

Summary
Tissue‐resident memory T (TRM) cells are CD8+ T lymphocytes that reside in the tissues, including tumours. This T cell subset possesses a magnitude of cytotoxicity, but its epigenetic regulation has not been studied. Here, we investigate the impact of perforin DNA methylation in TRM cells and correlate it with their functional potential. Fifty‐three urothelial urinary bladder cancer (UBC) patients were recruited prospectively. The DNA methylation status of the perforin gene (PRF1) locus in TRM cells was investigated by pyrosequencing. Flow cytometry with ViSNE analysis and in‐vitro stimulation were used to evaluate TRM cell phenotypes. We discovered that tumour TRM cells have low DNA methylation in the PRF1 locus (32·9% methylation), which corresponds to increased numbers of perforin‐expressing TRM cells. Surprisingly, programmed cell death 1 (PD‐1) expression is high in tumour TRM cells, suggesting exhaustion. Following interleukin‐15 and T cell receptor stimulation, perforin and T‐bet expressions are enhanced, indicating that TRM cells from tumours are not terminally exhausted. Moreover, a high number of TRM cells infiltrating the tumours corresponds to lower tumour stage in patients. In conclusion, TRM cells from UBC tumours are epigenetically cytotoxic with signs of exhaustion. This finding identifies TRM cells as potential new targets for cancer immunotherapy.
Keywords: CD8‐positive T lymphocytes, cell exhaustion, DNA methylation, perforin, tissue‐resident memory T cells, urinary bladder neoplasms
Introduction
Tissue‐resident memory T (TRM) cells are defined as a memory CD8+ T cell subset that does not recirculate. Consequently, TRM cells have a preferential localization in tissues, which has been demonstrated in cancer, infection, allergy and autoimmunity [1]. TRM cells are identified by their surface expression of CD103 (αEβ7 integrin), that binds to E‐cadherin expressed on epithelia, and CD69, which antagonizes sphingosine 1‐phosphate receptor 1 (S1PR1)‐mediated egress from tissues [2]. Both CD103 and CD69 contribute to anchoring of TRM cells in non‐lymphoid peripheral tissues [3]. Additionally, CD49a expression has been demonstrated to characterize TRM cells in the human epidermis [4].
The microenvironment in the tissues has been reported to induce the development of TRM cells. One signal reported to play a role is transforming growth factor (TGF)‐β1. TGF‐β1 induces CD103 expression through nuclear factor of activated T cells (NFAT)‐1 and suppressor of mothers against decapentaplegic (Smad)2/3 transcription factors binding to the promoter region of the ITGAE gene, encoding for CD103 [5]. Correspondingly, a similar effect has been demonstrated in TGF‐β‐rich tumours, in which TRM cells are consequently generated [6].
Reports have demonstrated that a high number of tumour‐infiltrating TRM cells is linked to improved prognosis in patients with urinary bladder, lung, cervical and ovarian cancers [7, 8, 9, 10]. The improved prognosis is due to the capacity of TRM cells to produce IFN‐γ upon in‐situ antigen recognition [11], resulting in the recruitment of circulating T cells from the blood [12]. Furthermore, the cytotoxic capacity of TRM cells against tumour cells is demonstrated by their production of the cytotoxic mediators, perforin, granzyme A, and granzyme B upon CD103/E‐cadherin ligation and T cell receptor (TCR) activation [8]. Notably, the cytotoxic activity of CD8+ T cells is initiated by perforin forming the pore that allows granzymes to enter and induce apoptosis of the tumour cells [13]. In contrast, TRM cells residing in the tumour tissue have been demonstrated to express programmed cell death 1 (PD‐1), suggesting exhaustion [14]. In this context, the phenotype of TRM cells displaying simultaneous cytotoxicity and exhaustion markers needs further study.
Here, we investigate the properties of perforin expression in TRM cells, starting at the epigenetic level by means of DNA methylation. DNA methylation is defined as methylation of the cytosine residue in a cytosine–phosphate–guanine (CpG) site [15]. It has been linked to gene silencing due to the inability of transcription factors to bind to the promoter of a particular gene [16]. Moreover, study of the DNA methylation profile from the enhancer region of a gene is more critical. This region contains abundant binding sites for key lineage transcription factors, and enhancer‐initiated transcription occurs as the most rapid transcriptional change when cells commence state differentiation [17]. Additionally, epigenetic modification of the lineage‐specific enhancer corresponds to lineage‐specific cell transcription [18].
In this study we focus upon urothelial urinary bladder cancer (UBC), which in the western world is the fourth most common cancer in men and ninth in women [19]. The non‐muscle‐invasive UBC is treated routinely using Bacillus Calmette–Guerin (BCG) instillation in the bladder, indicating a substantial level of immunogenicity in this cancer [20]. In this study we investigated the DNA methylation profile of the perforin gene enhancer on TRM cells lodged in the tumour from UBC patients. The study of the DNA methylation profile of this cell subset was accompanied by analysis of the phenotypical properties ex vivo and after in‐vitro stimulation.
Materials and methods
Patient characteristics and tissue collection
Fifty‐three patients diagnosed with UBC were recruited prospectively from nine hospitals in Sweden (Umeå University Hospital, Sundsvall‐Härnösand County Hospital, Västerås County Hospital, Jönköping/Ryhov County Hospital, Vrinnevi County Hospital in Norrköping, Linköping University Hospital, Östersund County Hospital, Uppsala University Hospital and Skellefteå County Hospital) between 2016 and 2018 (Supporting information, Table S1). All patients underwent diagnostic transurethral resection of the bladder (TUR‐B) and patients with muscle invasive tumours proceeded to radical cystectomy, with or without preceding neoadjuvant chemotherapy. Peripheral blood (PB) and tumour tissues were acquired at the time of TUR‐B, whereas sentinel nodes (SN) and PB were obtained at radical cystectomy. PB was collected in sodium heparin tubes (Greiner Bio‐One, Kremsmünster, Austria). SNs were identified perioperatively with detailed mapping, as described previously [21]. Briefly, radioactive tracer 99mTechnetium was injected into the area surrounding the tumour or tumour scar during the cystectomy operation. Lymphadenectomies were then carried out and the nodes radioactivity was measured by handheld Geiger counter. Tumour tissue and SN were divided into two parts: one part for routine pathological examination and one part for immunological analysis in this study. The tumour and SN for this study were immersed immediately in RPMI‐1640 medium (Sigma Aldrich, St Louis, MO, USA) in 4–8oC and all the samples were processed within 24 h. Buffy coat samples from healthy donors were acquired from Karolinska University Hospital blood bank, Sweden. Informed consent from all patients recruited in this study was obtained in accordance with the Declaration of Helsinki. The study was approved by the regional ethical board (EPN‐Stockholm, registration number: 2007/71‐31 with amendment 2017/190‐32).
Processing of samples
Peripheral blood mononuclear cells (PBMC) were obtained from whole blood samples of patients and healthy donors by density gradient separation using Ficoll‐Paque Plus (GE Healthcare, Chicago, IL, USA). Isolated PBMC were suspended in AIM‐V medium (Life Technologies, Paisley, UK).
Tumour‐infiltrating lymphocytes (TIL) were isolated from the tumour by slicing the collected tissue into small pieces. The small tissues were then transferred into the C‐tubes (Miltenyi Biotech, Bergisch Gladbach, Germany) containing 10 ml of AIM‐V medium with 1% DNAse I (Sigma Aldrich) and 1% collagenase/hyaluronidase (StemCell Technologies, Cambridge, UK). Tissues inside the C‐tubes were processed in GentleMACS (Miltenyi), according to the manufacturer’s protocol. The processed tissue was filtered through a 40‐μm cell strainer, washed and suspended in AIM‐V medium.
Lymphocytes from SN were isolated by gentle pressure homogenization through a 40‐μm cell strainer in AIM‐V medium. The cell suspension was washed and suspended in AIM‐V medium.
