

Abstract
Histones are positively charged nuclear proteins that facilitate packaging of DNA into nucleosomes common to all eukaryotic cells. Upon cell injury or cell signalling processes, histones are released passively through cell necrosis or actively from immune cells as part of extracellular traps. Extracellular histones function as microbicidal proteins and are pro‐thrombotic, limiting spread of infection or isolating areas of injury to allow for immune cell infiltration, clearance of infection and initiation of tissue regeneration and repair. Histone toxicity, however, is not specific to microbes and contributes to tissue and end‐organ injury, which in cases of systemic inflammation may lead to organ failure and death. This review details the processes of histones release in acute inflammation, the mechanisms of histone‐related tissue toxicity and current and future strategies for therapy targeting histones in acute inflammatory diseases.
Keywords: extracellular histones, immunothrombosis, inflammation, innate immunity
1. INTRODUCTION
Histones were first described by Albrecht Kossel in 1884 as histidine‐rich peptones derived from the nuclear component of avian red blood cells1; he was awarded the Nobel Prize in Physiology or Medicine for this and other work on the nucleus of cells in 1910. Histones are highly conserved across all eukaryotic cells,2 and act as nuclear chaperone proteins, interacting with nucleic acids due to their highly positive charge3 from lysine and arginine residues. Each nucleosome particle consists of 147 base pairs of DNA, wrapped in 1.7 turns around a protein octamer of core histones (H2A, H2B, H3 and H4), further compacted by linker histones (H1 and/or H5).4
Numerous post‐translational modifications of histones have been identified, including acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, ADP ribosylation, deimination and proline isomerization.5 In normal cell function, these alter the nature of the histone‐DNA interaction and allow transcription to occur. More recently, controlled histone degradation has been described in neutrophils leading to chromatin decondensation and release of genomic DNA laced with granular proteins as neutrophil extracellular traps (NETs).6, 7 These meshwork‐like structures promote intravascular thrombosis,8 limit spread of microorganisms, encourage cancer metastasis9 and cause direct injury to adjacent cells.10
This review details what is known about the role of histones as alarmins or DAMPs (damage‐associated molecular patterns), processes leading up to active histone release as principle components of NETs, mechanisms of injury related to extracellular histones and therapeutic strategies for histone detoxification in acute inflammatory conditions.
2. HISTONES AS DAMPS
Among the earliest recognized and better‐described ways in which histones exacerbate cellular injury is in their role as alarmins or DAMPs. Histones released passively from necrotic cells (or actively by other modes of cell death including NETosis) act on adjacent cells and circulating immune cells via pattern recognition receptors to effect specific biological activity. In in vivo systems, these effects can be difficult to study, as histones are co‐released with nuclear DNA and other nuclear DAMPs such as HMGB1 (high mobility group box protein 1), each with their individual activities. Indeed, the mechanism of cell necrosis has significant impact on the kinetics of nuclear DAMP release,11 and nuclear DAMPs acting as complexes have been reported to exert different activities compared to protein isolates.12 Furthermore, where purified histones injected into experimental animals are lethal within minutes,13 necrotic cell death releases nucleosomes (ie: histone‐DNA complexes) which overall appear to be less toxic.14 Indeed a study injecting similar doses of nucleosomes in mice makes no mention of toxicity,15 and others have demonstrated cofactors such as HMGB1 responsible for the immune‐stimulatory effects of nucleosomes.16 Only through the interplay of plasma proteases and nucleases including DNAse1 and factor VII activating protease does nucleosome decondensation occur17; however, this also degrades the histone component and limits cytotoxicity.18 These effects may have significant implications for in vitro signalling studies using recombinant proteins, as effects of isolated nucleosome components may not become apparent in this setup.
Fragments of cell membrane and nuclear proteins also interact with complement proteins and complement cascade regulators to facilitate cell turnover and clearance.19 An important regulator of nucleosome toxicity appears to be factor H of the family of complement regulator proteins. Factor H is actively internalized by apoptotic cells, where it leads to C3 complement activation and cell surface expression, as well as enhanced nucleosome clearance and phagocyte cytokine‐release response to nucleosomes.20 Cells undergoing secondary necrosis can thereby elicit a targeted pro‐inflammatory response.21
Once released from the nucleosome, extracellular histones exert their injurious effects in three ways summarized in Figure 1: (a) by acting as chemokines or inducing chemokine release; (b) by inducing cytokine release and/or apoptosis of adjacent cells and leukocytes; and (c) through direct cytotoxicity.
Figure 1.
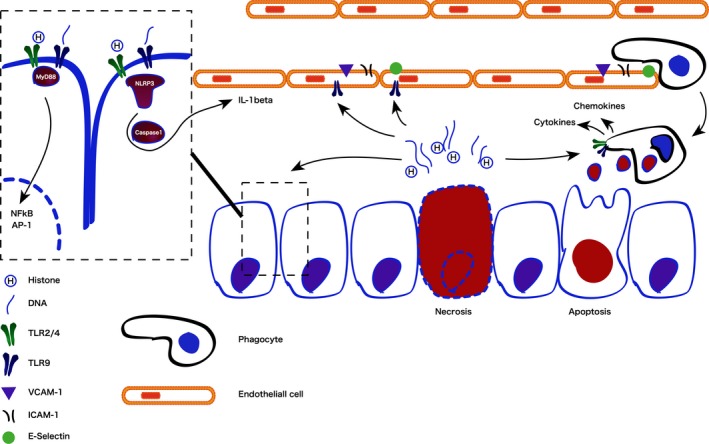
Immunostimulatory effects of passively released histones acting as damage‐associated molecular patterns
2.1. Chemoattractant effects
Histones are both directly chemoattractant, induce release of chemokines from adjacent cells and induce activation of vascular endothelium to promote adhesion and trans‐migration of leukocytes. Direct chemoattractant effects have been demonstrated in vitro using hepatocellular carcinoma cells. Histone‐induced migration of these cells is dependent on the activation of the MAPK/ERK/NF‐kB pathway via TLR4 (toll‐like receptor 4).22 Similarly, histones induce secretion of chemokines CXCL9 and CXCL10 from human monocytes and CXCL10 co‐localizes with extracellular histone H4 in necrotic (but not healthy) tissue.23 Extracellular histones H3 and H4 (but not H1 or H2A/H2B) also activate vascular endothelial cells to increase cell surface expression of E‐selectin, ICAM‐1 and VCAM‐1, thereby increasing leukocyte adhesion, rolling and transmigration in a TLR9‐dependent manner.24 In higher concentrations, these histones are toxic to the endothelium and represent a putative mechanism for pulmonary haemorrhage and ARDS in sepsis13 or pancreatitis.25 Histones acting on endothelial cells via TLRs 2 and 4 also activate NF‐kB and AP‐1 pathways to induce tissue factor expression,26 thereby creating a pro‐thrombotic milieu contributing to the microvascular thrombosis seen in many acute inflammatory diseases. Together, these effects describe the positive feedback loop that can lead to necroinflammation—where the death of relatively few cells induces further injury through inflammatory cell recruitment leading to organ failure, especially within the liver and/or kidney.27
2.2. Pattern recognition receptor responses
The intracellular signalling pathways of extracellular histones as DAMPs acting via TLRs2/4/9, MyD88, NF‐kB and the NLRP3 inflammasome have been well documented and recently reviewed.28, 29, 30 Functionally, histones injected into the renal artery of rats induced necroinflammation as well as IL‐6, TNF‐α and iNOS release.31 These effects were reduced in TLR2/4 knock‐out mice and more pronounced following LPS priming, which increased TLR2/4 mRNA transcription. Low doses of histone H3 (10 μg/mL) have been shown to induce release of IL‐6 and IL‐8 in ARPE‐19 cells, as well as lead to the phosphorylation of ERKs, p38 MAPK and JNK and inhibition of these kinases all resulted in reduced cytokine release.32 Higher doses (50 μg/mL), however, led to cell death in a manner that could not be inhibited using signalling kinase inhibitors. Histones also exacerbate ischaemia/reperfusion injury by a TLR9/MyD88‐dependent mechanism and enhance extracellular DNA‐mediated activation of TLR9 in immune cells.33 Further to their effect on TLRs, histones also appear to induce IL‐1β secretion and activation via an NLRP3/ASC/caspase1‐dependent mechanism, leading to neutrophil recruitment to sites of inflammation.34 Critically, induction of leukocyte cytokine production and release is not dependent on free, circulating histones; nuclear material within blebs from apoptotic cells can induce similar stimulatory effects within resident or infiltrating phagocytes.35
3. HISTONE PROCESSING AND ACTIVE RELEASE DURING NETOSIS
3.1. Signal recognition
A large number of different signals have been shown to be able to induce NET formation, including bacteria,36, 37 viruses,38 yeasts,39, 40 parasites,41 organic crystals,42 non‐organic matter,43 cytokines44 and cellular breakdown products including nuclear DAMPs.45, 46 In order to detect such a variety of signals, there is overlap and convergence of receptor pathways. This may explain some variability in early genetic knock out studies when defining which receptor is critical in mediating NET release. It would seem molecular pattern‐related NET release is mediated predominantly through TLRs 2, 4 and 9,31, 46, 47 immune complex‐related NET release is mediated via Fc receptors and MAC‐a48 and larger pathogens or inorganic matter lead to NETosis though size. The inability to phagocytose large particles within a given time appears to drive neutrophils to autodigest and release NETs in a process dependent on dectin‐1.49 While many signals leading to NETosis may make this an unlikely therapeutic target, it suggests that blocking destructive NETosis in sterile inflammation is possible without affecting a potentially beneficial antimicrobial response.
