. Because ETV7-expressing mouse pre-B cells proliferate twice as fast these cells without ETV7, we used Western blotting to show that these cells exhibited increased mTORC1 and mTORC2 signaling. Proliferation assays in the presence of the mTOR inhibitor rapamycin showed that the cells had become rapamycin-resistant and maintained mTORC1/2-like signaling. To explore whether ETV7 modifies the known mTOR complexes to induce rapamycin resistance, co-IP/Western blot studies of cell lysates of ETV7-expressing mouse pre-B cells and endogenous ETV7-expressing human cell lines (Karpas-299, K562, and OS-17) showed that ETV7 associated with mTOR in a complex that is distinct from mTORC1 or mTORC2, which we dubbed mTORC3. Next, we used IP/in vitro kinase assays, followed by phosphoprotein Western blotting, to verify that mTORC3 immunoprecipitated from Karpas-299 lysates phosphorylates a p70S6KThr389 peptide containing GST fusion protein in a rapamycin-insensitive but mTOR-competitive inhibitor–sensitive manner. We used gel filtration analysis combined with IP/Western blotting to show that mTORC3 was similar in size to mTORC2. Cell lines expressing endogenous or exogenous ETV7 were rapamycin-resistant and continued to proliferate at half-speed at high rapamycin concentrations. In contrast, a non–ETV7-expressing subclone or CRISPR-Cas9 ETV7 knockout clones of the Karpas-299 cell line became rapamycin-sensitive and proliferated at half-speed, a condition that was reversed upon introduction of exogenous ETV7. IP/Western blotting showed that rapamycin treatment of mTORC3-expressing cells resulted in destabilization of mTORC1 and mTORC2, while mTORC3 remained stable. Western blotting further confirmed that in the presence of high concentrations of rapamycin, mTOR1/2-like signaling remained active, while it was lost in the presence of the ATP-competitive inhibitor AZD-8055 at a concentration specific for mTOR kinase. Furthermore, Raptor or Rictor knockdown for 72 hours in Karpas-299 cells did not eliminate mTORC1- or mTORC2-like signaling, while it did in EW8 cells, which do not express ETV7/mTORC3. Also, Rictor knockout in Rictorf l/f l mouse embryonic fibroblasts (MEFs) did not eliminate mTORC2-like signaling.
To directly address mTORC3’s contribution to tumorigenesis in a mouse model, we created a transgenic mouse line carrying a single-copy human BAC integration encoding ETV7. These mice are not tumor-prone, but crossing them with the Ptch+/− model of RD increased tumor penetrance from 21 to 65% in 500 days. Cell lines isolated from these tumors express mTORC3 as determined by IP/Western blot analysis. We believe that future mTORC3 inhibitors will be valuable antitumor agents in tumors expressing ETV7.
Cell lines
Karpas-299, K-E7−, K-E7−–ETV7, K-E7Δ120/Δ154+60, U937, K562, RD, JR, and EW8 cells were maintained in RPMI-1640 (Corning) medium supplemented with 10% bovine calf serum (BCS) (Thermo Fisher Scientific), 50 mM GlutaMAX (Life Technologies) (RD and JR-1 with 1% l-glutamine rather than GlutaMAX), penicillin (100 U/ml), and streptomycin (100 μg/ml) (Life Technologies) in a humidified incubator at 37°C and 5% CO2. OS-17 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (cellgro) with the same additives, under the same conditions. MEFs were grown in DMEM supplemented with 10% fetal bovine serum (FBS), 50 mM GlutaMAX, 1 mM Na-pyruvate, 1× essential amino acids (Gibco, #11140-050), 55 μM β-mercaptoethanol, penicillin (100 U/ml), and streptomycin (100 μg/ml) at 3% O2 and 5% CO2. To maintain maximal proliferation, which is essential for maximal levels of mTORC3, all cells were harvested at 0.3 × 106 to 0.4 × 106 cells/ml or at 40 to 50% confluence for suspension and attaching cells lines, respectively. All mTORC3 IPs and Western blot analyses were carried out with cells growing in logarithmic phase for at least three passages. Cell concentration, viability, and average cell diameter were determined with a Vi-CELL Cell Viability Analyzer (Beckmann Coulter) using trypan blue exclusion. Primary mouse pre-B cells were obtained and maintained in culture as previously described (15), with bone marrow from femurs of 4- to 8-week-old C57BL/6/129svJ WT or Arf −/− mice. Red blood cells were lysed, and lineage depletion was performed with antibodies against B220, CD5, GR-1, and TER-119 (all from BD Pharmingen). The next day, lineage-negative cells were transduced with MSCV-IRES-GFP or MSCV-ETV7-IRES-GFP retroviral vectors at a multiplicity of infection (MOI) of 1 to 3 (17). Three days later, GFP-expressing cells were sorted by fluorescence-activated cell sorting and plated on S17 feeder cells in the presence of interleukin-7 (IL-7) (50 μg/ml) to induce B cell differentiation. Pre-B cell cultures were split twice weekly onto fresh S17 feeders in IL-7–containing medium to maintain low cell density and maximum proliferation.
Affymetrix array analysis
Low-passage Arf−/− vector and ETV7 pre-B cells were cultured in IL-7–containing medium (50 ng/ml) without feeders to a density of 0.5 × 106 cells/ml. RNA was extracted, and 100 ng of total RNA was processed using the Affymetrix Ambion WT protocol. Biotinylated complementary DNA (cDNA) (5 to 6 ng) was hybridized to the Mouse Gene 2.0 ST GeneChip, and signals were summarized using robust multi-array average algorithm (RMA) (Partek Genomics Suite v6.6). The results are the median of three samples each. EW8 cells expressing ETV7 or ETV7-KALK were grown to a density of 2 × 106 cells per T75 culture flask. RNA was extracted, and 100 ng was processed using the Affymetrix Ambion WT protocol. Biotinylated cDNA (5 ng) was hybridized to the Human Gene 2.0 ST GeneChip, and signals were summarized using RMA (Partek Genomics Suite v6). The results are the median of three samples each.
