

Abstract
In this paper we elaborate on recently developed molecular switch architectures and how these new systems can help with the realization of new functions and advancement of artificial molecular machines. Progress in chemically and photoinduced switches and motors is summarized and contextualized such that the reader may gain an appreciation for the novel tools that have come about in the past decade. Many of these systems offer distinct advantages over commonly employed switches, including improved fidelity, addressability, and robustness. Thus, this paper serves as a jumping-off point for researchers seeking new switching motifs for specific applications, or ones that address the limitations of presently available systems.
Keywords: molecular switches, molecular machines, photochromic compounds
Biology has set the stage for what can be accomplished with artificial molecular switches and machines (1, 2), and while molecular machinists take inspiration from it, and aspire to mimic the functions of machines found in nature, they are also pushing beyond what it can offer. Chemistry, the discipline that “creates its own object” per 19th-century French chemist Marcellin Berthelot, lends the toolsets to accomplish this lofty goal, as it enables the synthesis of structures limited only by imagination and tapping into increasingly complicated processes. There have been momentous advances in the field in the past decades, and strides have been taken to transform molecular switches into motors and machines, which is not a trivial task by any means (3). One of the biggest challenges in this respect is the fact that the tools available to the field are scarce and sometimes limited in capability. For example, until very recently there was only one family of photochromic motors (i.e., compounds that exhibit unidirectional 360° rotation) in the literature (4), while chemically activated ones have been even scarcer (5). Such limitations ultimately slow down progress and lead to a shortage of real-life applications (6). In this vein, the development of new molecular switches (i.e., what make molecular machines tick) that address the limitations of those present in the literature will open new research opportunities and venues.
In this paper, we will survey and evaluate new chemically and light-activated molecular switch architectures that have been developed in the past decade. The list of systems covered is not meant to be exhaustive (7–12). Historically prevalent switches, including interlocked molecules, will not be discussed as they have been recently reviewed elsewhere (13–18). Our focus will be on systems that bring unique dimensions to the field, including interswitch communication (19), new molecular motor designs (20–22), and novel photoswitching capabilities and properties (23–29), among others. Where possible, we will also show that new molecular architectures enable the design and synthesis of new molecular machines [e.g., a molecular robotic arm (30)] that otherwise would have been difficult to attain.
Chemically Activated Switches
Biology uses chemically responsive switches and machines to regulate a myriad of processes, including the creation of proton gradients, protein folding, and viral entry and translocation, among others (31). These biological processes offer excellent temporal control via well-regulated signaling cascades as well as fast and efficient switching sequences (32). In principle, artificial chemically activated switches can be used to mimic all these processes and expand their use in nonbiological areas. The bistability of these systems, their complete and reversible switching based on a myriad of benign and biocompatible inputs, which when selective allow for sensing, fatigue resistance, and use in intersystem communication are added benefits to their light-activated counterparts. One major drawback of such chemically activated systems, however, is the accumulation of sacrificial species in solution, a problem nature has solved through sequestration and excretion. Accumulation of waste products tends to reduce the efficiency of chemically activated switches over many cycles (33, 34). If we, as chemists, wish to mimic and eventually compete with biology then we need to address this issue, in addition to developing new chemically activated switches capable of complex functions. Here we survey new architectures of chemically activated molecular switches and the properties (e.g., switching efficiency, thermal stability, and switching cycles) needed to achieve these goals.
Redox-Regulated Nanoswitches.
In 2012 Schmittel et al. (35) reported a chemically activated switch capable of modulating catalysis of a Knoevenagel reaction. Their switch employs the HETPHEN system (two phenanthrolines bound to copper). In the initial state the biaryl lever binds to an attached Zn-porphyrin system. Upon addition of, and chelation to, Cu+ in the presence of phenanthroline, the lever rotates away from the Zn-porphyrin group. Treating with cyclam, which has a higher affinity to Cu+ than HETPHEN, returns the system to the initial state. A schematic representation of the switching cycles is shown in Fig. 1. The switch was used to control the Knoevenagel reaction between 4-nitrobenzaldehyde and diethyl malonate in the presence of piperidine. Briefly, when the switch is in the initial state, the Knoevenagel reaction occurs, as expected. Addition of Cu+, in the presence of phenanthroline, induces switching, opening a coordination site on Zn-porphyrin. Piperidine then coordinates to the Zn-porphyrin, rendering it incapable of catalysis. The system was reversibly toggled ON and OFF three times with no measurable loss in catalytic activity (23% yield per cycle).
Fig. 1.

Schematic representation of the signaling cascade by two redox-regulated nanoswitches.
Inspired by recent advances using switches in systems chemistry (19, 36), Schmittel et al. developed a two-switch signaling cascade (37). Both switches operate similar to the one described above and possess a Zn-porphyrin group and a phenanthroline derivative as binding sites. They differ, however, in their connecting levers. The first lever employs the HETPHEN system, while the second employs the HETTAP system (copper bound to phenanthroline and terpyridine, respectively). Each switch shows significantly different affinity for copper in its various oxidation states (i.e., the HETPHEN-based switch binds strongly to Cu+ while the HETTAP-based switch binds strongly to Cu2+). They showed that a solution of these two switches and copper could communicate with each other by changing the oxidation state of copper in solution via oxidation/reduction cycles. Briefly, adding Cu+ to a solution of both switches in their initial states (bound to Zn-porphyrin) triggers switching of the HETPHEN switch. In situ chemical oxidation to Cu2+ leads to migration of the metal to the HETTAP switch, resulting in the simultaneous switching of both systems. Reduction to Cu+ reverts the system to the previous state. Fig. 1 shows a schematic representation of this signaling cascade, and the detailed molecular structures and switching cycles are shown in SI Appendix, Fig. S1. Later, the system was made more complicated by introducing a third switch to the solution: a HETPHEN system bearing a ferrocene group (38). In this system Cu+ could be relayed between the two HETPHEN systems based on the oxidation state of the ferrocene, then to the HETTAP switch via oxidation of Cu+ to Cu2+.
More recently a three-stage switch was reported that can orthogonally control two different reactions (Knoevenagel and CuAAC “click”) (39). This idea was further expanded to include a fourth stage, enabling the control over two sequential reactions (CuAAC click and Michael addition) in one pot (40). These systems beautifully explore and elucidate the benefits of intersystem communication and its use in controlling catalysis. One shortcoming to consider in the future, though, is the elaborate structure of the switches. Avoiding the laborious multistep synthesis required to access them will accelerate progress in this arena.
Hydrazones.
The Aprahamian group developed a family of hydrazone-based chemically activated switches that show configurational change, rather than a conformational one, upon acid/base switching (41).
Structure property analysis led to the design of a switch having quinolinyl and pyridinyl rings as the stator and rotor parts, respectively (1-E) (42). This structural modification introduced an additional intramolecular H-bond to the system (compared with the parent systems employing naphthyl stators) that locks in the relative configuration and conformation of the rotor and stator. Addition of TFA (2.4 eq) to a solution of 1-E leads to 1-Z-H+ (configurational change), which transforms to 1-Z-H22+ (conformational change) upon further addition of TFA (Fig. 2). Addition of NEt3, to either 1-Z-H+ or 1-Z-H22+, affords the metastable Z configuration (1-Z) that thermally relaxes back to 1-E, albeit more slowly than the naphthalene-based parent compound. Through careful control of the amount of acid, this switch provides full fidelity of rotation around two independent axels (i.e., the C=N double and C–N single bonds). Leigh and coworkers (30) took advantage of this unique property of complete control over the configuration and conformation of a molecular switch to design a hydrazone-based robotic arm that transports cargo a distance of 2 nm. Additionally, Leigh and coworkers (43) recently used this same scaffold to build a molecular assembler capable of controlling the absolute stereochemical outcome of addition to an α,β-unsaturated ester. These last two examples showcase the importance of the development of new switch architectures for the proliferation of new molecular machines.