3.2. Signal transduction
Following signal detection, there are three critical steps leading to NET release: phagocyte oxidase/nicotinamide adenine dinucleotide phosphate‐oxidase (PHOX/NADPHO) activation, nuclear protease translocation and histone deimination (Figure 2).
Figure 2.
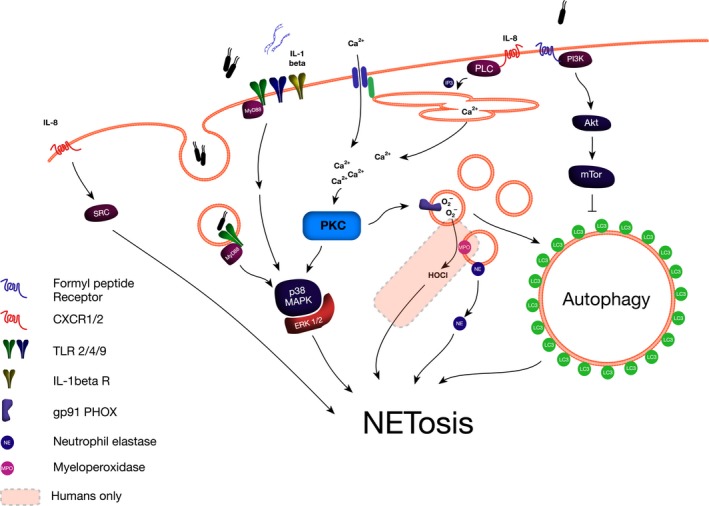
Signalling cascade leading to neutrophil extracellular trap release in murine and human neutrophils
Involvement of PHOX/NADPHO is illustrated by patients with chronic granulomatous disease, an inherited defect in PHOX activity, who are unable to produce NETs when stimulated with phorbol 12‐myristate 13‐acetate (PMA).50 This in turn leads to a clinical picture of recurrent and/or persistent infections, in particular with fungal pathogens. Impressively, there has been a successful report of gene therapy, where an 8‐year‐old boy was treated with a retroviral vector containing a functional gp91 (PHOX subunit) gene, resulting in neutrophils regaining the ability to NETose and leading to a termination of an intractable Aspergillus nidans infection.51 Experimentally, inhibition of NADPHO or myeloperoxidase (MPO) effectively inhibited NETosis stimulated by PMA, whereas inhibition of mitochondrial respiration or superoxide dismutase did not.52 PHOX/NADPHO is itself activated by protein kinase C (PKC). Pan‐activation of PKC isoforms using PMA or the di‐acyl glycerol analogue 1‐oleoyl‐2‐acetyl‐sn‐glycerol effectively stimulates NETosis.53 Specific inhibition of PKC isoform β inhibits both reactive oxygen species (ROS) production by PHOX and NETosis; however, there are conflicting reports on whether PKCζ is also able to inhibit NETosis.54 There are increasing reports of NADPHO‐independent NETosis, such as via the Rous sarcoma (src) kinase family in response to chemokine receptors (CXCR2) activation55 or via unspecified pathways following high‐dose uric acid stimulation.56 These reports highlight the deficiencies of investigating NETosis exclusively using PMA as the stimulant and demonstrate that while PKC activation is sufficient for the induction of NETosis, it is not the only pathway.
Histone deimination by peptidyl‐arginine transferase 4 (PAD4) is an essential step in NET release.57 PAD4 targets methyl‐arginine residues, reducing methylation and increasing citrullination on H4Arg3 and H3Arg2, 8 and 17 in HL‐60 cells58 over a time scale of 15 minutes to 2 hours, in a manner independent of caspase activity.59 These same post‐translational modifications are amongst the most immunogenic histone modifications seen in serum from patients with systemic lupus erythematosis,60 and levels of circulating nucleosomes and citrullinated histone H3 correlate with disease severity in acute inflammatory conditions including sepsis,61 trauma62 and pancreatitis.63 Genetic deletion of PAD4 leads to an inability of neutrophils to release NETs in response to calcium ionophore treatment or lipopolysaccharide (LPS),64 and pharmacological inhibition of PAD4 inhibits NET formation in murine and human neutrophils.65 Overexpression of PAD4, on the other hand, has been shown to cause histone hypercitrullination, nuclear decondensation and release of NET‐like structures in an osteosarcoma cell line.66
Nuclear translocation of granular proteases is the next step towards NET release. Neutrophil azoruphilic granules contain neutrophil elastase (NE), proteinase 3 (PR3) and cathepsin G (CG); however, only NE is translocated to the nucleus and neither inhibition of PR3 nor CG can prevent this translocation.7 Furthermore, the process does not appear to be mediated by fusion of granules with the nucleus, but rather NE dissociates from the granular membrane in a ROS‐dependent manner, before degrading cytosolic actin, arresting actin dynamics and translocating across the nuclear membrane using specific translocation mechanisms.67 Binding of nucleic acid by proteases initiates a process of degradation of nuclear binding proteins68 and controlled integration of MPO into the forming NET. Nuclear NE leads to early degradation of linker histone H1, followed by core histone H4 which coincides with nuclear chromatin decondensation.7 Histone H3 appears to be resistant to degradation in intact nuclei, but not in purified form, suggesting one of the purposes of post‐translational modification is to render histone H3 resistant to NE‐related degradation. This offers novel targets for therapy that have not yet been exploited.
The pathway described above is the best described due to the use of PMA as experimental stimulant of NETosis. In this experimental setup, the three steps are sequential; however, there have been recent reports of NET‐like structures being released rapidly (minutes), by budding of DNA/histone/protease‐containing vesicles from the nucleus followed by active exocytosis of NET‐containing vesicles.69, 70 This potentially bypasses most of the mechanisms described above and requires further study.
3.3. Autophagy
Although most studies support the conclusion that autophagy is essential for NETosis,71, 72 inhibition of mammalian target of Rapamycin (mTOR), a regulatory and inhibitory protein complex, has been reported to reduce NETosis stimulated by bacterial LPS.73 Stimulation of human neutrophils with vasculitis‐associated antibodies led to massive vacuolization, increased LC3BI degradation and could be reduced with the inhibitors of autophagy 3‐methyladenine (3MA) and LY294002.74 Similarly, LC3B containing vacuoles were observed preceding NETosis in LPS or septic plasma‐induced NETosis in human neutrophils which was also effectively inhibited by 3MA and bafilomycin A1.75 Knock down of phosphatase and tensin homologue deleted on chromosome 10 (PTEN), a potent regulator of autophagy, reduced PMA‐induced NETosis in HL‐60 cells and overexpression increased it.76 PKC has been shown to stimulate autophagy which in response to certain stimuli can be independent of mTOR, offering a potential explanation for this discrepancy.77 Figure 2 demonstrates how different stimuli resulting in NETosis can have differential effects on autophagy.