Isolation of K-E7−, K-E7Δ120/Δ154+60, and K-E7−–ETV7 cells
Karpas-299 cells were plated (103 cells per 10-cm plate) from a stationary culture in methyl cellulose in RPMI-1640, 10% BCS, 50 mM GlutaMAX, penicillin (100 U/ml), and streptomycin (100 μg/ml) in a humidified incubator at 37°C and 5% CO2. After 1 week, 10 colonies were picked and passaged in liquid culture for 2 weeks. One clone, K-E7−, grew slower than the rest and did not express ETV7, as tested by Western blotting. To restore ETV7 expression, K-E7− cells were transduced with pCL20c-TKp-ETV7-Actp-blasticidin lentivirus, selected on blasticidin (1 μg/ml) for 1 week, followed by single-cell plating as described above. Single colonies were expanded individually in liquid culture. One clone, K-E7−–ETV7, was used for experiments shown in Fig. 5 (A, B, and D).
To create mTORC3-negative Karpas-299 cells using CRISPR-Cas9, we replaced the Pci I–Xba I fragment of pX330 containing the U6 promoter and guide RNA scaffold with the Cla I–Sal I fragment of pEC-V25, containing the EBNA gene and the oriP replication origin of Epstein-Barr virus, producing the pX330-EBNA-Cas9 vector. Karpas-299 cells (106) were nucleofected (Amaxa, kit 5, program A-023) with pX330-EBNA-Cas9 together with an actin promoter blasticidin plasmid in a 6:1 molar ratio (total of 5 μg of DNA). Twenty-four hours later, cells were selected for 5 days with blasticidin (1 μg/ml) and were plated (1000 cells per 10-cm dish) in methylcellulose [1.5% methylcellulose, RPMI-1640, 10% FBS, uridine (7 μg/ml), and thymidine (0.04 μg/ml)]. Seven days later, 15 clones were picked, expanded in liquid culture, and tested for Cas9 expression on Western blot using FLAG antibody. All clones expressed Cas9 from the episomal vector at a similar level and were blasticidin-sensitive. To knock out mTORC3, one of the Karpas-299–EBNA–Cas9 clones (3 × 106 cells) was nucleofected with a pX330 plasmid containing an ETV7 guide sequence (GAGCACGGGTTCGAGAT↓GAA) targeting codon 82 in the ETV7 PNT domain together with the blasticidin plasmid in a 7.5:1 molar ratio (5 μg total). Blasticidin selection was done in a T75 culture flask with refeeding at day 4 and transfer to a T25 flask to keep the density of surviving cells ≥105/ml (that is, 300 cells/mm2). Lower densities resulted in loss of the culture upon loss of mTORC3. After 5 days, cells were transferred to nonselective medium and expanded for 4 weeks. After 3 weeks, the cells had adapted to mTORC3 loss and were viable at higher dilutions, which allowed single-cell cloning in methylcellulose. Ten clones were picked and tested for rapamycin (1000 ng/ml) resistance and all were sensitive. ETV7 exon 3 from genomic DNA of three of these clones was PCR-amplified using the following primers: forward, 5′-TTGCTGGTTGCAGGCATCC-3′; reverse, 5′-TGCGTTTCCCTGACCTGAG-3′. The PCR products were inserted in pGEM-T Easy, and the plasmid inserts of 10 bacterial colonies of each were sequenced using M13 forward and reverse primers. The compiled result of this analysis is shown in fig. S4B. Clone K-E7Δ120/Δ154+60 was used for the experiment shown in fig. S6B.
Western blotting and co-IP
Cells for Western blot analysis were lysed in 1× Cell Signaling lysis buffer [20 mM tris-HCl, (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and leupeptin (1 μg/ml) (Cell Signaling Technologies)] supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF). For co-IP experiments, cells were lysed in CHAPS lysis buffer [40 mM Hepes (pH 7.4), 1 mM EDTA, 120 mM NaCl, 10 mM sodium pyrophosphate, 10 mM β-glycerophosphate, 0.3% CHAPS, 50 mM NaF, 1.5 mM NaVO, 1 mM PMSF, and one tablet of EDTA-free protease inhibitors (Roche) per 10 ml of solution]. Samples were freeze-thawed three times, followed by centrifugation at 20,000g for 10 min at 4°C. Snap-frozen xenograft tissue samples were ground under liquid nitrogen and lysed in 1× Cell Signaling lysis buffer supplemented with 1 mM PMSF, spun through a QIAshredder column (Qiagen), and freeze-thawed three times. After centrifugation at 20,000g for 30 min at 4°C, the protein concentration in the supernatant was determined using a BCA protein assay kit (Bio-Rad). Per sample, 5 mg of protein was subjected to ETV7 IP. Antibody (2 μg) was added to 500 μl of lysate, and samples were rotated overnight at 4°C. Protein G–coated Dynabeads (Life Technologies) were added (10 μl per sample), and samples were rotated at 4°C for 90 min. Bead-antibody-protein complexes were captured using a DynaMag-2 magnet (Life Technologies) and washed four times with CHAPS lysis buffer. Antibody-protein complexes were retrieved from the beads by heating at 70°C in 1.25× LDS loading buffer (Life Technologies) in CHAPS lysis buffer and loaded on precast 4 to 12% bis-tris protein gels (Life Technologies). Proteins were transferred onto nitrocellulose membranes using the iBLOT system (Life Technologies) following the manufacturer’s protocol. Membranes were blocked with 5% milk and 0.1% Tween 20 in tris-buffered saline (TBS) and incubated with the appropriate antibodies in 5% bovine serum albumin in TBS with 0.1% Tween 20 overnight at 4°C. All primary antibody incubations were followed by incubation with secondary horseradish peroxidase (HRP)–conjugated antibody (Pierce) in 5% milk and 0.1% Tween 20 in TBS and visualized using SuperSignal West Pico or Femto Chemiluminescent Substrate (Pierce) on BioMax MR film (Kodak).
Antibodies
The following antibodies were used: anti-mTOR, anti-Raptor, anti-mSIN1, and anti-mLST8 antibodies (Bethyl Laboratories); anti-Frap and anti-DEK antibodies (Santa Cruz Biotechnology); anti-(p-)P70S6KThr389, anti−(p-)AKTThr308, anti−(p-)AKTSer473, anti−(p-)NDRGIThr346, anti−(p-)4E-BP1Thr37/46, anti−(p-)4E-BP1Ser65, anti−(p-)4E-BP1Thr70, anti−(p-)ERK1/2Thr202/Tyr204, anti-p62/SQSTM1, anti-IKBKE, anti−(p-)IKBKESer172, anti-TBK1, anti−(p-)TBK1Ser172, anti−(p-)Tyr, anti−(p-)GSK3-βSer9, anti−GSK3-β, anti-PTK2, anti−(p-)PTK2Tyr398, anti-GRB10, anti−(p-)GRB10Ser501/503, anti−4E-BP1, anti−4E-BP2, anti-eIF4E, and anti−FKBP-51 (Cell Signaling Technologies); anti–p-mTORSer2448 and anti-Rictor antibody (Millipore); anti-LAMP1 and anti–KI-67 (BD Pharmingen); anti–FLAG-M2 and anti-LC3BI/II (Sigma); anti-RUVBL2 (Protein Tech Group); and anti–FKBP-12 (Abcam). ETV7 rabbit polyclonal (15) was raised in the Grosveld laboratory, and generation of the rat monoclonal antibody (7E4) against a C-terminal peptide (FKDKRPEISP) was outsourced (Absea). Both antibodies were affinity-purified before use. Detection of ETV7 protein immunoprecipitated with 7E4 antibody on most immunoblots was done with a commercial ETV7 antibody (Sigma, #HPA 029033).
