

SUMMARY
Phagocyte microbiocidal mechanisms and inflammatory cytokine production are temporally coordinated, although their respective interdependencies remain incompletely understood. Here, we identify a nitric-oxide-mediated antioxidant response as a negative feedback regulator of inflammatory cytokine production in phagocytes. In this context, Keap1 functions as a cellular redox sensor that responds to elevated reactive nitrogen intermediates by eliciting an adaptive transcriptional program controlled by Nrf2 and comprised of antioxidant genes, including Prdx5. We demonstrate that engaging the antioxidant response is sufficient to suppress Toll-like receptor (TLR)-induced cytokine production in dendritic cells and that Prdx5 is required for attenuation of inflammatory cytokine production. Collectively, these findings delineate the reciprocal regulation of inflammation and cellular redox systems in myeloid cells.
In Brief
Following TLR ligation, phagocytes produce NO to elicit an antioxidant response that is mediated by Keap1. Graham et al. demonstrate that this antioxidant response, dependent on Prdx5, acts as a negative feedback loop to suppress TLR-induced cytokine and NO production.
Graphical Abstract
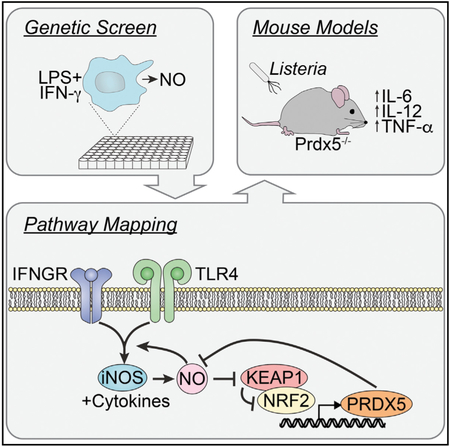
INTRODUCTION
Phagocytes utilize several effector mechanisms to combat foreign micro-organisms (Okabe and Medzhitov, 2016), including phagolysosomal killing (via low pH and hydrolytic enzymes), xenophagy, oxidative burst, and nitric oxide (NO) production (MacMicking, 2012). Upon exposure to microbial ligands and extrinsic cytokines, phagocytes express NOS2, which encodes iNOS and generates NO by oxidative deamination of arginine (Lowenstein et al., 1992; Moresco et al., 2011; Stuehr and Marletta, 1985; Xie et al., 1992). NO and downstream reactive nitrogen intermediates (RNIs) are toxic to microbes and host cells, a consequence of cysteine S-nitrosation of proteins, deamination of nucleic acids, and desaturation of lipids (Nathan and Cunningham-Bussel, 2013).
Several inflammatory stimuli induce transcriptional upregulation of NOS2, including Toll-like receptor (TLR) ligands and cytokines, such as interleukin-1b (IL-1b) (Gao et al., 1997; Lima-Junior et al., 2013; Lowenstein et al., 1993; Xie et al., 1993). The minimal requirement for transcriptional activation of the NOS2 promoter is nuclear factor κB (NF-κB), with synergy from Stat1 and Irf1 downstream of cytokine receptors (Farlik et al., 2010; Martin et al., 1994; Xie et al., 1993, 1994). Stimulation with lipopolysaccharide (LPS) and interferon (IFN)-γ reinforces transcriptional activation of the NOS2 promoter and results in elevated and sustained iNOS expression. Once expressed, iNOS forms a homodimer that utilizes nicotinamide adenine dinucleotide phosphate – reduced (NADPH) and oxygen to convert L-arginine to L-citrulline and NO (Bogdan, 2015; Hibbs et al., 1987; Stuehr, 1999). Although transcriptional regulation of NOS2 has been well characterized, little is known about negative regulators that fine-tune RNI production or how nitrosative stress impacts host inflammatory pathways.
The central role of RNIs in host defense was conclusively demonstrated with the generation of NOS2−/− mice (MacMicking et al., 1995; Wei et al., 1995), which were highly susceptible to infection with Listeria monocytogenes, exhibiting increased bacterial burden and decreased survival (MacMicking et al., 1995). NOS2 contributes to host defense against a variety of intracellular pathogens, including Leishmania major and Mycobacterium tuberculosis (MacMicking et al., 1997; Wei et al., 1995). Whereas murine studies demonstrated the importance of NOS2 in host defense, genome-wide association studies (GWASs) highlighted a more nuanced role for NOS2 in human disease. A SNP in the NOS2 promoter (C-1173T) was associated with protection from severe malaria through elevated RNI production (Anstey et al., 1996; Hobbs et al., 2002). GWASs in patients with very early onset inflammatory bowel disease (IBD) also identified SNPs, such as rs2297518, which marks an allele associated with higher RNI production (Dhillon et al., 2014). These human data demonstrate that RNI production is necessary for combating microbial infections, although it can be detrimental to hosts. Therefore, there is need to delineate how RNI production is regulated to optimize the balance between host defense and inflammatory pathologies.
NO also functions as a potent signaling molecule. Previous work showed that NO produced by endothelial nitric oxide synthase (eNOS) acts in a paracrine manner to induce smooth muscle relaxation and vascular dilation (Arnold et al., 1977; Furchgott and Zawadzki, 1980; Ignarro et al., 1987). NO functions as an intracellular second messenger controlling the antioxidant response and as a chemical modifier forming reversible posttranslational modifications on proteins to alter their signaling function (Buckley et al., 2003; Dhakshinamoorthy and Porter, 2004; Li et al., 2009; Nathan and Cunningham-Bussel, 2013). Given the diverse functions of RNIs in host defense, inflammation, and signal transduction, we sought to determine how phagocytes respond to RNI-mediated toxic insult and how RNIs impact inflammatory signaling cascades.
RESULTS
Functional Genetics Identifies Signaling Nodes Controlling Antimicrobial Defense
Phagocytes engage several innate antimicrobial defense mechanisms upon exposure to microbes. Parallel with iNOS expression, inflammatory cytokine production is transcriptionally induced upon TLR engagement. However, the interdependence of microbiocidal and inflammatory pathways is incompletely understood. To identify regulatory nodes controlling NO production, we performed a short hairpin RNA (shRNA) interference screen using candidate genes derived from loci implicated in autoimmune disease risk. We reasoned this candidate list, including genes in loci identified by IBD GWASs and associated with type 1 diabetes (T1D) and Mendelian inheritance of inflammatory pathologies (Graham et al., 2015), is likely to be enriched for key genes controlling fundamental pathways in immune responses. The arrayed screening library was comprised of ~2,000 shRNAs targeting 400 genes (Figure 1A; Graham et al.,2015). Following lentiviral delivery of shRNAs to bone-marrowderived dendritic cells (BMDCs), cultures were stimulated with LPS and IFN-γ to induce iNOS expression. NO production was assessed using the Griess assay, which measures the NO derivative nitrite. Hits were selected using a hit-calling algorithm that ranked Z scores for each shRNA individually (Figure 1B; Table S1). In the validation screen, we identified positive and negative regulators of RNI production (Figure 1C; Table S1). As expected, knockdown of known positive regulators of RNI production, Nos2 and Irf1, resulted in decreased RNI production (Figure 1C). We identified Keap1 as a regulator of iNOS that was required for RNI production. Under basal conditions, Keap1 functions as a redox sensor by recruiting a ubiquitinconjugating complex to Nrf2, leading to constitutive degradation (Gonzalez-Donquiles et al., 2017; Itoh et al., 1997, 1999; Kobayashi et al., 2004). Upon oxidative stress, oxidative modification of key cysteine residues on Keap1 induces a conformational change that releases Nrf2, allowing it to undergo nuclear translocation and initiate transcription of the antioxidant program (Kobayashi et al., 2006; Nguyen et al., 2005; Zhang and Hannink, 2003). Although it has been reported that RNIs induce antioxidant responses (Buckley et al., 2003; Dhakshinamoorthy and Porter, 2004; Li et al., 2009), our results suggest that induction of the antioxidant program potently inhibits RNI production as well. The antioxidant response could eliminate RNIs by “buffering” and/or exert a more direct effect on inflammatory pathways that inhibit expression of iNOS.
