

Abstract
The intrinsic humoral immunodeficiency of chronic lymphocytic leukemia (CLL) is often managed with immunoglobulin replacement therapy (IgRT) to maintain IgG levels in the low-normal range (6–8 g/L) but optimal targets for IgG and timing to commence IgRT are unclear. IgG levels fell near 6 g/L at rates of −0.85±0.14 g/L/year in 51 patients who required treatment for CLL within 4.5±0.4 years from initial diagnosis and − 0.27±0.04 g/L/year in 40 patients with progressive disease who remained untreated after 8.5±0.5 years. In contrast, endogenous IgG levels remained above 8 g/L in patients with highly indolent disease (n = 25) and TNFα and beta-2-microglobulin (β2M) in blood decreased when IgRT was used to increase IgG levels over 9 g/L. At 15 g/L but not 5 g/L, the IgRT product Hizentra® inhibited B cell receptor (BCR)-activation, TNFα production, and survival in vitro, particularly of CLL cells that spontaneously made little TNFα. These findings suggest deterioration of the humoral immune system is associated with progressive CLL and altering the dosing of IgRT to achieve higher than conventional IgG target levels may have therapeutic activity.
Keywords: Chronic lymphocytic leukemia, Immunoglobulin replacement therapy, IVIG, SCIG, Hypogammaglobulinemia, Immunodeficiency, Signal transduction, Cytokines, Toll-like receptors, Janus kinases, Bruton's tyrosine kinase
Graphical abstract
Highlights
-
•
Immunoglobulin levels decline at rates that reflect the clinical course of CLL.
-
•
IgG levels over 10 g/L achieved with replacement therapy are associated with evidence of disease control in vivo and inhibition of BCR-mediated activation of CLL cells in vitro.
-
•
Monitoring rates of decline of Ig levels in CLL patients gives biological information on disease severity.
-
•
Appropriate IgG target levels for immunoglobulin replacement therapy in CLL may be much higher than for patients with other immunodeficiencies.
Research in context.
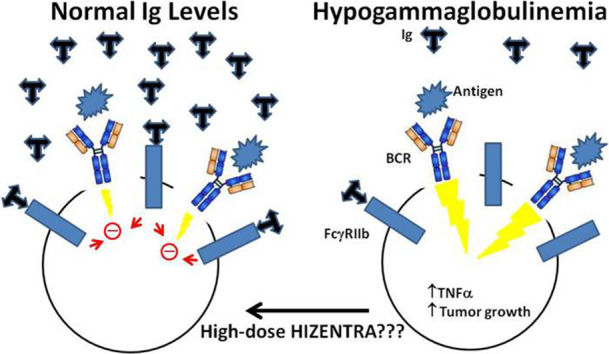
Chronic lymphocytic leukemia (CLL) is the commonest adult leukemia. Immunoglobulin deficiency is a unique and important problem in CLL that is often managed with immunoglobulin replacement therapy (IgRT). The mechanism of the immunodeficiency and optimal use of IgRT remain unclear. The results in this manuscript implicate defective transitional B cell development in the decline of Ig levels. Allowing IgG levels to fall below 8 g/L may promote growth of CLL cells and maintaining IgG levels close to the upper normal limit (15 g/L) with IgRT may slow disease progression. These findings have important implications for the care of CLL patients.
Alt-text: Unlabelled Box
1. Introduction
Hypogammaglobulinemia involving all three immunoglobulin (Ig) classes is intrinsic to Chronic Lymphocytic Leukemia (CLL) [14,35] and can cause significant morbidity, particularly from sino-pulmonary infections [48]. It is not entirely clear why hypogammaglobulinemia is so characteristic of CLL in the absence of immunosuppressive treatments like Fludarabine and Rituximab (Forconi et al., [14]). Explanations include direct inhibitory effects of leukemia cells on normal B cells and plasma cells or indirect effects on helper T cells [9,10,38].
Immunoglobulin replacement therapy (IgRT) is the major form of treatment for humoral immunodeficiency ([4,5]). The immunoglobulin product is precipitated from the blood of thousands of healthy donors and consists mainly of pooled IgG preparations. A number of commercial Ig formulations are available for injection as intravenous immunoglobulin (IVIG) or subcutaneous immunoglobulin (SCIG). These formulations use different small molecule stabilizers including sugars, polyols, and amino acids and contain different amounts of antibodies. Privigen® for intravenous use and Hizentra® for subcutaneous administration are made by CSL-Behring and stabilized with L-proline. Privigen® is 20% IgG and Hizentra® is 10% IgG ([4,5]). IgRT is usually prescribed at a dose of 0.4 g/kg monthly when IgG levels fall below 6 g/L in CLL patients with histories of infections. This approach decreases infection rates but does not affect the clinical course or improve survival [15,48]. The IgRT dose can be individualized to keep patients free of infections ([1, 4,5]) and it has been suggested that IgG levels >7.44 g/L provide the best protection from major infections [48]. However, optimal IgG target levels for IgRT have not been established in CLL.
Despite the importance of humoral immunodeficiency in CLL, little information is available about how Ig levels evolve in the absence of immunosuppressive drug treatment that might influence a decision to start IgRT. In this manuscript, the clinical records of 116 patients with serial Ig levels available over at least an 8 year period or until first treatment for symptomatic CLL were examined to provide such information. There is also little information about the effects of IgRT on CLL cells in vitro or in vivo that may help identify appropriate IgG target levels. To address this gap, records of 21 patients receiving IgRT were examined and experiments were performed to determine how IgRT affects CLL cells in vitro. The results suggest appropriate target levels for IgG in CLL may be much higher than achieved with conventional dosing.
2. Methods
The retrospective study population consisted of 116 patients attending the Sunnybrook CLL clinic in 2003, 2009, and 2013 with immunoglobulin quantification available from diagnosis for at least 5 years or until first treatment for CLL. CLL was diagnosed according to IWCLL criteria [19]. Twenty-five patients met criteria for indolent disease as described by Guarini [17] and 51 received treatment for CLL based on conventional criteria [19]. The remaining 40 patients were classed as “progressors”, implying most would eventually come to treatment. Information about demographics, prognostic markers [19], and treatments were obtained from electronic patient records. Serum beta-2-microglobulin (β2M), cytogenetics, and CD38 expression were obtained from pathology reports. Clinical staging was based on the Rai classification [19]. Ig measurements were performed by the clinical service laboratory at Sunnybrook. The last available Ig level or the level prior to treatment for CLL was used to calculate the rate of decline of the Ig class (Fig. 1). Patient characteristics are listed in Supplementary table 1.
Fig. 1.

