

Key Points
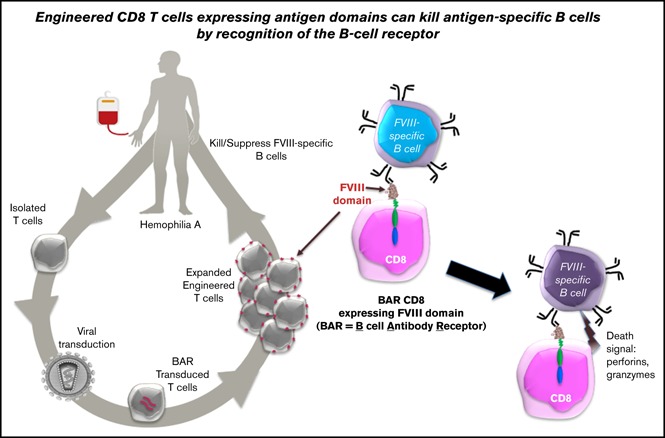
BAR CD8 T cells interact with and kill specific B cells.
Engineered CD8 T cells expressing antigen domains suppress B-cell responses to FVIII.
Abstract
Hemophilia A is an X-linked bleeding disorder caused by mutations in the factor VIII (FVIII) gene (F8). Treatment with recombinant or plasma-derived FVIII replacement therapy is standard therapy. A major problem in treating hemophilia A patients with therapeutic FVIII is that 20% to 30% of these patients produce neutralizing anti-FVIII antibodies (inhibitors) because they are not immunologically tolerant to this human protein. Hence, there is a need to establish tolerogenic protocols to FVIII epitopes. To specifically target FVIII-specific B cells, we engineered immunodominant FVIII domains (A2 and C2) as a chimeric antigen receptor expressed by both human and murine cytotoxic T cells. This FVIII domain engineered B-cell antibody receptor (BAR) that expresses T cells was capable of killing FVIII-reactive B-cell hybridomas in vitro and in vivo. Moreover, FVIII BAR CD8 T cells blocked the development of specific antibody from unimmunized spleen cells stimulated polyclonally with lipopolysaccharide in vitro. In addition, adoptive transfer of FVIII A2- and C2-BAR CD8 T cells significantly reduced the anti-FVIII antibody formation in hemophilic mice. These data suggest that BAR-engineered T cells are a promising approach for future prophylactic treatment for patients with severe hemophilia A who are at high risk of developing inhibitors.
Visual Abstract
Introduction
Hemophilia A (HA) is caused by mutations in the factor VIII (FVIII) gene (F8), which is located on the X chromosome. These patients have a deficiency of functional FVIII, which is critical for blood coagulation.1 Currently, the disorder can be treated with recombinant or plasma-derived FVIII replacement therapy. However, because patients are not immunologically tolerant to FVIII, an anti-drug antibody response can occur in up to 30% of the patients receiving replacement FVIII treatment.2,3 These immunoglobulin G (IgG) antibodies (called inhibitors) neutralize the function of therapeutic FVIII drugs.4 The standard treatment to reverse inhibitor formation consists of repeated high-dose infusions of FVIII, a process referred to as Immune Tolerance Induction (ITI).5 Despite some clinical success, immune tolerance induction is expensive and does not work for all patients with inhibitors, especially those with high inhibitor titers. Thus, alternative approaches for inducing tolerance in these patients or preventing inhibitor responses in the first place are an unmet need and of clinical importance.6
Our laboratory has previously demonstrated that expanded polyclonal human regulatory T cells (Tregs) can be rendered specific by transducing them to express T-cell receptor variable genes derived from a T-cell clone from an HA patient7 or chimeric antigen receptor single chain Fv (CAR scFv) obtained from a phage display library.8,9 Both of these engineered specific Tregs inhibit FVIII-specific T-cell proliferation and antibody formation in vitro and in vivo.8 As a further advance to render T cells specific and prevent inhibitor formation, we decided to target FVIII-binding B cells, the precursor for inhibitory antibodies. We theorized that expression of the FVIII antigen domains on the surface of a CD8 T cell would result in binding by FVIII-specific B cells. Thus, these antigen-expressing CD8 T cells can directly target and eliminate the FVIII-specific cells and would avoid nonspecific cytotoxicity.9 Herein, we present data supporting this hypothesis. We have now generated an engineered chimeric receptor, which we call a B-cell antibody-targeting receptor (BAR), analogous to a CAR. We demonstrate that FVIII-expressing BAR CD8 T cells are potent killers of FVIII-specific hybridomas and that these engineered T cells are also capable of eliminating FVIII-specific B cells in a dose-dependent manner in vitro and preventing anti-FVIII antibody formation in hemophilic mice in vivo.
