

Abstract
Cooperation between VEGFR2 and integrin αVβ3 is critical for neovascularization in wound healing, cardiovascular ischemic diseases, ocular diseases, and tumor angiogenesis. In the present study, we developed a rule- based computational model to investigate the potential mechanism by which the Src-induced integrin association with VEGFR2 enhances VEGFR2 activation. Simulations demonstrated that the main function of integrin is to reduce the degradation of VEGFR2 and hence stabilize the activation signal. In addition, receptor synthesis rate and recruitment from internal compartment were found to be sensitive determinants of the activation state of VEGFR2. The model was then applied to simulate the effect of integrin-binding peptides such as tumstatin and cilengitide on VEGFR2 signaling. Further, computational modeling proposed potential molecular mechanisms for the angiogenesis-modulating activity of other integrin-binding peptides. The model highlights the complexity of the crosstalk between αVβ3 integrin and VEGFR2 and the necessity of utilizing models to elucidate potential mechanisms in angiogenesis-modulating peptide therapy.
Keywords: VEGF, integrin signaling, computational model, mathematical model, systems biology
1. Introduction
Angiogenesis is the growth of new blood vessels from the preexisting vasculature, a process critical for both health and disease (Carmeliet, 2005). It plays crucial role in conditions such as tumor vascularization and metastasis (Folkman, 2002), wound healing (DiPietro, 2013), and diabetes induced ocular neovascularization and macular degeneration (Campochiaro, 2013). in diseases such as peripheral arterial disease (PAD) disruption of angiogenesis may be a factor contributing to the lack of blood flow to lower extremities (Annex, 2013). Angiogenesis is triggered by the binding of angiogenic growth factors such as vascular endothelial growth factor (VEGF) to their receptors in cooperation with transmembrane receptors known as integrins that facilitate cell-extracellular matrix (ECM) adhesion. VEGF and its cognate receptor vascular endothelial growth factor receptor 2 (VEGFR2) on vascular endothelial cells are the critical components of the angiogenic response. Binding of VEGF to VEGFR2 in cooperation with integrin signaling promotes vascular endothelial cell survival, motility, and proliferation, processes critical for the angiogenic response. There are several integrin heterodimers expressed on endothelial cells including αVβ3, aVp5, and α5β1. Among these, αVβ3 is the most prominent in angiogenesis with its expression upregulated in endothelial cells during wound healing, inflammation, and tumor angiogenesis (Shattil and Ginsberg, 1997; Stupack and Cheresh, 2004). Indeed, blocking αVβ3 with monoclonal antibodies or RGD peptides efficiently inhibits angiogenesis in different model systems (Cai and Chen, 2006; D’Andrea et al., 2006). Physical association between αVβ3 and various growth factor receptors such as platelet derived growth factor receptor (PDGFR), insulin growth factor receptor (IGFR), and VEGFR2 have been observed experimentally suggesting that αVβ3 and growth factor receptors are part of the same macromolecular signaling complex (Schneller et al., 1997; Soldi et al., 1999; Woodard et al., 1998). Given the significance of VEGF signaling in pathological angiogenesis (Nagy et al., 2007), the synergistic cooperation and physical association of αVβ3 and VEGFR2 is of particular interest (Somanath et al., 2009). Recent experimental evidence suggests the critical role of two tyrosine residues namely Y747 and Y759 on the β3 subunit of αVβ3 in pathological angiogenesis. Binding of αVβ3 to its ECM ligands leads to phosphorylation of these residues that is augmented by exposure to VEGF (Mahabeleshwar et al., 2006; West et al., 2012). Further, the mutant β3 knock-in mice models where the two tyrosines are changed to phenylalanines (Y747D/Y759F or DiYF strains) exhibit impaired angiogenic response (Mahabeleshwar et al., 2006; Mahabeleshwar et al., 2007). Subsequent investigation identified the kinase Src as the mediator between VEGFR2 and αVβ3 that phosphorylates the β3 subunit that results in cross-activation of the two receptors (Mahabeleshwar et al., 2007). Given this experimental evidence for the complex cross-interaction between the two receptors, it is not clear what mechanisms at the receptor level contribute to the enhancement of VEGFR2 activation signal upon its enhanced interaction with phosphorylated αVβ3 (pInt). Computational modeling may aid in clarifying and elucidating molecular mechanisms involved in augmenting VEGFR2 signaling by the integrin and offer new avenues of exploration for clinical applications. In particular, the development of integrin-biding peptides have already shown promise in anti-angiogenic therapeutic interventions (Barbhuiya et al., 2017; Buerkle et al., 2002; Lee et al., 2014; Lima e Silva et al., 2017; Maeshima et al., 2002; Maeshima et al., 2001; Rosea et al., 2014).
There are several computational models that focus solely on integrin clustering and downstream signaling (Cirit et al., 2010; Welf et al., 2012; Yee et al., 2008). These models however do not consider crosstalk between integrins and other receptor types. To our knowledge, there are no kinetic, ordinary differential equations-based models investigating the crosstalk between integrin signaling and receptor tyrosine kinases (RTK). The detailed model for the crosstalk between integrins and RTKs developed by Bauer et al. (Bauer et al., 2010) is a receptors. Here we develop a detailed kinetic rule-based computational model for the interaction of VEGFR2 with αVβ3 based on the experimental observations in endothelial cells. We incorporate Src activation downstream of VEGFR2 along with receptor association, internalization, and degradation. The use of rule-based approach implemented in the programming environment BioNetGen, allows for the incorporation in modular form of the available experimental evidence while taking into account the combinatorial complexity inherent in systems involving multi-receptor complex formation (Faeder et al., 2009; Hlavacek et al., 2006). This approach has been previously applied in the construction of detailed models of VEGF signaling (Bazzazi and Popel, 2017; Bazzazi et al., 2017; Bazzazi et al., 2018; Rohrs et al., 2016). We then utilized the model to investigate potential mechanisms by which αVβ3 association with VEGFR2 stabilizes VEGFR2 activation. We also applied the predictive model to perform simulations with classes of peptides inhibiting angiogenesis. Simulations demonstrate the complexity of the synergistic interaction between VEGFR2 and αVβ3 highlighting the crucial role of computational approaches in mechanism discovery and therapeutics.
2. Methods
The rules for the model were implemented in the BioNetGen environment(Faeder et al., 2009) which then automatically generated the reaction network and outputted the model into a MATLAB C programming language file (MEX). The supplementary methods section describes the species in the model and the biological rules involved with detailed description of the model.
The system of ordinary differential equations (ODEs) was simulated within the MATLAB (MathWorks, Natick, MA; 2015) environment using the SUNDIALS suite (Hindmarsh et al., 2005). The final model in the absence of the peptide consisted of 30 molecular species (number of differential equations) and 107 biochemical reactions. With peptide present, the model consisted of 55 species and 236 reactions. Global sensitivity analysis was performed using partial rank correlation coefficient (PRCC) (Marino et al., 2008). The parameter values were randomly chosen from a uniform distribution within a range 0.1 x fitted_value ≤ p ≤ 10 × fitted_value. The Systems Biology Markup Language (SBML) files are also provided as supplement.
For parameter fitting (parameters are listed in Table S1), we applied a direct search algorithm implemented in the MATLAB function patternsearch as part of the global optimization toolbox. All the pieces of data, including the surface and total receptor levels and downstream activation were fitted simultaneously in MATLAB. Data from the western blot images were extracted using the software imageJ (Schneider et al., 2012).