4. MOLECULAR BASIS OF HISTONE‐RELATED CELLULAR AND TISSUE INJURY
4.1. Effects of concentration and histone type on different cells and/or tissues
A wide variety of organisms actively release histones and histone degradation products as microbicidals (histone‐derived antimicrobial peptides; HDAP). Table 1 records a list of HDAPs, the species of origin and purported mechanism of antimicrobial action. The mechanisms of action appear to divide into membrane permeabilizing effects or DNA‐binding and disruption of transcription, which is why some HDAPs are also under investigation for the treatment of cancer. Full length core histones (H2A, H2B, H3 and H4) have shown antimicrobial activity in vitro78 in animal79 and human80 physiology and have the ability to neutralize bacterial endotoxin.
Table 1.
Summary of histone‐derived anti‐microbial peptides (HDAPs), their species of origin and mechanism of action
| Source histone | HDAP | Species of origin | Mechanism of action | References |
|---|---|---|---|---|
| H1 | Full length | Coho salmon (Oncorhynchus kisutch) | Synergism with flounder pleuricidin. Mechanism unknown | 81 |
| H2A | Hipposin | Atlantic halibut (Hippoglossus hippoglossus L.) | Membrane permeabilization | 82, 83 |
| Buforin I, II, III | Asian toad (Bufo bufo gargarizans) | DNA/RNA binding and disruption of cellular functions | 84, 85 | |
| Acipensins | Russian sturgeon (Acipenser gueldenstaedtii) | Outer membrane permeabilization | 86 | |
| Himanturin | Round whip ray (Himantura pastinacoides) | Unknown | 87 | |
| Abhesin | Disk abalone (Haliotis discus discus) | Unknown—possible inhibitor of transcription | 88 | |
| Parasin I | Catfish (Parasilurus asotus) | Membrane permeabilization | 89, 90 | |
| H4 | Full length | American cupped oysters (Crassostrea virginica) | Unknown | 91 |
| Histogranin | Cow (Bos taurus) | DNA gyrase inhibitor | 92 | |
| MrH4 | Freshwater giant prawn (Macrobrachium rosenbergii) | Unknown | 93 |
Cellular injury mediated by extracellular histones has been described experimentally or in human disease of the lung,94 heart,95 liver,96 kidney31 and vascular endothelium.62 Table 2 details the effects of extracellular histones observed in specific cell types.
Table 2.
Summary of effects of histones on different cell types of epithelial, endothelial and mesenchymal origin seen in in vitro and ex vivo experiments
| Cell type | Effects of histones in vitro or ex vivo | Effective therapies |
|---|---|---|
| Epithelial | ||
| A549,97, 98 BEAS‐2B,99 LA‐4,94 MLE‐12,94 mouse type II pneumocytes,98 L02 hepatocytes,100 CHO‐K1,101 CHO‐A745101 | Calcium influx, cytokine (IL‐1β, IL‐6, IL‐10, TNFa) production (A549, BEAS‐2B, LA‐4) and cell death (PI/LDH; all cell types) | Anti‐histone antibodies, APC, heparin, polysialic acid, CIINH |
| Pancreatic acinar cells102, 103 | Trypsin/Chymotrypsin activation, p‐STAT3/t‐STAT3 up‐regulation, cell death (PI) | Polysialic acid |
| HEK293,96, 104 parietal epithelial cells,105 podocytes105 | Up‐regulated TLR2 and TLR4 expression, APC generation | |
| Endothelial | ||
| HPMEC,62, 98 MLVEC106 | Cell death (PI/LDH) | Anti‐histone antibodies, APC, heparin, CIINH |
| HCAEC107 | Up‐regulation of tissue factor mRNA and extression and translation, NF‐kB/AP‐1 activation | |
| EA.hy926,13, 14, 26, 62, 108, 109 HUVEC13, 14, 62, 107, 109, 110, 111 | Calcium influx, IkB depletion, p38MAPK/NF‐kB/AP‐1 activation, tissue factor and vWF generation/release, cell death (PI/AnnexinV binding) | Anti‐histone antibodies, APC, heparin, polysialic acid, CRP, MBP‐p33, PTX3 |
| Glomerular endothelial cells105 | TNFa mRNA expression, cell death (MTT) | Anti‐histone antibodies |
| Mesenchymal | ||
| Murine cardiomyocytes,112 HL‐1 cardiomyocytes113 | Cytosolic ROS production, calcium entry, mitochondrial impairment, reduced contractility, cell death (PI) | |
| Peripheral neutrophils,46, 62 HL‐60101 | IL‐6 production, NETosis, cell death (PI) | Anti‐histone antibodies, IAIP, HMW‐HA |
| Peripheral monocytes,23, 114 MM6,115 U937,100 THP‐1114 | Cytokine production (IL‐1β, IL‐6, IL‐8, IL‐10, TNFa, CXCL10), cell death (PI/LDH), factor Xa/tissue factor generation | Anti‐histone antibodies, heparin, CRP |
| Murine peritoneal macrophages,116 RAW264.7,102, 107 Kupffer cells,33, 96 J774 macrophages105 | Inhibited clearance of other immune cells, HMGB1 secretion, TNFa production, increased tissue factor expression, vWF/angiopoietin‐2/P‐selectin release | APC |
| Human peripheral DCs,34 human monocyte‐derived DCs,117 murine BMDCs105, 118 | TNFa production, NLRP3 protein up‐regulation, mitochondrial membrane dysfunction | Anti‐histone antibodies, APC, heparin |
| Human peripheral lymphocytes119 | Apoptosis, p38‐MAPK phosphorylation, mitochondrial dysfunction, reduced bcl2 expression, caspase‐3 activation | |
| Platelets14, 101, 108, 120, 121, 122, 123 | Calcium influx, platelet aggregation, thrombin generation, P‐selectin/factor Va expression | APC, heparin, CRP, HAS, IAIP, HMW‐HA |
| Human erythrocytes47, 108, 124 | Haemolysis, procoagulant | APC, heparin, MBP‐p33 |
Breakdown of DNA in NETs with DNAse only partially ameliorates NET‐related toxicity, as it does not affect the histone component.125 Concentrations below 10 μg/mL seem to have a signalling function and can induce calcium transients in cells.126 Concentrations greater than 10 μg/mL (or 20 μg/mL in the presence of serum) induce cell death by an uncertain mechanism, which may involve the formation of non‐specific cationic pores in cell membranes.127, 128 Concentrations above 100 μg/mL cause rapid necrosis.
Core histones H3 and H4 are most frequently reported to increase in plasma from sepsis patients as well as experimental sepsis and therapeutic administration of antibodies to these histones improve outcomes in these models.102, 129, 130 It is conceivable that histone citrullination as described above renders these less susceptible to degradation and easier to detect, creating a publication bias. As core histones oligomerize readily with each other in solution131 and will surely rapidly do so upon histone release from any cell type, it is difficult to dissociate toxicity of individual histones from each other in biological systems. When used in in vitro studies, recombinant histones H2A and H2B were also able to induce cellular currents126 or activate thrombin.122
4.2. Calcium/ionic pore effects
The interaction of histones with cell membranes is heavily reliant on charge. Positively charged histones preferentially bind anionic phospholipids such as cardiolipin or phosphatidylserine, but not zwitterionic phospholipids like phosphatidylcholine.132 Furthermore, adding negative charge (eg, a phosphate head group as in phosphatidylinositol bis‐phosphate) increases the binding capacity of histones as measured by calorimetry.128 Histones have also been shown to expose phosphatidylserine on the surface of red blood cells in a dose‐dependent manner47; however, it is unclear whether this is as a result of altering flippase kinetics or via induction of apoptosis pathways. Once integrated, histones induce permeabilization of membranes to cations, disruptions of cellular calcium signalling112 and cell death by necrosis. Negatively charged acute‐phase proteins (such as C‐reactive protein, CRP),14 DNA,128 innate polysaccharides (heparin)120 or synthetic macromolecules126 compete with membrane phospholipids and prevent histone integration and toxicity. Bactericidal properties of histone fragments are dependent on their ability to form amphipathic α‐helices—potentially membrane spanning domains—however no such structural analyses have been performed on mammalian cells to date.89
4.3. Effects on coagulation
The ability of NETs and histones to influence the coagulation cascade and actually initiate venous thrombosis8, 133, 134 is the most recent detail in the emerging field of NETosis research. Clinically, circulating nucleosomes are independent prognostic markers of disseminated intravascular coagulopathy (DIC)135 and some countries, notably Japan, are actively promoting the use of anticoagulants as histone detoxification agents in DIC.136 Positive correlations between histone levels and coagulopathy can also be seen in trauma patients137 and patients with sepsis.129
Figure 3 summarizes the effect of histones and NETs on the coagulation cascade. Histones act synergistically to produce a profound pro‐coagulant drive. Histones are able to induce platelet aggregation and factor V/Va expression and prothrombinase activity, leading to thrombin activation independent of the intrinsic coagulation pathway.122 Histones also inhibit thrombomodulin and protein C activation,138 an effect most pronounced with histones H3 and H4, thus reducing a natural thrombin inhibitor system. Furthermore, histone H4 binding promotes thrombin autoactivation, probably by fixing the prothrombin molecule in a conformational state conducive to proteolytic attack.139 The only exception is linker histone H1, which has been shown to reduce thrombin activation and prolong clotting times140; this mechanism is likely insignificant in acute inflammation, as histone H1 is amongst the first nuclear proteins to be degraded in the process of NETosis.