Materials and methods
Mice
FVIII exon 16 knockout mice (E16) on a C57BL/6 background were used as the model for HA; these were derived from a colony belonging to Leon Hoyer, MD, at the American Red Cross.10,11 NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). They were maintained in sterile cages in a barrier facility. All mice used were 8 to 12 weeks old at the beginning of our experiments. Animal procedures were approved by the institutional Animal Care and Use Committee at the Uniformed Services University of the Health Sciences.
Cell lines
BO2C11, a human B-cell hybridoma specific for FVIII-C2 (IgG4 kappa) was derived from Epstein-Barr virus–transformed cells. This line was obtained from Jean-Marie Saint-Remy and Marc Jacquemin (Leuven, Belgium). The mouse B-cell hybridomas 3G6 (anti-FVIII-C2; IgG2a kappa) and 413 (anti-FVIII-A2; IgG1 kappa) were from Pete Lollar, MD (Emory University, Atlanta, GA), and 2JLO (anti-OVA; IgG1) was from John Kappler, MD (National Jewish Health, Denver, CO). All B-cell hybridoma lines were cultured with RPMI1640 plus 10% fetal bovine serum (FBS) at 37°C with 7% CO2.
BAR construction and retroviral production
BAR constructs were designed by using a complementary DNA (cDNA) sequence derived from GenBank for human FVIII-C2 and -A2 domains and for chicken ovalbumin (OVA). The transmembrane and intracellular region of human CD28 and the intracellular domain of human CD3ζ chain were derived from the nextprot database (nextprot.org) as previously described.8 To generate the FVIII C2-BAR construct, the C2-CD28-CD3ζ cDNA was synthesized by GenScript USA (Piscataway, NJ). The FVIII A2-BAR and the control OVA-BAR were generated by replacing the C2 sequence in the C2-BAR construct with an A2 or an OVA cDNA sequence, respectively. The constructs were cloned into a retroviral vector, pRetroX-IRES-ZsGreen (Clontech, Mountain View, CA); the retroviral particles were produced using a Phoenix-Ampho or Eco packaging system (Clontech) to transduce human and mouse T cells, respectively. The culture supernatant containing the retroviral particles was concentrated with Retro-X Concentrator (Clontech), aliquoted, and stored at −80°C until use.
Human CD8 T-cell isolation and activation
Human blood samples from healthy, anonymous donors ranging in age from 20 to 60 years were obtained with written consent from the Department of Transfusion Medicine, Clinical Center, National Institutes of Health (NIH) and were analyzed with the approval of the NIH Ethical Review Committee. Peripheral blood mononuclear cells (PBMCs) were isolated from mononuclear cell buffy coats using Ficoll-Hypaque (GE Healthcare Biosciences, Pittsburgh, PA) by density gradient centrifugation. CD8 T cells were enriched from PBMCs by positive selection using human CD8 MicroBeads (Miltenyi Biotec, Auburn, CA) according to the manufacturer’s instructions.
Isolated cells were activated for 24 hours with anti-CD3 (Clone OKT3; 5 μg/mL) and anti-CD28 (Clone CD28.2; 2.5 μg/mL) soluble antibody in complete media (RPMI 1640 with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mM nonessential amino acids, 50 μM β-mercaptoethanol, 1 mM sodium pyruvate, and N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [HEPES]) supplemented with human recombinant interleukin-2 (IL-2; 200 U/mL) (Teceleukin, NIH) at 37°C with 7% CO2.