Figure 1. Functional Genetics Identifies Signaling Nodes Controlling Antimicrobial Defense.

(A) Schematic overview of shRNA screen to identify novel regulatory nodes controlling NO production in BMDCs. After stimulation with LPS and IFN-γ, NO production was measured using the Griess assay. Results implicate a model in which NO acts on Keap1 to engage an anti-inflammatory negative feedback loop. Dashed horizontal line, viability cutoff; dashed vertical line, mean NO signal. Curved lines represent best fit ± 1 SD.
(B) Individual shRNAs were scored such that their nitrite signal was normalized to cell number for each well, and genes were subsequently ranked based on plate-wide Z scores for each corresponding shRNA. Small blue dots represent Z scores for individual hairpins. Larger black boxes represent mean Z scores for all hairpins targeting the given gene.
(C) Twenty-four hits from the primary screen were retested as described in (A) and (B). Individual shRNA-level scores were calculated as percent effect relative to controls (shRNAs targeting genes not present in the mouse genome). Each dot represents an individual shRNA. Bars represent the second best shRNA for each target gene. p values were calculated by Student’s t test comparing all shRNAs for a given gene relative to control shRNAs.
(D) Top scoring hits from the validation screen were selected for further testing in assays for inflammatory transcriptional responses. Candidate genes were knocked down in BMDCs, cells were stimulated with LPS and IFN-γ, and cytokine transcripts were measured relative to control shRNAs by Fluidigm.
See also Table S1.
Given that Keap1 knockdown potently inhibited RNI production in BMDCs, we sought to determine whether this effect was due to changes in Nos2 transcription. We performed a secondary assay on the top 5 scoring genes from the primary screen to validate their effects on Nos2 transcription and define their requirement for cytokine responses. Candidate genes were knocked down in BMDCs, and cells were stimulated with LPS and IFN-γ. Subsequent cytokine transcription was measured relative to control knockdown (Figure 1D). Knockdown of Dnmt3a, a DNA methyltransferase involved in epigenetic regulation, increased cytokine and chemokine transcription as expected. Knockdown of Tmem258 inhibited Nos2 and cytokine transcription, consistent with previous studies demonstrating that Tmem258 regulates tumor necrosis factor alpha (TNF-α) production in response to LPS (Parnas et al., 2015). Importantly, Keap1 knockdown dramatically impaired Nos2 transcription, suggesting that the antioxidant response acts upstream of RNI production by inhibiting Nos2 transcription. Moreover, Keap1 knockdown inhibited cytokine and chemokine transcription (Figure 1D). These results implicate Keap1-dependent redox regulation in differential regulation of TLR-induced inflammatory responses.
Reciprocal Regulation of Redox Pathways and Inflammatory Pathways
Our findings that Keap1 knockdown impairs RNI production and Nos2 transcription suggest a central role for the Keap1/Nrf2 axis in inflammatory regulation. To uncover the relationship between Keap1-dependent redox pathways and inflammatory cytokine responses, we derived a transcriptional signature defining the LPS-dependent antioxidant response in BMDCs using RNA sequencing (Table S2). We first identified differentially expressed genes in LPS-stimulated relative to unstimulated BMDCs, which included several well-characterized components of the LPS response (Il12a, Il10, and Csf2) and putative components of the antioxidant response (Gsst4 and Gsta1). Next, we identified differentially expressed genes upon knockdown of Keap1 relative to control hairpin in the absence of LPS. We reasoned that the overlapping gene set is enriched for genes in the Keap1-controlled arm of the TLR4 response (Figure 2A; Table S2). Enrichment results were represented as a network graph and implicate Keap1 in cellular metabolism and redox regulation during inflammation (Figure 2B). Consistent with the known role for Keap1 in Nrf2 regulation, Keap1 knockdown in BMDCs induced upregulation of several validated Nrf2 target genes (Malhotra et al., 2010).
Figure 2. Reciprocal Regulation of Redox Pathways and Inflammatory Pathways.

(A) To implicate a subset of LPS-induced genes controlled by the Keap1-Nrf2 axis, target genes were knocked down in BMDCs using lentiviral delivery of shRNAs, cells were stimulated with LPS and IFN-γ for 6 hr, and transcriptional profiling was performed using RNA sequencing. We queried overlap between LPS-induced genes (LPS versus unstim) and genes upregulated by Keap1 knockdown (shKeap1 versus shCtrl). Significance is indicated by Benjamini-Hochberg (BH) p value and Log2 fold change (FC). For each target gene, 2 distinct shRNAs were used, and each was performed in technical duplicate.
(B) Functional enrichment map of the LPS-induced Keap1-Nrf2 signature highlights pathways related to redox regulation and cellular metabolism. Enrichment results represented as a network graph, with nodes denoting gene sets and color intensity corresponding to gene set enrichment score (—log10[p value]). Edges denote the extent of mutually overlapping genes between gene sets using thickness, color intensity, and the displayed Jaccard coefficient (Anderson et al., 2011; Orvedahl et al., 2011).
(C and D) Target genes were knocked down in BMDCs using lentiviral delivery of shRNAs, cells were stimulated with LPS and IFN-γ for 6 hr, and transcriptional profiling was performed using RNA sequencing to analyze antioxidant (C) and cytokine (D) gene expression. Log2 expression values are expressed as signed difference ratios relative to shRNA and scaled by normalizing to maximum absolute deviation of each gene from the shRNA control (Ng and Xavier, 2011).
See also Table S2.
Having defined the antioxidant program following TLR engagement, we sought to establish whether this process requires RNIs. Nos2 knockdown is predicted to eliminate a major source of oxidative stress, thereby allowing for dissection of its contribution to the induction of the antioxidant program. We performed transcriptional profiling in BMDCs after knockdown of Nos2 or Keap1 and stimulation with LPS and IFN-γ (Figure 2C; Table S2). Keap1 knockdown resulted in the upregulation of some antioxidant genes, including members of the peroxiredoxin, thioredoxin, and glutathione systems (Figure 2C). In contrast, reduction of RNIs by Nos2 knockdown predominantly reduced expression of these antioxidant genes. These results suggest that Keap1 responds to RNI-dependent oxidative stress, leading to the induction of the antioxidant response.
Finally, we aimed to define the relationship between redox regulation and inflammatory responses. We impaired the antioxidant response with Prdx5 knockdown or induced the antioxidant response with Keap1 knockdown in BMDCs stimulated with LPS and IFN-γ and determined the impact on cytokine transcription (Figure 2D). Keap1 knockdown resulted in decreased cytokine expression (Il6, Il10, Il27, Ccl2, Ccl12, Cxcl2, and Cxcl10) relative to control knockdown, supporting the association of antioxidant responses with inhibition of inflammatory responses. Prdx5 knockdown, however, resulted in increased cytokine expression (Cxcl2, Ccl5, Ccl22, and Il15) relative to control knockdown. Thus, deficiency in the antioxidant response at the level of Prdx5 increased inflammatory cytokine and chemokine production. Together, these results reveal a reciprocal relationship between redox regulation and inflammatory pathways.
