

Shigella is an intracellular pathogen that invades the human host cell cytosol and exploits intracellular nutrients for growth, enabling the bacterium to create its own metabolic niche. For Shigella to effectively invade and replicate within the host cytoplasm, it must sense and adapt to changing environmental conditions; however, the mechanisms and signals sensed by S. flexneri are largely unknown. We have found that the secreted Shigella metabolism by-product formate regulates Shigella intracellular virulence gene expression and its ability to spread among epithelial cells. We propose that Shigella senses formate accumulation in the host cytosol as a way to determine intracellular Shigella density and regulate secreted virulence factors accordingly, enabling spatiotemporal regulation of effectors important for dampening the host immune response.
KEYWORDS: Shigella, formate, metabolism, virulence regulation
ABSTRACT
The intracellular human pathogen Shigella flexneri invades the colon epithelium, replicates to high cell density within the host cell, and then spreads to adjacent epithelial cells. When S. flexneri gains access to the host cytosol, the bacteria metabolize host cytosolic carbon using glycolysis and mixed acid fermentation, producing formate as a by-product. We show that S. flexneri infection results in the accumulation of formate within the host cell. Loss of pyruvate formate lyase (PFL; ΔpflB), which converts pyruvate to acetyl coenzyme A (CoA) and formate, eliminates S. flexneri formate production and reduces the ability of S. flexneri to form plaques in epithelial cell monolayers. This defect in PFL does not decrease the intracellular growth rate of S. flexneri; rather, it affects cell-to-cell spread. The S. flexneri ΔpflB mutant plaque defect is complemented by supplying exogenous formate; conversely, deletion of the S. flexneri formate dehydrogenase gene fdnG increases host cell formate accumulation and S. flexneri plaque size. Furthermore, exogenous formate increases plaque size of the wild-type (WT) S. flexneri strain and promotes S. flexneri cell-to-cell spread. We also demonstrate that formate increases the expression of S. flexneri virulence genes icsA and ipaJ. Intracellular S. flexneri icsA and ipaJ expression is dependent on the presence of formate, and ipaJ expression correlates with S. flexneri intracellular density during infection. Finally, consistent with elevated ipaJ, we show that formate alters S. flexneri-infected host interferon- and tumor necrosis factor (TNF)-stimulated gene expression. We propose that Shigella-derived formate is an intracellular signal that modulates virulence in response to bacterial metabolism.
INTRODUCTION
Shigella flexneri is an enteropathogenic subspecies of Escherichia coli that causes shigellosis, an acute mucosal inflammation resulting in severe bloody dysentery. After ingestion, Shigella traverses the digestive tract to the colon and crosses the colonic epithelium by exploiting M cells (1); the bacteria then invade the basolateral face of the epithelium using a contact-dependent type 3 secretion system (T3SS) encoded on a virulence plasmid, causing epithelial cells to engulf the bacteria. After Shigella enters the cell and escapes the host engulfment vacuole, it multiplies within the host cell cytoplasm and subsequently spreads to adjacent cells using the protein IcsA (also known as VirG), which catalyzes host actin synthesis, propelling the bacterium into neighboring cells (2, 3).
Expression of S. flexneri virulence genes inside the host epithelial cell is dynamic. Although initially required for invasion, T3SS genes are repressed upon entry into the host epithelial cell (4–6). The T3SS genes and additional cell-to-cell spread genes are later reactivated through an unidentified mechanism immediately prior to spread (6). S. flexneri expresses a suite of T3SS effectors to dampen the host response to cytosolic infection. The effectors IpgD, OspI, OspG, OspF, and IpaH work in concert to modulate inflammation (7, 8). Host intracellular trafficking, which alters both epithelial cell homeostasis and defense against cytosolic bacteria, is another target of S. flexneri-secreted effectors. The secreted effector IpaB redirects host cholesterol away from the Golgi apparatus, resulting in Golgi fragmentation (9). Likewise, the secreted effectors VirA and IpaJ alter Golgi apparatus-mediated cell trafficking (10, 11). IpaJ is a cysteine protease that alters N-myristoylation (11), ultimately inhibiting the trafficking of STING, a known host cytosolic sensor of S. flexneri infection (12, 13). This results in blocking the STING-mediated activation of the type I interferon response, including cytokines such as CXCL10 (11, 12).
S. flexneri differentially regulates over a quarter of its genes in the intracellular environment compared to S. flexneri grown in vitro, including many genes involved in central carbon metabolism (5, 14). S. flexneri tricarboxylic acid (TCA) cycle enzymes are repressed in the intracellular environment, whereas enzymes involved in glycolysis and mixed acid fermentation pathways are increased and necessary for virulence (15–17). Specifically, pflB, the gene encoding pyruvate formate lyase (PFL), and PFL-associated genes are upregulated in the host cell (5, 15). PFL converts pyruvate to acetyl coenzyme A (CoA), producing formate as a by-product. An S. flexneri ΔpflB mutant is defective in plaque formation; however, deletion of pflB does not impact S. flexneri growth rate within the host cell (15, 16).
The scope of this study was to determine the role of S. flexneri formate metabolism in virulence during the host-cytosolic phase of infection. We propose a model where Shigella-derived formate accumulates in the host cell cytoplasm and promotes the expression of IcsA and IpaJ, linking S. flexneri metabolism and intracellular cell density to intercellular spread and host-pathogen response.
RESULTS
Formate promotes S. flexneri plaque formation in Henle-407 monolayers.
One measure of S. flexneri virulence is the ability of S. flexneri to form plaques in cultured epithelial cell monolayers, which is contingent on both the ability of S. flexneri to replicate intracellularly and its ability to spread to adjacent host cells (18). Previous studies have shown that failure to form plaques, or formation of smaller-than-wild-type (WT) plaques, correlates with decreased virulence in animal models (18–20). An S. flexneri ΔpflB mutant forms smaller plaques in Henle-407 monolayers than does WT S. flexneri (15). Consistent with a previous study (16), the S. flexneri ΔpflB mutant showed no intracellular growth defect (see Fig. S1 in the supplemental material). Although not statistically different, the doubling time of the mutant was slightly shorter; as such, any effect of this small difference in growth over the time of plaque formation would be expected to lead to larger, rather than smaller, plaques. This suggested that formate, a product of PFL, secreted by S. flexneri promoted plaque formation independently of bacterial growth. If the plaque defect in the pflB mutant was due solely to the loss of formate, providing exogenous formate should rescue the ΔpflB plaque defect. At biological pH, formate cannot passively diffuse across bacterial or eukaryotic cell membranes (21). However, a previous study showed that exogenously supplemented formate could be taken up by cultured cells (22), and we confirmed that that Henle-407 cells take up radiolabeled formate when supplemented in tissue culture medium (Fig. S2). We supplemented tissue culture medium of cells infected with the S. flexneri PFL mutant (ΔpflB) with 20 mM formate after S. flexneri invasion and measured plaque formation. In the absence of exogenous formate, the average ΔpflB plaque size was approximately half the size of the WT strain (Fig. 1A), consistent with previous observations (14). However, with the addition of exogenous formate, the plaque size of the ΔpflB mutant was restored to WT size (Fig. 1A), suggesting that S. flexneri-derived formate promotes S. flexneri plaque formation. Additionally, the plaque size of the WT strain increased 2.3-fold in the presence of exogenous formate (Fig. 1A). The formate concentration used in these experiments is in the midrange of the dose-response curve (Fig. 1B) and is within the range found in the mammalian gastrointestinal tract (23–26).
FIG 1.

Formate-induced increase in S. flexneri cell-to-cell spread. (A) Wright-Giemsa stain of Henle-407 monolayers infected with S. flexneri and S. flexneri ΔpflB mutant with or without 20 mM formate. Plaque sizes were measured; an asterisk indicates statistical significance. (B) Plaque size of S. flexneri was measured when various concentrations of formate were added as supplements. The dotted line on the y axis indicates the plaque area when no formate was added as a supplement (0.1 mm2), while the dotted line on the x axis indicates the 50% effective concentration (28.4 mM). (C) Plaque size of S. flexneri was measured with 20 mM NaCl, formate, acetate, ethanol, or lactate; an asterisk indicates a statistically significant difference from MEM. Only formate was found to increase S. flexneri plaque size. (D) S. flexneri intracellular doubling time was measured in Henle-407 cells with or without 20 mM formate. (E) S. flexneri cell-to-cell spread was measured at 3 hpi with or without 20 mM formate; an asterisk indicates statistical significance.
S. flexneri intracellular doubling times of the WT and ΔpflB strains were measured in Henle-407 cells. No significant difference in doubling time was observed between the two strains. Download FIG S1, TIF file, 1.65 MB (1.7MB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
[14C]formate uptake was quantified in Henle-407 cells over the course of 48 hours. A significant increase in 14C cpm was observed over time. n = 3. Download FIG S2, TIF file, 0.85 MB (874.5KB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To determine if other fermentation by-products promote S. flexneri plaque formation, the medium of S. flexneri-infected Henle-407 cells was supplemented with 20 mM formate, acetate, ethanol, or lactate. With the exception of ethanol, these compounds were provided as supplements as sodium salts; thus, we included a control supplemented with 20 mM NaCl. Exclusively in the presence of formate, we observed a 2.7-fold increase in plaque area (Fig. 1C).
Although we cannot completely rule out an effect of formate on Henle-407 cells, experiments to find such an effect have been negative. Examination of the monolayers by light microscopy revealed no changes in morphology or cell density when Henle-407 cells were grown with 20 mM formate, and there was no effect on medium pH. Additionally, formate had no cytotoxic effects on Henle-407 cells, as measured by lactate dehydrogenase release (Fig. S3A), and formate did not alter the growth of Henle-407 cells (Fig. S3B).
(A) Uninfected Henle-407 cell cytotoxicity in the presence and absence of exogenous formate (20 mM) was determined by quantifying released lactate dehydrogenase (LDH) over 72 hours. We observed no significant difference in LDH release in the presence and absence of formate. (B) Henle-407 cells were plated in a 6-well plate and grown with and without formate. Cells were enumerated at 48 hours; no significant difference in Henle-407 cells per monolayer was observed. Download FIG S3, TIF file, 1.83 MB (1.8MB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Listeria monocytogenes is another pathogen that, similarly to Shigella, accesses the host cell cytosol and exploits host actin polymerization for cell-to-cell spread. We postulated that if formate was affecting host cell physiology to promote bacterial cell-to-cell spread, it would increase the plaque size of L. monocytogenes, as well as Shigella. However, supplementation with exogenous formate had no significant effect on the plaque size of L. monocytogenes in Henle-407 cell monolayers (Fig. S5), further supporting the hypothesis that enhanced spread of intracellular S. flexneri in the presence of formate is a response by the bacteria, rather than the host cell.