Fig. 2.

Four-step acid- and base-induced switching of hydrazone 1.
The same quinolinyl-based switch, when chelating Zn2+, triggers not only E → Z isomerization but also a concomitant release of the N–H proton (44). This coordination-coupled deprotonation (CCD) process is capable of acting as a signal from the switch to other species in solution and has been used to modulate fluorescence as well as catalyze imine hydrolysis in solution (19). Recently, a negative feedback loop (45) based on CCD was reported (Fig. 3). Briefly, below 20 mol % of Zn2+ the CCD process does not trigger the release of enough protons to deprotect a trityl-protected aminoquinoline found in the solution. Above 20 mol % the trityl group is cleaved and the obtained aminoquinoline reacts with pyridine carbaldehyde to form a tridentate imine. This new imine ligand outcompetes the hydrazone (2-Zn2+) for Zn2+ (3), driving the equilibrium of CCD product to the original hydrazone (2), effectively lowering the concentration of protons and shutting down further production of imine.
Fig. 3.

Synthetic negative feedback loop using hydrazone 2. Adapted with permission from ref. 45 (Copyright 2016, American Chemical Society).
Next, the hydrazone switch was used to tackle one of the major drawbacks associated with every chemically activated system: the accumulation of salts that degrade the efficacy of the system as the switching cycles proceed. To address this issue, the hydrazone switch was rendered water-soluble (4) and coupled with a reversible photoacid (34). Irradiation of a solution containing both switch and photoacid with blue light (419 nm) induces the release of a proton upon the conversion of MER to SPIRO, which triggers the switching of 4 from E to Z-H+ (Fig. 4). Equilibrating the solution under darkness for 150 min returns the system to its initial state. The advantage of this system is that the photoacid and its conjugate base are used to increase and decrease, respectively, the pH of the solution, thus eliminating the accumulation of deleterious salts! This system could be cleanly switched 100 times in methanol without any signs of degradation. While the use of photoacids is one way to limit the accumulation of sacrificial species, one could imagine that the integration of molecular switches with microfluidics (i.e., an open system vs. the closed ones predominantly used to date) can be another means to solve this issue.
Fig. 4.

Light-induced switching of 4 through the use of a photoacid (MER).
Biphenyl Motor.
One of the ultimate aims with molecular switches is to impose directionality into their motion, thus converting them into motors. Accomplishing this goal has been very challenging, especially in the context of chemically activated systems (46–48). Recently, Feringa and coworkers (5) developed such a motor based on an atropisomeric biphenyl (5) possessing a fluoro group and a chiral sulfone on one ring and a bromo substituent on the other. These groups were chosen to gain control over which ortho groups pass each other, and the direction of the crossing. The motor functions through orthogonal palladium functionalization of either the C–H [Pd(II)] or C–Br [Pd(0)] bond ortho to the aryl–aryl bond. The “one-pot” operation of the motor is shown in Fig. 5, and the conditions for sequential operation can be found in SI Appendix, Fig. S2. The C–H bond of (S,M)-5 is activated by reaction with Pd(II), which then coordinates to the sulfone. This Pd–S coordination dramatically reduces the rotational barrier, which allows the Br and F groups to pass each other. Fortuitously, this ring flip is thermodynamically favored (ca. 15 kJ⋅mol−1), thus driving the motor to (S,P)-5 after reforming the C–H bond by treating with sodium acetoxyborohydride. This first stage of the motor works in 45% yield. In the second stage, the C–Br bond of (S,P)-5 undergoes oxidative insertion in the presence of Pd(0); the Pd center coordinates to the chiral sulfone, lowering the barrier to rotation, thus allowing for the H and F groups to pass each other. After reforming the C–Br bond by reacting with N-bromosuccinimide, (S,M)-5 is obtained in 42% yield with ca. 11:1 diastereomeric selectivity, and the full 360° rotation is complete. The motor also works in a one-pot sequence, using the Pd(0) derived from the first step to fuel the second one. While the overall yield is low (19%, one-pot sequence), the fidelity of the motor is promising, and this example is an excellent proof of principle for the use of chemical input in controlling molecular motors.
Fig. 5.

One-pot operation of a Pd-fueled molecular motor. Conditions: (i) Pd(OAc)2 and TFA in 1,2-dichloroethane (1,2-DCE). (ii) Trans,trans-dibenzylideneacetone in 1,2-DCE followed by sodium triacetoxyborohydride. (iii) Tricyclohexylphosphine in THF. (iv) N-bromosuccinamide in DCM.
Photoswitches
Light offers several advantages over chemical stimuli, namely its spatial and temporal control, remote addressability, low toxicity, and ease of implementation. These promising attributes have led to the development of numerous photoswitching moieties for use in photopharmacology, sensing, energy production, and actuation (16–18, 49–54). Here we will focus on both T- and P-type photochromic compounds which undergo thermal- or photoinduced back-isomerization, respectively (55). Additionally, we will attempt to contextualize switches based on (i) ease of synthesis, (ii) switching efficiency [quantum yield and photostationary state (PSS)], (iii) addressability (band separation), (iv) thermal stability (half-life), and (v) photostability (switching cycles). Attention will also be given to systems where the activation wavelength is shifted to the red region, and in the case of azo compounds, are stable toward reduction by glutathione. These two points are important criteria for photoswitches being developed as part of the recent push toward photopharmacology (52, 53). For simplicity, each of the photoswitches discussed herein is presented in Fig. 6.
Fig. 6.

A summary of the discussed photoswitches.
Diarylethene.
Diarylethenes are classically known to undergo reversible intramolecular pericyclic reactions, yielding photoswitches with significant electronic differences between open and closed forms (17). Typically, these moieties consist of two five-membered heterocyclic aromatic rings (e.g., furan or thiophene) linked by ethene through their β-positions. By moving the linker to the α-position (6), Guo et al. (26) prevented pericyclization, forcing E/Z photoisomerization around the central double bond. The thermodynamically stable E isomer could be photoisomerized into the Z isomer with high efficiency using unfiltered “white” light (PSS of 100% Z with ΦE → Z undetermined), which reduces the moiety’s end-to-end distance by about 7 Å. The Z → E isomerization process stimulated with blue light was less effective (PSS of 60% E with ΦZ → E undetermined) but quantitative when induced with heat (100 °C). The switch exhibits good fatigue resistance with no appreciable degradation after 20 switching cycles, but quantification of its thermal half-life requires further investigation. The incorporation of thiophene units, a popular moiety in organic semiconductors, uniquely positions this switch for possible applications in switchable electronics.
Azo Derivatives.
The azobenzene moiety enjoys widespread popularity, owing to its ease of synthesis, large extinction coefficients, reasonable quantum yields, and appreciable change in end-to-end distance between the E and Z configurations, the latter being useful in actuation (16). However, the azo moiety suffers from incomplete photoswitching, frequent need for tissue-damaging UV light to induce switching, susceptibility to reduction by glutathione (GSH, a biologically relevant antioxidant), and concerns over it and its metabolites’ toxicity (51). Here, we discuss recent derivatizations of the azo functional group which depart from the traditional decorations on azobenzene.
