

Summary
Polymorphonuclear (PMN) leucocytes participate in acute inflammatory pathologies such as acute respiratory distress syndrome (ARDS) following traumatic injury and shock, which also activates the coagulation system systemically. Trauma can prime the PMN nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex for an enhanced respiratory burst, but the relative role of various priming agents in this process remains incompletely understood. We therefore set out to identify mediators of PMN priming during coagulation and trauma‐shock and determine whether PMN reactive oxygen species (ROS) generated in this manner could influence organ injury and coagulation. Initial experiments demonstrated that PMN are primed for predominantly extracellular ROS production by products of coagulation, which was abrogated by CD88/C5a receptor(C5aR) inhibition. The importance of this was highlighted further by demonstrating that known PMN priming agents result in fractionally different amounts of extracellular versus intracellular ROS release depending on the agent used. Plasma from trauma patients in haemodynamic shock (n = 10) also primed PMN for extracellular ROS in a C5a‐dependent manner, which correlated with both complement alternative pathway activation and thrombin generation. Furthermore, PMN primed by preincubation with products of blood coagulation directly caused loss of endothelial barrier function in vitro that was abrogated by C5aR blockade or NADPH oxidase inhibition. Finally, we show in a murine model of trauma‐shock that p47phox knock‐out (KO) mice with PMN incapable of generating ROS were protected from inflammatory end‐organ injury and activated protein C‐mediated coagulopathy. In summary, we demonstrate that trauma‐shock and coagulation primes PMN for predominantly extracellular ROS production in a C5a‐dependent manner that contributes to endothelial barrier loss and organ injury, and potentially enhances traumatic coagulopathy.
Keywords: coagulation, complement, endothelium, inflammation, NADPH oxidase, neutrophil, PMN, reactive oxygen species, trauma
Introduction
Polymorphonuclear (PMN) leucocytes, commonly referred to as neutrophils, the primary innate immunocyte involved in acute responses to infection and other pathophysiological insults, generate large amounts of bactericidal reactive oxygen species (ROS) via the NADPH oxidase complex 1. In addition to ingesting and killing pathogens, PMN can also undergo a process of ‘priming’, which results in partial release of proteases and other granule contents into the extracellular space, along with ‘preactivation’ of the NADPH oxidase 1, 2, 3. Primed PMN typically produce small to minimal amounts of ROS spontaneously, but upon exposure to an activating stimulus (e.g. N‐formyl peptides released by bacteria or from host mitochondria following trauma, or integrin engagement upon adhesion), primed PMN produce much larger amounts of ROS, with accelerated kinetics, compared to unprimed cells 3, 4, 5. PMN priming is thought to play a critical role in the development of organ injury/failure after major pathophysiological insults such as traumatic shock (trauma‐shock). The classic ‘two‐hit hypothesis’ of organ failure following trauma hypothesizes that PMN priming from the initial trauma followed by a second stimulus (e.g. surgery) activates the PMN to release excessive extracellular ROS and proteases, damaging adjacent non‐injured cells and leading to autoinflammatory organ injury 2, 6, 7. A significant number of cytokines and related molecules have been identified that are capable of inducing PMN priming, but the relative importance of the different priming agents in inducing organ injury after trauma‐shock has not been established firmly.
After major blunt or penetrating trauma, patients suffer from both tissue injury and hypovolaemic shock from blood loss. In patients with otherwise survivable injury, early death usually results from uncontrolled haemorrhage, due in part to trauma‐induced coagulopathy (TIC), while late death is usually due to inflammatory organ injury, microvascular thrombosis and/or infection due to the development of a profoundly immunosuppressive state 8, 9. The complex pathophysiology that underlies these clinical observations appears to be initiated by the effects of trauma‐shock on pathways that evolved originally to protect the host organism, including the innate immune response (e.g. PMN) to damage‐associated molecular patterns (DAMPs) and activation of both the coagulation and complement systems 10, 11, 12. Furthermore, the ability of PMN and the coagulation system to alter each other’s behaviour is probably paramount in trauma, where both PMN and the coagulation system are activated, but this has only recently received significant attention. Some of these recent findings include the observation that platelets are activated and thrombin is generated in response to neutrophil extracellular traps (NETs), and conversely that thrombin can lead to increased levels of the granule marker CD11b on the PMN surface 13, 14, 15. To delineate additional potential cross‐talk further between PMN inflammatory responses that drive the pathophysiology seen after trauma‐shock and the coagulation system, we hypothesized that specific molecules produced during the process of blood coagulation could directly prime PMN for ROS production and that the ROS released by PMN could damage the endothelium, cause organ injury and potentially alter coagulation after trauma‐shock.
Materials and Methods
Human blood products and patients
Blood products used in this study for PMN purification and ex‐vivo coagulation experiments were obtained (as described in more detail below) with written informed consent and approval from the Institutional Review Board at the Massachusetts institute of Technology. In clinical studies, whole blood was drawn into 3·2% citrate tubes from trauma patients in the emergency department (ED) and again 24 h later and platelet‐poor plasma generated and frozen at –80C, and stored at the US National Institutes of Health/US Department of Defense (NIH/DoD) Trans‐Agency Consortium for Trauma Induced Coagulopathy (TACTIC) repository for future use. The inclusion criteria of the studies from which the TACTIC samples were obtained were (a) adult trauma patients in shock, as defined by heart rate (HR) > 108 and systolic blood pressure (SBP) < 90, or a SBP < 70 without the tachycardia requirement (University of Pittsburgh Medical Center); and (b) adult trauma patients meeting full trauma team activation for blunt or penetrating trauma, excluding patients on anti‐coagulants, with pre‐existing coagulopathies or malignancies, dialysis patients, thermal injury or patients on immunosuppression (Mayo Clinic). These studies were both performed with approval from the respective Institutional Review Boards at the University of Pittsburgh Medical Center and the Mayo Clinic, and written informed consent for study participation was obtained on all trauma patients.
PMN preparation
Human PMN were prepared from healthy volunteer blood collected in 10 ml vacutainer tubes containing 18mg K2 ethylenediamine tetraacetic acid (EDTA) (Becton Dickinson, San Jose, CA, USA) via venipuncture of the antecubital veins. PMN were purified by centrifuging EDTA anti‐coagulated whole blood through a Ficoll‐Paque density gradient (GE Healthcare, Chicago, IL, USA) followed by red blood cell (RBC) sedimentation with 2% dextran w/v (Mr 450,000–650,000; Sigma, St Louis, MO, USA) in Dulbecco’s phosphate‐buffered saline (PBS) without divalent cations [Dulbecco’s phosphate buffered saline with divalent cations (DPBS–)] Thermo Fisher Scientific, Inc., Waltham, MA, USA) followed by hypotonic lysis, as described previously 2.
Coagulation and blood product preparation
Plasma was generated from whole blood collected in either EDTA or heparin tubes (final heparin concentration 15·8 units/ml) (Becton Dickinson) by centrifugation at 2000 × g for 10 min with collection of the straw‐coloured liquid fraction on top as plasma. Platelet‐poor plasma (PPP) was generated from whole blood collected in EDTA, heparin or 3·2% sodium citrate tubes and centrifuged at 3000 × g for 15 min, the top ¾ fraction of plasma was collected and then spun again at 3000 × g for an additional 15 min, with collection of the top ¾ fraction of this as PPP. Platelet depletion to < 10 × 103 platelets/ɑl was confirmed by manual counting with a haemocytometer.
Serum was generated by collection of healthy volunteer whole blood into uncoated 10 ml glass vacutainer tubes (Becton Dickinson) and clotted by incubation for 2 h at 37oC, then a 2‐h incubation on ice followed by centrifugation at 2000 × g for 10 min with recovery of the liquid product as serum.
Clotted plasma/PPP was generated by reversing EDTA anti‐coagulant with 15 µl of 9% CaCl2 (w/v in H2O) per ml plasma/PPP or by reversing heparin anti‐coagulant with 25 µl of 1% (w/v) protamine sulphate (Sigma) in Dulbecco’s PBS with divalent cations (DPBS++) per ml plasma/PPP, and thereafter was incubated and the liquid product collected in the same manner as for serum. All clotted products (serum, clotted plasma, clotted PPP) were controlled for anti‐coagulant by adding EDTA or heparin back to the collected liquid products of coagulation to allow for more appropriate comparison with unclotted plasma/PPP products.
