

Summary
The finding that dendritic cells (DCs) orchestrate innate and adaptive immune responses has stimulated research on harnessing DCs for developing more effective vaccines for DC therapy. The expression of cytomegalovirus (CMV) antigens in glioblastoma multiforme (GBM) presents a unique opportunity to target these viral proteins for tumour immunotherapy. Here, we demonstrate that Vγ9γδT cells, innate immune cells activated by zoledronate (Z) and Vα24 natural killer (Vα24NK) cells, innate/adaptive immune cells activated by α‐galactosylceramide (G) can link innate and adaptive immunities through cross‐talk with interferon (IFN) DCs from patients with glioblastoma multiforme (GBM) and healthy donors in a manner that can amplify the activation and proliferation of CMVpp65‐specific CD8+ T cells. The IFN DCs derived from patients with GBM used in this study express lower levels of programmed cell death ligand (PD)‐L1 and PD‐L2 and higher levels of C‐C receptor 7 (CCR7) than the most commonly used mature interleukin (IL)‐4 DCs. The expression level of programmed cell death 1 (PD‐1) on CD8+ T cells, including CMVpp65‐specific CD8+ T cells, expanded by IFN DCs pulsed with the CMVpp65‐peptide and Z plus G (IFN DCs/P+Z+G), was lower than that expanded by IFN DCs pulsed with the peptide alone (IFN DCs/P). Multi‐functional T cells, including human leucocyte antigen (HLA)‐A*0201‐restricted CMVpp65‐specific CD8+ T cells, Vγ9γδT cells and Vα24NKT cells, efficiently kill the HLA‐A*0201‐positive GBM cell line expressing CMVpp65 protein (T98G). These findings indicate that DC therapy using IFN DCs/P+Z+G and/or CTL therapy using CMVpp65‐specific CD8+ T cells expanded by IFN DCs/P+Z+G may lead to a good clinical outcome for patients with GBM.
Keywords: alpha‐galactosylceramide, CMVpp65‐specific CD8+ T cells, cytomegalovirus (CMV)pp65, glioblastoma multiforme, zoledronate
Introduction
Dendritic cells (DCs) are central players in the immune system and operate at the interface of innate and adaptive immunities. DCs have attracted much interest during the past decades for their use in therapeutic vaccination against cancer, owing to their professional antigen‐specific T cell immunity. Clinical trials have shown that the use of tumour antigen‐loaded DCs in cancer patients is safe; however, their potential in inducing anti‐tumour immunity to eradicate tumours is seen in only a minority of patients 1, 2. This underscores the necessity to redesign and optimize current procedures for DC vaccine manufacture.
For DC therapy that eradicates tumours successfully, it is crucial to marshal and orchestrate a vast array of effector cells, including adaptive immune cells such as tumour antigen‐specific CD8+ T and CD4+ helper T cells, innate immune cells such as γδT and natural killer (NK) cells and innate/adaptive immune cells, Vα24NKT cells 3, in which DCs play a pivotal role in bridging innate and adaptive immunities. Our clinical study using DCs pulsed with α‐galactosylceramide (G), which is a ligand of Vα24NKT cells for patients with melanoma, has demonstrated that the Vα24NKT cells activated by G‐pulsed DCs bridge innate and adaptive immunities to induce the activation and proliferation of interferon (IFN)‐γ‐producing CD8+ T cells following the activation of Vα24NK T and NK cells in vivo 4. Furthermore, an in‐vitro study has shown that the induction of tumour antigen‐specific CD8+ T cells was amplified by DCs pulsed with a tumour antigen and zoledronate (Z), in which Vγ9γδT cells expanded by Z function as T helper (Th) cells through the production of Th1 cytokines such as IFN‐γ and tumour necrosis factor (TNF)α 5, 6.
In this study, we aimed for a much stronger induction of tumour antigen‐specific CD8+ T cells. We speculated that DCs pulsed with a tumour antigen and Z+G may enhance the induction of tumour antigen‐specific CD8+ T cells through further expansion of not only Vγ9γδT cells, but also Vα24NKT cells.
The outcome of DC therapy depends upon the characteristics of DCs infused. The most widely adopted method of generating DCs of clinical use involves a 1‐week, two‐step in‐vitro culture. It requires incubation of monocytes with IL‐4 and granulocyte/machrophage‐colony stimulating factor (GM‐CSF) to obtain immature IL‐4‐induced DCs (IL‐4 DCs), followed by treatment with different maturation stimuli to obtain various mature IL‐4‐induced DCs (mIL‐4 DCs) 7, 8. In another method of DC preparation, it has been shown that monocytes cultured with GM‐CSF plus IFN‐α can be induced towards the DC lineage, so‐called IFN DCs, which highly express CD56 and CD14 molecules 9, 10, 11. Our previous in‐vitro study has shown that CD56high+IFN DCs possessing HLA‐A*0201 effectively induce melanoma‐associated antigen recognized by T cells (Mart1)‐modified melanoma peptide (A27L)‐specific CD8+ T cells in the presence of A27L and Z through preferential expansion of CD56+ Vγ9γδT cells, which are potent anti‐tumour effectors more capable of killing tumour cells than CD56‐Vγ9γδT cells 12. Taken together with these previous studies of DCs, Vγ9γδT cells and Vα24NKT cells, we highly expected that IFN DCs pulsed with a tumour antigen and Z+G enhance the induction of tumour antigen‐specific CD8+ T cells through the expansion of Vγ9γδT and Vα24NKT cells in vitro. Then, infusion of IFN DCs pulsed with a tumour antigen and Z plus G results in a better clinical outcome through the effective expansion of tumour antigen‐specific CD8+ T cells in vivo, in which Vγ9γδT and Vα24NKT cells function as adjuvants to enhance the expansion of tumour antigen‐specific CD8+ T cells.
Despite aggressive surgery, radiation and chemotherapy, treatment of patients with glioblastoma multiforme (GBM) is rarely curative, and conventional therapies are inherently non‐specific and damage surrounding normal tissues 13, 14. Immunotherapies including DC therapy provide a promising alternative owing to the intrinsic specificity and potentially long‐lasting effects of immune activation 15. In this study we chose pp65, an antigen from a common herpesvirus called CMVpp65, in GBM as the tumour antigen 16. Importantly, the ctyomegalovirus (CMV) antigens have been detected in cancellous areas of histological sections, but not in surrounding healthy tissues 17, 18.
Recently, various molecules that can modulate T cell receptor (TCR) signals have been identified. Multiple co‐stimulatory and inhibitory interactions regulate T cell responses to antigen‐presenting cells (APCs) such as DCs 19, 20, 21. Many ligands bind to multiple receptors, some of which deliver co‐stimulatory signals and others deliver inhibitory signals. In this study, we focused on the expression levels of programmed cell death ligand (PD)‐L1 and PD‐L2 on IFN DCs as well as the expression levels of programmed cell death (PD)‐1 and cytotoxic T lymphocyte antigen (CTLA)‐4 on CD8+ T cells, which are expanded by IFN DCs pulsed with the CMVpp65‐peptide alone (IFN DCs/P) and compared them with those on CD8+ T cells, which are expanded by IFN DCs pulsed with the peptide and Z+G (IFN DCs/P+Z+G).
Immunoinhibition renders T cells dysfunctional in the tumour microenvironment (TME), in which different immunosuppressive cell populations such as regulatory T cells (Tregs) and myeloid‐derived suppressor cells are involved 22. In this study, we also examined the induction of CD4+CD25+forkhead box protein 3 (FoxP3)+ Tregs 23 by IFN DCs/P versus IFN DCs/P+Z+G.