The bridged azobenzenes, diazocines, reintroduced by Herges and coworkers (7-10), differentiate themselves from previous systems through their strained structure, which results in a thermodynamic preference for the Z configuration over E (a reversal from traditional azobenzenes) (23, 56, 57). Thus, the switch undergoes end-to-end expansion upon isomerization, opposite from traditional azobenzenes, allowing it to access novel order/disorder transitions or actuation motions when properly incorporated into molecular scaffolds. The parent system, 7, boasts well separated n-π* bands (Δλmax = 86 nm) for the Z and E isomers which permits two-way switching using visible light with high PSSs and quantum yields (PSS385 of 92% E with ΦZ → E = 72% and PSS520 of 100% Z with ΦE → Z = 50%) (23). Samanta et al. (57) later elaborated on this switch with symmetric para-acetamido substitution (8), red-shifting the λmax of the E and Z isomers by 9 and 15 nm, respectively, while maintaining high PSSs. These derivatives demonstrated robust stability toward reduction by glutathione in the Z form and reasonable stability in the E configuration (τ1/2 ∼ 3 h at 25 °C, 5 mM GSH, pH 7.0). More recently, Hammerich et al. (56) documented further red-shifting of these switches through development of heterodiazocines (9 and 10). Although the n-π* band of the Z isomer remains around 400 nm, the λmax of the E configuration is shifted to 525 nm (compared with 490 nm for 7), allowing E → Z photoisomerization through irradiation of the band’s tail at 660 nm. The switching efficiencies of 9 and 10 were slightly lower than that of 7 for Z → E switching (PSS385/405 of 70–80% E) but remained high for E → Z isomerization (PSS530/660 of >99% Z) while quantum yields went unexamined. The stability of these newer derivatives to glutathione is not reported, and thus their potential biocompatibility for photopharmocology applications remains unknown. The reported diazocines appear to be resilient to multiple switching cycles. Encouragingly, their troublesome synthesis—yields for the azo-forming ring-closure step were ∼4% for 7—was recently addressed and improved with a solvent-free method to obtain 7 in moderate (51%) yields (58).
Alternatively, Aprahamian and coworkers (24, 59–61) explored BF2 coordination by quinolinyl and phenanthridinyl hydrazones as a means of red-shifting the activation wavelength of azo-compounds. Reasonable band separation between the E and Z isomers, coupled with low energy absorbance, enabled photoswitching from the thermodynamically stable E form to the metastable Z configuration with near-infrared (NIR, 710 nm) light! NIR switching was accessed through parasubstitution of the parent switch’s (11) benzene ring with electron-donating dialkyl amines (59). This substitution shortens the Z isomer’s thermal half-life (τ1/2) from 12.5 h for 11 down to 120 s for 14 (in deoxygenated samples). Extending the π-conjugation in the switch (15) was implemented as an alternative approach to red-shifting the absorption band of the parent system while maintaining longer τ1/2. This successful strategy was accompanied by an unexpected result: The Z → E isomerization rate was found to be concentration-dependent, allowing for its modulation by a factor of 1,800! Dynamic light scattering revealed the propensity of 15 toward aggregation (hydrodynamic radius, Rh, increases from 5 to 92 nm going from 1.0 × 10−4 to 8.3 × 10−3 M), which explains the slower isomerization rates at higher concentrations.
The azo-BF2 switches have variable PSS compositions owing in part to differences in their Z → E isomerization kinetics. For instance, switch 12 shows impressively efficient photoswitching (PSS490 of 92% E with ΦZ → E = 95% and PSS630 of 96% Z with ΦE → Z = 71%) while switches 13 and 14 isomerize too quickly to accurately determine the long-wavelength PSSs. As for switching in biologically relevant environments, complexes 13 and 14 can be switched in aqueous solvents (1:1 acetonitrile:PBS buffer); however, they undergo hydrolysis back to the hydrazone starting material with time. Encouragingly, complex 13 in its Z form showed minimal hydrolysis in acetonitrile:PBS buffer in the presence and absence of glutathione (59). This result indicates that further structural modification might stabilize the E isomer toward hydrolysis as well. If accomplished, the azo–BF2 complexes will be well positioned to be incorporated into the next generation of photoactive drugs.
Azoheteroarenes represent a nontrivial and exciting departure from the classic azobenzene framework, offering tunable thermal Z → E isomerization half-lives and activation wavelength(s). Fuchter and coworkers (25, 62) recently reintroduced nonsymmetrical azo switches composed of benzene and nitrogen-containing five-membered heteroaromatics. Arylazopyrazoles demonstrated long-lived metastable Z isomers and near-quantitative two-way switching with good quantum yields (ΦE → Z of 46–61% and ΦZ → E of 56–60%). Unfortunately, an inherent trade-off exists between the two features: 17 enjoys a long thermal half-life (τ1/2 ∼ 1,000 d) but diminished PSSs (PSS355 of >98% Z and PSS532 of 70% E). This result still represents an improvement over 1-methyl-5-phenylazoimidazole (PSS365 of 98% Z and PSS455 of 55% E with τ1/2 of 22 d) (63). Conversely, 18 adopts a twisted Z isomer because of steric destabilization from the “ortho”-methyl substituents, which truncates the half-life (10 d) but breaks symmetry, enabling access to 98% E at PSS532 (25). These conflicting features were later balanced in 19, which boasts a 74-d half-life and high PSSs (PSS355 of >98% Z and PSS532 of 97% E) (62). This system’s high addressability and thermal stability make it an ideal candidate for manipulating bulk material properties, such as cholesteric pitch, order/disorder in liquid crystalline (LC) elastomers, and so on.
However, Velasco and coworkers (64–66) developed asymmetric azoheteroarenes intending to reduce the Z → E thermal back-isomerization relaxation time. These systems employ donor and acceptor moieties on opposing sides of the azo linker that facilitate rapid isomerization (milliseconds to microseconds depending on solvent and substitution) (49, 67). The switches demonstrate reproducible isomerization after thousands of cycles and some derivatives have shown promising switching in PBS buffer. The efficiency (quantum yield, ΦE → Z) of these systems remains unclear and requires further exploration. Additionally, glutathione stability is unreported.
Imines.
Lehn (68) has long postulated the possibility of chiral imine motors based on orthogonal activation and relaxation modes, but these systems have only recently been realized (20, 21). The two- and four-stroke light-driven motors were developed using simple chiral imines with bulky hydrocarbons (i.e., Ph, tBu, and 1-naphthalene) on the N-side (rotor) and a dibenzo[a,d]-cycloheptene ring system on C-side (stator) (20). Four different four-stroke motors were developed in a single synthetic step from commercially available materials. Tracking the unidirectional molecular motion in these systems, however, is difficult, and thus the dibenzo[a,d]-cycloheptene stator was decorated with a methyl group to break symmetry, allowing full characterization of the four diastereoisomers of 20 (Fig. 7). The function of the motors relies on two orthogonal mechanisms: light-induced out-of-plane rotation around the C=N double bond and either thermal ring inversion (four-stroke) or inversion at imine nitrogen (two-stroke). For a four-stroke motor, two successive rotation–inversion cycles give rise to a full 360° rotation. Briefly, irradiation of 20-(P,cis) with >280-nm light affords 20-(M,trans), via out-of-plane rotation. Thermal ring inversion occurs over a 48-h incubation at room temperature to afford the 20-(P,trans) diastereoisomer. Irradiation with >280-nm light converts 20-(P,trans) to 20-(M,cis). Finally, the system undergoes thermal ring inversion at 25 °C over 48 h to bring the system back to the initial state. This system could be converted to a two-stroke motor by drastically increasing the energy barrier to ring inversion. This allows in-plane imine inversion to be the thermal relaxation pathway. Increasing the ring inversion barrier was achieved by expanding the dibenzo[a,d]-cycloheptene ring, effectively pushing the C–H bonds of the peripheral phenyl groups closer to the rotor, and using the bulky tBu group on the rotor. While in-plane imine inversion has been well-established (69), the out-of-plane rotation was only assumed, based on computation (70). Lehn and coworkers (21) later established, via the products of the photochemical ring expansion of cyclopropane rings in a simple camphor-based imine motor, that the rotation is indeed out-of-plane, and is biased to one direction at a 4:1 ratio. This structurally simple system expands the tools available to the community by offering a synthetically straightforward motor that might be used in driving self-assembled LC structures and polymeric assemblies out of equilibrium.