Purified coagulation factors, Russell Viper Venom Factor X Activating Enzyme (RVVX) and Russell Viper Venom Factor V Activating Enzyme (RVV) were all obtained from Haematologic Technologies, Inc. (Essex Junction, VT, USA). Factors Xa and Va were generated by incubation of factor X with RVVX and factor V with RVV for 2 h in sterile‐filtered DPBS++ with 0·01% bovine serum albumin (BSA) at 37oC in a 500 : 1 factor : activator ratio according to supplier instructions, where 0·12 µg/ml ZnCl2 was supplemented into the factor X + RVVX reaction. Factor IXa was generated by incubation of factor IX with activated factor XIa for 2 h in sterile‐filtered DPBS++ with 0·01% BSA at 37oC in a 500 : 1 factor IX : factor XIa ratio. Generation of activated factors and their corresponding activation peptides was confirmed by separating proteins via 10% sodium dodecyl page‐polyacrylamide gel electrophoresis (SDS‐PAGE) under non‐reducing conditions and staining with Coomassie SimplyBlueTM SafeStain (Thermo Fisher Scientific, Inc.). All purified and activated coagulation factors were used in PMN priming assays at concentrations equivalent to physiological levels of the preactivated factors (i.e. maximum physiological levels) 16.
Chemiluminescence measurements of ROS production
For measurements of PMN ROS production, PMN were resuspended in DPBS++ (for heparin‐based, purified priming agent‐based and purified coagulation factor experiments) or DPBS– (for EDTA and citrate‐based experiments) to 4 × 106 PMN/ml and were incubated with an equal volume of the designated blood product being tested to give 50% (v/v) blood product concentration during the PMN priming step/incubation or DPBS++ (for priming agent and coagulation factor experiments) for 30 min at 37oC. PMN were then centrifuged for 5 min at 250 × g at 4oC and washed twice with DPBS++ (for heparin‐based priming agents and purified coagulation factors) or DPBS– for EDTA and citrate followed by a final resuspension in DPBS++ (for all), equal to the initial volume. Of the resulting PMN, 75‐µl solutions were added to a 96‐well plate in duplicate and an equal volume of luminol solution (Sigma) in DPBS++ (final assay concentration of 150 βM luminol) was then added containing either horseradish peroxidase (HRP), final assay concentration (10 U/ml; Sigma) to specifically measure total ROS production 17, or containing a combination of superoxide dismutase SOD, final assay concentration 25U/ml (Sigma) and catalase final assay concentration 2000 U/ml (Sigma) to specifically measure intracellular ROS by quenching the extracellular ROS fraction, as has been described previously 18, 19. After a 5‐min incubation at 37oC with the designated luminol solution, either vehicle control or N‐formyl‐methionyl‐leucyl‐phenylalanine (fMLP, final assay concentration 100 nM; Sigma) were added and chemiluminescence events were measured over 10 min. Quantification of ROS was performed by integrating the area under the curve of chemiluminescence events over time, as described previously 20.
For experiments using healthy volunteers to measure the effects of different blood products on PMN priming, PMN were used with autologous blood products, and an internal positive priming control with the well‐known priming agent tumour necrosis factor (TNF)‐ɑ (20 ng/ml; R&D Systems, Minneapolis, MN, USA) was used to set the reference of normalization, such that comparison of priming was feasible across multiple different types of clotted and unclotted blood products. For experiments with the C5a receptor (C5aR) antagonist W‐54011 (Tocris, Minneapolis, MN, USA), PMN were preincubated for 10 min with 5 µM W‐54011 or vehicle control and then PMN were mixed with the designated blood products and experiments performed as already described. For experiments using a mouse anti‐human CD88 (C5a receptor 1) monoclonal antibody clone S5/1 (Bio‐Rad Laboratories) to block C5aR1, PMN were preincubated with 1 µM anti‐C5aR1 antibody or 1 µM IgG2a isotype control antibody (Bio‐Rad Laboratories, Hercules, CA, USA) 21 for 30 min at 37oC prior to performing the experiments in the fashion already described.
For experiments using traumatic shock patient citrate PPP (n = 10 patients) and comparison to healthy volunteer citrate PPP (n = 16), all PPP was heterologous to the PMN source, including the healthy PPP, in order to control for the fact that trauma PPP could not be tested with autologous PMN. When there was adequate trauma patient sample volume and available volunteers, each trauma sample underwent testing with two volunteers’ PMN and all ROS data was included from both. In these trauma versus healthy PPP experiments the reference point was set as the uninhibited (i.e. vehicle) +fMLP group and comparison made to each sample’s respective W‐54011/+fMLP group, with results reported as percentage change in ROS production due to C5aR inhibition with W‐54011 to reflect the degree of priming due to C5a in the samples.
Electric cell‐substrate impedance sensing co‐incubation assays for measurement of PMN effects on human umbilical vein endothelial cell monolayers
Pooled‐donor human umbilical vein endothelial cells (HUVEC) were obtained (Lonza Biologics, Inc., Portsmouth, NH, USA) and grown to confluence at passage 4 on 96‐well tissue culture plates containing interdigitated gold electrodes on the basal surface of each well underneath the cell layer (96W20idf; Applied BioPhysics, Inc., Troy, NY, USA). Resistance across the HUVEC monolayer was then measured continuously over time using an Electric Cell‐Substrate Impedance Sensing (ECIS) device (model Z‐Theta; Applied BioPhysics, Inc.). EGM‐2 medium (Lonza Biologics, Inc.) was used to grow the HUVEC to confluence, at which point the medium was switched to EBM‐2 basal medium (Lonza Biologics, Inc.) with no serum or growth factors added to the medium and the cells were allowed to acclimate and reach steady‐state resistance.
Once the HUVEC were stabilized in serum‐free growth factor‐free medium, PMN and blood products from healthy volunteers were made as already described and PMN were preincubated with one of the following: (a) 5 μM W‐54011 to inhibit C5aR signalling; (b) 10 μM diphenyleneiodium (DPI) (Sigma) to inhibit ROS production via the NADPH oxidase complex; or (c) vehicle control. After this preincubation with inhibitor or control, the PMN were incubated for 30 min at 37oC in an equal volume of heparin plasma or serum (heparinized after clotting to control for heparin in the plasma) [final blood product concentration during incubation of 50% (v/v)]. The PMN were then centrifuged at 250 × g for 5 min at 4oC and washed twice with DPBS–, resuspended in serum‐free growth factor‐free EBM‐2 medium, and placed in a Corning HTS Transwell 96‐well permeable support with a 0·4 µm pore size polycarbonate membrane (Corning Inc., New York, NY, USA) to a final concentration of 1 × 107 PMN/ml and resistance changes in the HUVEC monolayer were measured over time. This Transwell co‐incubation method where PMN and HUVEC were not in physical contact with one another allowed for evaluation of changes in endothelial barrier function resulting from any soluble PMN products (i.e. ROS) released in response to preincubation with various blood products, where the blood products themselves were washed away prior to PMN addition to the Transwell to prevent any confounding effect of the blood products directly on the HUVECs.
Animal trauma‐haemorrhage model of trauma‐induced coagulopathy and end‐organ injury
Murine experiments were performed with IACUC approval and in accordance with NIH guidelines using a literature‐established murine trauma‐haemorrhage model that is known to cause organ injury, activate the coagulation system and cause trauma‐induced coagulopathy 22, 23. Adult (8–12‐week‐old) male wild‐type (WT) and p47phox knock‐out (KO) mice (Taconic BioSciences, Rensselaer, NY, USA) on a C57Bl/6 background were compared, where p47phox KO mice have PMN that are unable to generate ROS via the NADPH oxidase complex due to the absence of the p47phox protein.