Figure 3.
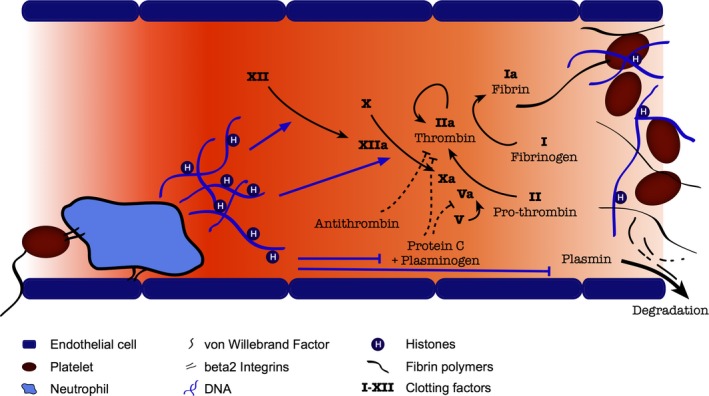
Interaction of histones and DNA with coagulation cascade to promote thrombosis
The presence of DNA in NETs also allows activation of the intrinsic coagulation pathway, demonstrated by NET‐enhanced thrombin generation in platelet‐poor plasma, reduced by factor XII/factor XI depletion or DNAse treatment.141 DNAse treatment in platelet‐rich plasma further increases thrombin generation, indicating differential effects of histones and NETs in different microenvironments. The addition of histones and DNA also increases fibrin fibre thickness, clot stability and delayed clot lysis142 as well as reducing anti‐thrombin‐mediated thrombin inactivation and plasmin activity.143 In in vivo systems, the interplay between von Willebrand factor (vWF), platelets and neutrophils anchors neutrophils to otherwise healthy vessel walls and permits NETing neutrophils to initiate clot formation,125, 144 with propagation that may occur or be enhanced by the mechanisms described above. Deficiencies in degradation of vWF produce clinical microangiopathies (eg, thrombotic thrombocytopenic purpura), the severity of which is also closely correlated with circulating NET components in humans.145
5. THERAPEUTIC STRATEGIES FOR HISTONE DETOXIFICATION IN PATHOLOGY
Histone toxicity is dependent on electrostatic membrane interaction with target cells. A number of innate and synthetic substances have demonstrated the ability to inhibit histone‐related toxicity based on surface charge alone, including plasma proteins (albumin,123 CRP14), polypeptides (polyglutamic acid126) and polysaccharides (heparin/heparanoids,111, 146 polysialic acid,147 bacterial O‐antigen148). Elevated histone‐degrading activated protein C (APC) levels are associated with better outcomes in sepsis115, 149 and trauma patients137; APC therapy is being evaluated for treatment of sepsis150 and pancreatitis.151 The effects of histones and NETs on the coagulation cascade can be overcome by therapy with thrombomodulin121 or tissue plasminogen activator (tPA),143 but the clot‐stabilizing effects of DNA in NETs must be overcome, which is well illustrated by the finding that DNAse therapy in addition to tPA is more effective than either therapy alone.152
In models of sterile and infective acute inflammatory diseases, administration of histones exacerbates end‐organ injury consistently (Table 3). Similarly, damaging effects are at least partly ameliorated by the application of histone‐targeted or histone‐specific therapies.
Table 3.
Summary of effects of extracellular histones observed in in vivo models
| Experimental model | Observations | Effective histone‐based treatment strategies | References |
|---|---|---|---|
| Sepsis | |||
| Bacterial lipopolysaccharide (1‐40 mg/kg i.p/i.v.) | Elevation of circulating histones (including cit‐H3), leukocyte/platelet depletion/DIC; lung: neutrophil margination; endothelial vacuolization, intra‐alveolar haemorrhage and thrombosis; renal: cytokine/chemokine release, tubular apoptosis, neutrophil infiltration, death | APC, anti‐histone antibodies (H1, H4, pan‐histone), heparin (unfractionated or anti‐thrombin activity depleted), PTX3, PLD2 inhibition | 13, 31, 95, 109, 111, 153, 154, 155, 156, 157, 158 |
| Caecal ligation and puncture | Elevation of circulating histones, leukocyte apoptosis; lung injury; reduced cardiac output, left ventricular stroke volume and blood pressure (systolic and diastolic); cytokine release and injury of liver, kidney and spleen; death | Neutrophil depletion, Complement (C5aR1/C5aR2) receptor knock‐out, anti‐histone antibody, non‐anticoagulant heparin, PAD4 inhibition (Cl‐amidine) | 13, 109, 112, 119, 154, 157, 159 |
| MRSA (1‐10 × 107 i.v.) | Bacterial dissemination in blood, liver, spleen, kidney and lung, with associated organ injury | Neutrophil depletion, unfractionated heparuin, DNAse I, vWF inhibition, PAD4 k/o, NE k/o or inhibition | 125 |
| Lung injury | |||
| Bacterial lipopolysaccharide (1‐40 μg/animal i.t.) | Elevation of circulating histones; Pulmonary neutrophil infiltration, NETosis, elevated NE activity, abnormal gas exchange; death | Anti‐H4, aspirin, tirofiban, DNAse I, neutrophil depletion, C5a k/o | 94, 97, 160 |
| Intra‐nasal influenza A virus (102 PFU) or Streptococcus pneumoniae (106 PFU) | Elevation of circulating histones; pulmonary chemokine/cytokine release and inflammatory infiltrate | C1 esterase inhibitor | 98 |
| Intra‐tracheal irritant (HCl, 2 μL/g i.t. 0.01‐0.5 mol/L; Bleomycin 2.5 U/kg i.t.) | Elevation of circulating histones and DNA complexes; elevated pulmonary MPO/LDH activity, neutrophil infiltration, inter‐ and intra‐ alveolar oedema, reduced arterial oxygenation | Anti‐H4, heparin (unfractionated or N‐acetyl), C1 esterase inhibitor | 98, 99, 161 |
| Liver injury | |||
| Ischaemia/reperfusion | Increase in hepatic H3 and H4 and cytokine release; increase in circulating histone‐DNA complexes | Anti‐H3/H4, PAD4 inhibitor | 33, 46, 162 |
| d‐galactosamine (300‐700 mg/kg i.p.) plus LPS (10‐40 mg/kg i.p.) | Hepatic leukocyte infiltration, hepatocellular apoptosis/necrosis; systemic cytokine release and transaminase elevation; death | Anti‐H4, antithrombin activity‐depleted heparin | 100, 163 |
| Acute pancreatitis | |||
| Caerulein (50 μg/kg/h × 4 or 12 i.p.) | Elevation of circulating histones; pancreatic necrosis | 163 | |
| Taurocholate (3.5%‐5% intra ductal) | Elevation of circulating and intra‐pancreatic histones and chemokines/cytokines; NETosis and inflammatory cell infiltrate within pancreas | Thrombin‐derived host defence peptides | 63, 103, 164 |
| l‐arginine (4 mg/kg i.p.) | Elevation of pancreatic histones, neutrophil infiltrate and oedema; pancreatic necrosis; death | Anti‐H3, thrombin‐derived host defence peptides | 102, 164 |
| Systemic administration of histones | |||
| Calf‐thymus histones (0.75‐75 mg/kg i.v.) | Platelet depletion, haemolysis, elevation of vWF, fibrin and thrombin as well as systemic cytokines; prolonged bleeding time; pulmonary oedema, haemorrhage and microvascular occlusion; death | Heparin (unfractionated or O‐desulfated), C‐reactive protein, soluble thrombomodulin, anti‐histone antibody | 14, 101, 104, 108, 110, 113, 120, 121, 165 |
| Recombinant H3 (25‐100 mg/kg) | Leukocyte and platelet depletion; liver injury; death | Heparin (unfractionated and/or low molecular weight) | 166 |
6. CONCLUSIONS
Histones and histone fragments are parts of an ancient antimicrobial mechanism conserved throughout eukaryotic species. In mammals, packaging of histones into NETs and interaction with the coagulation cascade presents an effective mechanism of limiting the spread of microorganisms and concentrating microbicidal peptides at a site of infection, but this comes at a cost of injury to adjacent tissue. In acute systemic inflammatory conditions, such as sepsis and trauma, systemic release of histones exacerbates micro‐circulatory thrombosis, worsens tissue perfusion and contributes significantly to organ injury. Recognition of this phenomenon may allow targeted therapy, limiting systemic injury and improving survival.