Cell sorting
For the initial DNA methylation site screening, perforin+ and perforin– CD8+ T cells were isolated from PBMC of healthy donors. First, bulk CD8+ T cells were sorted using EasySep Human CD8 Positive Selection Kit (StemCell Technologies). The selected CD8+ T cells were labelled with anti‐CD8 (clone RPA‐T8; BD Biosciences, San Jose, CA, USA) and anti‐CD56 (clone B159; BD Biosciences) antibodies, followed by fixation in HOPE I Fixation solution (DCS Innovative Diagnostik‐Systeme, Hamburg, Germany) overnight in 0–4oC. The next day, the cells were permeabilized with 0·1% saponin [w/v in phosphate‐buffered saline (PBS)] (Sigma Aldrich) and stained intracellularly with anti‐perforin antibody (clone δG9; BD Biosciences) for sorting by flow cytometry.
Bulk CD8+ T cells were presorted from patients’ PBMC, SN and tumour using the EasySep Human CD3 Positive Selection Kit (StemCell Technologies). The selected cells were subsequently sorted by flow cytometry after cell labelling with anti‐CD8 (clone RPA‐T8; BD Biosciences) and anti‐CD56 (clone B159; BD Biosciences) antibodies. Cells to be sorted were defined as CD8+CD56–.
For sorting of different memory CD8+ T cell subsets, CD8+ T cells were isolated initially from PBMC and tumour of UBC patients using the EasySep Human CD8 Positive Selection Kit. The cells were then labelled with anti‐CD8 (clone RPA‐T8; BD Biosciences), anti‐CD56 (clone B159; BD Biosciences), anti‐CD103 (clone Ber‐ACT8; BioLegend, San Diego, CA, USA), anti‐CCR7 (clone 150503; BD Biosciences) and anti‐CD45RA (clone HI100; BD Biosciences) antibodies and sorted by flow cytometry.
Cell sorting by flow cytometry was performed using the fluorescence‐activated cell sorter (FACS) Aria I instrument (BD Biosciences) and FACS Diva software (BD Biosciences).
DNA extraction and bisulfite conversion
DNA was extracted and bisulfite converted from frozen pellets of sorted cells using the EZ DNA Methylation Direct Kit (Zymo Research, Irvine, CA, USA), according to the manufacturer’s protocol.
TA cloning and bisulfite sequencing
Bisulfite‐converted DNA was amplified by polymerase chain reaction (PCR) using Platinum Taq DNA Polymerase High Fidelity (ThermoFisher, Fremont, CA, USA) in a T100 Thermal Cycler (Bio‐Rad). The primers used (Eurofins Genomics, Ebersberg, Germany) are listed in Supporting information, Table S2. PCR amplicons were gel‐purified using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA, USA). Next, the PCR amplicons were TA‐cloned into pCR4‐TOPO vector (ThermoFisher) and transformed into TOP10 Escherichia coli (ThermoFisher) by heat shock, according to the manufacturer’s protocol. Transformed cells were cultured on lysogeny broth (LB) plates with 50 μg/ml Kanamycin (Substrate Unit, Karolinska University Hospital) and incubated overnight at 37oC. The following day, 20 colonies were picked from each plate and cultured overnight in LB medium containing 50 μg/ml Kanamycin (Substrate Unit, Karolinska University Hospital) at 37oC at 210 rpm. Plasmid DNA was extracted using PureLink Quick Plasmid Miniprep Kit (ThermoFisher) and Sanger sequenced using T3 sequencing primer (Supporting information, Table S2) to establish CpG methylation profile on a single cell level.
Pyrosequencing
DNA amplification of the region containing PRF1 reporter CpG site [–1053 base pairs (bp) upstream of the transcription start site (TSS)] in the enhancer was performed with PCR using Jumpstart REDTaq DNA polymerase (Sigma Aldrich) in T100 Thermal Cycler (Bio‐Rad). One of the primers used for amplification was biotinylated (Biomers) (Supporting information, Table S2). Biotinylated PCR amplicons were purified using the Pyromark Q96 Vacuum workstation (Qiagen), followed by pyrosequencing using the Pyromark Q96 ID instrument (Qiagen) with Pyromark Gold Q96 reagent (Qiagen), according to the manufacturer’s protocol. Analysis of the data was performed using Pyromark Q96 software (Qiagen) to determine the CpG methylation profile.
Cell culture
For the 5‐azacytidine (5‐aza) experiment, CD8+ T cells from PB of healthy donors were cultured initially in vitro in AIM‐V medium supplemented with 200 IU/ml recombinant human interleukin (IL)‐2, 5 μg/ml plate‐coated anti‐CD3 and 1 μg/ml anti‐CD28 stimulating antibodies. After 48 h, 5‐aza was added to the culture at a final concentration of 2·5 μM/ml for 48 h. The cells were washed and fresh AIM‐V medium was added for another 48 h. The cells were kept at 37oC in 5% CO2 during the culture. At the end of culture, the cells were analysed by flow cytometry.
For the TRM cells activation assay, TRM cells were isolated from PB and tumour of patients. The cells were stimulated in vitro in AIM‐V medium supplemented with 20 ng/ml recombinant human IL‐15, 5 μg/ml plate‐coated anti‐CD3 and 1 μg/ml anti‐CD28 stimulating antibodies. The cells were incubated for 48 h at 37oC in 5% CO2, after which the cells were harvested and analysed by flow cytometry and pyrosequencing.
Flow cytometry
Single cell suspensions isolated from PB, SN and tumour were stained for surface and intracellular markers for flow cytometry analysis. Briefly, cells were stained with fixable live/dead dye (Life Technologies), followed by surface marker staining using anti‐CD8 (clone RPA‐T8; BD Biosciences), anti‐CD56 (clone B159; BD Biosciences), anti‐CD103 (clone Ber‐ACT8; BioLegend), anti‐CCR7 (clone 150503; BD Biosciences), anti‐CD45RA (clone HI100; BD Biosciences) and anti‐PD‐1 (clone EH12.2H7; BioLegend) antibodies. The cells were then fixated and permeabilized using the forkhead box P3 (FOXP3) transcription factor kit (eBioscience, San Diego, CA, USA). Next, the cells were stained for intracellular marker using: anti‐perforin (clone δG9; BD Biosciences), anti‐granzyme B (clone GB11; BD Biosciences), anti‐T‐bet (clone 4B10; BioLegend) and anti‐Ki‐67 (clone 20Raj1; eBioscience) antibodies. Isotype control was used to ascertain the correct gating for the following markers: perforin, granzyme B, T‐bet, Ki‐67 and PD‐1. Flow cytometry data were acquired using an LSR Fortessa instrument (BD Biosciences) and analysed with FlowJo version 10 (TreeStar, Inc., Ashland, OR, USA).
ViSNE
ViSNE was used as a bioinformatic tool to analyse high‐dimensional single cell data from flow cytometry. The Barnes–Hut Stochastic Neighbor Embedding (bh‐SNE) algorithm was used by the ViSNE tool to map the events. Samples utilized in the analysis were first gated manually using FlowJo version 10 for CD8+ T cells and the data were exported as flow cytometry standard (FCS) files with compensated parameters. The measured fluorescence intensity of the parameters included in the analysis was transformed using the arcsin function with a co‐factor of 150. Subsampling was generated randomly from each sample with a total number of cells varying between 2000 and 3000 cells/sample in order to adjust all groups of samples with the same comparable number of events. ViSNE analysis was performed using all parameters that were not subjected to manual gating of CD8+ T cell population. Analysis was perfomed using the tool CYT on the MATLAB (Mathworks) platform [22].
Statistical analysis
For comparison of the two independent groups, a two‐tailed independent t‐test was used for parametric data and the Mann–Whitney U‐test was used for non‐parametric data. One‐way analysis of variance (anova) was used to compare the means among three groups or more with parametric data, meanwhile the Kruskal–Wallis test was used when comparing non‐parametric data. Normality test was performed using the Kolmogorov–Smirnov test on all the data in this study; spss version 23 software (IBM, Armonk, NY, USA) was used for all the statistical analyses.