3. Results
3.1. Model construction
The rules implemented in BioNetGen generated chemical reactions based on the given biological rules for the interaction and activation of various species in the system. The rules and the species were derived according to the experimental evidence for the crosstalk between VEGFR2 and αVβ3 in (Mahabeleshwar et al., 2006; Mahabeleshwar et al., 2007; Somanath et al., 2009; West et al., 2012). Figure 1 summarizes the seed species and the main rules included in the model. As shown in Fig. 1A αVβ3 has a single binding domain, and a tyrosine modification site (Υβ3) on the cytoplasmic side. Notice that for simplicity αVβ3 is considered as a single agent and the two experimentally relevant tyrosine sites Y747 and Y759 (Mahabeleshwar et al., 2006) are lumped together as a single site called Υβ3. VEGFR2 and αVβ3 are assumed to be part of the same macromolecular complex denoted by the line connecting αVβ3 to VEGFR2 in Fig. 1A. There is indeed evidence for the physical interaction between the two receptors in non-phosphorylated state that is enhanced upon phosphorylation of β3 (West et al., 2012). VEGFR2 has a single binding site for VEGF, a tyrosine modification site (Y1175), and a binding domain (bd) for binding the phosphorylated form of Υβ3. The binding of VEGF to VEGFR2 is described by the rule in Fig. 1B as a one-to-one reaction based on the experimentally derived effective on and off kinetics (Stefanini et al., 2008; Yen et al., 2011). The rule for VEGFR2 modification on tyrosine 1175 (Y1175) is illustrated by Fig. 1C. Internalization, degradation, and recycling of the receptor complex are shown in Fig. 1D. Internalization also occurs for free VEGFR2. Src is activated downstream of phosphorylated VEGFR2 (for both surface-bound and internalized receptors) and by the autophosphorylation of tyrosine 419 (Y419) (Boggon and Eck, 2004). Dephosphorylation of Src is also included (Fig. 1E). Integrin activation is mediated by phosphorylation by Src as shown in Fig. 1F. Once phosphorylated on Υβ3, αVβ3 associates with VEGFR2 on a separate binding domain (see Fig. 1A) as in Fig. 1G. Fig. 1H illustrates the summary of the Src- mediated interaction between VEGFR2 and αVβ3. The values of the parameters in the model are included in supplementary Table 1. The final model consists of 30 species and 107 biochemical reactions with no peptide present, and 55 species and 236 reactions with peptide present. The list of generated species and reactions are included in the supplementary material along with The SBML file corresponding to the model is also provided in the supplement.
Figure 1. Rules and species for the interaction of VEGFR2 and αVβ3 integrin.
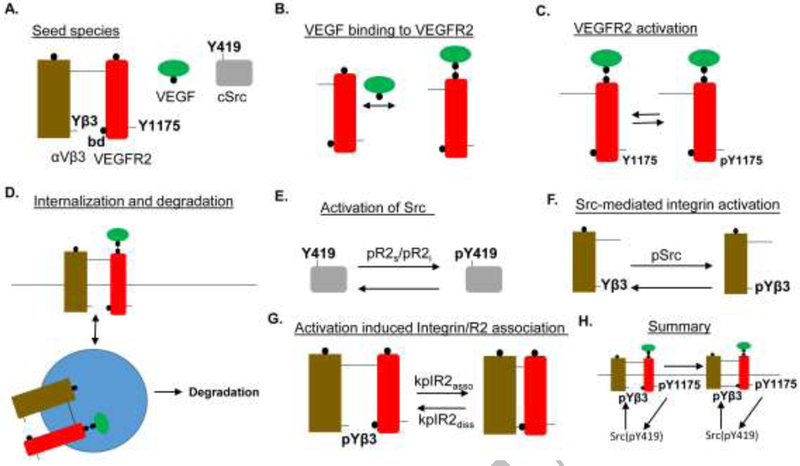
A. αVβ3 integrin has single binding site facing the extracellular matrix, and a tyrosine modification site (Υβ3) that is associated with VEGFR2 when phosphorylated. The integrin is pre-associated with VEGFR2 dimers. VEGFR2 contains the Y1175 tyrosine modification site that is autophosphorylated after VEGF binding to VEGFR2. VEGFR2 has a binding domain (bd) for binding phosphorylated Υβ3 or ρΥβ3. The ligand VEGF has a single binding site for binding VEGFR2 dimers. The kinase cSrc has a single tyrosine modification site (Y419) capable of being autophosphorylated. B. Rule for the binding of VEGF to VEGFR2. C. Activation of VEGFR2 by autophosphorylation on Y1175. D. Receptor complexes are internalized when VEGF is bound to VEGFR2 within the complex. Degradation occurs from the internalized compartment with first order kinetics. E. Src activation dependent on pVEGFR2 on surface and endosomes (pR2s and pR2i). F. Src-mediated phosphorylation of the integrin on Υβ3. G. The association of VEGFR2 binding domain (bd) with ρΥβ3, H. Summary of Src mediated cross-interaction between VEGFR2 and integrin.
3.2. Model parameters
The model parameters (see supplementary Table 1) are determined by fitting the model to a consistent set of data on VEGFR2 receptor dynamics and activation in human endothelial cells (Bruns et al., 2010; Ewan et al., 2006) along with the biological constraint that blocking activation-induced association of VEGFR2 and αVβ3 nearly abolishes VEGFR2 phosphorylation in accordance with the experimental data from DiYF knock-in mouse (Mahabeleshwar et al., 2006). The parameter values for the binding of VEGF to VEGFR2 were taken from (Yen et al., 2011). The VEGFR2 surface level was selected to be consistent with the flow cytometry-based measurements in (Imoukhuede and Popel, 2012; Imoukhuede and Popel, 2014). Normalized total VEGFR2 level is fitted to the data under different conditions in Fig. 2A-D. The experimental data involving cells exposed to cycloheximide (CHX) to block protein synthesis are utilized to estimate VEGFR2 synthesis rate (10−4 s−1) as in Fig . 2(B and C) . The case without any ligand is shown in Fig. 2D. Note that under baseline conditions there is reduction in surface receptors according to the experimental data. However, over sufficiently long time the receptor levels reach a non-zero steady state that is 74% of the initial receptor number (Fig. S1) The surface VEGFR2 level is fitted to the data points in (Ewan et al., 2006) (Fig. 2E). Corresponding phosphorylated VEGFR2 (pR2) level (blue) is shown in Fig. 2F along with the data points (red circles) derived from (Bruns et al., 2010). The constraining data points for the DiYF strain derived from (Mahabeleshwar et al., 2006) are shown in Fig. 2G (red circles) along with the model fit (blue). Preventing integrin phosphorylation blocks VEGFR2 activation. It is necessary that any model describing VEGFR2-integrin interaction take into account this crucial information as a starting point. The predicted pSrc (Fig. 2H) and plnt traces (Fig. 2l) are also shown.
Figure 2. Model parameterization and global sensitivity analysis.
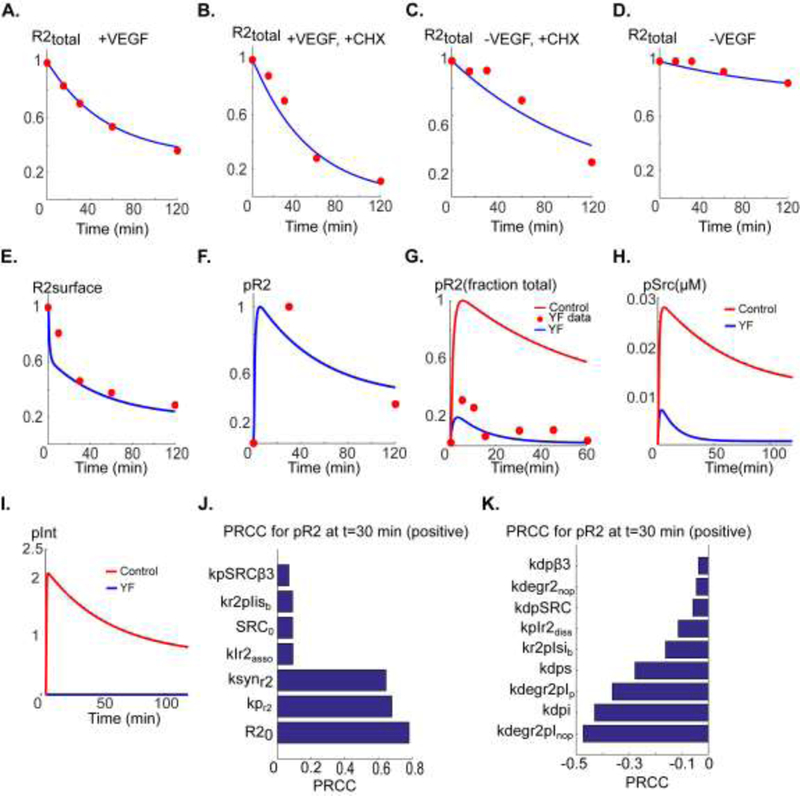
A. Total fraction VEGFR2 level (normalized to VEGFR2 at t=0) versus time in the presence of 50 ng/ml VEGF fitted to data in (Bruns et al., 2010) (red circles). B. Total VEGFR2 in the presence of VEGF and cycloheximide (CHX) fitted to the data in (Bruns et al., 2010). C. Total VEGFR2 in the presence of CHX with no VEGF included. D. Control decline in total VEGFR2 in the absence of VEGF fitted to the data in (Bruns et al., 2010). E. Surface level VEGFR2 fitted to the data in (Ewan et al., 2006). F. pR2 versus time fitted to the data in (Bruns et al., 2010). G. pR2 versus time for the case of Yβ3F mutation resulting in no activation-induced binding of αVβ3 to VEGFR2 fitted to the data from DiYF knock-in mouse model in (Mahabeleshwar et al., 2006) (red circles). The control trace is also shown (red trace). H. Predicted Src activation for the case Yβ3F mutation (blue) relative to control (red). I. Predicted integrin activation in Yb3F mutant (blue) relative to control (red). J. Ranking of the sensitive positively correlated parameters with pR2 at 30 min. K. Ranking of the negatively correlated parameters with PRCC.