Murine CD8 T-cell isolation and activation
Hemophilic (E16) splenocytes were isolated, and CD8+ T cells were enriched by negative selection using mouse CD8+ T-cell–negative selection kit (STEMCELL Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions. Mouse CD8 T cells were activated for 24 hours with anti-CD3 (Clone 145-2C11; 1 μg/mL) and anti-CD28 (Clone 37.51; 0.5 μg/mL) soluble antibody in complete media supplemented with recombinant IL-2 (30 U/mL) at 37°C with 7% CO2.
Retroviral transduction and expansion
Activated mouse or human CD8 T cells were transduced by spinfection, as described previously.7 BAR-transduced GFP+CD8+ T cells were sorted using FACSAria Fusion (BD Biosciences, Franklin Lakes, NJ). Sorted human T cells were expanded with anti-CD3 (Clone OKT3; 0.5 μg/mL) in the presence of 60 Gy–irradiated PBMCs and IL-2.12 Sorted mouse T cells were rested for 24 hours without IL-2 and then injected into animals.
Flow cytometry analysis and image acquisition
Isolated cells were washed (3% FBS in phenol red–free RPMI 1640) and stained with fluorochrome-conjugated antibodies for 20 minutes at 4°C. Antibodies used for flow cytometry analysis and sorting are listed in supplemental Table 1. For intracellular staining, cells were fixed with 4% paraformaldehyde for 7 minutes and permeabilized with 1× permeabilization buffer (eBioscience, Inc. Waltham, MA) overnight followed by staining with respective antibodies. Stained cells were acquired on an LSRII instrument (BD Biosciences) using BD FACSDiva software and were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR). For all analyses, dead cells were excluded by gating singlets followed by exclusion of cells that were positively stained with cell viability dye. For imaging, each sample was acquired by using the AMNIS brand ImageStream Mark II imaging flow cytometer (Millipore, Billerica, MA). Image analysis was performed using IDEAS image analysis software.
JAM cell death assay
BO2C11 target cells were labeled with 1 μCi/mL 3H thymidine (3H-TdR) for 48 hours. Cells were washed and cocultured with C2-BAR– or OVA-BAR–expressing CD8 T cells or untransduced T cells at various effector:target ratios. After 5 hours, the cells were harvested onto the fiberglass filters and were counted on a MicroBeta counter (Perkin Elmer, Boston, MA). Because only the target-cell DNA is labeled (and not the DNA of any of the death-inducing effector cells that might have been added), assessment of the amount of radiolabel taken up gives an accurate measure of the targets that survived. Percent-specific lysis was calculated as described previously.13
In vivo B-cell hybridoma killing in NSG mice
Male NSG mice were injected IV with 2 × 106 BO2C11 B-cell hybridoma cells on day 1. BO2C11 cells recognize an epitope in the FVIII-C2 domain.14 On day 5, 1 × 106 human CD8 T cells expressing C2-BAR or OVA-BAR were adoptively transferred IV. The animal’s body weight measurements were recorded every 2 days. A 20% gain or loss of body weight as the result of tumor growth was used as an end point for morbidity. In addition, an anti-OVA hybridoma (JLO) was transduced with a luciferase construct to facilitate tracking. The effect of OVA-BAR CD8s on these cells is shown in supplemental Figure 2.
In vitro naïve B-cell stimulation assay
Total splenic B cells were isolated using the mouse B-cell isolation kit (eBioscience, Inc. San Diego, CA). Naïve B cells were cultured with or without lipopolysaccharide (LPS; 1 μg/mL) for 48 hours for polyclonal activation.15,16 For BAR-mediated FVIII-specific naïve B-cell killing experiments, A2/C2-BAR or OVA-BAR CD8 T cells were mixed at a 5:1 ratio with isolated B cells, cultured in the presence of LPS for 48 hours, and assayed for antibody formation by enzyme-linked immunospot (ELISPOT) as described.8 Cells were washed, plated on FVIII (2 μg/mL) or OVA (10 µg/mL), or 2,4,6-trinitrophenyl (TNP)-bovine serum albumin (5 µg/mL)–coated ELISPOT plates (EMD Millipore, Billerica, MA) in duplicate and incubated overnight. The captured anti-FVIII antibodies were detected by alkaline phosphatase–conjugated goat anti-mouse IgM (H+L; Southern Biotech, Birmingham, AL) and developed with 1-Step NBT/BCIP substrate solution (Thermo Fisher, Waltham, MA). For IgG detection, horseradish peroxidase–conjugated rabbit anti-mouse IgG (H+L; Invitrogen, Carlsbad, CA) was used and developed with AEC substrate (BD Biosciences). Plates were imaged using the ImmunoSpot Reader (CTL, Cleveland, OH). ImmunoSpot Software using the SmartCount and Autogate functions automatically calculated the number of spots per well.
