

Summary
Growth factors induce and pattern sensory organs, but how their distribution is regulated by the extracellular matrix (ECM) is largely unclear. To address this question, we analyzed the diffusion behavior of Fgf10 molecules during sensory organ formation in the zebrafish posterior lateral line primordium. In this tissue, secreted Fgf10 induces organ formation at a distance from its source. We find that most Fgf10 molecules are highly diffusive and move rapidly through the ECM. We identify Anosmin1, which when mutated in humans causes Kallmann Syndrome, as an ECM protein that binds to Fgf10 and facilitates its diffusivity by increasing the pool of fast moving Fgf10 molecules. In the absence of Anosmin1, Fgf10 levels are reduced and organ formation is impaired. Global overexpression of Anosmin1 slows the fast moving Fgf10 molecules and results in Fgf10 dispersal. These results suggest that Anosmin1 liberates ECM-bound Fgf10 and shuttles it to increase its signaling range.
Introduction
During development tissues need to be patterned to give rise to different cell types. On a molecular level this is often achieved through graded signals that induce distinct cell identities at different concentrations. The graded distribution is frequently the result of locally produced signaling molecules with finite half-lives that diffuse through the extracellular space to establish concentration gradients (Kiecker and Lumsden, 2012). While our understanding of patterning signals is advanced, little is known about the mechanisms employed by the components of the extracellular matrix (ECM) to regulate the movement of individual signaling molecules and, thus, their local concentrations.
Fibroblast growth factors (FGFs) are a family of secreted signaling molecules, most of which bind to heparan sulfate (HS) in the ECM (Goetz and Mohammadi, 2013). During development, FGFs guide migrating cells, direct tissue patterning, induce cell proliferation and regulate cell differentiation (Turner and Grose, 2010). In order to accomplish these tasks, the levels and distribution of FGFs and, thus, FGF’s mobility in the extracellular space need to be tightly regulated.
FGF signaling in humans and animals is modulated by the extracellular matrix protein ANOS1 (Anosmin-1) (de Castro et al., 2013). ANOS1 contains a whey acidic protein (WAP)-like domain and four fibronectin type III (FN[III]) domains and is often coexpressed with FGF ligands in the embryo. Human patients with mutations in ANOS1 are anosmic and fail to undergo puberty due to erroneous olfactory axon targeting and GnRH neuron migration (Hardelin and Dodé, 2008). Similarly, animals lacking Anos1 show reduced neurite branching and cranial defects (Rugarli et al., 2002; Tecle et al., 2013; Yamada et al., 2012). In both humans and animals, these defects have been attributed to reduced FGF signaling due to the lack of Anos1-mediated FGF-FGF receptor complex formation based on in vitro studies (Díaz-Balzac et al., 2015; Hu et al., 2009; Yamada et al., 2012). Yet, how Anos1 regulates Fgf signaling in vivo remains largely elusive.
The development of the zebrafish posterior lateral line depends on Fgf signaling. The posterior lateral line is formed by a migrating primordium of about 140 cells that deposits five to seven cell clusters along the body of the embryo during its migration (Nogare and Chitnis, 2017). Each of these clusters differentiates into a neuromast, a specialized organ that senses water flow around the embryo. The rosette-shaped neuromasts start to differentiate while in the primordium and bud off from the rear of the primordium at regularly spaced intervals. The cells in the front of the primordium coexpress the FGF ligands fgf3 and fgf10a whereas the cells in the back of the primordium express the FGF receptor fgfr1a (Lecaudey et al., 2008; Matsuda and Chitnis, 2010; Nechiporuk and Raible, 2008). It is thought that Fgf secreted from the front of the primordium induces neuromast differentiation in the back of the primordium by diffusing to and concentrating in small, sealed-off extracellular spaces (microlumina) in the center of the forming neuromasts through an unknown mechanism (Durdu et al., 2014).
We used posterior lateral line development as a model system to examine the role of Anos1 in Fgf signaling. Our results indicate that Anos1 regulates the range of Fgf signaling in the primordium by promoting the diffusion of Fgf10a. Most Fgf10a molecules freely diffuse through the extracellular space but a small fraction is severely retarded, probably because it is bound to heparin sulfates in the ECM. We found that Anos1 binds to and co-diffuses with Fgf10a and thus increases the number of freely diffusing Fgf10a molecules. As a result, more Fgf10a produced in the front of the primordium diffuses to the rear, where it accumulates locally, induces neuromast formation and becomes trapped in the microlumina of these sensory organs. These results identify a role for ANOS1 as a modulator of the diffusion range of FGF in the extracellular matrix.
Results
Front-to-rear FGF diffusion promotes sensory organ formation
Sensory organ differentiation in the primordium depends on FGF signaling. Cells in the front of the primordium express the FGF ligands fgf3 and fgf10a. Additionally, one to two central cells in each forming neuromast express fgf10a (Lecaudey et al., 2008; Matsuda and Chitnis, 2010; Nechiporuk and Raible, 2008) (Fig. 1A, S1A). The FGF receptors fgfr1a and fgfr1b are expressed in the rear of the primordium (Fig. 1A, S1A) (Lecaudey et al., 2008; Nechiporuk and Raible, 2008). Loss of Fgf3 and Fgf10a function in the primordium results in loss of FGF signaling, a failure to form microlumina, absence of neuromasts and slower primordium migration (Fig. 1B, S1D) (Durdu et al., 2014; Lecaudey et al., 2008; Matsuda and Chitnis, 2010; Nechiporuk and Raible, 2008). Partial block of FGF signaling by reducing Fgf receptor activity or inducing the expression of a dominant-negative FGF receptor results in the formation of fewer neuromasts (Fig. 1B, D, Movie 1). Conversely, increasing FGF signaling by over-expressing Fgf10a results in the formation of extra neuromasts (Fig. 1B, D, Movie 1). Consistent with previous studies (Aman and Piotrowski, 2008; Durdu et al., 2014; Lecaudey et al., 2008; Matsuda and Chitnis, 2010; Nechiporuk and Raible, 2008), this indicates that FGF signaling induces neuromast formation in a dose-dependent manner.
Figure 1. Fgf ligand and receptor expression and function in sensory organ formation.
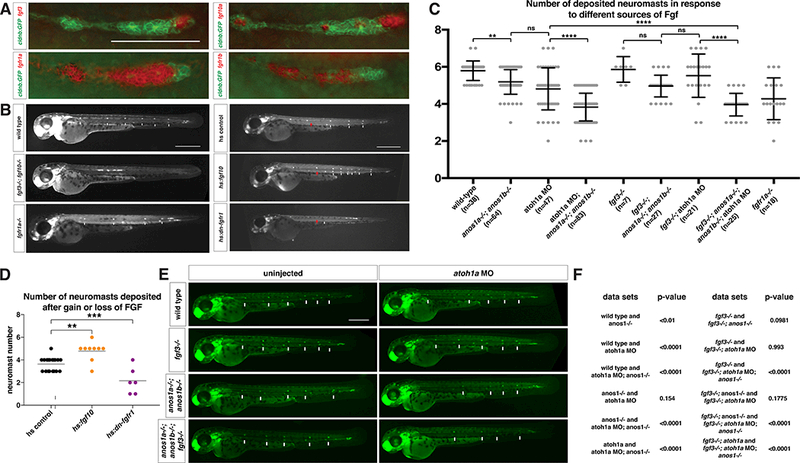
(A) fgf3, fgf10a, fgfr1a, and fgfr1b mRNA expression (red) in the primordium (green) at 32 hpf. Scale bar = 100 μm.