Rates of decline of immunoglobulin sub-classes in CLL patients along a spectrum of disease severity. A. CLL patients were classed as benign (black bars, n = 25), treated (gray bars, n = 51), or progressor (white bars, n = 40) as described in the materials and methods. Clinical characteristics of the three groups are shown in the bar graphs with units for each descriptor indicated along the Y-axis. B. Example of a “progressor” patient, showing concordant declines in IgA, IgM, and IgG levels with disease progression indicated by increases in blood lymphocyte and β2M levels. C–E. Average rates of decline (C), normalized rates of decline (D), and final levels (E) of Ig sub-classes along with standard errors are shown in the graphs. *, p < 0.05; ns = not significant.
For the observations in Fig. 2B, records of 119 consecutive patients attending the CLL clinic in 2017 were examined only for Ig levels at diagnosis and use of IgRT. The study was approved by the Sunnybrook Research Ethics Board.
Fig. 2.
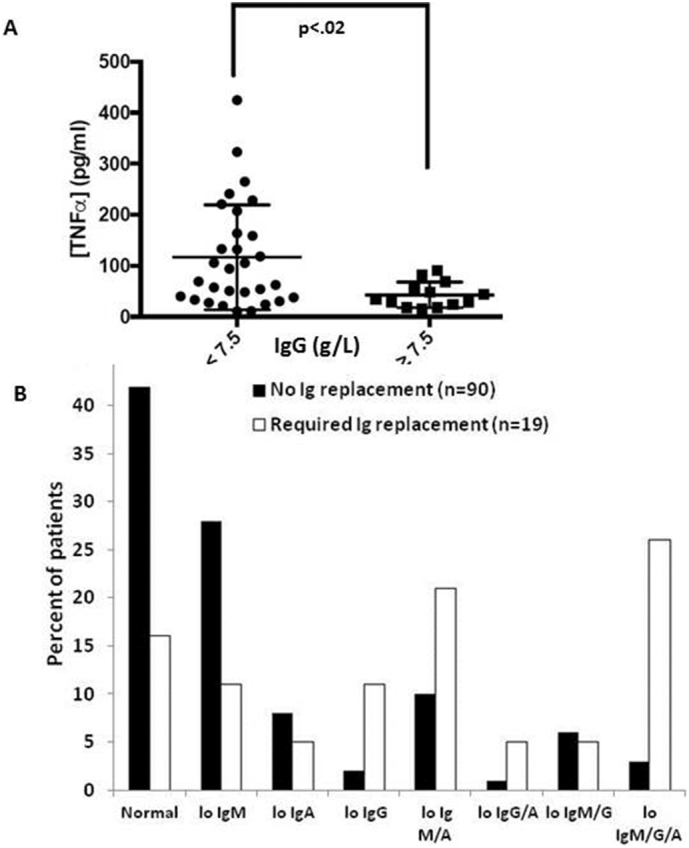
Relationship of blood immunoglobulin levels with TNFα and need for IgRT. A. Plasma was collected from 43 patients. Serum IgG was determined by the Sunnybrook Clinical Laboratory and plasma TNFα was measured by Multiplexing LASER Bead Technology. An iterative algorithm was used to group the samples, depending on whether they contained more or <7.5 g/L IgG. B. Ig levels at time of initial CLL diagnosis were recorded for 109 patients including 19 who were commenced ultimately on Ig RT. Low (lo) IgA, IgG, and IgM were defined as <0.7, 6.0, and 0.4 g/L, respectively.
Details about reagents, cell culture, flow cytometry [47], cytokine measurements [43], immunoblotting and statistical methods [29] are provided in the supplemental Methods (available on the Ebiomedicine Web site).
3. Results
3.1. Temporal changes in Ig classes in CLL patients
Records of 116 CLL patients on “watch and wait” management [19] with Ig quantification recorded over at least a 5-year period from time of diagnosis were studied to provide information about the evolution of IgG, IgM, and IgA levels during disease progression. No patient received IgRT in this time. Patients were classed into three groups based on the outcome at the end of the observation period. Twenty-five were classed as having “benign” disease [17] in that they did not progress beyond Rai stage 0/1, develop lymphocyte counts >30 × 109 cells/L, or require treatment in the observation period. Such patients may never need treatment for CLL in their life-time [17]. Fifty-one patients were treated for CLL in the study period on the basis of standard indications [19]. Their last recorded Ig quantification for the purposes of the study was taken just before beginning treatment. Remaining patients (n = 40) were classed as “progressors” as their lymphocyte counts increased to >30 × 109 cells/L, which often portends an eventual need for treatment although none was prescribed over the observation period.
The patient population is described in Fig. 1A and Supplementary table 1. Treated patients came to treatment within 4.7±0.4 years while progressors and benign patients were observed for 8.5±0.5 and 7.6±0.5 years, respectively, without any treatment. There was no difference in average ages among the different groups and a non-significant increase in proportion of males in the treated group. Rai stage at the end of the observation period was 0.2±0.1 for benign patients but increased to 2.3±0.3 and 3.5±0.1 for progressors and treated patients, respectively. Serum beta2-microglobulin (β2M) at the end of the observation period was higher in treated patients, and their CLL cells had higher CD38 expression and more high-risk genetic lesions (del11q22 and del17p) [19] than the other groups. Progressors were intermediate between benign and treated patients with respect to β2M levels, CD38 expression, and high-risk cytogenetic features (Fig. 1A).
Rates of change of blood lymphocytes were calculated by subtracting the initial lymphocyte count from the final count at the end of the observation period and dividing by the years of observation (Fig. 1A; bottom right panel). This rate was higher for treated patients (22.1±2.7 × 109 cells/L/year) and culminated in an average final lymphocyte count of 122.6±13.4 × 109 cells/L compared with final counts of 88.3±3.6 × 109 cells/L for progressors and only 11.7±1.4 × 109 cells/L for benign patients (Fig. 1A; top right panel).
3.2. Coordinated decline of Ig classes in patients with progressive disease
Initial inspection of the data suggested a parallel decline of all three classes of Ig with increases in β2M and circulating lymphocytes, as shown in the example (Fig. 1B). The approximately linear declines allowed a rate of change for each Ig class to be calculated for each patient by subtracting the initial amount of IgA, IgG, or IgM from the amount at the end and dividing by the length of the observation period. Rates of decline for IgA, IgG, and IgM were all greater in patients who went on to require treatment for their CLL (Fig. 1C) with highest absolute changes for IgG. However, normal levels are higher for IgG (6–16 g/L) than IgA (0.7–4 g/L) or IgM (0.4–2.5 g/L). To compare directly the rates of change of the different Ig classes, absolute values were normalized by dividing them by the midpoint value of the normal range (i.e. 11, 2.35, and 1.2 g/L for IgG, IgA, and IgM, respectively) (Fig. 1D). With this adjustment, there was no difference between the rates of change of IgG, IgA, and IgM for the patients who required treatment.