Anaesthesia was induced in an isoflurane chamber and the mice were then positioned supine on a plexiglass board and restrained with soft tape tethers to the limbs and tail with ongoing isoflurane anaesthesia via a nose cone. Normothermia was maintained using a thermal control unit and rectal temperature probe. Upon satisfactory positioning and anaesthesia, the abdomen and bilateral groins were shaved and prepped with betadine scrub ×3 and a 2‐cm midline laparotomy and bilateral groin incisions were made. The intra‐abdominal organs were inspected to confirm no inadvertent organ injuries, and then the laparotomy was closed with a 5‐0 prolene suture (Ethicon Inc., Somerville, NJ, USA) and the incision bathed in 1% lidocaine. The left femoral artery and right femoral vein were then cannulated with sterile PE‐10 tubing and the left femoral artery tubing was connected to a pressure transducer (TSD104A; Biopac Systems, Goleta, CA, USA) and amplifier (MP1004‐CE; Biopac Systems), and the groin cannulation sites were then bathed in 1% lidocaine solution (Hospira Inc., Lake Forest, IL, USA). Anaesthesia was then stopped and a baseline mean arterial pressure (MAP) > 90 mmHg was confirmed, at which point shock was induced by withdrawing an estimated 25% of total blood volume (calculated by multiplying body weight by 0·077, in ml) using a pipette to withdraw blood from the left femoral arterial catheter. The MAP was monitored continuously and aliquots of 60–70 μl of blood withdrawn as needed to maintain a goal MAP of 35 ± 5 mmHg for 60 min. Sham mice were treated in the same manner, except that they did not have any blood withdrawn to induce shock and thus underwent 60 min of board stress. Halfway through the 60‐min shock (or board stress) period, 1 mg/kg of filter‐sterilized [125I]‐labelled albumin (albumin and [125I] labelling kit from Thermo Fisher Scientific Inc. and labelled according to the manufacturer’s instructions) was administered via the right femoral vein catheter.
After the 60‐min shock (or board stress) period was complete, the mice were euthanized by isoflurane overdose and the laparotomy reopened for an immediate blood draw from the inferior vena cava (IVC) in a 9 : 1 ratio with 3·2% citrate (for thrombin–anti‐thrombin complex measurements) or 3·2% citrate containing 1 mM benzamidine (for activated protein C analysis), centrifuged to obtain PPP, and stored at –80 until use on enzyme‐linked immunosorbent assay (ELISA). The lungs were then harvested, vasculature flushed with saline, and lung tissue homogenized in saline for measurement of lung permeability to [125I]‐labelled albumin using a gamma counter and taking into account the plasma [125I]‐labelled albumin levels (counts/min).
ELISA measurement of complement fragments, thrombin generation (PF 1+2 and thrombin–anti‐thrombin) and activated protein C (aPC) levels
ELISA was performed according to the manufacturer’s instructions for the following: human C5a, Bb, and C4d levels (Quidel Corporation, San Diego, CA, USA); total thrombin generation from human samples by measuring PF1+2 levels (Lifespan Biosciences, Inc.); total thrombin generation in mouse samples by measuring thrombin–anti‐thrombin complex levels (Enzyme Research Laboratories Inc., South Bend, IN, USA). Mouse aPC levels were measured using a custom ELISA‐type assay developed in the laboratory of Dr Charles Esmon that is specific for the activated form of protein C, as described previously 23, 24.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 6.07 (GraphPad Software Inc., San Diego, CA). Data are expressed as mean ± standard error of the mean (s.e.m.). Significant differences between groups were determined using analysis of variance (anova) with Holm–Sidak’s correction for multiple comparisons, except for the C5a, Bb and C4d ELISA data for trauma patient PPP, which was not normally distributed and therefore compared using Kruskal–Wallis tests with Dunn’s correction for multiple comparisons. Spearman’s correlations were performed to determine relationships between complement fragment levels, PF1+2 levels and the ability to inhibit extracellular ROS using a C5aR inhibitor. Alpha was set at the 0·05 level.
Results
Products of coagulation prime PMN for early extracellular ROS generation in a cell‐ and platelet‐independent manner
Agents that prime the PMN NADPH oxidase for enhanced ROS production relative to agonists (e.g. fMLP) alone (Fig. 1a) vary considerably in the extent to which they enhance intracellular versus extracellular ROS following exposure to a second activating stimulus, with some priming agents leading to significantly larger proportions of extracellular ROS than others (Fig. 1b). To investigate a potential relationship between PMN priming and coagulation such as that seen in trauma, as well as the fractional distribution of extracellular versus intracellular ROS generated in response to any observed priming, we first explored whether the products of blood clotting ex vivo were sufficient to induce priming of PMN for enhanced ROS production. As shown in Fig. 1c,d, fMLP stimulation of resting human PMN incubated with unclotted blood plasma obtained from healthy volunteers showed minimal ROS production. In marked contrast, PMN incubated in serum from whole blood from the same donors that had been clotted ex vivo were primed for significant ROS production in response to fMLP. Furthermore, PMN priming similar to that seen by incubation of PMN with serum was also observed when PMN were incubated in supernatants from previously isolated plasma that was then clotted ex vivo (i.e. RBCs and leucocytes removed prior to clotting) (Fig. 1e). When platelets were also depleted from plasma to make PPP followed by clotting, the supernatants from clotted PPP also primed PMN in a similar fashion to serum (Fig. 1f). These results indicate that: (a) agents capable of priming PMN for large amounts of ROS can be generated by production of ‘serum’ from plasma upon blood clotting; and (b) the priming activity generated following ex‐vivo blood clotting does not require the presence of leucocytes, RBCs or platelets during the clotting process. Similar results were obtained regardless of the anti‐coagulant method used to prepare the plasma, as quantified in Fig. 1g,h. Furthermore, with respect to the fractional distribution of ROS, PMN priming by the products of coagulation led predominantly to generation of extracellular ROS, particularly at early times after stimulation (compare red and blue shaded curves in Fig. 1c–f), quantified at the 10‐min marker in Fig. 1i. Notably, even in the absence of fMLP, PMN incubated with clotted blood products generated more ROS over time compared to PMN incubated in unclotted blood products (cyan line in Fig1. c–f), although this was significantly less than that seen following fMLP stimulation.
Figure 1.
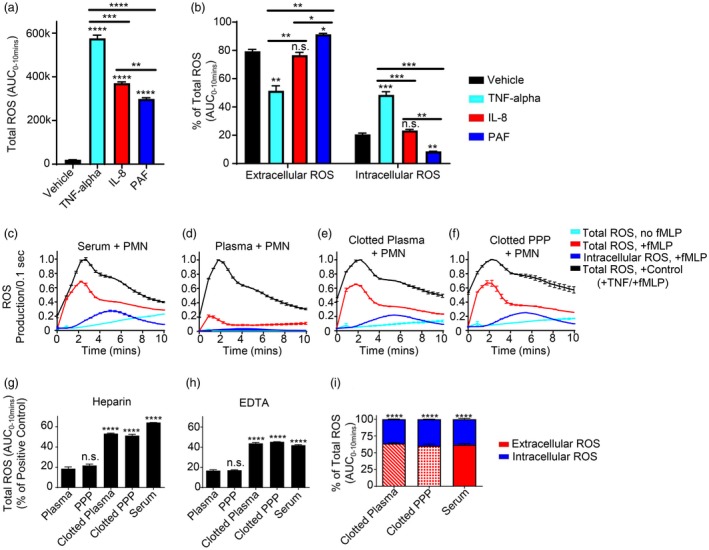
Polymorphonuclear leucocytes (PMN) are primed for early extracellular reactive oxygen species (ROS) generation by products of coagulation. (a,b) PMN incubated with vehicle control versus the designated known priming agents were measured for priming/ROS generation with addition of 100 nM N‐formylmethionine‐leucyl‐phenylalanine (fMLP) by luminol chemiluminescence with horseradish peroxidase (HRP) (total ROS generation) or superoxide dismutase (SOD) + catalase (intracellular ROS generation). Total and intracellular ROS generated over 10 min was then quantified by integrating the area under the curve (AUC) from 0 to 10 min (total ROS/luminescence generated from 0 to 10 min is shown in (a) to demonstrate the effect of fMLP alone versus priming agent + fMLP on total ROS generation), and extracellular and intracellular ROS was then expressed as a fraction (%) of total ROS generated (where extracellular ROS was measured indirectly by subtracting the intracellular fraction from the total ROS). PMN incubated in 50% (v/v) (c) serum, (d) plasma, (e) clotted plasma and (f) clotted platelet‐poor plasma (clotted PPP) were measured for priming/ROS generation without or with addition of 100 nM fMLP by luminol chemiluminescence with HRP (total ROS generation) or SOD + catalase (intracellular ROS generation). Each condition was normalized to an internal positive PMN priming control [20 ng/ml tumour necrosis factor (TNF)‐α + 100 nM fMLP] to allow for comparison between multiple different blood products. Total amounts of ROS generated for each condition were quantified by integrating the area under the curve (AUC) from 0 to 10 min for experiments performed using (g) heparin and (h) ethylenediamine tetraacetic acid (EDTA) as the initial anti‐coagulant and compared to their respective plasma group. (i) Extracellular versus intracellular ROS at 10 min was then quantified and expressed as a fraction (%) of total ROS generated, where extracellular ROS was measured indirectly by subtracting the intracellular fraction from the total ROS, and the difference between extracellular and intracellular amounts for each blood product were then compared within each group. Results reported as mean ± standard error of the mean (s.e.m.). Significance set at P ≤ 0·05. *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001; ****P ≤ 0·0001.