Results
Perforin expression is regulated by DNA methylation in the enhancer region
The enhancer region of the perforin coding gene, PRF1, was identified previously to be located upstream of the TSS (Fig. 1a), and regulates the expression of perforin [23]. This enhancer region is CpG‐rich, with 10 identified CpG sites (chr10: 72, 363, 525–72, 363, 863) (Fig. 1a). In order to investigate the evolutionary conservation of the regulatory element in this locus, we aligned human and mouse DNA sequences using the VISTA tool. We found that the enhancer region of the PRF1 was not conserved evolutionarily between the two species (< 70% conservation) (Fig. 1a). This indicated the greater importance of using a human model to explore DNA methylation of the PRF1 enhancer. Moreover, instead of using gene silencing analysis, we validated that DNA methylation regulated perforin expression, as displayed when treating CD8+ T cells with the demethylating agent, 5‐aza, which resulted in elevated perforin expression (Fig. 1b).
Figure 1.
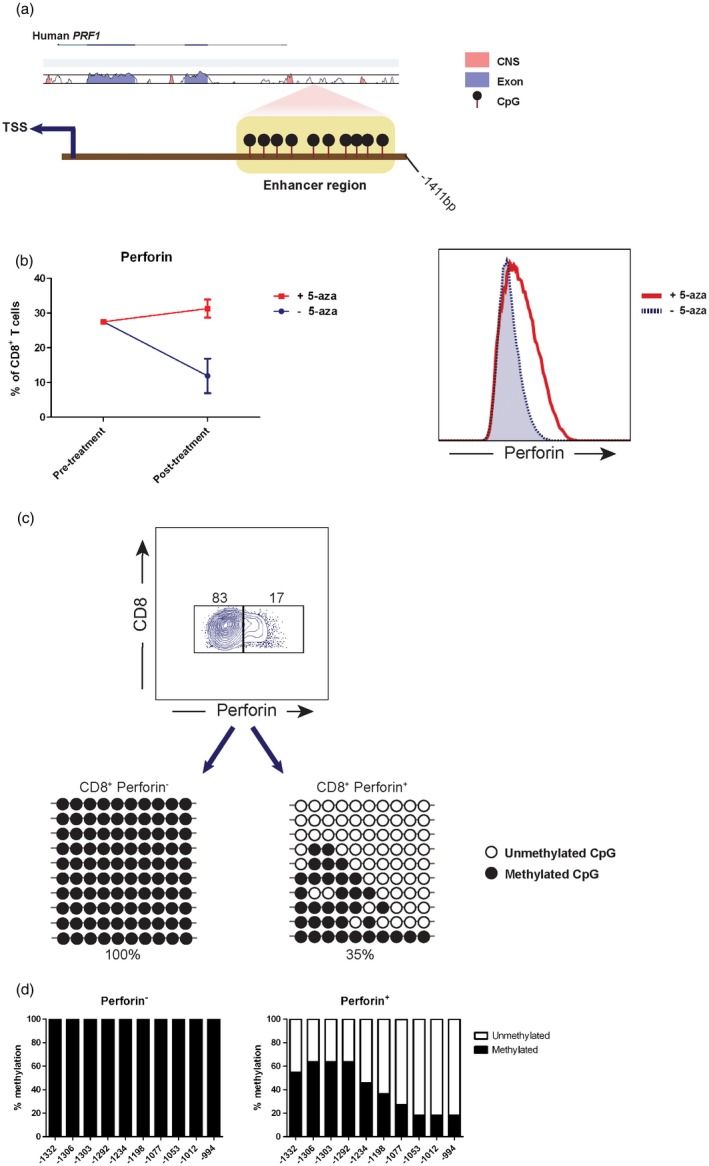
DNA methylation in the enhancer region regulates perforin transcription. (a) Promoter region sequence containing enhancer in the PRF1 gene was aligned between human and mouse using the VISTA tool. The conservation degree is indicated by the peaks, with coloured peaks (blue for exon and light red for the non‐coding sequence) indicating > 70% conservation. Ten cytosine–phosphate–guanine sites (CpGs) in the enhancer are displayed upstream from the transcription start site (TSS). (b) Sorted CD8+ T cells from healthy donors were cultured initially in the presence of 200 IU/ml recombinant human interleukin (IL)‐2, 5 μg/ml plate‐coated anti‐CD3 and 1 μg/ml anti‐CD28 stimulating antibodies in vitro. 5‐Azacytidine (5‐aza) was added to the culture at a final concentration of 2·5 μM/ml for 48 h, followed by culture medium replacement and incubation for another 48 h. Flow cytometry data demonstrating perforin expression pre‐ and post‐treatment are shown. (c) Perforin– and perforin+ CD8+ T cells were sorted. DNA was extracted and bisulfite‐converted from each cell subset, TA‐cloned to pCR4‐TOPO vector and transformed into TOP10 Escherichia coli. Colonies were picked from each cell subset and their DNA was sequenced to measure DNA methylation status of the 10 CpGs in the enhancer. The data display rows represent individually sequenced clone. (d) The bar graphs display total DNA methylation percentage in each CpG from clones in (c).
In order to identify CpGs responsible for regulating perforin transcription, we examined the methylation profile of the 10 CpGs identified in the enhancer region. Bisulfite‐converted DNA from perforin– and perforin+ CD8+ T cells obtained from healthy donors was TA‐cloned and sequenced in order to acquire the methylation status from a single cell level. We discovered that in the perforin– CD8+ T cells, all the CpGs were methylated completely, in contrast to perforin+ cells (35% total methylation) (Fig. 1c), indicating that CpG demethylation in this enhancer is crucial for perforin transcription. The three CpGs closest to the TSS (position –1053, –1012 and –994) were the least methylated in perforin+ CD8+ T cells (Fig. 1d). Correspondingly, CpG in position –1053 was chosen as the reporter for perforin expression.
CD8+ T cells possess distinct cytotoxicity in different tissues by epigenetic regulation
Next, we investigated the methylation profile of the PRF1 reporter CpG site (–1053) in CD8+ T cells isolated from UBC patients by pyrosequencing of the PCR‐amplified bisulfite‐converted target locus. We discovered no significant difference between CD8+ T cells from tumour (mean = 44·1%) and PBMC (mean = 46·6%) (P = 0·95) (Fig. 2a). In contrast, CD8+ T cells from SN had higher methylation in the PRF1 reporter CpG site (mean = 66·8%) compared to PBMC and tumour (both P < 0·05) (Fig. 2a).
Figure 2.
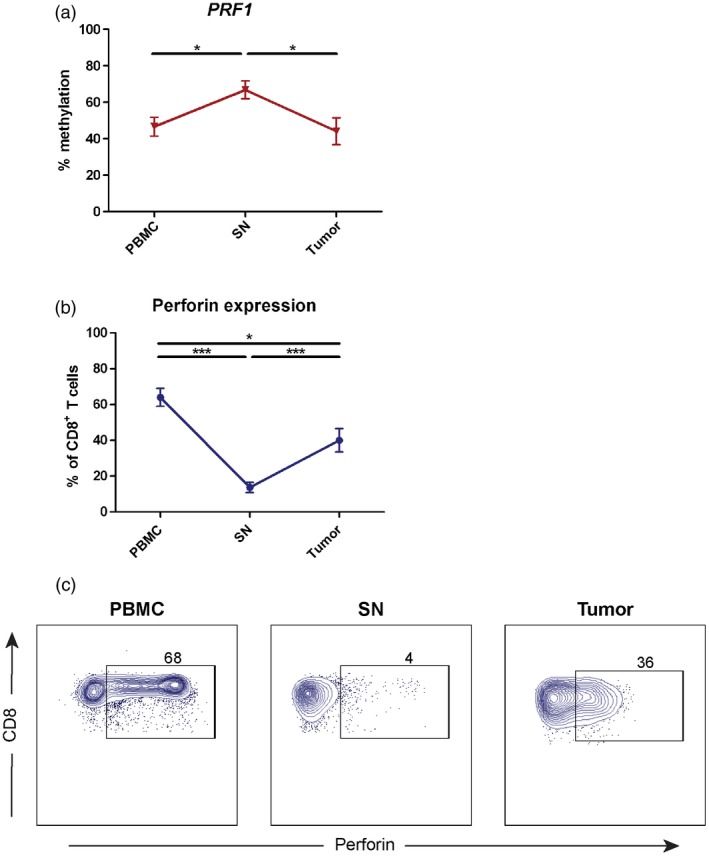
Distinct epigenetic regulation of perforin in CD8+ T cells from different tissues. (a) DNA methylation profile of PRF1 reporter cytosine–phosphate–guanine (CpG) site (–1053) was measured by pyrosequencing of polymerase chain reaction (PCR)‐amplified bisulfite‐converted DNA isolated from CD8+ T cells from peripheral blood mononuclear cells (PBMC), sentinel nodes (SN) and tumour (n = 15). The data are the mean with error bars indicating standard error of the mean (s.e.m.); one‐way analysis of variance (anova) was used as the statistical test. (b) The frequency of perforin+ CD8+ T cells from PBMC, SN and tumour (n = 25) was measured by flow cytometry. The data are the mean with error bars indicating s.e.m.; Kruskal–Wallis was used as the statistical test. (c) The expression of perforin in CD8+ T cells from different tissues (n = 25) were measured by flow cytometry. The gate was based on isotype control and the frequency of perforin+ cells was counted out of CD8+ T cells. Dot‐plots display representative data from one patient. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
To validate the DNA methylation status to the expression of perforin, we performed flow cytometry on CD8+ T cells from UBC patients. Correspondingly, the number of perforin‐expressing CD8+ T cells was nine times higher in tumour (P < 0·001) and 17 times higher in PBMC (P < 0·001) in comparison to SN (Fig. 2b and c). In addition, there was a significant difference between the frequency of perforin+ CD8+ T cells from tumour and PBMC (P = 0·01) (Fig. 2b). The findings indicate epigenetic regulation of the CD8+ T cell phenotype in different tissues with regard to the cytotoxic constituent perforin.