The role of formate in increasing Shigella plaque size could be due to faster growth of the bacteria or increased spread. There was no significant difference in S. flexneri intracellular doubling time between the presence and absence of exogenous formate (Fig. 1D). However, we observed an increase in S. flexneri cell-to-cell spread in the presence of exogenous formate (Fig. 1E), suggesting that formate promotes S. flexneri cell-to-cell spread, resulting in increased plaque size in Henle-407 monolayers.
S. flexneri has only one known formate transporter, FocA. FocA is a bidirectional formate transporter that directly interacts with PFL to export formate (27–29). FocA activity is dependent on pH, and when the pH is below 5.8, the function of FocA switches to a formate importer (28). focA is cotranscribed with pflB and expressed under aerobic or microaerobic conditions under the control of ArcA and the fumarate and nitrate reduction regulator FNR (30–32). We found that mutating S. flexneri focA reduced plaque size similarly to the ΔpflB mutant, likely due to reduced formate secretion. However, plaque size of the ΔfocA strain increased with exogenous formate (Fig. S4), indicating that S. flexneri formate import via FocA is dispensable for formate promotion of plaque size and suggesting that formate is sensed by S. flexneri outside the bacterial cytoplasm or, less probably, that formate is imported through an unidentified transporter.
Plaque size of S. flexneri WT, ΔpflB, and ΔfocA strains was measured; loss of focA, which transports formate, decreases plaque size similarly to the ΔpflB strain; however, formate significantly increases ΔfocA plaque size. An asterisk indicates statistical significance. Download FIG S4, TIF file, 1.58 MB (1.6MB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plaque size of Listeria monocytogenes was measured; formate has no significant effect on L. monocytogenes plaque size. Giemsa stains of plaques underneath corresponding measurements. Download FIG S5, TIF file, 3.50 MB (3.5MB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
pflB is required for S. flexneri formate secretion.
While S. flexneri metabolism is understudied, metabolism and mixed acid fermentation of the closely related bacterium E. coli have been characterized in detail, and the predominant E. coli fermentation products in vitro are formate, acetate, ethanol, and lactate (33). In limited oxygen, E. coli metabolizes carbon using glycolysis, producing pyruvate for mixed acid fermentation. Pyruvate is converted to lactate via lactate dehydrogenase or to acetyl-CoA via pyruvate dehydrogenase (PDH) or PFL; of these two enzyme complexes which generate acetyl-CoA, only PFL produces formate as a by-product. Acetyl-CoA is then converted to acetate or ethanol, while formate is predominantly secreted through one of two bidirectional transporters, FocA or FocB (28, 34). Cytoplasmic formate can be oxidized by the formate hydrogenlyase complex (FHL), while periplasmic formate can be oxidized by one of two formate dehydrogenase complexes (FDH-N or FDH-O). The genomes of E. coli and S. flexneri are highly similar, allowing us to project S. flexneri mixed acid fermentation (summarized in Fig. 2A). Of note, the S. flexneri locus containing focA and the PFL genes (including pflB) is highly conserved among sequenced Shigella species (Fig. S6). However, there are other notable differences in metabolism genes between E. coli and S. flexneri; S. flexneri has 58 metabolism-related pseudogenes compared to E. coli, including the putative formate transporter focB, and fdhF, a gene essential for the activity of FHL (35). We simulated S. flexneri metabolism in silico using a published S. flexneri genome-scale metabolic model (36, 37), which integrates S. flexneri genome data and enzyme stoichiometry of metabolic reactions into a metabolic network; constraint-based analysis is then applied to emulate metabolism maximizing biomass production (38, 39). To mimic intracellular conditions, we simulated a low-oxygen environment where carbon availability limits total growth. Under these conditions, S. flexneri is predicted to produce formate, acetate, and ethanol in silico at a ratio of approximately 3:2:1 and no lactate (Fig. 2A). The model predicted that 29.7% of total carbon available is converted to formate under the simulated conditions. This estimation is consistent with previous studies examining mixed acid fermentation in S. flexneri and E. coli (16, 40).
FIG 2.

(A) Model of S. flexneri fermentative metabolism. S. flexneri metabolism was simulated using a genome-scale metabolic network model; numbers indicate relative carbon flux. Under anoxic conditions, S. flexneri is predicted to secrete formate, acetate, and ethanol at a ratio of approximately 3:2:1. Formate is a by-product of PFL-mediated conversion of pyruvate to acetyl-CoA. (B) S. flexneri-secreted formate was measured in the supernatant from bacteria grown anaerobically in M63 medium. bd indicates “below detection.” No formate secretion was observed in the ΔpflB mutant. (C) Intracellular formate was measured in uninfected and infected Henle-407 cells at 3 hpi. Formate was normalized to the total number of Henle-407 cells. WT S. flexneri infection increased intracellular formate, while infection with the ΔpflB mutant did not.
(A) Schematic of the PFL locus of S. flexneri 2457T. (B) The PFL loci from 79 Shigella genomes were aligned, and phylogenetic analysis reveals that the PFL locus of S. flexneri 2457T is highly conserved from E. coli and among Shigella spp. S. flexneri 2457T is highlighted in green, while E. coli MG1655 is highlighted in blue. Other gastrointestinal pathogens are included for comparison. Download FIG S6, TIF file, 1.88 MB (1.9MB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The amount of formate secreted by the S. flexneri WT and ΔpflB mutant was quantified from cells grown in minimal medium, under the conditions simulated in the in silico model. After 18 h of growth, the concentration of formate secreted by the WT S. flexneri strain was 34.6 ± 0.5 mM, translating to a 38.4% conversion of available carbon to formate (Fig. 2B). In contrast, there was no detectable formate in supernatants from the ΔpflB mutant, confirming that PFL is essential for S. flexneri formate production. At host cytosolic pH, formate exists as a monovalent anion that cannot passively diffuse across bacterial or eukaryotic cellular membranes, with the exception of acidic compartments such as lysosomes (21); we therefore postulated that host cytosolic formate concentration would increase during S. flexneri infection due to S. flexneri metabolism and the spatial restriction imposed by the host cell membrane. We observed a 2.1-fold increase in intracellular formate of infected cells compared to uninfected cells (Fig. 2C). In contrast, Henle-407 cells infected with the S. flexneri ΔpflB mutant showed no increase in intracellular formate concentration.
FDH-N inhibits S. flexneri plaque formation.
S. flexneri encodes two periplasm-facing molybdoselenoformate dehydrogenase complexes, FDH-N and FDH-O, which couple formate oxidation to nitrate reduction. E. coli FDH-N, which catalyzes the conversion of periplasmic formate to H+ and CO2, is expressed during anaerobic growth and induced by nitrate in a NarL-dependent manner (41); FDH-O is active in the presence of oxygen and accounts for a smaller portion of the total formate dehydrogenase activity (42, 43). While S. flexneri PFL and FDH-N levels are elevated within an epithelial cell, expression of FDH-O-related genes is repressed (5, 15), suggesting that FDH-N is more important than FDH-O for formate metabolism by intracellular S. flexneri. If S. flexneri FDH-N is responsible for formate catabolism during intracellular growth, deletion of FDH-N would increase intracellular formate in infected Henle-407 cells and thus increase plaque size. We examined the plaque formation of S. flexneri ΔfdnG (catalytic subunit of FDH-N) and ΔfdoG (catalytic subunit of FDH-O) mutants. There was a 3.0-fold increase in plaque size of the S. flexneri ΔfdnG strain over WT (Fig. 3A). Furthermore, in the presence of exogenous formate the plaque size of the ΔfdnG strain increased 1.5-fold over the WT with formate. We measured formate levels of Henle-407 cells infected with S. flexneri WT and the ΔfdnG strain and found that formate was significantly higher in the ΔfdnG strain than the WT strain, consistent with reduced S. flexneri formate oxidation (Fig. 3B). These data indicate that FDH-N reduces S. flexneri spread, presumably by oxidizing formate derived from either S. flexneri or host metabolism and decreasing its concentration within the host cell cytosol. In contrast, the plaque size of the ΔfdoG strain was not significantly different from that of the WT strain, regardless of exogenous formate. This is consistent with the low expression of fdoG in intracellular bacteria (14). However, the ΔfdoG strain formed significantly fewer plaques than the WT strain, indicating that FDH-O, which is more highly expressed extracellularly, may be important for efficient invasion of host cells.
FIG 3.

(A) The ΔfdnG mutant forms larger plaques. Plaque size of S. flexneri WT, FDH-N mutant (ΔfdnG), or FDH-O mutant (ΔfdoG) was measured; loss of FDH-N, which oxidizes formate to CO2, increased plaque size. (B) Formate accumulates to higher levels in Henle-407 cells infected with the ΔfdnG mutant. Formate was normalized to the number of intracellular bacteria. An asterisk indicates statistical significance.
Formate alters S. flexneri virulence gene expression.
To identify S. flexneri genes affected by formate that could promote plaque formation, we determined formate-induced differences in the S. flexneri transcriptome using RNA sequencing (RNA-seq). We were able to map an average of 3 × 106 unique reads per sample to the S. flexneri genome, and we found that exogenous formate significantly altered the expression of four genes on the virulence plasmid (Table 1). The S. flexneri gene icsA, which recruits host N-WASP for actin-mediated intercellular spread, was upregulated 2.1-fold by formate. Additionally, two S. flexneri T3SS effectors were upregulated approximately 2-fold by formate: ipaJ and ipgD. Formate also altered the expression of the virulence plasmid maintenance gene ccdB and 13 S. flexneri chromosomal genes (Table 1); however, we focused on icsA, ipaJ, and ipgD, since these genes are known virulence determinants.
TABLE 1.
S. flexneri genes differentially regulated during infection of Henle-407 cells with exogenous 20 mM formate
| Carriage location and gene |
Function | Fold change |
|---|---|---|
| Virulence plasmid | ||
| ipaJ | Host protein N-myristoylation | 2.3 |
| icsA/virG | Actin-mediated intercellular spread | 2.1 |
| ipgD | Host phosphoinositide metabolism | 2.1 |
| ccdB | Plasmid maintenance | −2.1 |
| Chromosome | ||
| yjhA | Hypothetical protein | 3.3 |
| yccJ | Hypothetical protein | 2.5 |
| yhaH | Putative cytochrome | 2.3 |
| rfbE | Polysaccharide biosynthesis | 2.3 |
| melR | Regulator of melibiose operon | 2.1 |
| ybeK | Putative tRNA synthetase | 2.1 |
| yjhT | Hypothetical protein | 2.1 |
| yhiO | Hypothetical protein | 2.0 |
| proV | Osmotic regulation | −2.1 |
| Rnd | RNase D | −2.2 |
| S2640 | Hypothetical protein | −2.3 |
| yebJ | Hypothetical protein | −2.4 |
| ybaN | Hypothetical protein | −2.6 |
To validate the findings of our RNA-seq, we quantified S. flexneri icsA, ipaJ, and ipgD transcript levels during growth in vitro or in Henle-407 cells using quantitative reverse transcriptase PCR (RT-qPCR). We observed significant sample-to-sample variation both in vitro and in vivo; however, formate supplementation significantly increased S. flexneri icsA and ipaJ in vitro (Fig. 4). We also observed that exogenous formate increased intracellular S. flexneri ipaJ expression 1.5-fold, while the ΔpflB mutant showed a 1.3-fold decrease in ipaJ expression compared to WT (Fig. 4). Both exogenous formate and pflB had no significant impact on S. flexneri ipgD levels either in vitro or in Henle-407 cells (Fig. 4).
