

Abstract
Most cancer-related deaths come from metastasis. It was recently discovered that nanoparticles could inhibit cancer cell migration. While most researchers focus on single-cell migration, the effect of nanoparticle treatment on collective cell migration has not been explored. Collective migration occurs commonly in many types of cancer metastasis, where a group of cancer cells move together, which requires the contractility of the cytoskeleton filaments and the connection of neighboring cells by the cell junction proteins. Here, we demonstrate gold nanorods (AuNRs) and the introduction of near-infrared light could inhibit the cancer cell collective migration by altering the actin filaments and cell junctions with significantly triggered phosphorylation changes of essential proteins, using mass spectrometry-based phosphoproteomics. Further observation using super-resolution stochastic optical reconstruction microscopy (STORM) showed the actin cytoskeleton filament bundles were disturbed, which is difficult to differentiate under a normal fluorescence microscope. The decreased expression level of N-Cadherin junctions and morphological changes of tight junction protein zonula occludens 2 (ZO-2) were also observed. All these results indicate possible functions of the AuNRs treatments in regulating and remodeling the actin filaments and cell junction proteins, which contribute to decreasing cancer cell collective migration.
Keywords: collective cancer cell migration, metastasis, gold nanorods, plasmonic photothermal therapy, phosphoproteomics, STORM, super-resolution microscopy
For Table of Contents Only
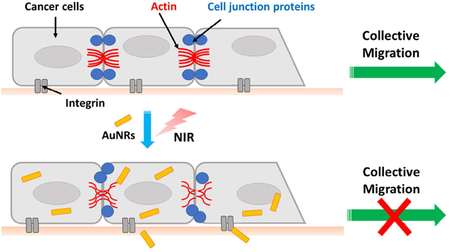
Metastasis is responsible for over 90% of cancer-related deaths.1 In order to initiate the metastasis, cancer cells must be equipped with the ability to migrate and invade the surrounding tissues, then intravasate to the microvasculature of the lymph and blood stream, and finally translocate to distant tissues and adapt in the microenvironment.1 However, past attempts to develop anti-metastasis drugs have not been efficacious in clinical trials.2 Recent advancements in nanomedicine provide new opportunities to avoid some drawbacks of commonly used cancer drugs, as nanoparticles can cross biological barriers, enter target cells with high selectivity, and function inside cell in a controlled manner.3–5 Nanoparticles have shown promise as anti-metastasis drug delivery vehicles targeting invasive or metastasized cancer cells,6–8 and they could even function as anti-metastasis drugs without drug loading.9–12 The optical and mechanical properties, such as plasmonic photothermal effect and high mechanical strength, as well as excellent biocompatibility of gold nanoparticles (AuNPs) make them very useful in attenuating cancer metastasis.13
Previously, we have developed cancer treatment using gold nanorods (AuNRs) for plasmonic photothermal therapy (PPTT). In PPTT, AuNRs that absorb the incident near infra-red (NIR) light to induce heat, and thereby could trigger tumor apoptosis.14,15 AuNRs-PPTT has been applied successfully on treating tumor bearing mice, cats and dogs. In these studies, we observed that animals with induced or spontaneous tumors were effectively cured with no tumor reoccurrence or metastasis.14,16,17 Our recent in vitro studies also revealed AuNPs and PPTT inhibit cancer cell migration and invasion.12,18 However, the mechanism of how AuNPs treatments inhibit cancer cell migration remains largely unresolved.
While the mechanism of nanoparticles on inhibiting the migration of single cells has been explored in the previous works, the mechanism regarding collective cell migration has rarely been studied. In collective cancer cell migration, a group of cancer cells migrate together, which might be a more efficient route for metastasis possibly due to a diverse cell population seeding other organs or the multicellular signal integration engaged.19 Collective cell migration has been widely observed in human cancers, especially in human epithelial cancers such as breast cancer and colon cancer.19, 20 It requires both the contractility of the cytoskeleton filaments and the active interactions of neighboring cells through the cell-cell junctions that connect the cytoskeleton of the neighboring cells.21 This process is highly dynamic and regulated by signal transduction through protein phosphorylation.22–24 Given their important roles, it is imperative to understand the signals evolved in the cytoskeleton filaments and cell-cell junctions shortly after AuNRs and PPTT stimulation for the rational design of effective strategies to inhibit cancer metastasis.
In the current study, we hypothesized that the integrin-targeting AuNRs and PPTT treatment could affect the cytoskeleton and cell junctions, due to their interactions and connections as a network, to result in the inhibition of collective cancer cell migration (as shown in Scheme 1 in the Experimental section). To test this hypothesis, quantitative mass spectrometry (MS)-based phosphoproteomics was employed to examine the signaling pathways upon the stimulation of AuNRs and PPTT. A primary signaling pathway map has been constructed to display a large number of identified alterations. Furthermore, super-resolution microscopy imaging techniques were used to visualize the changes of key cytoskeletal and cell junction proteins. Both phosphoproteomics and super-resolution imaging results indicated possible functions of the AuNRs and PPTT in regulating and changing the architecture of the cytoskeletal filaments and cell junctions, contributing to the inhibition of collective cancer cell migration.
Scheme 1.

Experimental design (A) and proposed mechanism (B) of AuNRs and PPTT in inhibiting cancer collective migration. Targeting integrin could affect the actin cytoskeleton and cell junctions to result in the inhibition of cancer cell collective migration. Phosphoproteomics and super-resolution fluorescence imaging, as well as Western blot, were the main experimental tools used in the current study.
RESULTS AND DISCUSSION
Gold Nanorods and NIR Light Attenuate the Migration and Invasion of Cancer Cells
The preparation of integrin targeted AuNRs was stated in our previous work.18 Briefly, AuNRs with a size of 25 (± 3) × 6 (± 2) nm (length × width) and an aspect ratio of 4.2 (Figure S1A, transmission electron microscopy (TEM) image) were synthesized using the seedless growth method.25 Optimal heat-generating efficacy in PPTT with these AuNRs has been demonstrated previously.26 To remove the cytotoxic cetyltrimethylammonium bromide (CTAB), the as synthesized AuNRs were washed twice with D.I. water. Then, the AuNRs were functionalized with polyethylene glycol thiol (PEG) and Arg–Gly–Asp (RGD) peptides to increase the biocompatibility.27 and obtain integrin targeting,28 respectively. The surface conjugations were confirmed by the red-shift of the longitudinal surface plasmon resonance (SPR) band (Figure S1B) and surface charge changes of the AuNRs (Figure S1C), consistent with the previous reports.18
The binding of RGD peptide to the cell surface integrin could enhance the endocytosis of AuNRs.29 The internalization of AuNRs within the cervical cancer cell line HeLa, was observed under a differential interference contrast (DIC) microscope (Figures 1A and B). DIC images indicate the AuNRs@RGD distribute spread the cytoplasm and the cell junction areas (Figure 1C). The z-scanning indicates the successful internalization of AuNPs inside cells after 24 h (Figure S2A–C). The cell viability (XTT) assay revealed that the cells remained viable and had similar proliferation rates after incubation with AuNRs and after PPTT for 24 h (Figure 1D). AuNRs@PEG was used as a “bare”, nonspecifically targeted AuNRs for control, as shown in Figure S3 (no cytotoxicity) and Figure S2B (cellular uptake not obvious), indicating the importance of RGD peptides to increase cellular uptake. In addition, no observable change of the apoptosis regulator Bcl-2-associated X (BAX) protein indicates no apoptosis after treatment (Figure 1E). We performed the same assays with the breast cancer cell line MCF-7, and similar results were obtained (Figure S2).
Figure 1.

Cellular uptake, cytotoxicity and motility upon AuNRs treatments. (A-B) Differential interference contrast (DIC) microscopic images of HeLa cells without (A) and with AuNRs@RGD after 24 h incubation (B). (C) DIC image of AuNRs@RGD distribute in the cell junction areas after 24 h incubation. The red arrows identify the locations of AuNRs. (D) Cell viability of HeLa cells after AuNRs and AuNRs+NIR treatments (n=3). (E) Western blotting for the BAX protein upon different treatments. (F and G) Scratch assay of HeLa cells (control, AuNRs treatment, and AuNRs+PPTT treatment) at 0 and 12 h (n=6). Student’s t test was used for statistical analysis. All values are expressed as means ± standard errors of the mean (SEM). ***p<0.001, **p< 0.01, *p<0.05. If not specified otherwise, “AuNRs” in all other figures means “AuNRs conjugated with RGD ligands”.