Redox Regulation of Metabolic Adaptation Associated with Inflammatory Signals
Having demonstrated Prdx5 is a central component of the antioxidant response, we hypothesized that defining the Prdx5 interactome may provide insight into its cellular function. To this end, we ectopically expressed Prdx5 in 293T cells and identified coimmunoprecipitated proteins by mass spectrometry (Figures 3A and 3B; Table S3). Prdx5 binding partners were enriched for mitochondrial proteins involved in nucleotide synthesis (Hprt1), lipid synthesis (Acly), and maintaining mitochondrial functions (Grpel1, Mthfd2, and Mthd1l). The Prdx5 interactome was also enriched for related antioxidant proteins, such as Txn, Prdx1, and Prdx3. These results support a role for Prdx5 in regulating mitochondrial metabolism.
Figure 3. Redox Regulation of Metabolic Adaptation Associated with Inflammatory Signals.

(A and B) HEK293T cells were transduced to ectopically express V5-tagged Prdx5. Interaction partners were immunoprecipitated, identified by mass spectrometry (A), and mitochondrial proteins were tabulated (B).
(C and D) Target genes were knocked down in BMDCs using lentiviral delivery of shRNAs. After stimulation with LPS and IFN-γ, BMDCs were lysed for metabolite profiling by mass spectrometry. (C) Pathway activity map illustrates perturbations in the urea cycle and TCA cycle in Nos2 knockdown (C), and fatty acid metabolism in Keap1 knockdown (D).
See also Tables S3 and S4 and Figures S1 and S3.
To illuminate how Prdx5 and redox pathways impact metabolic adaptation to inflammatory stimuli, we employed a metabolite profiling strategy. To this end, we knocked down Keap1 to induce the antioxidant response, Nos2 to eliminate a major source of oxidative stress induced by inflammatory stimuli, or Prdx5 to attenuate the antioxidant response in BMDCs. Cells were stimulated with LPS and IFN-γ and subjected to metabolite profiling (Figures 3C and 3D; Table S4). Nos2 knockdown significantly altered the urea cycle such that ratios of the iNOS byproducts citrulline and argininosuccinate were decreased. Additionally, Nos2 knockdown altered mitochondrial respiration, resulting in elevated tricarboxylic acid (TCA) activity, as indicated by depletion of citrate and accumulation of downstream intermediates isocitrate and malate. These findings are consistent with previous observations showing RNIs are necessary for activated dendritic cells to undergo a metabolic shift from oxidative phosphorylation to glycolysis (Everts et al., 2012; Krawczyk et al., 2010). Inhibition of RNI production precluded BMDCs from undergoing this metabolic shift, resulting in increased TCA activity, mitochondrial respiration, glycolysis, and ATP production (Figure S1). In contrast, induction of the antioxidant response by Keap1 knockdown was associated with metabolic conservation characterized by a reduction in mitochondrial respiration and glycolysis with a corresponding increase in ATP levels (Figure S1). These results suggest that Keap1 inhibition leads to ATP accumulation through reduced energy expenditure rather than increased oxidative phosphorylation, glycolysis, or fatty acid oxidation (Figure S1) and are consistent with findings in induced pluripotent stem cells, wherein Nrf2 drives a metabolic downregulation of oxidative phosphorylation (Hawkins et al., 2016). Finally, Prdx5 knockdown resulted in TCA dysfunction associated with accumulation of malate. These collective findings functionally link the antioxidant response with mitochondrial metabolism regulation during TLR stimulation.
Redox Regulation Attenuates Signaling Pathways Initiated by Microbial Ligands
Metabolite profiling suggested that redox pathways control immunometabolism. To test the hypothesis that Prdx5 deficiency impacts mitochondrial function, we generated Prdx5-deficient mice using standard gene-targeting approaches (Figure S2). All Prdx5−/− mice appeared healthy with no overt phenotypes or defects in immune system development. Numbers and percentages of B and T lymphocytes and myeloid cells in primary and secondary lymphoid organs were similar between Prdx5−/− and wild-type (WT) littermate controls (Figure S2).
We next monitored mitochondrial oxidative phosphorylation in BMDCs derived from Prdx5−/− mice or WT littermates by measuring the oxygen consumption rate (OCR). WT BMDCs exhibited a relatively high OCR at basal state indicative of active mitochondrial respiration (Figure 4A). Inhibition of ATP synthase with oligomycin (a complex V inhibitor) led to a reduction in OCR, which approximates the level of mitochondrial respiration associated with ATP production. Disruption of mitochondrial membrane potential upon carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) treatment elevated OCR to reflect maximal respiration, which was slightly higher than basal respiration, suggesting that WT BMDCs operate at near maximal respiration. Addition of antimycin A and rotenone (complex III and I inhibitors), which completely block the electron transport chain, resulted in a reduction in OCR to background levels, indicating very little extra-mitochondrial respiration in WT BMDCs without inflammatory stimuli. Stimulation of WT BMDCs with LPS and IFN-γ dramatically reduced basal and maximal mitochondrial respiration (Figure 4B). This response was completely reversed by iNOS inhibition with 1400W pretreatment (Garvey et al., 1997), indicating that RNI production inhibits mitochondrial respiration (Figure 4C), which is consistent with previous findings (Everts et al., 2012).
Figure 4. Redox Regulation Attenuates Signaling Pathways Initiated by Microbial Ligands.

(A–C) WT and Prdx5−/− BMDCs were unstimulated (A) or stimulated overnight (ON) with LPS and IFN-γ (B), with and without (C) 1400W pretreatment. Oxidative phosphorylation was monitored by the oxygen consumption rate (OCR) using the Seahorse Extracellular Flux Analyzer.
(D) Mitochondrial ROS production was measured using MitoSOX in WT and Prdx5−/− BMDCs at basal state and after ON stimulation with LPS and IFN-γ.
(E) Cellular oxidation state was measured using DCFDA in WT and Prdx5−/− BMDCs at basal state and after ON stimulation with LPS and IFN-γ, with and without 1400W pretreatment.
(F) Nitric oxide production was measured using the Griess assay in WT and Prdx5−/− BMDCs at basal state and after ON stimulation with LPS and IFN-γ, with and without tBHQ pretreatment.
(G) Cytokine production was measured by cytometric bead array in WT and Prdx5−/− BMDCs at basal state and after ON stimulation with LPS and IFN-γ, with and without tBHQ pretreatment.
(H) Serum cytokine response 36 (IL-6 and TNF-α) or 65 (IL-12 p40) hr after infection with Listeria monocytogenes.
*p < 0.05; **p < 0.01; ***p < 0.005; ns, not significant. See also Figures S1, S2, S3, S4, and S5.
After establishing that RNIs regulate mitochondrial respiration in WT BMDCs, we evaluated the requirement for Prdx5. Relative to WT, Prdx5−/− BMDCs exhibited a reduced basal OCR and a corresponding reduction in maximal respiration rate (Figure 4A). After stimulation with LPS and IFN-γ, Prdx5−/− and WT BMDCs showed similarly inactive mitochondrial respiration (Figure 4B). However, iNOS inhibition with 1400W pretreatment reversed RNI-dependent downregulation of mitochondrial respiration and restored maximal respiration in Prdx5−/− BMDCs to WT levels (Figure 4C). Together, these findings suggest that RNIs are a significant source of oxidative stress in the absence of Prdx5 and that Prdx5−/− BMDCs adapt to their impaired redox regulatory capacity by downregulating mitochondrial respiration. Notably, although Prdx5−/− BMDCs exhibited reduced levels of cellular ATP corresponding to decreased mitochondrial respiration, glycolysis and fatty acid oxidation were unperturbed (Figure S3).