CONFLICTS OF INTEREST
The authors confirm that there are no conflicts of interest.
AUTHOR CONTRIBUTIONS
This review was designed by P.S. W.H and P.S undertook a systematic and comprehensive review of the literature, with critical input from A.V.T, D.N.C and R.S. All authors contributed to the critical review, editing and final approval of the manuscript.
ACKNOWLEDGEMENTS
This work was supported by a Royal College of Surgeons of England Fellowship (P.S.); the UK/China Postgraduate Scholarship for Excellence (W.H.); the Medical Research Council (A.V.T., D.N.C., R.S.); and the Biomedical Research Unit funding scheme of the National Institute for Health Research (W.H., D.N.C., A.V.T., R.S.). Robert Sutton is an NIHR Senior Investigator.
Szatmary P, Huang W, Criddle D, Tepikin A, Sutton R. Biology, role and therapeutic potential of circulating histones in acute inflammatory disorders. J Cell Mol Med. 2018;22:4617–4629. 10.1111/jcmm.13797
REFERENCES
- 1. Kossel A. Ueber einen peptonartigen Bestandtheil des Zellkerns. Zschr Physiol Chem. 1884;8:511. [Google Scholar]
- 2. Talbert PB, Henikoff S. Histone variants—ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010;11:264‐275. [DOI] [PubMed] [Google Scholar]
- 3. Park YJ, Luger K. Histone chaperones in nucleosome eviction and histone exchange. Curr Opin Struct Biol. 2008;18:282‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251‐260. [DOI] [PubMed] [Google Scholar]
- 5. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693‐705. [DOI] [PubMed] [Google Scholar]
- 6. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532‐1535. [DOI] [PubMed] [Google Scholar]
- 7. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880‐15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cools‐Lartigue J, Spicer J, McDonald B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013. 10.1172/JCI67484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Allam R, Kumar SV, Darisipudi MN, Anders HJ. Extracellular histones in tissue injury and inflammation. J Mol Med (Berl). 2014;92:465‐472. [DOI] [PubMed] [Google Scholar]
- 11. Beyer C, Pisetsky DS. Modeling nuclear molecule release during in vitro cell death. Autoimmunity. 2013;46:298‐301. [DOI] [PubMed] [Google Scholar]
- 12. Chen R, Fu S, Fan XG, et al. Nuclear DAMP complex‐mediated RAGE‐dependent macrophage cell death. Biochem Biophys Res Commun. 2015;458:650‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu J, Zhang X, Pelayo R, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318‐1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abrams ST, Zhang N, Dart C, et al. Human CRP defends against the toxicity of circulating histones. J Immunol. 2013;191:2495‐2502. [DOI] [PubMed] [Google Scholar]
- 15. Gauthier VJ, Tyler LN, Mannik M. Blood clearance kinetics and liver uptake of mononucleosomes in mice. J Immunol. 1996;156:1151‐1156. [PubMed] [Google Scholar]
- 16. Urbonaviciute V, Furnrohr BG, Meister S, et al. Induction of inflammatory and immune responses by HMGB1‐nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007‐3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stephan F, Marsman G, Bakker LM, et al. Cooperation of factor VII‐activating protease and serum DNase I in the release of nucleosomes from necrotic cells. Arthritis Rheumatol. 2014;66:686‐693. [DOI] [PubMed] [Google Scholar]
- 18. Marsman G, von Richthofen H, Bulder I, et al. DNA and factor VII‐activating protease protect against the cytotoxicity of histones. Blood Adv. 2017;1:2491‐2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martin M, Blom AM. Complement in removal of the dead—balancing inflammation. Immunol Rev. 2016;274:218‐232. [DOI] [PubMed] [Google Scholar]
- 20. Martin M, Leffler J, Smolag KI, et al. Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ. 2016;23:903‐911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu X, Molinaro C, Johnson N, Casiano CA. Secondary necrosis is a source of proteolytically modified forms of specific intracellular autoantigens: implications for systemic autoimmunity. Arthritis Rheum. 2001;44:2642‐2652. [DOI] [PubMed] [Google Scholar]
- 22. Chen R, Xie Y, Zhong X, et al. Novel chemokine‐like activities of histones in tumor metastasis. Oncotarget. 2016;7:61728‐61740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Westman J, Papareddy P, Dahlgren MW, et al. Extracellular histones induce chemokine production in whole blood ex vivo and leukocyte recruitment in vivo. PLoS Pathog. 2015;11:e1005319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoo HJ, Lee JS, Kim JE, et al. Extracellular histone released from leukemic cells increases their adhesion to endothelium and protects them from spontaneous and chemotherapy‐induced leukemic cell death. PLoS ONE. 2016;11:e0163982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu T, Huang W, Szatmary P, et al. Accuracy of circulating histones in predicting persistent organ failure and mortality in patients with acute pancreatitis. Br J Surg. 2017;104:1215‐1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim JE, Yoo HJ, Gu JY, Kim HK. Histones induce the procoagulant phenotype of endothelial cells through tissue factor up‐regulation and thrombomodulin down‐regulation. PLoS ONE. 2016;11:e0156763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mulay SR, Linkermann A, Anders HJ. Necroinflammation in kidney disease. J Am Soc Nephrol. 2016;27:27‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Silk E, Zhao H, Weng H, Ma D. The role of extracellular histone in organ injury. Cell Death Dis. 2017;8:e2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen R, Kang R, Fan XG, Tang D. Release and activity of histone in diseases. Cell Death Dis. 2014;5:e1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Magna M, Pisetsky DS. The alarmin properties of DNA and DNA‐associated nuclear proteins. Clin Ther. 2016;38:1029‐1041. [DOI] [PubMed] [Google Scholar]
- 31. Allam R, Scherbaum CR, Darisipudi MN, et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol. 2012;23:1375‐1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kawano H, Ito T, Yamada S, et al. Toxic effects of extracellular histones and their neutralization by vitreous in retinal detachment. Lab Invest. 2014;94:569‐585. [DOI] [PubMed] [Google Scholar]
- 33. Huang H, Evankovich J, Yan W, et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll‐like receptor 9 in mice. Hepatology. 2011;54:999‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Allam R, Darisipudi MN, Tschopp J, Anders HJ. Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur J Immunol. 2013;43:3336‐3342. [DOI] [PubMed] [Google Scholar]
- 35. Wickman GR, Julian L, Mardilovich K, et al. Blebs produced by actin‐myosin contraction during apoptosis release damage‐associated molecular pattern proteins before secondary necrosis occurs. Cell Death Differ. 2013;20:1293‐1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, von Kockritz‐Blickwede M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun. 2010;2:576‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Young RL, Malcolm KC, Kret JE, et al. Neutrophil extracellular trap (NET)‐mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS ONE. 2011;6:e23637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saitoh T, Komano J, Saitoh Y, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus‐1. Cell Host Microbe. 2012;12:109‐116. [DOI] [PubMed] [Google Scholar]
- 39. Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006;8:668‐676. [DOI] [PubMed] [Google Scholar]
- 40. Bruns S, Kniemeyer O, Hasenberg M, et al. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 2010;6:e1000873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abi Abdallah DS, Lin C, Ball CJ, King MR, Duhamel GE, Denkers EY. Toxoplasma gondii triggers release of human and mouse neutrophil extracellular traps. Infect Immun. 2012;80:768‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schauer C, Janko C, Munoz LE, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. 2014;20:511‐517. [DOI] [PubMed] [Google Scholar]
- 43. Jhunjhunwala S, Aresta‐DaSilva S, Tang K, et al. Neutrophil responses to sterile implant materials. PLoS ONE. 2015;10:e0137550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rossaint J, Herter JM, Van Aken H, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap‐mediated sterile inflammation. Blood. 2014;123:2573‐2584. [DOI] [PubMed] [Google Scholar]
