

Abstract
Purpose of Review:
To succinctly summarize recent findings concerning dormancy regulating interactions between bone marrow stromal cells and disseminated tumor cells.
Recent Findings:
Recent studies have highlighted roles of the bone marrow microenviroment, including osteoblasts, mesenchymal stem cells (MSCs), and endothelial cells, in inducing or maintaining cancer cell dormancy. Key pathways of interest include: osteoblast induced transforming growth factor (TGF)-β2 signaling, transfer of MSC-derived exosomes containing dormancy inducing microRNA, cancer cell cannibalism of MSCs, and endothelial cell secretion of thrombospondin 1 (TSP1).
Summary:
The bone marrow is a common site of metastatic disease recurrence following a period of cancer cell dormancy. Understanding why disseminated tumor cells enter into dormancy and later resume cell proliferation and growth is vital to developing effective therapeutics against these cells. The bone marrow stroma and the various pathways through which it participates in crosstalk with cancer cells are essential to furthering understanding of how dormancy is regulated.
Keywords: cancer cell dormancy, bone marrow microenviroment, bone marrow stromal cells, osteoblasts, mesenchymal stem cells, endothelial cells
Introduction:
Although the numbers of long-term cancer survivors are rapidly increasing, many of them are still at risk of recurrence even after decades of disease-free status [1,2]. It has been suggested that very small numbers of cancer cells can escape from cytotoxic treatments and survive long periods of time either in primary or secondary sites [1,2]. These cells are not actively proliferating and are inherently resistent to cytotoxic treatments, such as chemotherapies and radiation, that target dividing cells [3]. To make matters worse, these occult cancer cells often remain dormant for a long period of time but are clinically undetectable. Cancer cell dormancy is currently a key obstacle in effectively treating cancer patients, due to the potential of treatment-resistant dormant cells to recur as lethal metastases [1,3].
Cancer dormancy is thought to be a reversible low metabolic state, since it is primarily characterized by: 1) prolonged cancer cell survival in cell cycle arrest (spanning from weeks to decades), and 2) their potential ability to leave this state and start proliferating again, ultimately leading to disease recurrence [4–7,2,1]. It has been suggested that the following three mechanisms are involved in the development of cancer dormancy: cellular dormancy, angiogenic dormancy, and immune-mediated dormancy [8,9]. Cellular dormancy is when disseminated tumor cells (DTCs) enter a state of quiescence or growth arrest in the G0 cell cycle phase due to a combination of external and/or internal cellular factors, such as upregulation of p27, a cell cycle inhibitor, to induce cycle arrest [8,6,3]. Angiogenic dormancy occurs when tumors lack sufficient vascularization, and consequently sufficient oxygen and nutrients, for further growth [8,9]. Immune-mediated dormancy is reliant upon cytotoxic activities of T cells and the innate immune system inhibiting tumor growth [8,10]. As noted above, the tumor microenvironment, such as endothelial cells and immune cells, are involved in the establishment of DTC dormancy.
The bone contains a rich tumor microenvironment and is a common site of cancer cell dissemination [1,6,11,12]. Indeed, DTCs within the bone marrow are often observed when cancer patients are initially diagnosed [2,1,8,3]. Most of these DTCs naturally die off or are eradicated by cytotoxic treatments. However, some of them can survive within the harsh bone marrow microenvironment, evade initial resection or treatment, lay dormant for years, and eventually regrow and develop into a full-blown recurrence [6,4]. Unfortunately, these recurrent tumors are very difficult to treat as they often acquire treatment resistance [6,11,12]. The ability of DTCs to enter and exit dormancy within the bone marrow is thought to be due in part to interactions with specific bone marrow microenvironment stromal cells: osteoblasts, osteoclasts, mesenchymal stem cells (MSCs), endothelial cells, etc. However, how these interactions collectively regulate cancer dormancy is not fully understood to date.
Effectively modeling dormant cancer cells in their microenvironment can be challenging. Cocultures are useful in studying the interactions between cancer and stromal cells; however, 2D cocultures of these cells do not adequately represent natural physiology [13,7]. This problem is being overcome through the use of 3D nonadherent cocultures [13]. The common strategies for measuring dormancy in cultured cells include proliferation assays, such as Ki-67 staining, fluorescent dye staining, and cell cycle analyses, where DNA and RNA markers are used to measure growth arrest at G0 phase [14,15]. With in vivo animal models, tumor growth over time and histology are typically used to determine if inoculated tagged cancer cells are dormant or actively growing [6,7]. A combination of in vitro and in vivo models are needed to adequately represent and study the dormancy phenotype [7].