IgG and IgM levels did not change significantly over the observation period in patients with a benign clinical course although there was a low decline (0.01±0.001 g/L/year) for IgA (Fig. 1D). Rates of decline of IgG, IgA, and IgM in patients with progressive disease that did not require treatment were lower than for patients who required treatment at the end of the observation period but higher than for benign patients and IgA levels appeared to decline somewhat faster than IgG (Fig. 1C,D).
Consistent with these rates of decline, Ig levels at the end of the observation period were significantly lower in progressor and treated patients compared to patients with benign disease (Fig. 1E). Notably, IgG levels that remained above 8 g/L were associated with a benign clinical course while IgG levels that fell near 6 g/L were associated with disease progression and need for treatment (Fig. 1E).
3.3. Inverse association of plasma TNFα and IgG levels
TNFα inhibits B cell development in the bone marrow [26] and blood TNFα is increased in CLL patients [12,13]. Higher TNFα levels are associated with more aggressive disease [12,13], although CLL cells with aggressive clinical behavior make less TNFα than cells from patients with indolent disease. [13,18,22]. TNFα in the plasma of an additional 43 patients was found to correlate inversely with IgG level at the same time-point. Average TNFα levels in patients with IgG levels <7.5 g/L were 120±21 g/L but only 30±10 g/L with IgG levels >7.5 g/L (Fig. 2A).
3.4. Ig levels at diagnosis in CLL patients
The observation that Ig levels declined inexorably in most CLL patients (Fig. 1C,D) coupled with the fact that asymptomatic CLL can go undetected for a long time [32] suggested many patients may be hypogammaglobulinemic by the time they are diagnosed officially with CLL [35]. Supportive treatment for symptomatic hypogammaglobulinemia involves IGRT with IVIG or SCIG ([48]; [4,5]; [25]). Records of 119 consecutive CLL patients were examined for an initial concomitant diagnosis of hypogammaglobulinemia (Fig. 2B). Nineteen patients received IgRT at some point in the disease course. Low IgG (lo IgG), IgA (lo IgA), and IgM (lo IgM) levels were defined as below the lower limit of normal (i.e. 6, 0.7, and 0.4 g/L for IgG, IgA, and IgM, respectively).
Ig levels were in the normal ranges at the initial diagnosis for most patients (Fig. 2B). However, over 25% of the patients had an isolated IgM deficiency and a smaller number had deficiencies in IgG or IgA alone or in more than one Ig class (Fig. 2B). Notably, subnormal Ig levels at time of diagnosis appeared to predict the need for subsequent IgRT that was prescribed according to accepted guidelines [25]. Only 16% of patients who required IgRT had Ig levels in the normal range at the initial CLL diagnosis. Defects in multiple Ig classes and especially pan-hypogammaglobulinemia were especially associated with subsequent need for IgRT (Fig. 2B).
3.5. Dose-dependent effect of IgRT on β2M and TNFα levels in vivo
IgRT is indicated for CLL patients with low IgG levels (generally <5 g/L) and a history of serious infections requiring antibiotics [25]. Trough levels achieved with conventional administration of IgRT are usually in the low-normal range. In the course of managing hypogammaglobulinemic CLL patients with IgRT, a relationship was noticed between the level of serum β2-microglobulin (β2M), a marker of CLL burden [45], and IgG level attained by IgRT as shown in the examples (Fig. 3A).
Fig. 3.

Plasma TNFα and β2-microglobulin (β2M) changes in CLL patients following institution of Ig replacement therapy. A. Examples of time courses of IgG and β2M levels along with lymphocyte numbers following institution of IgRT to IgG levels of ~ 6 g/L (upper panels) or 12 g/L (lower panels). B,C. TNFα levels were measured in plasma samples collected incidentally from 11 patients over a 3 year period (2015–17) prior to and at varying times after starting IVIG or Hizentra (B). β2M levels before and after starting IgRT were taken from the clinical records from these and an additional 10 patients (C). The time between the two plasma collections for each patient is indicated in the figure legends. Patient samples were grouped according to IgG level achieved by IgRT at time of sample collection, using 9 g/L as a cut-off. D. Summary graph of average change in serum β2M for each group indicating an IgG level > 9 g/L is significantly associated with a lowering of β2M levels. *, p < 0.05.
In some patients (Fig. 3A, top graphs) institution of IgRT with IVIG or SCIG was associated with an increase in β2M that was sometimes followed shortly by the need to start CLL therapy. In a minority of patients, β2M levels remained stable or even decreased somewhat upon institution of IgRT (Fig. 3A; bottom graphs). In one case, circulating lymphocytes declined steadily from 450 to 189 × 109 cells/L over 6 years with IgRT as the only deliberate therapeutic maneuver (Fig. 3A; right bottom graph, Pt. SS).
These different response patterns appeared to be associated with the magnitude of IgG level achieved with IgRT. In patients with apparent disease progression despite IgRT, IgG levels only increased into the 6–7 g/L range (Fig. 3A; top graphs). In patients on IgRT with apparently better disease control, IgG levels were maintained above 12 g/L (Fig. 3A; bottom graphs).
Given the apparent relationship of IgG to plasma TNFα (Fig. 2A), a panel of cytokines was measured in plasma samples of CLL patients that had incidentally been collected before and after commencing IgRT. Timing of the collections in relation to starting IgRT was not standardized as they had been obtained for other purposes over a three-year period. However, of the 11 paired samples available for analysis, TNFα levels increased after starting IgRT when replacement IgG levels were <9 g/L (Fig. 3B; left panel) but decreased in one patient when IgG levels reached >9 g/L (Fig. 3B; right panel). IL10 levels also increased in patients whose IgG levels were increased only below 9 g/L (Supplementary Fig. 1).
Due to the paucity of paired samples that had been cryopreserved and could be used to measure cytokines, the analysis was extended to include patients on IgRT with records of simultaneous IgG and β2M measurements (Fig. 3C). The rationale was that β2M levels are reduced by cytokine-signaling inhibitors and may serve as a partial surrogate for cytokine activity in CLL [3,16,45]. Sixteen patients were identified with IgG levels, measured within a few months of starting IgRT, that did not reach 9 g/L and five patients were identified with levels that became >9 g/L. Remarkably, serum β2M increased in the former group but declined in the latter (Fig. 3C). The average increase was 0.93±0.18 μg/L if IgG levels remained below 9 g/L but declined by 0.42±0.13 μg/L if IgG levels were above 9 g/L (Fig. 3D). Taken together, these observations suggested target IgG levels above those that prevent infections [48] might have anti-leukemia activity as indicated by lowered TNFα and β2M levels.