3.3. Sensitivity analysis
To determine sensitivity of pR2 to variations in model parameters and identify the most sensitive parameters determining VEGFR2 activation, global sensitivity analysis was performed using the partial rank correlation coefficient (PRCC) algorithm with Latin hypercube sampling (Marino et al., 2008). Top parameters with positive correlation (Fig. 2J) with pR2 are total VEGFR2 level (R20), autophosphorylation rate of R2 (kpr2), and the synthesis rate of R2 (ksynR2). Top parameters with negative correlation with pR2 (Fig. 2K) are the degradation rate of non-phosphorylated R2 that is associated with the phosphorylated integrin (parameter kdegr2pInoP), dephosphorylation rate of the internalized receptors (kdpi), and the degradation rate of phosphorylated receptors associated with phosphorylated integrin in the presence of a peptide (kdegr2pIP). These results imply that degradation rate of the receptors when associated with phosphorylated integrins is the crucial determinant of VEGFR2 activation. In other words, the model suggests that the mechanism by which integrin association promotes VEGFR2 activation is through stabilization of the VEGFR2 cellular level. This mechanism will be explored in more detail in the next section.
3.4. Src-mediated association of phosphorylated integrin with VEGFR2 promotes VEGFR2 activation by significantly lowering VEGFR2 degradation
Global sensitivity analysis identified the degradation rate of the integrin- associated receptors as the most sensitive parameter. Indeed, constraining the model with the experimental data revealed that the rate of degradation for VEGFR2 associated with pInt was ~400-fold lower (supplementary Table 1) than the receptor not associated with pint. As demonstrated in Fig. 3A, total R2 level drops sharply for the case of mutated integrin. Without association to activated integrin, total VEGFR2 drops by ~90% after 40 min. Maximum phosphorylated VEGFR2 (pR2) is relatively insensitive to the changes in degradation rate between pint-associated VEGFR2 (parameter kdegr2plnop) and VEGFR2 without plnt (parameter kdegr2lnop) with the basal value of 435-fold (kdegr2InoP/kdegr2pInoP ~435) as in Fig. 3B. The amplitude of pR2 is stable until the fold-change in degradation rate reaches 100, below which the amplitude drops sharply. The plateau-phase of pR2 (assayed at 60 min) is more sensitive to the changes in degradation rate as shown in Fig. 3C, implying that activation dependent association of αVβ3 with VEGFR2 helps stabilize the steady-state activation level of VEGFR2. Sample traces are shown in Fig. 3D (from the control trace in green to blue). The area under the curve (AUC) of pR2 versus kdegr2lnop/kdegr2plnop is also shown in Fig. S2A. The AUC of pR2 correlates well with pR2 at 60 min (Fig. S2B) indicating that AUC may also be used to investigate the effects of variations in these parameters.
Figure 3. Mechanism of activation-induced αVβ3 integrin and VEGFR2 cross-activation.
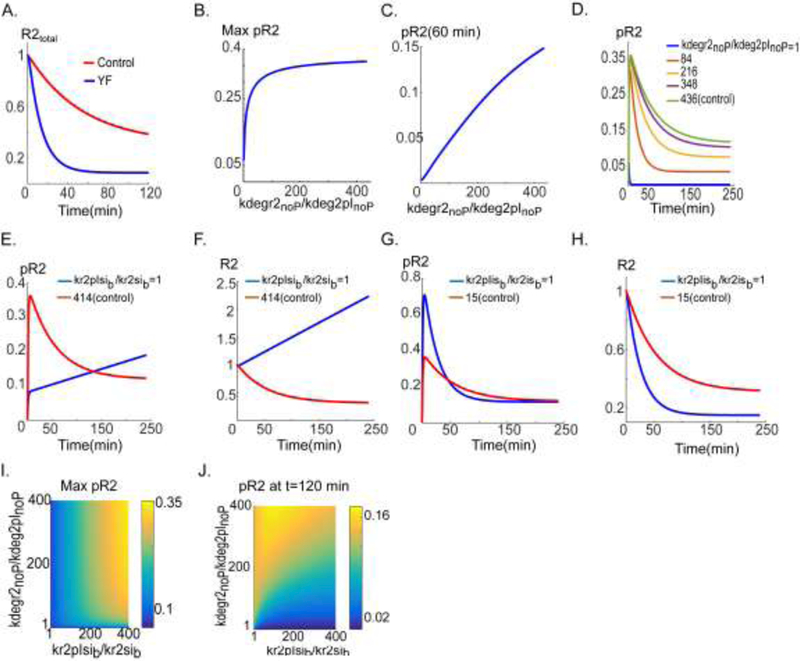
A. Total VEGFR2 (R2) versus time in control (red) and with Yβ3F mutation (blue). B. Maximum pR2 as a function of the ratio of the degradation rate parameters kdegr2nop (VEGFR2 degradation rate without association with ρΥβ3, no peptide (denoted by noP)) and kdegr2plnop (degradation rate when VEGFR2 associated with ρΥβ3, no peptide). C. pR2 at t=60 min as a function of the degradation rate ratio. D. pR2 traces for various values of the degradation ratio. E. pR2 versus time for the internalization rate parameter ratio, kr2sib (VEGFR2 internalization rate, not bound to ρΥβ3) and kr2plsib (VEGFR2 internalization rate when coupled to ρΥβ3). The traces are computed for control (red) and inhibited internalization case (blue). F. Total R2 traces. G. pR2 versus time trace for two values of the recycling rate parameter ratios namely, kr2isb (VEGFR2 recycling rate when not bound to ρΥβ3) and kr2plisb (VEGFR2 recycling rate when bound to ρΥβ3). H. Total R2 traces for the two parameter ranges. I. Two-parameter scan of max pR2 as a function of degradation and internalization parameters. J. pR2 at 120 min as a function of degradation and internalization.
This prediction from the simulations is entirely consistent with recent experimental evidence demonstrating that the inhibiting αVβ3 with a short peptide results in enhanced degradation of VEGFR2 that may contribute to the abolishing of VEGFR2 activation (Lima e Silva et al., 2017).