Therefore, this review seeks to provide a concise overview of the current knowledge of the stroma-cancer cell interactions that regulate cancer dormancy within the bone marrow. This will be accomplished by highlighting recent studies addressing the roles of individual classes of bone marrow stroma in this crosstalk, and then addressing the clinical significance of this research.
Osteoblasts:
Although the primary function of osteoblasts is new bone formation, they also participate in maintaining hematopoiesis homeostasis [16–21]. In the marrow, osteoblasts serve as the specific microenvironment for hematopoietic stem cells (HSCs), or the HSC niche, to control homing to the marrow, quiescence, proliferation, and differentiation of HSCs [16–21]. The HSC niche is also beneficial in recruiting DTCs to the bone marrow and promoting early colonization of the bone [22–24]. Osteoblasts secrete CXC-chemokine ligand 12 (CXCL12) that binds to the C-X-C motif chemokine receptor 4 (CXCR4) G-protein coupled receptor expressed on both HSCs and DTCs, promoting competitive cellular adhesion of both these cells in the bone marrow [25–28]. Moreover, DTCs hijack the functions of HSC niche that regulate HSC quiescence to become dormant. For example, transforming growth factor (TGF)-β expression in the HSC niche is considered a regulator of HSC quiescence, and can potentially regulate DTC quiescence [29,12].
Live cell imaging of C4–2B4 prostate cancer cells treated with conditioned media from differentiated and undifferentiated osteoblasts showed that mature osteoblasts release factors that promote cancer cell quiescence [12]. Specifically, TGF-β2 and growth differentiation factor (GDF)10 were identified as dormancy-promoting osteoblast-secreted proteins through gene array analysis of the conditioned media, that can promote cell cycle arrest by binding to the TGF-βRIII receptor expressed on the prostate cancer cell lines [12]. This binding activates p38 mitogen activated protein kinase (MAPK) to phosphorylate retinoblastoma (RB) at the S249/pT252 site [12]. This phosphorylation then upregulates p27 expression, and consequently inhibits cancer cell-cycle progression and division, indicative of dormancy [6,11,12]. Moreover, TGF-β2 dormancy signaling was linked to osteoblast-derived ligand growth arrest specific 6 (GAS6) and the tyrosine kinase receptor Axl expression in a coculture model of prostate cancer and osteoblasts [30]. GAS6 is expressed more highly from osteoblasts cocultured with prostate cancer cells, and this high expression correlates with increased prostate cancer cell survival and decreased proliferation [31,22]. In the coculture condition with osteoblasts, prostate cancer requires the GAS6/Axl axis to respond TGF-β2-derived dormancy stimulation [30]. Finally, TGF-β is known to be immunosuppresive and aids in immune system evasion of dormant cancer cells through the inhibition of cytotoxic cells of the innate and adaptive immune systems (e.g., T cells and natural killer (NK) cells) [11].
Osteoblasts and osteocytes also secrete leukemia inhibitory factor (LIF), and the importance of DTC LIF receptor (LIFR) expression in maintaining a dormant phenotype in breast cancer cells (e.g., MCF7) disseminated to the bone marrow has recently been illustrated in a mouse model. In this study, inoculated MCF7 cells with LIFR knockdown displayed increased levels of osteoclastogenesis and cancer cell proliferation [32]. As we will discuss in the following section, osteoclasts are known to be a proliferation inducer for dormant DTCs. LIFR is also involved in promoting osteoclastogenesis by inhibiting parathyroid hormone-related protein (PTHrP) expression in cancer [11]. PTHrP promotes pre-osteoclast maturation by enhancing the expression of receptor activator of nuclear factor κB ligand (RANKL) in osteoblasts [11].
The addition of other factors may be necessary to overcome the dormancy phenotype induced by osteoblasts. When breast cancer cell dormancy model cell lines, MDA-MB-231BRMS1 and MCF-7, were co-cultured in a 3D model with an osteoblast matrix, they remained dormant until tumor necrosis factor (TNF)-α and interleukin (IL)-1β were added to the culture [33]. These cytokines, which play a role in bone remodeling signaling, increased cancer cell proliferation by increasing the expression of prostaglandin E2 (PGE2). Cancer cell proliferation mediated by TNF-α and IL-1β was inhibited by blocking the cycloxygenase (COX) and PGE2 receptors [33].