Complement component C5a, but not coagulation factors or their activation peptides, is the principle PMN priming agent following ex‐vivo blood clotting
Multiple components of the coagulation cascade are known to modulate PMN functions, including fibrinopeptide B, a chemotactic agent 25, and thrombin via protease‐activated receptors 26. We therefore investigated whether any of the major activated coagulation factors or their activation peptides, that are produced during the clotting process, could prime PMN for ROS generation in response to fMLP stimulation. In these experiments, ROS production was assayed using luminol‐dependent chemiluminescence as described above, with TNF‐α (20 ng/ml) serving as an internal positive control for priming. As shown in Fig. 2a, no PMN priming was observed in response to maximum physiological concentrations of activated coagulation factors, their activation peptides or fibrinopeptides resulting from thrombin‐mediated fibrinogen cleavage. Another major protease system known to be present in the plasma component of blood is the complement system which, when activated, has known inflammatory signalling functions via its anaphylatoxins C3a and C5a, reviewed in 27. As seen in Fig. 2a, C3a was unable to prime PMN for ROS generation in response to fMLP, while C5a was a potent priming agent, consistent with numerous previous reports in the literature 28, 29, 30, 31. To determine whether C5a was the relevant priming agent following ex‐vivo blood coagulation, we investigated whether inhibition of CD88 C5a receptor‐1 (C5aR) signalling using a specific C5aR‐blocking monoclonal antibody or the C5aR antagonist W‐54011 could abrogate PMN priming for enhanced ROS production by products of coagulation (Fig. 2b–f). As seen in Fig. 2b–e, PMN priming by serum or clotted plasma was inhibited significantly by the C5aR blocking antibody (compare blue and red curves in panels b–d). Similar results were obtained using the C5aR antagonist W‐54011 (Fig. 2f). Both the C5aR blocking antibody and W‐54011 had no significant effect on PMN priming in the plasma controls, although very small amounts of non‐significant partial agonism were observed. To validate the potential role of C5a as the major PMN priming agent by products of coagulation an ELISA was performed (Fig. 2g), confirming the presence of elevated C5a levels in ex‐vivo clotted blood and clotted plasma compared to unclotted plasma (Fig. 2g). Importantly, both clotted plasma and serum had significant elevations in the complement component Bb relative to plasma (Fig. 2h), but not C4d (Fig. 2i), indicating activation of the alternative pathway during coagulation, but not the classical or lectin pathway.
Figure 2.
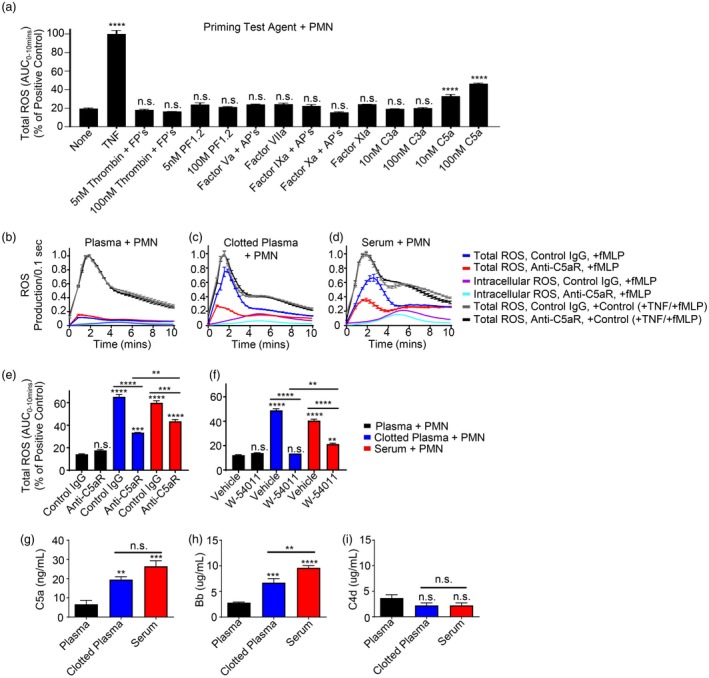
Complement component 5a (C5a) generated by the alternative pathway of complement during coagulation is responsible for priming of polymorphonuclear leucocytes (PMN) by products of coagulation. (a) Screen of potential PMN priming agents that could be responsible for the observed PMN priming by products of coagulation. PMN were preincubated with the specified agent(s), followed by addition of luminol + horseradish peroxidase (HRP) and then treatment with 100 nM N‐formylmethionine‐leucyl‐phenylalanine (fMLP). Total reactive oxygen species (ROS) production over 10 min was measured, with results normalized to an internal positive priming control [20 ng/ml tumour necrosis factor (TNF)‐α] and compared with fMLP alone (i.e. no priming agent, ‘none’) for reference. (b–f) PMN priming by products of coagulation [50% (v/v)] measured by total ROS generation in response to fMLP in the presence or absence of C5aR blocking antibody [versus immunoglobulin (Ig)G control] or the C5aR antagonist W‐54011 (versus vehicle control). Results normalized to an internal positive priming control (20 ng/ml TNF‐α). (g–i) Enzyme‐linked immunosorbent assay (ELISA) for complement fragments C5a, Bb, and C4d from n = 4 healthy volunteers. Results reported as mean ± standard error of the mean (s.e.m.). Significance was set at P ≤ 0·05. *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001; ****P ≤ 0·0001. FP = fibrinopeptides; PF1+2 = prothrombin fragment 1+2; AP = activation peptide.
Traumatic shock patient plasma primes PMN for extracellular ROS production in a C5a/alternative pathway‐dependent manner that correlates with thrombin generation
The observation that C5a generated during ex‐vivo coagulation primed PMN for extracellular ROS release led us to explore its potential clinical relevance in trauma patients with shock in whom the coagulation system is known to be activated systemically 11, 32. To accomplish this, citrate PPP from trauma patients in shock (n = 10, Table 1) was obtained upon patient arrival to the ED and again 24 h later and evaluated for its ability to prime resting PMN for ROS production in a C5a‐dependent manner by comparing vehicle controls with the C5aR antagonist W‐54011. Kinetic ROS production curves from an example patient and corresponding control are shown in Fig. 3a–c, demonstrating that while the healthy control PPP did not prime PMN, this traumatic shock patient’s PPP from the ED primed PMN for substantial extracellular ROS release, but by 24 h after admission this particular patient’s PPP no longer primed PMN. The amount of total, extracellular and intracellular ROS inhibited by W‐54011 treatment was quantified for all healthy and traumatic shock patient PPP groups and compared to determine the extent to which C5a was responsible for any observed PMN ROS production after incubation in the traumatic shock patients’ PPP (Fig. 3d–f). Overall (Fig. 3e), we found that traumatic shock patient PPP obtained from the ED and 24 h later both primed healthy resting PMN for extracellular ROS production in a manner that was inhibited significantly by C5aR antagonism with W‐54011, with somewhat less priming observed at the later (24‐h) time‐point ED versus healthy control (PPP: 25·4 ± 5·6% reduction in ROS, P = 0·0001; 24‐h versus healthy: 16·8 ± 5·5% reduction in ROS, P = 0·0074). In addition, an ELISA performed on traumatic shock patient PPP from the ED and 24‐h time‐points demonstrated significant elevations in C5a and Bb levels relative to healthy control PPP, but no change in C4d levels (Fig. 3h–j). PF1+2, a marker of total thrombin generation and activation of coagulation, demonstrated a trend towards elevated PF1+2 levels in trauma patient PPP relative to the healthy control PPP that did not reach significance when comparing absolute values ED versus healthy (P = 0·054; 24‐h versus healthy: P = 0·070). Importantly, however, there was a significant correlation between the amount of PMN extracellular ROS that could be inhibited by W‐54011 treatment and the levels of C5a, Bb and PF1+2 in the PPP. No such correlation was observed with C4d levels (Table 2. Furthermore, there was also a significant correlation between C5a levels and both Bb and PF1+2 levels (Table 2).