To evaluate the effects of AuNRs on cancer cell collective migration, we conducted a 2D scratch assay 30, 31 on the monolayers of MCF-7 and HeLa cells with or without the treatments. After introducing a “scratch” or “wound” into a cell culture, the cancer cells migrate collectively to the empty space, and images were captured immediately and 12 hours after the scratch of HeLa cells in Figure 1F (or 24 hours of MCF-7 cells in Figure S4). The statistics (Figure 1G) indicates that cells have exhibited significantly different wound-healing abilities in the control groups compared with those treated with AuNRs, while the introduction of NIR light to generate PPTT further decreases the wound-healing ability of cancer cells. If only treated with same dose of NIR light (no AuNRs added), no change in the cell viability and motility was observed (Figure S5). Our result shows both specific targeted AuNRs (AuNRs@RGD) and nonspecific targeted AuNRs (AuNRs@PEG, Figure S6) could inhibit collective cell migration to different extents, among which the AuNRs@RGD assisted PPTT is most effective.
Mass Spectrometry-Based Phosphoproteomic Analysis Reveals Perturbations of the Signal Transduction of Actin Network and Junction Proteins
To elucidate the effects of AuNRs and PPTT treatments on cytoskeleton filaments and cell junctions, we examined the phosphoproteomics of cancer cells using quantitative mass spectrometry (MS). A simplified experimental procedure is shown in Figure 2A (detailed and complete experimental procedure in the Methods section and Figure S7, including conditions of non-specific targeting AuNRs@PEG). Protein phosphorylation was identified and quantified in both HeLa and MCF-7 cells after incubation with AuNRs for 30 min or after AuNRs+PPTT treatment for 30 min. Three-plex dimethyl labeling was used for phosphoproteomic quantification, and titanium (IV) based immobilized metal ion affinity chromatography (Ti-IMAC) was used to enrich the phosphorylated peptides from the protein digest of cell lysate. The enriched phosphorylated peptides were analyzed by an on-line liquid chromatography-mass spectrometry (LC-MS) system. Three replications of each condition were conducted and about 1200 common phosphorylation sites (where the phosphorus group binds to the protein) were quantified. The clustering analysis (Figure S8) shows that the control and experimental groups were separately clustered with good reproducibility. Differential analysis identified proteins with significant changes in AuNRs-treated groups compared to the control group (Figure S9). The numbers of dysregulated phosphorylation sites of different treatments and their overlap in the Venn diagrams are shown in Figure S10. For instance, compared with the control group, the phosphorylation levels of 371 and 244 sites are significantly up- and down-regulated, respectively, for HeLa cells upon AuNRs treatment. Further changes from PPTT were observed, with 73 and 189 phosphorylated sites up- and down-regulated.
Figure 2.

Phosphoproteomics results. (A) Experimental workflow. Two comparisons were performed in data analysis. Comparison #1 (AuNRs vs. control): (B) Heatmap and (C) pathway analysis after AuNRs treatment. (D) Western blotting showing the altered phosphorylation sited in p120 Catenin (HeLa cells). (E) Altered phosphorylation sited in p120 Catenin (pS268) indicated by phosphoproteomics (HeLa cells). Comparison #2 (AuNRs + NIR vs. AuNRs): (F) Heatmap and (G) pathway analysis after AuNRs + NIR treatment. (H) Western blotting showing the altered phosphorylation sited in GSK3 (HeLa cells). (I) Altered phosphorylation sites GSK3 (pY216) indicated by phosphoproteomics (HeLa cells). Mean values in are shown in the heatmaps (n=3).
Proteins with their significantly altered phosphorylation sites are listed in heatmaps (Figure 2B for AuNRs and 2F for AuNRs+PPTT) and Table 1 (see Table S1 for more information). In order to understand the biological meanings of these phosphorylation changes, we performed pathway analysis (Figure 2C for AuNRs and 2G for AuNRs+PPTT), which revealed the significant perturbations to the signaling pathways related to the cytoskeleton and cell junctions. To further confirm the mass spectrometric results, the varied phosphorylated sites of p120 catenin (pS268) and glycogen synthase kinase (GSK3, pY216), which are highly related to cell adhesive junctions, and are regulators to actin cytoskeleton and microtubules,32 respectively, have been validated by Western blot results (Figure 2D, E, H, and J).
Table 1.
Selected significantly dysregulated phosphorylation sites of the cytoskeletal and junction proteins, specifying the phosphorylation sites and biological functions.
| Category | Protein | Protein Function | Phosphorylation Sites Altered | Phosphorylation Sites Function |
|---|---|---|---|---|
| Cytoskeleton | Paxillin | Form focal adhesions | pS303, pS302, pS106, pS85 | Increase of pS85 has an important function in cell adhesion 39, 40 |
| MYH9 | Form stress fibers and create a contraction force in cell migration 41 | pS1943 | pS1943 could alter cell motility 42 | |
| MLCP | pS299, pS445, pS871 | pS445 is closely related to cell adhesion 43 | ||
| MAP4 | Promotes microtubule assembly | pS1073, pS787, pS280, pS789 | pS1073 is related to cancer cell metastasis potential 44 and pS787 could promote tubulin polymerization 45 thereby changing the microtubule organization. | |
| Cells junctions | α-catenin | Form cell-cell adhesion complexes, anchoring actin cytoskeleton and interacting with cadherins46 | pT654, pS641, pT634, pS652, pS655 | S641 affects cell motility 47 |
| ZO-2 | Connect cytoskeletons of adjacent cells and act as barriers for the passage of molecules and ions 48 | pS966, pS986, pS978, pS266, pS986, pS1159, pS130 | No information found | |
| Vimentin | A hallmark protein of epithelial to mesenchymal transition (EMT), which is related to the increase of migration and invasive properties 49 | pS459, pS56, pT458 | pS56 was reported with the function of cytoskeleton reorganization 50 | |
| Keratin 18 | Keratin 18 and its filament partner keratin 8 are regarded as the most commonly found members of the intermediate filament family | pS34, pT65, pS420, pS42 | pS34 affects cell motility and cytoskeleton 51–54 |
We observed that our treatments can change the phosphorylation of the actin network (Table S1), including i) proteins forming the focal adhesions (FAs), such as paxillin, zyxin, vinculin; ii) the myosin related proteins, such as myosin-9 and myosin-light-chain phosphatase (MLCP); iii) the actin-binding proteins, such as filamin, cortactin and drebrin. Moreover, changes of cell junctions, such as tight junction proteins zonula occludens (ZO-1 and ZO-2) were also observed upon AuNRs stimulation. More changes were observed to ZO-2 after PPTT, indicating an enhanced perturbation in the tight junctions. In addition, cell junction protein catenins, including α-, β-, and p120 catenins, have altered phosphorylated sites upon treatment. Phosphorylation change of desmosomes junction related proteins, including desmoplakin, epiplakin, plectin, Keratin 18 and vimentin were observed. In addition, the phosphorylation of several microtubule (MT)-related proteins were changed, including microtubule associated proteins MAP4, microtubule associated protein 1B (MAP1B) and glycogen synthase kinase-3 alpha (GSK3A). Besides, Phosphorylation changes of protein kinases that could regulate the cytoskeleton filaments and cell motility were observed, such as RAF proto-oncogene serine/threonine-protein kinase (Raf1), mitogen-activated protein kinase kinase 2 (MAP2K2), cyclin-dependent kinase (CDK1), RAC-alpha serine/threonine-protein kinase (AKT1), etc.
Integrins are adhesive molecules located in the cell membrane and responsible of transporting signals and cell-cell communications.33 The ability of integrin-targeted AuNRs to alter the junction proteins is linked to the coordination and interdependence manner of integrin and cell junction to form adhesive networks, by connecting through the actin cytoskeleton and sharing common signaling molecules.34, 35 For instance, integrin-induced signaling molecules focal adhesion kinase (FAK) and paxillin regulate the N-cadherin junctions in Hela cells;36 α-catenin links cadherin to the actin cytoskeleton;37 and p120 catenin cooperates with cortactin to regulate lamellipodial dynamics and cell adhesion.38 Here, we observed possible signal cross-talk between the cytoskeleton and cell junctions, such as the altered phosphorylation of paxillin, α-, β-, and p120- catenin, as well as cortactin. Based on the phosphoproteomics results, a schematic diagram is constructed to show the signal transduction upon AuNRs and PPTT stimulation (Figure 3, and more details in Figure S11 and S12). By targeting integrins, our treatments induced the protein phosphorylation change of the downstream actin cytoskeletal and junction proteins.