Osteoclasts:
Just as osteoblasts and osteoclasts have opposing roles in bone turnover (bone formation and resorption, respectively), they also have opposing roles in dormancy regulation. While osteoblasts are primarily associated with dormancy initiation and maintenance, osteoclasts have been reported to play a role in an exit of DTCs from a dormant state and the induction of osteolytic metastases [6,34,11]. Indeed, high levels of vascular cell adhesion molecule 1 (VCAM1) by DTCs have also been correlated with early disease recurrence, due to the ability of VCAM1 to bind the integrin α4β1 on osteoclasts and promote their osteolytic activity [4]. A study using intravital two-photon microscopy monitored the proliferation of 5TGM1 myeloma cells over time by labeling with 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindodicarbocyanine (DiD) within murine long bones [34]. DiD is a fluorescent dye that binds the phospholipid membrane of cancer cells [35]. In non-proliferating cells, DiD expression remains high; however, in proliferating cells this expression will decrease overtime as the cells divide [35]. In this study, the Kal-Col-GFP murine model in which osteoblast-like cells express emerald GFP, was used to identify the osteoblastic endosteal niche. Myeloma cells colocalized with the endosteal niche and consistently highly expressed DiD (non-proliferating cells) [34]. Conversely, treatment of mice with the soluble form of RANKL to promote osteoclast maturation significantly decreased the number of DiD-expressing cells in the bone, suggesting that osteoclast activity can promote cellular proliferation of dormant cells [34].
Mesenchymal Stem Cells:
MSCs are a type of pluripotent mesenchymal stromal cell, resident within the bone marrow [4,36]. It has been established that crosstalk occurs between bone marrow MSCs and DTCs [15,13,36,4]. DTCs secrete factors, such as CXCL12, that act as an attractant for MSCs to initiate their migration to the cancer site, and the MSCs in turn modulate cancer cell growth and proliferation [37,36,4]. The exact pathways through which this crosstalk occurs, and the specific impacts it may have on dormancy are still being elucidated. One pathway currently of primary interest in the field is the transfer of dormancy initiating microRNA (miRNA) from MSCs to cancer cells through exosomes or extracellular vesicles. For example, in subpopulation of breast cancer cells, exosomes isolated from breast cancer-primed MSCs promoted cell cycle quiescence, ascertained through cell cycle analyses using propidium iodide (a DNA marker), 7-aminoactinomycin D (a DNA marker), and pyronin Y (a RNA marker) labels to track cells in G0 and G1 phase [15]. miR-222 and −223 were upregulated in these exosomes and antagomiR-222/223 could reverse the quiescent phenotype of breast cancer [15]. Additionally, antagomiR-222/223 treatment in a murine model increased the efficacy of carboplatin treatment in tumor-bearing mice inoculated with MSCs compared to control anti-miRNA-treated mice. Likewise, the extracellular vesicles produced by human MSCs reduced proliferation and sphere formation,indicative of dormancy, of MDA-MB-231 breast cancer cells in vitro [38]. These findings indicated that miRNA from MSC exosomes or extracellular vesicles plays roles in initiating cancer cell dormancy and developing subsequent chemotherapy resistance [15].
Another interesting phenomena and mechanism for MSCs to initiate cancer cell dormancy is cell cannibalism. A 3D hanging drop model containing MD-MBA-231 cells and bone marrow derived MSCs, showed that stressed cancer cells cannibalize surrounding MSCs before entering a state of increased survival, where viability of these cells persisted in nutrient-poor media in vitro [13]. Moreover, these cancer cells showed decreased tumorigenicity in mice following inoculation [13].