ETV7/mTOR in vitro association
Molar equivalents (100 ng of mTOR and 13 ng of ETV7) of purified mTOR and ETV7 (Origene) were mixed in 500 μl of CHAPS lysis buffer and incubated for 8 or 24 hours at 4°C while rotating. Control recombinant human RUVBL2 protein (not present in mTORC3) was purified from Escherichia coli in the Grosveld laboratory. Co-IP experiments were performed using 2 μg of appropriate antibodies.
MS of purified mTOR and ETV7 proteins
Purified human mTOR or ETV7 protein (1 μg; OriGene) was loaded on a precast 4 to 12% bis-tris protein gel and run shortly, and the protein bands were cut out. The proteins were reduced with dithiothreitol (DTT) and alkylated with iodoacetamide in situ to allow recovery of cysteine-containing peptides. The gel bands were washed, dried, and rehydrated with a buffer containing trypsin. The resulting peptides were diluted and analyzed by short-gradient liquid chromatography (LC)–MS/MS. Peptide samples were loaded on a nanoscale capillary reverse-phase C18 column by a high-performance LC system (Thermo EASY-nLC 1000) and eluted by a gradient (~90 min). Eluted peptides were ionized by electrospray ionization and detected by an in-line mass spectrometer (LTQ-Orbitrap Elite). MS spectra were collected first, and the top 20 abundant ions were sequentially isolated for MS/MS analysis. This process was cycled over the entire LC gradient, and more than 10,000 MS/MS spectra were acquired during a 90-min elution. Database searches were done with the Jump software suite for protein identification (57). All matched MS/MS spectra were filtered by mass accuracy and matching scores to eliminate false discoveries. Checking of the copurified proteins against the 1.1 database of nonspecific binding proteins (www.CRAPome.org suggests that all copurified proteins (table S1, G and H) are known contaminants of immune-purified protein preparations.
Subcellular fractionation IPs
Subcellular fractions were prepared using the Qproteome Cell Compartment Kit (Qiagen) with the following minor alterations: Per fractionation, 107 cells were used in one-third the buffer volume described in the Qproteome handbook. After fractionation, six individually isolated fractions were combined and subjected to co-IP in the presence of 0.3% CHAPS. From each co-IP input sample, a proportion was analyzed by Western blotting with anti-tubulin (cytoplasm), anti-LAMP1 (membrane), and anti-DEK (nucleus) antibodies to verify lack of cross-contamination between the fractions.
Superose 6 gel filtration
Whole-cell lysate was prepared from 2 × 107 Karpas-299 cells in 500 μl of CHAPS lysis buffer supplemented with 100 mM l-arginine, followed by four 10-min centrifugation runs at 20,000g at 4°C. Lysate was separated on a 20-ml Superose 6 gel filtration column (GE Healthcare) in phosphate-buffered saline using a UPC-900 AKTA FPLC system (Amersham). Fractions of 400 μl were collected into deep-well collection plates preloaded with 100 μl of 5× concentrated CHAPS lysis buffer. Collected fractions were analyzed for the presence of ETV7 and mTOR protein by Western blotting. Fractions 1 to 16 from identical but separate runs on the same column were immunoprecipitated with 3 μg of mTOR or ETV7 antibody per 250 μl of collected fraction. The Western blot of mTOR IPs was probed for mTOR, Rictor, Raptor, and ETV7, and that of ETV7 IPs was probed for mTOR, ETV7 and p–4E-BP1Ser37/46. The peak Raptor- and Rictor-containing fractions were used as molecular mass markers, given that mTORC1 measures 1 MDa and mTORC2 measures 1.3 MDa (4, 5). In addition, a size calibration standard (1.35 to 670 kDa; Gel Filtration Standard, Bio-Rad) was run to size lower molecular mass fractions.
Immunohistochemistry
Tissues were fixed in 10% formalin, processed, and embedded in paraffin. Thin (5 μm) sections were cut, dried overnight, and baked at 65°C for 30 min. Antigen retrieval was performed using citrate buffer at pH 6 (Sigma) for 15 min at 100°C. Endogenous peroxidase activity was blocked by incubating the slides in 3% peroxide in methanol for 5 min. All subsequent steps were intermitted by washing in TBS with 0.5% Tween 20. Endogenous biotin was blocked using an avidin/biotin blocking kit (Vector Labs), followed by a 30-min protein-blocking step with serum-free protein block (Life Technologies) at 37°C. Sections were incubated with anti-ETV7 antibody overnight at 4°C. For peptide competition, diluted antibody was incubated with ETV7 peptide at room temperature for 30 min before staining. Biotinylated secondary antibody (Vector Labs) was used at 6 μg/ml for 30 min at room temperature, followed by streptavidin-HRP (Dako) and 3,3′-diaminobenzidine (DAB) chromogen (Dako) following the manufacturer’s protocol. Images were captured using 200× magnification on a Nikon E800 microscope at the Cell Imaging Core Facility of St. Jude Children’s Research Hospital.