(B) Primordium position in embryos of indicated genotypes at 50 hpf. White arrows denote deposited neuromasts, arrowheads denote position of primordium and red arrows indicate position of primordium at the beginning of the heat shock. Scale bar = 500 μm.
(C) Plot of number of neuromasts for E and B. ** = p < 0.01, **** = p < 0.0001, ns = not significant. ANOVA p<0.0001.
(D) Quantification of the number of neuromasts deposited after heat-shock in embryos of the indicated genotypes. Black lines indicate the average. Each data point is an individual embryo. ** = p < 0.01, *** = p < 0.001. ANOVA p<0.0001.
(E) Neuromast deposition (arrows) and primordium migration (arrow head) in embryos of indicated genotype at 50 hpf. Scale bar = 500 μm.
(F) Tables of unpaired t-test with Welsh’s correction for pairwise comparisons of the indicated genotypes shown in F.
Due to the opposing expression patterns of the FGF ligands and receptors, it is thought that FGF diffuses from the front of the primordium to signal to cells in the rear of the primordium (Lecaudey et al., 2008; Nechiporuk and Raible, 2008). To directly test this idea, we asked whether FGF ligand from the front of the primordium is sufficient to induce sensory organ differentiation in the rear of the primordium. We inhibited fgf10a expression in the central cells by reducing the activity of the transcriptional factor atoh1a. This blocks the differentiation of the central cells (Fig. 2B) (Lecaudey et al., 2008; Matsuda and Chitnis, 2010). Loss of fgf10a expression in central cells was confirmed by in situ hybridization against fgf10a (Fig. S1B, S1C). We also removed the activity of Fgf3 because the central cells compensate for the loss of Fgf10a by up-regulating fgf3 expression (Matsuda and Chitnis, 2010). In these fgf3−/−; atoh1a MO embryos, sensory organs still differentiated similar to wild-type, fgf3 mutant or atoh1a MO-injected embryos (Fig. 1C, E). Thus, Fgf10a from the front diffuses to the rear to promote neuromast formation.
Figure 2.
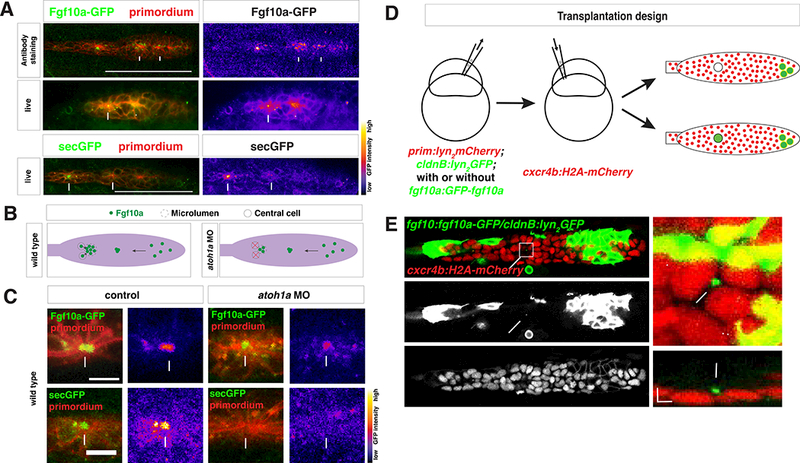
Secreted Fgf10a from the front diffuses to the back of the primordium.
(A) Top left, immunostaining of intracellular Fgf10a-GFP in the primordium. Top right, Fgf10a-GFP only. Arrows indicate intracellular Fgf10a-GFP accumulating at the apical constrictions near the midline of the primordium. Middle and lower left, live image of Fgf10a-GFP and secGFP, respectively, with membrane marker. Middle and lower right, false coloring of Fgf10a-GFP and secGFP signal, respectively. Arrows indicates Fgf10a- GFP and secGFP signal in microlumen and arrowhead indicates Fgf10a-GFP in a patch surrounding an apical constriction before microlumen formation. Scale bar = 100 μm.
(B) Schematic of Fgf10a protein production and transport into the microlumina in the primordium.
(C) Live images of microluminal Fgf10a-GFP with membrane marker and false coloring of Fgf10a-GFP signal only in embryos of indicated genotype. Arrows indicate Fgf10a- GFP signal in microlumina and arrowheads (left) indicate Fgf10a-GFP-producing central cells adjacent to the microlumen. Central cells are missing in embryos injected with atoh1a morpholino (right). Scale bar = 10 μm.
(D) Schematic representation of the mosaic analysis.
(E) Overview of mosaic primordium with cells of indicated genotypes on left. Square indicates enlarged region shown on right. Single slice of a Z-stack from Movie 3 is shown. Scale bar 25 μm. Close-up of forming neuromast on right. Maximum x,yprojection of Z-stack shown in Movie 3 on top right and maximum x,z-projection of Zstack shown in Movie 3 on bottom right. Scale bar 5 μm for x,y dimension and 10 μm for z dimension. Arrows indicate Fgf10a-GFP in microlumen, arrowhead indicates donorderived lateral line nerve underneath the primordium and double arrowhead indicates donor-derived skin cell on top of primordium. 3 primordia with Fgf10a-GFP secreting cells in the front and 5 primordia with non-Fgf10a-GFP secreting cells in the front were analyzed.
See also Figure S2 and Movie 2 and 3
We next assessed the distribution of Fgf10a in the primordium by expressing Fgf10a-GFP from a BAC transgene encompassing the fgf10a chromosomal locus (fgf10a:GFP-fgf10a) (Fig. S2A). This transgene recapitulated the endogenous fgf10a expression pattern and rescued primordium migration and neuromast formation in fgf3−/−; fgf10a−/− embryos (Fig. S2); fgf10a−/− embryos do not show a defect in primordium migration and neuromast formation due to compensation through Fgf3 up-regulation (Lecaudey et al., 2008). As a control, we generated an identical transgene that expresses secreted GFP (secGFP) from the fgf10a locus (fgf10a:secGFP, Fig. S2A). We found that Fgf10a-GFP was secreted apically from the front cells towards the midline of the primordium and concentrated above forming apical constrictions and in the microlumina of the maturing neuromasts (Fig. 2A, Movie 2). SecGFP also accumulated in microlumina of control embryos (Fig. 2A, Movie 2).
To determine the contribution of the front source and central cell source to Fgf10a in the microlumina, we blocked central cell formation. We found that in embryos lacking central cells, the accumulation of Fgf10a-GFP in microlumina was reduced to a third (Fig. 2B, 2C, S2E). In controls, secGFP accumulation in microlumina was completely abolished since GFP secreted from the front is not trapped (Fig. 2C). Using ubiquitously secreted mCherry, we confirmed that embryos with reduced Atoh1a function had intact microlumina (Fig. S4D). To confirm that Fgf10a-GFP produced at a distance can diffuse to and concentrate in microlumina, we used blastomere transplantation to generate mosaic primordia that contain Fgf10a-GFP-secreting cells in the front but not in a forming or deposited neuromast where central cells are located (Fig. 2E). In such mosaic primordia, Fgf10a-GFP from the front also accumulated in microlumina (Fig. 2E, Movie 3). Thus, luminal Fgf10a originates from two different sources—one source is the front of the primordium, where Fgf10a is secreted, diffuses to the rear, and becomes enclosed in microlumina, and the second source is the central cells, where Fgf10a is secreted directly into the adjacent microlumen.