Endothelial Cells:
As previously stated sufficient blood supply, typically provided through angiogenesis, is necessary to allow for cancer cell growth and progression [11]. Consequently, the absence or presence of angiogenesis can influence cancer cell entry into or exit from dormancy, respectively. The ability of angiogenesis to occur is in turn regulated by other components of the microenvironment. For example, a study of invasive ductal carcinoma (IDC) has shown that thrombospondin 1 (TSP1), a glycoprotein secreted from vascular endothelial cells that contains tryptophan-rich motifs, is associated with reduced IDC proliferation and cell cycle arrest at the G0/G1 phase in vitro [39,40]. More notably, this study identified a novel mechanism for IDC derived interferon-γ (IFN-γ) to promote IDC dormancy evasion by reducing tryptophan expression, and consequently decreasing TSP1 synthesis and total expression levels in a dose dependent manner [39]. However, endothelial secreted proteins are not always dormancy promoting. Secretion of TSP1 has also been reported to decrease when endothelial cells are activated, and the proliferation promoting factors TGF-β1 and periostin are instead secreted and encourage tumor growth [6,11]. As a result, DTC proliferation can be influenced based on their location near active or inactive endothelial cells.
Other Microenvironment Factors:
The bone marrow microenvironment contains an abundance of secreted factors that can inhibit DTC proliferation and promote entry into dormancy [6]. Although their primary sources have not been fully determined, all-trans retinoic acid, or atRA, and bone morphogenetic protein 7 (BMP-7) are both expressed in the bone marrow and are known to promote tumor dormancy [6,7]. atRA has been reported to increase DTC TGF-β2 expression and induce dormancy through the pathways previously discussed above (e.g., p38 and p27 activation) [6]. In addition to activating p38 MAPK signaling to inhibit proliferation, BMP-7, secreted from bone marrow stromal cells, has been shown to increase expression of N-myc downstream regulated gene 1 (NDRG1), a metastasis suppressor gene, and treatment of mice with BMP-7 suppresses cancer cell growth in the bone [6,41,42]. Notably, prostate cancer-derived secreted protein acidic and rich in cysteine (SPARC) induces bone marrow stromal expression of BMP-7 [42]. SPARC expression is upregulated in indolent cancer cells compared to aggressive cells isolated from murine bone following inoculation into the tibia [42]. Treatment of an in vivo mouse model with recombinant SPARC inhibited tumor growth in the bone [42].
Neurons can also modulate DTC dormancy. The role of sympathetic nervous system signaling through norepinephrine (NE) release in promoting dormancy exit has recently been appreciated [14]. NE promotes PC3 and DU145 human prostate cancer cell proliferation through binding to the β2-adrenergic receptors expressed on the prostate cancer cells, as well as reducing GAS6 expression by osteoblasts [14]. Prostate cancer cells treated in vitro with NE showed a dramatic increase in proliferation, measured with Ki-67 staining, that was reduced with the addition of a β-adrenergic receptor inhibitor [14]. In a novel ex vivo model, explanted femurs from mice with wild type or knockout GAS6 expression were injected with prostate cancer cells with and without NE [14]. After 48 hours, cell cycle phase of the cancer cells was determined with flow cytometry and showed significant increase in cells in the G2-M phase when treated with NE in GAS6+/+ femurs, indicative of cell cycle progression [14].
Clinical Relevance:
Cancer cell dormancy and disease recurrence in the bone marrow as metastases remain significant challenges in effectively treating cancer patients. Decades following initial treatment, dormant DTCs can resume proliferation into a potentially metastatic lesion [2]. Consequently, there is currently an abundant need for new therapeutics to treat these cancer patients with dormant DTCs [43]. Understanding the dormancy regulating interactions between DTCs and the surrounding bone marrow microenvironment will aid in devising more effective therapeutics that specifically target disseminated dormant cells, either to eradicate them through chemotherapeutics or immunotherapy, or to prevent them from exiting dormancy [6,5,43,44]. Manipulation of the microenvironment surrounding DTCs will alter their communication with these stromal cells and potentially make them more susceptible to treatment. One strategy for eliminating DTCs involves mobilizing them from the bone marrow, such as through the disruption of CXCL12/CXCR4 binding, to promote cell cycle progression and increase their susceptibility to established chemotherapeutics, although there is an inherent risk of inducing a potentially chemotherapeutic resistant recurrence with this strategy [3,45,44,46]. For example, preclinical and clinical studies are evaluating the efficacy of small molecules, peptides, and monoclonal antibodies in disrupting the CXCL12/CXCR4 axis in patients with acute myeloid leukemia in order to overcome resistance to chemotherapy [47]. To maintain DTCs in a dormant state would likely require administration of dormancy-permissive agents to patients (e.g., TSP1) to prevent the eventual switch to metastatic proliferation and disease recurrence [3,44]. However, the potential toxicities or off-target effects of chronic treatments to maintain dormancy will have to be carefully evaluated [44]. Alternatively, immunotherapy may be used in replacement of, or in conjunction with, chemotherapy to prevent tumor growth or eliminate DTCs [45]. This may be preferential to chemotherapy alone due to immunotherapy being dependent solely on immunogenecity of the DTCs and not on cell proliferation [45]. Further studies into the pathways and interactions regulating dormancy are needed to identify the most efficacious treatment strategy with the lowest patient risk. Additionally, this knowledge will: 1) provide greater insight into why some patients with DTCs never develop metastases, 2) provide strategies for identifying DTCs prone to resuming proliferation, and 3) potentially prevent overtreatment of patients who are at a lower risk of recurrence [4,6].