- 45. Tadie JM, Bae HB, Jiang S, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll‐like receptor 4. Am J Physiol Lung Cell Mol Physiol. 2013;304:L342‐L349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang H, Tohme S, Al‐Khafaji AB, et al. Damage‐associated molecular pattern‐activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62:600‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Semeraro F, Ammollo CT, Esmon NL, Esmon CT. Histones induce phosphatidylserine exposure and a procoagulant phenotype in human red blood cells. J Thromb Haemost. 2014;12:1697‐1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Behnen M, Leschczyk C, Moller S, et al. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcgammaRIIIB and Mac‐1. J Immunol. 2014;193:1954‐1965. [DOI] [PubMed] [Google Scholar]
- 49. Branzk N, Lubojemska A, Hardison SE, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. 2014;15:1017‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nishinaka Y, Arai T, Adachi S, Takaori‐Kondo A, Yamashita K. Singlet oxygen is essential for neutrophil extracellular trap formation. Biochem Biophys Res Commun. 2011;413:75‐79. [DOI] [PubMed] [Google Scholar]
- 51. Bianchi M, Hakkim A, Brinkmann V, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114:2619‐2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kirchner T, Moller S, Klinger M, Solbach W, Laskay T, Behnen M. The impact of various reactive oxygen species on the formation of neutrophil extracellular traps. Mediators Inflamm. 2012;2012:849136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gray RD, Lucas CD, Mackellar A, et al. Activation of conventional protein kinase C (PKC) is critical in the generation of human neutrophil extracellular traps. J Inflamm (Lond). 2013;10:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Neeli I, Radic M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front Immunol. 2013;4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Marcos V, Zhou Z, Yildirim AO, et al. CXCR2 mediates NADPH oxidase‐independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med. 2010;16:1018‐1023. [DOI] [PubMed] [Google Scholar]
- 56. Arai Y, Nishinaka Y, Arai T, et al. Uric acid induces NADPH oxidase‐independent neutrophil extracellular trap formation. Biochem Biophys Res Commun. 2014;443:556‐561. [DOI] [PubMed] [Google Scholar]
- 57. Neeli I, Radic M. Knotting the NETs: analyzing histone modifications in neutrophil extracellular traps. Arthritis Res Ther. 2012;14:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang Y, Li M, Stadler S, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol. 2008;180:1895‐1902. [DOI] [PubMed] [Google Scholar]
- 60. Liu CL, Tangsombatvisit S, Rosenberg JM, et al. Specific post‐translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res Ther. 2012;14:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hirose T, Hamaguchi S, Matsumoto N, et al. Presence of neutrophil extracellular traps and citrullinated histone H3 in the bloodstream of critically ill patients. PLoS ONE. 2014;9:e111755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abrams ST, Zhang N, Manson J, et al. Circulating histones are mediators of trauma‐associated lung injury. Am J Respir Crit Care Med. 2013;187:160‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ou X, Cheng Z, Liu T, et al. Circulating histone levels reflect disease severity in animal models of acute pancreatitis. Pancreas. 2015;44:1089‐1095. [DOI] [PubMed] [Google Scholar]
- 64. Martinod K, Demers M, Fuchs TA, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci USA. 2013;110:8674‐8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lewis HD, Liddle J, Coote JE, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. 2015;11:189‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Leshner M, Wang S, Lewis C, et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap‐like structures. Front Immunol. 2012;3:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase‐containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014;8:883‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Thomas MP, Whangbo J, McCrossan G, et al. Leukocyte protease binding to nucleic acids promotes nuclear localization and cleavage of nucleic acid binding proteins. J Immunol. 2014;192:5390‐5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kono M, Saigo K, Takagi Y, et al. Heme‐related molecules induce rapid production of neutrophil extracellular traps. Transfusion. 2014;54:2811‐2819. [DOI] [PubMed] [Google Scholar]
- 70. Pilsczek FH, Salina D, Poon KK, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus . J Immunol. 2010;185:7413‐7425. [DOI] [PubMed] [Google Scholar]
- 71. Itakura A, McCarty OJ. Pivotal role for the mTOR pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am J Physiol Cell Physiol. 2013;305:C348‐C354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Remijsen Q, Vanden Berghe T, Wirawan E, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21:290‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. McInturff AM, Cody MJ, Elliott EA, et al. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia‐inducible factor 1 alpha. Blood. 2012;120:3118‐3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tang S, Zhang Y, Yin SW, et al. Neutrophil extracellular trap formation is associated with autophagy‐related signalling in ANCA‐associated vasculitis. Clin Exp Immunol. 2015;180:408‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kambas K, Mitroulis I, Apostolidou E, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS ONE. 2012;7:e45427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Teimourian S, Moghanloo E. Role of PTEN in neutrophil extracellular trap formation. Mol Immunol. 2015;66:319‐324. [DOI] [PubMed] [Google Scholar]
- 77. Tan SH, Shui G, Zhou J, et al. Induction of autophagy by palmitic acid via protein kinase C‐mediated signaling pathway independent of mTOR (mammalian target of rapamycin). J Biol Chem. 2012;287:14364‐14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tagai C, Morita S, Shiraishi T, Miyaji K, Iwamuro S. Antimicrobial properties of arginine‐ and lysine‐rich histones and involvement of bacterial outer membrane protease T in their differential mode of actions. Peptides. 2011;32:2003‐2009. [DOI] [PubMed] [Google Scholar]
- 79. Drab T, Kracmerova J, Hanzlikova E, et al. The antimicrobial action of histones in the reproductive tract of cow. Biochem Biophys Res Commun. 2014;443:987‐990. [DOI] [PubMed] [Google Scholar]
- 80. Kim HS, Cho JH, Park HW, Yoon H, Kim MS, Kim SC. Endotoxin‐neutralizing antimicrobial proteins of the human placenta. J Immunol. 2002;168:2356‐2364. [DOI] [PubMed] [Google Scholar]
- 81. Patrzykat A, Zhang L, Mendoza V, Iwama GK, Hancock RE. Synergy of histone‐derived peptides of coho salmon with lysozyme and flounder pleurocidin. Antimicrob Agents Chemother. 2001;45:1337‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bustillo ME, Fischer AL, LaBouyer MA, Klaips JA, Webb AC, Elmore DE. Modular analysis of hipposin, a histone‐derived antimicrobial peptide consisting of membrane translocating and membrane permeabilizing fragments. Biochim Biophys Acta. 2014;1838:2228‐2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Birkemo GA, Luders T, Andersen O, Nes IF, Nissen‐Meyer J. Hipposin, a histone‐derived antimicrobial peptide in Atlantic halibut (Hippoglossus hippoglossus L.). Biochim Biophys Acta. 2003;1646:207‐215. [DOI] [PubMed] [Google Scholar]
- 84. Park CB, Kim HS, Kim SC. Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem Biophys Res Commun. 1998;244:253‐257. [DOI] [PubMed] [Google Scholar]
- 85. Cho JH, Sung BH, Kim SC. Buforins: histone H2A‐derived antimicrobial peptides from toad stomach. Biochim Biophys Acta. 2009;1788:1564‐1569. [DOI] [PubMed] [Google Scholar]
- 86. Shamova OV, Orlov DS, Balandin SV, et al. Acipensins—novel antimicrobial peptides from leukocytes of the Russian sturgeon Acipenser gueldenstaedtii . Acta Naturae. 2014;6:99‐109. [PMC free article] [PubMed] [Google Scholar]