The extracellular matrix protein Anosmin1 enhances FGF signaling
The extracellular protein ANOS1 modulates FGF signaling in many contexts (de Castro et al., 2013). In zebrafish, there are two ANOS1 paralogs, anos1a and anos1b (jointly referred to as anos1). Among other tissues, anos1a and anos1b are expressed in the migrating primordium (Ardouin et al., 2000; Yanicostas et al., 2008) (Fig. 3B, S3A), indicating that Anos1 could play a role in regulating FGF signaling during sensory organ formation in the primordium. To test this idea, we generated anos1 loss of function mutant alleles (Fig. S3D). These alleles disrupt a critical disulfide bond, truncate the protein, and exhibit mRNA non-sense mediated decay and thus are likely genetic null mutants (Fig. S3B). We assessed FGF signaling in anos1a; anos1b double mutant primordia in several ways.
Figure 3.
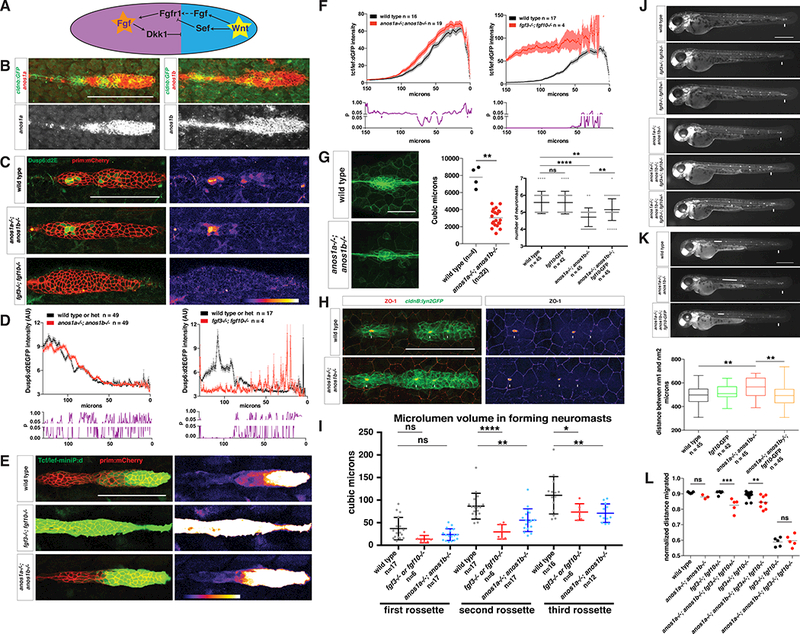
Anos1 enhances FGF signaling in the primordium.
(A) Schematic of the cross-repression between Wnt and FGF signaling in the primordium.
(B) Top, anos1a and anos1b mRNA expression in the primordium outlined by membrane GFP. Bottom, anos1a and anos1b mRNA expression only (white). Scale bar = 100 μm.
(C) dusp6:d2eGFP expression in the primordium and dusp6:d2eGFP expression only (fire heat map) for the genotypes indicated. Scale bar = 100 μm.
(D) Top, quantification of GFP intensity from dusp6:d2EGFP inside the primordium along the anterior-posterior axis. X-axis represents distance from the front of the primordium. Mean and SEM are shown. n indicates number of embryos. Bottom, plot of p values comparing the two genotypes in the graph above for each position along the X-axis.
(E) Wnt reporter readout at 36 hpf in the primordium on the left and Wnt reporter readout only (fire heatmap below). Scale bar = 100 μm.
(F) Quantification of Wnt reporter intensity. X-axis represents distance from the front of the primordium. Mean and SEM and are shown. n = number of individual embryos measured.
(G) Neuromast size and number defects in anos1 mutants. Neuromast 1 in embryos of the indicated genotype at 32 hpf (left). Scale bar = 50 μm. Quantification of neuromast 1 volume in embryos of indicated genotype at 32 hpf (middle). Horizontal lines are the mean and each data point is an individual embryo. Neuromast number in 42 hpf embryos of indicated genotype (right). Mean, SD and individual data points are shown. n.s.= p > 0.05, ** = p < 0.01., and **** = p < 0.0001. ANOVA p<0.0001.
(H) (Left) ZO-1 and GFP Immunostaining in the primordium of embryos of the indicated genotype at 36 hpf. (Right) ZO-1 only (fire look-up table). Arrows indicate ZO-1 signal at the center of apical constrictions. Scale bar = 100 μm.
(I) Quantification of microlumen volumes in forming neuromasts in embryos of indicated genotypes. Mean, SD and individual data points are shown. n.s.= p > or = 0.05, * = p < 0.05., ** = p < 0.01. and **** = p < 0.0001. ANOVA p<0.0001.
(J) Phenotype of embryos with reduced Fgf signaling with or without mutations in anos1a and anos1b at 50 hpf. Arrow indicates position of the primordium.
(K) Primordium and neuromast position in embryos of indicated genotypes at 50 hpf (left). Arrow denotes position of primordium. Double-headed arrow indicates spacing between neuromast 1 and 2. Scale bar = 500 μm. Box and whisker plot of the distance between neuromast 1 and 2 for the genotypes indicated (right). n is the number of individual embryos. ns = p >0.05, ** = p < 0.01. ANOVA p<0.01.
(L) Quantification of normalized distance migrated for the genotypes indicated. Mean, SD and individual data points are shown. n.s. = p > 0.05, ** = p < 0.01, *** = p < 0.001. ANOVA p<0.0001.
See also Figure S3.
First, we used the FGF signaling reporter dusp6:d2EGFP. Consistent with previous FGF gain and loss of function studies in other tissues (Molina et al., 2007), we found that the GFP fluorescence from the FGF reporter is absent in fgf3−/−; fgf10a−/− mutant primordia and more than doubled in fgf10a over-expressing primordia (Fig. 3C, 3D, 7E, 7F, S3H). This suggests that this transgene also reports FGF signaling in the primordium. Quantification of the FGF reporter fluorescence in anos1 mutant primordia showed mild reduction of FGF signaling in the rear of the primordium compared to wildtype embryos (Fig. 3C, D, S3H).
Figure 7.
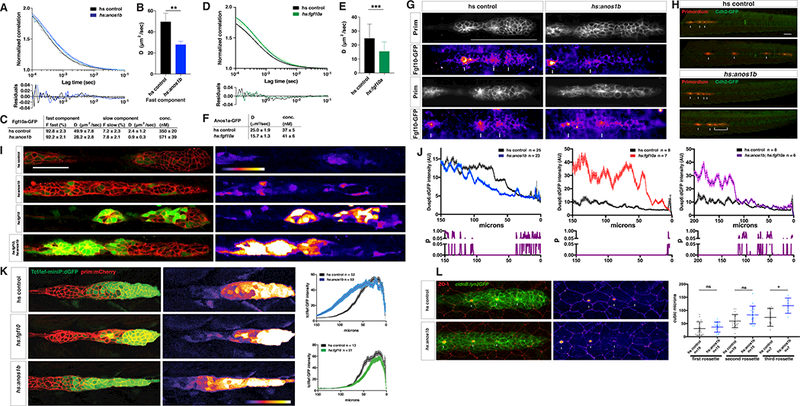
Anos1 over-expression blocks Fgf10a accumulation and signaling.