Human CMV (HCMV) is a ubiquitous opportunistic pathogen. Symptomatic HCMV infection occurs predominantly in immunocompromised hosts, such as patients after allogeneic haematopoietic stem cell transplantation (alloSCT), whereas symptomatic infection of healthy donors (HDs) is rare. Although inapparent CMV viraemia as a potential prestage of a manifest CMV system or an organ disease can be detected as early as 10–14 days after alloSCT and may last for several weeks, but usually resolves after an early pre‐emptive treatment with nucleoside anti‐viral agents such as ganciclovir 24, it is conceivable that infusions of CMV‐specific CD8+ T cells from allogenic HDs may decrease relapse risk in the patients who had alloSCT. Thus, we also analysed the ability of HD‐derived IFN DCs/P+Z+G.
The aims of this study were as follows:
To determine whether IFN DCs/P+Z+G derived from GBM patients can induce CMVpp65‐specific CD8+ T cells most extensively, as well as expanded Vγ9γδT and Vα24NKT cells, compared with IFN DCs/P, IFN DCs/P+Z or IFN DCs/P+G.
To assess whether the expression level of PD‐1 on CD8+ T cells, including CMVpp65‐specific CD8+ T cells expanded by IFN DCs/P+Z+G, is lower than that on the CD8+ T cell‐expanded IFN DCs/P.
To determine whether effector cells including HLA‐A*0201‐restricted CMVpp65‐specific CD8+ T, Vγ9γδT and Vα24NKT cells efficiently kill the HLA‐A*0201‐positive T98G cell line expressing CMVpp65 protein that was established from a patient with GBM 25, 26.
Materials and methods
Flow cytometry and reagents
Surface phenotypes were determined using an Epics XL MCL (Beckman Coulter, Brea, CA, USA). The following monoclonal antibodies (mAbs) were purchased from Beckman Coulter: anti‐CD3, anti‐CD4, anti‐CD8, anti‐Vγ9TCR, anti‐CD14, anti‐CD25, anti‐CD45, anti‐CD54, anti‐CD56, anti‐HLA‐DR, anti‐CD40, anti‐CD80, anti‐CD86, anti‐CD11c, anti‐CD36, mouse immunoglobulin (Ig)G1, mouse IgG2 and mouse IgG2b mAbs. Anti‐HLA‐class 1 and anti‐CCR7 mAbs were purchased from Beckton Dickinson (San Jose, CA, USA) and R&D Systems (Minneapolis, MN, USA), respectively. Anti‐TCR Vα24TCR and anti‐TCR Vβ11 mAbs were purchased from Beckman Coulter (Villepinte, France). Anti‐human CD273 (PD‐L2) and CD274 (PD‐L1) mAbs were purchased form eBioscience (San Diego, CA, USA). Anti‐human CD152 (CTLA‐4) and CD279 (PD‐1) mAbs were purchased from BioLegend (San Diego, CA, USA). Anti‐FoxP3 mAb for intracellular staining was purchased from BD Biosciences (Tokyo, Japan). All mAbs were conjugated with fluorescein isothiocyanate (FITC), phycoerythrin (PE), antigen‐presenting cells (APC), extracellular domain (ECD), proprotein convertase (PC)5 or PC7. Z was purchased from Novartis Pharmaceuticals (Basel, Switzerland) and G from Funakoshi Co. Ltd (Tokyo, Japan).
Generation of IFN DCs
Peripheral blood mononuclear cells (PBMCs) were isolated from four patients with GBM and three HDs. HLA types of these donors are listed in Table 1a (patients) and Table 1b (HDs). All subjects provided their written informed consent to use their PBMCs for research purpose. For cell culture in this study, AIM‐V medium (Invitrogen, Tokyo, Japan) containing 10% heat‐inactivated human AB serum was used. PBMCs separated by density gradient centrifugation with Lymphoprep (Nycomed, Asker, Norway) were suspended in the medium and then incubated in a flask (Corning Incorporated, Tokyo, Japan) for 1 h at 37oC. After removing non‐adherent cells containing CD8+ T cells, Vγ9γδT cells and Vα24NKT cells, monocytes obtained as adherent cells were cultured in the medium containing GM‐CSF (1000 U/ml; Primmune Inc., Kobe, Japan) and IFN‐α (1000 U/ml; Intron, MSD K.K., Chiyda‐ku, Japan) for 3 days to obtain IFN DCs. Alternatively, adherent cells were cultured for 5 days in the medium containing GM‐CSF (1000 U/ml) and IL‐4 (500 U/ml) to obtain IL‐4 DCs. The IL‐4 DCs were cultured for a further 2 days in the presence of TNF‐α (10 ng/ml; Reprotech Ltd, UK) to obtain mature IL‐4DCs (mIL‐4DCs).
Table 1.
Human leucocyte antigen (HLA)‐types of donors are listed from four patients with glioblatoma multiforme (GBM) (a) and three healthy donors (HDs) (b)
| (a) HLA‐type patients | |||
| Patient 1 | HLA‐A*0201/A*0101 | ||
| Patient 2 | HLA‐A*0201/A*3101 | ||
| Patient 3 | HLA‐A*0101 | ||
| Patient 4 | HLA‐A*2402/A*3002 | ||
| (b) HLA‐type healthy donors | |||
| HD‐1 | HLA‐A*0201/A*2402 | ||
| HD‐2 | HLA‐A*0201/A*2402 | ||
| HD‐3 | HLA‐A*0201 | ||
Induction of tumour antigen‐specific CD8+ T cells by IFN DCs
To investigate major histocompatibility complex (MHC)‐restricted immune responses mediated by CD8+ T cells, HLA‐A*0101‐restricted, CMVpp65‐modified 11‐mer synthetic peptides (YSEHPTFTSQY), HLA‐A*0201‐restricted, CMVpp65‐9‐mer synthetic peptides (NLVPMVATV) and HLA‐A*2402‐restricted, CMVpp65‐9‐mer synthetic peptides (VYALPLKML) (Proimmune, Oxford, UK) were used. Lymphocytes (2 × 106) as a non‐adherent cell fraction were cultured with autologous IFN DCs (2 × 105) under different conditions. IFN DCs were pulsed with the HLA‐A*0101‐, HLA‐A*0201‐ or HLA‐A*2402‐restricted P (5 μg/ml) for 2 h in serum‐free medium and then cultured with the lymphocytes possessing HLA‐A*0101, HLA‐A*0201 or HLA‐A*2402. In some experiments, Z (0.1 μM) and/or G (100 ng/ml) was added during the culture. The cultures were supplemented with 50 U/ml IL‐2 (Chiron Benelux B. V., Hengelo, the Netherlands) during culture. The percentage of CMVpp65‐specific CD8+ cells was assessed using a PE‐CMVpp65 pentamer (Proimmune) and FITC‐anti‐CD8 mAb (Beckton Dickinson).
Cytotoxicity assay
The T98G tumour cell line was purchased from the American Type Culture Collection (Manassas, VA, USA). Fluorochromasia cell‐mediated cytotoxicity assay was carried out using Terascan VPC (Minerva Tech., Fukuoka, Japan), according to the manufacturer’s instructions. Cells were labelled with calcein‐AM solution (Dojindo Laboratories, Kumamoto, Japan) and used as target cells at E/T rations of 0·6, 1·3, 2·5, 5, 10, 20 and 40 for 2 h and the cytotoxic activity was evaluated by the release of fluorochromasia into the medium 27.
Statistical analyses
The P‐values for analysis of the expressions of PD‐L1, PD‐L2 and CCR7 on DCs were calculated using paired/two‐tailed Student’s t‐test and considered highly significant at < 0·01. In some cases, the P‐values for analysis of the expression of CCR7 were calculated using Bonferroni’s multiple‐comparison test and considered highly significant at < 0·01.