3.6. Dose-dependent effects of SCIG on B cell-receptor-mediated CLL cell activation in vitro
The effect of different concentrations of the SCIG preparation Hizentra® on the activation of CLL cells was studied in vitro to try to understand why higher doses of IgRT might have a beneficial effect on the clinical course. Growth of CLL cells is thought to be driven mainly by signals from B cell receptors (BCRs) or toll-like receptors (TLRs), with clinically aggressive CLL cells responding more strongly to antigens and TLR-ligands [8,27,44]. The NFκB pathway conveys signals from both receptor types [27] and induces CD83 on the cell surface, which can be measured by flow cytometry to provide a simple readout of BCR- and TLR-activation [41]. As shown in the example in Fig. 4A, BCR-cross-linking with anti-IgM Fab fragments up-regulated CD83 (Fig. 4A; compare first and third panels from left). Hizentra® at 15 g/L strongly decreased CD83 expression, suggesting BCR-activation was inhibited (Fig. 4A; compare third and fourth panels from left). TLR7-signaling induced by Resiquimod caused significantly higher CD83 expression and was not inhibited as strongly by this dose of Hizentra® (Fig. 4A; compare fifth and sixth panels from left).
Fig. 4.

Dose-dependent inhibition of B cell receptor-signaling in CLL cells by Hizentra. Purified CLL cells were stimulated with anti-IgM antibodies (10 ng/mL) or the TLR7-agonist Resiquimod (1 μg/ml) in the presence and absence of Hizentra at the indicated concentrations. After 4 h, CD83 expression was measured by flow cytometry. A. An example for one patient sample is shown in the top histograms, with the numbers indicating mean fluorescence intensity (MFI) of CD83-staining. B,C. The graphs show results of IgM-cross-linking for 25 different patient samples. Individual values for each treatment condition are shown on the left (B) with averages and standard deviations on the right (C). Hizentra at 15 g/L significantly (p < .01) decreased BCR-activation in contrast to 4 g/L. D–F. After 30 min, phospho- and total AKT, phospho-PLCγ, and β-actin in 9 additional samples were measured by immunoblotting and quantified by densitometry. Representative immunoblots are shown (D). Relative densitometry values for each condition are shown with the lines indicating results from individual patient samples (E). Averages and standard errors are shown in the graphs (F). *, p < .05.
Based on these observations, the effects of Hizentra® on BCR-activation were studied in more detail in 25 additional patient samples, using final concentrations of 4 and 15 g/L to represent extremes of normal IgG values. Results for individual samples are shown in the left panel and summarized on the right (Fig. 4B). Hizentra® did not affect BCR-activation at 4 g/L but inhibited it significantly at 15 g/L (Fig. 4B).
BCR-cross-linking causes rapid phosphorylation of BCR-associated signaling molecules, particularly in the phospholipase C-γ2 (PLC-γ) and phosphoinositol-3-kinase (PI3K)/AKT pathways [46]. The effects of different concentrations of Hizentra on BCR-mediated phosphorylation of AKT and PLC-γ were examined by immunoblotting in 9 patient samples (Fig. 4D–F). Consistent with the in vivo observations (Fig. 3), Hizentra at 10 g/L often inhibited AKT and PLC-γ-phosphorylation (Fig. 4D–F). Consistent with the flow cytometric assay (Fig. 4B), the AKT-pathway was inhibited significantly by Hizentra at15 g/L.
3.7. Dose-dependent effect of Hizentra® on cytokine production by activated CLL cells
As the BCR-transcriptional program includes chemokine, cytokine, and growth factor genes, the effect of Hizentra® on 42 different proteins in culture supernatants from 4 different purified CLL cell samples activated by IgM-cross-linking was determined after 48 h (Supplementary Fig. 2). TNFα, PDGF-AA, CCL4 and CCL5 were especially changed by Hizentra®, which inhibited their spontaneous release in all patient samples and BCR-mediated production from most samples (Supplementary Fig. 2).
Given the effect on TNFα (Supplementary Fig. 2) and its importance in CLL [12], the consequences for TFNα production of different concentrations of Hizentra® were measured specifically in 10 additional patient samples. Results for individual samples are shown in Fig. 5A and summarized in Fig. 5B. High concentrations of Hizentra® (15 g/L) inhibited spontaneous and anti-IgM-mediated TNFα production more than low concentrations. TNFα production was even increased by low Hizentra® concentrations (5 g/L) (Fig. 5A) in some CLL samples that spontaneously made >38 pg/ml of TNFα. Such cells were designated “Group 1” cells. Patient samples with lower spontaneous TNFα production (<38 pg/ml) were designated “Group 2” cells. Group 2 cells were more sensitive to either Hizentra® concentration and high concentrations (15 g/L) reduced TNFα production from activated cells almost to background levels (Fig. 5A; bottom panel, Fig. 5B).
Fig. 5.

Hizentra inhibits TNFα production by resting and activated CLL cells in a dose- and disease subtype-dependent manner. TNFα was measured in the supernatants of cultures of CLL cells treated with or without anti-IgM antibodies in the presence or absence of Hizentra (5 or 15 g/L final concentration) (A,B) and the presence or absence of the BTK-inhibitor Ibrutinib (500 nM) (C). A. Individual results for 10 patient samples are graphed, grouped on the basis of spontaneous TNFα production of more or <38 pg/ml. B. TNFα levels in cultures of control and activated CLL cells in the presence of Hizentra were divided by the amounts in the control cultures without Hizentra. Averages and standard errors for each group are shown and indicate that 15 g/L Hizentra is more effective at preventing TNFα-production, especially by group 2 cells. C. Results in the presence of both Hizentra and Ibrutinib are shown for two patient samples. *, p < .05; **, p < .01.
3.8. Additive effects of Ibrutinib and Hizentra®
The above results (Supplementary Fig. 2, Fig. 5A,B) suggested spontaneous and BCR-activated production of TNFα by CLL cells could be inhibited by SCIG. The Bruton's tyrosine kinase (BTK) inhibitor Ibrutinib also lowers TNFα levels in CLL patients [34]. To determine if Hizentra® might inhibit BTK as well, the effect of combining Hizentra® and Ibrutinib on TNFα production by BCR-activated CLL cells was examined in two patient samples (Fig. 5C). As before (Supplementary Fig. 2, Fig. 5A,B), Hizentra® inhibited spontaneous and BCR-activated TNFα production, especially at 15 g/L (Fig. 5C, white bars). Ibrutinib strongly inhibited TNFα production in the presence and absence of Ig-cross-linking. Hizentra® increased the inhibitory effect of Ibrutinib (Fig. 5C), suggesting it acted through mechanisms other than inhibiting BTK.