Figure 3.

Schematic diagram of the signaling pathways that are engaged with the cytoskeleton and cell junctions upon the AuNRs and PPTT treatment. The blue and red “P”s indicate the altered phosphorylation level upon AuNRs treatment and PPTT treatment (AuNRs+NIR), respectively.
Super-Resolution Imaging for Confirming Disturbed Cytoskeletal and Cell Junction Proteins.
Collective cell migration requires the cells are effectively coupled by cell junctions, coordinating their actin dynamics and intracellular signaling thereby forming a functioning unit.20 The actin cytoskeletons of neighboring cells are coupled by the cell junctions. The drag force between the cells is provided by actomyosin contractility,55 which is important in maintaining effective cell junction and collective migration.56 Although the phosphorylation signal transduction takes place within a few minutes, the protein expression level may take hours to change. Therefore, to clearly observe the protein expression level changes, we monitored the actin filament structures after 24 hours of AuNRs incubation with or without PPTT (Figure 4). Under a normal fluorescence microscope, it is difficult to differentiate the changes of actin structure before and after treatments due to the insufficient resolution, as shown in Figure 4A–C. Stochastic optical reconstruction microscopy (STORM) provides superior spatial resolution than conventional fluorescence microscopy to reveal the detailed actin cytoskeletal structures (Figure S13). By using STORM, we observed the morphological changes of the circumferential actin filaments at the cell-cell junctions. Before AuNRs treatment, the well-aligned stress fibers (contractile actin bundles) are clearly visualized, with polymerized and stable structure (Figure 4D). However, after AuNRs treatment, the actin bundles became thinner, showing a clear sign of disturbance (Figure 4E). Furthermore, after NIR exposure, the circumferential actin filaments at cell junctions exhibited obvious changes (Figure 4F): the stress fibers were greatly decreased, while coil, depolymerized and reorganized structures appeared, which possibly indicated the heating effect on harming the actin filaments polymerization at the junction sites. In addition, the actin structure at the cell leading edges (filopodia and lamellipodia) was also imaged (Figure S14), and the observed decrease in stress fibers in the cell leading edges hinted a decrease in cell motility.
Figure 4.

STORM and epifluorescence images of actin filaments in the cell-cell junction upon different treatments: (A, D) Control; (B, E) AuNRs; (C, F) AuNRs + NIR. After NIR exposure, the actin filaments at cell junctions exhibited clearly altered morphology (scale bar = 5 μm).
We further examined the AuNRs and PPTT effects on cell junctions in faster-migrating HeLa cells and slower-migrating MCF-7 cells. Different cell lines could have highly diverse populations of cell junction proteins. The expression level of neural (N)-cadherin in HeLa was found to be much higher than that in the MCF-7 cells57 (not detectable in MCF-7 cells in our study). On the other hand, MCF-7 cells show significantly higher expression levels of tight junction proteins than HeLa cells (Figure S15). Therefore, we used HeLa cells as a model for studying the N-cadherin junction and MCF-7 cells for the tight junction.
The N-cadherin junction is well known to be highly expressed in many aggressive tumors and promote metastasis.58 It is reported that N-cadherin holds the cohesive cell clusters together, which tend to migrate persistently,59 playing a key role in collective migration.59, 60 The expression level of N-cadherin junction is largely known as a marker for cancer motility and invasiveness. We observed a decreased expression level of N-cadherin (Figure 5A–E, S16) upon the AuNRs treatments by fluorescence intensity and Western blot analysis.
Figure 5.

(A-C) Immunofluorescence images of N-cadherin in HeLa cells before (A) and after AuNRs (B) and AuNRs+PPTT (C) treatments (more images in Figure S14). The fluorescence intensities in these images are normalized together. (D) The fluorescence quantification of the N-cadherin (n=20 cells, ±SEM). (E) Western blot results also indicate a decreased expression level of N-cadherin after treatments. (F) Immunofluorescence images of tight junction protein ZO-2 in MCF-7 cells, before and after AuNRs or AuNRs+PPTT treatments. The morphology of ZO-2 change from a normal and continuous line-like structure in the control group to a discontinuous dot-like structure after treatments. The figures showed 3D scanning of ZO-2, where Layer 1 is close to the bottom of the cells, and Layer 3 is close to the top of the cells. Scale bar = 20 μm.
Tight junctions create strong intercellular links 61,62 at the invasion zone of tumors.20 During tumor development, tight junctions are remodeled, enabling cancer cells to adopt a migratory behavior.63, 64 It has been reported that tight junction protein ZO-1 can directly bind to integrin and regulate the mechanical properties of integrin-fibronectin links.65,66 In addition, the tight junction proteins ZO-1, ZO-2 and ZO-3 can bind to the cytoskeleton.67 Here, we studied the tight junction changes by labeling ZO-2. We observed the morphology of ZO-2 change from a normal and continuous line-like structure in the control group to a discontinuous dot-like structure after treatment, indicating possible impaired tight junctions (Figure 5F). If only treated with same dose of NIR light (no AuNRs added), no changes in the actin filaments, N-cadherins, and ZO-2 were observed (Figure S17).
This study differs from the previous works mainly in the following points: 1) Early signaling (30 min) was studied upon AuNRs and mild PPTT treatments using phosphoproteomics, while most of other work studied longer time scale, such as overnight or after several days.68, 69 2) The alterations of cell junction were reported here, while our previous work was focused on the cytoskeleton proteins after 24 h AuNRs and/or PPTT treatments.18 3) In addition, super-resolution imaging technique (STORM) revealed more detailed structural information on the effects of our treatment.
We have previously studied the PPTT for triggering apoptosis.14, 15 However, due to several reasons, such as the inhomogeneous distribution of AuNRs or the laser penetration ability, some locations within tumor might not generate apoptosis. In addition, it is possible for some cancer cells to develop thermal tolerance.70 For those cells that not able to receive enough dose or resistant to the treatment to cause apoptosis, their ability towards metastasis could decrease upon treatment.
Collective migration is widely observed in metastasis in vivo.71, 72 The relationship of cell mechanical properties (cell junction and adhesion, actomyosin contractility, geometry confinement, etc.) and cell collective migration in vivo has been reported previously.22, 73, 74 For instance, it has been reported that lipoma preferred partner (LPP), an actin-binding protein that could degrade N-cadherin in lung cancer, could inhibit collective cell migration during lung metastasis in mice model.75 Regarding our treatment, future studies on metastatic mice models will be performed.
As metastasis is a highly complex process, multiple factors, such as cytoskeleton, adhesion, extracellular matrix (ECM), tumor microenvironment, blood or lymphatic vessels, etc., will need to be considered for a comprehensive understanding of AuNRs-PPTT in inhibiting metastasis. Zhang et al. have shown that photodynamic therapy (using liposome with porphyrin-18) can greatly disturb the ECM, therefore decrease the attachment of the cells with the ECM and affect the actomyosin contractility.76 It will be interesting to look into how AuNRs and PPTT affect ECM, tumor microenvironment, blood or lymphatic vessels in future studies.
CONCLUSION
In this study, we investigated the mechanism of integrin-targeted AuNRs and PPTT in inhibiting collective cancer cell migration. Our phosphoproteomics results revealed the phosphorylation changes of many cytoskeletal and cell junction proteins, setting the foundation for current and future studies of the underlying mechanism at the molecular level. Using super-resolution fluorescence microscopy and Western blotting, we verified the changes to selected key proteins related to the actin cytoskeleton and cell junctions. The morphological changes of actin filaments and extensive phosphorylation changes to actin-associated proteins, such as filamin, paxillin, vinculin, zyxin, PAK, MLCP, MyHC, etc., upon integrin-targeted AuNRs and PPTT treatment also indicated weakened cell adhesion and stress fiber generation. Furthermore, in HeLa cells, we found a significantly lower expression level of N-cadherin, as well as the phosphorylation changes to α-, β- and p120-catenin that connect N-cadherin to the actin cytoskeleton, while in MCF-7 cells, a discontinuation and altered morphology of the tight junction protein ZO-2. All of the current experimental evidence has led to a proposed mechanism that the interactions between the integrin-targeted AuNRs and cells could trigger the phosphorylation changes of essential components associated with cytoskeleton filaments and cell-cell junctions, and cause their morphological or expression level changes, therefore inhibiting cancer collective migration. Further studies of the perturbations to individual related proteins will be carried out to provide a more complete understanding of the inhibition effect.
METHODS
Experimental Design.