Table 1.
Trauma patient demographics
| Variable | n (%) or median (IQR) |
|---|---|
| Total patients | n = 10 |
| Male | 6 (60%) |
| Female | 4 (40%) |
| Age (years) | 54 (37–67) |
| ISS | 23 (21–26) |
| Mortality at 30 days | 2 (20%) |
| Penetrating trauma | 1 (10%) |
| Blunt trauma | 9 (90%) |
| HR on scene | 118 (89–143) |
| HR in ED | 111 (77–147) |
| SBP on scene (mmHg) | 83 (64–97) |
| SBP in ED (mmHg) | 87 (76–105) |
| Shock index (HR/SBP) on scene | 1·4 (1·1–1·6) |
| Shock index (HR/SBP) in ED | 1·1 (0·9–2·0) |
| INR in ED | 1·2 (1·0–1·4) |
| Peak INR w/in 24 h | 1·3 (1·1–1·5) |
| Platelet count in ED | 211 (157–255) |
| RBC units transfused w/in 24 h | 5·5 (0·75–8·75) |
| Plasma units transfused w/in 24 h | 0·5 (0–4·5) |
| Platelet units transfused w/in 24 h | 0·5 (0–2) |
IQR = interquartile range; ISS = Injury Severity Score; HR = heart rate; ED = emergency department; SBP = systolic blood pressure; INR = ; RBC = red blood cells.
Figure 3.
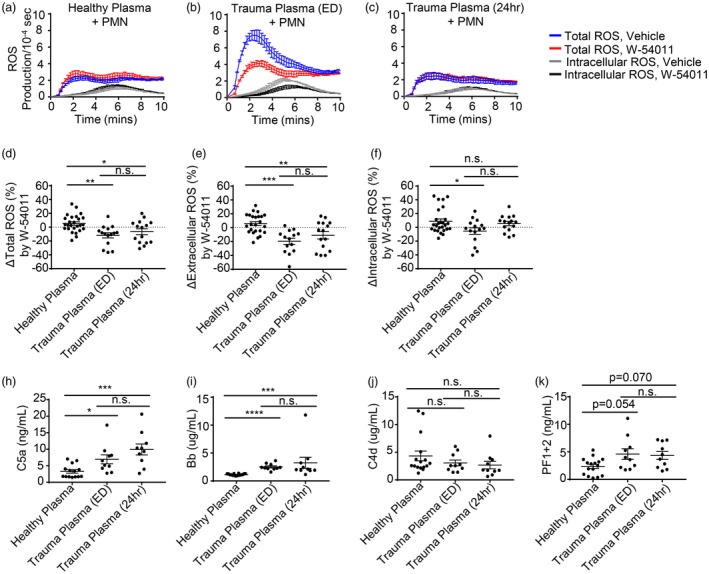
Trauma patient plasma primes polymorphonuclear leucocytes (PMN) for extracellular reactive oxygen species (ROS) production in a complement component 5a (C5a)‐dependent manner via complement alternative pathway activity. Kinetic curves of total and intracellular ROS generation from a representative experiment where healthy human PMN were pretreated with the C5aR antagonist W‐54011 or vehicle control and then incubated in 50% (v/v) (a) heterologous healthy citrate platelet‐poor plasma (PPP), (b) trauma patient citrate PPP drawn in the emergency department (ED) or (c) trauma patient citrate PPP drawn 24 h after traumatic injury, washed, and then stimulated with N‐formylmethionine‐leucyl‐phenylalanine (fMLP). (d–f) The % inhibition of total, extracellular and intracellular ROS generation over 10 min by the C5aR antagonist W‐54011 compared to vehicle controls for healthy and trauma patient citrate PPP. (h,i) Enzyme‐linked immunosorbent assay (ELISA) for levels of C5a, Bb, C4d and prothrombin fragment 1+2 (PF1+2) in healthy control and trauma patient citrate PPP. Results reported as mean ± standard error of the mean (s.e.m.). Significance was set at *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001; ****P ≤ 0·0001.
Table 2.
Correlation of trauma patient complement levels, thrombin generation and inhibition of polymorphonuclear leucocytes (PMN) extracellular reactive oxygen species (ROS) production by complement component 5a receptor (C5aR) antagonism
| ΔExtracellular ROS | C5a level | C4d level | Bb level | PF1+2 level | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| r | P‐value | r | P‐value | r | P‐value | r | P‐value | r | P‐value | |
| ΔExtracellular ROS | 0·309 | 0·027 | 0·033 | 0·815 | 0·464 | < 0·001 | 0·371 | 0·006 | ||
| C5a level | 0·309 | 0·027 | 0·198 | 0·254 | 0·626 | < 0·001 | 0·350 | 0·039 | ||
| C4d level | 0·033 | 0·815 | 0·198 | 0·254 | 0·185 | 0·278 | 0·063 | 0·714 | ||
| Bb level | 0·464 | < 0·001 | 0·626 | < 0·001 | 0·185 | 0·278 | 0·271 | 0·109 | ||
| PF1+2 level | 0·371 | 0·006 | 0·350 | 0·039 | 0·063 | 0·714 | 0·271 | 0·109 | ||
PF1+2 = prothrombin fragment 1+2.
PMN primed by products of coagulation are sufficiently active to cause loss of endothelial barrier function in vitro via C5a‐dependent ROS release
The potential importance of PMN priming by C5a present in products of coagulation was next evaluated using electrical cell‐substrate impedance sensing (ECIS) assays to evaluate whether soluble products released by PMN in response to C5a levels seen in clotted blood products were sufficient to compromise endothelial barrier function in culture. In these experiments, PMN were preincubated in a given blood product, washed with HUVEC culture medium (serum‐free, growth hormone‐free) to remove the blood product, and then added to a Transwell filter placed over a layer of confluent HUVEC cells on an ECIS plate that measures electrical impedance and resistance electrically across the monolayer. This set‐up allows evaluation of the effects of PMN release products on the endothelium, as no direct contact can occur between PMN and HUVEC due to the presence of the Transwell filter. As seen in Fig. 4a,b, PMN that were preincubated in serum led to significant losses of endothelial barrier function, while PMN preincubated in plasma did not cause any changes in endothelial resistance during a 12‐h time–course (compare red and black traces). Resistance just prior to PMN addition and then at the 6‐ and 12‐h time‐points after PMN addition are quantified in Fig. 4b. Importantly, either blockade of PMN C5aR signalling with W‐54011 during serum incubation, or inhibition of ROS generation by the PMN NADPH oxidase complex with DPI, led to abrogation of the observed endothelial barrier loss caused by PMN priming.
Figure 4.
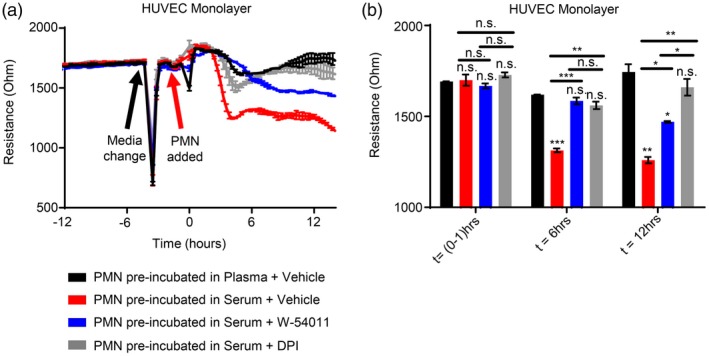
Polymorphonuclear leucocyte (PMN) reactive oxygen species (ROS) released in response to complement component 5a (C5a) generated by the complement alternative pathway during coagulation degrades endothelial barrier function. (a) Kinetic curves and (b) multiple comparisons at discrete time points of electric cell‐substrate impedance sensing (ECIS) assays. Human PMN were first incubated in 50% (v/v) plasma or serum without or with pretreatment of PMN with W‐54011 or diphenyleneiodium (DPI), then washed with cell culture medium, and subsequently placed in a Transwell filter over confluent human umbilical vein endothelial cell (HUVEC) monolayers grown on ECIS plates to measure endothelial barrier function. Vehicle control experiments without PMN shown in Figure S1. Results reported as mean ± standard error of the mean (s.e.m.). Significance was set at P ≤ 0·05. *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001; ****P ≤ 0·0001.