Given that Prdx5 deficiency impaired cellular antioxidant function and reduced mitochondrial respiration, we sought to quantify mitochondrial reactive oxygen species (mROS). WT and Prdx5−/− BMDCs were stimulated with LPS and IFN-γ, and mROS production was measured using MitoSOX, a superoxide-specific fluorescent probe that localizes to mitochondria (Figure 4D). WT BMDCs showed relatively high basal mROS levels, which decreased after stimulation. Compared to WT, Prdx5−/− BMDCs exhibited lower mROS production at basal state and after stimulation. Although these results suggest that Prdx5 deficiency does not result in elevated mROS, they do not directly evaluate the overall cellular redox state. To this end, we utilized 2’,7’-dichlorodihydrofluorescein diacetate (DCFDA), a redox-sensitive probe that detects several reactive oxygen and nitrogen intermediates. Cellular redox states were similar between unstimulated WT and Prdx5−/− BMDCs (Figure 4E). After stimulation with LPS and IFN-γ, Prdx5−/− BMDCs showed elevated cellular oxidative state relative to WT BMDCs (Figure 4E). This increase in oxidative stress was reduced to WT levels after iNOS inhibition with 1400W pretreatment, indicating that RNI production underlies the increase in overall oxidative stress in stimulated Prdx5−/− BMDCs.
We leveraged the Prdx5−/− mouse model to determine the impact of elevated oxidative stress and NO production on inflammatory responses. First, we quantified RNI production in Prdx5−/− BMDCs; after stimulation with LPS and IFN-γ, Prdx5−/− BMDCs produced significantly higher levels of nitrite relative to WT BMDCs, indicating elevated RNI synthesis (Figure 4F). To determine whether increasing the antioxidant response is capable of bypassing the requirement for Prdx5 in limiting RNI production, we pretreated BMDCs with tert-butylhydroquinone (tBHQ), an electrophile that acts on Keap1 to induce the antioxidant response (Kobayashi et al., 2006; Zhang and Hannink, 2003). As expected, tBHQ dramatically reduced NO production in both WT and Prdx5−/− BMDCs (Figure 4F). Thus, a deficiency in the antioxidant response (Prdx5−/−) resulted in increased RNI production, whereas induction of antioxidant activity (tBHQ) inhibited RNI production. Next, we sought to determine whether the antioxidant response also inhibits inflammatory cytokine production. WT and Prdx5−/− BMDCs were stimulated with LPS and IFN-γ, and cytokine production (IL-6, IL-12 p40, and TNF-α) was measured by cytometric bead array. After stimulation, Prdx5−/− BMDCs produced significantly more cytokines compared to WT BMDCs (Figure 4G). With tBHQ pretreatment, IL-12 p40 production decreased in both WT and Prdx5−/− BMDCs, whereas IL-6 production decreased only in Prdx5−/− BMDCs. We obtained similar results using ML334, a non-electrophilic compound that directly disrupts the Keap1/ Nrf2 interaction (Figure S4; Hu et al., 2013). By inducing the antioxidant response with tBHQ, the effect of Prdx5 deficiency was partially rescued, consistent with the notion that upregulating additional thioredoxin and glutathione systems engage compensatory mechanisms. This suggests redundancy among peroxiredoxin family members. More importantly, it demonstrates the striking potency by which antioxidant pathways function in a negative feedback loop to inhibit inflammatory cytokine production.
Given the central observation in vitro that Prdx5−/− BMDCs exhibited increased cytokine production in response to the bacterial ligand LPS, we sought to monitor the analogous in vivo acute response to a bacterial pathogen (Listeria monocytogenes). At 36 hr post-infection, we observed a significant increase in TNF-α and a trend toward increased IL-6 in the serum of Prdx5−/− mice relative to littermate controls (Figure 4H). At 65 hr post-infection, IL-12 p40 was significantly elevated in Prdx5−/− mice (Figure 4H). We did not, however, observe significant effects on bacterial burden in the spleen or liver of Prdx5−/− mice relative to controls (Figure S5). These findings highlight the specific role of Prdx5 in tempering the inflammatory response in vivo and are consistent with our in vitro findings demonstrating elevated IL-6, TNF-α, and IL-12 p40 in Prdx5−/− BMDCs stimulated with LPS (Figure 4G). While the phenotype of Prdx5−/− mice in vivo is not severe, this is perhaps expected based on the known redundancy of Prdx family members. Thus, our findings point to Prdx5 as being an important contributor to a broader antioxidant response, likely elicited by additional components of the thioredoxin and glutathione systems.
Redox Regulation Signatures in Biopsies from Patients with Active IBD
Our findings indicate inflammatory stimuli induce oxidative stress via RNIs, and phagocytes adapt to this insult by eliciting an antioxidant response that dampens inflammatory cytokine production. To determine whether antioxidant responses are induced in IBD patients, we mined publicly available expression data from intestinal biopsies from IBD patients with active disease, IBD patients in remission, and healthy controls (Planall et al., 2013; Vanhove et al., 2015). We performed a differential expression analysis to determine whether the antioxidant signature we identified (Figure 2A) was altered between active disease state and healthy controls (Figure 5A). We identified enrichment for the antioxidant signature (Keap1-inhibition genes) in samples from IBD patients with active disease relative to healthy controls (Figure 5B). This pattern was replicated across independent datasets (Figures 5B-5D), indicating that the oxidative stress response is engaged in the active disease state. Thus, chronic inflammation associated with IBD elicits an antioxidant response, which is likely an important cytoprotective response engaged to limit and resolve inflammation.
Figure 5. A Transcriptional Signature Associated with Oxidative Stress Is Enriched in Inflamed Tissue from IBD Patients.

(A) Publicly available (GEO) microarray datasets relating to ulcerative colitis (UC) and Crohn’s disease (CD) were analyzed for an over-representation of a Keap1-inhibition signature gene set assembled from differential expression analysis of RNA sequencing (RNA-seq) data (Figure 2). In 3 of the 4 IBD datasets analyzed, Keap1-inhibition genes were over-represented in comparisons of diseased versus healthy and inflamed versus uninflamed tissue, with the most significant results appearing in comparisons of UC patients with healthy controls. The genes contributing to these overlaps are presented as a word cloud with the size of the word denoting how many public datasets these genes were found enriched.
(B–D) Z-transformed expression values for Keap-1 inhibition signature genes also differentially expressed in GSE59071 (Vanhove et al., 2015) CD samples (B) or UC samples (C). Z-transformed expression values for Keap-1 inhibition signature genes also differentially expressed in GSE38713 (Planell et al., 2013) (D).