Conclusions:
There is much that still remains unknown about the mechanisms driving DTC dormancy and subsequent disease relapse in the bone marrow, and new discoveries in this field can be potentially groundbreaking. For example, it has only recently been shown through a genome wide short hairpin RNA screen that MSK1 gene expression in estrogen receptor-positive breast cancer inhibits metastatic progression, allowing for potentially novel identification of patients with dormant DTCs at a high or low risk or metastatic relapse based on this gene expression and treatment customization [48]. While the gene alteration or mutation itself can drive cancer cells into a dormancy state, current evidence also supports the role of the tumor microenvironment, including stromal cells, in regulating both of these processes through crosstalk with the DTCs [6] (Figure 1). As we have highlighted in this review, osteoblasts and MSCs have a role in initiating dormancy, while osteoclasts are primarily associated with metastasis progression [34]. Endothelial cells are able to promote both dormancy and renewed cancer cell growth depending on the activation status of the cells [6,11]. Further studies are needed in order to take full advantage of the crosstalk with resident bone marrow stromal cells to therapeutically target and/or monitor DTCs prior to metastatic recurrence, potentially preventing disease relapse. Relapse prevention will dramatically improve patient quality of life and overall survival.
Figure 1. The roles of the interactions between bone marrow resident disseminated tumor cells (DTCs) and bone marrow stroma in cancer cell dormancy and recurrence.
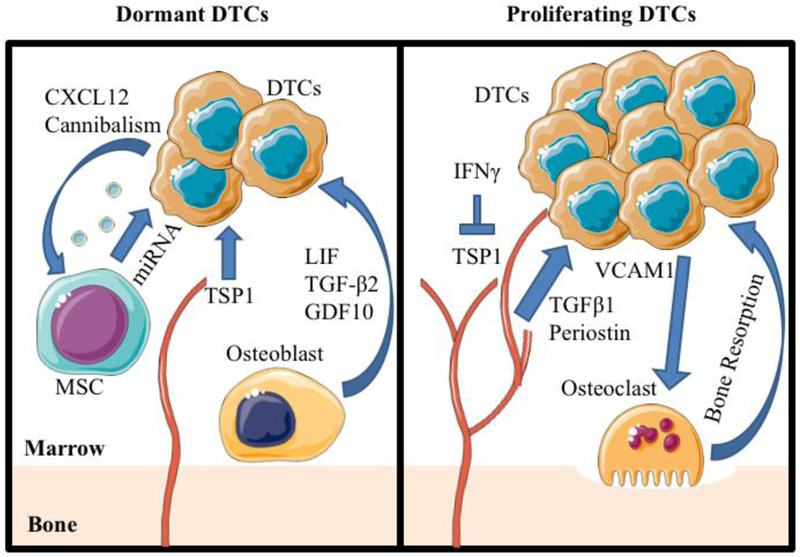
Left Panel: Mesenchymal stem cells (MSCs) are mobilized to DTCs through CXC-chemokine ligand 12 (CXCL12) and release exosomes containing dormancy promoting miRNA or are cannibalized by the DTCs. Inactive endothelial cells release thrombospondin 1 (TSP1) to promote dormancy and reduce proliferation of DTCs. Osteoblasts secrete transforming growth factor (TGF)-β2 and growth differentiation factor 10 (GDF10) to inhibit cellular proliferation, and express leukemia inhibitory factor (LIF) that is recognized by LIF receptor (LIFR) on dormant DTCs.
Right Panel: DTCs express interferon-γ (IFN- γ) that inhibits TSP1 expression in endothelial cells. Active endothelial cells secrete TGF-β1 and periostin to promote DTC proliferation. DTCs secrete vascular cell adhesion protein 1 (VCAM1) to promote maturation and activation of osteoclasts. Graphics adapted from Servier Medical Art (https://smart.servier.com/).