PMN ROS generated in response to trauma‐shock is a critical mediator of end‐organ injury and coagulopathy in vivo
Given our finding that traumatic shock patient PPP primes resting PMN for extracellular ROS, we next asked whether PMN ROS generated during trauma‐shock had a role in end‐organ injury and coagulation in vivo, using a well‐established mouse model of trauma‐haemorrhagic shock that causes lung injury and trauma‐induced coagulopathy (TIC) 23, 31. To elucidate specifically the contribution of PMN‐derived ROS, these experiments were conducted comparing WT control mice to p47phox KO mice that lack a functional NADPH oxidase, resulting in an absence of PMN ROS production 33.
As shown in Fig. 5a, p47phox KO mice subjected to the trauma‐haemorrhagic shock model were protected significantly from lung injury, as measured by the [125I]‐radiolabelled albumin lung permeability assay when compared to WT mice (P < 0·001), where lung capillary permeability to proteins is a principle component of ARDS, a severe form of inflammatory lung injury and failure 34. In contrast, the lung permeability of the p47phox KO mice was essentially identical to that of the mock‐injured controls. Furthermore, with respect to effects of PMN ROS on coagulation, p47phox KO mice had significantly lower aPC levels compared to WT mice (P = 0·026), suggesting protection from traumatic coagulopathy in this TIC model (Fig. 5b). Finally, while a non‐significant trend towards lower total thrombin generation was also observed in p47phox KO mice compared to WT mice (P = 0·078) (Fig. 5c), it is noteworthy that the WT mice had a significant elevation in total thrombin generation compared to sham treated controls (P = 0·015), while the p47phox KO mice did not (P = 0·153) (Fig. 5c).
Figure 5.
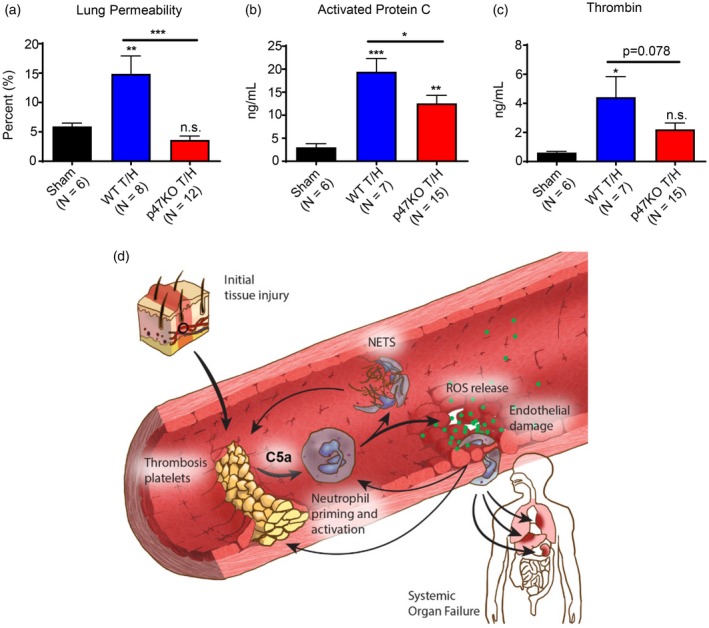
p47phox knock‐out (KO) mice with polymorphonuclear leucocytes (PMN) unable to generate reactive oxygen species (ROS) via the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex are protected from inflammatory lung injury and coagulopathy in a trauma‐haemorrhage (T/H) model. C57Bl/6 wild‐type (WT) and p47phox KO mice underwent laparotomy and haemorrhagic shock via a controlled femoral arterial bleed to a MAP of 35 ± 5 mmHg for 60 min, followed by euthanasia, inferior vena cava (IVC) blood draw and organ harvest. Sham mice underwent all the same procedures including femoral artery cannulation; however, no shock was induced during the 60 min prior to euthanasia. Mice were then evaluated for organ injury via (a) [125I]‐labelled albumin lung permeability assay, and coagulopathy via enzyme‐linked immunosorbent assay (ELISA) for (b) activated protein C levels and (c) thrombin–anti‐thrombin complex levels as a surrogate for total thrombin generation. Results reported as mean ± standard error of the mean (s.e.m.). Significance was set at P ≤ 0·05. *P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001; ****P ≤ 0·0001 (modified from original work by Iris Fung and reproduced from 53).
Discussion
Trauma is the leading cause of death in the United States from ages 1 to 44 years, responsible for more than 180 000 deaths and $406 billion in health‐care costs annually 35, 36. Death after traumatic injury often involves organ failure and sepsis resulting from inflammatory organ injury and immune dysfunction, and in many cases surgical bleeding and coagulopathy is also a major contributor to death 8, 9, 37. While the importance of PMN priming and activation in contributing to end‐organ injury after trauma‐shock and other illnesses (e.g. sepsis) has been recognized previously, a complete understanding of the underlying mechanisms responsible for priming after trauma‐shock is lacking. A plethora of cytokines, chemokines and DAMPs are released into the circulation after trauma 38, but the relative importance of different cytokines in the PMN priming process after trauma‐shock and haemorrhage is unclear. In the current study, we explored possible links between the activation of blood clotting after trauma‐shock and subsequent inflammatory tissue injury mediated though PMN priming, where priming refers specifically to a state where PMN have an enhanced respiratory burst after exposure to a second activating stimulus 3, 4, 5, 39.
Our initial studies showed that products produced by blood coagulation ex vivo led to robust priming of the PMN NADPH oxidase complex predominantly for early extracellular ROS release in response to fMLP. This observation was independent of the presence or absence of platelets and blood cells during the coagulation process, and led to priming and total ROS release approaching 60% of that observed in the internal positive control (20 ng/ml TNF‐ɑ), which corresponds to a level of TNF‐β approximately 100‐fold higher than levels seen typically in septic shock 40.
Surprisingly, a focused screen of activated coagulation factors and peptides relevant to in‐vivo clotting, including fibrinopeptides, failed to demonstrate priming of PMN for enhanced ROS production (Fig 2. This was somewhat unexpected, as: (a) many known PMN chemotactic agents also prime PMN for increased ROS production, yet no priming was observed for the known PMN chemotactic protein fibrinopeptide B 25; and (b) protease‐activated receptors, e.g. PAR‐2 on PMN, which are activated by thrombin cleavage, have been shown to elevate levels of the granule marker CD11b on the PMN surface 15. Instead, the only signalling molecule we identified in our targeted screen of plasma proteases, co‐factors and activation peptides that could prime PMN specifically for ROS release was C5a, which arose, at least in part, through activation of the complement alternative pathway.
Loss of endothelial barrier function is a key component of organ injury after physiological insults such as trauma and sepsis, reviewed in 41, and complement has been implicated in organ injury/failure after trauma in several context‐dependent manners 12, 42, 43, 44, 45, 46. Building on this, we attempted to show that C5a generated during the process of unregulated blood clotting could cause PMN to release soluble mediators that provoke endothelial barrier loss as an ex‐vivo surrogate for organ injury. This is highly relevant to major trauma, where coagulation is no longer locally regulated but instead systemically active 11, and is also particularly relevant to surgical and traumatic bleeding into body cavities/potential spaces where large collections of blood undergo clotting in the extravascular space (e.g. traumatic haemothorax, haemoperitoneum, etc.) much as they would ex vivo in a tube. Using a Transwell filter ECIS assay to measure endothelial barrier resistance while preventing physical contact between PMN and confluent HUVEC during co‐incubation, we found that C5a generated during unregulated clotting of whole blood caused PMN to release extracellular ROS that, in turn, directly caused a loss in endothelial barrier function. These observations linking PMN ROS to endothelial barrier loss were supported further by our finding of a decrease in lung capillary permeability to albumin in vivo in p47phox‐null mice subjected to trauma with haemorrhagic shock.