DISCUSSION
Here, we demonstrate a reciprocal relationship between antioxidant and inflammatory responses. Our findings indicate inflammatory stimuli induce oxidative stress via RNIs, and phagocytes adapt to this insult by eliciting an antioxidant response to dampen inflammatory cytokine production. Several lines of evidence demonstrate the critical role for RNI production in host defense against intracellular pathogens (Karupiah et al., 1993; MacMicking et al., 1995, 1997; Stuehr and Marletta, 1985; Wei et al., 1995), and emerging evidence indicates a broader role for iNOS in shaping the microbiome. In particular, iNOS was shown to be selectively required for the outgrowth of commensal Enterobacteriaceae that exploit nitrate respiration during intestinal inflammation (Winter et al., 2013). Conversely, commensals produce electrophilic metabolites that engage the host antioxidant response. Peptostreptococcus rusellii reduce susceptibility to epithelial injury in mice and contain a unique biosynthetic gene cluster enabling production of the tryptophan metabolite, indoleacrylic acid (Wlodarska et al., 2017). This electrophilic compound directly engages the Keap1/Nrf2-dependent antioxidant response to limit inflammatory cytokine production in macrophages (Wlodarska et al., 2017).
Beyond its impact on commensal and pathogenic microbes, iNOS expression during inflammation delivers a toxic insult to the host. Previous studies documented a significant increase of iNOS expression in inflamed intestinal tissue of IBD patients (Hofseth et al., 2003). These observations are consistent with our transcriptomic analyses, which reveal an oxidative stress signature in intestinal biopsies from IBD patients (Figure 5). In chronic inflammation, the antioxidant response can mitigate tissue damage but may be insufficient to resolve inflammation on its own. In fact, chronic inflammation and RNI exposure have been shown to drive tumorigenesis in mouse models of colon cancer (Meira et al., 2008; Wang et al., 2017). Thus, the antioxidant response induced by RNIs plays an important protective role against chronic inflammation. The Keap1/Nrf2 axis also controls cellular adaptation to oxidative stress in cancer, neurodegenerative diseases, and liver disease (Chen et al., 2017; Eriksson et al., 2015; Kerr et al., 2017; Prigge et al., 2017; Romero et al., 2017).
The results of our genetic screen provide insight into the immunoregulatory function of the antioxidant response (Figure 1). Specifically, we demonstrate that TLR stimulation in dendritic cells induces the Keap1-mediated antioxidant response in an RNI-dependent manner (Figure 2) and that Keap1 knockdown or small-molecule inhibition of the Keap1-Nrf2 interaction was sufficient to inhibit TLR-driven cytokine production (Figure 4). These findings provide mechanistic insight into recent studies evaluating the role of Keap1 and Nrf2 in models of pathological inflammation (Suzuki and Yamamoto, 2017). Induction of Nrf2 by systemic Keap1 knockdown ameliorated autoimmune symptoms in Treg-deficient Scurfy mice (Suzuki et al., 2017). Nrf2-deficient mice were shown to be highly susceptible to lethal endotoxin challenge and exhibited elevated serum cytokine levels relative to WT mice (Thimmulappa et al., 2006). In a model of sepsis induced by cecal ligation puncture, myeloid-specific Nrf2 deletion exacerbated, and myeloid-specific Keap1 deletion protected against, sepsis (Kong et al., 2011). Likewise, Nrf2-deficient mice exhibited more severe intestinal pathology compared to WT in the dextran sulfate sodium (DSS) model of epithelial injury (Khor et al., 2006). These findings are consistent with the notion that the antioxidant response initiated by Keap1 dissociation from Nrf2 reduces inflammatory responses in vivo. In fact, disruption of the Keap1-Nrf2 complex with the electrophilic compound dimethyl fumarate or the protein-protein interaction inhibitor CPUY192018 exhibited protective effects in DSS colitis (Liu et al., 2016; Lu et al., 2016). Our work illuminates the molecular functions of the antioxidant response by defining the Keap1-associated LPS response and identifying Prdx5 as a key component of the antioxidant response required for suppressing TLR signaling in dendritic cells. Ectopic expression of Prdx5 in RAW 264.7 macrophages was sufficient to reduce LPS-induced RNI and IL-6 production (Figure S5). These findings suggest that Prdx5 is an important anti-inflammatory Nrf2 target gene that may act in parallel with a previously described mechanism in which Nrf2 represses IL-6 transcription by directly binding its promoter (Kobayashi et al., 2016).
Prdx5 is an atypical 2-Cys peroxiredoxin family member that functions within the thioredoxin system to scavenge ROS, such as superoxide and hydrogen peroxide (Rhee and Kil, 2015). We show that Prdx5−/− dendritic cells exhibit elevated oxidative stress that is ameliorated by iNOS inhibition (Figure 4). Further, increased NO production in Prdx5−/− dendritic cells correlated with cellular redox state (Figure 4). Peroxiredoxins have been shown to accept SNO modifications on their reactive cysteines (Engelman et al., 2013), and the thioredoxin system has emerged as an important regulator in the cellular catabolism of SNO during nitrosative stress (Kronenfeld et al., 2015). Elevated NO has been previously shown to function as a signaling modifier that generates SNO modifications on key signaling molecules. For example, RNIs inhibit NLRP3 inflammasome activation (Mao et al., 2013) by thiol nitrosation (Mishra et al., 2013), resulting in reduced IL-1b secretion. In addition, RNIs are particularly effective at inhibiting enzymes with catalytic cysteines, such as phosphatases. Oxidation of the Ptpn1 (Ptp1b) catalytic cysteine reduces its tyrosine phosphatase activity (Salmeen et al., 2003; van Montfort et al., 2003). This cysteine has been subsequently shown to accept SNO modifications from endogenously produced cellular RNIs (Hsu and Meng, 2010; Hsu et al., 2016). Studies in macrophages identified Ptpn1 as a critical negative regulator of TLR signaling (Xu et al., 2008), and we identified Ptpn1 as a component of the antioxidant response (Table S2). Together, our findings support a model in which (1) TLR signaling induces RNIs and cytokines; (2) RNIs inhibit phosphatases to potentiate TLR signaling; (3) RNIs act on Keap1 to induce the Nrf2-mediated antioxidant response; (4) antioxidant proteins, such as Prdx5, scavenge RNIs; and finally (5) phosphatases resume inhibition of the TLR pathway when RNI-mediated negative feedback is relieved.
EXPERIMENTAL PROCEDURES
NO Screen
The NO screen was performed using bone marrow harvested from C57/BL6 mice. Bone marrow was seeded at a concentration of 6 × 104 cells in 50 μL media (DMEM with 10% fetal bovine serum [FBS], GlutaMAX 1 × [Thermo Fisher Scientific], minimal essential medium (MEM) non-essential amino acids 1 × [Thermo Fisher Scientific], penicillin-streptomycin 100 U/mL and 100 μg/mL, respectively [Thermo Fisher Scientific], and GM-CSF [4% TOPO-derived media]) in a 96-well, flat, clear bottom, black plate. On day 2 after seeding, cells were transduced with 50 μL shRNA lentivirus. On day 5, cells were selected with 3 μg/mL puromycin (InvivoGen) for an additional 2 days. On day 7, cells were stimulated overnight: media was replaced with 200 μL media supplemented with 10 ng/mL IFN-γ (Peprotech) and 50 ng/mL LPS (InvivoGen). Supernatant was collected for Griess assay. Remaining supernatant was discarded, and adherent cells in 96-well plate were collected for cell viability assay using Alamar blue.
Secondary Cytokine Screen
Hits from the NO screen were knocked down in BMDCs as described above and stimulated with LPS and IFN-γ. Cells were collected, and total RNA was extracted (QIAGEN RNeasy Mini kit). Cytokine transcriptional analysis was performed as previously described (Graham et al., 2015). Briefly, total RNA was reverse transcribed using the qScript Supermix (VWR), and target specific pre-amplification was performed by standard PCR. Custom primers specific for cytokine genes were utilized for pre-amplification and followed by qPCR (EvaGreen Supermix; Bio-Rad). Transcript quantification was performed with the BioMark HD system (Fluidigm).