Acknowledgements:
This work is directly supported by Department of Defense (W81XWH-14-1-0403, Y. Shiozawa; W81XWH-17-1-0541, Y. Shiozawa), the Wake Forest Baptist Comprehensive Cancer Center Internal Pilot Funding (Y. Shiozawa), and the Wake Forest School of Medicine Internal Pilot Funding (Y. Shiozawa). Y Shiozawa is supported as the Translational Research Academy which is supported by the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, through Grant Award Number UL1TR001420. This work is also supported by the National Cancer Institute’s Cancer Center Support Grant award number P30CA012197 issued to the Wake Forest Baptist Comprehensive Cancer Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute.
Footnotes
Compliance with Ethical Guidelines
Conflict of Interest
Brooke Widner, Sun H. Park, Matthew R. Eber, and Yusuke Shiozawa declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References:
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Gomis RR, Gawrzak S (2016) Tumor cell dormancy. Mol Oncol 10.1016/j.molonc.2016.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao XL, Zhang M, Tang YL, Liang XH (2017) Cancer cell dormancy: mechanisms and implications of cancer recurrence and metastasis. Onco Targets Ther 10:5219–5228. 10.2147/OTT.S140854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Toom EE, Verdone JE, Pienta KJ (2016) Disseminated tumor cells and dormancy in prostate cancer metastasis. Curr Opin Biotechnol 40:9–15. 10.1016/j.copbio.2016.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graham N, Qian BZ (2018) Mesenchymal Stromal Cells: Emerging Roles in Bone Metastasis. Int J Mol Sci 19 (4). 10.3390/ijms19041121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marx V (2018) How to pull the blanket off dormant cancer cells. Nat Methods 15 (4):249–252. 10.1038/nmeth.4640 [DOI] [PubMed] [Google Scholar]
- 6.Linde N, Fluegen G, Aguirre-Ghiso JA (2016) The Relationship Between Dormant Cancer Cells and Their Microenvironment. Adv Cancer Res 132:45–71. 10.1016/bs.acr.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gay LJ, Malanchi I (2017) The sleeping ugly: Tumour microenvironment’s act to make or break the spell of dormancy. Biochim Biophys Acta 1868 (1):231–238. 10.1016/j.bbcan.2017.05.002 [DOI] [PubMed] [Google Scholar]
- 8.Sosa MS, Bragado P, Aguirre-Ghiso JA (2014) Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14 (9):611–622. 10.1038/nrc3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeh AC, Ramaswamy S (2015) Mechanisms of Cancer Cell Dormancy--Another Hallmark of Cancer? Cancer Res 75 (23):5014–5022. 10.1158/0008-5472.CAN-15-1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Senft D, Ronai ZA (2016) Immunogenic, cellular, and angiogenic drivers of tumor dormancy--a melanoma view. Pigment Cell Melanoma Res 29 (1):27–42. 10.1111/pcmr.12432 [DOI] [PubMed] [Google Scholar]
- 11.Dittmer J (2017) Mechanisms governing metastatic dormancy in breast cancer. Semin Cancer Biol 44:72–82. 10.1016/j.semcancer.2017.03.006 [DOI] [PubMed] [Google Scholar]
- 12.Yu-Lee LY, Yu G, Lee YC, Lin SC, Pan J, Pan T, Yu KJ, Liu B, Creighton CJ, Rodriguez-Canales J, Villalobos PA, Wistuba II, de Nadal E, Posas F, Gallick GE, Lin SH (2018) Osteoblast-secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the TGFbetaRIII-p38MAPK-pS249/T252RB pathway. Cancer Res 10.1158/0008-5472.CAN-17-1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartosh TJ, Ullah M, Zeitouni S, Beaver J, Prockop DJ (2016) Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). Proc Natl Acad Sci U S A 113 (42):E6447–E6456. 10.1073/pnas.1612290113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Decker AM, Jung Y, Cackowski FC, Yumoto K, Wang J, Taichman RS (2017) Sympathetic Signaling Reactivates Quiescent Disseminated Prostate Cancer Cells in the Bone Marrow. Mol Cancer Res 15 (12):1644–1655. 10.1158/1541-7786.MCR-17-0132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bliss SA, Sinha G, Sandiford OA, Williams LM, Engelberth DJ, Guiro K, Isenalumhe LL, Greco SJ, Ayer S, Bryan M, Kumar R, Ponzio NM, Rameshwar P (2016) Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res 76 (19):5832–5844. 10.1158/0008-5472.CAN-16-1092 [DOI] [PubMed] [Google Scholar]