(A) Top, normalized autocorrelation curves (gray and light blue) and 2-component fits (black and blue) for Fgf10a-GFP in the indicated genotypes. Bottom, residuals of the fits.
(B) Bar graph of the diffusion coefficient of the fast component in the genotypes indicated. Error bars represent SD. ** = p < 0.01.
(C) Table of fitted values. F represents proportion of molecules in each component and D is the diffusion coefficient.
(D) Top, normalized autocorrelation curves (gray and light green) and 1-component fits (black and green) for Anos1a-GFP in the indicated genotypes. Bottom, residuals of the fits.
(E) Bar graph of the diffusion coefficient in the genotypes indicated. Error bars represent SD. *** = p < 0.001.
(F) Table of fitted diffusion coefficients (D).
(G) Maximum intensity projection of the apical half of two heat-shocked control (left) and two heat-shocked hsp70:anos1b (right) live embryos transgenic for prim:lyn2mCherry and fgf10a:GFP-fgf10. Prim panel shows membrane outline of the primordium. Arrows in Fgf10a-GFP panels indicate patches of extracellular Fgf10a-GFP surrounding apical constrictions. These patches are present in 10 out of 10 heat-shocked control embryos, but absent or strongly reduced in 5 out of 8 heat-shocked hsp70:anos1b embryos (top right) and slightly reduced in the remaining 3 out of 8 heat-shocked hsp70:anos1b embryos (bottom right). Warmer colors represent higher GFP fluorescence intensities. Scale bar = 100 μm.
(H) Maximum projections of Z-stacks of primordia in embryos of indicated genotype one hour and ten hours after heat shock. Images are stills from Movie 7.
(I) dusp6:d2EGFP expression (green) in the primordium (red) and dusp6:d2EGFP expression alone (fire heat map) for the genotypes indicated. Scale bar = 50 μm. Fire heat map scale is shown below, with warmer colors indicating higher GFP fluorescence intensity.
(J) Top, quantification of GFP intensity from dusp6:d2EGFP in the primordium along the anterior-posterior axis. X-axis represents distance from the front of the primordium. Mean and SEM shown. n = number of embryos. Bottom, plot of p values comparing the two genotypes in the graph above for each position along the X-axis.
(K) Left, Wnt reporter readout at 36 hpf in the primordium of heat-shocked embryos of the indicated genotypes. Middle, Wnt reporter readout only. Scale bar = 100 μm. Right, quantification of Wnt reporter intensity. X-axis represents distance from the front of the primordium. Mean and SEM shown. n = number of embryos.
(L) Left, immunostaining against ZO-1 and GFP in the primordium of 36 hpf embryos. Images are maximum projections. Middle, ZO-1 only (fire look-up table). Arrows indicate ZO-1 signal at the center of apical constrictions. Scale bar = 100 μm. Right, quantification of microlumen volumes in forming neuromasts. Horizontal lines are the mean, vertical lines indicate the SD and each data point is an individual embryo. n.s.= p > or = 0.05, * = p < 0.05. ANOVA p<0.0001.
See also Figure S7 and Movie 6 to 8.
Second, we assessed FGF signaling through its effect on Wnt signaling. FGF signaling in the rear and Wnt signaling in the front of the primordium cross-repress each other such that reduced FGF signaling in the rear results in an expansion of the Wnt signaling domain in the front of the primordium (Fig. 3A) (Aman and Piotrowski, 2008). To quantify Wnt signaling in the primordium, we used the Wnt reporter Tcf/LefminiP:dGFP (Shimizu et al., 2012). The Wnt reporter responded in a graded manner to FGF levels. The activation domain expanded from the front towards the rear of the primordium with decreasing fgf3 and fgf10a gene copy numbers until it was fully activated in the entire primordium in fgf3−/−; fgf10a−/− double mutant embryos (Fig. 3E, F, S3E). The activation domain contracted to the first 50 μm of the front of the primordium in Fgf10a over-expressing embryos (Fig. 7K). Using this reporter, we found that the Wnt signaling domain expanded about 10 mm from the front towards the rear of the primordium in anos1 mutant primordia compared to wild-type embryos (Fig. 3E, F). This is similar to fgf10a mutants (Fig. S3E).
Third, we assessed Fgf signaling through sensory organ size and deposition, both of which depend on FGF signaling. Reduced FGF signaling leads to smaller and fewer neuromasts with delayed deposition while increased FGF signaling results in larger and more neuromasts with more frequent deposition (Fig. 1B, D) (Durdu et al., 2014; Lecaudey et al., 2008; Nechiporuk and Raible, 2008). We found that neuromasts are smaller in anos1 mutant embryos than in wild-type embryos (Fig. 3G). We also found that microlumina are smaller in anos1 mutant embryos, similar to fgf3 and fgf10a mutant embryos, and that their formation is slightly delayed (Fig. 3H, I). Finally, the spacing between the initial neuromasts was increased and frequently one neuromast was missing (Fig. 3G, K, S3C) while primordium migration and body size were only mildly affected in anos1 mutant embryos compared to wild-type embryos (Fig. S3F). This is in contrast to anos1a morpholino knockdown experiments, in which the primordium stalled (Yanicostas et al., 2008). Since anos1a and anos1b single mutants did not display a primordium migration defect (Fig. S3C) and anos1a−/−; anos1b−/− double mutants showed only a mild defect (Fig. 3L, S3F, Movie 4), this discrepancy is probably due to the error-prone nature of morpholino-mediated knockdowns (Kok et al., 2015) rather than the absence of genetic compensation in morpholino-mediated knockdown (Rossi et al., 2015).
Next, we asked if anos1 genetically interacts with fgf3 and fgf10. We found that the mild primordium migration defect in anos1a−/−; anos1b−/− double mutant embryos was enhanced when we further lowered FGF signaling by also reducing the copy numbers of fgf3 and fgf10a. This frequently caused the primordium to stall prematurely (Fig. 3J, L). However, loss of Anos1 did not enhance the migration phenotype in embryos completely lacking FGF signaling (fgf3−/−; fgf10a−/−; anos1a−/−; anos1b−/− quadruple mutants, Fig. 3L), suggesting that Anos1 acts solely through FGF in this context. Importantly, the delayed deposition of the initial neuromasts and neuromast number in anos1 mutant embryos was rescued when we added an extra copy of fgf10a from a transgene (Fig. 3G, K). In wild-type embryos, adding an additional copy of fgf10a did not affect neuromast size, number or deposition frequency, probably because FGF levels were not raised sufficiently to induce an FGF over-expression phenotype (Fig. S3G). Consistent with the olfactory axon migration defect in humans with Kallman Syndrome (Schwanzel-Fukuda et al., 1989; Shetty, 2015), we also observed mismigrated olfactory axons in anos1 mutants (Fig. S3I, S3J).
Together these data suggest that Anos1 enhances FGF signaling in the primordium to promote neuromast formation.