According to parameter values, the two other parameters that exhibit the largest relative change are the internalization rates of pint-associated VEGFR2 (kr2plsib) and VEGFR2 not associated with pint (kr2lsib). The basal value for fold-change in VEGFR2 internalization rate is ~414 (kr2pIsib/kr2Isib ~414). The consequence of modifying the internalization ratio relative to the control value is investigated in Fig. 3 (E and F). While the amplitude of pR2 is compromised by the changes in the internalization rate, VEGFR2 activation is robust over the long term and pR2 reaches a similar level as control in ~150 min (Fig. 3E). The reason for robust activation of pR2 over the long term is that R2 synthesis counteracts the negative effect of blocking receptor internalization. The simulated total VEGFR2 over time for the two cases is shown in Fig. 3F. The steady-state levels of pR2 and surface R2 for the case kr2pIsib/kr2Isib=1 are shown in Fig. S3A and Fig. S3B respectively. The changes in recycling rates of pInt-associated VEGFR2 (kr2pIisb) and VEGFR2 without pint (kr2Iisb) are investigated in Fig. 3 (G and H). The basal value is 15-fold (kr2pIisb/kr2Iisb ~15) implying that the rate of recycling from internalized endosome to plasma membrane is 15-fold faster for VEGFR2 associated with phosphorylated integrin. As demonstrated in Fig. 3G, this parameter however does not seem to significantly affect VEGFR2 activation. Importantly, the plateau-phase of pR2 is not affected by the changes in the recycling rate. Total R2 traces are shown in Fig. 3H illustrating that increased recycling of the pInt-associated VEGFR2 stabilizes the total receptor level. Twodimensional parameter scans with the internalization and the degradation ratios are performed to investigate the contribution of each parameter as shown in Fig. 3(I and J). The scans determine the range of parameter values that result in robust VEGFR2 activation. Overall, simulations here suggest that stabilization of VEGFR2 receptor by Src-phosphorylated integrin (by lowering the degradation rate of VEGFR2) is sufficient to explain the observed enhancement of VEGFR2 phosphorylation signal. These model findings point at the crucial effect of integrin signaling on VEGFR2 internalization and recycling. This is consistent with experiments demonstrating that low concentration of αVβ3 agonist leads to rapid increase in VEGFR2 surface levels through Rab-4 dependent recycling and recruitment pathway (Reynolds et al., 2009). Our model does not explicitly include Golgi resident receptors, but the parameter ksynR2 is a phenomenological representation of the effect of synthesis and recruitment of the receptors residing within the internalized compartments. Next section considers the implications of modifying ksynR2 on VEGFR2 activation and explores the application of the model to integrin-binding peptides that have been shown to modulate protein synthesis and VEGFR2 recruitment from internalized compartments.
3.5. VEGFR2 synthesis rate and recruitment as critical modulators of VEGFR2 signaling: implications for integrin-binding peptides cilengitide and tumstatin
An important finding of the model is that VEGFR2 activation is very sensitive to the parameter ksynR2 that represents overall increase in surface VEGFR2 level (synthesis rate and recruitment from internal compartments) as demonstrated by global sensitivity analysis (Fig. 2J). To our knowledge, this is the first model that specifically includes the CHX-based data in (Bruns et al., 2010) to estimate the synthesis rate of VEGFR2. Interestingly, some of the short peptides identified as anti-angiogenic also modulate protein synthesis rate and recruitment. Specifically, the RGD motif containing peptide cilengitide has been shown to increase steady-state VEGFR2 level at low concentrations in Rab4-dependent recycling pathway that recruits receptors from internal compartments to the cell surface (up to 20 nM as in (Reynolds et al., 2009)). This paradoxical increase in VEGFR2 surface level might have contributed to the failure of cilengitide as an angiogenesis inhibitor in glioblastoma clinical trials (Stupp et al., 2014). To evaluate whether increase in VEGFR2 results in downstream effects on VEGFR2 phosphorylation, Src, and integrin activation, we simulate the effect of cilengitide in our model and assess the sensitivity of VEGFR2, Src, and integrin activation to cilengitide as illustrated in Fig. 4. The experimental data showed a two-fold increase in VEGFR2 levels at low concentrations of cilengitide (Reynolds et al., 2009). The corresponding ksynR2 value (this is the parameter that phenomenologically describes the overall increase in VEGFR2 levels) that results in experimentally observed two-fold increase in steady-state level of VEGFR2 is shown in Fig. 4A (red circle, ksynR2~6×10−4 s−1). The fractional pR2 at 240 min (Fig. 4B) and maximum pR2 (Fig. 4C) are also shown. Both the steady-state pR2 (pR2 at 240 min in Fig. 4B) and maximum pR2 (Fig. 4C) are sensitive to increases in ksynR2 relative to the control fitted value. Specifically for cilengitide, pR2 is strongly enhanced as shown in Fig. 4D relative to the control. Simulation in Fig. 4D implies that the enhancement in VEGFR2 activation in response to low doses of cilengitide should be observed after ~60 min of application. The AUC of pR2 is correlated with variations in ksynR2 and is also correlated with the value of pR2 at 240 min (Fig. S2C and S2D). Application of cilengitide powerfully increases pR2 level and abolishes the decay phase of the pR2 signal. This is achieved by increasing the total R2 level (Fig. 4E). Strong enhancement is also predicted for Src activation (Fig. 4F) and phosphorylated integrin (Fig. 4G). This demonstrates that the effects of cilengitide on angiogenesis are amplified by signaling to downstream targets by Src and integrin activation. Sample pR2 traces for various values of ksynR2 are shown in Fig. 4H further demonstrating the high sensitivity of pR2 to perturbations in ksynR2.
Figure 4. Simulating the effect of an integrin-binding peptide cilengitide (or similar) on VEGR2 and αVβ3 signaling.
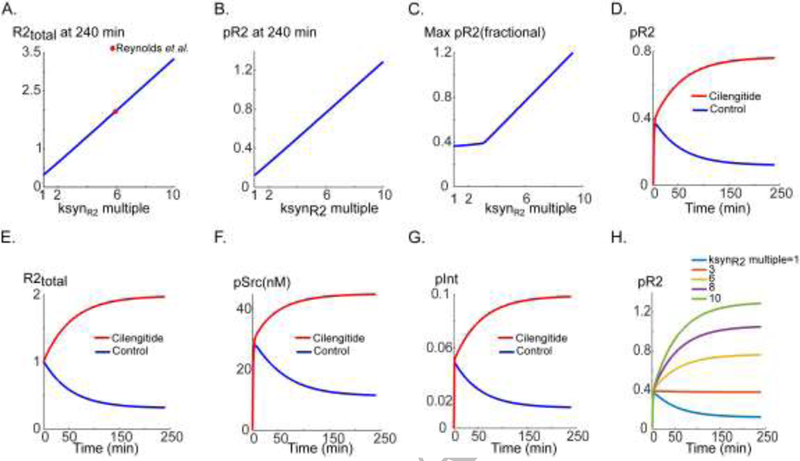
A. Steady-state total R2 versus the synthesis rate (blue) and the value of synthesis rate parameter corresponding to the experimental observation in Reynolds et al. (red circle). B. pR2 versus ksynR2. C. Max pR2 versus ksynR2. D. pR2 versus time for control (blue) and cilengitide treatment (simulated by increasing the synthesis rate, red). E. Total R2 in response to cilengitide (red) and control (blue). F. pSrc versus time for cilengitide (red) and control (blue). G. αVβ3 integrin phosphorylation for cilengitide (red) and control (blue). H. pR2 traces for a wide range of increase in receptor synthesis rate parameter ksynR2.
We next investigate the effect of inhibiting the synthesis rate that may be achieved by the application of tumstatin (a large fragment derived from type IV collagen) or a short peptide derived from tumstatin (Eikesdal et al., 2008; Maeshima et al., 2002). According to the experimental data, tumstatin inhibits R2 synthesis by about 50% (Maeshima et al., 2002). According to Fig. 5A (red circle indicating data point), a 50% reduction in ksynR2 would result in a steady-state R2 level that is about half the control value. Reduction in ksynR2 is predicted to linearly decrease the steady-state pR2 (assayed by computing pR2 at 240 min, Fig. 5B) without affecting the amplitude of the signal (maximum pR2 as in Fig. 5C). The corresponding effect of tumstatin on pR2 is simulated in Fig. 5D relative to control (blue versus red); the major effect is on the steady-state level of pR2. The mechanism is the reduction in R2 levels as shown in Fig. 5E. Tumstatin also inhibits Src activation (Fig. 5F) and integrin phosphorylation (Fig. 5G) which may further amplify its anti-angiogenic effects. Sample pR2 traces corresponding to various values of the parameter ksynR2 are shown in Fig. 5H demonstrating the lowering of the steady-state pR2. An interesting model prediction is that complete blockade of VEGFR2 synthesis abolishes the steady-state pR2 (purple versus blue in Fig. 5H).