The experiment is based on our hypothesized that integrin-targeting AuNRs and PPTT treatment could affect the cytoskeleton and cell junctions, thus results in the inhibition of cancer cell collective migration. To test this hypothesis, phosphoproteomics was performed to understand the signal transduction among the integrin, cytoskeleton and cell junctions. Super-resolution imaging tools, as well as Western blot, were used to observe the changes of the actin cytoskeleton and cell junctions.
Materials.
Dulbecco’s modified Eagle’s medium (DMEM), phosphate buffered saline (PBS), fetal bovine serum (FBS), antibiotic/antimycotic solution, and 0.25% trypsin/2.2 mM EDTA solution were purchased from VWR. Methoxypolyethylene glycol-thiol (mPEG-SH, MW 5000) was purchased from Laysan Bio, Inc. Cell penetrating peptide RGD (RGDRGDRGDRGDPGC) was purchased from GenScript, Inc. Mammalian cell protease inhibitors and phosphatase inhibitors were purchased from Roche Applied Sciences, and sequencing grade trypsin was purchased from Promega. Tetrachloroauric acid trihydrate (HAuCl4.3H2O), ascorbic acid, cetyltrimethylammonium bromide (CTAB), AgNO3, NaBH4, 4-(2- hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), NaCl, sodium deoxycholate, sodium dodecyl sulfate (SDS), paraformaldehyde, glutaraldehyde, formaldehyde-D2 (DCDO), sodium cyanoborohydride (NaBH3CN), formic acid (FA), trypsin (TPCK treated), iodoacetamide (IAA), dithiothreitol (DTT), trifluoroacetic acid (TFA) and triethylammonium bicarbonate buffer (TEAB), Triton X-100, 2-(N-Morpholino)ethanesulfonic acid hemisodium salt (MES), NaCl, EGTA, glucose, MgCl2, NaBH4, BSA, Anti-BAX and anti-beta-actin primary antibody, (H+L) HRP conjugate, Alexa 647-phalloidin, 100 mM Tris pH 8.0, glucose oxidase, catalase, β-mercaptoethanol were purchased from Sigma (St. Louis, MO). Urea were from Shanghai Sangon Biotech (Shanghai, China). BCA protein assay kit was from Beyotime Institute of Biotechnology (Shanghai, China). HPLC-grade acetonitrile (ACN) was from Merck (Darmstadt, Germany). Fused silica capillaries with dimensions of 75 and 200 μm i.d. were obtained from Yongnian Optical Fiber Factory (Hebei, China). C18 AQ beads (3 and 5 μm, 120 Å) were purchased from Daiso (Osaka, Japan). Anti-ZO-2 (Cell Signaling Technology) and Anti-N-Cadherin (ABclonal) Alexa Fluor-568 conjugated anti-rabbit IgG (H+L) (Abcam), goat anti-rabbit IgG Antibody. All the water used in experiments was purified with a Milli-Q system from Millipore (Milford, MA).
Instrumentation.
AuNRs were imaged using a JEOL 100CX-2 transmission electron microscope (TEM) microscope, with their average size being measured by ImageJ software (NIH). UV−vis spectra were obtained by an Ocean Optics HR4000CG UV-NIR spectrometer. A Nikon Eclipse 80i upright microscope and a back-illuminated scientific complementary metal oxide semiconductor (sCOMS) camera (Dhyana 400BSI, Tucsen) were used to record high magnification (up to 200 ×) differential interference contrast (DIC) images. Phosphoproteomics analysis was performed on a hybrid dual-cell quadrupole linear ion trap − Orbitrap mass spectrometer LTQ Orbitrap Elite (Thermo Fisher) with XCalibur 3.0.63 software. An 808 nm cw laser (0.7 W/cm2) was used for PPTT. STORM imaging was conducted on modified Zeiss Axiovert 100 TV microscope equipping with a high sensitive back-illuminated sCOMS camera (Dhyana 95, Tucsen).
Synthesis, Conjugation and Characterization of AuNRs.
AuNRs with an average size of 25 × 6 μm (length × width) were synthesized using a seedless growth method according to our previous reports 18, 25. Briefly, 5 mL of 1.0 mM HAuCl4 was added to a solution containing 5 mL of 0.2 M cetyltrimethylammonium bromide (CTAB), 250 μL of 4.0 mM AgNO3, and 8 μL of 37% HCl. Then, 70 μL of 78.8 mM ascorbic acid was added, followed by immediate injection of 15 μL of 0.01M of ice-cold NaBH4. The solution was left undisturbed for 12 hours. To remove extra cytotoxic CTAB, the AuNRs were centrifuged at 21000 g for 1 hour and dispersed in DI water, followed by a second centrifugation at 19000 g for 40 min. The sizes and homogeneity of the AuNRs were measured by TEM. AuNRs were then conjugated with surface ligands PEG and RGD. For first-step preparation of AuNRs@PEG, mPEG-SH (1 mM in H2O) was added to the nanoparticles overnight to achieve about 1000 ligands per AuNR. Then, RGD (1 mM) was added to achieve 10000 molar excess per AuNR. The solution was allowed to shake overnight at room temperature. Excess of ligands were removed by centrifugation. UV-vis spectrometer and zetasizer were used to test the successful conjugation of the ligands.
Cell Culture, AuNRs Treatments, and PPTT.
HeLa and MCF-7 cells were grown in Dulbecco’s modified Eagles’ medium DMEM containing 10% (v/v) fetal bovine serum and 1% antibiotic solution at 37 °C in a humidified incubator under 5% CO2. Cells were cultured for 24 hours followed by incubation with AuNRs (5 nM) for 24 hours. Then, a cw 808 nm laser (0.75 W/cm2) was applied to the cells for 2 minute. The temperature range of the photothermal effect mediated by AuNRs is 42 ±1 °C.
Toxicity and Uptake of AuNRs to Cancer Cells.
In order to examine the nanoparticle cytotoxicity in cells, XTT assay was performed. The uptake of AuNRs to HeLa and MCF-7 cells was visualized under a DIC microscope. Plasmonic AuNRs can be easily discerned from the cellular features as they appeared with high DIC contrast at/near SPR wavelength.
Measuring Cell Migration Speed upon AuNRs Treatment.
The 2D scratch assay was performed according to previous report.31 For measuring cell migration rate, a scratch assay will be used where cells will be cultured in a 6 well plate to form a confluent monolayer. A p200 pipet tip will be used to scrape the cell monolayer in a straight line to create an empty gap. Then the cells will be allowed for migration into the gap and imaged to track their migration rates. The cells were imaged on an inverted Nikon Eclipse Ti-E microscope using bright field microscopy. A Nikon Plan Fluor 10 × objective (Numerical aperture: 0.30, working distance: 16.0 mm) and a 12 V/100 W halogen lamp as light source was used. The output power of the light source was kept constant for all the imaging experiments and the exposure time of 30 ms was used to provide optimal contrast and brightness. Images were then recorded by a sCOMS camera (Dhyana 400BSI, Tucsen).
Super-resolution Imaging Setup:
The STORM imaging system was integrated into an inverted microscope (Zeiss Axiovert 100 TV, Jena, Germany). 405 nm and 660 nm lasers (Newport Excelsior one 405 nm, 200 mW, Irvine, CA; Laser Quantum Gem 660, 200 mW, Stockport, Cheshire, England) were collimated into a single light path after the beam expander (Thorlabs BE03M-A, Newton, NJ) with 3 × magnification. Collimation of multicolor lasers was done by using a dichroic mirror (Thorlabs, DMLP425T), thus allowing simultaneous illumination of the sample at multi-wavelengths. Uniblitz mechanical shutters (Vincent Associates, LS2Z2, Rochester, NY) in front of each laser were used to control the illumination conditions, either pulsed or continuous illumination profiles. The collimated light was expanded by a telescope of a pair of achromatic lenses (Thorlabs, AC127–025-A & AC254–150-A) and then focused at the back focal plane of a high refractive index oil immersion objective (Olympus, 60X Oil, N.A. 1.49) using another achromatic lens (Thorlabs, AC508–300-A). The incident angle of illumination light is controlled by the lateral shift of the light path, through a three-dimensional stage (Sigma KOKI, SGSP-20–20, Tokyo, Japan), before entering the objective. A multi-edge beam splitter (Semrock, DC-405-388-543-635, Rochester, NY) was used to reflect the light into the working objective to excite the sample. The emission light is collected by the same objective. After the tube lens, provided with the microscope, a pair of relay lenses (Thorlabs, AC127–125-A & AC127–150-A) was used to focus emission light onto an sCMOS chip (Tucsen, Dhyana 95) enabling a pixel size of ~110 nm. A combination of filters (Semrock, 664 nm RazorEdge long-pass edge filter (LP02–664RU-25), 658 nm StopLine single-notch filter (NF03–658E-25), 708/75 nm BrightLine single-band bandpass filter (FF01–708/75–25)) were inserted in front of the camera to reduce the background noise. Both epi-fluorescence images and STORM images were performed using the customized system.