Tissue‐resident memory T cells in tumour are cytotoxic but exhausted
To explore further potential phenotypical differences between CD8+ T cells from tumour and PBMC, not revealed by DNA methylation of the PRF1 locus (Fig. 2a), we characterized memory CD8+ T cell subsets using flow cytometry and ViSNE analysis. Tissue‐resident memory T (TRM) cells, based on CD103 expression (Fig. 3a), were discovered to be increased significantly in the tumour (mean = 56·8%) compared to PBMC (mean = 2·7%) (P < 0·0001) (Fig. 3b and c). Interestingly, a small number of TRM cells could be observed both in the blood (PBMC) of UBC patients and healthy individuals (Supporting information, Fig. S1).
Figure 3.
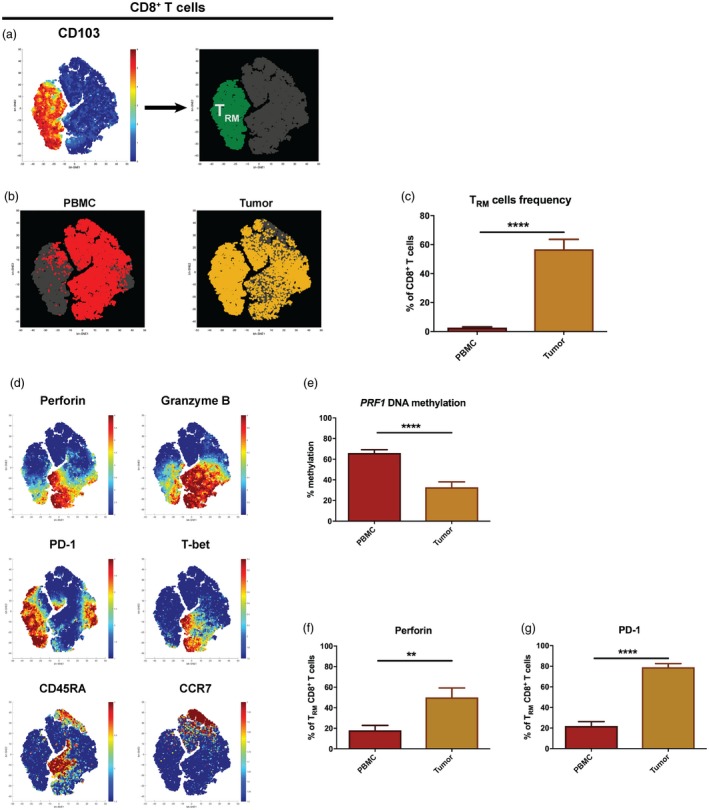
Cytotoxic tissue‐resident memory T cells in tumour despite of exhaustion. (a) ViSNE analysis was performed from flow cytometry data of CD8+ T cells from peripheral blood mononuclear cells (PBMC) and tumour (n = 8). Tissue‐resident memory T (TRM) cells are indicated in green based on CD103 expression. (b) ViSNE analysis of CD8+ T cells derived from PBMC and tumour (n = 8) were plotted (red = PBMC and yellow = tumour). (c) TRM cell frequency in PBMC and tumour (n = 13) were measured by flow cytometry and counted out of total CD8+ T cells. The bar graph shows the mean with error bars indicating standard error of the mean (s.e.m.). Independent t‐test was used as statistical test. (d) The parameter expressions, in which the algorithm of ViSNE in (a,b) was calculated, are displayed with colour indicator representing fluorescence intensity (n = 8). (e) DNA methylation profile of PRF1 reporter cytosine–phosphate–guanine site (CpG) site (–1053) was measured by pyrosequencing of polymerase chain reaction (PCR)‐amplified bisulfite‐converted DNA isolated from TRM cells of PBMC and tumour (n = 10). The bar graph shows the mean with error bars indicating s.e.m.; independent t‐test was used as statistical test. (f,g) Fraction of perforin+ TRM cells or programmed cell death 1 (PD‐1)+ TRM cells in PBMC and tumour (n = 8) were measured by flow cytometry. The bar graph shows the mean with error bars indicating s.e.m.; independent t‐test was used as statistical test. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
Furthermore, DNA methylation of the PRF1 reporter CpG site (–1053) in tumour‐derived TRM cells was decreased twofold compared to PBMC (P < 0·0001) (Fig. 3e). Consequently, perforin‐expressing TRM cells were increased in tumours (P = 0·008) (Fig. 3d and f). In addition, we found that the fraction of TRM cells expressing granzyme B was increased in the tumours compared to PBMC (P < 0·001) (Fig. 3d, Supporting information, Fig. S2a).
The tumour contained a significantly higher number of TRM cells that expressed the exhaustion marker PD‐1 compared to PBMC (mean tumour = 79% versus mean PBMC = 21·9%) (P < 0·0001) (Fig. 3d and g). Additionally, T‐bet expression was low in tumours (Fig. 3d, Supporting information, Fig. S2b), suggesting exhaustion of tumour‐derived TRM cells.
Tissue‐resident memory T cells can be activated despite signs of exhaustion
In order to investigate the functional capacity of TRM cells despite the signs of exhaustion (high PD‐1 and low T‐bet expression), we activated TRM cells from tumour and PBMC with IL‐15 in the presence of anti‐CD3 and anti‐CD28 stimulating antibodies. Following 48 h of stimulation, the number of perforin‐expressing TRM cells from the tumour increased twofold compared to PBMC (P = 0·008) (Fig. 4a and b), with no difference in the proportion of granzyme B+, PD‐1+ and T‐bet+ cells (Supporting information, Figure S3). No prominent change in the DNA methylation of PRF1 signature locus was observed (Supporting information, Fig. S1).
Figure 4.