- 87. Sathyan N, Philip R, Chaithanya ER, Anil Kumar PR, Antony SP. Identification of a histone derived, putative antimicrobial peptide Himanturin from round whip ray Himantura pastinacoides and its phylogenetic significance. Results Immunol. 2012;2:120‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. De Zoysa M, Nikapitiya C, Whang I, Lee JS, Lee J. Abhisin: a potential antimicrobial peptide derived from histone H2A of disk abalone (Haliotis discus discus). Fish Shellfish Immunol. 2009;27:639‐646. [DOI] [PubMed] [Google Scholar]
- 89. Koo YS, Kim JM, Park IY, et al. Structure‐activity relations of parasin I, a histone H2A‐derived antimicrobial peptide. Peptides. 2008;29:1102‐1108. [DOI] [PubMed] [Google Scholar]
- 90. Park IY, Park CB, Kim MS, Kim SC. Parasin I, an antimicrobial peptide derived from histone H2A in the catfish, Parasilurus asotus . FEBS Lett. 1998;437:258‐262. [DOI] [PubMed] [Google Scholar]
- 91. Dorrington T, Villamil L, Gomez‐chiarri M. Upregulation in response to infection and antibacterial activity of oyster histone H4. Fish Shellfish Immunol. 2011;30:94‐101. [DOI] [PubMed] [Google Scholar]
- 92. Lemaire S, Trinh TT, Le HT, et al. Antimicrobial effects of H4‐(86‐100), histogranin and related compounds—possible involvement of DNA gyrase. FEBS J. 2008;275:5286‐5297. [DOI] [PubMed] [Google Scholar]
- 93. Chaurasia MK, Palanisamy R, Bhatt P, et al. A prawn core histone 4: derivation of N‐ and C‐terminal peptides and their antimicrobial properties, molecular characterization and mRNA transcription. Microbiol Res. 2015;170:78‐86. [DOI] [PubMed] [Google Scholar]
- 94. Bosmann M, Grailer JJ, Ruemmler R, et al. Extracellular histones are essential effectors of C5aR‐ and C5L2‐mediated tissue damage and inflammation in acute lung injury. FASEB J. 2013;27:5010‐5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Alhamdi Y, Abrams ST, Cheng Z, et al. Circulating histones are major mediators of cardiac injury in patients with sepsis. Crit Care Med. 2015;43:2094‐2103. [DOI] [PubMed] [Google Scholar]
- 96. Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011;187:2626‐2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Saffarzadeh M, Juenemann C, Queisser MA, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. 2012;7:e32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wygrecka M, Kosanovic D, Wujak L, et al. Anti‐histone properties of C1 esterase inhibitor protect against lung injury. Am J Respir Crit Care Med. 2016;196:186‐199. [DOI] [PubMed] [Google Scholar]
- 99. Zhang Y, Zhao Z, Guan L, et al. N‐acetyl‐heparin attenuates acute lung injury caused by acid aspiration mainly by antagonizing histones in mice. PLoS ONE. 2014;9:e97074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wen Z, Lei Z, Yao L, et al. Circulating histones are major mediators of systemic inflammation and cellular injury in patients with acute liver failure. Cell Death Dis. 2016;7:e2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chaaban H, Keshari RS, Silasi‐Mansat R, et al. Inter‐alpha inhibitor protein and its associated glycosaminoglycans protect against histone‐induced injury. Blood. 2015;125:2286‐2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kang R, Zhang Q, Hou W, et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology. 2014;146:1097‐1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Merza M, Hartman H, Rahman M, et al. Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology. 2015;149:1920‐1931.e8. [DOI] [PubMed] [Google Scholar]
- 104. Kowalska MA, Zhao G, Zhai L, et al. Modulation of protein C activation by histones, platelet factor 4, and heparinoids: new insights into activated protein C formation. Arterioscler Thromb Vasc Biol. 2014;34:120‐126. [DOI] [PubMed] [Google Scholar]
- 105. Kumar SV, Kulkarni OP, Mulay SR, et al. Neutrophil extracellular trap‐related extracellular histones cause vascular necrosis in severe GN. J Am Soc Nephrol. 2015;26:2399‐2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhang Y, Guan L, Yu J, et al. Pulmonary endothelial activation caused by extracellular histones contributes to neutrophil activation in acute respiratory distress syndrome. Respir Res. 2016;17:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yang X, Li L, Liu J, Lv B, Chen F. Extracellular histones induce tissue factor expression in vascular endothelial cells via TLR and activation of NF‐kappaB and AP‐1. Thromb Res. 2016;137:211‐218. [DOI] [PubMed] [Google Scholar]
- 108. Westman J, Smeds E, Johansson L, et al. Treatment with p33 curtails morbidity and mortality in a histone‐induced murine shock model. J Innate Immun. 2014;6:819‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Daigo K, Nakakido M, Ohashi R, et al. Protective effect of the long pentraxin PTX3 against histone‐mediated endothelial cell cytotoxicity in sepsis. Sci Signal. 2014;7:ra88. [DOI] [PubMed] [Google Scholar]
- 110. Lam FW, Cruz MA, Parikh K, Rumbaut RE. Histones stimulate von Willebrand factor release in vitro and in vivo. Haematologica. 2016;101:e277‐e279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wang F, Zhang N, Li B, et al. Heparin defends against the toxicity of circulating histones in sepsis. Front Biosci (Landmark Ed). 2015;20:1259‐1270. [DOI] [PubMed] [Google Scholar]
- 112. Kalbitz M, Grailer JJ, Fattahi F, et al. Role of extracellular histones in the cardiomyopathy of sepsis. FASEB J. 2015;29:2185‐2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Alhamdi Y, Zi M, Abrams ST, et al. Circulating histone concentrations differentially affect the predominance of left or right ventricular dysfunction in critical illness. Crit Care Med. 2016;44:e278‐e288. [DOI] [PubMed] [Google Scholar]
- 114. Gould TJ, Lysov Z, Swystun LL, et al. Extracellular histones increase tissue factor activity and enhance thrombin generation by human blood monocytes. Shock. 2016;46:655‐662. [DOI] [PubMed] [Google Scholar]
- 115. Ekaney ML, Otto GP, Sossdorf M, et al. Impact of plasma histones in human sepsis and their contribution to cellular injury and inflammation. Crit Care. 2014;18:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Friggeri A, Banerjee S, Xie N, et al. Extracellular histones inhibit efferocytosis. Mol Med. 2012;18:825‐833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Donis‐Maturano L, Sanchez‐Torres LE, Cerbulo‐Vazquez A, et al. Prolonged exposure to neutrophil extracellular traps can induce mitochondrial damage in macrophages and dendritic cells. Springerplus. 2015;4:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hsu LW, Chen CL, Nakano T, et al. The role of a nuclear protein, histone H1, on signalling pathways for the maturation of dendritic cells. Clin Exp Immunol. 2008;152:576‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Liu ZG, Ni SY, Chen GM, et al. Histones‐mediated lymphocyte apoptosis during sepsis is dependent on p38 phosphorylation and mitochondrial permeability transition. PLoS ONE. 2013;8:e77131. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 120. Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood. 2011;118:3708‐3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Nakahara M, Ito T, Kawahara K, et al. Recombinant thrombomodulin protects mice against histone‐induced lethal thromboembolism. PLoS ONE. 2013;8:e75961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet‐dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118:1952‐1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lam FW, Cruz MA, Leung HC, Parikh KS, Smith CW, Rumbaut RE. Histone induced platelet aggregation is inhibited by normal albumin. Thromb Res. 2013;132:69‐76. [DOI] [PubMed] [Google Scholar]
- 124. Rosenbluh J, Hariton‐Gazal E, Dagan A, Rottem S, Graessmann A, Loyter A. Translocation of histone proteins across lipid bilayers and Mycoplasma membranes. J Mol Biol. 2005;345:387‐400. [DOI] [PubMed] [Google Scholar]
- 125. Kolaczkowska E, Jenne CN, Surewaard BG, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Gamberucci A, Fulceri R, Marcolongo P, Pralong WF, Benedetti A. Histones and basic polypeptides activate Ca2+/cation influx in various cell types. Biochem J. 1998;331(Pt 2):623‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kleine TJ, Lewis PN, Lewis SA. Histone‐induced damage of a mammalian epithelium: the role of protein and membrane structure. Am J Physiol. 1997;273:C1925‐C1936. [DOI] [PubMed] [Google Scholar]