Results
Phenotypical analysis of IFN DCs derived from monocytes as adherent cells in the presence of IFN‐α and GM‐CSF
To characterize surface antigens on IFN DCs, in comparison with those on mIL‐4 DCs, IFN DCs and mIL‐4 DCs were obtained from four patients with GBM and three HDs. As representative results from patient 1, the IFN DCs showed an almost identical phenotypical pattern to mIL‐4 DCs expressing high levels of HLA class I, HLA‐DR, CD80, CD86, CD40, CD54, CD11c and CD36 (data not shown), but not CD56, CD14, PD‐L1, PD‐L2 and CCR7 (Fig. 1a). The expression levels of CD56 and CD14 on IFN DCs from patients with GBM were significantly higher than those on mIL‐4 DCs (data not shown), as reported previously in patients with various cancers 12, 28. Phenotypical patterns of IFN DCs from HDs are very similar to those from patients regarding the expression levels of HLA‐class I, HLA‐DR, CD80, CD86, CD40, CD54, CD11c, CD36, CD56, CD14, PD‐L1, PD‐L2 and CCR7, as described previously 28.
Figure 1.
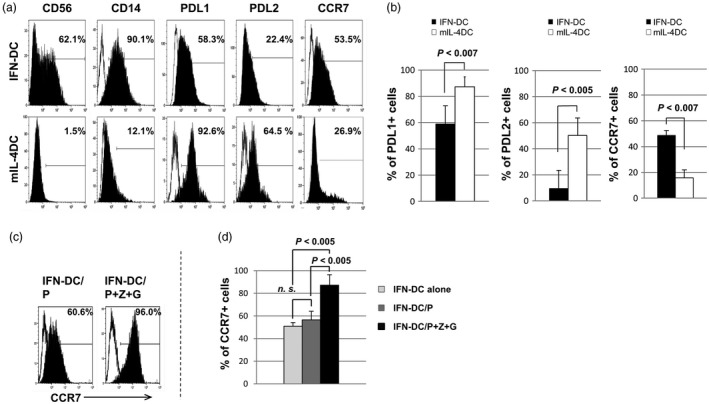
Phenotypical patterns of interferon dendritic dells (IFN DCs) versus mature interleukin (mIL)‐4 DCs. (a) Representative data on phenotypical patterns of CD56, CD14, programmed death ligand 1 (PD)‐L1, PD‐L2 and C‐C receptor 7 (CCR7) on IFN DC and mIL‐4 DCs from patient 1 with glioblatoma multiforme (GBM). (b) Statistical analysis using of data from four patients with GBM using the paired/two‐tailed Student’s t‐test showed that the percentages of PD‐L1+ cells and PD‐L2+ cells among IFN DCs were significantly lower than those among mIL‐4 DCs, and the percentage of CCR7+ cells among IFN DCs was significantly higher than those among mIL‐4 DCs. (c) The expression level of CCR7 on IFN DCs from patient 1 gradually increased following addition of the CMV66‐peptide (P) alone, and the addition of the cytomegalovirus (CMV)pp65‐peptide (P) and zoledronate (Z) plus α‐galactosylceramide (G). (d) Statistical analysis of data from four patients with GBM using Bonferroni’s multiple‐comparison test showed that the percentage of CCR7+ cells among IFN DCs/P was not significantly (n.s.) higher than that among IFN DCs alone. The percentage of CCR7+ cells among IFN DCs/P+Z+G was significantly higher than that among IFN DCs alone (P < 0·005) and was significantly higher than that among IFN DCs/P (P < 0·005).
The expression levels of PD‐L1 and PD‐L2, which are considered to mediate inhibitory signals predominantly towards T cells 29 on IFN DCs, were significantly lower than those on mIL‐4 DCs (Fig. 1b), indicating that IFN DCs have a higher ability to expand T cells than mIL‐4 DCs. Furthermore, the expression level of lymph node homing receptor CCR7 on IFN DCs was significantly higher than that on mIL‐4 DCs (Fig. 1b). The expression level of CCR7 on IFN DCs/P+Z+G was significantly higher than that on IFN DCs/P (Fig. 1c,d). These results suggest that IFN DCs pulsed with a tumour antigen and Z+G have a higher ability to migrate to the lymph nodes, in comparison with IFN DCs pulsed with a tumour antigen.
Effective induction of CMVpp65‐specific CD8+ T cells by IFN DCs/P+Z+G derived from patients with GBM
The induction of CMVpp65‐specific CD8+ T cells by autologous IFN DCs was examined under four different conditions: (1) IFN DCs/P, (2) IFN DCs/P+Z, (3) IFN DCs/P+G and (4) IFN DCs/P+Z+G. The case of patient 1 possessing HLA‐A*0201 and HLA‐A*0101 before culture each population is shown in Fig. 2a. Before culture, percentages of HLA‐A*0201‐restsricted CMVpp65‐specific CD8+ T cells and HLA‐A*0101‐restricted CMVpp65‐specific CD8+ T cells among CD8+ T cells were 0·62 and 0·17%, respectively, and percentages of CD8+ T cells, Vγ9γδT cells and Vα24NKT cells among lymphocytes were 30·00, 1·71 and 0·06%, respectively. After culture for 10 days, the results are shown in Fig. 2b. Remarkably, IFN DCs expanded most CMVpp65‐specific CD8+ T cells extensively when pulsed with HLA‐A*0201‐restricted P and Z+G, compared with other different conditions mentioned above. In addition, using IFN DCs/P+Z+G, expansions of Vγ9γδT and Vα24NKT cells were also observed (Fig. 2b).
Figure 2.
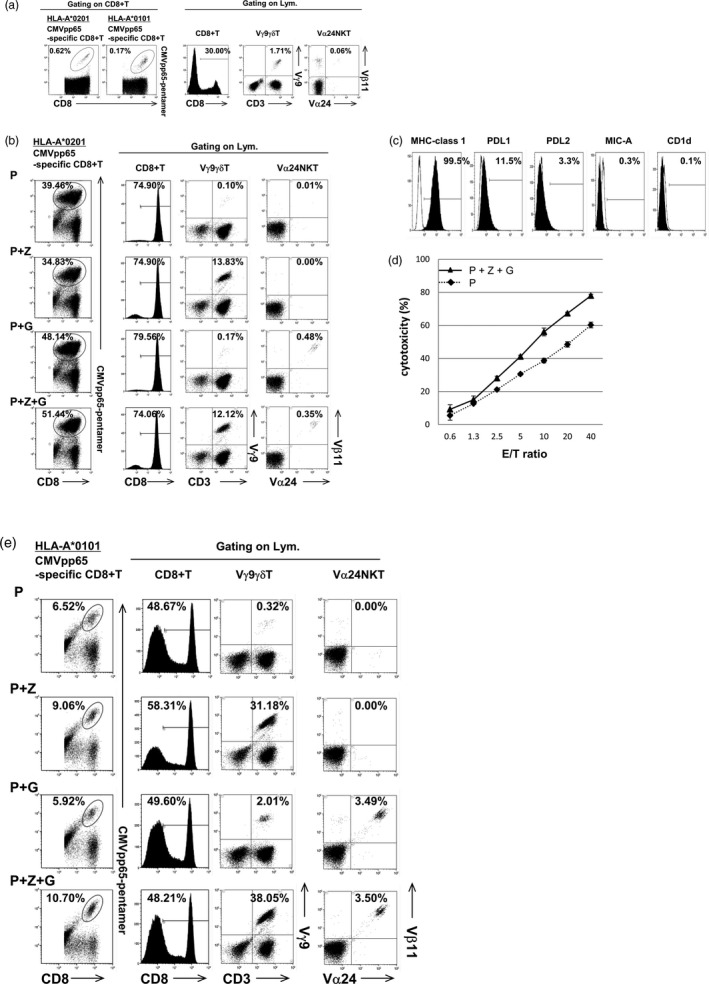
Effective induction of cytomegalovirus (CMV)pp65‐specific CD8+ T cells by interferon dendritic dells/peptide and zoledronate plus α‐galactosylceramide (IFN DCs/P+Z+G) derived from patients with glioblastoma multiforme (GBM). (a) Each cell population was shown in the case of patient 1 possessing human leucocyte antigen (HLA)‐A*0201 and HLA‐A*0101, before culture. (b) After culture, IFN DCs from patient 1 induced CMVpp65‐specific CD8+ T cells most extensively when pulsed with HLA‐A*0201‐restricted P and Z+G in addition to the expansion of Vγ9γδT and Vα24NKT cells, in comparison with the other three different conditions (P, P+Z and P+G). (c) Expression levels of major histocompatibility complex (MHC) class I, programmed cell death ligand 1 (PD‐L1), PD‐L2, MHC class I‐related chain A (MIC‐A) and CD1d on the T98G tumour cell line established from a patient with GBM used as target cells are shown. (d) The effector cells including CD8+ T cells containing CMVpp65‐specific CD8+ T, Vγ9γδT and Vα24NKT cells (as shown in Fig. 2b with P+Z+G) killed the T98G tumour cell line more efficiently in a dose‐dependent manner, in comparison with the effector cells including mainly CD8+ T cells containing CMVpp65‐specific CD8+ T cells (as shown in Fig. 2b with P alone). (e) After culture, IFN DCs from patient 1 induced CMVpp65‐specific CD8+ T cells most extensively when pulsed with HLA‐A*0101‐restricted P plus Z+G, in addition to the expansion of Vγ9γδT and Vα24NKT cells, in comparison with the other three different conditions (P, P+Z and P+G).