Griess Assay
Supernatants from stimulated cells were collected to measure nitrite concentrations using the Griess assay. Griess reagent was made by mixing 0.1% N-1-naphthyl-ethylenediamine HCl with 1.32% sulfanilamide/60% acetic acid (all reagents from Sigma-Aldrich) at a 1:1 ratio. Subsequently, 75 μL of supernatant was mixed with 75 μL Griess reagent in each well of a 96-well, flat, clear bottom, black plate. Absorbance at 520 nm was read using the BioTek SynergyH4 microplate reader.
RNA Sequencing
Knockdown of the indicated genes was performed as described above in BMDCs, where 2 independent shRNA sequences targeting a given gene were considered biological replicates and technical duplicates were performed for each biological replicate. After a 6-hr stimulation with LPS and IFN-γ, BMDCs were collected and lysed. RNA sequencing was performed as previously described (Shishkin et al., 2015). Briefly, total RNA was purified, fragmented, and depleted of genomic DNA. Barcoded adaptors were ligated to individual samples prior to sample pooling. Next, cDNA synthesis, mRNA degradation, and 3’ adaptor ligation were performed to create Illumina cDNA libraries. After amplification with sequencing adaptors, RNA sequencing analysis was performed.
Metabolic Profiling
Knockdown of indicated genes was performed as described above in BMDCs. Cells were collected after overnight stimulation with LPS and IFN-γ. Metabolic profiling was performed as previously described (Graham et al., 2015). Briefly, polar metabolites were profiled in the positive ion MS mode using a liquid chromatography (LC) system coupled to a 4000 QTRAP mass spectrometer (AB SCIEX) equipped with an electrospray ionization source. Analyses of polar metabolites using negative ion mode MS were performed using an ACQUITY UPLC (Waters) coupled to a 5500 QTRAP triple quadrupole mass spectrometer (AB SCIEX). Raw liquid chromatography-mass spectrometry (LC-MS) data were processed using MultiQuant software and used to integrate chromatographic peaks. The processed data were manually reviewed for quality of integration and compared against known standards to confirm metabolite identities.
Immunoprecipitation/Mass Spectrometry
Prdx5 was ectopically expressed in HEK293T cells as previously described (Graham et al., 2016). Briefly, cDNA constructs were cloned as PCR products into pCMV (Invitrogen) using Gibson assembly. HEK293T cells (ATCC) were transfected with Lipofectamine 2000 (Thermo Fisher Scientific) as recommended by the manufacturer. Transfected cells were lysed in 1% Triton X-100, PBS (pH 7.4). V5-Prdx5 was immunoprecipitated with anti-V5 magnetic agarose beads (MBL International) with rotation at 4°C for 4 hr. Samples were trypsin digested on beads, desalted, and labeled with iTRAQ reagents according the manufacturer’s instructions (AB Sciex). Reconstituted peptides were separated on an online nanoflow EASY-nLC 1000 UHPLC system (Thermo Fisher Scientific) and analyzed on a benchtop Orbitrap Q Exactive mass spectrometer (Thermo Fisher Scientific). Mass spectra were processed using the Spectrum Mill software (Agilent Technologies), and peptide identifications were searched against the human Uniprot database.
MitoSOX Assay
To measure mitochondrial ROS production, we performed the MitoSOX assay. BMDCs were plated at a concentration of 3.0 × 105/well in 300 μL media in a 48-well, flat tissue, culture-treated plate. Media was supplemented with stimuli, and cells were stimulated overnight as noted. The following day, cells were washed with PBS, and 5 μM MitoSOX(C43H43N3IP; Thermo Fisher Scientific) in phenol-free media was added for 20 min at 37°C. Cells were washed twice with cold PBS (Sigma-Aldrich), incubated with 100 μL TrypLE express enzyme buffer (Thermo Fisher Scientific); 200 μL media was added, and cells were scraped off and collected. BMDCs were analyzed using the Accuri flow cytometer (BD Biosciences).
DCFDA
The DCFDA assay was performed to measure overall cellular oxidative stress. BMDCs were plated at a concentration of 3.0 × 105/ well in 300 μL media in a 48-well, flat tissue, culture-treated plate. Media was supplemented with stimuli, and cells were cultured overnight as noted. The following day, cells were washed with PBS (Sigma-Aldrich), and 5 μM CM-H2DCFDA (Thermo Fisher Scientific) in phenol-free media was added to the cells for 20 min at 37°C. Cells were washed twice with cold PBS 1 ×, incubated with 100 μL TrypLE express enzyme buffer (Thermo Fisher Scientific); 200 μL media was added, and cells were scraped off and collected. BMDCs were analyzed using the Cytoflex S (Beckman Coulter).
Cytometric Bead Array Assay
To measure cytokines from serum and supernatant samples, the cytometric bead assay Flex Set (BD Biosciences) was used according to the manufacturer’s instructions. Cytokines measured included IL-6, IL-12 p40, IL-10, TNF-α, and MCP1. Samples were run on the Cytoflex S (Beckman Coulter) and normalized to cell count using CyQUANT (Thermo Fisher Scientific).
Statistical Methods
Statistical procedures specific for each technology are described above. For comparisons between two groups, unpaired Student’s t tests (two-tailed) or non-parametric tests were employed. See figure legends for additional details.
Supplementary Material
Highlights.