FIG 4.
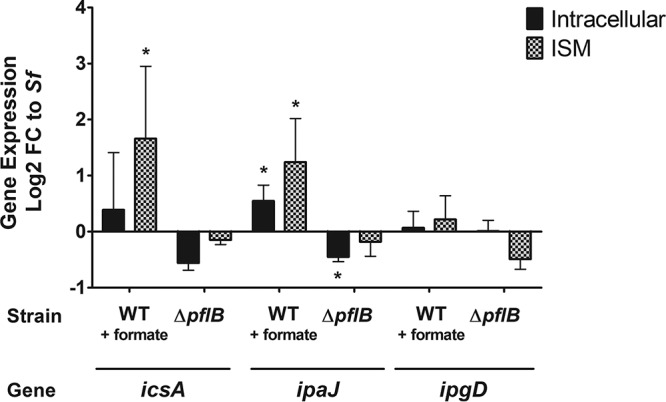
Formate increases the expression of icsA and ipaJ. S. flexneri icsA, ipaJ, and ipgD expression in Henle-407 cells at 3 hpi (black bars) or ISM (checkered bars) was independently determined by RT-qPCR. Log2 fold change is relative to WT S. flexneri. An asterisk indicates statistical significance.
Formate increases surface S. flexneri IcsA.
S. flexneri uses polarly localized IcsA to recruit host N-WASP, catalyzing host actin synthesis and propelling the bacterium through the host cell cytosol (44, 45). ΔicsA mutants cannot spread and do not form plaques in Henle-407 monolayers (2, 46); therefore, to confirm that S. flexneri icsA is differentially regulated by formate, S. flexneri IcsA was visualized in infected Henle-407 cells by fluorescence staining and microscopy. IcsA labeled with fluorescein isothiocyanate (FITC) appeared as green, U-shaped foci on the micrographs that corresponded with the poles of intracellular bacteria, which were stained blue with 4′,6-diamidino-2-phenylindole (DAPI) (Fig. 5). As expected, no green foci were observed in cells infected with the ΔicsA mutant. We observed more S. flexneri with IcsA staining (45.8% ± 0.5%) in infected Henle-407 cells supplemented with formate than in those without formate supplementation (22.0% ± 6%), and the foci appeared brighter. In contrast, a smaller percentage of the intracellular S. flexneri ΔpflB mutant showed IcsA staining (7.0% ± 1.7%), and the foci were dimmer than with the WT strain. These findings together suggest that formate increases the levels of S. flexneri IcsA in infected Henle-407 cells.
FIG 5.

Formate increases S. flexneri IcsA in infected Henle-407 cells. Polarly localized IcsA (green) was visualized in intracellular S. flexneri (blue) using fluorescence microscopy. Fluorescence images were overlaid on a phase-contrast micrograph. We observed more polar IcsA WT S. flexneri grown with formate and lower polar IcsA levels in the ΔpflB mutant than in the WT strain.
ipgD is required for formate-mediated plaque formation.
S. flexneri IpgD is a secreted PtdIns(4,5)P2 4-phosphatase that produces PtdIns5P in the host cell; increased PtdIns5P has widespread effects on the host, including alteration of host membrane tension, activation of host Akt kinase, inhibition of T-cell migration, inhibition of host extracellular ATP (eATP) secretion, activation of host ARF6, and altered host Ca2+ signaling (8, 47–50). To determine if ipgD is involved in the S. flexneri response to formate, we measured the plaque size of an S. flexneri ΔipgD mutant in Henle-407 monolayers with exogenous formate. Compared to the S. flexneri WT strain, the ΔipgD mutant formed smaller plaques (Fig. 6). Importantly, the ΔipgD mutant showed no significant increase in plaque size when the tissue culture medium was supplemented with 20 mM formate. When S. flexneri ipgD was complemented on a plasmid, the formate-mediated increase in plaque size was restored (Fig. 6). This suggests that ipgD is required for the formate-mediated increase in S. flexneri plaque size.
FIG 6.
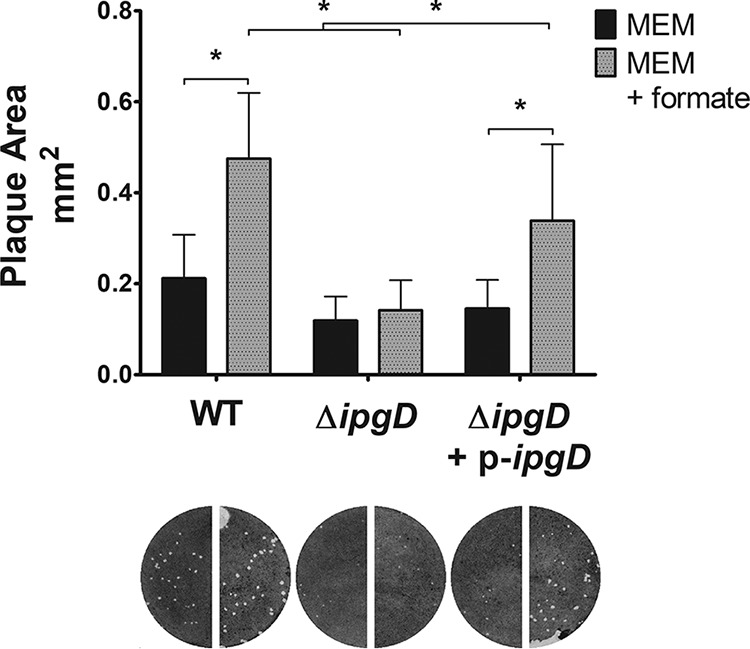
The ipgD mutant does not increase plaque size with formate. Plaque size of S. flexneri WT and ΔipgD mutant was measured in cultured cells in the presence or absence of exogenous formate. (An asterisk indicates statistical significance.) The ΔipgD mutant did not increase plaque size in the presence of exogenous formate, and this phenotype is complemented by providing ipgD on a plasmid.
ipaJ contributes to plaque size increase by formate.
IpaJ is an S. flexneri-secreted cysteine protease that disrupts the host Golgi apparatus by altering host protein N-myristoylation (11, 12, 51). We examined how the plaque size of an S. flexneri ΔipaJ mutant changes in response to exogenous formate. We found that in the absence of exogenous formate, the mean plaque size of the ΔipaJ mutant was not significantly different from that of the WT strain (Fig. 7), consistent with previous reports (52). The ΔipaJ mutant response to formate was diminished compared to WT, with the mutant forming smaller plaques than the WT strain when both were supplemented with exogenous formate; this phenotype was complemented by providing ipaJ on a plasmid (Fig. 7). These results indicate that ipaJ contributes to the formate-mediated increase in S. flexneri plaque size.
FIG 7.
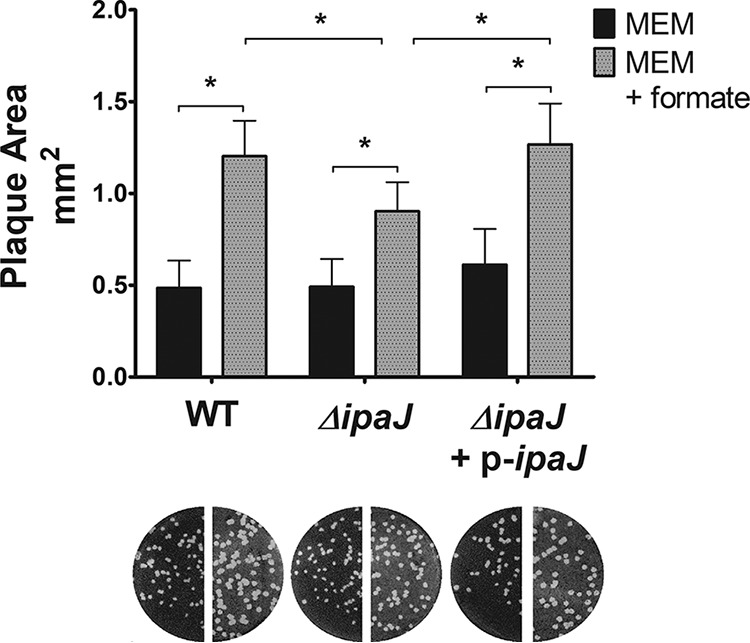
The ipaJ mutant has reduced response to formate. Plaque size of S. flexneri WT and ΔipaJ mutant was measured in cultured cells in the presence or absence of exogenous formate. (An asterisk indicates statistical significance.) The difference in plaque size caused by the addition of exogenous formate is significantly less in the ΔipaJ mutant than in the WT strain, and this phenotype is complemented by providing ipaJ on a plasmid.
ipaJ expression is dependent on S. flexneri cell density and formate.
Because S. flexneri secretes formate in the host cytosol as a by-product of carbon metabolism, and this formate accumulates in the host cytosol of S. flexneri-infected Henle-407 cells (Fig. 2C), we expected that formate-induced virulence gene expression would increase as S. flexneri intracellular density increased, as formate is produced from bacterial metabolism. To test this, we constructed an ipaJ transcriptional reporter, in which the promoter region of ipaJ was fused to gfp on a plasmid, and ipaJ expression was visualized using fluorescence microscopy. We observed increased fluorescence in the intracellular S. flexneri WT cells compared to the ΔpflB mutant (Fig. 8A); likewise, formate supplementation of Henle-407 cells infected with WT S. flexneri increased fluorescence. Green fluorescent protein (GFP) fluorescence was then measured in single bacterial cells from fluorescence micrographs. We observed a 1.8-fold increase in gfp of S. flexneri when exogenous formate was added as a supplement (Fig. 8B), similar to the changes in ipaJ levels observed in previous experiments (Table 1 and Fig. 4). Likewise, we observed a 1.3-fold decrease of ipaJ expression in the ΔpflB mutant compared to the WT strain. We then measured the number of intracellular S. flexneri bacteria and the two-dimensional cell area of Henle-407 cells stained with wheat germ agglutinin (WGA) and regressed mean ipaJ expression of S. flexneri within individual Henle-407 cells on intracellular S. flexneri density. ipaJ expression positively correlated with S. flexneri intracellular density (Fig. 8C), and when cells were supplemented with exogenous formate, ipaJ expression levels were constitutively elevated regardless of intracellular S. flexneri density. Furthermore, ipaJ expression of the ΔpflB mutant was lower than that of the WT strain, regardless of intracellular S. flexneri density. These data indicate that intracellular ipaJ expression is dependent on the intracellular density of S. flexneri cells, and this regulation is positively associated with formate accumulation.