To verify our own pentamer results, we assessed the antigen‐specific cytotoxic activity of lymphocytes containing 51·44% HLA‐A*0201‐restricted CMVpp65‐specific CD8+ T cells, 12·12% Vγ9γδT cells and 0·35% Vα24NKT cells with P+Z+G (Fig. 2b with P+Z+G) versus that of lymphocytes containing 39·46% HLA‐A*0201‐restricted CMVpp65‐specific CD8+ T cells, 0·10% Vγ9γδT cells and 0·01% Vα24NKT cells (Fig. 2b with P). Expression levels of MHC class 1, PD‐L1, PD‐L2, MHC class I‐related chain A (MIC‐A), which is a ligand for NKG2D on Vγ9γδ T cells, and CD1d, which is a ligand for Vα24NK T cells, on the T98G tumour cell line used as target cells were 99·5, 11·5, 3·3, 0·3 and 0·1%, respectively (Fig. 2c).
As shown in Fig. 2d, the effector cells including CD8+ T cells containing CMVpp65‐specific CD8+ T, Vγ9γδ T and Vα24NK T cells (Fig. 2b with P+Z+G) killed the T98G cell line more efficiently in a dose‐dependent manner in comparison with the effector cells including mainly CD8+ T cells containing CMVpp65‐specific CD8+ T cells (Fig. 2b with P). This killing activity was not enhanced in the presence of anti‐PD‐L1 and/or anti‐PD‐L2m Ab, owing presumably to the low expression levels of PD‐L1 and PD‐L2 on the T98G cell line, as shown in Fig. 2c.
Furthermore, we conducted experiments using IFN DCs from patient 1 pulsed with the HLA‐A*0101‐restricted P. The results were similar to those from patient 1 pulsed with HLA‐A*0201‐restricted P, showing that IFN DCs expanded CMVpp65‐specific CD8+ T cells most extensively when pulsed with HLA‐A*0101‐restricted P and Z+G, in addition to the expansions of Vγ9γδT and Vα24NKT cells, compared with the other three different conditions mentioned above (Fig. 2e). We conducted further experiments using IFN DCs from another HLA‐A*0201‐positive patient 2 and observed that the percentage of HLA‐A*0201‐restricted CMVpp65‐specific CD8+ T cells among the expanded CD8+ T cells expanded by IFN DCs pulsed with HLA‐A*0201‐restricted P+Z+G (40·37%) was higher than that expanded by IFN DCs pulsed with P (22·34%). In another HLA‐A*0101‐positive patient 3 using the HLA‐A*0101‐restricted P, the induction of CMVpp65‐specific CD8+ T cells by IFN DCs/P+Z+G was also higher than that by IFN DCs/P (21·61% for IFN DCs/P+Z+G versus 15·43% for IFN DCs/P). We also tested HLA‐A*2402‐positive patient 4 using the HLA‐A*2402‐restricted peptide. The results showed that the induction of CMVpp65‐specific CD8+ T cells by IFN DCs/P+Z+G was also higher than that by IFN DCs/P (24·30% for IFN DCs/P+Z+G versus 14·31% for IFN DCs/P). Taken together, the induction of CMVpp65‐specific CD8+ T cells by IFN DCs/P+Z+G was higher than that by IFN DCs/P in all cases using HLA‐A*0201‐restricted, HLA‐A*0101‐restricted and HLA‐A*2402‐restricted P. Note that remarkable expansion of Vγ9γδT and Vα24NKT cells was observed in all patients using IFN DCs pulsed with P+Z+G.
For all patients, it was observed that the total number of lymphocytes expanded by IFN DCs/P+Z+G was larger than that expanded by IFN DCs/P (Table 2: experiments 1–5). This resulted in the higher induction of CMVpp65‐specific CD8+ T cells using IFN DCs/P+Z+G, in comparison with the use of IFN DCs/P (Table 2: experiments 1–5). Note that in the cases of stimulation by IFN DCs/P+Z+G from patient 1, the total number of lymphocytes expanded was larger than those of stimulation by DCs under the other three conditions: with P, P+Z and P+G (Table 2: experiments 1 and 2). This provided the highest induction of CMVpp65‐specific CD8+ T cells in the case of using IFN DCs/P+Z+G. Furthermore, using IFN DCs/P+Z+G for five experiments from four patients, the number of Vγ9γδT cells among lymphocytes expanded was eight‐ to 198‐fold larger than that in using IFN DCs/P (Table 3). Also, the number of Vα24NKT cell‐expanded IFN DCs/P+Z+G was larger than that expanded by IFN DCs/P, although the number was small in any conditions (data not shown). Moreover, the percentage of CD4+CD25+FoxP3+ Tregs among lymphocytes following culture with IFN DCs was examined. The results show that the percentage of CD4+CD25+FoxP3+ Tregs among lymphocytes following culture with IFN DCs/P+Z+G was lower than that following culture with IFN DCs/P in patients 1, 2 and 4 tested so far (Fig. 3a). Statistical analysis showed that the CD4+CD25+FoxP3+ Tregs among lymphocytes following culture with IFN DCs/P+Z+G was significantly lower than the percentage of the Tregs among lymphocytes following culture with IFN DCs/P (4·22 ± 2·00% versus 5·05 ± 1·93%, P < 0·01) (Fig. 3b).
Table 2.
Numbers of total lymphocytes and cytomegalovirus (CMV)pp65‐specific CD8+ T cells following culture with interferon dendritic dells (IFN DCs) derived from patients with GBM before and after culture under different conditions [with the CMVpp65‐peptide alone (P), the peptide with Z (P+Z), the peptide with G (P+G) and the peptide and Z+G (P+Z+G)]
| Patients | Peptide used | Culture | Agents | No. of total lym. | No. of CMV‐specific CD8+ T | ||
|---|---|---|---|---|---|---|---|
| A*0201 | A*0101 | A*2402 | |||||
| Experiment 1 | A*0201 | Before | None | 2.0 × 106 | 37.2 × 102 | n.t. | n.t. |
| Patient 1 | After | P | 14.0 × 106 | 41.5 × 105 | n.t. | n.t. | |
| P+Z | 11.2 × 106 | 29.2 × 105 | n.t. | n.t. | |||
| P+G | 10.9 × 106 | 41.7 × 105 | n.t. | n.t. | |||
| P+Z+G | 14.1 × 106 | 53.7 × 105 | n.t. | n.t. | |||
| Experiment 2 | A*0101 | Before | None | 2.0 × 106 | n.t. | 10.2 × 102 | n.t. |
| Patient 1 | After | P | 6.8 × 106 | n.t. | 21.5 × 104 | n.t. | |
| P+Z | 9.8 × 106 | n.t. | 51.7 × 104 | n.t. | |||
| P+G | 6.8 × 106 | n.t. | 19.9 × 104 | n.t. | |||
| P+Z+G | 11.5 × 106 | n.t. | 56.8 × 104 | n.t. | |||
| Experiment 3 | A*0201 | Before | None | 2.0 × 106 | 8.2 × 102 | n.t. | n.t. |
| Patient 2 | After | P | 9.8 × 106 | 5.1 × 105 | n.t. | n.t. | |
| P+Z+G | 12.4 × 106 | 22.7 × 105 | n.t. | n.t. | |||
| Experiment 4 | A*0101 | Before | None | 2.0 × 106 | n.t. | 4.1 × 102 | n.t. |
| Patient 3 | After | P | 7.2 × 106 | n.t. | 22.4 × 104 | n.t. | |
| P+Z+G | 10.6 × 106 | n.t. | 67.3 × 104 | n.t. | |||
| Experiment 5 | A*2402 | Before | None | 2.0 × 106 | n.t. | n.t. | 2.3 × 102 |
| Patient 4 | After | P | 10.1 × 106 | n.t. | n.t. | 35.7 × 104 | |
| P+Z+G | 12.8 × 106 | n.t. | n.t. | 80.5 × 104 | |||
Table 3.