- 16.Wu JY, Scadden DT, Kronenberg HM (2009) Role of the osteoblast lineage in the bone marrow hematopoietic niches. J Bone Miner Res 24 (5):759–764. 10.1359/jbmr.090225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neiva K, Sun YX, Taichman RS (2005) The role of osteoblasts in regulating hematopoietic stem cell activity and tumor metastasis. Braz J Med Biol Res 38 (10):1449–1454. doi:/S0100-879X2005001000001 [DOI] [PubMed] [Google Scholar]
- 18.Taichman RS, Emerson SG (1998) The role of osteoblasts in the hematopoietic microenvironment. Stem Cells 16 (1):7–15. 10.1002/stem.160007 [DOI] [PubMed] [Google Scholar]
- 19.Wilson A, Trumpp A (2006) Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol 6 (2):93–106. 10.1038/nri1779 [DOI] [PubMed] [Google Scholar]
- 20.Yin T, Li L (2006) The stem cell niches in bone. J Clin Invest 116 (5):1195–1201. 10.1172/JCI28568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrison SJ, Scadden DT (2014) The bone marrow niche for haematopoietic stem cells. Nature 505 (7483):327–334. 10.1038/nature12984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shiozawa Y, Eber MR, Berry JE, Taichman RS (2015) Bone marrow as a metastatic niche for disseminated tumor cells from solid tumors. Bonekey Rep 4:689 10.1038/bonekey.2015.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, Kim JK, Patel LR, Ying C, Ziegler AM, Pienta MJ, Song J, Wang J, Loberg RD, Krebsbach PH, Pienta KJ, Taichman RS (2011) Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest 121 (4):1298–1312. 10.1172/JCI43414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, Zhao H, Zhao Z, Du S, Tao J, Lee B, Westbrook TF, Wong ST, Jin X, Rosen JM, Osborne CK, Zhang XH (2015) The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 27 (2):193–210. 10.1016/j.ccell.2014.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK (2002) Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res 62 (6):1832–1837 [PubMed] [Google Scholar]
- 26.Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414 (6859):105–111. 10.1038/35102167 [DOI] [PubMed] [Google Scholar]
- 27.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410 (6824):50–56. 10.1038/35065016 [DOI] [PubMed] [Google Scholar]
- 28.Sugiyama T, Kohara H, Noda M, Nagasawa T (2006) Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25 (6):977–988. 10.1016/j.immuni.2006.10.016 [DOI] [PubMed] [Google Scholar]
- 29.Blank U, Karlsson S (2015) TGF-beta signaling in the control of hematopoietic stem cells. Blood 125 (23):3542–3550. 10.1182/blood-2014-12-618090 [DOI] [PubMed] [Google Scholar]
- 30.Yumoto K, Eber MR, Wang J, Cackowski FC, Decker AM, Lee E, Nobre AR, Aguirre-Ghiso JA, Jung Y, Taichman RS (2016) Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow. Sci Rep 6:36520 10.1038/srep36520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Decker AM, Jung Y, Cackowski F, Taichman RS (2016) The role of hematopoietic stem cell niche in prostate cancer bone metastasis. J Bone Oncol 5 (3):117–120. 10.1016/j.jbo.2016.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson RW, Finger EC, Olcina MM, Vilalta M, Aguilera T, Miao Y, Merkel AR, Johnson JR, Sterling JA, Wu JY, Giaccia AJ (2016) Erratum: Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat Cell Biol 18 (11):1260 10.1038/ncb3433 [DOI] [PubMed] [Google Scholar]
- 33.Sosnoski DM, Norgard RJ, Grove CD, Foster SJ, Mastro AM (2015) Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin Exp Metastasis 32 (4):335–344. 10.1007/s10585-015-9710-9 [DOI] [PubMed] [Google Scholar]