FIG 8.

Expression of ipaJ in the intracellular environment increases in the presence of formate and is influenced by bacterial cell density and formate concentration in the Henle-407 cells. (A) The ipaJ promoter was fused to gfp (green), and infected Henle-407 cells were visualized using fluorescence microscopy. Nuclei were stained with DAPI (blue), and Henle-407 membranes were stained with wheat germ agglutinin (red). Asterisks indicate infected cells. The ΔpflB mutant displayed lower gfp intensity, while formate increased gfp intensity in WT. (B) ipaJ expression was measured by quantitative fluorescence microscopy. Dotted lines indicate median gfp intensity. An asterisk indicates statistical significance compared to each of the other two medians. Formate increased ipaJ expression 1.8-fold. (C) ipaJ expression of intracellular S. flexneri was regressed on S. flexneri density. The slope of WT S. flexneri ipaJ expression (R2 = 0.7) was significantly different from that of WT plus formate (R2 = 0.4) and the ΔpflB strain (R2 = 0.4).
Altering formate results in modified host response of S. flexneri-infected Henle-407 cells.
S. flexneri infection elicits a strong inflammation response from the host cell (8, 53). To counteract this, S. flexneri employs IpaJ and IpgD, which dampen host interferon- and tumor necrosis factor (TNF)-stimulated genes (8, 11, 12). Because ipgD and ipaJ contribute to the S. flexneri response to formate (Fig. 6 and 7), we hypothesized that supplementation with exogenous formate would result in altered host immune response to S. flexneri infection. We selected 4 host genes known to be affected by either ipgD or ipaJ: CXCL10, interleukin-8 (IL-8), TNF-α, and TNFAIP3 (8, 11). We found that exogenous formate lowered the levels of CXCL10, IL-8, and TNFAIP3 in S. flexneri-infected Henle-407 cells (Fig. 9A, black bars), while in uninfected cells, exogenous formate had no effect on CXCL10, IL-8, TNF-α, or TNFAIP3 expression (1.0-, 1.1-, 0.9-, and 1.1-fold change, respectively).
FIG 9.

Formate inhibited interferon- and TNF-stimulated gene expression levels in infected Henle-407 and CoN-841 cells. (A) Host gene expression of infected Henle-407 cells (black bars) and CoN-841 cells (gray checkered bars) was determined by RT-qPCR. (B) CXCL10 transcript levels of Henle-407 cells infected with different S. flexneri strains were determined by RT-qPCR. Log2 fold change is relative to host cells infected with WT S. flexneri in the absence of exogenous formate, and an asterisk indicates statistical significance compared to WT infection.
Henle-407 cells, a HeLa derivative cell line, have altered immune pathways relating to S. flexneri infection response. For example, Henle-407 cells do not express the connexin hemichannels involved in secretion of eATP, a potent inflammatory signal (54). Therefore, we analyzed S. flexneri-infected CoN-841 cells grown with or without exogenous formate and quantified CXCL10, IL-8, TNF-α, or TNFAIP3 expression levels. We observed more gene expression variation in infected CoN-841 cells than in Henle-407 cells, and TNF-α was the most strongly repressed gene in infected CoN-841 cells supplemented with exogenous formate (Fig. 9A, checkered bars), suggesting that the specific genes and the dynamics of the host immune response in regard to formate-regulated S. flexneri effectors vary in a different human cell line.
To confirm the effect of formate on S. flexneri ipaJ, we examined host CXCL10 expression in Henle-407 cells infected with the ΔpflB and ΔipaJ mutant strains. In contrast to formate lowering CXCL10 expression in cells infected with WT S. flexneri, CXCL10 expression was increased in Henle-407 cells infected with the S. flexneri ΔpflB mutant, and this difference was abated when exogenous formate was provided as a supplement (Fig. 9B). Similarly to the ΔpflB mutant, CXCL10 expression was elevated in Henle-407 cells infected with the S. flexneri ΔipaJ mutant, and formate reduced this response.
DISCUSSION
Formate is an important biomolecule for both human and bacterial metabolism. In bacteria, formate is a by-product of pyruvate formate lyase (PFL)-mediated conversion of pyruvate to acetyl-CoA. In addition to PFL, Shigella encodes a pyruvate dehydrogenase (PDH) complex that converts pyruvate to acetyl-CoA under aerobic conditions but does not produce formate. An S. flexneri ΔaceE mutant defective in PDH has an intracellular growth defect, indicating that PDH is utilized for acetyl-CoA generation within the host cell (16). Eukaryotic cells contain low levels of cytosolic oxygen, a condition that permits the activity of both PFL and PDH, which explains why the S. flexneri ΔpflB mutant intracellular growth rate is unaffected despite missing this source of acetyl-CoA (16).
Eukaryotic cells metabolize formate via one-carbon metabolism for purine biosynthesis, among other things (55). At biological pH, formate cannot passively cross bacterial or eukaryotic cell membranes (21); however, extracellular formate exchange occurs in cultured human cells (56–58), presumably facilitated through the formate transporter SLC26A6 (56, 59, 60). Likewise, formate is shuttled between the mitochondria and eukaryotic cytosol through an unknown mechanism (55). It is unclear how Henle-407 cells react to the influx of cytosolic formate from S. flexneri infection. We predict that a portion of S. flexneri-derived host cytosolic formate is oxidized by S. flexneri FDH-N (Fig. 3), a second portion is converted to purines by host one-carbon metabolism, and a third portion accumulates in the host cytosol.
Formate turnover has been previously linked to the intracellular growth phase of S. flexneri (5, 15). Here, we demonstrate that it is not the metabolism of formate but formate itself that promotes S. flexneri plaque formation by increasing intercellular spread via IcsA and modulating the host response to S. flexneri infection. While PFL can be reversed to convert formate to pyruvate for metabolism (61), formate increases ΔpflB mutant plaque size, indicating that formate is not used solely as a substrate for PFL to produce pyruvate for growth (Fig. 2B). And, while formate oxidation is a potential source of NADH, we also show that formate oxidation is dispensable for intracellular S. flexneri metabolism, since exogenous formate does not increase S. flexneri growth rate within the host cell (Fig. 1C) and knocking out FDH-N promotes S. flexneri plaque formation (Fig. 4). Unlike the closely related E. coli, Shigella spp. (including S. flexneri) have a mutated FHL, suggesting an evolutionary selection against Shigella formate oxidation (35); however, formate dehydrogenases contribute to the fitness of closely related E. coli in the lumen during inflammation-associated colon dysbiosis (62), possibly explaining why S. flexneri maintains its formate dehydrogenases.
Although it is clear that extracellular formate can enter Henle-407 cells when provided as an exogenous supplement, as shown by uptake of radiolabeled formate (see Fig. S2 in the supplemental material) and alterations in S. flexneri gene expression (Table 1; Fig. 4 and 8), we believe that Shigella-derived formate is sufficient to differentially regulate expression related to intercellular spread and host immune dampening because we observe these phenotypes in S. flexneri formate metabolism mutants. We propose a model in which, following entry into the host cell cytoplasm, S. flexneri begins to replicate and metabolize host sugars and pyruvate, producing and secreting formate as a by-product. Formate accumulates in the infected cell cytosol rapidly due to the spatial constrictions of the host cell membrane and induces icsA to promote S. flexneri intercellular spread. Formate also induces the expression of ipaJ and possibly ipgD to alter the host immune response to S. flexneri infection. Because formate regulates S. flexneri icsA and ipaJ expression in vitro (Fig. 4), S. flexneri likely possesses a formate sensory mechanism; this S. flexneri formate sensor would likely act at the outer membrane or periplasm, as formate increases plaque size of the ΔfocA formate transport mutant (Fig. S2). One possible sensing mechanism is the BarA-UvrY two-component regulatory system, which senses formate and short-chain fatty acids to modulate the activity of the Csr regulatory system in E. coli (63) and regulates virulence in S. flexneri and Salmonella enterica (25, 64). However, the fact that acetate fails to increase plaque size indicates that BarA-UvrY is not the sensory mechanism; consistent with this, we found that S. flexneri ΔbarA or ΔuvrY mutants still increase plaque size in response to formate (Fig. S5).
Previous studies demonstrate that there is spatiotemporal regulation of S. flexneri-secreted effectors during host cell infection (5, 6, 65–67). Our data indicate that formate is an S. flexneri signal to differentially regulate icsA and ipaJ expression in the host cell, and this regulation is correlated with increased intracellular S. flexneri density. The dynamic nature of formate-mediated S. flexneri virulence gene regulation could explain the variation in both host and S. flexneri gene expression that we observed in response to formate (Fig. 4 and 9) and why there were inconsistencies such as formate-altered S. flexneri ipgD expression (Table 1 and Fig. 4). The production of formate is not essential to S. flexneri virulence, evidenced by the fact that the ΔpflB mutant is still able to form plaques, albeit smaller than those of the WT. Rather, formate-mediated regulation of icsA/ipaJ appears to be a way to fine-tune expression in response to intracellular bacterial density or to facilitate spread once a threshold density has been achieved. There is currently a high degree of interest in the complex relationship between metabolism and pathogenesis (68, 69), and the concept of a metabolic by-product regulating bacterial virulence is not new; one example is indole, a by-product of tryptophan hydrolysis that regulates virulence phenotypes and has been reported as a social signal in enteric microbial communities (70–72). It is easy to draw comparisons between indole and formate signaling; however, one notable difference between indole and formate signaling is that indole is diffusible across cellular membranes while formate is not (73), allowing cytosolic formate to accumulate during intracellular infection.
As the bacterial load within a host cell increases, so does the need of the bacteria to dampen the host immune response in order to evade host cytosolic defenses such as autophagy, as well as systemic defenses such as neutrophil recruitment and inflammation. Consistent with other pathogens that suppress host immune response, immunomodulating effectors secreted by a single S. flexneri cell, which are costly to produce, benefit the entire intracellular S. flexneri population (74). Thus, we consider modulating host response to be a cooperative social behavior which conveys a benefit to S. flexneri intracellular populations. If formate sensing is social in nature, it could convey local information about both intracellular S. flexneri cell density and S. flexneri spatial constraint, as formate accumulation differs in the intracellular and extracellular environments. It should also be noted that inflammation in the colon results in higher levels of formate in the lumen (59). During Shigella infection, the Shigella-induced inflammatory response could lead to increased formate in the lumen and subsequently in the epithelial cells, promoting bacterial spread and production of anti-inflammatory effectors. A more complete understanding of the role of formate production and sensing in infected host cells will provide a better understanding of this complex host-pathogen interaction.