Briefly, cells were cultured in an 8-well glass chamber (ibidi) and washed once with pre-warmed PBS buffer (Invitrogen). Cells were then fixed and permeabilized with 0.3% glutaraldehyde (Sigma) and 0.25% Triton X-100 (Sigma) in a cytoskeleton buffer containing 10 mM MES pH 6.1 (Sigma), 150 mM NaCl (Sigma), 5 mM EGTA (Sigma), 5 mM glucose (Sigma), and 5 mM MgCl2 (Sigma). Freshly prepared 0.1% NaBH4 (Sigma) in a PBS buffer was used to reduce the autofluorescence background generated during the cell fixation. The cells were then washed with a PBS buffer three times followed by applying a blocking buffer (3% BSA (Sigma) + 0.2% Triton-X100 in PBS buffer) for 60 min. To label the actin, cells were stained with 0.5 μM Alexa 647-phalloidin (Invitrogen) in a PBS buffer, wrapped with aluminum foil to protect from light and incubated at 4° C overnight. Remove the staining solution and briefly wash once with a PBS buffer. Immediately mount the sample for STORM imaging in an imaging buffer containing 100 mM Tris pH 8.0 (Invitrogen), 10 mM NaCl (Sigma), 0.5mg/mL glucose oxidase (Sigma), 40μg/mL catalase (Sigma), 10% (w/v) glucose (Sigma) and 1% (v/v) β-mercaptoethanol (Sigma) for STORM imaging.
STORM Imaging Data Processing.
In our experiments, an imaging sequence of 30,000–40,000 frames recorded at 60 Hz was used to reconstruct a high resolution STORM image. Within each frame, individual molecules identified were fit by an elliptical Gaussian function for determining their centroid positions. Molecules that were too dim, too wide or too elliptical to yield high localization accuracy were eliminated in order to generate high resolution images. Furthermore, positions for those molecules that were appealing continuously in several imaging frames were determined using the weighted centroid positions in all consecutive frames. To generate the super-resolution images, molecular positions were assigned as one point and their sizes were rendered as a normalized 2D Gaussian distribution. The width of 2D rendered spot depends the localization accuracy calculated from the number of photons detected for that localization event. The reconstructed STORM images have a pixel size of 10 nm.
Sample Preparation for Phosphoproteomics Experiment.
Cells were cultured in 100 mm dishes (Corning). The cells were then harvested for MS analysis, with a final confluence about 80–90%. After AuNRs treatment for 30 min, cells were washed twice with PBS before directly adding the lysis buffer (50 mM HEPES (pH =7.4), 150 mM NaCl, 0.1% SDC, 10 units/mL benzonase, protease inhibitor cocktail and phosphatase inhibitors) to the cells followed by scraping and collecting the cell lysate on ice. Lysates were vortexed and sonicated on ice, followed by centrifugation at 18000 g for 20 min at 4 °C to remove cell debris. The proteins in the supernatant were precipitated by adding 4 × excess volumes of ice-cold precipitation solvents (acetone: ethanol: acetic acid=50:50:0.1) and kept at −20 °C for overnight. The proteins were obtained after centrifugation, and were re-dissolved in 8 M urea and 50 mM HEPES (pH=8). The protein concentration was determined by Bradford assay. For mass spectrometry analysis, the disulfide bonds of proteins were firstly reduced by 1 mM dithiothreitol (DTT), followed by alkylation with 5.5 mM iodoacetamide. Then, trypsin (1:50 w/w) was used for protein digestion overnight 77.
Stable-isotope dimethyl labeling was performed according to previous reports 78. Briefly, for light, intermediate and heavy dimethyl labeling, 4 μL of CH2O (4%, v/v), CD2O (4%, v/v) or 13CD2O (4%, v/v) was added into 100 μg cell protein digest, respectively. Then 4 μL of freshly prepared NaBH3CN (0.6 M), NaBH3CN (0.6 M), and NaBD3CN (0.6 M) was added. The mixtures were then incubated for 1 h at room temperature for labeling reaction. For quenching the reaction, 16 μL of ammonia (1%, v/v) and 8 μL formic acid (5% v/v) were successively added.
Phosphorylation enrichment was conducted according to previous reports by using Ti4+-IMAC microspheres after dimethyl labeling 79. Briefly, the microspheres were suspended in the sample loading buffer containing 80% (vol/vol) ACN and 6% (v/v) TFA, and mixed with protein digest with a ratio of 10:1 (w/w), followed by violent vibration for 30 min. After removing the supernatant by centrifugation, the microspheres were washed with washing buffer 1 (50% (v/v) ACN, 6% (v/v) TFA containing 200 mM NaCl) and washing buffer 2 (30% (v/v) ACN and 0.1% (v/v) TFA) for 20 min, respectively. Finally, the phosphopeptides were eluted by adding 10% (v/v) ammonia-water and lyophilized to powder for following analysis.
RPLC-MS/MS Analysis for Quantitative Phosphoproteomics.
LTQ-Orbitrap Elite (Thermo Scientific) coupled with Dionex UltiMate 3000 RSLCnano system (Thermo Scientific) was used for all proteomic analyses. The lyophilized phosphopeptide samples were re-dissolved in aqueous solution with 1% FA and loaded onto a 4 cm × 200 μm i.d. C18 trap column packed with C18 AQ beads (5 μm, 120 Å) and separated by a 50 cm × 75 μm i.d C18 (5 μm, 120 Å) capillary column kept in 50 °C with a flow rate 300 nL/min. Aqueous solution with 0.1% FA (solvent A) and 80% ACN with 0.1% FA (solvent B) were used for the reversed phase (RP) binary gradient separation, and the RP binary gradient was set as: from 0–3% solvent B in 3 min, from 3–30% solvent B in 135 min, from 30–45% solvent B in 15 min, from 45%−100% solvent B in 2 min, after flush with 100% solvent B for 11 min the whole system was equilibrated by using solvent A for 13 min. The MS full scan was acquired from m/z 350 to 1650 in an LTQ-Orbitrap Elite with a mass resolution of 60 000 at m/z 400, and the MS/MS scan was acquired in ion trap. All MS and MS/MS spectra were acquired in the data dependent analysis (DDA) mode, in which the 20 most intense ions in the MS scan were selected for MS/MS scan by collision induced dissociation (CID) with the normalized collision energy at 35%. The dynamic exclusion function was: repeat count 1, repeat duration 30 s, and exclusion duration 90 s.
Phosphoproteomics Data Processing.
MS data were processed using MaxQuant (version 1.5.3.30, http://www.maxquant.org/) using Andromeda as search engine against the Uniprot human protein database (69712 sequences, downloaded from http://www.uniprot.org/) with precursor mass tolerance of 4.5 ppm and fragment mass deviation of 0.5 Da. Variable modifications consisted of methionine oxidation, acetylation of protein N-term and phosphorylation (STY). Fixed modification contained cysteine carbamidomethylation. Trypsin was set as specific proteolytic enzyme. Peptides with a minimum of six amino acids and a maximum of two missed cleavages were allowed for the analysis. For peptide and protein identification, the false discovery rate (FDR) cutoffs were both set to 0.01. Triplets were selected as the quantification mode with the dimethyl Lys 0 and N-term 0 as light labels, dimethyl Lys 4 and N-term 4 as median labels and dimethyl Lys 8 and N-term 8 as heavy labels. All other parameters are the default setting in MaxQuant.
Bioinformatics Analysis.
Bioinformatics analysis of phosphoproteomics study was performed. Three biological replications for each condition (control, AuNRs@RGD, AuNRs@RGD+NIR) in MCF7 and HeLa cells were conducted. Raw data from phosphoproteomics was normalized using supervised normalization of the microarray (SNM) 80. In the SNM procedure, variance due to biological replicates was adjusted by setting them as variables in the model. Variance explained by different experimental treatments (control, AuNRs@RGD, and AuNRs@RGD+NIR) was fitted as a biological variable in the model. Hierarchical clustering was done with statistical software R. Phosphoproteomics data were log2-transformed before analysis of variance (ANOVA), which was used to detect differential phosphorylated proteins between two treatment groups (e.g., AuNRs@RGD vs. AuNRs@RGD+NIR), with treatment conditions set as fixed effects. P value threshold at 0.1 was set to select differential phosphorylated proteins. The proteins identified as being affected were subjected to pathway analysis using the MetaCore pathway analysis software (“MetaCore from Thomson Reuters”).