The specific importance of C5a in trauma‐shock‐induced PMN priming was then investigated using platelet‐poor plasma from trauma patients who presented in shock (Table 1), where we found that C5a levels were elevated sufficiently in these patients to prime PMN for extracellular ROS generation (Fig. 3) that correlated with both thrombin generation and alternative complement pathway activity (Table 2). Reciprocally, we also observed, using an in‐vivo murine model of trauma‐shock 22, 23, that the ROS released from PMN following trauma and haemorrhagic shock contributed directly to lung injury and coagulopathy (Fig. 5). These findings suggest that trauma‐shock primes PMN for extracellular ROS release, probably due, at least in part, to C5a generation, and that this may be an important mechanism involved in the development of inflammatory organ injury and possibly also coagulopathy, as illustrated in Fig. 5d.
The existence of such a link is supported by several other studies, particularly those from Huber‐Lang and colleagues 42, 43, 47. Links between complement pathway activation and C5a generation with modulation of PMN‐mediated inflammation and dysfunction of haemostasis had been elucidated previously in a variety of models of sepsis, with early PMN activation and late PMN dysfunction 48, 49, 50. The extent to which different pathways contribute to C5a production after trauma, however, remains an area for additional investigation. In a murine study of immune‐mediated lung injury, for example, Huber‐Lang et al. showed that C5a was generated in both a C3‐dependent and ‐independent manner 51. These authors suggested further that the C3‐independent component of C5 convertase activity, which was observed primarily in C3 null animals, arose from thrombin cleavage of C5, as the extent of lung damage that they observed was reduced significantly by ATIII or hirudin addition. Further work by Amara et al. has clearly established a potential role for coagulation and fibrinolytic (e.g. plasmin) proteases in the activation of complement proteins 47, and alternative complement pathway activation has been reported previously in trauma and burn patients 12, 52. These findings are consistent with ours, where we found that elevated C5a levels in plasma from our trauma‐shock patients correlated significantly with both alternative pathway activation (Bb levels) and thrombin generation (PF1+2 levels) and, importantly, had the ability to prime naive PMN for extracellular ROS production.
There are several limitations to our study. First, the majority of our experiments were conducted ex vivo in idealized systems that do not necessarily directly extrapolate to in‐vivo findings, indicating that further in‐vivo studies will be needed in the future. Secondly, while we focused on PMN‐derived ROS, a variety of other bioactive mediators are released from primed and activated PMN that could contribute further to tissue injury and coagulation changes, including proteases and other hydrolases. Thirdly, while our murine model used trauma‐shock to study the in‐vivo importance of PMN ROS for development of inflammatory organ injury and effects on the coagulation system (where the mouse trauma‐shock model was chosen specifically because our trauma patients were in shock), we were limited in our sample measurements and were unable to measure C5a levels to corroborate our C5a findings further in the human trauma samples. Finally, we acknowledge that our trauma samples were from a limited number of patients and the results must therefore be interpreted with caution and replicated in larger studies.
In summary, we have shown that trauma‐shock and products of coagulation prime PMN for pathological extracellular ROS release in a C5a‐dependent manner that, in trauma patients in shock, correlates with both thrombin generation and alternative pathway activation. Using a mouse model of trauma and haemorrhagic shock to mimic our patients’ clinical state, we showed that ROS released by PMN contributes directly to lung injury and aPC‐mediated traumatic coagulopathy. These findings, in combination with the work of others, suggest that a critical link exists between complement and PMN ROS in the development of organ injury and coagulation changes after trauma. Future studies and computational models are needed to elucidate further the connections between complement, coagulation and inflammation at the systems biology level.
Acknowledgements
This work was supported by NIH Grants UM1‐HL120877 (M. B. Y., M. J. C., B. S. Z., M. S. P., J. L. S. and M. D. N.), F32‐HL134244 (C. D. B.) and L30‐GM120751 (C. D. B.); and DoD Peer Reviewed Medical Research Program, Contract Number W81XWH‐16‐1‐0464 (M. B. Y.). We gratefully acknowledge Lucia Suarez‐Lopez, Ian Cannell, Lambertus van de Kooij, and Brian Joughin for helpful suggestions throughout the course of this work; Jacob Kim, Joseph Immermann and Timothy Halling for technical support
Disclosure
The authors have no conflicts of interest to report for this manuscript.
Supporting information
Fig. S1. Media, treatment and blood product controls for ECIS PMN‐HUVEC Transwell experiments. Vehicle (DMSO), W‐54011 and DPI effects on HUVEC monolayer barrier function compared to EBM2 alone, as measured by both capacitance (a,b) and resistance (c,d). Effects of plasma or serum when added to EBM2 media (no PMN) (e).
References
- 1. El‐Benna J, Dang PM, Yaffe MB. The phagocyte NADPH oxidase: structure and assembly of the key multicomponent enzyme of innate immunity In: Bradshaw RA, Stahl PD, eds. Encyclopedia of cell biology, 3. Oxford: Academic Press; 2016:702–9. [Google Scholar]
- 2. Hsu AT, Barrett CD, DeBusk GM et al Kinetics and role of plasma matrix metalloproteinase‐9 expression in acute lung injury and the acute respiratory distress syndrome. Shock 2015;44:128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. El‐Benna J, Dang PM, Gougerot‐Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunol 2008;30:279–89. [DOI] [PubMed] [Google Scholar]
- 4. Condliffe AM, Kitchen E, Chilvers ER. Neutrophil priming: pathophysiological consequences and underlying mechanisms. Clin Sci 1998;94:461–71. [DOI] [PubMed] [Google Scholar]
- 5. Guthrie LA, McPhail LC, Henson PM, Johnston RB Jr. Priming of neutrophils for enhanced release of oxygen metabolites by bacterial lipopolysaccharide. Evidence for increased activity of the superoxide‐producing enzyme. J Exp Med 1984;160:1656–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Partrick DA, Moore FA, Moore EE, Barnett CC Jr, Silliman CC. Neutrophil priming and activation in the pathogenesis of postinjury multiple organ failure. New Horiz 1996;4:194–210. [PubMed] [Google Scholar]
- 7. Botha AJ, Moore FA, Moore EE, Peterson VM, Silliman CC, Goode AW. Sequential systemic platelet‐activating factor and interleukin 8 primes neutrophils in patients with trauma at risk of multiple organ failure. Br J Surg 1996;83:1407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vanzant EL, Lopez CM, Ozrazgat‐Baslanti T et al Persistent inflammation, immunosuppression, and catabolism syndrome after severe blunt trauma. J Trauma Acute Care Surg 2014;76:21–9; discussion 9–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brohi K, Cohen MJ, Davenport RA. Acute coagulopathy of trauma: mechanism, identification and effect. Curr Opin Crit Care 2007;13:680–5. [DOI] [PubMed] [Google Scholar]
- 10. Zhang Q, Raoof M, Chen Y et al Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010;464:104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yanagida Y, Gando S, Sawamura A et al Normal prothrombinase activity, increased systemic thrombin activity, and lower antithrombin levels in patients with disseminated intravascular coagulation at an early phase of trauma: comparison with acute coagulopathy of trauma‐shock. Surgery 2013;154:48–57. [DOI] [PubMed] [Google Scholar]
- 12. Ganter MT, Brohi K, Cohen MJ et al Role of the alternative pathway in the early complement activation following major trauma. Shock 2007;28:29–34. [DOI] [PubMed] [Google Scholar]