Numbers of total lymphocytes and Vγ9γδT cells following culture with interferon dendritic dells (IFN DCs) derived from patients with glioblatoma multiforme (GBM) before and after culture under different conditions [with the cytomegalovirus (CMV)pp65‐peptide alone (P) and the peptide and Z+G (P+Z+G)]
| Patients | Peptide used | Culture | Agents | No. of Vγ9γδT |
|---|---|---|---|---|
| Experiment 1 | A*0201 | Before | None | 3·4 × 104 |
| Patient 1 | After | P | 1·4 × 104 | |
| P+Z+G | 170·9 × 104 | |||
| Experiment 2 | A*0101 | Before | None | 3·4 × 104 |
| Patient 1 | After | P | 2·2 × 104 | |
| P+Z G | 437·6 × 104 | |||
| Experiment 3 | A*0201 | Before | None | 3·7 × 104 |
| Patient 2 | After | P | 27·8 × 104 | |
| P+Z+G | 232·0 × 104 | |||
| Experiment 4 | A*0101 | Before | None | 4·1 × 104 |
| Patient 3 | After | P | 6·1 × 104 | |
| P+Z+G | 406·6 × 104 | |||
| Experiment | A*2402 | Before | None | 4·1 × 104 |
| Patient 4 | After | P | 7·5 × 104 | |
| P+Z+G | 462·2 × 104 |
Figure 3.
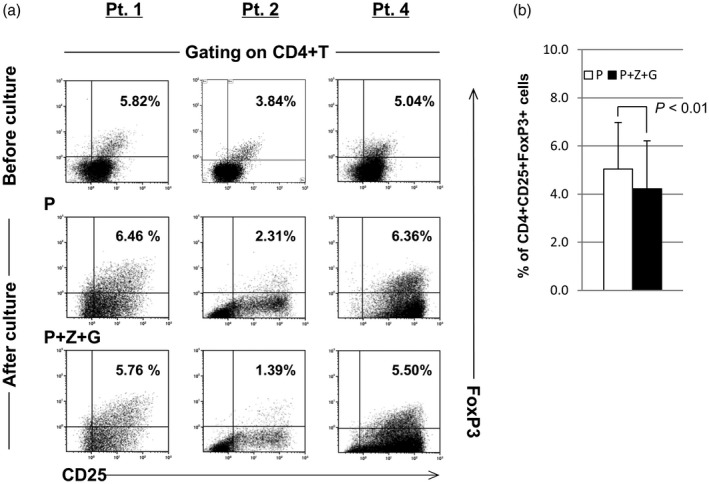
(a) The percentage of CD4+CD25+forkhead box protein 3 (FoxP3)+ regulatory T cells (Tregs) among lymphocytes following culture with interferon dendritic dells/peptide and zoledronate plus α‐galactosylceramide (IFN DCs/P+Z+G) from patients (patients 1, 2 and 4) was lower than that following culture with IFN DCs/P. (b) Statistical analysis showed that the percentage of CD4+CD25+FoxP3+ Tregs among lymphocytes following culture with IFN DCs/P+Z+G from patients was significantly lower than the percentage of CD4+CD25+FoxP3+ Tregs among lymphocytes following culture with IFN DCs/P (4·22±2·00% versus 5·05±1·93%, P < 0·01).
Immune checkpoint blockade with monoclonal antibodies against the inhibitory immune receptors of CTLA‐4 and PD‐1 has emerged as a successful treatment approach for patients with advanced cancer 30. Considering the use of a combination of immunotherapies such as DC and CTL therapy with monoclonal antibodies against CTLA‐4 and PD‐1, we determined the expression levels of PD‐1 and CTLA‐4 on CD8+ T cells including CMVpp65‐specific CD8+ T cells using IFN DCs from patients 1, 2, 3 and 4. Before culture for patients, the expression levels of PD‐1 for four patients and CTLA‐4 for three patients on CD8+ T cells were lower than 1% (Fig. 4a). Following culture with IFN DCs under two conditions (with P and with P+Z+G), the expression level of PD‐1 on CD8+ T cells including CMVpp65‐specific CD8+ T cells increased markedly, in which the expression level of PD1 on CD8+ T cells following stimulation by IFN DCs/P was higher than that following stimulation by IFN DCs/P+Z+G (for patient 1, 72·91 versus 65·78%; for patient 2, 63·61 versus 40·72%; for patient 3, 61·10 versus 54·74%; for patient 4, 77·21 versus 58·31%) (Fig. 4a). Statistical analysis showed that the expression level of PD1 on CD8+ T cells following stimulation by IFN DCs/P+Z+G was statistically lower than that following stimulation by IFN DCs/P (54·89 ± 9·09% versus 68·71 ± 6·59%, P < 0·05), as shown in Fig. 4b. In contrast, the expression level of CTLA‐4 on CD8+ T cells, including CMVpp65‐specific CD8+ T cells, was less than 3% under both conditions (Fig. 4a). Taken together, these results suggest that among different conditions, IFN DCs/P+Z+G in combination with anti‐PD‐1 mAb may be able to induce a large number of PD‐1‐negative CD8+ T cells in vitro and in vivo.
Figure 4.
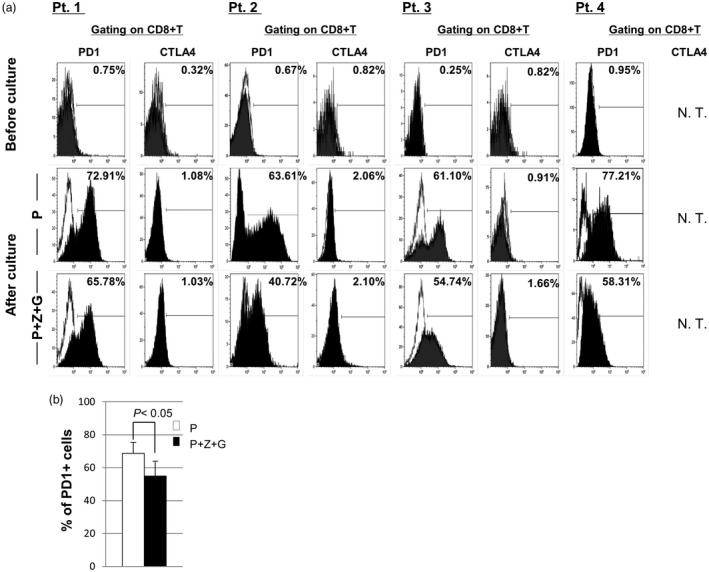
The expression levels of programmed cell death 1 (PD)‐1 and cytotoxic T lymphocyte (CTLA)‐4 on CD8+ T cells before and after culture with patient (patients 1, 2, 3 and 4)‐derived interferon dendritic dells/peptide (IFN DCs/P) versus patient‐derived IFN DCs/P+Z+G. (a) The expression levels of PD‐1 and CTLA‐4 on CD8+ T cells including cytomegalovirus (CMV)pp65‐specific CD8+ T cells before culture were less than 1%. After culture, the expression level of PD‐1 on CD8+ T cells increased intensively, but the expression levels of CTLA‐4 were less than 2·1% under both conditions (IFN DCs/P and IFN DCs/P+Z+G). (b) The expression level of PD‐1 on CD8+ T cells following stimulation by IFN DCs/P+Z+G was significantly lower than that following stimulation by IFN DCs/P (54·89±9·09% for IFN DCs/P+Z+G versus 68·71±6·59% for IFN DCs/P, P < 0·05).