- 128. Lete MG, Sot J, Ahyayauch H, et al. Histones and DNA compete for binding polyphosphoinositides in bilayers. Biophys J. 2014;106:1092‐1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wildhagen KC, Wiewel MA, Schultz MJ, et al. Extracellular histone H3 levels are inversely correlated with antithrombin levels and platelet counts and are associated with mortality in sepsis patients. Thromb Res. 2015;136:542‐547. [DOI] [PubMed] [Google Scholar]
- 130. Li Y, Liu Z, Liu B, et al. Citrullinated histone H3: a novel target for the treatment of sepsis. Surgery. 2014;156:229‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ramakrishnan V. Histone structure and the organization of the nucleosome. Annu Rev Biophys Biomol Struct. 1997;26:83‐112. [DOI] [PubMed] [Google Scholar]
- 132. Pereira LF, Marco FM, Boimorto R, et al. Histones interact with anionic phospholipids with high avidity; its relevance for the binding of histone‐antihistone immune complexes. Clin Exp Immunol. 1994;97:175‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Brill A, Fuchs TA, Savchenko AS, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10:136‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. von Bruhl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Kim JE, Lee N, Gu JY, Yoo HJ, Kim HK. Circulating levels of DNA‐histone complex and dsDNA are independent prognostic factors of disseminated intravascular coagulation. Thromb Res. 2015;135:1064‐1069. [DOI] [PubMed] [Google Scholar]
- 136. Iba T, Gando S, Thachil J. Anticoagulant therapy for sepsis‐associated disseminated intravascular coagulation: the view from Japan. J Thromb Haemost. 2014;12:1010‐1019. [DOI] [PubMed] [Google Scholar]
- 137. Kutcher ME, Xu J, Vilardi RF, Ho C, Esmon CT, Cohen MJ. Extracellular histone release in response to traumatic injury: implications for a compensatory role of activated protein C. J Trauma Acute Care Surg. 2012;73:1389‐1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin‐dependent protein C activation. J Thromb Haemost. 2011;9:1795‐1803. [DOI] [PubMed] [Google Scholar]
- 139. Barranco‐Medina S, Pozzi N, Vogt AD, Di Cera E. Histone H4 promotes prothrombin autoactivation. J Biol Chem. 2013;288:35749‐35757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kheiri SA, Fasy TM, Billett HH. Effects of H1 histones and a monoclonal autoantibody to H1 histones on clot formation in vitro: possible implications in the antiphospholipid syndrome. Thromb Res. 1996;82:43‐50. [DOI] [PubMed] [Google Scholar]
- 141. Gould TJ, Vu TT, Swystun LL, et al. Neutrophil extracellular traps promote thrombin generation through platelet‐dependent and platelet‐independent mechanisms. Arterioscler Thromb Vasc Biol. 2014;34:1977‐1984. [DOI] [PubMed] [Google Scholar]
- 142. Longstaff C, Varju I, Sotonyi P, et al. Mechanical stability and fibrinolytic resistance of clots containing fibrin, DNA, and histones. J Biol Chem. 2013;288:6946‐6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Varju I, Longstaff C, Szabo L, et al. DNA, histones and neutrophil extracellular traps exert anti‐fibrinolytic effects in a plasma environment. Thromb Haemost. 2015;113:1289‐1298. [DOI] [PubMed] [Google Scholar]
- 144. Slaba I, Wang J, Kolaczkowska E, McDonald B, Lee WY, Kubes P. Imaging the dynamic platelet‐neutrophil response in sterile liver injury and repair in mice. Hepatology. 2015;62:1593‐1605. [DOI] [PubMed] [Google Scholar]
- 145. Fuchs TA, Kremer Hovinga JA, Schatzberg D, Wagner DD, Lammle B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood. 2012;120:1157‐1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Freeman CG, Parish CR, Knox KJ, et al. The accumulation of circulating histones on heparan sulphate in the capillary glycocalyx of the lungs. Biomaterials. 2013;34:5670‐5676. [DOI] [PubMed] [Google Scholar]
- 147. Merza M, Hartman H, Rahman M, et al. Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology. 2015;149:1920‐1931.e8. [DOI] [PubMed] [Google Scholar]
- 148. Chaput C, Spindler E, Gill RT, Zychlinsky A. O‐antigen protects gram‐negative bacteria from histone killing. PLoS ONE. 2013;8:e71097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zeerleder S, Stephan F, Emonts M, et al. Circulating nucleosomes and severity of illness in children suffering from meningococcal sepsis treated with protein C. Crit Care Med. 2012;40:3224‐3229. [DOI] [PubMed] [Google Scholar]
- 150. Zhang Z. The efficacy of activated protein C for the treatment of sepsis: incorporating observational evidence with a Bayesian approach. BMJ Open. 2015;5:e006524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Babu BI, Genovese T, Mazzon E, et al. Recombinant human activated protein C (Xigris) attenuates murine cerulein‐induced acute pancreatitis via regulation of nuclear factor kappaB and apoptotic pathways. Pancreas. 2012;41:619‐628. [DOI] [PubMed] [Google Scholar]
- 152. Ge L, Zhou X, Ji WJ, et al. Neutrophil extracellular traps in ischemia‐reperfusion injury‐induced myocardial no‐reflow: therapeutic potential of DNase‐based reperfusion strategy. Am J Physiol Heart Circ Physiol. 2015;308:H500‐H509. [DOI] [PubMed] [Google Scholar]
- 153. Li Y, Liu B, Fukudome EY, et al. Identification of citrullinated histone H3 as a potential serum protein biomarker in a lethal model of lipopolysaccharide‐induced shock. Surgery. 2011;150:442‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Wildhagen KC, Garcia de Frutos P, Reutelingsperger CP, et al. Nonanticoagulant heparin prevents histone‐mediated cytotoxicity in vitro and improves survival in sepsis. Blood. 2014;123:1098‐1101. [DOI] [PubMed] [Google Scholar]
- 155. Iba T, Miki T, Hashiguchi N, Tabe Y, Nagaoka I. Combination of antithrombin and recombinant thrombomodulin modulates neutrophil cell‐death and decreases circulating DAMPs levels in endotoxemic rats. Thromb Res. 2014;134:169‐173. [DOI] [PubMed] [Google Scholar]
- 156. Kusano T, Chiang KC, Inomata M, et al. A novel anti‐histone H1 monoclonal antibody, SSV monoclonal antibody, improves lung injury and survival in a mouse model of lipopolysaccharide‐induced sepsis‐like syndrome. Biomed Res Int. 2015;2015:491649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Lee SK, Kim SD, Kook M, et al. Phospholipase D2 drives mortality in sepsis by inhibiting neutrophil extracellular trap formation and down‐regulating CXCR2. J Exp Med. 2015;212:1381‐1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Kawai C, Kotani H, Miyao M, et al. Circulating extracellular histones are clinically relevant mediators of multiple organ injury. Am J Pathol. 2016;186:829‐843. [DOI] [PubMed] [Google Scholar]
- 159. Biron BM, Chung CS, O'Brien XM, Chen Y, Reichner JS, Ayala A. Cl‐amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immun. 2017;9:22‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Caudrillier A, Kessenbrock K, Gilliss BM, et al. Platelets induce neutrophil extracellular traps in transfusion‐related acute lung injury. J Clin Invest. 2012;122:2661‐2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Zhang Y, Wen Z, Guan L, et al. Extracellular histones play an inflammatory role in acid aspiration‐induced acute respiratory distress syndrome. Anesthesiology. 2015;122:127‐139. [DOI] [PubMed] [Google Scholar]
- 162. Huang H, Nace GW, McDonald KA, et al. Hepatocyte‐specific high‐mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high‐mobility group box 1 in cellular protection. Hepatology. 2014;59:1984‐1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Wen Z, Liu Y, Li F, et al. Circulating histones exacerbate inflammation in mice with acute liver failure. J Cell Biochem. 2013;114:2384‐2391. [DOI] [PubMed] [Google Scholar]
- 164. Merza M, Rahman M, Zhang S, et al. Human thrombin‐derived host defense peptides inhibit neutrophil recruitment and tissue injury in severe acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2014;307:G914‐G921. [DOI] [PubMed] [Google Scholar]
- 165. Michels A, Albanez S, Mewburn J, et al. Histones link inflammation and thrombosis through the induction of Weibel‐Palade body exocytosis. J Thromb Haemost. 2016;14:2274‐2286. [DOI] [PubMed] [Google Scholar]
- 166. Iba T, Hashiguchi N, Nagaoka I, Tabe Y, Kadota K, Sato K. Heparins attenuated histone‐mediated cytotoxicity in vitro and improved the survival in a rat model of histone‐induced organ dysfunction. Intensive Care Med Exp. 2015;3:36. [DOI] [PMC free article] [PubMed] [Google Scholar]