Western-blot Analysis.
Cells were lysed in RIPA buffer (20 mM Tris pH 7.4, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS) supplemented with protease inhibitors (Sigma-Aldrich) and phosphatase inhibitors (25 mM sodium fluoride, 10 mM sodium pyrophosphate, 50 mM β-glycerophosphate, 1 mM sodium orthovanadate). Protein concentrations were measured by BCA assay (Pierce), and equal amounts of protein were loaded on a SDS-PAGE gel. After SDS-PAGE, the resulting gels were transferred to PVDF membranes (Millipore) by Bio-Rad trans blot turbo (Bio-Rad). Afterwards, the membranes were treated with blocking buffer (5% BSA in TBS (20 mM Tris, 150 mM NaCl)). The primary antibodies p120 catenin (pS268), GSK3 (pY216), N-Cadherin, and BAX were incubated with the membranes for different sets of experiments overnight in 4° C with shaking, followed by adding the secondary antibodies (Goat Anti-Rabbit IgG Antibody, (H+L) HRP conjugate, purchased from Millipore Sigma). Blots were washed three times for 10 m in TBS after primary and secondary antibodies.
Immunofluorescence Labeling and Confocal Microscopy.
Cells were cultured on 8 well μ-Slide with glass bottom (Ibidi). After treatment, cells were fixed in 3% Paraformaldehyde/0.1% Glutaraldehyde for 7 min at room temperature, followed by treated with 0.1 % (m/v) NaBH4 for 7 min and the wash three times with PBS. Cells were then blocked with 3% (w/v) BSA and 0.5% (v/v) Triton-X100 in PBS for 30 minutes at room temperature with mild shaking. Primary antibody was diluted to a working concentration in a blocking solution, and incubated at 4 °C overnight. After three times washing with PBS, secondary antibody (Goat Anti-Rabbit IgG H&L (Alexa Fluor® 568) from abcam) was added for 1 h, followed by wash 3 X with PBS before mounting with Prolong Gold (Invitrogen). Images were taken with a Zeiss LSM 700–405 confocal microscopes.
Statistical Information.
For the other experiments in this study if not mentioned, two-tailed t-tests were performed and the differences between data sets were considered significant when P < 0.05.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Eric Snider for his great help in Western blot analysis, Cecily Ritch for her help in cell culture, Hashem Mohilldean for his help in analyzing the scratch assay results, and Dr. Xiao Kuang for several discussions. We also thank Erin Bryant for their critical proofreading the English of the manuscript. M.A.E. acknowledges National Science Foundation Division of Chemistry (CHE) Grant 1608801 for its support of this work; F.W. acknowledges support from the National Natural Science Foundation of China (21675152) and the Youth Innovation Promotion Association CAS (2014164); and N.F. acknowledge funding support from NIH Grant 1R01GM115763.
Funding Sources
M.A.E. acknowledges National Science Foundation Division of Chemistry (CHE) Grant 1608801 for its support of this work; F.W. acknowledges support from the National Natural Science Foundation of China (21675152) and the Youth Innovation Promotion Association CAS (2014164); and N.F. acknowledge funding support from NIH Grant 1R01GM115763.
Publisher's Disclaimer: “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts.
ASSOCIATED CONTENT
Supporting Information.
The Supporting Information is available free of charge via the Internet at http://pubs.acs.org. Information of AuNR synthesis and characterization, cytotoxicity of non-specifically targeted AuNRs, tests on different cell lines, data analysis for phosphoproteomics, high resolution images of actin and cell junctions of different conditions, list of selected proteins with their altered phosphorylation sites, etc (PDF)
REFERENCES
- (1).Chaffer CL; Weinberg RA, A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [DOI] [PubMed] [Google Scholar]
- (2).Gandalovičová A; Rosel D; Fernandes M; Veselý P; Heneberg P; Čermák V; Petruželka L; Kumar S; Sanz-Moreno V; Brábek J, Migrastatics-Anti-Metastatic and Anti-Invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Murphy CJ; Gole AM; Stone JW; Sisco PN; Alkilany AM; Goldsmith EC; Baxter SC, Gold Nanoparticles in Biology: Beyond Toxicity to Cellular Imaging. Acc. Chem. Res 2008, 41, 1721–1730. [DOI] [PubMed] [Google Scholar]
- (4).Petros RA; DeSimone JM, Strategies in the Design of Nanoparticles for Therapeutic Applications. Nat. Rev. Drug Discov 2010, 9, 615–627. [DOI] [PubMed] [Google Scholar]
- (5).Peer D; Karp JM; Hong S; Farokhzad OC; Margalit R; Langer R, Nanocarriers as an Emerging Platform for Cancer Therapy. Nat. Nanotechnol 2007, 2, 751–760. [DOI] [PubMed] [Google Scholar]
- (6).Zhao Y; Alakhova DY; Kabanov AV, Can Nanomedicines Kill Cancer Stem Cells? Adv. Drug Deliv. Rev 2013, 65, 1763–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yang S; Gao H, Nanoparticles for Modulating Tumor Microenvironment to Improve Drug Delivery and Tumor Therapy. Pharmacol. Res 2017. 126, 97–108. [DOI] [PubMed] [Google Scholar]
- (8).Mu Q; Wang H; Zhang M, Nanoparticles for Imaging and Treatment of Metastatic Breast Cancer. Expert Opin. Drug Deliv 2017, 14, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tay CY; Cai P; Setyawati MI; Fang W; Tan LP; Hong CH; Chen X; Leong DT, Nanoparticles Strengthen Intracellular Tension and Retard Cellular Migration. Nano Lett 2014, 14, 83–88. [DOI] [PubMed] [Google Scholar]
- (10).Soenen SJ; Nuytten N; De Meyer SF; De Smedt SC; De Cuyper M, High Intracellular Iron Oxide Nanoparticle Concentrations Affect Cellular Cytoskeleton and Focal Adhesion Kinase-Mediated Signaling. Small 2010, 6, 832–842. [DOI] [PubMed] [Google Scholar]
- (11).Zhou T; Yu M; Zhang B; Wang L; Wu X; Zhou H; Du Y; Hao J; Tu Y; Chen C; Wei T, Inhibition of Cancer Cell Migration by Gold Nanorods: Molecular Mechanisms and Implications for Cancer Therapy. Adv. Funct. Mater 2014, 24, 6922–6932. [Google Scholar]
- (12).Ali M; Wu Y; Ghosh D; Do B; Chen K; Dawson M; Fang N; Sulchek T; El-Sayed M, Nuclear Membrane-Targeted Gold Nanoparticles Inhibit Cancer Cell Migration and Invasion. Acs Nano 2017, 11, 3716–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schroeder A; Heller DA; Winslow MM; Dahlman JE; Pratt GW; Langer R; Jacks T; Anderson DG, Treating Metastatic Cancer with Nanotechnology. Nat. Rev. Cancer 2011, 12, 39–50. [DOI] [PubMed] [Google Scholar]
- (14).Ali MR; Rahman MA; Wu Y; Han T; Peng X; Mackey MA; Wang D; Shin HJ; Chen ZG; Xiao H; Wu R; Tang Y; Shin DM; El-Sayed MA, Efficacy, Long-Term Toxicity, and Mechanistic Studies of Gold Nanorods Photothermal Therapy of Cancer in Xenograft Mice. Proc. Natl. Acad. Sci. U S A 2017, 114, E3110–E3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Abadeer N; Murphy C, Recent Progress in Cancer Thermal Therapy Using Gold Nanoparticles. J. Phys. Chem. C 2016, 120, 4691–4716. [Google Scholar]