Phagocyte NO production induces a Keap1-mediated antioxidant response
The antioxidant response acts as a negative feedback regulator of inflammation
The antioxidant protein Prdx5 is required to suppress cytokine production
Prdx5−/− dendritic cells exhibit elevated NO production and oxidative stress
ACKNOWLEDGMENTS
This work was funded by The Leona M. and Harry B. Helmsley Charitable Trust and NIH grants (DK043351 and DK097485) to R.J.X. G.J.J. was supported by a Ford Foundation Fellowship and an NIH T32 grant (2T32DK007191-42). We thank Moran Yassour, Isabel Latorre, Elizabeth Creasey, Natalia B. Nedelsky, and Theresa Reimels for technical expertise.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the data reported in this paper is GEO: GSE115553.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, five figures, and four tables and can be found with this article online at https://doi.org/10.1016Zj.celrep.2018.06.081.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A, et al. (2011). Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet 43, 246–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstey NM, Weinberg JB, Hassanali MY, Mwaikambo ED, Manyenga D, Misukonis MA, Arnelle DR, Hollis D, McDonald MI, and Granger DL (1996). Nitric oxide in Tanzanian children with malaria: inverse relationship between malaria severity and nitric oxide production/nitric oxide synthase type 2 expression. J. Exp. Med. 184, 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold WP, Mittal CK, Katsuki S, and Murad F (1977). Nitric oxide activates guanylate cyclase and increases guanosine 3’:5’-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. USA 74, 3203–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan C (2015). Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol. 36, 161–178. [DOI] [PubMed] [Google Scholar]
- Buckley BJ, Marshall ZM, and Whorton AR (2003). Nitric oxid estimulates Nrf2 nuclear translocation in vascular endothelium. Biochem. Biophys. Res. Commun. 307, 973–979. [DOI] [PubMed] [Google Scholar]
- Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R, and Gu W (2017). NRF2 is a major target of ARF in p53-independent tumor suppression. Mol. Cell 68, 224–232.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhakshinamoorthy S, and Porter AG (2004). Nitric oxide-induced transcriptional up-regulation of protective genes by Nrf2 via the antioxidant response element counteracts apoptosis of neuroblastoma cells. J. Biol. Chem. 279, 20096–20107. [DOI] [PubMed] [Google Scholar]
- Dhillon SS, Mastropaolo LA, Murchie R, Griffiths C, Thöni C, Elkadri A, Xu W, Mack A, Walters T, Guo C, et al. (2014). Higher activity of the inducible nitric oxide synthase contributes to very early onset inflammatory bowel disease. Clin. Transl. Gastroenterol. 5, e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman R, Weisman-Shomer P, Ziv T, Xu J, Arnér ESJ, and Benhar M (2013). Multilevel regulation of 2-Cys peroxiredoxin reaction cycle by S-nitrosylation. J. Biol. Chem. 288, 11312–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson S, Prigge JR, Talago EA, Arnér ESJ, and Schmidt EE (2015). Dietary methionine can sustain cytosolic redox homeostasis in the mouse liver. Nat. Commun. 6, 6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts B, Amiel E, van der Windt GJW, Freitas TC, Chott R, Yarasheski KE, Pearce EL, and Pearce EJ (2012). Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120, 1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlik M, Reutterer B, Schindler C, Greten F, Vogl C, Müller M, and Decker T (2010). Nonconventional initiation complex assembly by STAT and NF-kappaB transcription factors regulates nitric oxide synthase expression. Immunity 33, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott RF, and Zawadzki JV (1980). The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288, 373–376. [DOI] [PubMed] [Google Scholar]
- Gao J, Morrison DC, Parmely TJ, Russell SW, and Murphy WJ (1997). An interferon-gamma-activated site (GAS) is necessary for full expression of the mouse iNOS gene in response to interferon-gamma and lipopolysaccharide. J. Biol. Chem. 272, 1226–1230. [DOI] [PubMed] [Google Scholar]
- Garvey EP, Oplinger JA, Furffne ES, Kiff RJ, Laszlo F, Whittle BJ, and Knowles RG (1997). 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 272, 4959–4963. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Donquiles C, Alonso-Molero J, Fernandez-Villa T, Vilorio-Marqués L, Molina AJ, and Martin V (2017). The NRF2 transcription factor plays a dual role in colorectal cancer: A systematic review. PLoS ONE 12, e0177549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DB, Becker CE, Doan A, Goel G, Villablanca EJ, Knights D, Mok A, Ng ACY, Doench JG, Root DE, et al. (2015). Functional genomics identifies negative regulatory nodes controlling phagocyte oxidative burst. Nat. Commun. 6, 7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DB, Lefkovith A, Deelen P, de Klein N, Varma M, Boroughs A, Desch AN, Ng ACY, Guzman G, Schenone M, et al. (2016). TMEM258 is a component of the oligosaccharyltransferase complex controlling ER stress and intestinal inflammation. Cell Rep. 17, 2955–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins KE, Joy S, Delhove JMKM, Kotiadis VN, Fernandez E, Fitzpatrick LM, Whiteford JR, King PJ, Bolanos JP, Duchen MR, et al. (2016). NRF2 orchestrates the metabolic shift during induced pluripotent stem cell reprogramming. Cell Rep. 14, 1883–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs JB Jr., Taintor RR, and Vavrin Z (1987). Macrophage cytotoxicity: role for L-arginine deiminase and imino nitrogen oxidation to nitrite. Science 235, 473–476. [DOI] [PubMed] [Google Scholar]
- Hobbs MR, Udhayakumar V, Levesque MC, Booth J, Roberts JM, Tkachuk AN, Pole A, Coon H, Kariuki S, Nahlen BL, et al. (2002). A new NOS2 promoter polymorphism associated with increased nitric oxide production and protection from severe malaria in Tanzanian and Kenyan children. Lancet 360, 1468–1475. [DOI] [PubMed] [Google Scholar]
- Hofseth LJ, Saito S, Hussain SP, Espey MG, Miranda KM, Araki Y, Jhappan C, Higashimoto Y, He P, Linke SP, et al. (2003). Nitric oxide induced cellular stress and p53 activation in chronic inflammation. Proc. Natl. Acad. Sci. USA 100, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M-F, and Meng T-C (2010). Enhancement of insulin responsiveness by nitric oxide-mediated inactivation of protein-tyrosine phosphatases. J. Biol. Chem. 285, 7919–7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M-F, Pan K-T, Chang F-Y, Khoo K-H, Urlaub H, Cheng C-F, Chang G-D, Haj FG, and Meng T-C (2016). S-nitrosylation of endogenous protein tyrosine phosphatases in endothelial insulin signaling. Free Radic. Biol. Med. 99, 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Magesh S, Chen L, Wang L, Lewis TA, Chen Y, Khodier C, Inoyama D, Beamer LJ, Emge TJ, et al. (2013). Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med. Chem. Lett. 23, 3039–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ, Buga GM, Wood KS, Byrns RE, and Chaudhuri G (1987). Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 84, 9265–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et al. (1997). An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236, 313–322. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, and Yamamoto M (1999). Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karupiah G, Xie QW, Buller RM, Nathan C, Duarte C, and MacMicking JD (1993). Inhibition of viral replication by interferon-gamma-induced nitric oxide synthase. Science 261, 1445–1448. [DOI] [PubMed] [Google Scholar]
- Kerr F, Sofola-Adesakin O, Ivanov DK, Gatliff J, Gomez Perez-Nievas B, Bertrand HC, Martinez P, Callard R, Snoeren I, Cocheme HM, et al. (2017). Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet. 13, e1006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor TO, Huang M-T, Kwon KH, Chan JY, Reddy BS, and Kong A-N (2006). Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 66, 11580–11584. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang M-I, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, and Yamamoto M (2004). Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Kang M-I, Watai Y, Tong KI, Shibata T, Uchida K, and Yamamoto M (2006). Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 26, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, and Yamamoto M (2016). Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 7, 11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Thimmulappa R, Craciun F, Harvey C, Singh A, Kombairaju P, Reddy SP, Remick D, and Biswal S (2011). Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am. J. Respir. Crit. Care Med. 184, 928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, and Pearce EJ (2010). Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenfeld G, Engelman R, Weisman-Shomer P, Atlas D, and Benhar M (2015). Thioredoxin-mimetic peptides as catalysts of S-denitrosylation and anti-nitrosative stress agents. Free Radic. Biol. Med. 79, 138–146. [DOI] [PubMed] [Google Scholar]