In vivo prevention of anti-FVIII antibody response in FVIII knockout mice
Female E16 mice (n = 5 per group, age matched) were adoptively transferred with 1 × 106 fluorescence-activated cell sorted syngeneic CD8 T cells expressing either A2/C2- or OVA-BAR. Within 24 hours, the mice were subcutaneously immunized (base of tail) with 2 µg FVIII in incomplete Freund’s adjuvant. To test the persistence of FVIII-specific B-cell clearance, mice were boosted IV with 1 µg FVIII at week 10. The mice were bled once per week beginning at 2 weeks after the initial immunization, and the anti-FVIII antibody levels were followed using an FVIII enzyme-linked immunosorbent assay as described previously.7 To test the secondary response in vitro, at 2 weeks after the last FVIII administration, CD138-depleted splenocytes from individual immunized mice were isolated and cultured for 6 days in the presence of FVIII (1 μg/mL). FVIII-specific antibody-secreting cells were enumerated by ELISPOT assay. Cells were washed, plated on FVIII (2 μg/mL)-coated ELISPOT plates (EMD Millipore, Billerica, MA) in duplicate, and incubated overnight. The captured anti-FVIII antibodies were detected by horseradish peroxidase–conjugated rabbit anti-mouse IgG (H+L, Invitrogen) and developed with AEC substrate (BD Biosciences).
Statistical analysis
Statistical analyses were performed using Prism software 174 (v6.0, GraphPad Software, La Jolla, CA). Either Student t test or one-way analysis of variance (ANOVA) was chosen to evaluate the significance of the in vitro ELISPOT counts by FVIII BAR CD8 T cells. P < .05 was considered statistically significant. Log-rank (Mantel-Cox) test was chosen to compare survival curves in experiments with NSG mice. Using SPSS, dose-dependent responses were analyzed using two-way ANOVA followed by post hoc polynomial contrasts to test for trend.
Results
Generation and function of BAR CD8 T cells
Based on the success of CD19 CAR, we hypothesized that we could target FVIII-specific B cells by expressing the major antigenic epitopes that are recognized by these B cells. Therefore, we developed BAR constructs to express either immunodominant FVIII A2 or C2 domains (or full-length OVA as control) to replace the scFv on the extracellular surface of the murine and human CD8 T cells (Figure 1A-B). Retroviral transduction of bicistronic vectors demonstrated that up to 45% of CD8 T cells were green fluorescent protein–positive; surface staining with monoclonal anti-FVIII A2 (413) and C2 (3G6) and anti-OVA antibodies confirmed the expression and folding of the BAR on the surface of the transduced murine CD8 T cells (Figure 1C-D). Our laboratory previously demonstrated that transduced CD4 T effector cells and Tregs proliferate upon stimulation through the BAR.17 Similarly, stimulation through the BAR with platebound anti-A2, anti-C2, or anti-OVA led to the upregulation of cytotoxic markers such as granzyme B, perforin, and interferon-γ in the transduced CD8 T cells (Figure 1D-E).
Figure 1.