- (16).Ali MR; Ibrahim IM; Ali HR; Selim SA; El-Sayed MA, Treatment of Natural Mammary Gland Tumors in Canines and Felines Using Gold Nanorods-Assisted Plasmonic Photothermal Therapy to Induce Tumor Apoptosis. Int. J. Nanomedicine 2016, 11, 4849–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dickerson E; Dreaden E; Huang X; El-Sayed I; Chu H; Pushpanketh S; McDonald J; El-Sayed M, Gold Nanorod Assisted Near-Infrared Plasmonic Photothermal Therapy (PPTT) of Squamous Cell Carcinoma in Mice. Cancer Lett 2008, 269, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ali MRK; Wu Y; Tang Y; Xiao H; Chen K; Han T; Fang N; Wu R; El-Sayed MA, Targeting Cancer Cell Integrins Using Gold Nanorods in Photothermal Therapy Inhibits Migration Through Affecting Cytoskeletal Proteins. Proc. Natl. Acad. Sci. U S A 2017, 114, E5655–E5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Clark AG; Vignjevic DM, Modes of Cancer Cell Invasion and the Role of the Microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22. [DOI] [PubMed] [Google Scholar]
- (20).Ilina O; Friedl P, Mechanisms of Collective Cell Migration at a Glance. J. Cell. Sci 2009, 122 (Pt 18), 3203–3208. [DOI] [PubMed] [Google Scholar]
- (21).Ladoux B; Mège RM, Mechanobiology of Collective Cell Behaviours. Nat. Rev. Mol. Cell Biol 2017, 18, 743–757. [DOI] [PubMed] [Google Scholar]
- (22).De Pascalis C; Etienne-Manneville S, Single and Collective Cell Migration: the Mechanics of Adhesions. Mol. Biol. Cell 2017, 28, 1833–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bertocchi C; Vaman Rao M; Zaidel-Bar R, Regulation of Adherens Junction Dynamics by Phosphorylation Switches. J. Signal. Transduct 2012, 2012, 125295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ritchie SM; Battery NH, Phosphorylation and the Cytoskeleton. Springer: 1996. [Google Scholar]
- (25).Ali MRK; Snyder B; El-Sayed MA, Synthesis and Optical Properties of Small Au Nanorods Using a Seedless Growth Technique. Langmuir 2012, 28, 9807–9815. [DOI] [PubMed] [Google Scholar]
- (26).Mackey MA; Ali MR; Austin LA; Near RD; El-Sayed MA, The Most Effective Gold Nanorod Size for Plasmonic Photothermal Therapy: Theory and in Vitro Experiments. J. Phys. Chem. B 2014, 118, 1319–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Prencipe G; Tabakman SM; Welsher K; Liu Z; Goodwin AP; Zhang L; Henry J; Dai H, PEG Branched Polymer for Functionalization of Nanomaterials with Ultralong Blood Circulation. J. Am. Chem. Soc 2009, 131, 4783–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ruoslahti E; Pierschbacher MD, Arg-Gly-Asp: a Versatile Cell Recognition Signal. Cell 44, 517–518. [DOI] [PubMed] [Google Scholar]
- (29).Kim Y-H; Jeon J; Hong SH; Rhim W-K; Lee Y-S; Youn H; Chung J-K; Lee MC; Lee DS; Kang KW; Nam J-M, Tumor Targeting and Imaging Using Cyclic RGDPEGylated Gold Nanoparticle Probes with Directly Conjugated Iodine-125. Small 2011, 7, 2052–2060. [DOI] [PubMed] [Google Scholar]
- (30).Grada A; Otero-Vinas M; Prieto-Castrillo F; Obagi Z; Falanga V, Research Techniques Made Simple: Analysis of Collective Cell Migration Using the Wound Healing Assay. J. Invest. Dermatol 2017, 137, e11–e16. [DOI] [PubMed] [Google Scholar]
- (31).Liang CC; Park AY; Guan JL, In Vitro Scratch Assay: A Convenient and Inexpensive Method for Analysis of Cell Migration in Vitro. Nat. Protoc 2007, 2, 329–333. [DOI] [PubMed] [Google Scholar]
- (32).Sun T; Rodriguez M; Kim L, Glycogen Synthase Kinase 3 in the World of Cell Migration. Dev. Growth Differ 2009, 51, 735–742. [DOI] [PubMed] [Google Scholar]
- (33).Juliano RL, Signal Transduction by Cell Adhesion Receptors and the Cytoskeleton: Functions of Integrins, Cadherins, Selectins, and Immunoglobulin-Superfamily Members. Annu. Rev. Pharmacol. Toxicol 2002, 42, 283–323. [DOI] [PubMed] [Google Scholar]
- (34).Weber GF; Bjerke MA; DeSimone DW, Integrins and Cadherins Join Forces to Form Adhesive Networks. J. Cell Sci 2011, 124 (Pt 8), 1183–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mui KL; Chen CS; Assoian RK, The Mechanical Regulation of Integrin-Cadherin Crosstalk Organizes Cells, Signaling and Forces. J. Cell Sci 2016, 129, 1093–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Yano H; Mazaki Y; Kurokawa K; Hanks SK; Matsuda M; Sabe H, Roles Played by a Subset of Integrin Signaling Molecules in Cadherin-Based Cell-Cell Adhesion. J. Cell Biol 2004, 166, 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Desai R; Sarpal R; Ishiyama N; Pellikka M; Ikura M; Tepass U, Monomeric A-Catenin Links Cadherin to the Actin Cytoskeleton. Nat. Cell Biol 2013, 15, 261–273. [DOI] [PubMed] [Google Scholar]
- (38).Boguslavsky S; Grosheva I; Landau E; Shtutman M; Cohen M; Arnold K; Feinstein E; Geiger B; Bershadsky A, P120 Catenin Regulates Lamellipodial Dynamics and Cell Adhesion in Cooperation with Cortactin. Proc. Natl. Acad. Sci. U S A 2007, 104, 10882–10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Efimov A; Schiefermeier N; Grigoriev I; Ohi R; Brown MC; Turner CE; Small JV; Kaverina I, Paxillin-Dependent Stimulation of Microtubule Catastrophes at Focal Adhesion Sites. J. Cell Sci 2008, 121 (Pt 2), 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kwak TK; Lee MS; Ryu J; Choi YJ; Kang M; Jeong D; Lee JW, Cell Adhesion-Dependent Serine 85 Phosphorylation of Paxillin Modulates Focal Adhesion Formation and Haptotactic Migration via Association with the C-Terminal Tail Domain of Talin. J. Biol. Chem 2012, 287, 27499–27509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Murrell M; Oakes PW; Lenz M; Gardel ML, Forcing Cells into Shape: the Mechanics of Actomyosin Contractility. Nat. Rev. Mol. Cell Biol 2015, 16, 486–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Dulyaninova NG; House RP; Betapudi V; Bresnick AR, Myosin-IIA Heavy-Chain Phosphorylation Regulates the Motility of MDA-MB-231 Carcinoma Cells. Mol. Biol. Cell 2007, 18, 3144–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zagórska A; Deak M; Campbell DG; Banerjee S; Hirano M; Aizawa S; Prescott AR; Alessi DR, New Roles for the LKB1-NUAK Pathway in Controlling Myosin Phosphatase Complexes and Cell Adhesion. Sci. Signal 2010, 3, ra25. [DOI] [PubMed] [Google Scholar]
- (44).Wang YT; Tsai CF; Hong TC; Tsou CC; Lin PY; Pan SH; Hong TM; Yang PC; Sung TY; Hsu WL; Chen YJ, An Informatics-Assisted Label-Free Quantitation Strategy that Depicts Phosphoproteomic Profiles in Lung Cancer Cell Invasion. J Proteome Res 2010, 9, 5582–5597. [DOI] [PubMed] [Google Scholar]
- (45).Kitazawa H; Iida J; Uchida A; Haino-Fukushima K; Itoh TJ; Hotani H; Ookata K; Murofushi H; Bulinski JC; Kishimoto T; Hisanaga S, Ser787 in the Proline-Rich Region of Human MAP4 is a Critical Phosphorylation Site that Reduces its Activity to Promote Tubulin Polymerization. Cell Struct. Funct 2000, 25, 33–39. [DOI] [PubMed] [Google Scholar]
- (46).Hartsock A; Nelson WJ, Adherens and Tight Junctions: Structure, Function and Connections to the Actin Cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ji H; Wang J; Nika H; Hawke D; Keezer S; Ge Q; Fang B; Fang X; Fang D; Litchfield DW; Aldape K; Lu Z, EGF-Induced ERK Activation Promotes CK2-Mediated Disassociation of Alpha-Catenin from Beta-Catenin and Transactivation of Beta-Catenin. Mol. Cell 2009, 36, 547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zihni C; Mills C; Matter K; Balda MS, Tight Junctions: from Simple Barriers to Multifunctional Molecular Gates. Nat. Rev. Mol. Cell Biol 2016, 17, 564–580. [DOI] [PubMed] [Google Scholar]