Figure 5. Simulating the effect of an integrin-binding peptide tumstatin (or similar) on VEGFR2 and αVβ3 signaling.
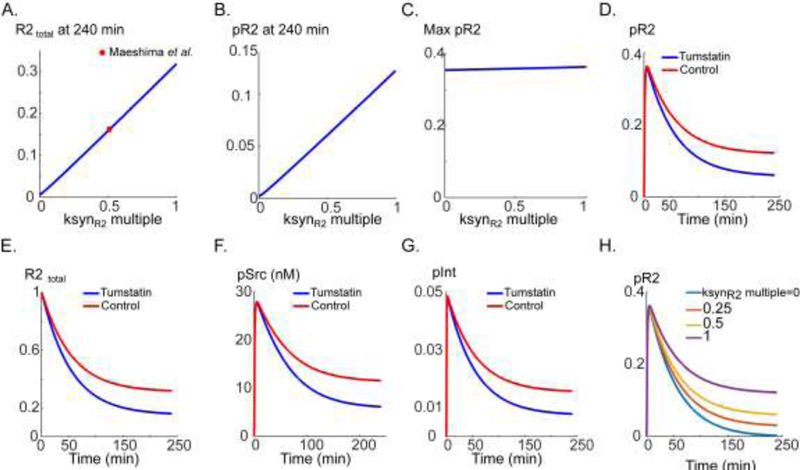
A. Steady-state total R2 (blue) and the ksynR2 value corresponding to the experimental data in (Maeshima et al., 2002) (red circle). B. Steady-state pR2 at 240 min as a function of ksynR2. C. Max pR2 as a function of ksynR2. D. pR2 versus time trace for tumstatin (red) and control (blue). E. The corresponding total R2 for tumstatin (red) and control (blue). F. Src activation in response to tumstatin (red) and control (blue). G. Integrin phosphorylation trace in response to tumstatin (red) and control (blue). H. pR2 traces in response to decreasing values of ksynR2.
In summary, our simulations here demonstrate the crucial role of VEGFR2 synthesis rate and recruitment in modulating VEGFR2 activation and signaling to Src and integrins, highlighting the importance of considering this parameter when designing peptide modulators of angiogenesis for therapeutic applications.
3.6. αVβ3-binding peptide inhibition of VEGFR2 and integrin via the acceleration of VEGFR2 degradation
Cilengitide and tumstatin are examples of αVβ3 binding peptides developed as inhibitors of angiogenesis (Rosca et al., 2011). Our laboratory has developed a class of mimetic integrin-binding peptides derived from non-collagenous domain of type IV collagen (Rosca et al., 2012). Peptides from this class show activity in inhibiting angiogenesis and lymphangiogenesis as well as tumor growth and metastasis in mouse xenograft models (Barbhuiya et al., 2017; Lee et al., 2014; Rosea et al., 2014), and in mouse and rabbit models of ocular neovascularization (Lima e Silva et al., 2017). This class has been shown to impact VEGFR2 degradation (Lima e Silva et al., 2017); however, the experiments did not clarify whether the effects on VEGFR2 degradations are sufficient to explain the inhibitory effects of the peptides. The model here demonstrates that the acceleration of VEGFR2 degradation may be sufficient to explain the anti- angiogenic effects of the peptide or at least major portion of the inhibitory responses. Utilizing computational model like the present one should go a long way towards a quantitative mechanistic description of the complex signal transduction that involves multiple molecular players. In the simulations below, we refer to a representative peptide from this class. Experimentally, incubation of the cells with 50 DM of this peptide inhibits phosphorylation of several RTKs including VEGFR2 (Lee et al., 2014; Rosca et al., 2014). We postulated that the mechanism of action is through acceleration of the degradation of VEGFR2- integrin complex when bound to the peptide. This is schematically illustrated in Fig. 6A. The parameter of interest is the degradation rate of plnt-associated non- phosphorylated receptor (kdegr2plnop) This parameter was the most negatively correlated parameter in our global sensitivity analysis (Fig. 2K). The amplitude of the pR2 signal decreases as a function of this parameter as in Fig. 6B. A 40-fold increase in degradation rate by the peptide is sufficient to significantly reduce the pR2 signal within 10 min consistent with the observed experimental data (Lima e Silva et al., 2017; Rosca et al., 2014). Corresponding pR2 time traces are shown in Fig 6D indicating that a 5-fold change in the degradation rate (yellow trace) would essentially inhibit the steady-state pR2 signal. Similarly, Src activation is predicted to decrease monotonically as a function of enhanced degradation as a result of exposure to peptide (Fig. 6E). Sample traces for pSrc are shown in Fig. 6F. Integrin phosphorylation is also predicted to be inhibited by the same mechanism (Fig. 6G). Peptide is therefore able to inhibit VEGFR2-αVβ3 crossactivation via acceleration of VEGFR2 degradation without affecting any other node in the pathway. Indeed, the model shows that enhanced degradation is sufficient to explain the observed experimental results with peptides within this class.
Figure 6. Simulating the effect of an integrin-binding peptide SP2043 (or similar) on VEGFR2 and αVβ3 signaling.
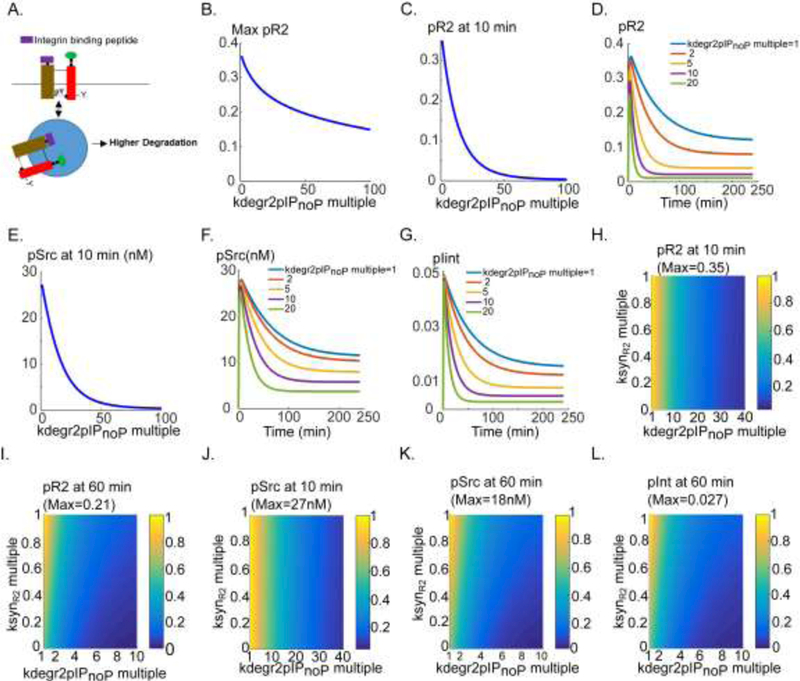
A. Schematic diagram describing the peptide action in the model. B. max pR2 as a function of increase in degradation rate when peptide bound to integrin. C. pR2 at 10 min as a function of degradation rate. D. pR2 traces versus time for various values of the degradation rate. E. Src activation at 10 min. F. pSrc traces for a range of degradation rate values. G. Traces of pint versus time for degradation rate values. H. Two- parameter scan of max pR2 as a function of degradation rate and synthesis rate ksynR2. I. Two-parameter scan of pR2 at 60 min. J. pSrc at 10 min as a function of synthesis rate and the degradation rate. K. Two-parameter scan of pSrc at 60 min. L. Two-parameter scan of pint at 60 min.