Design and properties of BARs. (A) Schematic representation of the BAR constructs containing FVIII-A2 or FVIII-C2 or OVA. (B) Representation of BAR-expressing T cells engaging with the antigen-specific B cell through their surface BCR. (C) Green fluorescent protein (GFP) expression levels in transduced mouse CD8 T cells at day 7. Inlet picture shows the fluorescent imaging of cells in culture 72 hours after transduction. (D) Single-cell imaging analysis and expression of CD8 on the surface and intracellular localization of GFP, interferon-γ (IFN-γ), perforin, and granzyme B (Gzym B) 6 hours after stimulation with anti-A2 or anti-C2 antibodies in the presence of protein transport inhibitor. Far right panel indicates the overlay of GFP, granzyme B, and perforin channels. (E) Flow cytometry analysis of C2-BAR and OVA-BAR CD8 T cells after stimulation with respective antibodies and increase in cytolytic granule proteins, such as granzyme B and perforin. Hu, human; IRES, internal ribosome entry site; PE, phycoerythrin.
Taken together, the results indicate that FVIII-specific BAR T cells can signal through the BAR, indicating that BAR-expressing CD8 T cells have the potential to target and kill the antigen-specific B cells. This hypothesis was formally tested using FVIII-specific hybridomas as targets. Killing of target cells was measured by FVIII-specific ELISPOT assay of antibody secretion, as well as by the JAM assay (reduction of thymidine-labeled cells) by A2- or C2-BAR in a dose-dependent manner (Figure 2A-D). As a further test of the cytotoxic potential of the BAR CD8 T cells to eliminate specific B cells, we injected NSG mice with BO2C11 hybridoma cells (2 × 106) and measured anti-FVIII antibody secretion in serum to confirm growth of these transformed cells (Figure 2E). On days 5 to 6, 1 × 106 BAR CD8 T cells were injected, and the mice were weighed every 2 days. As shown in Figure 2F, 4 of 5 mice injected with C2-BAR CD8 T cells survived past day 60, whereas recipients given OVA-BAR CD8 T cells (or phosphate-buffered saline) developed lymphomas and did not survive.
Figure 2.

Specific cytotoxicity of BAR CD8s in vitro and in vivo. (A-B) Quantification of FVIII-specific spots formed by 3G6 and 413 hybridomas after coculture with BAR effector CD8 T cells (P = .0157). (C) Dose-dependent killing of target cells (BO2C11) by C2-BAR–expressing human CD8 T cells. (D) Loss of FVIII-specific IgG antibody secretion by BO2C11 cells at increasing effector:target (E:T) ratio of C2-BAR human CD8 T cells compared with controls (P < .05). (E) Schematic diagram of adoptive cell transfer into NSG mice and sample collection. (F) Kaplan-Meier survival analysis of NSG mice injected with BO2C11 hybridoma cells (n = 5). Log-rank (Mantel-Cox) test P = .0104. CPM, counts per minute; hCD8+, human CD8+; SD, standard deviation; SEM, standard error of the mean.
A2/C2-BAR CD8 T cells can kill FVIII-specific B cells from unimmunized mice
To demonstrate the ability of the BAR CD8 T cells to kill antigen-specific B cells, we used B-cell activation with LPS, which leads to polyclonal IgM secretion,15 and measured responses to FVIII, OVA, and the TNP hapten after 48 to 72 hours. Splenic B cells from the naïve FVIII−/− E16 mice were stimulated with the LPS (1 µg/mL) for 48 hours in the presence of A2- and C2-BAR CD8 cells (1:1) (referred as A2/C2), or OVA-BAR mouse CD8 (mCD8) T cells. Compared with the OVA-BAR–treated group, the addition of A2/C2-BAR CD8 T cells led to depletion of anti-FVIII–specific antibody secretion by LPS-stimulated naïve B cells (Figure 3A). Upon coculture with A2-BAR alone or C2-BAR alone or A2/C2-BAR CD8 T cells, we observed a dose-dependent decrease in FVIII-specific antibody-secreting cells (ASCs; Figure 3B). A2-BAR or C2-BAR CD8 T cells alone led to a partial reduction in ASCs, presumably reflecting activation of B cells of other FVIII specificities; in contrast, the combination of both A2- and C2-BAR CD8s led to almost complete reduction in ASCs. These results indicate that A2/C2-BAR CD8 T cells have the potential to cover the major immunogenic epitopes and prevent anti-FVIII antibody formation. The specificity of the A2/C2-BAR mCD8 T cells to kill only specific target cells was confirmed by the fact that no reduction of anti-TNP or anti-OVA ASCs was observed in the presence of A2/C2-BAR CD8 cells (Figure 3C). However, OVA-specific ASCs were reduced when OVA-BAR CD8 T cells were added to LPS-activated B cells, thus confirming the specificity of the BAR-expressing CD8 T cells (Figure 3C).