- (49).Mendez MG; Kojima S; Goldman RD, Vimentin Induces Changes in Cell Shape, Motility, and Adhesion During the Epithelial to Mesenchymal Transition. FASEB J 2010, 24, 1838–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Li QF; Spinelli AM; Wang R; Anfinogenova Y; Singer HA; Tang DD, Critical Role of Vimentin Phosphorylation at Ser-56 by P21-Activated Kinase in Vimentin Cytoskeleton Signaling. J. Biol. Chem 2006, 281, 34716–34724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Flitney EW; Kuczmarski ER; Adam SA; Goldman RD, Insights into the Mechanical Properties of Epithelial Cells: The Effects of Shear Stress on the Assembly and Remodeling of Keratin Intermediate Filaments. FASEB J 2009, 23, 2110–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Kakade PS; Budnar S; Kalraiya RD; Vaidya MM, Functional Implications of O-Glcnacylation-Dependent Phosphorylation at a Proximal Site on Keratin 18. J. Biol. Chem 2016, 291, 12003–12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ku NO; Michie S; Resurreccion EZ; Broome RL; Omary MB, Keratin Binding to 14–3-3 Proteins Modulates Keratin Filaments and Hepatocyte Mitotic Progression. Proc. Natl. Acad. Sci. U S A 2002, 99, 4373–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Stumptner C; Omary MB; Fickert P; Denk H; Zatloukal K, Hepatocyte Cytokeratins are Hyperphosphorylated at Multiple Sites in Human Alcoholic Hepatitis and in a Mallory Body Mouse Model. Am. J. Pathol 2000, 156, 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Pandya P; Orgaz JL; Sanz-Moreno V, Actomyosin Contractility and Collective Migration: May the Force be With You. Curr. Opin. Cell Biol 2017, 48, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Mège RM; Gavard J; Lambert M, Regulation of Cell-Cell Junctions by the Cytoskeleton. Curr. Opin. Cell Biol 2006, 18, 541–548. [DOI] [PubMed] [Google Scholar]
- (57).Hazan RB; Kang L; Whooley BP; Borgen PI, N-Cadherin Promotes Adhesion Between Invasive Breast Cancer Cells and the Stroma. Cell Adhes. Commun 1997, 4, 399–411. [DOI] [PubMed] [Google Scholar]
- (58).Hazan RB; Phillips GR; Qiao RF; Norton L; Aaronson SA, Exogenous Expression of N-Cadherin in Breast Cancer Cells Induces Cell Migration, Invasion, and Metastasis. J. Cell Biol 2000, 148, 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Shih W; Yamada S, N-Cadherin-Mediated Cell-Cell Adhesion Promotes Cell Migration in a Three-Dimensional Matrix. J. Cell Sci 2012, 125, 3661–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Shih W; Yamada S, N-Cadherin as a Key Regulator of Collective Cell Migration in a 3D Environment. Cell Adh. Migr 2012, 6, 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Martin TA; Jiang WG, Tight Junctions and Their Role in Cancer Metastasis. Histol. Histopathol 2001, 16, 1183–1195. [DOI] [PubMed] [Google Scholar]
- (62).Singh AB; Sharma A; Dhawan P, Claudin Family of Proteins and Cancer: an Overview. J. Oncol 2010, 2010, 541957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mandicourt G; Iden S; Ebnet K; Aurrand-Lions M; Imhof BA, JAM-C Regulates Tight Junctions and Integrin-Mediated Cell Adhesion and Migration. J. Biol. Chem 2007, 282, 1830–1837. [DOI] [PubMed] [Google Scholar]
- (64).Karagiannis GS; Schaeffer DF; Cho CK; Musrap N; Saraon P; Batruch I; Grin A; Mitrovic B; Kirsch R; Riddell RH; Diamandis EP, Collective Migration of Cancer-Associated Fibroblasts is Enhanced by Overexpression of Tight Junction-Associated Proteins Claudin-11 and Occludin. Mol. Oncol 2014, 8, 178–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).González-Tarragó V; Elosegui-Artola A; Bazellières E; Oria R; Pérez-González C; Roca-Cusachs P, Binding of ZO-1 to α5β1 Integrins Regulates the Mechanical Properties of A5β1-Fibronectin Links. Mol. Biol. Cell 2017, 28, 1847–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Tuomi S; Mai A; Nevo J; Laine JO; Vilkki V; Ohman TJ; Gahmberg CG; Parker PJ; Ivaska J, Pkcepsilon Regulation of an Alpha5 Integrin-ZO-1 Complex Controls Lamellae Formation in Migrating Cancer Cells. Sci. Signal 2009, 2, ra32. [DOI] [PubMed] [Google Scholar]
- (67).Fanning AS; Van Itallie CM; Anderson JM, Zonula Occludens-1 and −2 Regulate Apical Cell Structure and the Zonula Adherens Cytoskeleton in Polarized Epithelia. Mol. Biol. Cell 2012, 23, 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Bhattacharya S; Ahir M; Patra P; Mukherjee S; Ghosh S; Mazumdar M; Chattopadhyay S; Das T; Chattopadhyay D; Adhikary A, Pegylated-Thymoquinone-Nanoparticle Mediated Retardation of Breast Cancer Cell Migration by Deregulation of Cytoskeletal Actin Polymerization Through Mir-34a. Biomaterials 2015, 51, 91–107. [DOI] [PubMed] [Google Scholar]
- (69).Zhang Y; Hu L; Yu D; Gao C, Influence of Silica Particle Internalization on Adhesion and Migration of Human Dermal Fibroblasts. Biomaterials 2010, 31, 8465–8474. [DOI] [PubMed] [Google Scholar]
- (70).Ali MR; Ali HR; Rankin CR; El-Sayed MA, Targeting Heat Shock Protein 70 Using Gold Nanorods Enhances Cancer Cell Apoptosis in Low Dose Plasmonic Photothermal Therapy. Biomaterials 2016, 102, 1–8. [DOI] [PubMed] [Google Scholar]
- (71).Barriga EH; Franze K; Charras G; Mayor R, Tissue Stiffening Coordinates Morphogenesis by Triggering Collective Cell Migration in vivo. Nature 2018, 554, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Cai D; Dai W; Prasad M; Luo J; Gov NS; Montell DJ, Modeling and Analysis of Collective Cell Migration in an in vivo Three-Dimensional Environment. Proc. Natl. Acad. Sci. U S A 2016, 113, E2134–E2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Trepat X; Wasserman M; Angelini T; Millet E; Weitz D; Butler J; Fredberg J, Physical Forces During Collective Cell Migration. Nat. Phys 2009, 5, 426–430. [Google Scholar]
- (74).Vedula SR; Leong MC; Lai TL; Hersen P; Kabla AJ; Lim CT; Ladoux B, Emerging Modes of Collective Cell Migration Induced by Geometrical Constraints. Proc. Natl. Acad. Sci. U S A 2012, 109, 12974–12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Kuriyama S; Yoshida M; Yano S; Aiba N; Kohno T; Minamiya Y; Goto A; Tanaka M, LPP Inhibits Collective Cell Migration During Lung Cancer Dissemination. Oncogene 2016, 35, 952–964. [DOI] [PubMed] [Google Scholar]
- (76).Zhang D; Feng F; Li Q; Wang X; Yao L, Nanopurpurin-Based Photodynamic Therapy Destructs Extracellular Matrix Against Intractable Tumor Metastasis. Biomaterials 2018, 173, 22–33. [DOI] [PubMed] [Google Scholar]
- (77).Choudhary C; Kumar C; Gnad F; Nielsen ML; Rehman M; Walther TC; Olsen JV; Mann M, Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [DOI] [PubMed] [Google Scholar]
- (78).Boersema PJ; Raijmakers R; Lemeer S; Mohammed S; Heck AJ, Multiplex Peptide Stable Isotope Dimethyl Labeling for Quantitative Proteomics. Nat. Protoc 2009, 4, 484–494. [DOI] [PubMed] [Google Scholar]
- (79).Wu Y; Wang FJ; Liu ZY; Qin HQ; Song CX; Huang JF; Bian YY; Wei XL; Dong J; Zou HF, Five-Plex Isotope Dimethyl Labeling for Quantitative Proteomics. ChemComm 2014, 50, 1708–1710. [DOI] [PubMed] [Google Scholar]
- (80).Mecham BH; Nelson PS; Storey JD, Supervised Normalization of Microarrays. Bioinformatics 2010, 26, 1308–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.