3.9. Effect of Hizentra® on activated CLL cell death in vitro
CLL cells undergo spontaneous death, especially in serum-containing media [20,23], that can be prevented by signals through the BCR (Fig. 6A; compare first and second panels from the left). Hizentra® had variable effects on spontaneous and activated cell death that also appeared to be related to baseline production of TNFα. Examples are shown in Fig. 6A and the results are summarized in Fig. 6B. In “Group 1” samples with spontaneous TNFα secretion >38 pg/ml, Hizentra® had little effect on death and even appeared to increase relative survival (i.e. percent 7AAD− cells in the presence of anti-IgM and/or Hizentra® divided by percent 7AAD− cells in control cultures) (Fig. 6A; top panels and Fig. 6B; white bars). In contrast, Hizentra® increased the death of “Group 2” CLL cells with low spontaneous release of TNFα (<38 pg/ml) independent of Ig-cross-linking (Fig. 6A; bottom panel, Fig. 6B; black bars).
Fig. 6.

Hizentra decreases survival of activated CLL cells from group 2 patients. Purified CLL cells were cultured in AIM-V serum-free media in the presence or absence of anti-IgM antibodies with or without Hizentra at a final concentration of 5 or 15 g/L. Percentages of viable cells that exclude the dye 7AAD were measured by flow cytometry after 5 days. A. Examples are shown in the dot-plots with the numbers representing percentages of viable 7AAD− cells. B. The summary graph shows the average and standard error of the relative survival of 6 Group 1 patient samples and 4 group 2 samples. Relative survival is defined as the percentage of 7AAD− cells with Hizentra divided by the percentage in control cultures without Hizentra. Groups 1 and 2 represent CLL cells that spontaneously make > or <38 pg/ml of TNFα, respectively. The results indicate group 2 samples are most sensitive to Hizentra, especially at 15 g/L. C. Schematic diagram suggesting that high-normal IgG levels may help control BCR-signaling in CLL cells. *;p < 0.05.
4. Discussion
The observations in this manuscript suggest: 1. IgG, IgA, and IgM levels fall steadily over time except in CLL patients with truly indolent disease. Rates of decline are faster for patients with aggressive disease who will require treatment (Fig. 1). 2. Plasma TNFα levels are inversely correlated with IgG (Fig. 2A). 3. IgRT that achieves IgG levels >9 g/L is associated with biochemical evidence of disease control in vivo (Fig. 3). 4. IgG concentrations at the upper limit of normal (over 10 g/L) inhibit BCR-activation and TNFα production and can increase cell death in vitro (Fig. 4, Fig. 5, Fig. 6A), especially in CLL cells with low spontaneous production of TNFα.
Except for patients who will never need treatment, the fact that Ig levels appear to decline in a coordinated manner (Fig. 1) suggests many patients will have experienced significant deterioration from normal by the time they receive a diagnosis of CLL. Such patients may then have “relative” hypogammaglobulinemia despite Ig levels in the low-normal range (Fig. 2B). Low IgM levels in many CLL patients at diagnosis (Fig. 2B) may not be a selective deficiency but simply reflect the narrow normal range for IgM compared to IgA and IgG, making it easier to decline below the lower limit. The rate of decline of Ig levels (Fig. 1C) might have prognostic importance in the practical management of CLL patients, much like the lymphocyte doubling time (LDT) [31].
The etiology of hypogammaglobulinemia in CLL is multi-factorial and has been ascribed to various mechanisms including defective helper T cell function and Fas-mediated killing of plasma cells by CLL cells [9,10,21,38]. The main source of immunoglobulin in CLL patients is normal B cells as paraproteins are not routinely made by CLL cells [28]. However, numbers of normal B cells, defined as CD19+CD5− blood lymphocytes, did not exhibit an obvious relationship with Ig levels at the time of the CLL diagnosis (Supplementary Fig. 3). The observation of a parallel decline in all three Ig classes (Fig. 1) suggests a major defect may occur at the transitional stage of B cell development [42]. Transitional cells exit the bone marrow to develop in the periphery and ultimately give rise to memory and plasma cells that make all classes of immunoglobulins. Studies based on quantifying kappa-deleting recombination excision circles (KRECs) concluded newly produced B lymphocytes are deficient even at early clinical stages of CLL [33]. It is not clear why transitional B cells should be impaired in the bone marrow of CLL patients. TNFα can inhibit human B cell development at the transitional stage [26] and the results in Fig. 2A suggest plasma TNFα correlates inversely with IgG in CLL patients. While TNFα in blood may originate from other cells and the correlation may simply reflect higher levels in patients with more advanced disease that independently develop hypogammaglobulinemia [12], perhaps bone marrow CLL cells that secrete TNFα suppress B cell development in the manner of senescent B cells that make TNFα and are implicated in the humoral immunodeficiency of aging [36].
IgRT is prescribed for patients with hypogammaglobulinemia and a history of infections because it can prevent subsequent ones [15,48]. The dose of IVIG or SCIG for secondary immunodeficiency is generally the equivalent of 0.4 g/kg/month but can be titrated upward in the event of breakthrough infections ([4,5]; [1]). It is not clear if this dose is optimal for CLL patients. A review of local patient records suggests a replacement dose of 0.4 g/kg/month usually limits IgG trough levels to the low-normal range (i.e. 6–8 g/L) (Fig. 3), which is associated with evidence of biochemical progression (Fig. 3A,C) and increased plasma TNFα (Fig. 3B). However, endogenous IgG levels that remain above 8 g/L are associated with an indolent clinical course (Fig. 1E) and maintenance of IgG levels in the high-normal range (9–12 g/L) with IgRT in the absence of a paraprotein is associated with biochemical evidence of disease control (Fig. 3C,D).