Fig. 7.

The sequential operation of a four-stroke imine-based motor 20.
Acylhydrazones and Hydrazones.
Although acylhydrazone and hydrazone moieties find usage within a variety of supramolecular chemistry niches, their usage as configurational photoswitches is relatively novel. Inspired by reports from Lehn and coworkers (71–73), Van Dijken et al. (74) studied a library of acylhydrazone photoswitches (40-plus analogs) and demonstrated their tunability. The favorable features among the reported switches include ease of synthesis, negative photochromism, high degrees of band separation (up to 109 nm), tunable τ1/2 (from minutes to days), and respectable quantum yields (ΦE → Z = 30–40%). The switches also demonstrate robust fatigue resistance despite the need for UV-light switching. In an elegant manifestation of the potential of acylhydrazones, Leigh and coworkers (75) employed pyridyl-acylhydrazones in switchable enantioselective catalysis. The switchable cinchona alkaloid-squaramide catalysts were readily toggled ON/OFF either through their photoisomerization or heating with catalytic amounts of TFA. This report encourages further development of acylhydrazones in switchable catalytic systems so that two switchable catalysts may be used simultaneously in one pot to switch between the production of opposing enantiomers.
Departing from acylhydrazones, Qian et al. (27) recently reported bistable photochromic hydrazone photoswitches whose thermal isomerization half-lives extend up to 2,700 y! Tuning the system’s H-bond acceptor(s) provided the key to photoswitching. For instance, hydrazones possessing a pyridyl rotor do not efficiently photoisomerize; however, when the pyridyl moiety in the rotor is exchanged for a phenyl ring (21) isomerization takes place (PSS340 of 76% Z with ΦE → Z = 10.2% and PSS442 of 95% E with ΦZ → E = 1.7%)! Interestingly the Z isomer is the thermodynamically stable configuration in these switches, again showcasing the importance of the intramolecular H-bond in determining switch properties. By substituting the phenyl group in the stator for a quinolinyl moiety (22), the isomerization barrier going from E to Z increased, resulting in an enhancement in τ1/2 from 255 to 2,700 y. This enhancement came with a switching efficiency penalty (PSS340 of 76% Z with ΦE → Z = 2.4% and PSS442 of 91% E with ΦZ → E = 0.3%). Both 21 and 22 are photostable and retain their switching capability in polar protic and aprotic solvents, and in the presence of either acid or base.
Hemithioindigo and Derivatives.
Hemithioindigo derivatives, consisting of thioindigo moieties bound to a stilbene fragment through a central C=C bond, also undergo E/Z photoisomerization. Although these compounds have been known for decades, their development recently experienced a resurgence (28). Hemithioindigo boasts visible/visible E/Z photoswitching, tunable thermal half-lives and acceptable quantum yields (ΦZ → E = 14–23% and ΦE → Z = 5–33%) and PSSs, as well as straightforward syntheses. Perhaps the largest detriment to hemithioindigo switches is their propensity to undergo intermolecular [2+2] cycloaddition at the central C=C bond under concentrated conditions (76).
Dube and coworkers (77–79) computationally and experimentally explored numerous derivatives having para-substituents of varying electron-donor strength on the stilbene fragment, allowing them to define key structure/property relationships. For instance, sterically congested stilbene fragments (twisted in the S0 state) bearing strong electron-donating groups (23 and 24) form twisted intramolecular charge-transfer states in polar solvents (78), lowering their ΦZ → E (a 31- and 220-fold reduction from cyclohexane to DMSO, respectively). This demonstrates the potential to control deexcitation pathways (nonproductive single-bond rotation or double-bond isomerization) based on local polarity. However, derivatives with highly twisted ground-state geometries and weak electron donors (25 and 26) do not show solvent-dependent photoisomerization.
Derivatizing the thioindigo fragment with electron-donating groups offers an alternative avenue for red-shifting absorbance profiles while maintaining thermal stability, characteristics that are typically in conflict (80). Incorporating an NMe2 group into the thioindigo fragment (27) demonstrated this property as both the E and Z λmax shifted by about 50 nm. This structural change also enabled dual mode switching through (i) protonation and (ii) Z/E photoisomerization, allowing for basic logic functionality (80). However, and as with most of the reported hemithioindigos discussed above, fatigue resistance must be considered for the successful implementation of these switches.
Recently, Zweig and Newhouse (81) substituted the traditional phenyl-based stilbene fragment for α-pyrrole, inverting the thermodynamic minimum from the Z to E configuration thanks to the stabilizing H-bonding interaction in the E isomer. This feature led to negative photochromism in these systems while pyrrole’s electron-rich nature red-shifted the absorbance of both the E and Z isomers (E shifted much more, widening Δλmax). These switches display very good addressability well into the visible range with high PSSs [e.g., the α′-(p-OMePh) substituted derivative showcases a PSS623 of 97% Z and PSS460 of 99% E] and acceptable Z → E quantum yields (ΦZ → E = 13% and ΦE → Z unreported). This derivative retains its photoswitchability after 20 cycles, and its E isomer is stable to glutathione. Strong H-bond-accepting solvents [DMSO, dimethylformamide (DMF), and NMP], however, disrupt Z → E photoisomerization and addition of water to DMF solutions further impairs Z → E switching.
By introducing steric congestion into the hemithioindigo architecture, Dube and coworkers (22, 82) synthesized a notable rotary motor (28-30). For motor 28, the Z-(S)-(P) isomer is the global thermodynamic minimum, whereas E-(S)-(P) is the metastable state, while E-(S)-(M) and Z-(S)-(M) are fleeting and undergo rapid helical inversion to the (meta)stable forms (Fig. 8). Motors 29 and 30 have reversed global and metastable minima but still rotate in the same direction (clockwise) during isomerization (Fig. 8). Motors 28 and 30 invert too quickly from Z-(S)-(M) to Z-(S)-(P) to probe the former configuration, but 29, with its sterically cumbersome iso-propyl substituent, decelerates the isomerization rate, permitting the observation of all four states at −105 °C. This property enabled 1H NMR spectroscopy characterization of each of the four states shown in Fig. 8 and confirmed the motor’s unidirectional motion. Impressively, the motors could be driven with visible light (460–470 nm) with rotational frequencies ranging from 20 Hz to 1 kHz.
Fig. 8.