- 34.Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, Kaplan W, Paton-Hough J, Fellows C, Pettitt JA, Neil Dear T, Van Valckenborgh E, Baldock PA, Rogers MJ, Eaton CL, Vanderkerken K, Pettit AR, Quinn JM, Zannettino AC, Phan TG, Croucher PI (2015) Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun 6:8983 10.1038/ncomms9983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yumoto K, Berry JE, Taichman RS, Shiozawa Y (2014) A novel method for monitoring tumor proliferation in vivo using fluorescent dye DiD. Cytometry A 85 (6):548–555. 10.1002/cyto.a.22434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cammarota F, Laukkanen MO (2016) Mesenchymal Stem/Stromal Cells in Stromal Evolution and Cancer Progression. Stem Cells Int 2016:4824573 10.1155/2016/4824573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi Y, Du L, Lin L, Wang Y (2017) Tumour-associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat Rev Drug Discov 16 (1):35–52. 10.1038/nrd.2016.193 [DOI] [PubMed] [Google Scholar]
- 38.Vallabhaneni KC, Penfornis P, Xing F, Hassler Y, Adams KV, Mo YY, Watabe K, Pochampally R (2017) Stromal cell extracellular vesicular cargo mediated regulation of breast cancer cell metastasis via ubiquitin conjugating enzyme E2 N pathway. Oncotarget 8 (66):109861–109876. 10.18632/oncotarget.22371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopes-Bastos B, Jin L, Ruge F, Owen S, Sanders A, Cogle C, Chester J, Jiang WG, Cai J (2017) Association of breast carcinoma growth with a non-canonical axis of IFNgamma/IDO1/TSP1. Oncotarget 8 (49):85024–85039. 10.18632/oncotarget.18781Paper introduces a new mechanism for IFNγ to regulate dormancy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young GD, Murphy-Ullrich JE (2004) The tryptophan-rich motifs of the thrombospondin type 1 repeats bind VLAL motifs in the latent transforming growth factor-beta complex. J Biol Chem 279 (46):47633–47642. 10.1074/jbc.M404918200 [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, Pai SK, Liu W, Fukuda K, Chambers C, Wilber A, Watabe K (2011) Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med 208 (13):2641–2655. 10.1084/jem.20110840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma S, Xing F, Liu Y, Wu K, Said N, Pochampally R, Shiozawa Y, Lin HK, Balaji KC, Watabe K (2016) Secreted Protein Acidic and Rich in Cysteine (SPARC) Mediates Metastatic Dormancy of Prostate Cancer in Bone. J Biol Chem 291 (37):19351–19363. 10.1074/jbc.M116.737379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hurst RE, Bastian A, Bailey-Downs L, Ihnat MA (2016) Targeting dormant micrometastases: rationale, evidence to date and clinical implications. Ther Adv Med Oncol 8 (2):126–137. 10.1177/1758834015624277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghajar CM (2015) Metastasis prevention by targeting the dormant niche. Nat Rev Cancer 15 (4):238–247. 10.1038/nrc3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manjili MH (2014) The inherent premise of immunotherapy for cancer dormancy. Cancer Res 74 (23):6745–6749. 10.1158/0008-5472.CAN-14-2440 [DOI] [PubMed] [Google Scholar]
- 46.Price TT, Burness ML, Sivan A, Warner MJ, Cheng R, Lee CH, Olivere L, Comatas K, Magnani J, Kim Lyerly H, Cheng Q, McCall CM, Sipkins DA (2016) Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci Transl Med 8 (340):340ra373 10.1126/scitranslmed.aad4059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cho BS, Kim HJ, Konopleva M (2017) Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: from bench to bedside. Korean J Intern Med 32 (2):248–257. 10.3904/kjim.2016.244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gawrzak S, Rinaldi L, Gregorio S, Arenas EJ, Salvador F, Urosevic J, Figueras-Puig C, Rojo F, Del Barco Barrantes I, Cejalvo JM, Palafox M, Guiu M, Berenguer-Llergo A, Symeonidi A, Bellmunt A, Kalafatovic D, Arnal-Estape A, Fernandez E, Mullauer B, Groeneveld R, Slobodnyuk K, Stephan-Otto Attolini C, Saura C, Arribas J, Cortes J, Rovira A, Munoz M, Lluch A, Serra V, Albanell J, Prat A, Nebreda AR, Benitah SA, Gomis RR (2018) MSK1 regulates luminal cell differentiation and metastatic dormancy in ER(+) breast cancer. Nat Cell Biol 20 (2):211–221. 10.1038/s41556-017-0021-z [DOI] [PubMed] [Google Scholar]