An explanation for these clinical findings is provided by the observations that IgG at sufficiently high concentrations inhibits BCR-activation and cytokine production and sometimes kills CLL cells in vitro (Fig. 4, Fig. 5, Fig. 6). In addition to inhibiting antigen-driven BCR-signaling represented by Ig-cross-linking (Fig. 4), IgRT may also affect antigen-independent cell-autonomous BCR-signaling that has been implicated in the pathogenesis of CLL ([11]; [30]). Hizentra inhibited spontaneous release of TNFα by CLL cells (Supplementary Fig. 2; Fig. 5) that is ibrutinib-sensitive (Fig. 5C) and may be due to autonomous BCR-signaling. Given that BCR-signaling drives CLL progression [8], these results suggest maintaining plasma IgG levels near 15 g/L with higher doses of IgRT may have anti-tumor activity. Indeed, it was shown almost 35 years ago that IVIG did have an anti-CLL effect when administered every 3 weeks, which would lead to higher trough levels than a conventional modern schedule of every 4 weeks [6]. IgG concentrations below 8 g/L that are associated with disease progression (Fig. 1E, Fig. 3) can sometimes even increase AKT- and PLCγ-phosphorylation (Fig. 4D,E), cytokine production (Fig. 5) and survival of activated CLL cells in vitro (Fig. 6), consistent with a tumor-promoting effect.
Why some patients attain higher IgG levels than others with the same dosing strategy (Fig. 3) is unclear. Patient SS (Fig. 3A, right lower graph) was maintained on IVIG but other patients with trough levels higher than 9 g/L (Fig. 3A,B,C) were using SCIG. One possibility is that CLL cells bind IgG and lower free IgG levels in the blood. In this case, circulating CLL numbers should correlate inversely with IgG levels achieved by IgRT. However, changes in IgG levels following institution of IgRT did not exhibit an obvious relationship with blood lymphocytes (Supplementary Fig. 4). Potentially such variables as patient technique or decreased clearance of injected IgG molecules account for these variations.
The mechanism by which high IgG concentrations inhibit BCR-signaling in CLL cells is unclear. In normal human B cells, IVIG can bind the inhibitory Fc receptor CD32B, which recruits phosphatases such as SHIP-1 to prevent BCR-mediated activation of kinase cascades [40]. CD32B is the only Fc receptor expressed by CLL cells [7]. Consistent with this, the CD32 antibody AT10 [37] partially reversed the inhibitory effects of Hizentra at 15 g/L on BCR-mediated expression of CD83 in some patients (Supplementary Fig. 5). IgRT formulations also contain sialyated IgG proteins that can potentially bind CD22 on CLL cells and down-regulate BCR-signaling as described in normal human B cells ([39]).
While inhibition of BCR-signaling provides a plausible mechanism to justify using higher doses of IgRT, the effects likely also depend on the underlying biology of the CLL cells. For example, Group 2 CLL cells that produce little spontaneous TNFα appear to be more sensitive to IgRT than group 1 cells that make more spontaneous TNFα as suggested by greater inhibition of BCR-mediated TNFα-production and death in vitro (Fig. 5, Fig. 6). However, the ability of these in vitro assays to predict subsequent responses to high-dose IgRT in vivo is not clear. It is also not clear if conventional prognostic factors can predict responses to IgRT. Patients classified as “<9 g/L” or “<9 g/L” (Fig. 3B,C) were not obviously different in terms of these factors. Although it has been reported that lack of spontaneous production of TNFα by CLL cells is associated with clinically aggressive behavior [13,18,22], there are no obvious clinical characteristics that distinguish group 1 and group 2 patients (Fig. 5, Fig. 6), possibly due to the cut-off used for the classification. Studies with more patient samples and a prospective clinical trial are needed to determine if these in vitro assays and conventional prognostic and biologic properties of CLL cells can determine if some patients are more appropriate to receive high-dose IgRT than others.
Taken together, these observations suggest a model whereby progressive hypogammaglobulinemia in CLL patients constitutes a “feed-forward” oncogenic event by removing inhibitory effects on BCR-signaling once IgG levels fall below 8 g/L (Fig. 1, Fig. 3, Fig. 6C). This effect of the humoral immunodeficiency may be compounded by gut microbial products such as endotoxin that can leak through the intestinal wall and drive the growth of CLL cells by activating TLRs and BCRs, as described in common variable immunoglobulin deficiency patients [2]. Accordingly, institution of IgRT (either IVIG or SCIG) to maintain IgG levels near 15 g/L in CLL patients on a “watch and wait” management strategy whose IgG levels have fallen below 8 g/L (Fig. 1E) may have disease modifying activity resembling other BCR-signaling inhibitors such as Ibrutinib in some patients (Fig. 5, Fig. 6). This conclusion is limited by the retrospective nature of the study. For example, failure to reach IgG levels above 9 g/L with conventional Ig replacement in some patients may simply reflect rapid catabolism of IgG and represent an epiphenomena of more aggressive disease that is independent of the IgG level. A randomized prospective interventional clinical trial with Ig replacement dosed to IgG target level is needed to confirm the potential importance of maintaining high IgG levels in CLL patients. Note that increased toxicity from high IgG levels for prolonged periods of time in CLL patients is not anticipated based on the tolerability of maintenance therapies in neurological diseases that use doses of 1 g/kg administered every three weeks [24].
Authorship contributions
DS conceived the project and wrote the manuscript. YS, RV, JH, and GW performed in vitro studies and helped write manuscript. AZ and PN provided reagents and helped design experiments.
Disclosure of conflicts of interest
DS acknowledges funding from CSLberhing to support the studies reported here.
Role of the funding source
The funding source had no role in the study design, in the collection, analysis, and interpretation of data, in the writing of the report, and in the decision to submit the article for publication.
Acknowledgements
This work was supported by CIHR grant #374817, the Leukemia and Lymphoma Society of Canada, and CSL Behring Canada. We thank Dr. Mel Berger (CSL Behring) for helpful advice.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2018.08.045.