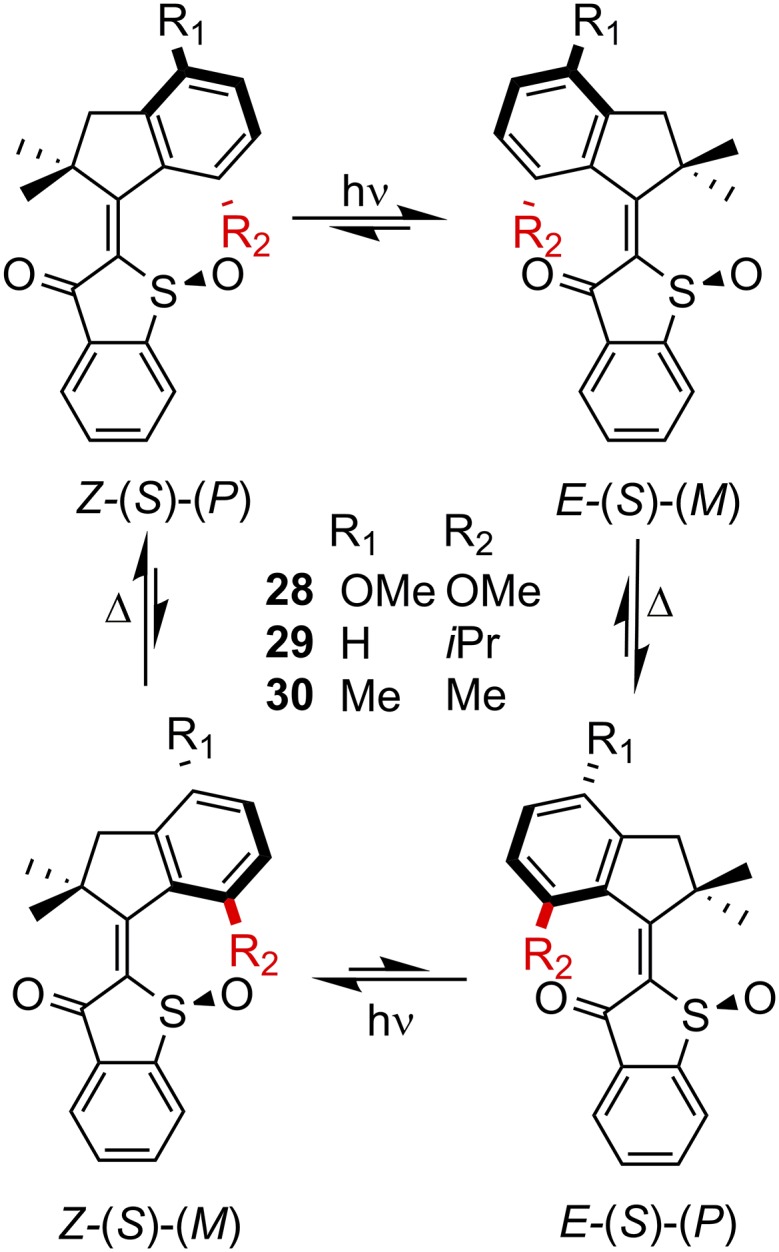
A molecular motor based on a hemithioindigo photoswitch. For clarity, only the S enantiomers are shown. R2 is colored red for easier viewing.
Donor–Acceptor Stenhouse Adducts.
Donor–acceptor Stenhouse adducts (DASAs) consist of an amine donor fragment separated from a Meldrum’s or barbituric acid-derived acceptor by a triene bridge (generalized structures and switching cycles are provided in SI Appendix, Fig. S3). Read de Alaniz and coworkers (29, 83) reported the “first-generation” DASA photoswitches (SI Appendix, Fig. S3) by following a high-yielding two-step synthesis. These derivatives undergo reversible switching (photocyclization/thermal ring-opening) in aromatic solvents (toluene, benzene, or xylenes) but fail to thermally ring-open in polar protic media (methanol or water) or photoswitch in halogenated solvents [dichloromethane (DCM), CHCl3, or chlorobenzene]. These negatively photochromic systems are highly colored in their open form and become colorless upon cyclization. Feringa and coworkers (84, 85) showed that upon irradiation DASAs follow a three-step photoswitching mechanism, beginning with a photoinduced Z → E isomerization of the central double bond, followed by thermal single bond rotation which permits the final and rate-limiting thermal conrotatory 4π electrocyclization from open (triene) to cyclic (cyclopentenone) forms. Reversion to the open form is achieved thermally (τ1/2 is seconds to minutes, depending on the solvent and switch substituents) and has not yet been accessed photochemically.
By substituting the alkyl amine donor for aniline (n = 0), indoline (n = 1), or tetrahydroquinoline (n = 2) derivatives, the Read de Alaniz and Beves groups (86, 87) introduced “second-generation” DASA photoswitches (SI Appendix, Fig. S3) which circumvent some of the solvent-dependency issues. However, and as recently noted in a detailed review about DASAs (88), even these improved switches suffer from drawbacks ranging from limited structural scope to poor reversible switching and fatigue resistance. If these issues are resolved by future investigations, then the switching between the hydrophobic triene and hydrophilic zwitterionic cyclopentenone forms presents an opportunity to devising new molecular machines that can be used in creating out-of-equilibrium solution gradients. Steps toward this goal have been made recently, as Lerch et al. (89) creatively exploited this quality to develop a multiphotochromic system that acts as a switchable phase transfer agent.
Conclusion
The recent surge in the development of chemically and photochemically activated switches bodes well for the future of molecular machines. Not only do these new systems expand the toolbox available to the community, but they also address the shortcomings of currently available switches, and in doing so pave the way for unprecedented functions and opportunities. Additionally, this burgeoning activity is broadening the participation in the field, bringing in new and diverse expertise and perspectives, which will expand the range of influence of molecular switches and machines. We recognize these new developments as exciting and heralding a new era that will bring with it new possibilities. We are already seeing the first glimpses of how molecular switches can be used in systems chemistry and expect to see more examples in the future. The use of a cohort of switches/machines, working together to control catalysis, feedback loops, chemical gradients, and equilibria will bring us closer to the myriad of functions present in biology. The introduction of flow systems to remove waste, compartmentalization to isolate events, and better control over switching events and timing through interswitch communication is needed to reach this goal. We also expect to see a rise in efforts to incorporate molecular switches and machines into LCs, LC elastomers, metal-organic frameworks, and 3D-printed polymers/objects. This combination will allow for the transduction of molecular-level motions into macroscopic events (i.e., actuation) and result in environment-responsive bulk materials. We look forward to all of the new opportunities that “switching beyond the molecule” will bring forth!
Supplementary Material
Acknowledgments
J.D.H. thanks the Dartmouth College Society of Fellows for their generous support. This work was supported by Army Research Office Grant W911NF-15-1-0587.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1714499115/-/DCSupplemental.