We next consider the inhibition of protein synthesis (discussed in Fig. 5) in concert with enhanced receptor degradation in the presence of the peptide to quantitatively evaluate how the inhibition of synthesis might augment the effect on degradation. The representative mimetic peptide considered here and tumstatin peptide are both derived from type IV collagen and it is therefore reasonable to also postulate inhibition of protein synthesis by the same mimetic peptide. The two-parameter scan of pR2 at 10 min is shown in Fig. 6H. Inhibiting synthesis rate does not appreciably influence the profile of pR2 inhibition at 10 min. However, the inhibition of the plateau phase by enhanced receptor degradation is augmented by the inhibition of synthesis as in Fig 6I. Indeed, 50% inhibition of the synthesis rate and 5-fold increase in receptor degradation are sufficient to result in 90% inhibition of pR2 signal at 60 min. This implies that the effects on synthesis rate and receptor degradation may work in concert to inhibit VEGFR2 signaling at steady-state level. The computational predictions for the inhibition profile of pSrc at 10 and 60 min are shown in Fig. 6 (panels J and K respectively). Similar inhibitory profile is observed for the phosphorylated integrin (Fig. 6L). Taken together, our simulations demonstrate quantitatively that the effect of peptide on receptor stability (manifested as causing enhanced degradation within the endosomes) is the dominant explanation for the inhibitory effects on VEGFR2 and integrin signaling. The postulated inhibition of synthesis may work as a global inhibition mechanism to augment the inhibitory effects. The simulations highlight the need for cautious interpretation of the experimental data with integrin-binding peptides that should take into account the potential inhibitory long-term effects on protein synthesis.
4. Discussion
This study focused on developing a mechanistic rule-based model to investigate the mechanism by which the experimentally observed Src-dependent crosstalk between αVβ3 integrin and VEGFR2 promote VEGFR2 activation. Protein kinase Src acts as a positive feedback to sustain both VEGFR2 and integrin activation. The main findings are as follows: 1) association of the Src- phosphorylated integrins with VEGFR2 stabilizes VEGFR2 by reducing the degradation rate of the receptors; 2) VEGFR2 synthesis rate and recruitment (determined by fitting to the data obtained in cells exposed to CHX) is a crucial parameter in determining VEGFR2 activation and any perturbation in this parameter by integrin-binding peptides such as cilengitide and tumstatin should profoundly perturb VEGF and integrin signaling; 3) both the amplitude and the steady-state VEGFR2 activation are very sensitive to increases in receptor synthesis rate and recruitment, while the inhibition of synthesis predominantly affects the steady-state VEGFR2 activation, highlighting the necessity of carefully assessing the effect of potential inhibitors of angiogenesis on synthesis before further deployment in clinical trials; 4) the mimetic peptide representing a class of integrin-binding peptides inhibits VEGF signaling by increasing the degradation rate of the receptor consistent with experimental findings (Lima e Silva et al., 2017). Any potential effect on protein synthesis would contribute to the global inhibition of VEGF and integrin signaling.
The pro-angiogenic properties of cilengitide at low concentrations has complicated its applicability as an anti-angiogenic agent in cancer; however, considering the sensitivity of VEGFR2 activation to increases in synthesis rate, there is a potential in the use of cilengitide (at carefully calibrated low concentrations) in ischemic diseases such as peripheral arterial disease (PAD) where the goal is to enhance angiogenesis to promote circulation to the lower extremities (Annex, 2013; Annex and Beller, 2016; Clegg et al., 2017; Collinson and Donnelly, 2004). Since currently there are no curative and effective treatment options for advanced PAD, insights based on computer simulations may highlight and identify potential avenues for the development of novel pro- angiogenic therapies in this disease based on short peptides such as cilengitide. As for model limitations, a major drawback of the model is that the focus here has been on a single receptor type, namely VEGFR2 and described receptor activation with a basic one-to-one interaction with VEGF ligand. We ignored the effect of other VEGF receptors such as neuropilin-1 (NRP1) and vascular endothelial growth factor receptor 1 (VEGFR1) on VEGFR2 activation and internalization. Detailed computational models considering VEGFR1, VEGFR2, and NRP1 signaling have been developed in recent years (Bazzazi and Popel, 2017; Bazzazi et al., 2018; Clegg and Mac Gabhann, 2017; Clegg et al., 2017; Clegg and Mac Gabhann, 2015; Mac Gabhann and Popel, 2007; Mac Gabhann and Popel, 2008; Mac Gabhann et al., 2010; Tan et al., 2013a; Tan et al., 2013b; Vempati et al., 2014; Wu et al., 2009) but very few studies have considered interactions with integrins (Bauer et al., 2010). The advantage of utilizing a rule-based environment is that it allows for modular incorporation of different receptor types and convenient extension of the model and signaling pathway.
Another limitation of the current model is that integrin clustering is not included. Clustering might substantially augment the results predicted in this study through mechanisms described in a previous integrin model (Welf et al., 2012). It seems reasonable to assume that the basic cross-activation mechanism between αVβ3 and VEGFR2 should not change with integrin clustering and the outcome of clustering would be the amplification of the signal from the membrane. Expanding the model to include integrin clustering may prove useful in identifying additional mechanisms that contribute to the potentiation of VEGFR2 activation. However, the purpose of the study here was to construct a simpler model incorporating essential features of VEGFR2 and integrin signaling in order to identify mechanisms that would explain the observed experimental data. A further extension of the model may involve the addition of downstream crosstalk between integrin and VEGFR2 signals as in (Bauer et al., 2010). A more sophisticated description of synthesis and receptor recruitment from the Golgi apparatus to the plasma membrane according to the experimental data in (Manickam et al., 2011) may also yield a more realistic model without changing the essential features described in this study.
5. Conclusions
Overall, even within the simplifying assumptions of our current model and the phenomenological description of receptor synthesis and recruitment, a focused attention to a consistent set of experimental data in endothelial cells allowed us to propose specific molecular mechanism for the αVβ3 activation of VEGFR2 that was consistent with the experimental findings. The simulations also provided new insight for novel therapeutic intervention and drug repurposing within the context of PAD utilizing low concentrations of cilengitide, targeting the integrin αVβ3- VEGFR2 axis.
Supplementary Material
Highlights.
A mechanistic computational model is developed for the SRC-mediated interaction of VEGFR2 and integrins
Simulations demonstrate that integrin association with VEGFR2 enhances VEGFR2 activation by reducing its degradation rate and by stabilizing the activation signal
Computer simulations shed light on the potential mechanisms for modulation of angiognesis by integrin binding peptides
Acknowledgement
This work was supported by the National Institutes of Health grants R01HL101200, R21EY026148, and R01CA138264 (ASP). We thank Drs. Kairbaan Hodivala-Dilke, Adam C. Mirando and Niranjan B. Pandey for insightful discussions related to the angiogenesis-modulating integrin-binding peptides.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contribution
HB and ASP conceived and designed the study. YZ implemented an initial version of the model in MATLAB and carried out simulations and contributed to the analysis of the data and discussion. MJ contributed to the analysis of the data and discussion. HB expanded and implemented the model in BioNetGen and carried out extensive simulations and analysis of the results. All authors contributed to the writing of the paper.
Disclosure of Potential Conflicts of Interest
ASP is a co-founder and serves as the CSO of AsclepiX Therapeutics, LLC. The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict of interest policies.