Appendix A. Supplementary data
Supplementary material
References
- 1.Agostini C., Blau I.W., Kimby E., Plesner T. Prophylactic immunoglobulin therapy in secondary immune deficiency - an expert opinion. Expert Rev Clin Immunol. 2016;12:921–926. doi: 10.1080/1744666X.2016.1208085. [DOI] [PubMed] [Google Scholar]
- 2.Berbers R.M., Nierkens S., van Laar J.M., Bogaert D., Leavis H.L. Microbial dysbiosis in common variable immune deficiencies: Evidence, causes, and consequences. Trends Immunol. 2017;38:206–216. doi: 10.1016/j.it.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Berrebi A., Bassous L., Haran M., Shtalrid M., Shvidel L. The significance of elevated beta 2-microglobulin in chronic lymphocytic leukemia (CLL): evidence of in vitro secretion following activation of CLL cells. Leuk Res. 2010;34:e248–e249. doi: 10.1016/j.leukres.2010.03.027. [DOI] [PubMed] [Google Scholar]
- 4.Berger M. Choices in IgG replacement therapy for primary immune deficiency diseases: subcutaneous IgG vs. intravenous IgG and selecting an optimal dose. Curr Opin Allergy Clin Immunol. 2011;11:532–538. doi: 10.1097/ACI.0b013e32834c22da. [DOI] [PubMed] [Google Scholar]
- 5.Berger M. L-proline-stabilized human IgG: Privigen®10% for intravenous use and Hizentra®20% for subcutaneous use. Immunotherapy. 2011;3:163–176. doi: 10.2217/imt.10.108. [DOI] [PubMed] [Google Scholar]
- 6.Besa E.C. Use of intravenous immunoglobulin in chronic lymphocytic leukemia. Am J Med. 1984;76:209–218. doi: 10.1016/0002-9343(84)90344-9. [DOI] [PubMed] [Google Scholar]
- 7.Bosch R., Mora A., Vicente E.P., Ferrer G., Jansà S., Damle R. FcgammaRIIb expression in early stage chronic lymphocytic leukemia. Leuk Lymphoma. 2017;58:2642–2648. doi: 10.1080/10428194.2017.1307981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burger J.A., Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. 2013;34:592–601. doi: 10.1016/j.it.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cantwell M., Hua T., Pappas J., Kipps T.J. Acquired CD40-ligand deficiency in chronic lymphocytic leukemia. Nat Med. 1997;3:984–989. doi: 10.1038/nm0997-984. [DOI] [PubMed] [Google Scholar]
- 10.Cerutti A., Kim E.C., Shah S., Schattner E.J., Zan H., Schaffer A. Dysregulation of CD30+ T cells by leukemia impairs isotype switching in normal B cells. Nat Immunol. 2001;2:150–156. doi: 10.1038/84254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dühren-Von Minden M., Übelhart R., Schneider D., Wossning T., Bach M., Buchner M. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489:309–312. doi: 10.1038/nature11309. [DOI] [PubMed] [Google Scholar]
- 12.Ferrajoli A., Keating M.J., Manshouri T., Giles F.J., Dey A., Estrov Z. The clinical significance of tumor necrosis factor-alpha plasma level in patients having chronic lymphocytic leukemia. Blood. 2002;100:1215–1219. [PubMed] [Google Scholar]
- 13.Foa R., Massaia M., Cardona S., Tos A.G., Bianchi A., Attisano C. Production of tumor necrosis factor-alpha by B-cell chronic lymphocytic leukemia cells: a possible regulatory role of TNF in the progression of the disease. Blood. 1990;76:393–400. [PubMed] [Google Scholar]
- 14.Forconi F., Moss P. Perturbation of the normal immune system in patients with CLL. Blood. 2015;126:573–581. doi: 10.1182/blood-2015-03-567388. [DOI] [PubMed] [Google Scholar]
- 15.Gale R.P., Chapel H.M., Bunch C., Rai K.R., Foon K., Courter S.G. Intravenous immunoglobulin for the prevention of infection in chronic lymphocytic leukemia. A randomized, controlled clinical trial. N Engl J Med. 1988;319:902–907. doi: 10.1056/NEJM198810063191403. [DOI] [PubMed] [Google Scholar]
- 16.Gobin S.J., Biesta P., van den Elsen P.J. Regulation of human beta 2-microglobulin transactivation in hematopoietic cells. Blood. 2003;101:3058–3064. doi: 10.1182/blood-2002-09-2924. [DOI] [PubMed] [Google Scholar]
- 17.Guarini A., Gaidano G., Mauro F.R., Capello D., Mancini F., De Propris M.S. Chronic lymphocytic leukemia patients with highly stable and indolent disease show distinctive phenotypic and genotypic features. Blood. 2003;102:1035–1041. doi: 10.1182/blood-2002-12-3639. [DOI] [PubMed] [Google Scholar]
- 18.Hahn T., Kusminsky G., Bassous L., Berrebi A. Tumor necrosis factor production in B cell chronic lymphocytic leukemia. Leuk Lymphoma. 1991;4:117–121. doi: 10.3109/10428199109068054. [DOI] [PubMed] [Google Scholar]
- 19.Hallek M., Cheson B.D., Catovsky D., Caligaris-Cappio F., Dighiero G., Döhner H. Guidelines for diagnosis, indications for treatment, response assessment and supportive management of chronic lymphocytic leukemia. Blood. 2018;131:2745–2760. doi: 10.1182/blood-2017-09-806398. [DOI] [PubMed] [Google Scholar]
- 20.Hammond C., Shi Y., Mena J., Tomic J., Cervi D., He L. Effect of serum and antioxidants on the immunogenicity of protein kinase C-activated chronic lymphocytic leukemia cells. J Immunother. 2005;28:28–39. doi: 10.1097/00002371-200501000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Hersey P., Wotherspoon J., Reid G., Gunz F.W. Hypogammaglobulinaemia associated with abnormalities of both B and T lymphocytes in patients with chronic lymphatic leukaemia. Clin Exp Immunol. 1980;39:698–707. [PMC free article] [PubMed] [Google Scholar]
- 22.Hulkkonen J., Vilpo J., Vilpo L., Hurme M. Diminished production of interleukin-6 in chronic lymphocytic leukaemia (B-CLL) cells from patients at advanced stages of disease. Br J Haematol. 1998;100:478–483. doi: 10.1046/j.1365-2141.1998.00595.x. [DOI] [PubMed] [Google Scholar]
- 23.Kay N.E., Shanafelt T.D., Strege A.K., Lee Y.K., Bone N.D., Raza A. Bone biopsy derived marrow stromal elements rescue chronic lymphocytic leukemia B-cells from spontaneous and drug induced cell death and facilitates an "angiogenic switch". Leuk Res. 2007;31:899–906. doi: 10.1016/j.leukres.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuwabara S., Misawa S., Mori M., Iwai Y., Ochi K., Suzuki H. Glovenin-I MMN Study Group. Intravenous immunoglobulin for maintenance treatment of multifocal motor neuropathy: a multi-center, open-label, 52-week phase 3 trial. J Peripher Nerv Syst. 2018;23:115–119. doi: 10.1111/jns.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lachance S., Christofides A.L., Lee J.K., Sehn L.H., Ritchie B.C., Shustik C. A Canadian perspective on the use of immunoglobulin therapy to reduce infectious complications in chronic lymphocytic leukemia. Curr Oncol. 2016;23:42–51. doi: 10.3747/co.23.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lebien T.W. Fates of human B-cell precursors. Blood. 2000;96:9–23. [PubMed] [Google Scholar]