mTORC3 IP/kinase assay
Kinase assays were performed as previously described (58) with minor modifications. Kinase buffer consisted of 25 mM Hepes at pH 7.4 and 50 mM KCl. Just before use, 10 mM MgCl2, 10 nM of the PP1/2 inhibitor calyculin A, 0.5 mM DTT, and 1 μM of the MEK1/2 inhibitor UO126 were added. Karpas-299 cells (2 × 107) in logarithmic phase (0.5 × 106/ml) were lysed in CHAPS lysis buffer. Co-IP was performed using 2 μg of the appropriate antibodies. Captured antibody-protein complexes were preincubated with inhibitors where indicated [FKBP-12/rapamycin (37 μg/ml of FKBP-12 and 10 μM rapamycin), OSI-27 (10 μM), or AZD-8055 (10 μM)] for 30 min at 30°C while shaking. Purified recombinant GST-P70S6KThr389 peptide (80 ng; Millipore) was added to preincubated antibody-protein complexes in the presence of 250 μM ATP and incubated for 30 min at 30°C in a final volume of 30 μl. Kinase reactions were terminated by adding LDS loading buffer to 100 μl, of which 20 μl was loaded on precast 4 to 12% bis-tris gels (Life Technologies). The kinase substrates were visualized using phospho-specific antibodies against p-P70S6KThr389, and a GST antibody (EMD Millipore) was used to control for equal loading.
mTOR inhibition
Cells were plated 24 hours before treatment at low density (0.05 × 106/ml) to allow for rapid division and maximal mTORC3 expression. Cells were harvested and replated in fresh medium containing escalating amounts (0.1, 0.3, 1, 3, 10, 30, 100, 300, and 1000 nM) of rapamycin. Dose-escalation studies were performed in triplicate and terminated when untreated control cultures reached three population doublings. Cell counts and viability for all cultures were determined using a Vi-CELL Cell Viability Analyzer (Beckmann Coulter) using trypan blue exclusion.
Combined NPM-ALK and mTOR inhibition
Karpas-299 cells were plated 24 hours before alectinib (Selleckchem) treatment at 0.05 × 106 cells/ml. Alectinib dose-escalation studies (0.1, 0.3, 1, 3, 10, 30, 100, 300, and 1000 nM) were performed in triplicate and terminated when untreated control cultures reached three population doublings. Given that 10 nM alectinib was the maximum concentration at which Karpas-299 proliferation was not inhibited, cells were treated with 10 nM alectinib and an escalating dose of rapamycin (0.1, 1, 10, 100, and 100 ng/ml) in triplicate and terminated when 10 nM alectinib alone control cultures reached three population doublings. Cell counts and viability were determined using a Vi-CELL Cell Viability Analyzer using trypan blue exclusion.
Raptor/Rictor knockdown
EW8 cells expressing GFP alone or GFP and exogenous ETV7 and Karpas-299 cells were transduced with pLKO1-puro vectors encoding shRNA against either Raptor (Addgene vector 21339) or Rictor (Addgene vector 21341) or a scrambled shRNA (Addgene vector 1864) (58). Forty-eight hours after transduction, cells were selected using puromycin (1 μg/ml) (Sigma) for 72 hours. Selected cells were harvested for Western blot analysis and co-IP of mTOR complexes as described above.
Rictor knockout in mouse Rictor fl/fl MEFs
MEFs were isolated from day 13.5 Rictorfl/fl mouse embryos (Jax #020649) and cultured in DMEM, 10% FBS, 5% CO2, and 3% O2. Cells were transduced with pCL20c-TKp-ETV7-Actp-blasticidin or pCL20c-TKp-Actp-blasticidin empty vector and selected on blasticidin (1 μg/ml) for 5 days. Vector cells, ETV7 cells, and untransduced cells (2 × 106) were transduced with a MOI of 200 with adenovirus GFP-iCre (Vector Biolabs #1776) overnight, followed by cell lysis 72 hours later and Western blot analysis.
ETV7 shRNA knockdown
shRNA vectors were constructed by cloning the inducible shRNA MIR30 cassette from pTRIPZ constructs (Open Biosystems) into the pCL20c lentiviral backbone. The shRNA constructs consisted of a sense (ETV7: ACGCCACTATTATAAGCTTAAT; nontargeting: TCTCGCTTGGGCGAGAGTAAG) and antisense sequence separated by a loop (TAGTGAAGCCACAGATGTA). Mouse actin promoter GFP was cloned 3′ of the shRNA–red fluorescent protein (RFP) cassette to allow selection of vector-containing cells. Addition of doxycycline induces expression of RFP in-frame with the shRNA mir30 backbone. For time-lapse imaging, GFP+ DAOY cells, which grow attached and express endogenous ETV7/mTORC3, were plated at 200 cells/cm2 on four-well multichamber coverslip slides (Thermo Fisher Scientific) in medium containing doxycycline (1 μg/ml) 48 hours before imaging. Forty fields per well were followed for 60 hours using 400× magnification on a Marianas Inverted Spinning Disk Confocal Microscope. Imaging was performed in a humidified chamber at 37°C and 5% CO2 in the live-cell imaging core of St. Jude Children’s Research Hospital.
Mice
RP11-978H23 BAC DNA (BACPAC Resources Centre), containing the human ETV7 gene locus, was microinjected into the pronucleus of fertilized FVB mouse oocytes and transferred to pseudo pregnant CD1 fosters. Tail snip DNA of offspring was genotyped using primers specific for exons 1 and 8 of human ETV7. Samples positive for both PCRs were subjected to PCR screening of the upstream and downstream sequences of ETV7, as well as the first and last exons of all open reading frames present within the RP11-918H23 BAC. When ETV7 was present (ETV7TG+/−), a second biopsy was subjected to fluorescent in situ hybridization using a fluorescein isothiocyanate–labeled RP11-918H23 probe by the Cytogenetics Core of St. Jude Children’s Hospital to determine copy number and potential mosaicism of the founders. ETV7TG+/− mice were backcrossed with C57B6 mice for six generations and were then crossed with Ptch1+/− mice (43). Littermates were monitored for tumor formation for 500 days. Mice presenting with masses were observed for lethargy and sacrificed when moribund, or if they displayed symptoms such as paralysis or when the tumor volume reached 20% of body mass in accordance with guidelines approved by the Institutional Animal Care and Use Committee of St. Jude Children’s Research Hospital.
To generate ETV7TGΔ9+/− mice, 50 fertilized eggs of an ETV7TG+/− × C57B6 cross were injected with Cas9 RNA (200 ng/ml) and single-guide RNA (50 ng/ml), both transcribed in vitro, targeting codon 82 of ETV7 exon 3 (guide sequence: GAGCACGGGTTCGAGAT↓GAA) following the methods of Yang et al. (59). The Cas9 RNA was capped and polyadenylated using the vaccinia RNA capping system (NEB) and E. coli poly(A) polymerase (NEB). Tail DNA of five offspring was checked for ETV7 exon 3 deletions using the surveyor assay kit (Transgenomic). Two pups contained an ETV7 exon 3 mutation, one of which consisted of a 9–base pair (bp) deletion encoding a protein missing the sequence E81M82N83 as confirmed by DNA sequence analysis. Combined RT-PCR/DNA sequence analysis of bone marrow RNA confirmed that this mouse expressed ETV7Δ9 mRNA only. This mouse was crossed with Ptch+/− mice to yield Ptch+/−/ETV7TG-Δ9 mice. Littermates were monitored for tumor formation for 365 days.