- Li C-Q, Kim MY, Godoy LC, Thiantanawat A, Trudel LJ, and Wogan GN (2009). Nitric oxide activation of Keap1/Nrf2 signaling in human colon carcinoma cells. Proc. Natl. Acad. Sci. USA 106, 14547–14551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva ALN, Mineo TWP, Gutierrez FRS, Bellio M, Bortoluci KR, Flavell RA, et al. (2013). Inflammasome-derived IL-1β production induces nitric oxidemediated resistance to Leishmania. Nat. Med. 19, 909–915. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhou W, Zhang X, Lu P, Du Q, Tao L, Ding Y, Wang Y, and Hu R (2016). Dimethyl fumarate ameliorates dextran sulfate sodium-induced murine experimental colitis by activating Nrf2 and suppressing NLRP3 inflammasome activation. Biochem. Pharmacol. 112, 37–49. [DOI] [PubMed] [Google Scholar]
- Lowenstein CJ, Glatt CS, Bredt DS, and Snyder SH (1992). Cloned and expressed macrophage nitric oxide synthase contrasts with the brain enzyme. Proc. Natl. Acad. Sci. USA 89, 6711–6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein CJ, Alley EW, Raval P, Snowman AM, Snyder SH, Russell SW, and Murphy WJ (1993). Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc. Natl. Acad. Sci. USA 90, 9730–9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M-C, Ji J-A, Jiang Y-L, Chen Z-Y, Yuan Z-W, You Q-D, and Jiang Z-Y (2016). An inhibitor of the Keap1-Nrf2 protein-protein interaction protects NCM460 colonic cells and alleviates experimental colitis. Sci. Rep. 6, 26585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking JD (2012). Interferon-inducible effector mechanisms in cellautonomous immunity. Nat. Rev. Immunol. 12, 367–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, et al. (1995). Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 81, 641–650. [DOI] [PubMed] [Google Scholar]
- MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, and Nathan CF (1997). Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. USA 94, 5243–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, and Biswal S (2010). Global mapping of binding sites for Nrf2 identifies novel targets in cell survival responsethrough ChIP-seq profiling and networkanalysis. Nucleic Acids Res. 38, 5718–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, He Z, Zeng Y, Hu Y, Sun S, et al. (2013). Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 23, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Nathan C, and Xie QW (1994). Role of interferon regulatoryfactor 1 in induction of nitric oxide synthase. J. Exp. Med. 180, 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meira LB, Bugni JM, Green SL, Lee C-W, Pang B, Borenshtein D, Rickman BH, Rogers AB, Moroski-Erkul CA, McFaline JL, et al. (2008). DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Invest. 118, 2516–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra BB, Rathinam VAK, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, and Sassetti CM (2013). Nitric oxide controls the immunopathology oftuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1 β. Nat. Immunol. 14, 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moresco EMY, LaVine D, and Beutler B (2011). Toll-like receptors. Curr. Biol. 21, R488–R493. [DOI] [PubMed] [Google Scholar]
- Nathan C, and Cunningham-Bussel A (2013). Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 13, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A, and Xavier RJ (2011). Leucine-rich repeat (LRR) proteins: integrators of pattern recognition and signaling in immunity. Autophagy 7, 1082–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Sherratt PJ, Nioi P, Yang CS, and Pickett CB (2005). Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem. 280, 32485–32492. [DOI] [PubMed] [Google Scholar]
- Okabe Y, and Medzhitov R (2016). Tissue biology perspective on macrophages. Nat. Immunol. 17, 9–17. [DOI] [PubMed] [Google Scholar]
- Orvedahl A, Sumpter R Jr., Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, et al. (2011). Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ, Przybylski D, Platt RJ, Tirosh I, Sanjana NE, et al. (2015). A genomewide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 162, 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planell N, Lozano JJ, Mora-Buch R, Masamunt MC, Jimeno M, Ordás I, Esteller M, Ricart E, Piqué JM, Panés J, and Salas A (2013). Transcriptional analysis of the intestinal mucosa of patients with ulcerative colitis in remission reveals lasting epithelial cell alterations. Gut 62, 967–976. [DOI] [PubMed] [Google Scholar]
- Prigge JR, Coppo L, Martin SS, Ogata F, Miller CG, Bruschwein MD, Orlicky DJ, Shearn CT, Kundert JA, Lytchier J, et al. (2017). Hepatocyte hyperproliferation upon liver-specific co-disruption of thioredoxin-1, thioredoxin reductase-1, and glutathione reductase. Cell Rep. 19, 2771–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG, and Kil IS (2017). Multiplefunctionsand regulation ofmammalian peroxiredoxins. Annu. Rev. Biochem. 86, 749–775. [DOI] [PubMed] [Google Scholar]
- Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sanchez-Rivera FJ, et al. (2017). Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 23, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmeen A, Andersen JN, Myers MP, Meng T-C, Hinks JA, Tonks NK, and Barford D (2003). Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423, 769–773. [DOI] [PubMed] [Google Scholar]
- Shishkin AA, Giannoukos G, Kucukural A, Ciulla D, Busby M, Surka C, Chen J, Bhattacharyya RP, Rudy RF, Patel MM, et al. (2015). Simultaneousgeneration of many RNA-seq libraries in a single reaction. Nat. Methods 12, 323–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuehr DJ (1999). Mammalian nitricoxidesynthases. Biochim. Biophys.Acta 1411, 217–230. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ, and Marletta MA (1985). Mammalian nitrate biosynthesis: mouse macrophages produce nitrite and nitrate in response to Escherichia coli lipopolysaccharide. Proc. Natl. Acad. Sci. USA 82, 7738–7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, and Yamamoto M (2017). Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J. Biol. Chem. 292, 16817–16824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Murakami S, Biswal SS, Sakaguchi S, Harigae H, Yamamoto M, and Motohashi H (2017). Systemic activation of NRF2 alleviates lethal autoimmune inflammation in scurfy mice. Mol. Cell. Biol. 37, e00063–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, and Biswal S (2006). Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest. 116,984–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Montfort RLM, Congreve M, Tisi D, Carr R, and Jhoti H (2003). Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 423, 773–777. [DOI] [PubMed] [Google Scholar]
- Vanhove W, Peeters PM, Staelens D, Schraenen A, Van der Goten J, Cleynen I, De Schepper S, Van Lommel L, Reynaert NL, Schuit F, et al. (2015). Strong upregulation ofAIM2 and IFI16 inflammasomes in the mucosa of patients with active inflammatory bowel disease. Inflamm. Bowel Dis. 21, 2673–2682. [DOI] [PubMed] [Google Scholar]
- Wang C, Gong G, Sheh A, Muthupalani S, Bryant EM, Puglisi DA, Holcombe H, Conaway EA, Parry NAP, Bakthavatchalu V, et al. (2017). Interleukin-22 drives nitric oxide-dependent DNA damage and dysplasia in a murine model of colitis-associated cancer. Mucosal Immunol. 10, 1504–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, Xu D, Müller W, Moncada S, and Liew FY (1995). Altered immune responses in mice lacking inducible nitric oxide synthase. Nature 375, 408–411. [DOI] [PubMed] [Google Scholar]
- Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, Laughlin RC, Gomez G, Wu J, Lawhon SD, et al. (2013). Host derived nitrate boosts growth of E. coli in the inflamed gut. Science 339, 708–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarska M, Luo C, Kolde R, d’Hennezel E, Annand JW, Heim CE, Krastel P, Schmitt EK, Omar AS, Creasey EA, et al. (2017). Indoleacrylic acid produced by commensal peptostreptococcus species suppresses inflammation. Cell Host Microbe 22, 25–37.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Ding A, Troso T, and Nathan C (1992). Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science 256, 225–228. [DOI] [PubMed] [Google Scholar]
- Xie QW, Whisnant R, and Nathan C (1993). Promoterofthe mouse geneen coding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J. Exp. Med. 177,1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie QW, Kashiwabara Y, and Nathan C (1994). Role oftranscription factor NF-kappa B/Rel in induction of nitric oxide synthase. J. Biol. Chem. 269,4705–4708. [PubMed] [Google Scholar]
- Xu H, An H, Hou J, Han C, Wang P, Yu Y, and Cao X (2008). Phosphatase PTP1B negatively regulates MyD88- and TRIF-dependent proinflammatory cytokine and type I interferon production in TLR-triggered macrophages. Mol. Immunol. 45, 3545–3552. [DOI] [PubMed] [Google Scholar]
- Zhang DD, and Hannink M (2003). Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 23, 8137–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.