MATERIALS AND METHODS
Media and growth conditions.
Bacterial strains, plasmids, and primers used in this study are listed in Table S1 in the supplemental material. S. flexneri was cultured on tryptic soy broth agar plates with 0.01% (wt/vol) Congo red (TSBA-CR), and red colonies were selected to ensure the presence of the S. flexneri virulence plasmid. Overnight bacterial cultures were grown in Luria-Bertani (LB) broth at 30°C, subcultured 1:100, and grown at 37°C to an optical density at 650 nm (OD650) of 0.5 to 1.0 (mid-log phase) prior to infection. Deoxycholate (DOC) was added as a supplement at 0.1% (wt/vol) where indicated to increase efficiency of invasion of Henle-407 cultured cells (75). Antibiotics were added as supplements where indicated at the following concentrations: 25 μg/ml ampicillin, 50 μg/ml kanamycin, 6 μg/ml chloramphenicol, and 20 μg/ml gentamicin. Sodium formate was added as a supplement where indicated at 20 mM.
(A) Human tissue culture lines and bacterial strains used in this study, along with their respective sources. (B) Bacterial plasmids used in this study. (C) Oligonucleotide sequences of primers for cloning, sequencing, and RT-qPCR used in this study. Download Table S1, DOCX file, 0.02 MB (22.5KB, docx) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Henle-407 cells (ATCC intestinal 407, CCL-6) and CoN-841 cells (ATCC CCD 841, CRL-1790) were cultured in minimal essential medium (MEM; Gibco 61100-087) supplemented with 10% (vol/vol) Bacto tryptone phosphate broth (Difco), 10% (vol/vol) fetal bovine serum (Gibco 16140-071), 2 mM glutamine, and nonessential amino acids (Gibco 11140-050). Henle-407 and CoN-841 cells were grown at 37°C with 5% CO2.
Construction of S. flexneri mutants and plasmids.
Strains, primers, and plasmids used in this study are listed in Table S1. The ΔfdnG, ΔfdoG, and ΔfocA mutants were created using P1 bacteriophage transduction of the genes from E. coli strains JW1470, JW3865, and JW0887 (respectively) from the Keio collection (76). The ΔipaJ mutant was generated using a modified method of Datsenko and Wanner (77, 78). Overlap extension PCR was used to fuse 399 bp upstream and 383 bp downstream of ipaJ to each side of the chloramphenicol resistance cassette of pKD3 (77). This product was amplified by PCR using the ipaJKO-1 and ipaJKO-4 primers, ethanol precipitated, and brought to a concentration of >500 ng/μl in water. S. flexneri containing the plasmid pKD46 (77) was grown to mid-log phase in 25 ml LB with no NaCl at 30°C, and then 2 mM arabinose was added and the culture was brought to 37°C for 30 min. Cells were pelleted and resuspended in 500 μl warm water, mixed with 10 μl ipaJ knockout (KO) PCR product, and immediately electroporated. Mutants were selected on TSBA-CR with chloramphenicol. All mutants were verified by PCR and DNA sequencing at the University of Texas at Austin DNA sequencing facility.
The ipaJ complement plasmid (pBK24) was constructed by amplifying the ipaJ locus beginning ∼400 bp upstream of the annotated ipaJ gene and ending at the ipaJ stop codon from S. flexneri DNA by PCR with primers ipaJ-30c-fw and ipaJ-30c-rv, containing 5′ KpnI and XbaI sites, respectively. After restriction digest, the product was ligated into the corresponding sites of the plasmid pWKS30 (79). The ipaJ-gfp transcriptional reporter plasmid (pBK25) was constructed by amplifying the ipaJ promoter region, beginning ∼500 bp upstream of ipaJ and ending at the ipaJ start codon, by PCR using primers ipaJ-gfp-fw and ipaJ-gfp-rv containing 5′ SmaI and XbaI sites. After restriction digest, the product was ligated into the corresponding sites of the plasmid pLR29 (14). Both plasmids were confirmed by PCR and DNA sequencing at the University of Texas at Austin DNA sequencing facility.
In silico analysis of S. flexneri metabolism.
In silico metabolism simulations were performed using a previously published S. flexneri genome-scale metabolic network reconstruction (36) and OptFlux (37). We removed external boundary metabolites and used the core biomass production as our objective function and the pFBA simulation method. To simulate anaerobic growth in M63 medium, default environmental conditions were used, except that the lower bound of O2 exchange was set to 0, the lower bound of nicotinate exchange was set to −0.162, and the lower bounds of glucose and pyruvate exchange were set to −10. Total biomass production was limited by carbon availability under these simulated conditions.
Tissue culture assays.
Plaque assays were performed as described previously (18) with modifications (17). Bacteria at mid-log growth were centrifuged and resuspended in sterile saline to a final concentration of 5 × 104 CFU/ml, and 100 μl of this suspension was added to Henle-407 monolayers grown to confluence. Plates were centrifuged for 10 min at 1,000 × g and incubated for 60 min at 37°C and 5% CO2. Monolayers were then washed four times with phosphate-buffered saline (PBS) and incubated in MEM supplemented with 0.2% glucose, gentamicin, and formate where indicated for either 48 or 72 h. Monolayers were stained with Wright-Giemsa stain (Camco) and imaged using an Alpha Innotech AlphaImager (Protein Simple), and plaque area was measured using ImageJ (80). Statistical significance was determined by analysis of variance (ANOVA) with a Bonferroni posttest, P < 0.05 (GraphPad Prism); figures are representative of at least 2 independent experiments.
For L. monocytogenes plaque assays, L. monocytogenes was grown in brain heart infusion (BHI) broth for approximately 18 h at 30°C without shaking. One milliliter of culture was centrifuged at 13,000 × g for 2 min, and then the supernatant was removed and the pellet was resuspended in 1 ml PBS. Two microliters of L. monocytogenes suspension was added to Henle-407 monolayers grown in a 6-well plate and gently shaken for 1 min. The plate was incubated for 1 h at 37°C and 5% CO2. The monolayers were then washed four times with PBS and incubated in MEM supplemented with 0.2% glucose, gentamicin (10 μg/ml), and formate where indicated for 48 to 72 h. Monolayers were stained with Wright-Giemsa stain (Camco) and imaged using an Alpha Innotech AlphaImager (Protein Simple), and plaque area was measured using ImageJ (80). Statistical significance was determined by Student’s t test (P < 0.05; GraphPad Prism); figures are representative of 2 independent experiments.
To determine bacterial doubling time, Henle-407 monolayers in 6-well plates were infected as described for plaque assays with S. flexneri grown in deoxycholate and added at a multiplicity of infection (MOI) of 20. At 1 h postinfection (hpi) and every 30 min thereafter, one well was trypsinized and Henle-407 cells were then lysed in 1% deoxycholate, diluted, and spot plated on TSBA-CR. Doubling time (r) was determined using the formula t × log2/(log y2 − log y1), where t is time (150 min), y1 is Shigella at 1 hpi, and y2 is Shigella at 3.5 hpi (n = 2).
Cell-to-cell spread assays were performed as previously described (81). S. flexneri was added to Henle-407 cells at approximately 65% confluence at an MOI of 10. Cells were stained with Wright-Giemsa stain at 3 hpi, and cell-to-cell spread rates were calculated by counting approximately 100 infected Henle-407 cells adjacent to other Henle-407 cells. The proportion of S. flexneri spreading events was scored by determining if any Henle-407 cells adjacent to an infected cell also contained 3 or more internal bacterial cells. Statistical significance was determined by Student’s t test (n = 3, P < 0.05).
Formate-induced cytotoxicity was determined by measuring secreted lactate dehydrogenase of Henle-407 cells supplemented with 20 mM formate every 24 h over 3 days using a lactate dehydrogenase assay (Sigma MAK066) according to the manufacturer’s instructions. Statistical significance was determined using ANOVA with a Bonferroni posttest.
Formate uptake.
The formate uptake assay was modified from a previous study (22). Briefly, Henle-407 cells were cultured in 30- by 10-mm plates, and 20 μM [14C]formate (Moravek) was added to the tissue culture medium. Over the course of 48 h, cells were washed with PBS, trypsinized, and counted. Cells were then lysed directly in Optiphase HiSafe 3 (PerkinElmer). Counts per minute (cpm) was determined by scintillation, and counts were normalized to cell number and are relative to untreated cell lysates.
Formate quantification.
For supernatant formate quantification, overnight cultures of S. flexneri WT and the indicated mutants were diluted 1:1,000 in M63 medium [100 mM KH2PO4, 15 mM (NH4)2SO4, 1.8 μM FeSO4, 1 mM MgSO4, 162.5 nM nicotinic acid, 10 mM glucose, 10 mM pyruvate]. Cultures were grown for approximately 18 h at 37°C in sealed anaerobe jars containing anaerobic gas packs (BD 260651). The OD650 was recorded to determine bacterial growth. Cultures were then centrifuged, supernatants were filter sterilized prior to analysis using a formate assay (Sigma MAK059) according to the manufacturer’s instructions, and samples were diluted 1:100 in formate assay buffer. Statistical significance was determined by ANOVA with a Bonferroni posttest (P < 0.05, n = 3) using GraphPad Prism.
For intracellular formate quantification, Henle-407 cells were infected with S. flexneri grown with deoxycholate as described above at an MOI of 5. At 5 hpi, Henle-407 monolayers were trypsinized and Henle-407 cells were counted using a Countess II cell counter (ThermoFisher). Where indicated, bacteria were quantified by plating. Cell pellets were then resuspended in 1 ml cold methanol-water (50:50) and incubated for 20 min on ice. Five hundred microliters of cold chloroform was added, and the mixture was vortexed and then centrifuged at 13,000 × g for 15 min. The aqueous phase was transferred to a new tube, liquid was evaporated using a vacuum manifold, and then samples were rehydrated in 100 μl formate assay buffer. Formate was quantified using the assay described above, and statistical significance was determined by either ANOVA with a Bonferroni posttest (P < 0.05, n = 3) or Student’s t test (P < 0.05, n = 3). Results are representative of 2 independent experiments.
Phylogenetic analysis.
For the phylogenetic analysis of the PFL locus, the PFL locus was identified in the 79 Shigella complete genomes currently represented in the NCBI database. The sequences were aligned using Geneious and had 96.2% identical sites. The PFL loci from other related gastrointestinal pathogens were included for comparison. A tree was built using the Geneious Tree Builder, using the Tamura-Nei model and neighbor joining method, with global alignment with free end gaps and a cost matrix at 51% similarity. The tree was visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree).
RNA extraction.