Histology
Tumors were dissected from surrounding tissues, fixed in 10% neutral buffered formalin, and paraffin-embedded. Tumors were diagnosed independently by two veterinarian pathologists at St. Jude. For diagnostic immunohistochemistry, 4- to 6-μm serial sections were deparaffinized in xylene and rehydrated by incubation in graded alcohol solutions. Heat-induced epitope retrieval (HIER) was performed using a decloaking chamber (BioCare Medical). After HIER, all steps were performed on an autostainer (Thermo) at room temperature with rinses in TBS and 0.05% Tween 20 between steps. Endogenous peroxidases were blocked with 3% hydrogen peroxide. Following antibody staining, DAB (Thermo Shandon) was used as the chromogenic substrate, and sections were counterstained with Mayer’s hematoxylin (Thermo Shandon). For labeling of MSA (Dako) and α-SMA (Dako), the primary antibody was biotinylated using the ARK Kit (Dako) following the manufacturer’s instructions. HIER was performed with citrate buffer (pH 6) (Invitrogen) for 15 min at 110°C. Sections were incubated with the biotinylated primary antibody at 1:50 for 30 min, followed by the HRP-labeled streptavidin method (Thermo Shandon). For labeling of desmin, HIER was performed with citrate buffer (pH 6) (Invitrogen) for 15 min at 110°C. Sections were blocked with Background Sniper for 30 min (BioCare Medical) and incubated with primary antibody (1:600, 30 min; Thermo Fisher Scientific) and with HRP-labeled Rodent-on-Rodent polymer (BioCare Medical) for 30 min. For Ki67 staining, the same procedure was followed (60 min, primary antibody, 1:200; Thermo Shandon). Before myogenin staining, HIER was performed with Target Retrieval (pH 9) (Dako) for 15 min at 110°C. Sections were blocked with Rodent Block (Quanto Kit, BioCare Medical) for 30 min, incubated with primary antibody (1:100, 30 min; Dako), followed by incubation with HRP-labeled Mouse-on-Mouse polymer (BioCare Medical) for 30 min. Staining using antibodies against ETV7 and p–4E-BP1 was performed as described above.
Isolation of primary murine Ptch1+/−/ETV7TG ERMS tumor cell lines
For isolation of cell lines from primary murine ERMS, tumors were dissected and rinsed in Hanks’ balanced salt solution (HBSS) with calcium and magnesium (cellgro). Ten–cubic millimeter pieces of tumor were minced using scalpels in HBSS solution and transferred to 1.5 ml of snap cap tubes, supplemented with liberase TL enzyme blend (Roche) and incubated at 37°C for 45 min with vigorous mixing every 15 min. The resulting cell suspensions were passed over 70-μm cell strainers and spun at 350g for 5 min. Cell pellets were resuspended in DMEM supplemented with 10% BCS, 2 mM GlutaMAX, 25 mM Hepes (Gibco), penicillin (100 U/ml), and streptomycin (100 μg/ml), and incubated in a humidified incubator at 37°C and 5% CO2. Twenty-four hours after isolation, medium was changed and cells were split (1 to 3) when the culture was subconfluent. Cell counts and viability for all cultures were determined using a Vi-CELL Cell Viability Analyzer. Isolated cell lines were plated on multiwell slides and stained for muscle-specific markers to ensure their identity.
qRT-PCR of RNA from the RD, JR, and 293T cell lines
Total RNA was isolated from 2 × 106 RD, JR, and 293T cells. First-strand cDNA was synthesized using the Invitrogen SuperScript Kit using oligo-dT priming. qPCR was performed using the CYBR Green PCR Master Mix from ABI using ETV7 primers (forward, TGAAGCTCAAATTAACCTGCT; reverse, TCCTCCCT GCTCCACAGT) and hypoxanthine-guanine phosphoribosyltransferase primers (Bio-Rad PrimePCR primer no. qHsaCID0016375). Data are means ± SEM of three independent experiments.
Generation of ETV7 mutants
The PNT and ETS domains of ETV6 and ETV7 are highly homologous (fig. S4). On the basis of the 3D structure of the ETV6 pointed domain (60), which contains two interaction surfaces (called ML and EH), indicated in red and yellow in fig. S4, respectively, we introduced mutations in ETV7 PNT sequence that coincide with the ETV6 ML surface (ΔE81MN83 = Δ9; ΔM82NGRALCIL90 = Δ27).
We created these deletion mutants by using double-stranded oligonucleotides (Integrated DNA Technologies) spanning the sequence between the Sac II and Apa I sites (149 bp) in the ETV7 cDNA containing Δ9-bp (ΔE79MN81) or Δ27-bp (Δ M80 NGRALCIL88) deletions (fig. S4). After cloning into the Sac II and Apa I sites of pCL20c-TKp-ETV7-Actp-blasticidin, the presence of the mutations was verified by DNA sequence analysis (pCL20c-TKp-ETV7-PNTΔ9-Actp-blasticidin and pCL20c-TKp-ETV7-PNTΔ27-Actp-blasticidin). The ETV-KALK mutation has been described before (17), and the ETV7-KALK Eco RI fragment was shuttled into the Eco RI site of pCL20c-TKp-ActP-blasticidin.
EW8 cells were transduced with each of the pCL20c-ETV7-Actp-blasticidin lentiviral vectors (ETV7-WT, ETV7-PNTΔ9, ETV7-PNTΔ27, and ETV7-KALK) and selected for 1 week with blasticidin (1 μg/ml). Resistant cells were expanded for 3 weeks to obtain maximally proliferating cultures, which were then tested for rapamycin resistance by plating the cells at a density of 2 × 104 per well in a six-well plate. Twenty-four hours later, cultures were treated with rapamycin at 0, 0.1, 0.3, 1, 3, 10, 100, and 1000 ng/ml for 4 days. After cell lysis in hypotonic buffer, nuclei were counted using a Vi-CELL Viability Analyzer.
Statistical analysis
Data were expressed as means ± SD or SEM of at least three independent experiments and evaluated using Student’s t test for unpaired samples.