Figure 3.

Effect of A2/C2-BAR CD8 T cells on naïve B cells. (A) Significant loss of IgM+ FVIII-specific spot formation with LPS stimulation of naïve B cells in A2/C2-BAR–treated group compared with control OVA-BAR or no T-cell groups (P = .0209). (B) Dose-dependent effect of A2-BAR, C2-BAR, or A2/C2-BAR CD8 T cells on the LPS-stimulated naïve B cells to form IgM+ FVIII-specific spots compared with the OVA-BAR group (P = .001). (C) TNP-specific IgM (top) or OVA-specific IgM (bottom) secretion by the LPS-stimulated naïve B cells in the presence of either OVA- or A2/C2-BAR–expressing T cells.
A2/C2-BAR mCD8 T cells reduce antibody formation in FVIII-immunized hemophilic mice
We hypothesized that injecting A2/C2-BAR mCD8 T cells could prevent anti-FVIII antibody formation in vivo. To test this, we injected E16 FVIII−/− mice with 1 × 106 A2/C2-BAR or OVA-BAR mCD8 T cells on day 0, and then immunized the mice with FVIII (1 µg in incomplete Freund’s adjuvant subcutaneously) followed by measurement of anti-FVIII antibody once per week. FVIII-specific antibody titers rose after immunization in the OVA-BAR–injected group, but recipients of A2/C2-BAR CD8 T cells had low to negligible titers (Figure 4A-B). Thus, A2/C2-BAR CD8 T cells can prevent the formation of anti-FVIII from both naïve cells stimulated with LPS in vitro and prophylactically blocked antibody formation in vivo.
Figure 4.

Effect of BAR CD8 T cells on the antibody response to FVIII in vivo. (A) Schematic diagram of immunization and sample collection with n = 5 per group of A2/C2-BAR– or OVA-BAR–injected animals. (B) Anti-FVIII concentrations in serum at different time points after injecting BAR T cells and FVIII immunization. P < .05 at weeks 4, 5, 9, and 10; P < .01 at week 12. (C) FVIII-specific IgG spot formation by naïve B cells isolated from mouse injected with A2/C2-BAR or phosphate-buffered saline upon coculture with FVIII (1 μg/mL) for 5 days (P = .0087). Bars indicate standard error. IFA, incomplete Freund’s adjuvant; rFVIII, recombinant FVIII; s.c., subcutaneously.
To investigate the long-term protective effect of A2/C2-BAR CD8 T cells, we boosted the animals at week 10 and measured the anti-FVIII titers. The A2/C2-BAR–treated group maintained low titers, whereas OVA-BAR–treated mice had significantly increased high anti-FVIII titers (Figure 4C). At the end of the study, spleen cells from these mice were cultured in vitro with FVIII to examine the effect on B-cell memory. No anti-FVIII IgG ASCs were observed when the splenocytes from mice injected with A2/C2-BARs were tested compared with the controls (Figure 4D). These results confirm that A2/C2-BAR–transduced CD8 T cells are capable of preventing anti-FVIII antibody formation for the long term in vivo, probably by eliminating the precursors of FVIII-specific memory B cells.
Discussion
Targeting the inhibitor-producing FVIII-specific B cells without compromising protective humoral immunity is ideal for treating patients with HA in whom an unwanted B-cell response to therapeutic FVIII is a major challenge. Our BAR CD8 approach provides a potential solution to targeting these FVIII-specific B cells and reducing unwanted B-cell responses.