For in vitro RNA extractions, S. flexneri was grown in intracellular salts medium (ISM) (4) supplemented with 10 mM pyruvate, RDM supplement (82), 20 mM bicarbonate, and 100 μM nitrate. Five-milliliter cultures were grown statically at 37°C with 5% CO2 for 7 h. One milliliter of cold RNA-Stay (95% ethanol, 5% phenol) was added to each culture, and the entire volume was pelleted by centrifugation. One milliliter of cold RNA-Bee (Amsbio) was pipetted over the pellet. For RNA extractions of S. flexneri-infected Henle-407 cells, the Henle-407 cells were infected as described above with S. flexneri WT and mutants grown in deoxycholate and added at an MOI of 20. At 3 hpi, monolayers were washed once with PBS, and 1 ml of cold RNA-Bee (Amsbio) was pipetted over the monolayer. The cell lysates from either bacterial pellets or Henle-407 monolayers were transferred to new tubes, and 200 μl chloroform was added. The mixture was vortexed and after 5 min on ice was centrifuged at 13,000 × g, and the upper aqueous phase was transferred to a new tube. RNA was then precipitated with isopropanol, treated with DNase I (Invitrogen), and solubilized in water.
cDNA library generation and RNA-seq.
RNA from infected Henle-407 cells grown with and without formate and a mock uninfected sample was used to generate cDNA by the Genomic Sequencing and Analysis Facility (GSAF) at the University of Texas at Austin. Single samples from each condition tested were analyzed. RNA was checked for quality using a Bioanalyzer (Agilent); samples had an RNA integrity number (RIN) of >9. One microgram of total RNA was used for ribosome depletion using the Ribo-Zero Gold rRNA removal kit (Illumina) according to the manufacturer’s instructions to eliminate both eukaryotic host rRNA and Shigella rRNA. The depleted RNA was fragmented to ∼200 bp, and cDNA was generated using the NEBNext Ultra II directional RNA kit. Samples were barcoded, and single-end sequencing (75 cycles) of cDNA libraries was performed using a NextSeq 500 system (Illumina) at the GSAF. Sequencing generated 1.1 × 108 total reads for infected Henle-407 cells, and 1.0 × 108 total reads for infected Henle-407 cells grown with formate.
Read mapping and gene expression analysis.
Sequence reads were aligned to the S. flexneri 2457T genome (GenBank accession no. AE014073.1) and the S. flexneri virulence plasmid pCP301 (GenBank accession no. AF386526.1) using CLC Genomics Workbench (Qiagen). Multimapped reads were excluded, as were genes containing reads that mapped to the S. flexneri genome or virulence plasmid in the mock treatment (listed in Table S2). Values of reads per kilobase per million (RPKM) were used for expression analysis, and genes with an RPKM value of <20 (arbitrary cutoff) were excluded. A Baggerley proportions test with a false-discovery rate correction was used to determine statistical significance (P < 0.05). Genes with a fold change greater than 2 were considered significantly different. A complete list of read mappings can be found in Table S2.
(A) Complete mapping information for S. flexneri genes induced by formate in the host cell listed in Table 1. (B) Mapping of all intracellular S. flexneri genes grown with and without formate. (C) Eighty-seven S. flexneri genes were mapped in mock-treated samples, to determine human RNA that maps to S. flexneri genes (false positives). Download Table S2, XLSX file, 1.5 MB (1.5MB, xlsx) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Quantitative reverse transcriptase PCR.
RNA was quantified using a ND-1000 spectrophotometer (NanoDrop), and a total of 1 μg RNA was used with a high-capacity cDNA reverse transcription kit (Applied Biosystems). Two microliters of cDNA was then used as the template for SYBR green quantitative PCR (qPCR) (Applied Biosystems) using a ViiA 7 real-time PCR system (Applied Biosystems) at the University of Texas at Austin GSAF. Human gene expression was normalized to actB, and Shigella gene expression was normalized to rssA. All primers used for RT-qPCR are listed in Table S1. Statistical significance was determined using Student’s t test of threshold cycle (ΔCT) values compared to the S. flexneri WT-infected Henle-407 cells or S. flexneri WT grown in ISM.
Fluorescence microscopy.
For IcsA labeling, Henle-407 cells were grown on glass coverslips in 6-well plates and infected with the S. flexneri WT, ΔpflB, or ΔicsA strain grown with deoxycholate (DOC) at an MOI of 1, as described above. At 3 hpi, cells were washed with PBS and then fixed with formaldehyde. Cells were then permeabilized with 0.5% Triton X-100, blocked with 5% bovine serum albumin (BSA) in PBS, and then incubated overnight with rabbit polyclonal antibody to IcsA (rabbit no. 5) provided by Edwin Oaks (Walter Reed Army Institute of Research) diluted 1:50 in PBS with 2.5% BSA. Cells were then washed with 0.05% Tween 20 in PBS and then blocked with 5% BSA in PBS for 30 min. The cells were then incubated with goat anti-rabbit IgG-FITC (sc-2012; Santa Cruz Biotechnology) diluted 1:1,000 and DAPI in PBS with 2.5% BSA for 2 h and then washed with 0.05% Tween 20 in PBS. Coverslips were then mounted on slides with Prolong Diamond antifade mountant (ThermoFisher). Images were then acquired using an Olympus BX41 microscope (100× objective) with a DP73 digital camera (Olympus) and processed using cellSens software (Olympus) or using a Zeiss LSM 710 confocal microscope (63× objective) at the University of Texas Microscopy and Imaging Facility. All exposures were identical, and images were processed in Photoshop (Adobe) to enhance contrast; all images were processed identically. Micrographs are representative of at least 3 independent experiments. To determine the proportion of intracellular bacteria with polar IcsA, the total number of bacteria and the number with polar IcsA staining were counted. Results shown are means ± standard deviations (SD) for the percent IcsA-positive bacteria in at least 4 fields.
S. flexneri strains containing the ipaJ-gfp reporter were grown with deoxycholate and ampicillin and diluted to an MOI of 1. Infections were carried out as described above, and at 3 hpi, cells were washed with PBS and formaldehyde fixed. Fixed cells were stained with wheat germ agglutinin (WGA)-Alexa Fluor 555 at 1 μg/ml. Slides were mounted with Vectashield with DAPI (Vector Laboratories), and images were acquired using an Olympus BX41 microscope (100× objective) with a DP73 digital camera (Olympus) and processed using cellSens software (Olympus). Acquisition of FITC (gfp) was fixed at 300 ms. Two-dimensional Henle-407 cell area was measured from the WGA membrane-labeled red channel, and gfp intensity of individual bacteria was quantified from the green channel using ImageJ (80). Background gfp fluorescence was measured in portions of each cell without bacteria and subtracted from individual bacterial cell fluorescence. A D’Agostino-Pearson normality test indicated that measurements did not conform to a Gaussian distribution. Therefore, statistical significance of gfp differences was determined by a Kruskal-Wallis test with a Dunn posttest (P < 0.05, n > 300). Regressions were performed using GraphPad Prism (n = 12 to 20), and slopes were found to be significantly different (P < 0.05).
Plaque size of S. flexneri WT, ΔbarA, and ΔuvrY strains was measured; formate significantly increases WT plaque size 2.2-fold, increases ΔbarA mutant plaque size 1.8-fold, and increases ΔuvrY mutant plaque size 2.0-fold. An asterisk indicates statistical significance. Download FIG S7, TIF file, 2.71 MB (2.7MB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Accession number.
Sequencing data were deposited in GEO under accession number GSE119622.
ACKNOWLEDGMENTS
We acknowledge A. R. Mey for reading and editing the manuscript and E. L. Bruger (ORCID: 0000-0002-6369-4180) and M. Shin for thoughtful discussions. We also thank C. F. Lesser for providing the ΔipgD mutant, M. B. Goldberg for providing the ipgD complement plasmid, and E. V. Oaks and R. W. Kaminski for IcsA antiserum.
This work was funded by Public Health Service grant AI16935 from the National Institutes of Health to S.M.P.
Footnotes
Citation Koestler BJ, Fisher CR, Payne SM. 2018. Formate promotes Shigella intercellular spread and virulence gene expression. mBio 9:e01777-18. https://doi.org/10.1128/mBio.01777-18.