Anos1 is required for luminal accumulation of FGF from the front of the primordium
Our observations indicate that Anos1 enhances FGF signaling in the rear of the primordium. As indicated by previous reports (Díaz-Balzac et al., 2015; Hu et al., 2009; Yamada et al., 2012), Anos1 could enhance FGF signaling by facilitating the formation of the FGF ligand-receptor complex. Alternatively, Anos1 could affect how much FGF ligand reaches the rear of the primordium. Using a BAC transgene that encompasses the anos1a chromosomal locus (anos1a:GFP-anos1a) (Fig. S4A), we found that Anos1a is expressed throughout the primordium and is concentrated in the microlumina of neuromasts (Fig. 4A, S4B, C, Movie 5). This localization pattern is consistent with both the ideas that Anos1 interacts with the FGF receptors or regulates the accumulation of FGF ligand in the rear.
Figure 4.
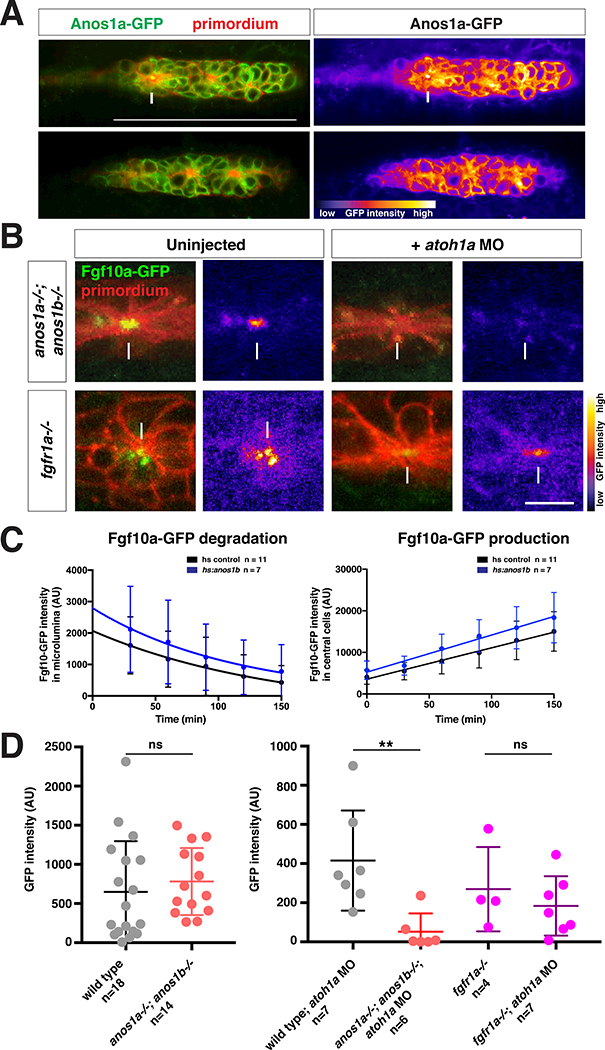
Anos1 is required for luminal accumulation of FGF10a.
(A) Left, Anos1a-GFP protein (green) in the primordium marked with prim:lyn2mCherry (red). Right, Anos1a-GFP only in false colors. Top, arrow indicates Anos1a-GFP in microlumen. Bottom, arrowhead indicates Anos1a-GFP signal above apical constrictions.
(B) Live images of microluminal Fgf10a-GFP with membrane marker and false coloring of Fgf10a-GFP signal only in embryos of indicated genotypes. Arrows indicate Fgf10a- GFP signal in microlumina and arrowheads (left) indicate Fgf10a-GFP-producing central cells adjacent to the microlumen. Central cells are missing in embryos injected with atoh1a morpholino (right). Scale bar = 10 μm.
(C) Total Fgf10a-GFP fluorescence intensity in the microlumen (left) and central cells (right) when secretion is blocked with Brefeldin A. Error bars indicate the SD. Fgf10a- GFP degradation was fitted to a one-phase decay model with half life values of 102 min (95% CI 33 min to 197 min) for heat-shock control embryos and 79 min (95% CI 25 min to 157 min) for anos1b-over-expressing embryos. Fgf10a-GFP production was fitted to a linear model with a production rate of 75.2 ± 8.2 min−1 for heat-shock control embryos and a production rate of 89.2 ± 11.8 min−1 for anos1b-over-expressing embryos.
(D) Total Fgf10a-GFP intensity in mature microlumina at the fourth apical constriction from the front in uninjected embryos (left) and embryos injected with atoh1a morpholino (right). Mean, SD and individual data points are shown. n.s. = p > 0.05, ** = p < 0.01. ANOVA p<0.05.
See also Figure S4 and Movie 4 and 5.
To distinguish between these possibilities, we first asked whether Anos1 activity is required to accumulate FGF from the front of the primordium in the microlumina in the primordium’s rear. For this, we blocked fgf10a expression in the central cells by reducing Atoh1a activity (Fig. 2C arrowheads) - such that the only Fgf10a source is the Fgf10a secreted from the front of the primordium - and analyzed the distribution of Fgf10a-GFP in wild-type or anos1 mutant embryos lacking Atoh1a activity. In contrast to wild-type embryos in which Fgf10a-GFP from the front still accumulates in microlumina (Fig. 2C), Fgf10a-GFP fails to accumulate in microlumina when Anos1 was genetically removed (Fig. 4B, D). Also, Fgf10a-GFP accumulated to similar levels in microlumina of wild-type and anos1 mutant embryos when central cell production of Fgf10a-GFP was not blocked (Fig. 4D). In these genetic scenarios, lumen integrity was not affected (Fig. S4D), demonstrating that the absence of Fgf10a-GFP in anos1 mutants with reduced Atoh1a function was not due to Fgf10a-GFP leakage from incompletely sealed-off microlumina. Moreover, the accumulation of Fgf10a-GFP from the front of the primordium in microlumina did not require Fgfr1a (Fig. 4B, D), indicating that Anos1 affects the transport of Fgf10a-GFP largely independently of the Fgf receptors in the primordium.
Next, we asked whether loss of Anos1 activity on sensory organ formation is enhanced when Fgf10a secretion from the central cells and compensation through Fgf3 are both blocked. For this, we injected anos1a−/−; anos1b−/−; fgf3−/− triple mutant embryos with atoh1a morpholino. In these embryos, we found that the primordia deposited fewer neuromasts compared to control embryos that have functional Anos1 but do not secrete Fgf10a from central cells (reduced atoh1a activity, Fig. 1C, E) or that have functional Anos1 but do not secrete either Fgf3 or Fgf10a from central cells (reduced atoh1a activity and fgf3 mutant, Fig. 1C, E).
Together, these observations indicate that Anos1 is required for the microluminal accumulation of Fgf10a-GFP secreted from the front of the primordium, and if redundant Fgf sources are removed, lack of Anos1 results in impaired sensory organ formation. This implies an additional role for Anos1 in the regulation of FGF distribution besides its known role in facilitating FGF ligand-receptor complex formation (Díaz-Balzac et al., 2015; Hu et al., 2009; Yamada et al., 2012).
Anos1 increases the pool of fast diffusing Fgf10a molecules
Anos1 could regulate the distribution of Fgf10a to increase its microluminal accumulation in the rear of the primordium in three ways. First, it could increase the stability/half-life of Fgf10a and, thus, its overall levels. Second, Anos1 could trap Fgf10a above forming neuromasts to locally enrich the growth factor. Third, it could facilitate the diffusion of Fgf10a from the front to the rear such that more Fgf10a reaches the rear of the primordium.