Supplementary Material
Acknowledgments
We thank M. van der Ent for providing purified RuvBL2 protein; G. Germain for generating the OS-17 cell line from OS-17 xenografts; C. Morton for providing the Karpas-299 and EW8 cell lines and the BT28, BT39, and BT41 xenograft samples and sections; J. Rehg for diagnosis of ERMS tumors; C. Fraga for help with immunohistochemistry; C. Carella for performing experiments that initiated this study; J. Sublett for isolation and microinjection of ETV7 BAC DNA; A. Marshall and I. Lagutina for technical assistance and helpful discussions regarding muscle cell culture; S. Savage and M. Anderson for assistance with necropsy; P. Johnson for help with immunohistochemistry; A. Interiano for help with immunofluorescence; M. Hatley for providing JR-1 and RD cDNA; H. Chi and Y.-G. Han for providing Rictorfl/fl mice; D. Sabatini for providing scrambled, Raptor, and Rictor knockdown lentiviral vectors via Addgene (vector #21339, #21341, and #1864); F. Zhang for providing pX330 via Addgene (#42230); and S. d’Azzo for helpful suggestions. We apologize to investigators in the mTOR field whose work could not be cited due to space constraints. Funding: This work was supported in part by the Van Vleet Foundation of Memphis and grants R01CA-72999, RO1CA-77776 (to G.C.G.), and RO1CA-165995 (to P.J.H.); the Cancer Center Core Grant (CA021765); and the American Lebanese Syrian Associated Charities. Author contributions: F.C.H. designed and performed experiments. R.I.K.G. designed and performed experiments and wrote parts of the paper. B.P.O., I.E., and L.P. provided technical assistance. M.C. developed the pre-B cell cultures and performed the Affymetrix analysis. L.J. performed pathological analysis of mice. D.F. analyzed St. Jude ERMS RNA sequence data. P.J.H. collaborated during the initial phase of the experiments. G.C.G. conceived the project, designed and supervised experiments, and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/9/eaar3938/DC1
Fig. S1. ETV7 is expressed in pediatric malignancies.
Fig. S2. PI3K and PTK2 signaling and anti–p-tyr Western blot of cell lysate of vector and ETV7 mouse pre-B cells.
Fig. S3. ETV7 expression in human pediatric tumor xenografts.
Fig. S4. Sequence comparison of ETV6 and ETV7 PNT domains and the position of Δ9, Δ27, Δ120/Δ115, Δ120/Δ159/+60 mutations in the ETV7 PNT domain, and the KALK mutation in the ETV7 ETS domain.
Fig. S5. mTOR localization and assembly.
Fig. S6. Effects of mTOR inhibitors and mTORC3 loss on mTOR signaling.
Fig. S7. Effect of ALK inhibition on p-AKTSer473 and relative FK506-binding protein expression in Karpas-299 cells.
Fig. S8. mTORC3 kinase is insensitive to Raptor or Rictor knockdown or Rictor knockout.
Fig. S9. ERMS-specific markers in Ptch+/−/ETVTG+/− tumors are preserved in Ptch+/−/ETVTG+/− cell lines.
Fig. S10. Whole phospho-p70S6KThr389 and p70S6K Western blots relating to Figs. 1C and 5D.
Table S1. Expression effects of ETV7.
Movie S1. Induction of non-targeting ETV7shRNA in human DAOY medulloblastoma cells.
Movie S2. Induction of targeting ETV7shRNA in human DAOY medulloblastoma cells.
REFERENCES AND NOTES
- 1.Saxton R. A., Sabatini D. M., mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oh W. J., Wu C.-c., Kim S. J., Facchinetti V., Julien L.-A., Finlan M., Roux P. P., Su B., Jacinto E., mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 29, 3939–3951 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zinzalla V., Stracka D., Oppliger W., Hall M. N., Activation of mTORC2 by association with the ribosome. Cell 144, 757–768 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Aylett C. H. S., Sauer E., Imseng S., Boehringer D., Hall M. N., Ban N., Maier T., Architecture of human mTOR complex 1. Science 351, 48–52 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Baretić D., Berndt A., Ohashi Y., Johnson C. M., Williams R. L., Tor forms a dimer through an N-terminal helical solenoid with a complex topology. Nat. Commun. 7, 11016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang H., Rudge D. G., Koos J. D., Vaidialingam B., Yang H. J., Pavletich N. P., mTOR kinase structure, mechanism and regulation. Nature 497, 217–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chresta C. M., Davies B. R., Hickson I., Harding T., Cosulich S., Critchlow S. E., Vincent J. P., Ellston R., Jones D., Sini P., James D., Howard Z., Dudley P., Hughes G., Smith L., Maguire S., Hummersone M., Malagu K., Menear K., Jenkins R., Jacobsen M., Smith G. C. M., Guichard S., Pass M., AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 70, 288–298 (2010). [DOI] [PubMed] [Google Scholar]
- 8.García-Martínez J. M., Moran J., Clarke R. G., Gray A., Cosulich S. C., Chresta C. M., Alessi D. R., Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem. J. 421, 29–42 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thoreen C. C., Kang S. A., Chang J. W., Liu Q., Zhang J., Gao Y., Reichling L. J., Sim T., Sabatini D. M., Gray N. S., An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284, 8023–8032 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armengol G., Rojo F., Castellví J., Iglesias C., Cuatrecasas M., Pons B., Baselga J., Ramón y Cajal S., 4E-binding protein 1: A key molecular “funnel factor” in human cancer with clinical implications. Cancer Res. 67, 7551–7555 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Dowling R. J. O., Topisirovic I., Alain T., Bidinosti M., Fonseca B. D., Petroulakis E., Wang X., Larsson O., Selvaraj A., Liu Y., Kozma S. C., Thomas G., Sonenberg N., mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328, 1172–1176 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Livingstone M., Bidinosti M., Rapamycin-insensitive mTORC1 activity controls eIF4E:4E-BP1 binding. F1000Res. 1, 4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tamburini J., Green A. S., Bardet V., Chapuis N., Park S., Willems L., Uzunov M., Ifrah N., Dreyfus F., Lacombe C., Mayeux P., Bouscary D., Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood 114, 1618–1627 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Potter M. D., Buijs A., Kreider B., van Rompaey L., Grosveld G. C., Identification and characterization of a new human ETS-family transcription factor, TEL2, that is expressed in hematopoietic tissues and can associate with TEL1/ETV6. Blood 95, 3341–3348 (2000). [PubMed] [Google Scholar]