The inhibitor response to FVIII is T-cell dependent in both humans and mice.18,19 The major epitopes recognized by these inhibitors are the A2 and C2 domains of FVIII.20 By generating A2/C2-BAR CD8 T cells, we were able to eliminate the anti-FVIII antibody formation because the major of inhibitory antibodies are against these 2 domains. In this article, we also demonstrate the specificity and precision of these BAR-expressing T cells because only specific naïve or memory B cells that express surface Ig receptor for FVIII are targeted and killed. That is, there is no effect on irrelevant (eg, TNP-specific) B cells. This is a result of the ability of the BAR-expressing CD8 T cells to kill B lymphocytes through their recognition by the B-cell receptor (BCR). Whether this approach will work on long-lived plasma cells is unknown because these cells express little or no surface Ig. It is likely that additional treatments targeting plasma cells (such as using bortezomib) may be necessary.
A similar strategy can also be applied to the autoimmune diseases in which antibody formation is problematic. For example, Ellebrecht et al21 have demonstrated that engineered human cytotoxic T cells that express the major pemphigus antigen desmoglein 3 (Dsg3) can specifically target Dsg3-specific hybridoma cells, even in the presence of circulating antibodies in NSG mice. We have confirmed this with the killing of FVIII-specific hybridomas. Because we used an immunocompetent HA mouse model, our data have extended this approach and confirmed that the infusion of FVIII BAR CD8 T cells can prevent the formation of anti-FVIII inhibitors. We have also shown that BAR CD8 T cells can be effective in the presence of up to 10 Bethesda units monoclonal antibodies to FVIII, at least in vitro. Whether they can function and survive in vivo in mice that produce high-titered inhibitors remains to be determined.
Advanced design strategies such as the Tet-On system inducible CARs, GoCAR-Ts, and O2-sensing CARs are easily applicable to the BAR design.22-24 To ensure safety and reduce the risk of insertional mutagenesis, suicide genes or small drug (rimiducid)–mediated deletion of the T cells can also be applied to our BAR concept.25 Gene editing tools such as CRISPR/CAS9 or TALENs can be used to eliminate major histocompatibility complex and endogenous receptors and thereby create universal donors and avoid both rejection and graft-versus-host reactions. Future studies will involve testing the ability of BAR CD8 T cells to kill FVIII-specific B cells after the B-cell memory response has been established. As discussed, the BAR T-cell approach was designed to target only antigen-specific B cells in which a BCR is present. Thus, the BAR approach will not target terminally differentiated plasma cells, which no longer express the surface BCR. Other complementary therapies will need to be used to target these long-lived plasma cells.
In summary, we report a novel engineered BAR T-cell receptor approach for specifically targeting antigen-specific B cells in vivo. The BAR CD8 T-cell therapy can be applicable to other antidrug antibody formation as well, in which antibody formation to therapeutic drugs is a major problem.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Kathleen Pratt, Ai-Hong Zhang, Maha Abdeladhim, Yongchan Kim, and Patrick Adair, for reviewing the manuscript and Birgit M. Reipert (Baxalta Innovation GmbH, Vienna, Austria) for providing recombinant FVIII.
Supported in part by a Pfizer ASPIRE award and by National Heart, Lung, and Blood Institute, National Institutes of Health grants R21 HL127495 and R01 HL061883 (D.W.S.).
Authorship
Contribution: K.P. and D.W.S. designed the experiments; K.P. performed the research; and K.P. and D.W.S. wrote and edited the manuscript.
Conflict-of-interest disclosure: D.W.S. and K.P. have a provisional patent filed via The Henry Jackson Foundation on “Chimeric Antigen Receptors to Arm Cytotoxic T Cells to Destroy Antigen-Specific B Cells” with laboratory members Aihong Zhang and Yongchan Kim.
Correspondence: David W. Scott, Department of Medicine, Uniformed Services University of Health Science, A3069, 4301 Jones Bridge Rd, Bethesda, MD 20814; e-mail: david.scott@usuhs.edu.