REFERENCES
- 1.Wassef JS, Keren DF, Mailloux JL. 1989. Role of M cells in initial antigen uptake and in ulcer formation in the rabbit intestinal loop model of shigellosis. Infect Immun 57:858–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernardini ML, Mounier J, d'Hauteville H, Coquis-Rondon M, Sansonetti PJ. 1989. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci U S A 86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldberg MB, Theriot JA. 1995. Shigella flexneri surface protein IcsA is sufficient to direct actin-based motility. Proc Natl Acad Sci U S A 92:6572–6576. doi: 10.1073/pnas.92.14.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Headley VL, Payne SM. 1990. Differential protein expression by Shigella flexneri in intracellular and extracellular environments. Proc Natl Acad Sci U S A 87:4179–4183. doi: 10.1073/pnas.87.11.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lucchini S, Liu H, Jin Q, Hinton JCD, Yu J. 2005. Transcriptional adaptation of Shigella flexneri during infection of macrophages and epithelial cells: insights into the strategies of a cytosolic bacterial pathogen. Infect Immun 73:88–102. doi: 10.1128/IAI.73.1.88-102.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell-Valois F-X, Schnupf P, Nigro G, Sachse M, Sansonetti PJ, Parsot C. 2014. A fluorescent reporter reveals on/off regulation of the Shigella type III secretion apparatus during entry and cell-to-cell spread. Cell Host Microbe 15:177–189. doi: 10.1016/j.chom.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Ashida H, Mimuro H, Sasakawa C. 2015. Shigella manipulates host immune responses by delivering effector proteins with specific roles. Front Immunol 6:219. doi: 10.3389/fimmu.2015.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puhar A, Tronchere H, Payrastre B, Nhieu GT, Sansonetti PJ. 2013. A Shigella effector dampens inflammation by regulating epithelial release of danger signal ATP through production of the lipid mediator PtdIns5P. Immunity 39:1121–1131. doi: 10.1016/j.immuni.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 9.Mounier J, Boncompain G, Senerovic L, Lagache T, Chretien F, Perez F, Kolbe M, Olivo-Marin JC, Sansonetti PJ, Sauvonnet N. 2012. Shigella effector IpaB-induced cholesterol relocation disrupts the Golgi complex and recycling network to inhibit host cell secretion. Cell Host Microbe 12:381–389. doi: 10.1016/j.chom.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 10.Dong N, Zhu Y, Lu Q, Hu L, Zheng Y, Shao F. 2012. Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell 150:1029–1041. doi: 10.1016/j.cell.2012.06.050. [DOI] [PubMed] [Google Scholar]
- 11.Burnaevskiy N, Fox TG, Plymire DA, Ertelt JM, Weigele BA, Selyunin AS, Way SS, Patrie SM, Alto NM. 2013. Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor IpaJ. Nature 496:106–109. doi: 10.1038/nature12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. 2015. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18:157–168. doi: 10.1016/j.chom.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng Z, Wei C, Guan K, Yuan Y, Zhang Y, Ma S, Cao Y, Wang F, Zhong H, He X. 2016. Bacterial E3 ubiquitin ligase IpaH4.5 of Shigella flexneri targets TBK1 to dampen the host antibacterial response. J Immunol 196:1199–1208. doi: 10.4049/jimmunol.1501045. [DOI] [PubMed] [Google Scholar]
- 14.Runyen-Janecky LJ, Payne SM. 2002. Identification of chromosomal Shigella flexneri genes induced by the eukaryotic intracellular environment. Infect Immun 70:4379–4388. doi: 10.1128/IAI.70.8.4379-4388.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pieper R, Fisher CR, Suh M-J, Huang S-T, Parmar P, Payne SM. 2013. Analysis of the proteome of intracellular Shigella flexneri reveals pathways important for intracellular growth. Infect Immun 81:4635–4648. doi: 10.1128/IAI.00975-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kentner D, Martano G, Callon M, Chiquet P, Brodmann M, Burton O, Wahlander A, Nanni P, Delmotte N, Grossmann J, Limenitakis J, Schlapbach R, Kiefer P, Vorholt JA, Hiller S, Bumann D. 2014. Shigella reroutes host cell central metabolism to obtain high-flux nutrient supply for vigorous intracellular growth. Proc Natl Acad Sci U S A 111:9929–9934. doi: 10.1073/pnas.1406694111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waligora EA, Fisher CR, Hanovice NJ, Rodou A, Wyckoff EE, Payne SM. 2014. Role of intracellular carbon metabolism pathways in Shigella flexneri virulence. Infect Immun 82:2746–2755. doi: 10.1128/IAI.01575-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oaks EV, Wingfield ME, Formal SB. 1985. Plaque formation by virulent Shigella flexneri. Infect Immun 48:124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fisher CR, Davies NM, Wyckoff EE, Feng Z, Oaks EV, Payne SM. 2009. Genetics and virulence association of the Shigella flexneri sit iron transport system. Infect Immun 77:1992–1999. doi: 10.1128/IAI.00064-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cersini A, Salvia AM, Bernardini ML. 1998. Intracellular multiplication and virulence of Shigella flexneri auxotrophic mutants. Infect Immun 66:549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saleh AM, Rudnick H, Aronson PS. 1996. Mechanism of H+-coupled formate transport in rabbit renal microvillus membranes. Am J Physiol 271:F401–F407. doi: 10.1152/ajprenal.1996.271.2.F401. [DOI] [PubMed] [Google Scholar]
- 22.Patel H, Pietro ED, MacKenzie RE. 2003. Mammalian fibroblasts lacking mitochondrial NAD+-dependent methylenetetrahydrofolate dehydrogenase-cyclohydrolase are glycine auxotrophs. J Biol Chem 278:19436–19441. doi: 10.1074/jbc.M301718200. [DOI] [PubMed] [Google Scholar]
- 23.Laerke HN, Jensen BB. 1999. D-tagatose has low small intestinal digestibility but high large intestinal fermentability in pigs. J Nutr 129:1002–1009. doi: 10.1093/jn/129.5.1002. [DOI] [PubMed] [Google Scholar]
- 24.Laerke HN, Jensen BB, Højsgaard S. 2000. In vitro fermentation pattern of D-tagatose is affected by adaptation of the microbiota from the gastrointestinal tract of pigs. J Nutr 130:1772–1779. doi: 10.1093/jn/130.7.1772. [DOI] [PubMed] [Google Scholar]
- 25.Huang Y, Suyemoto M, Garner CD, Cicconi KM, Altier C. 2008. Formate acts as a diffusible signal to induce Salmonella invasion. J Bacteriol 190:4233–4241. doi: 10.1128/JB.00205-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zoetendal EG, Raes J, van den Bogert B, Arumugam M, Booijink CC, Troost FJ, Bork P, Wels M, de Vos WM, Kleerebezem M. 2012. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J 6:1415–1426. doi: 10.1038/ismej.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Huang Y, Wang J, Cheng C, Huang W, Lu P, Xu YN, Wang P, Yan N, Shi Y. 2009. Structure of the formate transporter FocA reveals a pentameric aquaporin-like channel. Nature 462:467–472. doi: 10.1038/nature08610. [DOI] [PubMed] [Google Scholar]
- 28.Lu W, Du J, Schwarzer NJ, Gerbig-Smentek E, Einsle O, Andrade SL. 2012. The formate channel FocA exports the products of mixed-acid fermentation. Proc Natl Acad Sci U S A 109:13254–13259. doi: 10.1073/pnas.1204201109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doberenz C, Zorn M, Falke D, Nannemann D, Hunger D, Beyer L, Ihling CH, Meiler J, Sinz A, Sawers RG. 2014. Pyruvate formate-lyase interacts directly with the formate channel FocA to regulate formate translocation. J Mol Biol 426:2827–2839. doi: 10.1016/j.jmb.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sawers G, Suppmann B. 1992. Anaerobic induction of pyruvate formate-lyase gene expression is mediated by the ArcA and FNR proteins. J Bacteriol 174:3474–3478. doi: 10.1128/jb.174.11.3474-3478.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sawers G. 1993. Specific transcriptional requirements for positive regulation of the anaerobically inducible pfl operon by ArcA and FNR. Mol Microbiol 10:737–747. doi: 10.1111/j.1365-2958.1993.tb00944.x. [DOI] [PubMed] [Google Scholar]
- 32.Partridge JD, Sanguinetti G, Dibden DP, Roberts RE, Poole RK, Green J. 2007. Transition of Escherichia coli from aerobic to micro-aerobic conditions involves fast and slow reacting regulatory components. J Biol Chem 282:11230–11237. doi: 10.1074/jbc.M700728200. [DOI] [PubMed] [Google Scholar]
- 33.Clark DP. 1989. The fermentation pathways of Escherichia coli. FEMS Microbiol Rev 5:223–234. [DOI] [PubMed] [Google Scholar]
- 34.Beyer L, Doberenz C, Falke D, Hunger D, Suppmann B, Sawers RG. 2013. Coordination of FocA and pyruvate formate-lyase synthesis in Escherichia coli demonstrates preferential translocation of formate over other mixed-acid fermentation products. J Bacteriol 195:1428–1435. doi: 10.1128/JB.02166-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei J, Goldberg MB, Burland V, Venkatesan MM, Deng W, Fournier G, Mayhew GF, Plunkett G, Rose DJ, Darling A, Mau B, Perna NT, Payne SM, Runyen-Janecky LJ, Zhou S, Schwartz DC, Blattner FR. 2003. Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect Immun 71:2775–2786. doi: 10.1128/IAI.71.5.2775-2786.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monk JM, Charusanti P, Aziz RK, Lerman JA, Premyodhin N, Orth JD, Feist AM, Palsson BO. 2013. Genome-scale metabolic reconstructions of multiple Escherichia coli strains highlight strain-specific adaptations to nutritional environments. Proc Natl Acad Sci U S A 110:20338–20343. doi: 10.1073/pnas.1307797110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rocha I, Maia P, Evangelista P, Vilaca P, Soares S, Pinto JP, Nielsen J, Patil KR, Ferreira EC, Rocha M. 2010. OptFlux: an open-source software platform for in silico metabolic engineering. BMC Syst Biol 4:45. doi: 10.1186/1752-0509-4-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oberhardt MA, Palsson BO, Papin JA. 2009. Applications of genome-scale metabolic reconstructions. Mol Syst Biol 5:320. doi: 10.1038/msb.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feist AM, Herrgard MJ, Thiele I, Reed JL, Palsson BO. 2009. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol 7:129–143. doi: 10.1038/nrmicro1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu J, Shimizu K. 2004. The effect of pfl gene knockout on the metabolism for optically pure D-lactate production by Escherichia coli. Appl Microbiol Biotechnol 64:367–375. doi: 10.1007/s00253-003-1499-9. [DOI] [PubMed] [Google Scholar]
- 41.Wang H, Gunsalus RP. 2003. Coordinate regulation of the Escherichia coli formate dehydrogenase fdnGHI and fdhF genes in response to nitrate, nitrite, and formate: roles for NarL and NarP. J Bacteriol 185:5076–5085. doi: 10.1128/JB.185.17.5076-5085.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Darwin A, Tormay P, Page L, Griffiths L, Cole J. 1993. Identification of the formate dehydrogenases and genetic determinants of formate-dependent nitrite reduction by Escherichia coli K12. J Gen Microbiol 139:1829–1840. doi: 10.1099/00221287-139-8-1829. [DOI] [PubMed] [Google Scholar]