References
- 1.Schliwa M, editor. Molecular Motors. 1st Ed Wiley-VCH; Weinheim, Germany: 2003. [Google Scholar]
- 2.Feringa BL, Browne WR, editors. Molecular Switches. 2nd Ed Wiley-VCH; Weinheim, Germany: 2011. [Google Scholar]
- 3.Kassem S, et al. Artificial molecular motors. Chem Soc Rev. 2017;46:2592–2621. doi: 10.1039/c7cs00245a. [DOI] [PubMed] [Google Scholar]
- 4.Koumura N, Zijlstra RW, van Delden RA, Harada N, Feringa BL. Light-driven monodirectional molecular rotor. Nature. 1999;401:152–155. doi: 10.1038/43646. [DOI] [PubMed] [Google Scholar]
- 5.Collins BSL, Kistemaker JCM, Otten E, Feringa BL. A chemically powered unidirectional rotary molecular motor based on a palladium redox cycle. Nat Chem. 2016;8:860–866. [Google Scholar]
- 6.Coskun A, Banaszak M, Astumian RD, Stoddart JF, Grzybowski BA. Great expectations: Can artificial molecular machines deliver on their promise? Chem Soc Rev. 2012;41:19–30. doi: 10.1039/c1cs15262a. [DOI] [PubMed] [Google Scholar]
- 7.Jones IM, Hamilton AD. Designed molecular switches: Controlling the conformation of benzamido-diphenylacetylenes. Org Lett. 2010;12:3651–3653. doi: 10.1021/ol101397y. [DOI] [PubMed] [Google Scholar]
- 8.Jones IM, Lingard H, Hamilton AD. pH-dependent conformational switching in 2,6-benzamidodiphenylacetylenes. Angew Chem Int Ed Engl. 2011;50:12569–12571. doi: 10.1002/anie.201106241. [DOI] [PubMed] [Google Scholar]
- 9.Jones IM, Hamilton AD. Anion-dependent switching: Dynamically controlling the conformation of hydrogen-bonded diphenylacetylenes. Angew Chem Int Ed Engl. 2011;50:4597–4600. doi: 10.1002/anie.201100144. [DOI] [PubMed] [Google Scholar]
- 10.Hatano S, Horino T, Tokita A, Oshima T, Abe J. Unusual negative photochromism via a short-lived imidazolyl radical of 1,1′-binaphthyl-bridged imidazole dimer. J Am Chem Soc. 2013;135:3164–3172. doi: 10.1021/ja311344u. [DOI] [PubMed] [Google Scholar]
- 11.Fujita K, Hatano S, Kato D, Abe J. Photochromism of a radical diffusion-inhibited hexaarylbiimidazole derivative with intense coloration and fast decoloration performance. Org Lett. 2008;10:3105–3108. doi: 10.1021/ol801135g. [DOI] [PubMed] [Google Scholar]
- 12.Yamaguchi T, Kobayashi Y, Abe J. Fast negative photochromism of 1,1′-binaphthyl-bridged phenoxyl-imidazolyl radical complex. J Am Chem Soc. 2016;138:906–913. doi: 10.1021/jacs.5b10924. [DOI] [PubMed] [Google Scholar]
- 13.Erbas-Cakmak S, Leigh DA, McTernan CT, Nussbaumer AL. Artificial molecular machines. Chem Rev. 2015;115:10081–10206. doi: 10.1021/acs.chemrev.5b00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pezzato C, Cheng C, Stoddart JF, Astumian RD. Mastering the non-equilibrium assembly and operation of molecular machines. Chem Soc Rev. 2017;46:5491–5507. doi: 10.1039/c7cs00068e. [DOI] [PubMed] [Google Scholar]
- 15.Stoddart JF. Mechanically interlocked molecules (MIMs)-molecular shuttles, switches, and machines (nobel lecture) Angew Chem Int Ed Engl. 2017;56:11094–11125. doi: 10.1002/anie.201703216. [DOI] [PubMed] [Google Scholar]
- 16.Bandara HMD, Burdette SC. Photoisomerization in different classes of azobenzene. Chem Soc Rev. 2012;41:1809–1825. doi: 10.1039/c1cs15179g. [DOI] [PubMed] [Google Scholar]
- 17.Irie M, Fukaminato T, Matsuda K, Kobatake S. Photochromism of diarylethene molecules and crystals: Memories, switches, and actuators. Chem Rev. 2014;114:12174–12277. doi: 10.1021/cr500249p. [DOI] [PubMed] [Google Scholar]
- 18.Klajn R. Spiropyran-based dynamic materials. Chem Soc Rev. 2014;43:148–184. doi: 10.1039/c3cs60181a. [DOI] [PubMed] [Google Scholar]
- 19.Ray D, Foy JT, Hughes RP, Aprahamian I. A switching cascade of hydrazone-based rotary switches through coordination-coupled proton relays. Nat Chem. 2012;4:757–762. doi: 10.1038/nchem.1408. [DOI] [PubMed] [Google Scholar]
- 20.Greb L, Lehn JM. Light-driven molecular motors: Imines as four-step or two-step unidirectional rotors. J Am Chem Soc. 2014;136:13114–13117. doi: 10.1021/ja506034n. [DOI] [PubMed] [Google Scholar]
- 21.Greb L, Eichhöfer A, Lehn JM. Synthetic molecular motors: Thermal N inversion and directional photoinduced C=N bond rotation of camphorquinone imines. Angew Chem Int Ed Engl. 2015;54:14345–14348. doi: 10.1002/anie.201506691. [DOI] [PubMed] [Google Scholar]
- 22.Guentner M, et al. Sunlight-powered kHz rotation of a hemithioindigo-based molecular motor. Nat Commun. 2015;6:8406. doi: 10.1038/ncomms9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siewertsen R, et al. Highly efficient reversible Z-E photoisomerization of a bridged azobenzene with visible light through resolved S(1)(n π*) absorption bands. J Am Chem Soc. 2009;131:15594–15595. doi: 10.1021/ja906547d. [DOI] [PubMed] [Google Scholar]
- 24.Yang Y, Hughes RP, Aprahamian I. Visible light switching of a BF2-coordinated azo compound. J Am Chem Soc. 2012;134:15221–15224. doi: 10.1021/ja306030d. [DOI] [PubMed] [Google Scholar]
- 25.Weston CE, Richardson RD, Haycock PR, White AJP, Fuchter MJ. Arylazopyrazoles: Azoheteroarene photoswitches offering quantitative isomerization and long thermal half-lives. J Am Chem Soc. 2014;136:11878–11881. doi: 10.1021/ja505444d. [DOI] [PubMed] [Google Scholar]
- 26.Guo X, Zhou J, Siegler MA, Bragg AE, Katz HE. Visible-light-triggered molecular photoswitch based on reversible E/Z isomerization of a 1,2-dicyanoethene derivative. Angew Chem Int Ed Engl. 2015;54:4782–4786. doi: 10.1002/anie.201410945. [DOI] [PubMed] [Google Scholar]
- 27.Qian H, Pramanik S, Aprahamian I. Photochromic hydrazone switches with extremely long thermal half-lives. J Am Chem Soc. 2017;139:9140–9143. doi: 10.1021/jacs.7b04993. [DOI] [PubMed] [Google Scholar]
- 28.Wiedbrauk S, Dube H. Hemithioindigo–An emerging photoswitch. Tetrahedron Lett. 2015;56:4266–4274. [Google Scholar]
- 29.Helmy S, et al. Photoswitching using visible light: A new class of organic photochromic molecules. J Am Chem Soc. 2014;136:8169–8172. doi: 10.1021/ja503016b. [DOI] [PubMed] [Google Scholar]
- 30.Kassem S, Lee ATL, Leigh DA, Markevicius A, Solà J. Pick-up, transport and release of a molecular cargo using a small-molecule robotic arm. Nat Chem. 2016;8:138–143. doi: 10.1038/nchem.2410. [DOI] [PubMed] [Google Scholar]