Effective induction of CMVpp65‐specific CD8+ T cells by IFN DCs/P+Z+G derived from HDs
We further tested the induction ability of CMV‐specific CD8+ T cells using HLA‐A*0201‐positive IFN DCs from three HDs. The results are similar to those from the patients; that is, the ability of IFN DCs/P+Z+G to induce CMV‐specific CD8+ T cells was higher than that of IFN DCs/P [Fig. 5a (HD1) and Fig. 5b (HD2)]. In the case of HD3, no induction of CMV‐specific CD8+ T cells by IFN DCs under any conditions was observed, but the induction level of Vγ9γδT and Vα24NKT cells increased in using IFN DCs/P+Z+G (data not shown). It is important to note that for three HDs, the percentage of CD4+CD25+FoxP3+ Tregs among lymphocytes following their co‐culture with IFN DCs/P+Z+G was lower than that following their co‐culture with IFN DCs/P (Fig. 5c). Similar to the results for patients as shown in Table 2, the number of CMV‐specific CD8+ T cells expanded by HD‐derived IFN DCs/P+Z+G was larger than those expanded by IFN DCs/P (data not shown).
Figure 5.
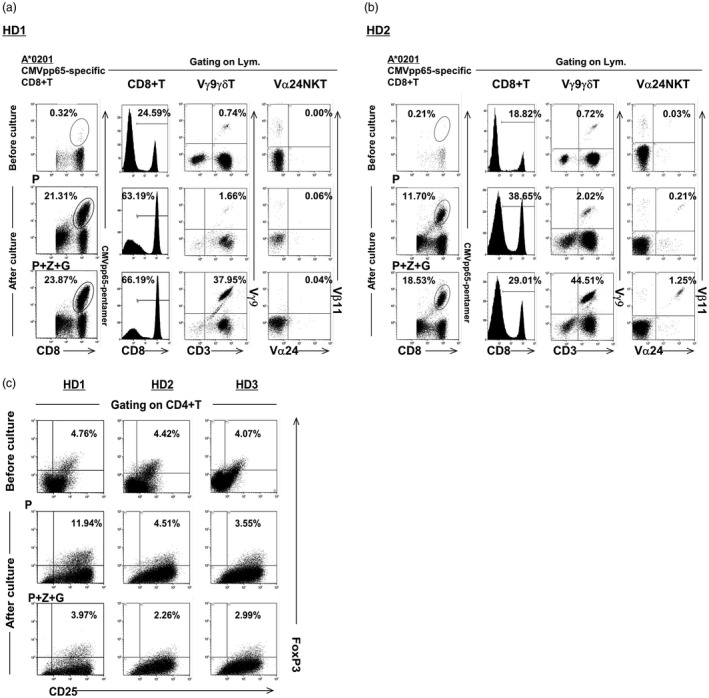
Effective induction of cytomegalovirus (CMV)pp65‐specific CD8+ T cells by healthy donor (HD)‐derived interferon dendritic dells/peptide and zoledronate plus α‐galactosylceramide (IFN DCs/P+Z+G). (a) The ability of IFN DCs from human leucocyte antigen (HLA)‐A*0201‐positive HD‐1 pulsed with HLA‐A*0201‐restricted P and Z+G to induce CMV‐specific CD8+ T cells was higher than that of IFN DCs/P. Using IFN DCs/P+Z+G, the expansions of Vγ9γδT and Vα24NKT cells were also observed. (b) The ability of IFN DCs from HLA‐A*0201‐positive HD2 pulsed with HLA‐A*0201‐restricted P+Z+G to induce CMV‐specific CD8+ T cells was higher than that of IFN DCs/P. Using IFN DCs/P+Z+G, expansions of Vγ9γδT and Vα24NKT cells were also observed. (c) The percentage of CD4+CD25+forkhead box protein 3 (FoxP3)+regulatory T cells (Tregs) among lymphocytes from three HLA‐A*0201‐positive HDs (HD‐1, H‐2 and HD‐3) following co‐culture with autologous IFN DCs pulsed with HLA‐A*0201‐restricted P+Z+G) was lower than that following co‐culture with autologous IFN DCs/P.
Discussion
In this study, we demonstrate that IFN DCs/P+Z+G from patients with GBM and HD cross‐talk with CMVpp65‐specific CD8+ T cells, Z‐activated Vγ9γδT cells and G‐activated Vα24NK T cells in a manner that amplifies the activation and proliferation of CMVpp65‐specific CD8+ T cells. This has a number of important implications, some of which may have a significant impact on the use of Z and G in the setting of cancer therapy and represents a best‐case scenario for immunotherapies, such as DC and CTL therapies, because a large number of tumour antigen‐specific T cells may then be available in vitro and in vivo. As noted, the expansion ability of CMVpp65‐specific CD8+ T cells by IFN DCs pulsed with HLA‐A*0201‐restricted P and Z+G was stronger than that by IFN DCs pulsed with HLA‐A*0101‐restricted P and Z+G, due presumably to a higher frequency of HLA‐A*0201‐restricted CMVpp65‐specific CD8+ T cells before culture (0·62% for HLA‐A*0201‐restricted versus 0·17% for HLA‐A*0101‐restricted peptide), as shown in Fig. 2a.
The use of IFN DCs in this study had some advantages compared with mIL‐4 DCs. Our results showed that the expression levels of PD‐L1 and PD‐L2 on IFN DCs were significantly lower than those on mIL‐4 DCs (Fig. 1b). This indicates that inhibitory signals from IFN DCs to T cells are weaker than those from mIL‐4 DCs, which may be associated with the effective induction of tumour antigen‐specific CD8+ T cells by IFN DCs in vitro and in vivo.
The expression of CCR7 on DCs is essentially required for DC trafficking from peripheral tissues to draining lymph nodes, constituting a central determinant of DC migration and function 31. In this study, IFN DCs/P+Z+G expressed CCR7 highly (Fig. 1c,d). This indicates a high trafficking activity of IFN DCs/P+Z+G across endothelial cells to lymph nodes, where they interact with T cells, including antigen‐specific CD8+ T, Vγ9γδT and Vα24NKT cells.
Current immunotherapies focus on promising a strong anti‐tumour T cell response by altering the T cells themselves, either by increasing tumour‐specific T cell frequency by adoptive cell transfer 32 with or without DCs, increasing the reactivity of responding cells by engineering of high‐avidity TCRs specific for tumours 33, or by eliciting more potent T cells by blockade of inhibitory co‐stimulatory molecules such as CTLA‐4 or PD‐1 30. Ipilimumab (anti‐CTLA‐4 mAb) is the first anti‐cancer agent associated with a documented improved overall survival benefit in patients with advanced melanoma. Many tumour responses achieved by PD‐1 and PD‐L1 inhibition were durable and were seen in a higher proportion of patients with melanoma than observed typically with ipilimumab 34. Nivolumab was the first mAb targeting PD‐1 to show significant clinical activity in unresectable or metastatic melanoma, non‐small‐cell lung carcinoma and metastatic renal cell carcinoma 35.