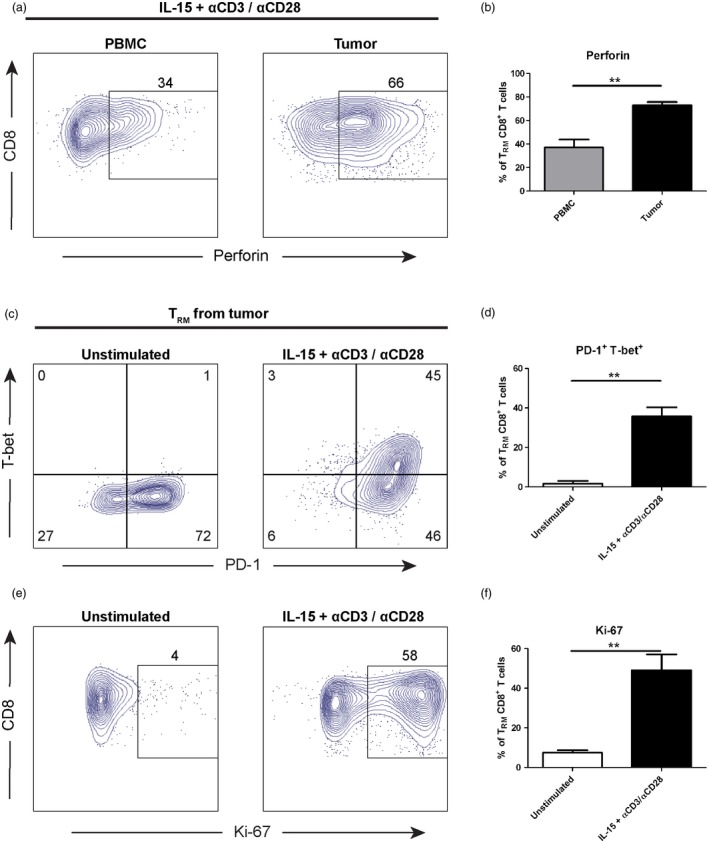
Tissue‐resident memory T cells in tumour are not terminally exhausted. (a) Tissue‐resident memory T (TRM) cells from peripheral blood mononuclear cells (PBMC) and tumour (n = 3) were activated using 20 ng/ml recombinant human interleukin (IL)‐15, 5 μg/ml plate‐coated anti‐CD3 and 1 μg/ml anti‐CD28 stimulating antibodies for 48 h. The expression of perforin post‐stimulation was measured by flow cytometry. The gate was based on isotype control and the frequency of perforin+ cells was counted out of TRM cells. Dot‐plots display representative data from one patient. (b) The frequency of perforin+ TRM cells in PBMC and tumour (n = 3) from (a) is shown. The bar graph demonstrate the mean with error bars indicating standard error of the mean (s.e.m.); independent t‐test was used as statistical test. (c) The expression of T‐bet and programmed cell death 1 (PD‐1) in TRM cells from tumour (n = 3) were measured by flow cytometry post‐culture, comparing unstimulated and stimulated groups. The gate was based on isotype control. Dot‐plots display representative data from one patient. (d) The frequency of PD‐1+ T‐bet+ TRM cells in unstimulated and stimulated groups (n = 3) from (c) is shown. The bar graph shows the mean with error bars indicating s.e.m.; independent t‐test was used as statistical test. (e) The same as in (c), but the dot‐plots show the expression of Ki‐67 in TRM cells (n = 3). (f) Same as in (d), but the graph shows the frequency of Ki‐67+ TRM cells from (e) (n =3). *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
Moreover, the frequency of TRM cells expressing PD‐1+ T‐bet+ increased 21 times following stimulation (P = 0·002) (Fig. 4c and d). Additionally, Ki‐67 expression was elevated significantly in stimulated TRM cells, demonstrated by a 6·6 times increase after IL‐15 and T cell receptor (TCR)‐stimulation (P = 0·007) (Fig. 4e and f). Taken together, PD‐1‐expressing TRM cells from the tumour possessed great cytotoxic and proliferative potential following activation of the cells, demonstrating that they are not terminally exhausted.
Tissue‐resident memory T cells are the most dominant subset in the tumour
As we have demonstrated the cytotoxic and functional capacity of TRM cells when they resided in the tumour, the next step was to compare the phenotype of TRM cells to other memory cell subsets present. ViSNE analysis was performed on total CD8+ T cells isolated from the tumour. As mentioned earlier, TRM cells co‐expressed CD49a and CD69 alongside CD103 and constituted the dominant memory cell subset in the tumour (P < 0·001) (Fig. 5a and b). However, other memory cell subsets were also present, such as central memory T (TCM) cells (CD103–CD45RA–CCR7+, mean = 4·25%), effector memory T (TEM) cells (CD103–CD45RA–CCR7–, mean = 27·3%) and effector memory T cells with CD45RA up‐regulation (TEMRA) (CD103–CD45RA+CCR7–, mean = 8·7%) (Fig 5a and b).
Figure 5.
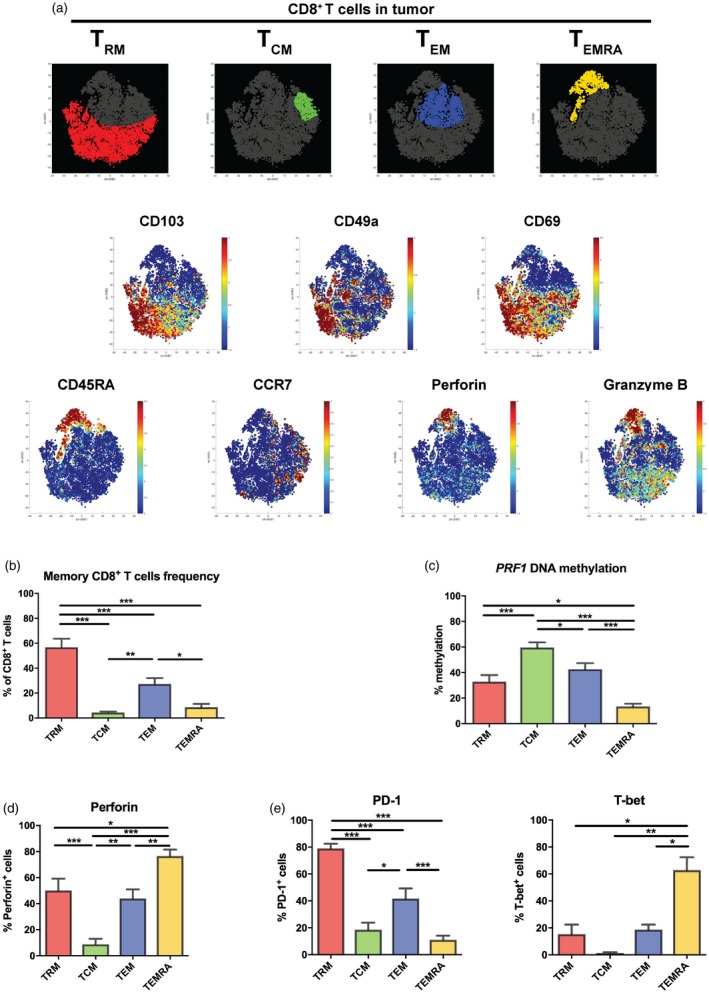
Tumour tissue is highly infiltrated by tissue‐resident memory T cells. (a) ViSNE analysis was performed on tumour CD8+ T cells data measured by flow cytometry (n = 4). Distinct CD8+ T cell memory subsets were plotted [red = tissue‐resident memory T (TRM), green = TCM, blue = TEM and yellow = TEMRA]. The parameter expressions, in which the algorithm of ViSNE was calculated, are displayed with colour indicator representing fluorescence intensity. (b) The frequency of memory CD8+ T cell subsets were measured by flow cytometry and counted out of total CD8+ T cells (n = 13). The bar graph shows the mean with error bars indicating standard error of the mean (s.e.m.); one‐way analysis of variance (anova) was used as statistical test. (c) DNA methylation profile of PRF1 reporter cytosine–phosphate–guanine (CpG site) (–1053) was measured by pyrosequencing of polymerase chain reaction (PCR)‐amplified bisulfite‐converted DNA isolated from different tumour memory CD8+ T cell subsets (n = 10). The bar graph shows the mean with error bars indicating s.e.m.; one‐way anova was used as statistical test. (d,e) Fraction of perforin+, programmed cell death 1 (PD‐1)+ or T‐bet+ in different memory CD8+ T cell subsets were measured by flow cytometry (n = 8). The bar graph shows the mean with error bars indicating s.e.m.; one‐way anova was used as statistical test. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
When investigating the DNA methylation profile of the PRF1 reporter CpG site (–1053) in each cell subset, TRM cells and TEM cells demonstrated intermediate methylation (mean = 32·9 and 42·6%, respectively) with significant lower to TCM cells, but higher when compared to TEMRA cells (Fig. 5c). This corresponded inversely with the number of perforin+ cells among TRM and TEM cells, which was significantly higher compared to TCM cells (P < 0·01) and lower than terminally differentiated TEMRA cells (P < 0·05) (Fig. 5d). This expression pattern was also observed for granzyme B (Supporting information, Fig. S5).