To test whether Anos1 stabilizes Fgf10a, we blocked secretion of Fgf10-GFP from central cells using Brefeldin A and measured the degradation rate of luminal Fgf10a-GFP in embryos over-expressing anos1b from a heat shock promoter and in heat-shocked control embryos. We found that the degradation rate of luminal Fgf10a-GFP was similar in anos1b over-expressing and control embryos (Fig. 4C), indicating that Anos1 does not affect the stability of Fgf10a significantly.
To determine whether Anos1 traps Fgf10a or facilitates its diffusion, we asked how Anos1 affects the diffusivity of Fgf10a-GFP using fluorescence correlation spectroscopy (FCS, Fig. 5A). FCS measurements yield information about the absolute concentration, the diffusion coefficients and the number of species with different diffusivities (Ries and Schwille, 2012). These measurements were performed in the microlumina because the extracellular space above the primordium is too small for reliable measurements. It is likely that Fgf10a diffusion in the microlumina approximates Fgf10a diffusion above the primordium. Consistent with a previous study on Fgf8-GFP diffusion (Yu et al., 2009), we found that Fgf10a-GFP also diffused with two different mobilities in wild-type embryos. Most Fgf10a-GFP molecules (88.2 %) diffused rapidly (40.2 μm2/s) as monomers, while a small fraction (11.8 %) of Fgf10a-GFP diffused slowly (1.7 μm2/s) (Fig. 5B, 5D, 5F,S5A, S5E-K). The diffusion coefficient of the fast population is typical for a freely diffusing globular protein of Fgf10a-GFP’s molecular weight (Phillips et al., 2012), suggesting that most Fgf10a molecules move freely through the extracellular space. In contrast, the 20-fold lower diffusion coefficient of the slow population suggests that about 10 % of Fgf10a is bound to much larger complexes, such as HS, in the extracellular space (Bishop et al., 2007; Li et al., 2016; Makarenkova et al., 2009; Yu et al., 2009).
Figure 5.
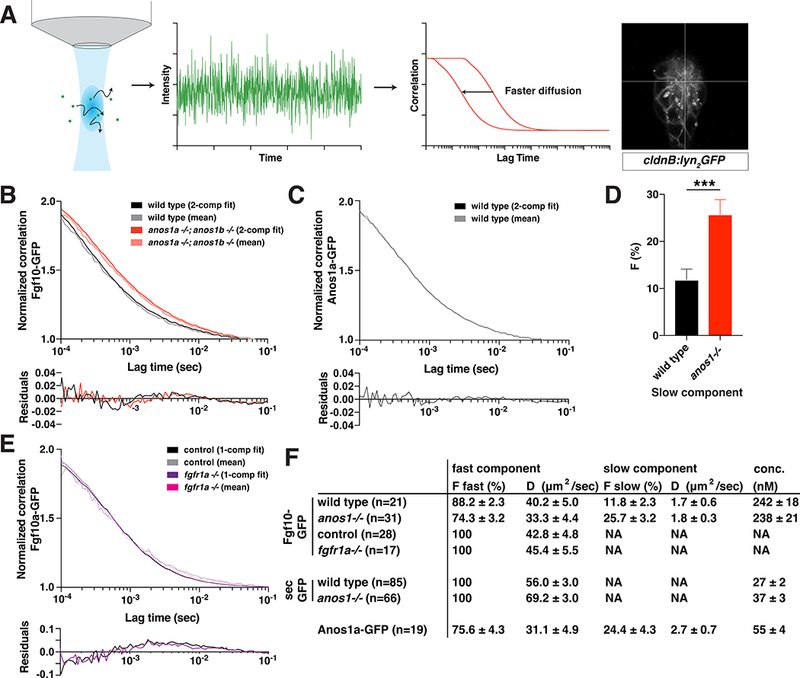
Anos1 enhances Fgf10a diffusion by increasing the pool of fast-diffusing Fgf10a molecules.
(A) Schematic of FCS measurements. Left, fluorescent molecules diffuse through a diffraction-limited spot. Arrows indicate diffusion. Middle left, fluorescence intensity fluctuations generated by molecules diffusing into and out of the confocal volume. Middle right, autocorrelation is calculated as the correlation between fluorescence intensity at time = t and time = t + τ. The X-axis represents τ, the lag time, and the Y-axis represents the correlation G(τ).Right, cross-hair indicating the position in the microlumen (white) where the FCS measurements were performed.
(B) Top, autocorrelation curves normalized to 50 μsec and 2-component fits for Fgf10a-GFP in the indicated genotypes. Bottom, residuals of the fits.
(C) Top, autocorrelation curves normalized to 50 μsec and 2-component fits for Anos1a-GFP in wild-type embryos. Bottom, residuals of the fits.
(D) Bar graph of the fraction of molecules in the slow component in the genotypes indicated. Error bars represent SD. *** = p < 0.001.
(E) Top, autocorrelation curves normalized to 50 μsec and 1-component fits for Fgf10a- GFP in the indicated genotypes. Controls include fgfr1a−/+ and wild-type sibling embryos. Bottom, residuals of the fits.
(F) Table of fitted values. F represents proportion of molecules in each component and D is the diffusion coefficient. The anos1a−/−; anos1b−/− genotype is abbreviated as anos1−/− in D and F.
See also Figure S5.
In the absence of Anos1 activity, the fraction of slow moving Fgf10a-GFP molecules more than doubled (25.7 %) at the expense of the fraction of fast moving Fgf10a-GFP molecules (74.3 %, Fig. 5B, D, F). The diffusivity of the two populations was little affected and Fgf10a-GFP remained monomeric (Fig. 5F, S5E-K). Control measurements showed that secreted GFP diffused as one species in microlumina with a diffusion coefficient of 56.0 μm2/sec characteristic for freely diffusing GFP (Dayel et al., 1999; Yu et al., 2009) and its diffusivity was not affected much by Anos1 activity (Fig. 5F, S5B). These observations suggest that Anos1 shifts the equilibrium between the fast and slow diffusing Fgf10a molecules towards faster diffusing molecules and hence promotes overall Fgf10a diffusivity. Consistent with this idea, we found that most Anos1a molecules (75.6 %) are also highly diffusive (31.1 um2/s), which suggests that most Anos1 molecules are not tethered to the ECM (Fig. 5C, F, S5F, S5G). Importantly, the overall diffusivity of Fgf10a-GFP was not affected by Fgfr1a (Fig. 5E, S5D). Together, these observations indicate that Anos1 facilitates the diffusion of Fgf10a rather than trapping it.
Anos1 and Fgf10a are in a complex and diffuse rapidly together
Anos1 could increase the pool of fast diffusing Fgf10a molecules by competing with Fgf10a for binding sites in the ECM or by binding and shuttling Fgf10a. To distinguish between these possibilities, we asked whether Anos1a and Fgf10a diffuse together using fluorescence cross-correlation spectroscopy (FCCS). FCCS measurements provide information about the movement of two molecules moving together in a complex, the fraction of the molecules in the complex and the diffusivity of the molecules in the complex (Bacia et al., 2006; Foo et al., 2012; Schwille et al., 1997; Shi et al., 2009; Sudhaharan et al., 2009). To increase the signal-to-noise ratio, we performed these measurements in the early embryo rather than in the microlumen, and we used elevated laser intensities (Fig. 6A). Because high laser intensities bleach fluorescent proteins as they traverse the confocal volume and thus overestimate the diffusion coefficients (Fig. 6C) and underestimate the number of co-diffusing molecules (Fig. 6B), we also performed these experiments at lower laser powers with a decreased signal to noise (Fig. 6D, E).