- 31.Knipe PC, Thompson S, Hamilton AD. Ion-mediated conformational switches. Chem Sci (Camb) 2015;6:1630–1639. doi: 10.1039/c4sc03525a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol. 2011;7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balzani V, Credi A, Venturi M. Light powered molecular machines. Chem Soc Rev. 2009;38:1542–1550. doi: 10.1039/b806328c. [DOI] [PubMed] [Google Scholar]
- 34.Tatum LA, Foy JT, Aprahamian I. Waste management of chemically activated switches: Using a photoacid to eliminate accumulation of side products. J Am Chem Soc. 2014;136:17438–17441. doi: 10.1021/ja511135k. [DOI] [PubMed] [Google Scholar]
- 35.Schmittel M, De S, Pramanik S. Reversible ON/OFF nanoswitch for organocatalysis: Mimicking the locking and unlocking operation of CaMKII. Angew Chem Int Ed Engl. 2012;51:3832–3836. doi: 10.1002/anie.201108089. [DOI] [PubMed] [Google Scholar]
- 36.Silvi S, et al. A simple molecular machine operated by photoinduced proton transfer. J Am Chem Soc. 2007;129:13378–13379. doi: 10.1021/ja0753851. [DOI] [PubMed] [Google Scholar]
- 37.Pramanik S, De S, Schmittel M. Bidirectional chemical communication between nanomechanical switches. Angew Chemie Int Ed Engl. 2014;53:4709–4713. doi: 10.1002/anie.201400804. [DOI] [PubMed] [Google Scholar]
- 38.Pramanik S, De S, Schmittel M. A trio of nanoswitches in redox-potential controlled communication. Chem Commun (Camb) 2014;50:13254–13257. doi: 10.1039/c4cc05773b. [DOI] [PubMed] [Google Scholar]
- 39.De S, Pramanik S, Schmittel M. A toggle nanoswitch alternately controlling two catalytic reactions. Angew Chem Int Ed Engl. 2014;53:14255–14259. doi: 10.1002/anie.201408457. [DOI] [PubMed] [Google Scholar]
- 40.Gaikwad S, Goswami A, De S, Schmittel M. A metalloregulated four-state nanoswitch controls two-step sequential catalysis in an eleven-component system. Angew Chem Int Ed Engl. 2016;55:10512–10517. doi: 10.1002/anie.201604658. [DOI] [PubMed] [Google Scholar]
- 41.Landge SM, Aprahamian I. A pH activated configurational rotary switch: Controlling the E/Z isomerization in hydrazones. J Am Chem Soc. 2009;131:18269–18271. doi: 10.1021/ja909149z. [DOI] [PubMed] [Google Scholar]
- 42.Su X, Aprahamian I. Switching around two axles: Controlling the configuration and conformation of a hydrazone-based switch. Org Lett. 2011;13:30–33. doi: 10.1021/ol102422h. [DOI] [PubMed] [Google Scholar]
- 43.Kassem S, et al. Stereodivergent synthesis with a programmable molecular machine. Nature. 2017;549:374–378. doi: 10.1038/nature23677. [DOI] [PubMed] [Google Scholar]
- 44.Su X, Robbins TF, Aprahamian I. Switching through coordination-coupled proton transfer. Angew Chem Int Ed Engl. 2011;50:1841–1844. doi: 10.1002/anie.201006982. [DOI] [PubMed] [Google Scholar]
- 45.Pramanik S, Aprahamian I. Hydrazone switch-based negative feedback loop. J Am Chem Soc. 2016;138:15142–15145. doi: 10.1021/jacs.6b10542. [DOI] [PubMed] [Google Scholar]
- 46.Kelly TR, De Silva H, Silva RA. Unidirectional rotary motion in a molecular system. Nature. 1999;401:150–152. doi: 10.1038/43639. [DOI] [PubMed] [Google Scholar]
- 47.Lin Y, Dahl BJ, Branchaud BP. Net directed 180° aryl–aryl bond rotation in a prototypical achiral biaryl lactone synthetic molecular motor. Tetrahedron Lett. 2005;46:8359–8362. [Google Scholar]
- 48.Fletcher SP, Dumur F, Pollard MM, Feringa BL. A reversible, unidirectional molecular rotary motor driven by chemical energy. Science. 2005;310:80–82. doi: 10.1126/science.1117090. [DOI] [PubMed] [Google Scholar]
- 49.Bléger D, Hecht S. Visible-light-activated molecular switches. Angew Chem Int Ed Engl. 2015;54:11338–11349. doi: 10.1002/anie.201500628. [DOI] [PubMed] [Google Scholar]
- 50.Kathan M, Hecht S. Photoswitchable molecules as key ingredients to drive systems away from the global thermodynamic minimum. Chem Soc Rev. 2017;46:5536–5550. doi: 10.1039/c7cs00112f. [DOI] [PubMed] [Google Scholar]
- 51.Velema WA, Szymanski W, Feringa BL. Photopharmacology: Beyond proof of principle. J Am Chem Soc. 2014;136:2178–2191. doi: 10.1021/ja413063e. [DOI] [PubMed] [Google Scholar]
- 52.Broichhagen J, Frank JA, Trauner D. A roadmap to success in photopharmacology. Acc Chem Res. 2015;48:1947–1960. doi: 10.1021/acs.accounts.5b00129. [DOI] [PubMed] [Google Scholar]
- 53.Lerch MM, Hansen MJ, van Dam GM, Szymanski W, Feringa BL. Emerging targets in photopharmacology. Angew Chem Int Ed Engl. 2016;55:10978–10999. doi: 10.1002/anie.201601931. [DOI] [PubMed] [Google Scholar]
- 54.Russew MM, Hecht S. Photoswitches: From molecules to materials. Adv Mater. 2010;22:3348–3360. doi: 10.1002/adma.200904102. [DOI] [PubMed] [Google Scholar]
- 55.Bouas-Laurent H, Dürr H. Organic photochromism. Pure Appl Chem. 2001;73:639–665. [Google Scholar]
- 56.Hammerich M, et al. Heterodiazocines: Synthesis and photochromic properties, trans to cis switching within the bio-optical window. J Am Chem Soc. 2016;138:13111–13114. doi: 10.1021/jacs.6b05846. [DOI] [PubMed] [Google Scholar]
- 57.Samanta S, Qin C, Lough AJ, Woolley GA. Bidirectional photocontrol of peptide conformation with a bridged azobenzene derivative. Angew Chem Int Ed Engl. 2012;51:6452–6455. doi: 10.1002/anie.201202383. [DOI] [PubMed] [Google Scholar]
- 58.Moormann W, Langbehn D, Herges R. Solvent-free synthesis of diazocine. Synthesis. 2017;49:3471–3475. [Google Scholar]
- 59.Yang Y, Hughes RP, Aprahamian I. Near-infrared light activated azo-BF2 switches. J Am Chem Soc. 2014;136:13190–13193. doi: 10.1021/ja508125n. [DOI] [PubMed] [Google Scholar]
- 60.Qian H, Wang Y-Y, Guo D-S, Aprahamian I. Controlling the isomerization rate of an azo-BF2 switch using aggregation. J Am Chem Soc. 2017;139:1037–1040. doi: 10.1021/jacs.6b10982. [DOI] [PubMed] [Google Scholar]
- 61.Qian H, Shao B, Aprahamian I. Visible-light fluorescence photomodulation in azo-BF2 switches. Tetrahedron. 2017;73:4901–4904. [Google Scholar]
- 62.Calbo J, et al. Tuning azoheteroarene photoswitch performance through heteroaryl design. J Am Chem Soc. 2017;139:1261–1274. doi: 10.1021/jacs.6b11626. [DOI] [PubMed] [Google Scholar]