The fraction of PD‐1‐expressing cells was the highest among TRM cells when compared to other cell subsets (P < 0·001) (Fig. 5e, left panel), which resulted consequently in a significantly lower expression of T‐bet (P = 0·014 TRM versus TEMRA) (Fig. 5e, right panel). We conclude that the TRM cells are dominant in the tumours demonstrating higher cytotoxic quality compared to the other cell subsets.
High infiltration of tumour tissue‐resident memory T cells corresponds with lower tumour stage
Because TRM cells were demonstrated to have positive cytotoxic properties, we correlated the proportion of different memory CD8+ T cell subsets infiltrating the tumour to clinical tumour–node–metastasis (TNM) stage in UBC patients. We observed that in the patients with stage I (T1 N0 M0), there was a trend towards dominant TRM cell infiltration in the tumour (Fig. 6a). Notably, patient 48 had a lower TRM cell number, similar to high‐stage tumour. Hypothetically, it was due to the high‐risk stage I tumour (cT1 + carcinoma in situ) in this patient. In contrast, in patients with muscle invasive tumour, i.e. stage II (T2 N0 M0) and stage III (T3 N0 M0), the trend demonstrated a lower fraction of TRM cells in the tumour (Fig. 6a).
Figure 6.
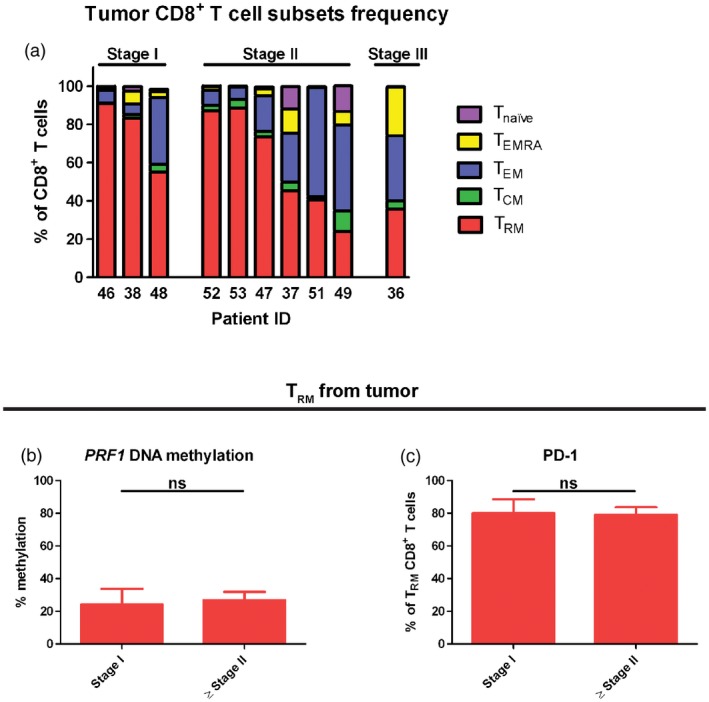
The increased number of tissue‐resident memory T (TRM) cells in tumours correlates with lower tumour stage. (a) The proportion of distinct memory CD8+ T cell subsets in the tumour was measured by flow cytometry and displayed from each urinary bladder cancer (UBC) patient (n = 10). Patients were grouped based on the tumour–node–metastasis (TNM) tumour stage judged clinically. Stage I = T1 N0 M0, stage II = T2 N0 M0 and stage III = T3 N0 M0. (b) DNA methylation profile of PRF1 reporter cytosine–phosphate–guanine (CpG) site (–1053) was measured by pyrosequencing of polymerase chain reaction (PCR)‐amplified bisulfite‐converted DNA isolated from tumour TRM cells and compared between patients with stage I and ≥ stage II (n = 10). The bar graph shows the mean with error bars indicating standard error of the mean (s.e.m.); Mann–Whitney was used as statistical test. (c) The frequency of programmed cell death 1 (PD‐1)+ TRM cells from tumour was measured by flow cytometry and compared between patients with stage I and ≥ stage II (n = 10). The bar graph shows the mean with error bars indicating s.e.m.; Mann–Whitney was used as statistical test.
Furthermore, when comparing DNA methylation in the PRF1 locus, there was no significant difference between patients with non‐muscle invasive (stage I) and muscle‐invasive (≥ stage II) tumours (P = 0·571) (Fig. 6b). In addition, the PD‐1‐expressing TRM cell number was not seen to be different between the two stage groups (P = 1·0) (Fig. 6c). This suggests that the tumour TRM cells have similar phenotypes among tumour stages.
Discussion
Here, for the first time to our knowledge, we demonstrated that phenotypically exhausted TRM cells are epigenetically available for the transcription of perforin and harbour effector functions, which may correlate with a less advanced tumour stage. Our findings suggest that TRM cells residing in the tumour tissue of urothelial UBC have low DNA methylation in the reporter CpG site located in the enhancer of the PRF1 locus (Fig. 3e). This discovery has a solid correlation with the perforin protein expression inside the cells (Fig. 3f) and renders the possibility to activate tumour TRM cells (Fig. 4). Moreover, a high number of TRM cells infiltrating the tumour corresponds to a lower tumour stage in UBC patients (Fig. 6a). Epigenetic profiling of TRM cells may help us in revealing mechanisms that can be targeted to reverse exhaustion and enhance cytotoxic potency in future cancer immunotherapy regimens.
The rationale for investigating the enhancer region of the PRF1 gene relies on the fact that the regulation by enhancers are proven to be highly dynamic upon CD8+ T cell activation and differentiation [24]. Furthermore, the transcription of PRF1 is regulated mainly by the enhancer located at position –1kb from the TSS, which has binding sites for transcription factors related to T cell activation, such as signal transducer and activator of transcription (STAT)‐5, nuclear factor kappa B (NF‐κB) and activator protein 1 (AP‐1) [25]. Hence, it is compelling to examine the DNA methylation status of the CpG signature site in the enhancer region, where we found a good correlation with perforin expression (Fig. 2). However, we observed that PRF1 methylation was equivalent in PBMC and tumour CD8+ T cells, but the perforin protein expression is lower in the tumour (Fig. 2). This discrepancy may be due to additional mechanisms that regulate perforin expression, such as microRNAs [26] or histone modifications.
It is widely acknowledged that DNA methylation has a critical role in gene expression and specific cell lineage maintenance. Therefore, DNA methylation establishes a stable epigenetic mark which can be inherited through successive cell divisions [27]. In the context of CD8+ T cell differentiation, a shift in DNA methylation is not a straightforward phenomenon once CD8+ T cells have retained their specific memory subset. Accordingly, effector‐associated loci, such as PRF1, have a stably unchanged DNA methylation in peripheral blood‐derived naive, TCM and TEM CD8+ T cells, even after three rounds of cell division by ex‐vivo stimulation using homeostatic cytokines IL‐7 and IL‐15 [28].
These insights prompted us to study DNA methylation of the PRF1 locus in tissue‐resident memory T (TRM) cells, which have unique transcriptional signatures compared to TCM and TEM cells [3]. TRM cells are defined as memory CD8+ T cells that reside in the non‐lymphoid tissues and have a distinct functional profile [29]. Here, we demonstrated that the PRF1 locus of TRM cells was hypomethylated compared to TCM and TEM cells from the tumour (Fig. 5c), which corresponded to an increased fraction of perforin‐expressing TRM cells (Fig. 5d). Although we could see that terminally differentiated TEMRA cells displayed the lowest DNA methylation in the PRF1 locus, leading to consequently higher perforin expression compared to TRM cells, we know that TEMRA cells are short‐lived in contrast to TRM cells [30]. Moreover, long‐lived TRM cells are demonstrated to elicit more potent local immune protection in the tissue against infection [31].