- 15.Cardone M., Kandilci A., Carella C., Nilsson J. A., Brennan J. A., Sirma S., Ozbek U., Boyd K., Cleveland J. L., Grosveld G. C., The novel ETS factor TEL2 cooperates with Myc in B lymphomagenesis. Mol. Cell. Biol. 25, 2395–2405 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carella C., Potter M., Bonten J., Rehg J. E., Neale G., Grosveld G. C., The ETS factor TEL2 is a hematopoietic oncoprotein. Blood 107, 1124–1132 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawagoe H., Potter M., Ellis J., Grosveld G. C., TEL2, an ETS factor expressed in human leukemia, regulates monocytic differentiation of U937 cells and blocks the inhibitory effect of TEL1 on ras-induced cellular transformation. Cancer Res. 64, 6091–6100 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Ross M. E., Mahfouz R., Onciu M., Liu H.-C., Zhou X., Song G., Shurtleff S. A., Pounds S., Cheng C., Ma J., Ribeiro R. C., Rubnitz J. E., Girtman K., Williams W. K., Raimondi S. C., Liang D.-C., Shih L.-Y., Pui C.-H., Downing J. R., Gene expression profiling of pediatric acute myelogenous leukemia. Blood 104, 3679–3687 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Ross M. E., Zhou X., Song G., Shurtleff S. A., Girtman K., Williams W. K., Liu H.-C., Mahfouz R., Raimondi S. C., Lenny N., Patel A., Downing J. R., Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood 102, 2951–2959 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Neale G., Su X., Morton C. L., Phelps D., Gorlick R., Lock R. B., Reynolds C. P., Maris J. M., Friedman H. S., Dome J., Khoury J., Triche T. J., Seeger R. C., Gilbertson R., Khan J., Smith M. A., Houghton P. J., Molecular characterization of the pediatric preclinical testing panel. Clin. Cancer Res. 14, 4572–4583 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson M. C., Fuller C., Hogg T. L., Dalton J., Finkelstein D., Lau C. C., Chintagumpala M., Adesina A., Ashley D. M., Kellie S. J., Taylor M. D., Curran T., Gajjar A., Gilbertson R. J., Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J. Clin. Oncol. 24, 1924–1931 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Matos J. M., Witzmann F. A., Cummings O. W., Schmidt C. M., A pilot study of proteomic profiles of human hepatocellular carcinoma in the United States. J. Surg. Res. 155, 237–243 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H.-C., Appeddu P. A., Isoda H., Guan J.-L., Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J. Biol. Chem. 271, 26329–26334 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Chiang G. G., Abraham R. T., Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J. Biol. Chem. 280, 25485–25490 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Waskiewicz A. J., Johnson J. C., Penn B., Mahalingam M., Kimball S. R., Cooper J. A., Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol. Cell. Biol. 19, 1871–1880 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scheper G. C., Morrice N. A., Kleijn M., Proud C. G., The mitogen-activated protein kinase signal-integrating kinase Mnk2 is a eukaryotic initiation factor 4E kinase with high levels of basal activity in mammalian cells. Mol. Cell. Biol. 21, 743–754 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pyronnet S., Imataka H., Gingras A.-C., Fukunaga R., Hunter T., Sonenberg N., Human eukaryotic translation initiation factor 4G (eIF4G) recruits Mnk1 to phosphorylate eIF4E. EMBO J. 18, 270–279 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.United Kingdom Cancer Cytogenetics Group (UKCCG) , Translocations involving 12p in acute myeloid leukemia: Association with prior myelodysplasia and exposure to mutagenic agents. Genes Chromosomes Cancer 5, 252–254 (1992). [DOI] [PubMed] [Google Scholar]
- 29.So L., Lee J., Palafox M., Mallya S., Woxland C. G., Arguello M., Truitt M. L., Sonenberg N., Ruggero D., Fruman D. A., The 4E-BP–eIF4E axis promotes rapamycin-sensitive growth and proliferation in lymphocytes. Sci. Signal. 9, ra57 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yanagiya A., Suyama E., Adachi H., Svitkin Y. V., Aza-Blanc P., Imataka H., Mikami S., Martineau Y., Ronai Z. A., Sonenberg N., Translational homeostasis via the mRNA cap-binding protein, eIF4E. Mol. Cell 46, 847–858 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu Y., Yoon S.-O., Poulogiannis G., Yang Q., Ma X. M., Villén J., Kubica N., Hoffman G. R., Cantley L. C., Gygi S. P., Blenis J., Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332, 1322–1326 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Reilly K. E., Rojo F., She Q.-B., Solit D., Mills G. B., Smith D., Lane H., Hofmann F., Hicklin D. J., Ludwig D. L., Baselga J., Rosen N., mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nojima H., Tokunaga C., Eguchi S., Oshiro N., Hidayat S., Yoshino K.-i., Hara K., Tanaka N., Avruch J., Yonezawa K., The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 278, 15461–15464 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Kim C. A., Phillips M. L., Kim W., Gingery M., Tran H. H., Robinson M. A., Faham S., Bowie J. U., Polymerization of the SAM domain of TEL in leukemogenesis and transcriptional repression. EMBO J. 20, 4173–4182 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hollenhorst P. C., McIntosh L. P., Graves B. J., Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 80, 437–471 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ali S. M., Sabatini D. M., Structure of S6 kinase 1 determines whether raptor-mTOR or rictor-mTOR phosphorylates its hydrophobic motif site. J. Biol. Chem. 280, 19445–19448 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Shin S., Wolgamott L., Tcherkezian J., Vallabhapurapu S., Yu Y., Roux P. P., Yoon S.-O., Glycogen synthase kinase-3β positively regulates protein synthesis and cell proliferation through the regulation of translation initiation factor 4E-binding protein 1. Oncogene 33, 1690–1699 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Cross D. A. E., Alessi D. R., Cohen P., Andjelkovich M., Hemmings B. A., Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789 (1995). [DOI] [PubMed] [Google Scholar]