- 63.Wendler T, Schütt C, Näther C, Herges R. Photoswitchable azoheterocycles via coupling of lithiated imidazoles with benzenediazonium salts. J Org Chem. 2012;77:3284–3287. doi: 10.1021/jo202688x. [DOI] [PubMed] [Google Scholar]
- 64.Garcia-Amorós J, Díaz-Lobo M, Nonell S, Velasco D. Fastest thermal isomerization of an azobenzene for nanosecond photoswitching applications under physiological conditions. Angew Chem Int Ed Engl. 2012;51:12820–12823. doi: 10.1002/anie.201207602. [DOI] [PubMed] [Google Scholar]
- 65.Garcia-Amorós J, R Castro MC, Coelho P, M Raposo MM, Velasco D. New heterocyclic systems to afford microsecond green-light isomerisable azo dyes and their use as fast molecular photochromic switches. Chem Commun (Camb) 2013;49:11427–11429. doi: 10.1039/c3cc46736h. [DOI] [PubMed] [Google Scholar]
- 66.Garcia-Amorós J, Castro MC, Coelho P, Raposo MM, Velasco D. Fastest non-ionic azo dyes and transfer of their thermal isomerisation kinetics into liquid-crystalline materials. Chem Commun (Camb) 2016;52:5132–5135. doi: 10.1039/c6cc00403b. [DOI] [PubMed] [Google Scholar]
- 67.García-Amorós J, Velasco D. Recent advances towards azobenzene-based light-driven real-time information-transmitting materials. Beilstein J Org Chem. 2012;8:1003–1017. doi: 10.3762/bjoc.8.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lehn J-M. Conjecture: Imines as unidirectional photodriven molecular motors-motional and constitutional dynamic devices. Chemistry. 2006;12:5910–5915. doi: 10.1002/chem.200600489. [DOI] [PubMed] [Google Scholar]
- 69.Knorr R, Ruhdorfer J, Mehlstäubl J, Böhrer P, Stephenson DS. Demonstration of the nitrogen inversion mechanism of imines in a Schiff base model. Chem Ber. 1993;126:747–754. [Google Scholar]
- 70.Tavernelli I, Röhrig UF, Rothlisberger U. Molecular dynamics in electronically excited states using time-dependent density functional theory. Mol Phys. 2005;103:963–981. [Google Scholar]
- 71.Chaur MN, Collado D, Lehn JM. Configurational and constitutional information storage: Multiple dynamics in systems based on pyridyl and acyl hydrazones. Chemistry. 2011;17:248–258. doi: 10.1002/chem.201002308. [DOI] [PubMed] [Google Scholar]
- 72.Vantomme G, Lehn JM. Photo- and thermoresponsive supramolecular assemblies: Reversible photorelease of K+ ions and constitutional dynamics. Angew Chem Int Ed Engl. 2013;52:3940–3943. doi: 10.1002/anie.201210334. [DOI] [PubMed] [Google Scholar]
- 73.Vantomme G, Hafezi N, Lehn J-M. A light-induced reversible phase separation and its coupling to a dynamic library of imines. Chem Sci. 2014;5:1475–1483. [Google Scholar]
- 74.van Dijken DJ, Kovaříček P, Ihrig SP, Hecht S. Acylhydrazones as widely tunable photoswitches. J Am Chem Soc. 2015;137:14982–14991. doi: 10.1021/jacs.5b09519. [DOI] [PubMed] [Google Scholar]
- 75.De Bo G, Leigh DA, McTernan CT, Wang S. A complementary pair of enantioselective switchable organocatalysts. Chem Sci. 2017;8:7077–7081. doi: 10.1039/c7sc02462b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seki T, Tamaki T, Yamaguchi T, Ichimura K. Photochromism of hemithioindigo derivatives. II. Photochromic behaviors in bilayer membranes and related systems. Bull Chem Soc Jpn. 1992;65:657–663. [Google Scholar]
- 77.Maerz B, et al. Making fast photoswitches faster–Using Hammett analysis to understand the limit of donor-acceptor approaches for faster hemithioindigo photoswitches. Chemistry. 2014;20:13984–13992. doi: 10.1002/chem.201403661. [DOI] [PubMed] [Google Scholar]
- 78.Wiedbrauk S, et al. Twisted hemithioindigo photoswitches: Solvent polarity determines the type of light-induced rotations. J Am Chem Soc. 2016;138:12219–12227. doi: 10.1021/jacs.6b05981. [DOI] [PubMed] [Google Scholar]
- 79.Wiedbrauk S, et al. Ingredients to TICT formation in donor substituted hemithioindigo. J Phys Chem Lett. 2017;8:1585–1592. doi: 10.1021/acs.jpclett.7b00371. [DOI] [PubMed] [Google Scholar]
- 80.Kink F, Collado MP, Wiedbrauk S, Mayer P, Dube H. Bistable photoswitching of hemithioindigo with green and red light: Entry point to advanced molecular digital information processing. Chemistry. 2017;23:6237–6243. doi: 10.1002/chem.201700826. [DOI] [PubMed] [Google Scholar]
- 81.Zweig JE, Newhouse TR. Isomer-specific hydrogen bonding as a design principle for bidirectionally quantitative and redshifted hemithioindigo photoswitches. J Am Chem Soc. 2017;139:10956–10959. doi: 10.1021/jacs.7b04448. [DOI] [PubMed] [Google Scholar]
- 82.Huber LA, et al. Direct observation of hemithioindigo-motor unidirectionality. Angew Chem Int Ed Engl. 2017;56:14536–14539. doi: 10.1002/anie.201708178. [DOI] [PubMed] [Google Scholar]
- 83.Helmy S, Oh S, Leibfarth FA, Hawker CJ, Read de Alaniz J. Design and synthesis of donor-acceptor Stenhouse adducts: A visible light photoswitch derived from furfural. J Org Chem. 2014;79:11316–11329. doi: 10.1021/jo502206g. [DOI] [PubMed] [Google Scholar]
- 84.Lerch MM, Wezenberg SJ, Szymanski W, Feringa BL. Unraveling the photoswitching mechanism in donor-acceptor Stenhouse adducts. J Am Chem Soc. 2016;138:6344–6347. doi: 10.1021/jacs.6b01722. [DOI] [PubMed] [Google Scholar]
- 85.Di Donato M, et al. Shedding light on the photoisomerization pathway of donor-acceptor Stenhouse adducts. J Am Chem Soc. 2017;139:15596–15599. doi: 10.1021/jacs.7b09081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hemmer JR, et al. Tunable visible and near infrared photoswitches. J Am Chem Soc. 2016;138:13960–13966. doi: 10.1021/jacs.6b07434. [DOI] [PubMed] [Google Scholar]
- 87.Mallo N, et al. Photochromic switching behaviour of donor-acceptor Stenhouse adducts in organic solvents. Chem Commun (Camb) 2016;52:13576–13579. doi: 10.1039/c6cc08079k. [DOI] [PubMed] [Google Scholar]
- 88.Lerch MM, Szymański W, Feringa BL. The (photo)chemistry of Stenhouse photoswitches: Guiding principles and system design. Chem Soc Rev. 2018;47:1910–1937. doi: 10.1039/c7cs00772h. [DOI] [PubMed] [Google Scholar]
- 89.Lerch MM, Hansen MJ, Velema WA, Szymanski W, Feringa BL. Orthogonal photoswitching in a multifunctional molecular system. Nat Commun. 2016;7:12054. doi: 10.1038/ncomms12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.