In cancer, TRM cells are reported to delay tumour growth and prolong survival [32]. Additionally, TRM cells are necessary for the efficacy of cancer vaccine [11, 33]. The protective effect of TRM cells in cancer is linked to their high degree of cytotoxicity, which is correlated with the magnitude of their perforin and granzyme expression [14]. In spite of expressing cytotoxic mediators, TRM cells in the tumour express the exhaustion markers PD‐1, T cell immunoglobulin and mucin domain 3 (TIM‐3) and lymphocyte‐activation gene 3 (LAG‐3) [34]. In accordance with these previous findings, in this study we have demonstrated a high number of PD‐1+ TRM cells from the tumour (Fig. 3g) and a low fraction of T‐bet+ cells in this subset (Supporting information, Fig. S2b). However, when we activated TRM cells from tumours using the homeostatic cytokine IL‐15 and T cell receptor (TCR) stimulation in vitro, we observed that the dominant PD‐1+ TRM cells began to gain T‐bet expression (Fig. 4c and d). Consequently, this resulted in elevated perforin and granzyme B expression in TRM cells from tumour and increased proliferation, i.e. elevated Ki‐67 (Fig. 4e and f). Taken together, this indicates that the tumour‐derived TRM cells are not terminally exhausted. Consistent with our data, skin TRM cells have been described to have high expression of PD‐1 and other co‐inhibitory molecules after viral infection, thus suggesting exhaustion. Surprisingly, following antigenic challenge, in‐situ proliferation of TRM cells, marked by up‐regulation of Ki‐67, occurs [35]. Therefore, we may speculate that the TRM cell is a memory CD8+ T cell subset with an outstanding potential for cytotoxicity and is not terminally exhausted.
The mechanism behind the co‐expression of exhaustion markers and cytotoxic mediators seen in TRM cells may be explained as part of T cell activation [36]. We know that PD‐1 expression in CD8+ T cells results from chronic antigenic exposure in the tumour tissue [37]. However, recent evidence describes that in the case of chronic infection, permanent PD‐1 expression in TRM cells is imprinted epigenetically and is antigen‐independent [38, 39]. Additionally, PD‐1 signalling has been demonstrated to be important for antigen‐independent maintenance of memory CD8+ T cells [40]. Therefore, we may speculate that PD‐1 expression is a mere TRM cell marker that does not affect cell functionality, and may be seen as an engagement marker with tumour cells. Accordingly, it was seen in our data that PD‐1 expression was similar at different stages of the tumour (Fig. 6c).
Notably, our finding indicated that TRM cells present in the peripheral circulation had the capacity to increase their expression of granzyme B, PD‐1 and T‐bet, following IL‐15 and TCR stimulation. The results were comparable to tumour TRM cells (Supporting information, Fig. S3). It is important to note that in this context PD‐1 acts as an indicator of T cell activation instead of exhaustion [41]. Therefore, TRM cells from the circulation may have a promising feature of activation following TCR engagement and possibly CD103/E‐cadherin ligation once they lodge in the tumour tissue [6, 42, 43]. However, their specificity towards distinct tumour antigens needs to be investigated further.
The limitation of this study was that we correlated TRM cell numbers with the tumour stage in only 10 patients. However, from this observation we could recapitulate the trend in which high TRM cell infiltration in the tumours corresponded to less advanced tumour stage, suggesting the potential anti‐tumour capacity of TRM cells. Therefore, further correlation between patients’ clinical prognosis with TRM cell numbers is required in a larger cohort.
In conclusion, epigenetic profiling adds to our understanding about TRM cell phenotypes and functionality. These findings help us to recognize the cytotoxic potential of TRM cells and therefore provide us with new strategies for cancer immunotherapy.
Disclosure
None.
Author contributions
C. A. H., A. S. and O. W. designed the study, developed the methodology, wrote and reviewed the manuscript. C. A. H, E. A. B., A. B. and S. B. performed the experiments. M. J., F. A., T. J., Y. H., F. A., K. P., B. H., K. R. and A. S. provided clinical samples and patients’ clinical information. A. S. assembled the clinical information database of all patients. C. A. H. and S. B. performed data analysis. H. G. supervised the study.
Supporting information
Fig S1. A small fraction of TRM cells is present in the blood of UBC patients and healthy individuals. ViSNE analysis was performed on PBMC CD8+ T cells data measured by flow cytometry (n = 7). Tissue‐resident memory T (TRM) cells are indicated in green based on CD103 expression. PBMC CD8+ T cells from healthy donors and UBC patients were plotted (red = healthy donors and yellow = UBC patients).
Fig S2. Cytotoxic tissue‐resident memory T cells in tumour despite of exhaustion. (a) The frequency of granzyme B+ TRM cells in PBMC and tumour (n = 8) were measured by flow cytometry and counted out of TRM cells. The bar graph shows means with error bars indicating SEM. Independent t‐test was used as statistical test. (b) Same as in (a), but the data show T‐bet+ TRM cell frequency (n = 8). Mann‐Whitney was used as statistical test. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
Fig S3. TRM cells from blood can potentially be activated. (a) The frequency of granzyme B+ TRM cells from PBMC and tumour (n = 3) in unstimulated and stimulated condition with IL‐15, anti‐CD3 and anti‐CD28 stimulating antibodies were measured by flow cytometry and counted out of TRM cells. The bar graph shows means with error bars indicating SEM. Independent t‐test was used as statistical test. (b)(c) Same as in (a), but the data show the frequency of PD‐1+ TRM cells or T‐bet+ TRM cells (n = 3). *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
Fig S4. DNA methylation of PRF1 locus does not change following activation. DNA methylation profile of PRF1 reporter CpG site (‐1053) was measured by pyrosequencing of PCR‐amplified bisulfite‐converted DNA isolated from pre‐culture, unstimulated and stimulated (IL‐15 + αCD3 + αCD28) tumour TRM cells. The data was acquired from one representative patient.
Fig S5. TRM cells are cytotoxic in the tumour. The fraction of tumour‐derived memory CD8+ T cell subsets expressing granzyme B were measured by flow cytometry (n = 8). The bar graph shows means with error bars indicating SEM. One‐way ANOVA was used as statistical test. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
Acknowledgements
This paper was supported by the Swedish Cancer Society (Cancerfonden), the Regional Research Council in Uppsala‐Örebro region, Sweden (RFR in Uppsala‐Örebro), the Swedish Research Council funding for clinical research in medicine (ALF) in Västerbotten, VLL, Sweden, the Cancer Research Foundation in Norrland, Umeå, Sweden and the Stiftelsen Emil Anderssons fond för medicinsk forskning, Sundsvall, Sweden. The authors acknowledge research nurses Britt‐Inger Dahlin and Kerstin Almroth (Department of Surgical and Perioperative Sciences, Urology and Andrology, Umeå University) for their great assistance in this work.