- 43.Abaibou H, Pommier J, Benoit S, Giordano G, Mandrand-Berthelot MA. 1995. Expression and characterization of the Escherichia coli fdo locus and a possible physiological role for aerobic formate dehydrogenase. J Bacteriol 177:7141–7149. doi: 10.1128/jb.177.24.7141-7149.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldberg MB. 2001. Actin-based motility of intracellular microbial pathogens. Microbiol Mol Biol Rev 65:595–626. doi: 10.1128/MMBR.65.4.595-626.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suzuki T, Sasakawa C. 2001. Molecular basis of the intracellular spreading of Shigella. Infect Immun 69:5959–5966. doi: 10.1128/IAI.69.10.5959-5966.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rossi RM, Yum L, Agaisse H, Payne SM. 2017. Cardiolipin synthesis and outer membrane localization are required for Shigella flexneri virulence. mBio 8:e01199-17. doi: 10.1128/mBio.01199-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niebuhr K, Giuriato S, Pedron T, Philpott DJ, Gaits F, Sable J, Sheetz MP, Parsot C, Sansonetti PJ, Payrastre B. 2002. Conversion of PtdIns(4,5)P2 into PtdIns(5)P by the S. flexneri effector IpgD reorganizes host cell morphology. EMBO J 21:5069–5078. doi: 10.1093/emboj/cdf522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pendaries C, Tronchere H, Arbibe L, Mounier J, Gozani O, Cantley L, Fry MJ, Gaits-Iacovoni F, Sansonetti PJ, Payrastre B. 2006. PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J 25:1024–1034. doi: 10.1038/sj.emboj.7601001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garza-Mayers AC, Miller KA, Russo BC, Nagda DV, Goldberg MB. 2015. Shigella flexneri regulation of ARF6 activation during bacterial entry via an IpgD-mediated positive feedback loop. mBio 6:e02584-14. doi: 10.1128/mBio.02584-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun CH, Wacquier B, Aguilar DI, Carayol N, Denis K, Boucherie S, Valencia-Gallardo C, Simsek C, Erneux C, Lehman A, Enninga J, Arbibe L, Sansonetti P, Dupont G, Combettes L, Tran Van Nhieu G. 2017. The Shigella type III effector IpgD recodes Ca2+ signals during invasion of epithelial cells. EMBO J 36:2567–2580. doi: 10.15252/embj.201696272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Slagowski NL, Kramer RW, Morrison MF, LaBaer J, Lesser CF. 2008. A functional genomic yeast screen to identify pathogenic bacterial proteins. PLoS Pathog 4:e9. doi: 10.1371/journal.ppat.0040009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buysse JM, Dunyak DS, Hartman AB, Venkatesan MM. 1997. Identification and molecular characterization of a 27 kDa Shigella flexneri invasion plasmid antigen, IpaJ. Microb Pathog 23:357–369. doi: 10.1006/mpat.1997.0164. [DOI] [PubMed] [Google Scholar]
- 53.Perry AK, Chen G, Zheng D, Tang H, Cheng G. 2005. The host type I interferon response to viral and bacterial infections. Cell Res 15:407–422. doi: 10.1038/sj.cr.7290309. [DOI] [PubMed] [Google Scholar]
- 54.Elfgang C, Eckert R, Lichtenberg-Fraté H, Butterweck A, Traub O, Klein RA, Hülser DF, Willecke K. 1995. Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J Cell Physiol 129:805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brosnan ME, Brosnan JT. 2016. Formate: the neglected member of one-carbon metabolism. Annu Rev Nutr 36:369–388. doi: 10.1146/annurev-nutr-071715-050738. [DOI] [PubMed] [Google Scholar]
- 56.Aronson PS. 2006. Essential roles of CFEX-mediated Cl-oxalate exchange in proximal tubule NaCl transport and prevention of urolithiasis. Kidney Int 70:1207–1213. doi: 10.1038/sj.ki.5001741. [DOI] [PubMed] [Google Scholar]
- 57.Meiser J, Vazquez A. 2016. Give it or take it: the flux of one-carbon in cancer cells. FEBS J 283:3695–3704. doi: 10.1111/febs.13731. [DOI] [PubMed] [Google Scholar]
- 58.Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, Vafai SB, Cox AG, Marutani E, Ichinose F, Goessling W, Regev A, Carr SA, Clish CB, Mootha VK. 2016. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 5:e10575. doi: 10.7554/eLife.10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Z, Petrovic S, Mann E, Soleimani M. 2002. Identification of an apical Cl−/HCO3− exchanger in the small intestine. Am J Physiol Gastrointest Liver Physiol 282:G573–G579. doi: 10.1152/ajpgi.00338.2001. [DOI] [PubMed] [Google Scholar]
- 60.Nozawa T, Sugiura S, Hashino Y, Tsuji A, Tamai I. 2004. Role of anion exchange transporter PAT1 (SLC26A6) in intestinal absorption of organic anions. J Drug Target 12:97–104. doi: 10.1080/10611860410001693742. [DOI] [PubMed] [Google Scholar]
- 61.Zelcbuch L, Lindner SN, Zegman Y, Vainberg Slutskin I, Antonovsky N, Gleizer S, Milo R, Bar-Even A. 2016. Pyruvate formate-lyase enables efficient growth of Escherichia coli on acetate and formate. Biochemistry 55:2423–2426. doi: 10.1021/acs.biochem.6b00184. [DOI] [PubMed] [Google Scholar]
- 62.Hughes ER, Winter MG, Duerkop BA, Spiga L, Furtado de Carvalho T, Zhu W, Gillis CC, Büttner L, Smoot MP, Behrendt CL, Cherry S, Santos RL, Hooper LV, Winter SE. 2017. Microbial respiration and formate oxidation as metabolic signatures of inflammation-associated dysbiosis. Cell Host Microbe 21:208–219. doi: 10.1016/j.chom.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chavez RG, Alvarez AF, Romeo T, Georgellis D. 2010. The physiological stimulus for the BarA sensor kinase. J Bacteriol 192:2009–2012. doi: 10.1128/JB.01685-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gore AL, Payne SM. 2010. CsrA and Cra influence Shigella flexneri pathogenesis. Infect Immun 78:4674–4682. doi: 10.1128/IAI.00589-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Demers B, Sansonetti PJ, Parsot C. 1998. Induction of type III secretion in Shigella flexneri is associated with differential control of transcription of genes encoding secreted proteins. EMBO J 17:2894–2903. doi: 10.1093/emboj/17.10.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Enninga J, Mounier J, Sansonetti P, Tran Van Nhieu G. 2005. Secretion of type III effectors into host cells in real time. Nat Methods 2:959–965. doi: 10.1038/nmeth804. [DOI] [PubMed] [Google Scholar]
- 67.Ehsani S, Santos JC, Rodrigues CD, Henriques R, Audry L, Zimmer C, Sansonetti P, Tran Van Nhieu G, Enninga J. 2012. Hierarchies of host factor dynamics at the entry site of Shigella flexneri during host cell invasion. Infect Immun 80:2548–2557. doi: 10.1128/IAI.06391-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rohmer L, Hocquet D, Miller SI. 2011. Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol 19:341–348. doi: 10.1016/j.tim.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olive AJ, Sassetti CM. 2016. Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat Rev Microbiol 14:221–234. doi: 10.1038/nrmicro.2016.12. [DOI] [PubMed] [Google Scholar]
- 70.Ahmer BM. 2004. Cell-to-cell signalling in Escherichia coli and Salmonella enterica. Mol Microbiol 52:933–945. doi: 10.1111/j.1365-2958.2004.04054.x. [DOI] [PubMed] [Google Scholar]
- 71.Lee JH, Lee J. 2010. Indole as an intercellular signal in microbial communities. FEMS Microbiol Rev 34:426–444. doi: 10.1111/j.1574-6976.2009.00204.x. [DOI] [PubMed] [Google Scholar]
- 72.Kim J, Park W. 2015. Indole: a signaling molecule or a mere metabolic byproduct that alters bacterial physiology at a high concentration? J Microbiol 53:421–428. doi: 10.1007/s12275-015-5273-3. [DOI] [PubMed] [Google Scholar]
- 73.Pinero-Fernandez S, Chimerel C, Keyser UF, Summers DK. 2011. Indole transport across Escherichia coli membranes. J Bacteriol 193:1793–1798. doi: 10.1128/JB.01477-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rundell EA, McKeithen-Mead SA, Kazmierczak BI. 2016. Rampant cheating by pathogens? PLoS Pathog 12:e1005792. doi: 10.1371/journal.ppat.1005792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pope LM, Reed KE, Payne SM. 1995. Increased protein secretion and adherence to HeLa cells by Shigella spp. following growth in the presence of bile salts. Infect Immun 63:3642–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zurawski DV, Mitsuhata C, Mumy KL, McCormick BA, Maurelli AT. 2006. OspF and OspC1 are Shigella flexneri type III secretion system effectors that are required for postinvasion aspects of virulence. Infect Immun 74:5964–5976. doi: 10.1128/IAI.00594-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang RF, Kushner SR. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195–199. doi: 10.1016/0378-1119(91)90366-J. [DOI] [PubMed] [Google Scholar]
- 80.Rasband WS. 1997. ImageJ. US National Institutes of Health, Bethesda, MD: http://imagej.nih.gov/ij/. [Google Scholar]
- 81.Carpenter CD, Cooley BJ, Needham BD, Fisher CR, Trent MS, Gordon V, Payne SM. 2014. The Vps/VacJ ABC transporter is required for intercellular spread of Shigella flexneri. Infect Immun 82:660–669. doi: 10.1128/IAI.01057-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sharma AK, Payne SM. 2006. Induction of expression of hfq by DksA is essential for Shigella flexneri virulence. Mol Microbiol 62:469–479. doi: 10.1111/j.1365-2958.2006.05376.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S. flexneri intracellular doubling times of the WT and ΔpflB strains were measured in Henle-407 cells. No significant difference in doubling time was observed between the two strains. Download FIG S1, TIF file, 1.65 MB (1.7MB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
[14C]formate uptake was quantified in Henle-407 cells over the course of 48 hours. A significant increase in 14C cpm was observed over time. n = 3. Download FIG S2, TIF file, 0.85 MB (874.5KB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Uninfected Henle-407 cell cytotoxicity in the presence and absence of exogenous formate (20 mM) was determined by quantifying released lactate dehydrogenase (LDH) over 72 hours. We observed no significant difference in LDH release in the presence and absence of formate. (B) Henle-407 cells were plated in a 6-well plate and grown with and without formate. Cells were enumerated at 48 hours; no significant difference in Henle-407 cells per monolayer was observed. Download FIG S3, TIF file, 1.83 MB (1.8MB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plaque size of S. flexneri WT, ΔpflB, and ΔfocA strains was measured; loss of focA, which transports formate, decreases plaque size similarly to the ΔpflB strain; however, formate significantly increases ΔfocA plaque size. An asterisk indicates statistical significance. Download FIG S4, TIF file, 1.58 MB (1.6MB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plaque size of Listeria monocytogenes was measured; formate has no significant effect on L. monocytogenes plaque size. Giemsa stains of plaques underneath corresponding measurements. Download FIG S5, TIF file, 3.50 MB (3.5MB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Schematic of the PFL locus of S. flexneri 2457T. (B) The PFL loci from 79 Shigella genomes were aligned, and phylogenetic analysis reveals that the PFL locus of S. flexneri 2457T is highly conserved from E. coli and among Shigella spp. S. flexneri 2457T is highlighted in green, while E. coli MG1655 is highlighted in blue. Other gastrointestinal pathogens are included for comparison. Download FIG S6, TIF file, 1.88 MB (1.9MB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Human tissue culture lines and bacterial strains used in this study, along with their respective sources. (B) Bacterial plasmids used in this study. (C) Oligonucleotide sequences of primers for cloning, sequencing, and RT-qPCR used in this study. Download Table S1, DOCX file, 0.02 MB (22.5KB, docx) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Complete mapping information for S. flexneri genes induced by formate in the host cell listed in Table 1. (B) Mapping of all intracellular S. flexneri genes grown with and without formate. (C) Eighty-seven S. flexneri genes were mapped in mock-treated samples, to determine human RNA that maps to S. flexneri genes (false positives). Download Table S2, XLSX file, 1.5 MB (1.5MB, xlsx) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plaque size of S. flexneri WT, ΔbarA, and ΔuvrY strains was measured; formate significantly increases WT plaque size 2.2-fold, increases ΔbarA mutant plaque size 1.8-fold, and increases ΔuvrY mutant plaque size 2.0-fold. An asterisk indicates statistical significance. Download FIG S7, TIF file, 2.71 MB (2.7MB, tif) .
Copyright © 2018 Koestler et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.