In this study, we confirmed that the expression level of PD‐1 on CD8+ T cells expanded by IFN DCs/P+Z+G was significantly lower than that on CD8+ T cells expanded by IFN DCs/P and that the expression level of CTLA‐4 on CD8+ T cells was very low under both conditions (Fig. 4a,b). Under consideration of DC, therapy together with an immunocheckpoint blockade, our results suggest that in the case of DC therapy using IFN DCs/P+Z+G, administration of a small amount of anti‐PD‐1mAb such as nivolumab, not anti‐CTL‐A4 mab such as ipilimumab, may result in higher induction of PD‐1‐negative CMV‐specific CD8+ T cells in vivo.
In a previous study, CMV‐specific T cells were expanded using PBMCs from patients with GBM and infused into the patients 36. T cells, including CMV‐specific T cells, were prepared by culturing PBMC with autologous PBMCs presensitized with the CMV‐peptide and IL‐2. Then, 25 to 40 × 106 autologous CMV‐specific T cells/infusion were infused. This previous study suggests that combination therapy with autologous CMV‐specific T cells and chemotherapy is a safe novel treatment and may provide clinical benefits to patients with recurrent GBM. Considering CTL therapy using CMVpp65‐specific CD8+ T cells expanded by IFN DCs/P+Z+G, more than 30 × 106 of CMVpp65‐specific CD8+ T cells will be harvested easily when 20–30 × 106 lymphocytes are cultured with IFN DCs/P+Z+G. Usually, 36·0 ± 10·3 × 106 lymphocytes are collected from 50 ml of peripheral blood (our own observation; n = 71). Therefore, it is not difficult to prepare a sufficient number of CMV‐specific CD8+ T cells per injection for patients with GBM when lymphocytes are cultured with IFN DCs/P+Z+G. Furthermore, in our study using IFN DCs/P+Z+G, a 500–2700‐fold expansion of CMVpp65‐specific T cells was completed (Table 2), which is much higher than using DCs transfected with CMVpp65‐RNA, as reported previously 37.
A recent study provides strong evidence supporting the potential of combinatorial targeted and/or immunotherapeutic regimens in patients with GBM that may change patient outcome 38, 39. Our results suggest that a combination of immune checkpoint inhibitors, particularly anti‐PD‐1 mAb, and DC therapy as well as CTL therapy, leads to a better clinical outcome for patients with GBM. In future, the therapeutic pillar of checkpoint blockade will probably keep its central role in cancer immunotherapy. Therefore, adaptive immunotherapies such as DC and CTL therapy combined with checkpoint blockade will play important roles in treatment of cancer patients, including GBM patients.
Identification of the molecular drivers of T cell dysfunction is essential for continued progress of cancer research and therapy. Among different immunosuppressive cell populations in the TME, Tregs accumulate frequently in tumour microenvironment and even represent the major populations of infiltrating CD4+ T cells 22. Depletion of Tregs or disruption of Treg differentiation may restore anti‐tumour T cell responses and immune surveillance against cancer. In this study, the induction of CD4+CD25+FoxP3+ Tregs among the lymphocytes expanded by IFN DCs/P+Z+G decreased following stimulation by IFN DCs/P+Z+G in comparison with stimulation by IFN DCs/P (Fig. 3a,b). These results indicate that the adjuvant effect of Vα24NKT and Vγ9γδT cells may be associated partially with the decrease in the percentage of CD4+CD25+FoxP3+ Tregs among the lymphocytes expanded. Taken together, our results suggest that DC therapy using DCs pulsed with tumour antigen and Z+G is a promising approach which results in the increase in the number of tumour antigen‐specific CD8+ T cells and in the decrease in that of CD4+CD25+FoxP3+ Tregs, although the more precise phenotypical and functional analysis of Tregs are warranted, as shown in the previous study 40.
In this study, the results of HDs are very similar to those of patients, showing that a large number of CMVpp65‐specific CD8+ T cells from HDs were harvested following activation of lymphocytes with autologous IFN DCs pulsed with the CMVpp65‐peptide and Z+G in vitro. Thus, infusion of the CMVpp65‐specific CD8+ T cells expanded by IFN DCs derived from allogenic HDs to patients with haematological malignancy who received alloSCT may decrease the relapse risk, although side effects should be considered carefully.
The adjuvant effects of a combination of Z‐activated Vγ9γδT cells with G‐activated Vα24NKT cells on antigen‐specific CD8+ T cells were similar when stimulator IFN DCs were washed to remove extracellular tumour antigens and Z+G, as when lymphocytes were cultured with IFN DCs pulsed with the tumour antigen and Z+G. These observations support the possible therapeutic applications of IFN DCs pulsed with a tumour antigen and Z+G. The observed favourable in‐vitro expansion of CMVpp65‐specific CD8+ T cells in response to DCs with IL‐2 indicates that additional, small amounts of exogenous IL‐2 may be required to maximize the immunotherapeutic benefits of DCs pulsed with a tumour antigen and Z+G in clinical settings.
Furthermore, it has been shown recently that, similarly to DCs, Vγ9γδT cells present antigens to αβ T cells 41, 42. It is conceivable the Vγ9γδT cells activated in vivo, for example, by infused DCs pulsed with a tumour antigen and Z+G, might play a role as APCs in the uptake of apoptotic bodies from tumour cells. They subsequently induce tumour antigen‐specific CD8+ T cells in vivo in which Vα24NKT cells may function as helper T cells producing large amounts of IFN‐γ, as shown in our previous clinical study 4. Note that the concurrent activation and proliferation of Vγ9γδT cells by Z is MHC‐unrestricted 43 and that Vα24NKT cells recognize G presented by non‐polymorphic CD1d molecules 3. Therefore, the adjuvant effect of the combination of Z and G is theoretically applicable to all cancer patients.
In summary, our study demonstrates the adjuvant effect of a combination of Z and G, resulting in the expansions of Vγ9γδT and Vα24NKT cells upon induction of antigen‐specific CD8+ T cells using IFN DCs/P+Z+G. This effect may be a physiologically and therapeutically important bridge between innate and adaptive immunities through DCs. Our results are consistent with accumulating evidence from clinical and laboratory studies, indicating that heterogeneous activation of lymphocytes shapes the responses to vaccination. Furthermore, our observations are anticipated to facilitate the design of more effective immune therapy strategies, particularly DC therapy combined with immunocheckpoint blockades such as anti‐PD1 mAbs for patients with GBM.
Disclosure
The authors have declared no conflicting interests.
Author contributions
Y. E. performed the experiments, analysed the data and prepared figures, H. T. designed the experiments, M.Y. analysed the data and prepared figures, X. D. designed the experiments, A. N. conceived the study and designed the experiments and M.N. conceived the study, performed the experiments, analysed the data and wrote the manuscript.