References
- 1. Mueller SN, Mackay LK. Tissue‐resident memory T cells: local specialists in immune defence. Nat Rev Immunol 2016;16:79–89. [DOI] [PubMed] [Google Scholar]
- 2. Watanabe R, Gehad A, Yang C et al Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med 2015;7. doi: 10.1126/scitranslmed.3010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mackay LK, Rahimpour A, Ma JZ et al The developmental pathway for CD103(+)CD8(+) tissue‐resident memory T cells of skin. Nat Immunol 2013;14:1294–301. [DOI] [PubMed] [Google Scholar]
- 4. Cheuk S, Schlums H, Serezal IG et al CD49a expression defines tissue‐resident CD8(+) T cells poised for cytotoxic function in human skin. Immunity 2017;46:287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mokrani M, Klibi J, Bluteau D, Bismuth G, Mami‐Chouaib F. Smad and NFAT pathways cooperate to induce CD103 expression in human CD8 T lymphocytes. J Immunol 2014;192:2471–79. [DOI] [PubMed] [Google Scholar]
- 6. Le Floc'h A, Jalil A, Vergnon I et al. alpha(E)beta(7) integrin interaction with E‐cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med 2007;204:559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang B, Wu SX, Zeng H et al CD103(+) tumor infiltrating lymphocytes predict a favorable prognosis in urothelial cell carcinoma of the bladder. J Urol 2015;194:556–62. [DOI] [PubMed] [Google Scholar]
- 8. Djenidi F, Adam J, Goubar A et al CD8(+) CD103(+) tumor‐infiltrating lymphocytes are tumor‐specific tissue‐resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol 2015;194:3475–86. [DOI] [PubMed] [Google Scholar]
- 9. Komdeur FL, Prins TM, van de Wall S et al CD103+tumor‐infiltrating lymphocytes are tumor‐reactive intraepithelial CD8+T cells associated with prognostic benefit and therapy response in cervical cancer. Oncoimmunology 2017;6: e1338230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Webb JR, Milne K, Watson P, deLeeuw RJ, Nelson BH. Tumor‐infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high‐grade serous ovarian cancer. Clin Cancer Res 2014;20:434–44. [DOI] [PubMed] [Google Scholar]
- 11. Nizard M, Roussel H, Diniz MO et al Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun 2017;8. doi: 10.1038/ncomms15221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol 2013;14:509–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Diff 2010;17:616–23. [DOI] [PubMed] [Google Scholar]
- 14. Ganesan AP, Clarke J, Wood O et al Tissue‐resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol 2017;18:940–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet 2016;17:487–500. [DOI] [PubMed] [Google Scholar]
- 16. Schubeler D. Function and information content of DNA methylation. Nature 2015;517:321–26. [DOI] [PubMed] [Google Scholar]
- 17. Arner E, Daub CO, Vitting‐Seerup K et al Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 2015;347:1010–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8+ T cell differentiation. NatRev Immunol 2018;18: 340–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burger M, Catto JW, Dalbagni G, et al Epidemiology and risk factors of urothelial bladder cancer. Eur Urol 2013;63:234–41. [DOI] [PubMed] [Google Scholar]
- 20. Redelman‐Sidi G, Glickman MS, Bochner BH. The mechanism of action of BCG therapy for bladder cancer‐a current perspective. Nat Rev Urol 2014;11:153–62. [DOI] [PubMed] [Google Scholar]
- 21. Rosenblatt R, Johansson M, Alamdari F et al Sentinel node detection in muscle‐invasive urothelial bladder cancer is feasible after neoadjuvant chemotherapy in all pT stages, a prospective multicenter report. World J Urol 2017;35:921–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Amir ED, Davis KL, Tadmor MD et al viSNE enables visualization of high dimensional single‐cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 2013;31:545– 52.+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu QJ, Wu AL, Ray D et al DNA methylation and chromatin structure regulate T cell perforin gene expression. J Immunol 2003;170:5124–32. [DOI] [PubMed] [Google Scholar]
- 24. He B, Xing SJ, Chen CY et al CD8(+) T cells utilize highly dynamic enhancer repertoires and regulatory circuitry in response to infections. Immunity 2016;45:1341–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pipkin ME, Rao A, Lichtenheld MG. The transcriptional control of the perforin locus. Immunol Rev 2010;235:55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Trifari S, Pipkin ME, Bandukwala HS et al. MicroRNA‐directed program of cytotoxic CD8+ T‐cell differentiation. Proc Natl Acad Sci USA 2013;110:18608–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cedar H, Bergman Y. Programming of DNA methylation patterns. Ann Rev Biochem 2012;81:97–117. [DOI] [PubMed] [Google Scholar]
- 28. Abdelsamed HA, Moustaki A, Fan YP et al Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med 2017;214:1593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sathaliyawala T, Kubota M, Yudanin N et al Distribution and compartmentalization of human circulating and tissue‐resident memory T cell subsets. Immunity 2013;38:187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Ann Rev Immunol 2004;22:745–63. [DOI] [PubMed] [Google Scholar]
- 31. Mackay LK, Stock AT, Ma JZ et al Long‐lived epithelial immunity by tissue‐resident memory T (T‐RM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci USA 2012;109:7037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Milner JJ, Toma C, Yu BF et al Runx3 programs CD8(+) T cell residency in non‐lymphoid tissues and tumours. Nature 2017;552:253–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Granier C, Blanc C, Karaki S, Tran T, Roussel H, Tartour E. Tissue‐resident memory T cells play a key role in the efficacy of cancer vaccines. Oncoimmunology 2017;6. doi: 10.1080/2162402X.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Webb JR, Milne K, Nelson BH. PD‐1 and CD103 are widely coexpressed on prognostically favorable intraepithelial CD8 T cells in human ovarian cancer. Cancer Immunol Res 2015;3:926–35. [DOI] [PubMed] [Google Scholar]
- 35. Park SL, Zaid A, Hor JL et al Local proliferation maintains a stable pool of tissue‐resident memory T cells after antiviral recall responses. Nat Immunol 2018;19:183–91. [DOI] [PubMed] [Google Scholar]
- 36. Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity 2018;48:202–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat RevImmunol 2015;15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Youngblood B, Oestreich KJ, Ha SJ et al Chronic virus infection enforces demethylation of the locus that encodes PD‐1 in antigen‐specific CD8(+) T cells. Immunity 2011;35:400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shwetank Abdelsamed HA, Frost EL, Schmitz HM, Mockus TE, Youngblood BA, Lukacher AE. Maintenance of PD‐1 on brain‐resident memory CD8 T cells is antigen independent. Immunol Cell Biol 2017;95:953–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yuzefpolskiy FM, Penny LA, Kalia V, Sarkar S. Signaling through PD‐1 on CD8 T cells is critical for antigen‐independent maintenance of immune memory. Immunology 2016. J Immunol 2016;196(Suppl 1):129.6. [Google Scholar]
- 41. Legat A, Speiser DE. Pircher H, Zehn D, Marraco SAF. Inhibitory receptor expression depends more dominantly on differentiation and activation than ‘exhaustion’ of human CD8 T cells. Front Immunol 2013;4:455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Le Floc'h A, Jalil A, Franciszkiewicz K, Validire P, Vergnon I, Mami‐Chouaib F. Minimal engagement of CD103 on cytotoxic T lymphocytes with an E‐cadherin‐Fc molecule triggers lytic granule polarization via a phospholipase C gamma‐dependent pathway. Cancer Res 2011;71:328–38. [DOI] [PubMed] [Google Scholar]
- 43. Gauthier L, Corgnac S, Boutet M et al. Paxillin binding to the cytoplasmic domain of CD103 promotes cell adhesion and effector functions for CD8(+) resident memory T cells in tumors. Cancer Res 2017;77:7072–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. A small fraction of TRM cells is present in the blood of UBC patients and healthy individuals. ViSNE analysis was performed on PBMC CD8+ T cells data measured by flow cytometry (n = 7). Tissue‐resident memory T (TRM) cells are indicated in green based on CD103 expression. PBMC CD8+ T cells from healthy donors and UBC patients were plotted (red = healthy donors and yellow = UBC patients).
Fig S2. Cytotoxic tissue‐resident memory T cells in tumour despite of exhaustion. (a) The frequency of granzyme B+ TRM cells in PBMC and tumour (n = 8) were measured by flow cytometry and counted out of TRM cells. The bar graph shows means with error bars indicating SEM. Independent t‐test was used as statistical test. (b) Same as in (a), but the data show T‐bet+ TRM cell frequency (n = 8). Mann‐Whitney was used as statistical test. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
Fig S3. TRM cells from blood can potentially be activated. (a) The frequency of granzyme B+ TRM cells from PBMC and tumour (n = 3) in unstimulated and stimulated condition with IL‐15, anti‐CD3 and anti‐CD28 stimulating antibodies were measured by flow cytometry and counted out of TRM cells. The bar graph shows means with error bars indicating SEM. Independent t‐test was used as statistical test. (b)(c) Same as in (a), but the data show the frequency of PD‐1+ TRM cells or T‐bet+ TRM cells (n = 3). *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
Fig S4. DNA methylation of PRF1 locus does not change following activation. DNA methylation profile of PRF1 reporter CpG site (‐1053) was measured by pyrosequencing of PCR‐amplified bisulfite‐converted DNA isolated from pre‐culture, unstimulated and stimulated (IL‐15 + αCD3 + αCD28) tumour TRM cells. The data was acquired from one representative patient.
Fig S5. TRM cells are cytotoxic in the tumour. The fraction of tumour‐derived memory CD8+ T cell subsets expressing granzyme B were measured by flow cytometry (n = 8). The bar graph shows means with error bars indicating SEM. One‐way ANOVA was used as statistical test. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.