References
- 1.Scott DW, Pratt KP, Miao CH. Progress toward inducing immunologic tolerance to factor VIII. Blood. 2013;121(22):4449-4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scott DW. Why do immunology research in hemophilia? Cell Immunol. 2016;301:1. [DOI] [PubMed] [Google Scholar]
- 3.Gouw SC, van den Berg HM, Oldenburg J, et al. . F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis. Blood. 2012;119(12):2922-2934. [DOI] [PubMed] [Google Scholar]
- 4.Pratt KP. Inhibitory antibodies in hemophilia A. Curr Opin Hematol. 2012;19(5):399-405. [DOI] [PubMed] [Google Scholar]
- 5.Mancuso ME, Cannavò A. Immune tolerance induction in hemophilia. Clin Invest (Lond). 2015;5(3):321-335. [Google Scholar]
- 6.Sherman A, Biswas M, Herzog RW. Innovative approaches for immune tolerance to factor VIII in the treatment of hemophilia A. Front Immunol. 2017;8:1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim YC, Zhang AH, Su Y, et al. . Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood. 2015;125(7):1107-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon J, Schmidt A, Zhang AH, Königs C, Kim YC, Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood. 2017;129(2):238-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parvathaneni K, Abdeladhim M, Pratt KP, Scott DW. Hemophilia A inhibitor treatment: the promise of engineered T-cell therapy. Transl Res. 2017;187:44-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qian J, Collins M, Sharpe AH, Hoyer LW. Prevention and treatment of factor VIII inhibitors in murine hemophilia A. Blood. 2000;95(4):1324-1329. [PubMed] [Google Scholar]
- 11.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH Jr. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10(1):119-121. [DOI] [PubMed] [Google Scholar]
- 12.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26(4):332-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matzinger P. The JAM test. A simple assay for DNA fragmentation and cell death. J Immunol Methods. 1991;145(1-2):185-192. [DOI] [PubMed] [Google Scholar]
- 14.Spiegel PC Jr, Jacquemin M, Saint-Remy JM, Stoddard BL, Pratt KP. Structure of a factor VIII C2 domain-immunoglobulin G4kappa Fab complex: identification of an inhibitory antibody epitope on the surface of factor VIII. Blood. 2001;98(1):13-19. [DOI] [PubMed] [Google Scholar]
- 15.Andersson J, Sjöberg O, Möller G. Induction of immunoglobulin and antibody synthesis in vitro by lipopolysaccharides. Eur J Immunol. 1972;2(4):349-353. [DOI] [PubMed] [Google Scholar]
- 16.Gronowicz ES, Doss C, Assisi F, Vitetta ES, Coffman RL, Strober S. Surface Ig isotypes on cells responding to lipopolysaccharide by IgM and IgG secretion. J Immunol. 1979;123(5):2049-2056. [PubMed] [Google Scholar]
- 17.Zhang AH, Yoon J, Kim YC, Scott DW. Targeting antigen-specific B cells using antigen-expressing transduced regulatory T cells. J Immunol. 2018;201(5):1434-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian J, Borovok M, Bi L, Kazazian HH Jr, Hoyer LW. Inhibitor antibody development and T cell response to human factor VIII in murine hemophilia A. Thromb Haemost. 1999;81(2):240-244. [PubMed] [Google Scholar]
- 19.Bray GL, Kroner BL, Arkin S, et al. . Loss of high-responder inhibitors in patients with severe hemophilia A and human immunodeficiency virus type 1 infection: a report from the Multi-Center Hemophilia Cohort Study. Am J Hematol. 1993;42(4):375-379. [DOI] [PubMed] [Google Scholar]
- 20.Green D. Factor VIII inhibitors: a 50-year perspective. Haemophilia. 2011;17(6):831-838. [DOI] [PubMed] [Google Scholar]
- 21.Ellebrecht CT, Bhoj VG, Nace A, et al. . Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353(6295):179-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350(6258):aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Juillerat A, Marechal A, Filhol JM, et al. . An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. 2017;7:39833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakemura R, Terakura S, Watanabe K, et al. . A Tet-On inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol Res. 2016;4(8):658-668. [DOI] [PubMed] [Google Scholar]
- 25.Duong MT, Collinson-Pautz MR, Foster AE, Bayle JH, Spencer DM. Uni-CIDeCAR-T cells: MyD88/CD40-enhanced, Ab-directed CAR incorporating the CaspaCIDe safety switch [abstract]. Cancer Immunol Res. 2016;4. Abstract A057. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.