References
- 1. Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol 2005; 5:296–306. [DOI] [PubMed] [Google Scholar]
- 2. Conrad C, Nestle FO. Dendritic cell‐based cancer therapy. Curr Opin Mol Ther 2003; 5:405–12. [PubMed] [Google Scholar]
- 3. Spada FM, Koezuka Y, Porcelli SA. CD1d‐restricted recognition of synthetic glycolipid antigens by human natural killer T cells. J Exp Med 1998; 188:1529–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nieda M, Okai M, Tazbirkova A et al. Therapeutic activation of Vα24+Vβ11+NK T cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood 2004; 103:383–89. [DOI] [PubMed] [Google Scholar]
- 5. Takahara M, Miyai M, Tomiyama M, Mutou M, Nicol AJ, Nieda M. Copulsing tumor antigen‐pulsed dendritic cells with zoledronate efficiently enhance the expansion of tumor antigen specific CD8+T cells via Vγ9γδT cell activation. J Leuk Biol 2008; 83:742–54. [DOI] [PubMed] [Google Scholar]
- 6. Castella B, Riganti C, Fior F et al. Immune modulation by zoledronic acid in human myeloma: an advantageous cross‐talk between Vγ9Vδ2T cells, αβCD8+T cells, regulatory T cells, and dendritic cells. J Immunol 2011; 187:1578–90. [DOI] [PubMed] [Google Scholar]
- 7. Nestle FO, Alijagic S, Gillet M et al. Vaccination of melanoma patients with peptide‐ or tumor lysate‐pulsed dendritic cells. Nat Med 1998; 4:328–32. [DOI] [PubMed] [Google Scholar]
- 8. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/machrophage colony‐stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med 1994; 179:1109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paquette RL, Hsu NC, Kiertscher SM et al. Interferon‐alpha and granulocyte–macrophage colony‐stimulating factor differentiate peripheral blood monocytes into potent antigen‐presenting cells. J Leuk Biol 1998; 64:358–67. [DOI] [PubMed] [Google Scholar]
- 10. Farkas A, Kemeny L. Interferon‐α in the generation of monocyte‐derived dendritic cells: recent advances and implications for dermatology. Br J Dermatol 2011; 165:247–54. [DOI] [PubMed] [Google Scholar]
- 11. Gruenbacher G, Gander H, Rahm A, Nussbaumer W, Romani N, Thurnher M. CD56+ human blood dendritic cells effectively promote Th1‐type γδT cells responses. Blood 2009; 114:4422–31. [DOI] [PubMed] [Google Scholar]
- 12. Nieda M, Terunuma H, Eiraku Y, Deng X, Nicol AJ. Effective induction of melanoma‐antigen‐specific CD8+T cells via Vγ9γδT cell expansion by CD56high+ interferon‐γ‐induced dendritic cells. Exp Dermatol 2015; 24:35–41. [DOI] [PubMed] [Google Scholar]
- 13. Imperato JP, Paleologos NA, Vick NA. Effects of treatment on long‐term survivors with malignant astrocytomas. Ann Neurol 1990; 28:818–822. [DOI] [PubMed] [Google Scholar]
- 14. Stupp R, Mason WP, van den Bent MJ et al . Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987–996. [DOI] [PubMed] [Google Scholar]
- 15. Prins RM, Cloughesy TF, Liau LM. Induction of cytomegarovirous‐specific anti‐tumor immunity after autologous tumor lysate‐pulsed dendritic cell vaccination in a patient with glioblasotma. New Engl J Med 2008; 359:539–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sabado RL, Bhardwaj N. Dendritic cell vaccines on the move. Nature 2015; 519:300–301. [DOI] [PubMed] [Google Scholar]
- 17. Cobbs CS, Harkins L, Samanta M et al. Human cytomegalovirous infection and expression in human malignant glioma. Cancer Res 2002; 62:3347–50. [PubMed] [Google Scholar]
- 18. Mitchell DA, Xie W, Schmittling R et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblatoma. Neuro Oncol 2008; 10:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carreno BM, Collins M. The B7 family of ligands and its receptor: new pathways for costimualtion and inhibition of immune responses. Ann Rev Immunol 2002; 20:29–53. [DOI] [PubMed] [Google Scholar]
- 20. Carreno BM, Collins M. BTLA: a new inhibitory receptor with a B7‐like ligand. Trends Immunol 2003; 24:524–527. [DOI] [PubMed] [Google Scholar]
- 21. Yamazaki T, Akiba H, Iwai H et al. Expression of programmed death 1 ligands by murine T cells and APCs. J Immunol 2002; 169:5538–5545. [DOI] [PubMed] [Google Scholar]
- 22. Speise ED, Ho PC, Verdeil H. Regulatory circuits of T cell function in cancer. Nat Rev Immunol 2016; 16:599–611. [DOI] [PubMed] [Google Scholar]
- 23. Dumitriu IE, Dunbar DR, Howie SE, Sethi T, Gregory CD. Human dendritic cells produce TGF‐β1 under the influence of lung carcinoma cells and prime the differentiation of CD4+CD25+FoxP3+ regulatory T cells. J Immunol 2009; 182:2795–2807. [DOI] [PubMed] [Google Scholar]
- 24. Boeckh M, Nichols WG, Papanicolaou G, Rubin R, Wingard JR, Zaia J. Cytomegalovirus in hematopoietic stem cell transplant recipients: current status, known challenges, and future strategies. Biol Blood Marrow Transplant 2003; 9:543–558. [DOI] [PubMed] [Google Scholar]
- 25. Ueda R, Ohkusu‐Tsukada K, Fusaki N et al . Identification of HLA‐A2‐ and A24‐restricted T‐cell epitopes derived from SOX6 expressed in glioma stem cells for immunotherapy. Int J Cancer 2010; 126:919–929. [DOI] [PubMed] [Google Scholar]
- 26. Luo MH, Fortunato EA. Long term infection and shedding of human cytomegalovirus in T98G glioblastoma cells. J Virol 2007; 81:10424–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sasawatari S, Tadaki T, Isogai M, Takahara M, Nieda M, Kakimi K. Efficient priming and expansion of antigen‐specific CD8+T cells by a novel cell‐based artificial APC. Immunol Cell Biol 2006; 84:512–21. [DOI] [PubMed] [Google Scholar]
- 28. Nieda M, Terunuma H, Eiraku Y, Deng X, Nicol AJ. Professional expansion of Th1 type‐CD56+Vγ9γδT cells by CD56+ Vγ9γδT cells by CD56high+IFNγ‐induced dendritic cells derived from cancer patients in vitro and in vivo . Int J Immunol Immunother 2015; 2:1. [Google Scholar]
- 29. Blank C, Mackensen A. Contribution of the PD‐L1/PD‐1 pathway to T‐cell exhaustion: an update on implications for chronic infections and tumor evasion. CancerImmunol Immunother 2007; 56:739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ott PA, Hodi FS, Robert C. CTLA‐4 and PD‐1/PD‐L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res 2013; 19:5300–5309. [DOI] [PubMed] [Google Scholar]
- 31. Worbs T, Hammerschmidt S, Foerster R. Dendritic cell migration in health and disease. Nat Rev Immunol 2017; 17:30–48. [DOI] [PubMed] [Google Scholar]
- 32. Dudly ME, Wunderlich JR, Robbins PF et al . Cancer regression and autoimmunity in patients with antitumor lymphocytes. Science 2002; 298:850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park TS, Rosenberg SA, Morgan RA. Treating cancer with genetically engineered T cells. Trends Biotechnol 2011; 29:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hodi FS, O’Day SJ, McDermott DF et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen L, Han X. Anti‐PD‐1/PD‐L1 therapy of human cancer: past, present, and future. J Clin Invest 2015; 125:3384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schuessler A, Smith C, Beagley L et al . Autologous T‐cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res 2014; 74:3466–76. [DOI] [PubMed] [Google Scholar]
- 37. Nair SK, De Leon G, Boczkowski D et al. Recognition and killing of autologous, primary glioblastoma tumor cells by human cytomegalovirus pp65‐specific cytotoxic T cells. Clin Cancer Res 2014; 20:2684–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shernberg A, Marabelle A, Massard C et al. What’s next in glioblastoma treatment: tumor‐targeted or immune targeted therapies? Bull Cancer 2016. May; 103:484–98. [DOI] [PubMed] [Google Scholar]
- 39. Maxwell R, Jackson CM, Lim M. Clinical trials investigating immune checkpoint blockade in glioblasotma. Curr Treat Options Oncol 2017; 18:51. doi: 10.1007/s11864‐017‐0492‐y. [DOI] [PubMed] [Google Scholar]
- 40. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res 2017; 27:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brandes M, Willimann K, Moser B. Professional antigen‐presentation function by human γδT cells. Science 2005; 309:264–68. [DOI] [PubMed] [Google Scholar]
- 42. Muto M, Baghdadi M, Maekawa R, Wada H, Seino K. Myeloid molecular characterization of human γδT cells support their acquisition of tumor antigen‐presenting capacity. Cancer Immunol Immunother 2015; 64:941–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanaka Y, Morita CT, Tanaka Y, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non‐peptide antigens recognized by human γδT cells. Nature 1995; 375:155–58. [DOI] [PubMed] [Google Scholar]