References
- Annex BH, 2013. Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol 10, 387–96, doi: 10.1038/nrcardio.2013.70. [DOI] [PubMed] [Google Scholar]
- Annex BH, Beller GA, 2016. Towards the Development of Novel Therapeutics for Peripheral Artery Disease. Trans Am Clin Climatol Assoc 127, 224–234. [PMC free article] [PubMed] [Google Scholar]
- Barbhuiya M, Mirando AC, Simons B, Lemtiri-Chlieh G, Green JJ, Popel AS, Pandey NB, Tran PT, 2017. Therapeutic potential of an anti-angiogenic multimodal biomimetic peptide in hepatocellular carcinoma. Oncotarget 8, 101520–101534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer AL, Jackson TL, Jiang Y, Rohlf T, 2010. Receptor cross-talk in angiogenesis: mapping environmental cues to cell phenotype using a stochastic, Boolean signaling network model. J Theor Biol 264, 838–46, doi: 10.1016/j.jtbi.2010.03.025. [DOI] [PubMed] [Google Scholar]
- Bazzazi H, Popel AS, 2017. Computational investigation of sphingosine kinase 1 (SphK1) and calcium dependent ERK1/2 activation downstream of VEGFR2 in endothelial cells. PLoS Comput Biol 13, e1005332, doi: 10.1371/journal.pcbi.1005332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzazi H, Isenberg J, Popel A, 2017. Inhibition of VEGFR2 activation and its downstream signaling to ERK1/2 and calcium by Thrombospondin-1 (TSP1): In silico investigation. Frontiers in Physiology 8, 48, doi: 10.3389/fphys.2017.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzazi H, Zhang Y, Jafarnejad M, Isenberg JS, Annex BH, Popel AS, 2018. Computer Simulation of TSP1 Inhibition of VEGF-Akt-eNOS: An Angiogenesis Triple Threat. Frontiers in Physiology 9, 644, doi: 10.3389/fphys.2018.00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggon TJ, Eck MJ, 2004. Structure and regulation of Src family kinases. Oncogene 23, 7918–27, doi: 10.1038/sj.one.1208081. [DOI] [PubMed] [Google Scholar]
- Bruns AF, Herbert SP, Odell AF, Jopling ΗM, Hooper NM, Zachary IC, Walker JH, Ponnambalam S, 2010. Ligand-stimulated VEGFR2 signaling is regulated by co-ordinated trafficking and proteolysis. Traffic 11, 161–74, doi: 10.1111/j.1600-0854.2009.01001.x. [DOI] [PubMed] [Google Scholar]
- Buerkle MA, Pahernik SA, Sutter A, Jonczyk A, Messmer K, Dellian M, 2002. Inhibition of the alpha-nu integrins with a cyclic RGD peptide impairs angiogenesis, growth and metastasis of solid tumours in vivo. Br J Cancer 86, 788–95, doi: 10.1038/sj.bjc.6600141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Chen X, 2006. Anti-angiogenic cancer therapy based on integrin alphavbeta3 antagonism. Anticancer Agents Med Chem 6, 407–28. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, 2013. Ocular neovascularization. J Mol Med (Berl) 91, 311–21, doi: 10.1007/s00109-013-0993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, 2005. Angiogenesis in life, disease and medicine. Nature 438, 932–6, doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- Cirit M, Krajcovic M, Choi CK, Welf ES, Horwitz AF, Haugh JM, 2010. Stochastic model of integrin-mediated signaling and adhesion dynamics at the leading edges of migrating cells. PLoS Comput Biol 6, e1000688, doi: 10.1371/journal.pcbi.1000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg LE, Mac Gabhann F, 2017. A computational analysis of in vivo VEGFR activation by multiple co-expressed ligands. PLoS Comput Biol 13, e1005445, doi: 10.1371/journal.pcbi.1005445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg LE, Ganta VC, Annex BH, Mac Gabhann F, 2017. Systems Pharmacology of VEGF165b in Peripheral Artery Disease. CPT Pharmacometrics Syst Pharmacol, doi: 10.1002/psp4.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg LW, Mac Gabhann F, 2015. Site-Specific Phosphorylation of VEGFR2 Is Mediated by Receptor Trafficking: Insights from a Computational Model. PLoS Comput Biol 11, e1004158, doi: 10.1371/journal.pcbi.1004158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinson DJ, Donnelly R, 2004. Therapeutic angiogenesis in peripheral arterial disease: can biotechnology produce an effective collateral circulation? Eur J Vasc Endovasc Surg 28, 9–23, doi: 10.1016/j.ejvs.2004.03.021. [DOI] [PubMed] [Google Scholar]
- D’Andrea LD, Del Gatto A, Pedone C, Benedetti E, 2006. Peptide-based molecules in angiogenesis. Chem Biol Drug Des 67, 115–26, doi: 10.1111/j.1747-0285.2006.00356.x. [DOI] [PubMed] [Google Scholar]
- DiPietro LA, 2013. Angiogenesis and scar formation in healing wounds. Curr Opin Rheumatol 25, 87–91, doi: 10.1097/BOR.0b013e32835b13b6. [DOI] [PubMed] [Google Scholar]
- Eikesdal HP, Sugimoto H, Birrane G, Maeshima Y, Cooke VG, Kieran M, Kalluri R, 2008. Identification of amino acids essential for the antiangiogenic activity of tumstatin and its use in combination antitumor activity. Proc Natl Acad Sci U S A 105, 15040–5, doi: 10.1073/pnas.0807055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewan LC, Jopling HM, Jia H, Mittar S, Bagherzadeh A, Howell GJ, Walker JH, Zachary IC, Ponnambalam S, 2006. Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic 7, 1270–82. [DOI] [PubMed] [Google Scholar]
- Faeder JR, Blinov ML, Hlavacek WS, 2009. Rule-based modeling of biochemical systems with BioNetGen. Methods Mol Biol 500, 113–67, doi: 10.1007/978-1-59745-525-1_5. [DOI] [PubMed] [Google Scholar]
- Folkman J, 2002. Role of angiogenesis in tumor growth and metastasis. Semin Oncol 29, 15–8, doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- Hindmarsh AC, Brown PN, Grant KE, Lee SLR,S, Shumaker DE., Woodward CS., 2005. SUNDIALS: Suite of Nonlinear and Differential/Algebraic Equation Solvers. ACM Trans Math Softw 31, 363–396. [Google Scholar]
- Hlavacek WS, Faeder JR, Blinov ML, Posner RG, Hucka M, Fontana W, 2006. Rules for modeling signal-transduction systems. Sci STKE 2006, re6, doi: 10.1126/stke.3442006re6. [DOI] [PubMed] [Google Scholar]
- Imoukhuede PI, Popel AS, 2012. Expression of VEGF receptors on endothelial cells in mouse skeletal muscle. PLoS One 7, e44791, doi: 10.1371/journal.pone.0044791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoukhuede PI, Popel AS, 2014. Quantitative fluorescent profiling of VEGFRs reveals tumor cell and endothelial cell heterogeneity in breast cancer xenografts. Cancer Med 3, 225–44, doi: 10.1002/cam4.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Lee SJ, Koskimaki JE, Han Z, Pandey NB, Popel AS, 2014. Inhibition of breast cancer growth and metastasis by a biomimetic peptide. Sci Rep 4, 7139, doi: 10.1038/srep07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima e Silva R, Kanan Y, Mirando AC, Kim J, Shmueli RB, Lorenc VE, Fortmann SD, Sciamanna J, Pandey NB, Green JJ, Popel AS, Campochiaro PA, 2017. Tyrosine kinase blocking collagen IV-derived peptide suppresses ocular neovascularization and vascular leakage. Sci Transl Med 9, pii: eaai8030, doi: 10.1126/scitranslmed.aai8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Gabhann F, Popel AS, 2007. Dimerization of VEGF receptors and implications for signal transduction: a computational study. Biophys Chem 128, 125–39, doi: 10.1016/j.bpc.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Gabhann F, Popel AS, 2008. Systems biology of vascular endothelial growth factors. Microcirculation 15, 715–38, doi: 10.1080/10739680802095964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Gabhann F, Qutub AA, Annex BH, Popel AS, 2010. Systems biology of pro-angiogenic therapies targeting the VEGF system. Wiley Interdiscip Rev Syst Biol Med 2, 694–707, doi: 10.1002/wsbm.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima Y, Sudhakar A, Lively JC, Ueki K, Kharbanda S, Kahn CR, Sonenberg N, Hynes RO, Kalluri R, 2002. Tumstatin, an endothelial cell- specific inhibitor of protein synthesis. Science 295, 140–3, doi: 10.1126/science.1065298. [DOI] [PubMed] [Google Scholar]