- 39.Staber P. B., Vesely P., Haq N., Ott R. G., Funato K., Bambach I., Fuchs C., Schauer S., Linkesch W., Hrzenjak A., Dirks W. G., Sexl V., Bergler H., Kadin M. E., Sternberg D. W., Kenner L., Hoefler G., The oncoprotein NPM-ALK of anaplastic large-cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood 110, 3374–3383 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Schreiber K. H., Ortiz D., Academia E. C., Anies A. C., Liao C.-Y., Kennedy B. K., Rapamycin-mediated mTORC2 inhibition is determined by the relative expression of FK506-binding proteins. Aging Cell 14, 265–273 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahajan K., Mahajan N. P., PI3K-independent AKT activation in cancers: A treasure trove for novel therapeutics. J. Cell. Physiol. 227, 3178–3184 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarbassov D. D., Ali S. M., Kim D.-H., Guertin D. A., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M., Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Goodrich L. V., Milenković L., Higgins K. M., Scott M. P., Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109–1113 (1997). [DOI] [PubMed] [Google Scholar]
- 44.Hinson A. R. P., Jones R., Crose L. E. S., Belyea B. C., Barr F. G., Linardic C. M., Human rhabdomyosarcoma cell lines for rhabdomyosarcoma research: Utility and pitfalls. Front. Oncol. 3, 183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Falcon B. L., Barr S., Gokhale P. C., Chou J., Fogarty J., Depeille P., Miglarese M., Epstein D. M., McDonald D. M., Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res. 71, 1573–1583 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bashir T., Cloninger C., Artinian N., Anderson L., Bernath A., Holmes B., Benavides-Serrato A., Sabha N., Nishimura R. N., Guha A., Gera J., Conditional astroglial Rictor overexpression induces malignant glioma in mice. PLOS ONE 7, e47741 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masri J., Bernath A., Martin J., Jo O. D., Vartanian R., Funk A., Gera J., mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 67, 11712–11720 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Yori J. L., Lozada K. L., Seachrist D. D., Mosley J. D., Abdul-Karim F. W., Booth C. N., Flask C. A., Keri R. A., Combined SFK/mTOR inhibition prevents rapamycin-induced feedback activation of AKT and elicits efficient tumor regression. Cancer Res. 74, 4762–4771 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cussac D., Greenland C., Roche S., Bai R.-Y., Duyster J., Morris S. W., Delsol G., Allouche M., Payrastre B., Nucleophosmin-anaplastic lymphoma kinase of anaplastic large-cell lymphoma recruits, activates, and uses pp60c-src to mediate its mitogenicity. Blood 103, 1464–1471 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Poe J. C., Minard-Colin V., Kountikov E. I., Haas K. M., Tedder T. F., A c-Myc and surface CD19 signaling amplification loop promotes B cell lymphoma development and progression in mice. J. Immunol. 189, 2318–2325 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smithson L. J., Gutmann D. H., Proteomic analysis reveals GIT1 as a novel mTOR complex component critical for mediating astrocyte survival. Genes Dev. 30, 1383–1388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bendell J. C., Kelley R. K., Shih K. C., Grabowsky J. A., Bergsland E., Jones S., Martin T., Infante J. R., Mischel P. S., Matsutani T., Xu S., Wong L., Liu Y., Wu X., Mortensen D. S., Chopra R., Hege K., Munster P. N., A phase I dose-escalation study to assess safety, tolerability, pharmacokinetics, and preliminary efficacy of the dual mTORC1/mTORC2 kinase inhibitor CC-223 in patients with advanced solid tumors or multiple myeloma. Cancer 121, 3481–3490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mateo J., Olmos D., Dumez H., Poondru S., Samberg N. L., Barr S., Van Tornout J. M., Jie F., Sandhu S., Tan D. S., Moreno V., LoRusso P. M., Kaye S. B., Schöffski P., A first in man, dose-finding study of the mTORC1/mTORC2 inhibitor OSI-027 in patients with advanced solid malignancies. Br. J. Cancer 114, 889–896 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calimeri T., Ferreri A. J. M., m-TOR inhibitors and their potential role in haematological malignancies. Br. J. Haematol. 177, 684–702 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Huang S., Yang Z. J., Yu C., Sinicrope F. A., Inhibition of mTOR kinase by AZD8055 can antagonize chemotherapy-induced cell death through autophagy induction and down-regulation of p62/sequestosome 1. J. Biol. Chem. 286, 40002–40012 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Preuss E., Hugle M., Reimann R., Schlecht M., Fulda S., Pan-mammalian target of rapamycin (mTOR) inhibitor AZD8055 primes rhabdomyosarcoma cells for ABT-737-induced apoptosis by down-regulating Mcl-1 protein. J. Biol. Chem. 288, 35287–35296 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X., Li Y., Wu Z., Wang H., Tan H., Peng J., JUMP: A tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol. Cell. Proteomics 13, 3663–3673 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M., Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Yang H., Wang H., Shivalila C. S., Cheng A. W., Shi L., Jaenisch R., One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marchler-Bauer A., Derbyshire M. K., Gonzales N. R., Lu S., Chitsaz F., Geer L. Y., Geer R. C., He J., Gwadz M., Hurwitz D. I., Lanczycki C. J., Lu F., Marchler G. H., Song J. S., Thanki N., Wang Z., Yamashita R. A., Zhang D., Zheng C., Bryant S. H., CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/9/eaar3938/DC1
Fig. S1. ETV7 is expressed in pediatric malignancies.
Fig. S2. PI3K and PTK2 signaling and anti–p-tyr Western blot of cell lysate of vector and ETV7 mouse pre-B cells.
Fig. S3. ETV7 expression in human pediatric tumor xenografts.
Fig. S4. Sequence comparison of ETV6 and ETV7 PNT domains and the position of Δ9, Δ27, Δ120/Δ115, Δ120/Δ159/+60 mutations in the ETV7 PNT domain, and the KALK mutation in the ETV7 ETS domain.
Fig. S5. mTOR localization and assembly.
Fig. S6. Effects of mTOR inhibitors and mTORC3 loss on mTOR signaling.
Fig. S7. Effect of ALK inhibition on p-AKTSer473 and relative FK506-binding protein expression in Karpas-299 cells.
Fig. S8. mTORC3 kinase is insensitive to Raptor or Rictor knockdown or Rictor knockout.
Fig. S9. ERMS-specific markers in Ptch+/−/ETVTG+/− tumors are preserved in Ptch+/−/ETVTG+/− cell lines.
Fig. S10. Whole phospho-p70S6KThr389 and p70S6K Western blots relating to Figs. 1C and 5D.
Table S1. Expression effects of ETV7.
Movie S1. Induction of non-targeting ETV7shRNA in human DAOY medulloblastoma cells.
Movie S2. Induction of targeting ETV7shRNA in human DAOY medulloblastoma cells.