Figure 6.
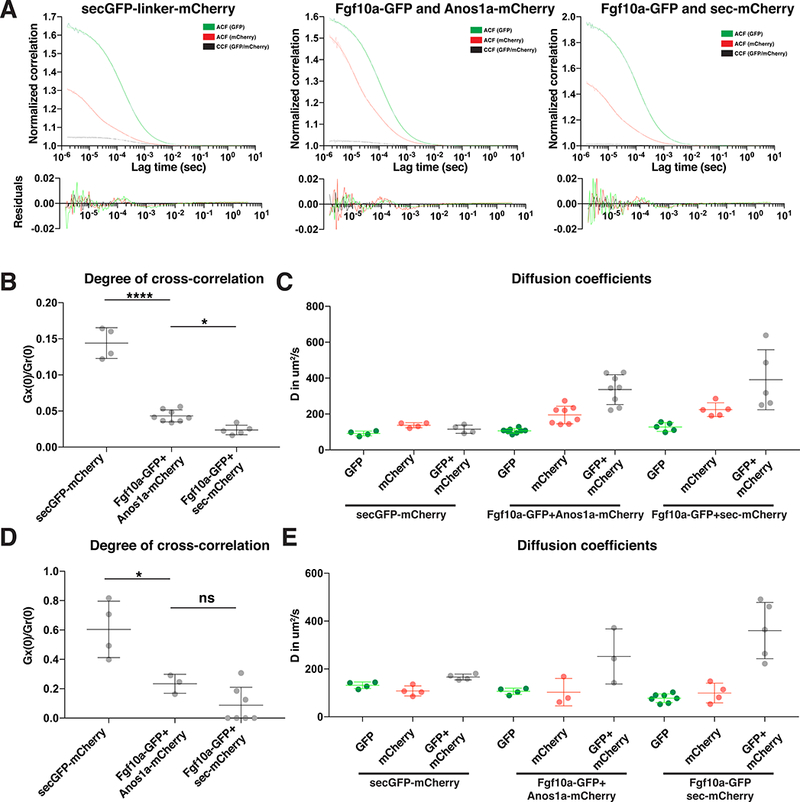
Anos1 and Fgf10a co-diffuse.
(A) Normalized autocorrelation curves and cross-correlation curves for secreted GFPmCherry dimers (left), Fgf10a-GFP and Anos1a-mCherry (middle) and Fgf10a-GFP and secreted mCherry (right) of FCCS measurements with high laser power. Bottom, residuals of the one-component fits.
(B) Plot of the degree of cross-correlations of the individual FCCS measurements for secreted GFP-mCherry dimers (left), Fgf10a-GFP and Anos1a-mCherry (middle) and Fgf10a-GFP and secreted mCherry (right) of FCCS measurements with high laser power. Mean, SD and individual data points are shown. * = p < 0.05, **** = p < 0.0001. ANOVA p<0.0001.
(C) Plot of the diffusion coefficients of the individual FCCS measurements with high laser powers for secreted GFP-mCherry dimers (left), Fgf10a-GFP and Anos1a-mCherry (middle) and Fgf10a-GFP and secreted mCherry (right). Mean, SD and individual data points are shown.
(D) Same plot as in (A) but for FCCS measurements with low laser power. Mean, SD and individual data points are shown. * = p < 0.05, ns = p > 0.05. ANOVA p=0.0002.
(E) Plot of the diffusion coefficients of the individual FCCS measurements with low laser powers for secreted GFP-mCherry dimers (left), Fgf10a-GFP and Anos1a-mCherry (middle) and Fgf10a-GFP and secreted mCherry (right). Horizontal lines indicate the average, vertical lines the SD and dots individual data points.
See also Figure S6.
This analysis showed that at least 12 % of Fgf10a-GFP diffused together with Anos1a-mCherry for higher laser power FCCS measurements (Fig. 6 A, B). Noisier lower laser intensity FCCS measurements with reduced fluorescent protein bleaching indicate that the fraction of Fgf10a-GFP co-diffusing with Anos1a-mCherry is 28 % (Fig. 6D). Compared to the mobilities of the slow and fast moving Fgf10a-GFP and Anos1amCherry populations in the microlumen, the mobility of the Fgf10a-GFP/Anos1amCherry complex is more comparable to the fast moving Fgf10a-GFP and Anos1amCherry populations (Fig. 5F, 6C, E, S6D). Importantly, control measurements showed that secreted GFP-mCherry dimers co-diffused rapidly in the early embryo (Fig. 6A, C, S6A, S6D) and that Fgf10a-GFP and secreted mCherry diffused freely and largely independently of each other (Fig. 6A, C, S6B, S6D). Thus, a fraction of Fgf10a is in a fast diffusing complex with Anos1a, suggesting that Anos1 does not block Fgf10a binding sites in the ECM but rather binds to Fgf10a.
Excess Anos1 slows the diffusion of the fast Fgf10a population and disperses Fgf10a
If Anos1 binds to a subset of Fgf10a molecules, liberates them from the ECM and then locally increases the overall diffusivity of Fgf10a, then globally overexpressing Anos1 should increase the fraction of Anos1-bound Fgf10a, reduce the ECM-bound Fgf10a pool and enhance Fgf10a diffusion and dispersal throughout the embryo. To test these predictions, we used FCS to measure the mobilities of Fgf10a-GFP and Anos1a-GFP in the microlumina of embryos ubiquitously over-expressing Anos1b or Fgf10a from a heat-shock promoter. Compared to heat-shocked control embryos, the fraction of Fgf10a-GFP molecules in the fast moving populations did not increase in Anos1b overexpressing embryos (92.8 % and 92. 2 %, respectively, Fig. 7 A, C, S7A). However, Anos1b over-expression caused the fast moving Fgf10a-GFP molecules to move slower; the diffusion coefficient of the fast Fgf10a-GFP population was reduced from 49.9 μm2/s to 28.2 μm2/s (Fig. 7B, S7D). Assuming a spherical shape, this reduction in diffusivity indicates a five- to six-fold size increase of the fast moving Fgf10a-GFP particles. Anos1b over-expression had little effect on the diffusivity of the slow moving population (Fig. 7C). Together, this resulted in overall slower Fgf10a-GFP diffusion similar to the loss of Anos1 activity (Fig. 7A). Intriguingly, the decrease in the diffusivity of Fgf10a-GFP in Anos1b over-expressing embryos was mirrored by a decrease of Anos1a-GFP diffusivity in Fgf10a over-expressing embryos. In Fgf10a over-expressing embryos, the Anos1a-GFP diffusion coefficient was reduced from 25.0 μm2/s to 15.7 μm2/s (Fig. 7D-F, S7B, S7D). Assuming a spherical shape, this reduction in diffusivity indicates a four-fold size increase of the Anos1a-GFP particles, which is similar to the size increase of the Fgf10a-GFP particles upon Anos1b over-expression (Fig. 7B, E).