- 13. Massberg S, Grahl L, von Bruehl ML et al Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 2010;16:887–96. [DOI] [PubMed] [Google Scholar]
- 14. Gould TJ, Vu TT, Swystun LL et al Neutrophil extracellular traps promote thrombin generation through platelet‐dependent and platelet‐independent mechanisms. Arterioscler Thromb Vasc Biol 2014;34:1977–84. [DOI] [PubMed] [Google Scholar]
- 15. Howells GL, Macey MG, Chinni C et al Proteinase‐activated receptor‐2: expression by human neutrophils. J Cell Sci 1997;110:881–7. [DOI] [PubMed] [Google Scholar]
- 16. Baynes J, Dominiczak MH. Medical biochemistry, 4th edn. New York: Elsevier; 2014. [Google Scholar]
- 17. Ellson CD, Davidson K, Ferguson GJ, O'Connor R, Stephens LR, Hawkins PT. Neutrophils from p40phox–/– mice exhibit severe defects in NADPH oxidase regulation and oxidant‐dependent bacterial killing. J Exp Med 2006;203:1927–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lundqvist H, Dahlgren C. Isoluminol‐enhanced chemiluminescence: a sensitive method to study the release of superoxide anion from human neutrophils. Free Rad Biol Med 1996;20:785–92. [DOI] [PubMed] [Google Scholar]
- 19. Bedouhene S, Moulti‐Mati F, Hurtado‐Nedelec M, Dang PM, El‐Benna J. Luminol‐amplified chemiluminescence detects mainly superoxide anion produced by human neutrophils. Am J Blood Res 2017;7:41–8. [PMC free article] [PubMed] [Google Scholar]
- 20. Brown GE, Stewart MQ, Bissonnette SA, Elia AEH, Wilker E, Yaffe MB. Distinct ligand‐dependent roles for p38 MAPK in priming and activation of the neutrophil NADPH oxidase. J Biol Chem 2004;279:27059–68. [DOI] [PubMed] [Google Scholar]
- 21. Sumichika H, Sakata K, Sato N et al Identification of a potent and orally active non‐peptide C5a receptor antagonist. J Biol Chem 2002;277:49403–7. [DOI] [PubMed] [Google Scholar]
- 22. Wang P, Ba ZF, Burkhardt J, Chaudry IH. Trauma‐hemorrhage and resuscitation in the mouse: effects on cardiac output and organ blood flow. Am J Physiol 1993;264:H1166–73. [DOI] [PubMed] [Google Scholar]
- 23. Chesebro BB, Rahn P, Carles M et al Increase in activated protein C mediates acute traumatic coagulopathy in mice. Shock 2009;32:659–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li W, Zheng X, Gu J et al Overexpressing endothelial cell protein C receptor alters the hemostatic balance and protects mice from endotoxin. J Thromb Haemost 2005;3:1351–9. [DOI] [PubMed] [Google Scholar]
- 25. Senior RM, Skogen WF, Griffin GL. Wilner GD. Effects of fibrinogen derivatives upon the inflammatory response. Studies with human fibrinopeptide B. J Clin Invest 1986;77:1014–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coughlin SR. Protease‐activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost 2005;3:1800–14. [DOI] [PubMed] [Google Scholar]
- 27. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol 2010;11:785–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zimmerli W, Reber AM, Dahinden CA. The role of formylpeptide receptors, C5a receptors, and cytosolic‐free calcium in neutrophil priming. J Infect Dis 1990;161:242–9. [DOI] [PubMed] [Google Scholar]
- 29. Bender JG, McPhail LC, Van Epps DE. Exposure of human neutrophils to chemotactic factors potentiates activation of the respiratory burst enzyme. J Immunol 1983;130:2316–23. [PubMed] [Google Scholar]
- 30. Wrann CD, Winter SW, Barkhausen T, Hildebrand F, Krettek C, Riedemann NC. Distinct involvement of p38‐, ERK1/2 and PKC signaling pathways in C5a‐mediated priming of oxidative burst in phagocytic cells. CellImmunol 2007;245:63–9. [DOI] [PubMed] [Google Scholar]
- 31. Howard BM, Miyazawa BY, Dong W et al The tissue factor pathway mediates both activation of coagulation and coagulopathy after injury. J Trauma Acute Care Surg 2015;79:1009–13; discussion 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gando S, Kameue T, Nanzaki S, Hayakawa T, Nakanishi Y. Participation of tissue factor and thrombin in posttraumatic systemic inflammatory syndrome. Crit Care Med 1997;25:1820–6. [DOI] [PubMed] [Google Scholar]
- 33. Jackson SH, Gallin JI, Holland SM. The p47phox mouse knock‐out model of chronic granulomatous disease. J Exp Med 1995;182:751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Han S, Mallampalli RK. The acute respiratory distress syndrome: from mechanism to translation. J Immunol 2015;194:855–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Centers for Disease Control NCfIPaC. Web‐based Injury Statistics Query and Reporting System (WISQARS) . Available at: https://www.cdc.gov/injury/wisqars/leadingcauses.html (accessed 14 February 2017).
- 36. Finkelstein E, Corso PS, Miller TR. The incidence and economic burden of injuries in the United States, vol. xiii Oxford, New York: Oxford University Press; 2006: 187. [Google Scholar]
- 37. Keel M, Trentz O. Pathophysiology of polytrauma. Injury 2005;36:691–709. [DOI] [PubMed] [Google Scholar]
- 38. Namas R, Ghuma A, Hermus L et al The acute inflammatory response in trauma/hemorrhage and traumatic brain injury: current state and emerging prospects. Libyan J Med 2009;4:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yaffe MB, Xu J, Burke PA, Forse RA, Brown GE. Priming of the neutrophil respiratory burst is species‐dependent and involves MAP kinase activation. Surgery 1999;126:248–54. [PubMed] [Google Scholar]
- 40. Pinsky MR, Vincent JL, Deviere J, Alegre M, Kahn RJ, Dupont E. Serum cytokine levels in human septic shock. Relation to multiple‐system organ failure and mortality. Chest 1993;103:565–75. [DOI] [PubMed] [Google Scholar]
- 41. Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Exp Rev Mol Med 2009;11:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Burk AM, Martin M, Flierl MA et al Early complementopathy after multiple injuries in humans. Shock 2012;37:348–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Flierl MA, Perl M, Rittirsch D et al The role of C5a in the innate immune response after experimental blunt chest trauma. Shock 2008;29:25–31. [DOI] [PubMed] [Google Scholar]
- 44. Mulligan MS, Schmid E, Beck‐Schimmer B et al Requirement and role of C5a in acute lung inflammatory injury in rats. J Clin Invest 1996;98:503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Weigelt JA, Chenoweth DE, Borman KR, Norcross JF. Complement and the severity of pulmonary failure. J Trauma 1988;28:1013–9. [DOI] [PubMed] [Google Scholar]
- 46. Solomkin JS, Cotta LA, Satoh PS, Hurst JM, Nelson RD. Complement activation and clearance in acute illness and injury: evidence for C5a as a cell‐directed mediator of the adult respiratory distress syndrome in man. Surgery 1985;97:668–78. [PubMed] [Google Scholar]
- 47. Amara U, Flierl MA, Rittirsch D et al Molecular intercommunication between the complement and coagulation systems. J Immunol 2010;185:5628–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guo RF, Ward PA. Role of C5a in inflammatory responses. Ann Rev Immunol 2005;23:821–52. [DOI] [PubMed] [Google Scholar]
- 49. Huber‐Lang M, Sarma VJ, Lu KT et al Role of C5a in multiorgan failure during sepsis. J Immunol 2001;166:1193–9. [DOI] [PubMed] [Google Scholar]
- 50. Till GO, Ward PA. Oxygen radicals in complement and neutrophil‐mediated acute lung injury. J Free Radic Biol Med 1985;1:163–8. [DOI] [PubMed] [Google Scholar]
- 51. Huber‐Lang M, Sarma JV, Zetoune FS et al Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med 2006;12:682–7. [DOI] [PubMed] [Google Scholar]
- 52. Gelfand JA, Donelan M, Burke JF. Preferential activation and depletion of the alternative complement pathway by burn injury. Ann Surg 1983;198:58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barrett CD, Yaffe MB. Neutrophils, inflammation and innate immunity in trauma‐induced coagulopathy In: Gonzalez E, Moore HB, Moore EE. eds. Trauma induced coagulopathy. New York: Springer Scientific, 2016:149–65. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Media, treatment and blood product controls for ECIS PMN‐HUVEC Transwell experiments. Vehicle (DMSO), W‐54011 and DPI effects on HUVEC monolayer barrier function compared to EBM2 alone, as measured by both capacitance (a,b) and resistance (c,d). Effects of plasma or serum when added to EBM2 media (no PMN) (e).