- 27.Mansouri L., Papakonstantinou N., Ntoufa S., Stamatopoulos K., Rosenquist R. NFκB activation in chronic lymphocytic leukemia: a point of convergence of external triggers and intrinsic lesions. Semin Cancer Biol. 2016;39:40–48. doi: 10.1016/j.semcancer.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Maurer M., Cerhan J., Katzmann J., Link B., Allmer C., Zent C. Monoclonal and polyclonal serum free light chains and clinical outcome in chronic lymphocytic leukemia. Blood. 2011;118:2821–2826. doi: 10.1182/blood-2011-04-349134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCaw L., Shi Y., Wang G., Li Y.J., Spaner D.E. Low density lipoproteins amplify cytokine-signaling in chronic lymphocytic leukemia cells. EBioMedicine. 2017;15:24–35. doi: 10.1016/j.ebiom.2016.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minici C., Gounari M., Übelhart R., Scarfò L., Dühren-Von Minden M., Schneider D. Distinct homotypic B-cell receptor interactions shape the outcome of chronic lymphocytic leukaemia. Nat Commun. 2017;8 doi: 10.1038/ncomms15746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molica S., Alberti A. Prognostic value of the lymphocyte doubling time in chronic lymphocytic leukemia. Cancer. 1987;60:2712–2716. doi: 10.1002/1097-0142(19871201)60:11<2712::aid-cncr2820601122>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 32.Moreira J., Rabe K.G., Cerhan J.R., Kay N.E., Wilson J.W., Call T.G. Infectious complications among individuals with clinical monoclonal B-cell lymphocytosis (MBL): a cohort study of newly diagnosed cases compared to controls. Leukemia. 2013;27:136–141. doi: 10.1038/leu.2012.187. [DOI] [PubMed] [Google Scholar]
- 33.Motta M., Chiarini M., Ghidini C., Zanotti C., Lamorgese C., Caimi L. Quantification of newly produced B and T lymphocytes in untreated chronic lymphocytic leukemia patients. J Transl Med. 2010;8:111. doi: 10.1186/1479-5876-8-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niemann C.U., Herman S.E., Maric I., Gomez-Rodriguez J., Biancotto A., Chang B.Y. Disruption of in vivo chronic lymphocytic leukemia tumor-microenvironment interactions by ibrutinib - findings from an investigator initiated phase 2 study. Clin Cancer Res. 2015;22:1572–1582. doi: 10.1158/1078-0432.CCR-15-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parikh S.A., Leis J.F., Chaffee K.G., Call T.G., Hanson C.A., Ding W. Hypogammaglobulinemia in newly diagnosed chronic lymphocytic leukemia: Natural history, clinical correlates, and outcomes. Cancer. 2015;121:2883–2891. doi: 10.1002/cncr.29438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ratliff M., Alter S., Frasca D., Blomberg B.B., Riley R.L. In senescence, age-associated B cells secrete TNFalpha and inhibit survival of B-cell precursors. Aging Cell. 2013;12:303–311. doi: 10.1111/acel.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roghanian A., Teige I., Mårtensson L., Cox K.L., Kovacek M., Ljungars A. Antagonistic human FcγRIIB (CD32B) antibodies have anti-tumor activity and overcome resistance to antibody therapy in vivo. Cancer Cell. 2015;27:473–488. doi: 10.1016/j.ccell.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 38.Sampalo A., Brieva J.A. Humoral immunodeficiency in chronic lymphocytic leukemia: role of CD95/CD95L in tumoral damage and escape. Leuk Lymphoma. 2002;43:881–884. doi: 10.1080/10428190290017033. [DOI] [PubMed] [Google Scholar]
- 39.Séïté J.F., Cornec D., Renaudineau Y., Youinou P., Mageed R.A., Hillion S. IVIg modulates BCR signaling through CD22 and promotes apoptosis in mature human B lymphocytes. Blood. 2010;116:1698–1704. doi: 10.1182/blood-2009-12-261461. [DOI] [PubMed] [Google Scholar]
- 40.Seite J.F., Hillion S., Harbonnier T., Pers J.O. Review: intravenous immunoglobulin and B cells: when the product regulates the producer. Arthritis Rheumatol. 2015;67:595–603. doi: 10.1002/art.38910. [DOI] [PubMed] [Google Scholar]
- 41.Shi Y., White D., He L., Miller R.L., Spaner D.E. Toll-like receptor-7 tolerizes malignant B cells and enhances killing by cytotoxic agents. Cancer Res. 2007;67:1823–1831. doi: 10.1158/0008-5472.CAN-06-2381. [DOI] [PubMed] [Google Scholar]
- 42.Simon Q., Pers J.O., Cornec D., Le Pottier L., Mageed R.A., Hillion S. In-depth characterization of CD24(high)CD38(high) transitional human B cells reveals different regulatory profiles. J Allergy Clin Immunol. 2016;137:1577–1584. doi: 10.1016/j.jaci.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 43.Spaner D.E., Wang G., McCaw L., Li Y., Disperati P., Cussen M.A. Activity of the janus kinase inhibitor Ruxolitinib in chronic lymphocytic leukemia: results of a phase II trial. Haematologica. 2016;101:e192–e195. doi: 10.3324/haematol.2015.135418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tarnani M., Laurenti L., Longo P.G., Piccirillo N., Gobessi S., Mannocci A. The proliferative response to CpG-ODN stimulation predicts PFS, TTT and OS in patients with chronic lymphocytic leukemia. Leuk Res. 2010;34:1189–1194. doi: 10.1016/j.leukres.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 45.Thompson P.A., O'Brien S.M., Xiao L., Wang X., Burger J.A., Jain N. beta-microglobulin normalization within 6 months of ibrutinib-based treatment is associated with superior progression-free survival in patients with chronic lymphocytic leukemia. Cancer. 2016;122:565–573. doi: 10.1002/cncr.29794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toapanta F.R., Bernal P.J., Sztein M.B. Diverse phosphorylation patterns of B cell receptor-associated signaling in naïve and memory human B cells revealed by phosphoflow, a powerful technique to study signaling at the single cell level. Front Cell Infect Microbiol. 2012;2:128. doi: 10.3389/fcimb.2012.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tomic J., Lichty B., Spaner D.E. Aberrant interferon-signaling is associated with aggressive chronic lymphocytic leukemia. Blood. 2011;117:2668–2680. doi: 10.1182/blood-2010-05-285999. [DOI] [PubMed] [Google Scholar]
- 48.Visentin A., Compagno N., Cinetto F., Imbergamo S., Zambello R., Piazza F. Clinical profile associated with infections in patients with chronic lymphocytic leukemia. Protective role of immunoglobulin replacement therapy. Haematologica. 2015;100:e515–e518. doi: 10.3324/haematol.2015.126763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material