- Maeshima Y, Yerramalla UL, Dhanabal M, Holthaus KA, Barbashov S, Kharbanda S, Reimer C, Manfredi M, Dickerson WM, Kalluri R, 2001. Extracellular matrix-derived peptide binds to alpha(v)beta(3) integrin and inhibits angiogenesis. J Biol Chem 276, 31959–68, doi: 10.1074/jbc.M103024200. [DOI] [PubMed] [Google Scholar]
- Mahabeleshwar GH, Feng W, Phillips DR, Byzova TV, 2006. Integrin signaling is critical for pathological angiogenesis. J Exp Med 203, 2495–507, doi: 10.1084/jem.20060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahabeleshwar GH, Feng W, Reddy K, Plow EF, Byzova TV, 2007. Mechanisms of integrin-vascular endothelial growth factor receptor crossactivation in angiogenesis. Circ Res 101, 570–80, doi: 10.1161/CIRCRESAHA.107.155655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manickam V, Tiwari A, Jung JJ, Bhattacharya R, Goel A, Mukhopadhyay D, Choudhury A, 2011. Regulation of vascular endothelial growth factor receptor 2 trafficking and angiogenesis by Golgi localized t-SNARE syntaxin 6. Blood 117, 1425–35, doi: 10.1182/blood-2010-06-291690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Hogue IB, Ray CJ, Kirschner DE, 2008. A methodology for performing global uncertainty and sensitivity analysis in systems biology. J Theor Biol 254, 178–96, doi: 10.1016/j.jtbi.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JA, Dvorak AM, Dvorak HF, 2007. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol 2, 251–75, doi: 10.1146/annurev.pathol.2.010506.134925. [DOI] [PubMed] [Google Scholar]
- Reynolds AR, Hart IR, Watson AR, Welti JC, Silva RG, Robinson SD, Da Violante G, Gourlaouen M, Salih M, Jones MC, Jones DT, Saunders G, Kostourou V, Perron-Sierra F, Norman JC, Tucker GC, Hodivala-Dilke KM, 2009. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat Med 15, 392–400, doi: 10.1038/nm.1941. [DOI] [PubMed] [Google Scholar]
- Rohrs JA, Sulistio CD, Finley SD, 2016. Predictive model of thrombospondin-1 and vascular endothelial growth factor in breast tumor tissue. Npj Systems Biology And Applications 2, 16030, doi:10.1038/npjsba.2016.30 10.1038/npjsba.2016.30https://www.nature.com/articles/npisba201630#supplementary-informationhttps://www.nature.com/articles/npisba201630#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca EV, Koskimaki JE, Pandey NB, Tamiz AP, Popel AS, 2012. Structure-activity relationship study of collagen-derived anti-angiogenic biomimetic peptides. Chem Biol Drug Des 80, 27–37, doi: 10.1111/j.1747-0285.2012.01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca EV, Koskimaki JE, Rivera CG, Pandey NB, Tamiz AP, Popel AS, 2011. Anti-angiogenic peptides for cancer therapeutics. Curr Pharm Biotechnol 12, 1101–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca EV, Penet MF, Mori N, Koskimaki JE, Lee E, Pandey NB, Bhujwalla ZM, Popel AS, 2014. A biomimetic collagen derived peptide exhibits anti-angiogenic activity in triple negative breast cancer. PLoS One 9, e111901, doi: 10.1371/journal.pone.0111901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW, 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneller M, Vuori K, Ruoslahti E, 1997. Alphavbeta3 integrin associates with activated insulin and PDGFbeta receptors and potentiates the biological activity of PDGF. EMBO J 16, 5600–7, doi: 10.1093/emboj/16.18.5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil SJ, Ginsberg MH, 1997. Perspectives series: cell adhesion in vascular biology. Integrin signaling in vascular biology. J Clin Invest 100, 1–5, doi: 10.1172/JC1119500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldi R, Mitola S, Strasly M, Defilippi P, Tarone G, Bussolino F, 1999. Role of alphavbeta3 integrin in the activation of vascular endothelial growth factor receptor-2. EMBO J 18, 882–92, doi: 10.1093/emboj/18.4.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanath PR, Malinin NL, Byzova TV, 2009. Cooperation between integrin alphavbeta3 and VEGFR2 in angiogenesis. Angiogenesis 12, 177–85, doi: 10.1007/s10456-009-9141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanini MO, Wu FT, Mac Gabhann F, Popel AS, 2008. A compartment model of VEGF distribution in blood, healthy and diseased tissues. BMC Syst Biol 2, 77, doi: 10.1186/1752-0509-2-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupack DG, Cheresh DA, 2004. Integrins and angiogenesis. Curr Top Dev Biol 64, 207–38, doi: 10.1016/S0070-2153(04)64009-9. [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, Aldape KD, Lhermitte B, Pietsch T, Grujicic D, Steinbach JP, Wick W, Tarnawski R, Nam DH, Hau P, Weyerbrock A, Taphoorn MJ, Shen CC, Rao N, Thurzo L, Herrlinger U, Gupta T, Kortmann RD, Adamska K, McBain C, Brandes AA, Tonn JC, Schnell O, Wiegel T, Kim CY, Nabors LB, Reardon DA, van den Bent MJ, Hicking C, Markivskyy A, Picard M, Weller M, European Organisation for R, Treatment of C, Canadian Brain Tumor C, team C. s., 2014. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071–22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 15, 1100–8, doi: 10.1016/S1470-2045(14)70379-1. [DOI] [PubMed] [Google Scholar]
- Tan WH, Popel AS, Mac Gabhann F, 2013a. Computational model of VEGFR2 pathway to ERK activation and modulation through receptor trafficking. Cell Signal 25, 2496–510, doi: 10.1016/j.cellsig.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan WH, Popel AS, Mac Gabhann F, 2013. b. Computational Model of Gab1/2- Dependent VEGFR2 Pathway to Akt Activation. PLoS One 8, e67438, doi: 10.1371/journal.pone.0067438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vempati P, Popel AS, Mac Gabhann F, 2014. Extracellular regulation of VEGF: isoforms, proteolysis, and vascular patterning. Cytokine Growth Factor Rev 25, 1–19, doi: 10.1016/j.cytogfr.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welf ES, Naik UP, Ogunnaike BA, 2012. A spatial model for integrin clustering as a result of feedback between integrin activation and integrin binding. Biophys J 103, 1379–89, doi: 10.1016/j.bpj.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West XZ, Meller N, Malinin NL, Deshmukh L, Meller J, Mahabeleshwar GH, Weber ME, Kerr BA, Vinogradova O, Byzova TV, 2012. Integrin beta3 crosstalk with VEGFR accommodating tyrosine phosphorylation as a regulatory switch. PLoS One 7, e31071, doi: 10.1371/journal.pone.0031071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard AS, Garcia-Cardena G, Leong M, Madri JA, Sessa WC, Languino LR, 1998. The synergistic activity of alphavbeta3 integrin and PDGF receptor increases cell migration. J Cell Sci 111 ( Pt 4), 469–78. [DOI] [PubMed] [Google Scholar]
- Wu FT, Stefanini MO, Mac Gabhann F, Kontos CD, Annex BH, Popel AS, 2009. Computational kinetic model of VEGF trapping by soluble VEGF receptor- 1: effects of transendothelial and lymphatic macromolecular transport. Physiol Genomics 38, 29–41, doi: 10.1152/physiolgenomics.00031.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee KL, Weaver VM, Hammer DA, 2008. Integrin-mediated signalling through the MAP-kinase pathway. IET Syst Biol 2, 8–15, doi: 10.1049/iet-syb:20060058. [DOI] [PubMed] [Google Scholar]
- Yen P, Finley SD, Engel-Stefanini MO, Popel AS, 2011. A two-compartment model of VEGF distribution in the mouse. PLoS One 6, e27514, doi: 10.1371/journal.pone.0027514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.