Next, we asked whether Fgf10a-GFP still localizes above forming neuromasts in embryos over-expressing Anos1b from a heat-shock promoter. In most heat-shocked control embryos, Fgf10a-GFP was visible as a patch directly above forming but not enclosed microlumina of immature neuromasts (Fig. 7D, Movie 6). In contrast, Fgf10a- GFP was reduced or failed to concentrate above forming neuromasts in embryos that over-expressed Anos1b (Fig. 7G, Movie 6). In both scenarios, Fgf10a-GFP transcription in the central cell, Fgf10a-GFP secretion into the microlumen, Fgf10a-GFP luminal degradation, Fgf10a-GFP luminal levels and lumen integrity - judged by retention of GFP secreted from the central cells - were not altered in microlumina in the rear of the primordium, which had already been enclosed at the time Anos1b over-expression was induced (Fig. 4C, S7E-S7I).
Together, these observations suggest that excess Anos1 leads to clustering of freely diffusing Fgf10a into oligomeric, likely Anos1-containing, complexes. However, it does not decrease the pool of slow moving, presumably ECM-bound, Fgf10a further than wild-type levels of Anos1 do. Moreover, excess Anos1 blocks the accumulation of Fgf10a in patches above forming neuromasts but not in enclosed microlumina, consistent with the idea that excess Anos1 results in the dispersal of the growth factor in the embryo.
Excess Anos1 blocks Fgf signaling and neuromast formation in the front of the primordium
Anos1 over-expression blocks the accumulation of Fgf10a above forming neuromasts and, therefore, it should block FGF signaling, microlumen assembly and neuromast formation in the front of the primordium and, as a consequence, primordium migration (Aman and Piotrowski, 2008; Lecaudey et al., 2008). We assessed FGF signaling in the primordium of Anos1b over-expressing embryos using the FGF-signaling reporter dusp6:d2EGFP and, as an indirect readout for FGF signaling, the Wnt reporter Tcf/Lef-miniP:dGFP. Compared to control embryos, the FGF signaling domain in the back of the primordium was shifted rearwards by 35 μm in Anos1b over-expressing embryos, a distance roughly equal to the diameter of a forming neuromast (Fig. 7I, J, S3H). Complementary to the retraction of the FGF-signaling domain, the Wnt-signaling domain in the front expanded by a similar distance of 30 μm towards the rear of the primordium in Anos1b over-expressing embryos compared to control embryos (Fig. 7K).
Next, we analyzed microlumen assembly in the primordium of Anos1b overexpressing embryos using the apically localized tight junction-associated protein and luminal marker ZO-1. In Anos1b over-expressing embryos, ZO-1 accumulation at the site of the first newly formed microlumen was shifted rearward by the length of one neuromast compared to control embryos, such that the first microlumen was positioned where the second microlumen is normally found (Fig. 7L, S7C). Consistent with this, the size of the first, second and third microlumen was increased in Anos1b over-expressing embryos compared to the size of the corresponding microlumen in control embryos (Fig. 7L). This indicates that the first microlumen was lost in Anos1b over-expressing embryos.
We tested this idea further by asking whether Anos1b over-expression would dissolve forming microlumina using time-lapse microscopy of the luminal marker Cadherin-2-GFP (Cdh2-GFP). This analysis showed that upon Anos1b over-expression, assembling microlumina in the front of the primordium dissolved while mature microlumina in the rear of the primordium were not affected (Fig. 7H, Movie 7). Microlumen maturation was not affected in control embryos (Fig. 7H, Movie 7). Consistent with the requirement of FGF signaling for migration (Aman and Piotrowski, 2008; Lecaudey et al., 2008; Nechiporuk and Raible, 2008), Anos1b over-expression also caused the primordium to stall (Fig. S7J, Movie 7 and 8).
In vitro studies suggested that excess Anos1 binds the FGF receptor and blocks its activation (Hu et al., 2009). This would imply that Anos1 over-expression could block FGF signaling and microlumen formation in the front of the primordium by blocking FGF receptor activation rather than dispersing the growth factor. To test this possibility, we analyzed the activation of the FGF-signaling reporter dusp6:d2EGFP in embryos over-expressing Anos1b, Fgf10a or both Anos1b and Fgf10a together from heat shock promoters. We found that globally over-expressing Fgf10a alone or together with Anos1b increased FGF signaling in the rear of the primordium to similar levels. FGF signaling was not increased in heat shocked control embryos and was decreased in Anos1b only over-expressing embryos (Fig. 7I, J, S3H). This suggests that Anos1 does not block FGF receptor activation.
Computational Modeling
Our experimental results suggest that localized production of FGF and Anos1 and their interactions with HS expressed on the surfaces of the cells of the primordium shape the spatial characteristics of these molecules’ distributions as they diffuse in and around the primordium. To assess whether these effects can indeed account for the observed distribution of FGF and its dependence on Anos1 expression levels, we developed a computational model using the Simmune software (Angermann et al., 2012; Cheng et al., 2014; Zhang et al., 2013) (Fig. 8A, S8)
Figure 8.
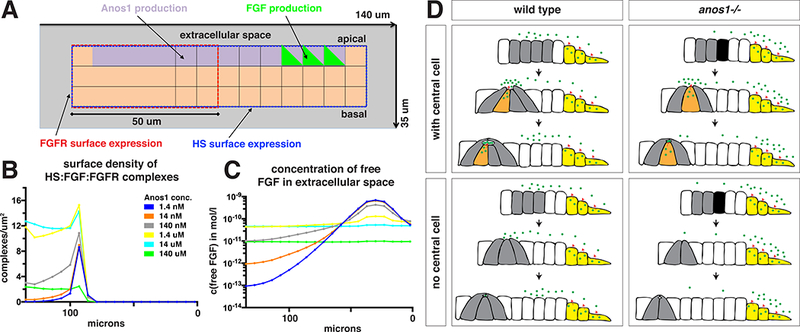
Computational analysis of model for FGF shuttling by Anos1.
(A) Schematic showing a cut through the 3-dimensionally modeled primordium to indicate which cell positions express which surface receptors or secrete Anos1 or FGF.
(B) Computationally modeled density of the FGF/FGFR/HS complex on the primordium’s surface as a function of different Anos1 concentrations.
(C) Computationally modeled concentration of free FGF in the extracellular space above the apical side of the primordium as a function of different Anos1 concentrations. 0 microns indicate the front of the modeled primordium in Fig. 8A.
(D) Summary of how Anos1 affects Fgf10a ligand distribution. Black arrows indicate the progression of time. Green dots indicate Fgf10a and red arrows indicate sites of Fgf10a secretion.
See also Figure S8.
The biochemical model consists of FGF, the FGF receptor FGFR, surface bound HS and Anos1. The model assumes that FGF can bind to its receptor and, with a secondary binding site, to HS and that the trimeric complex FGFR:FGF:HS has a greater stability than the dimeric complexes FGFR:FGF and HS:FGF as suggested previously (Belov and Mohammadi, 2013). Since Anos1 does not seem to bind to HS (Fig. 6), our model further assumes that Anos1 binds to the binding site of FGF that the growth factor uses for its interaction with HS (see methods and Fig. S8).
In our simulations, we varied the production rate of Anos1 over six orders of magnitude and thus modulated the Anos1 concentration around the primordium. The simulation results illustrate that competition between Anos1 and HS for the shared binding site on FGF can influence the retention of FGF by HS and, thus, its spatial concentration profile and availability for complex formation with the FGF receptor (Fig. 8B).
The assumption of cooperative binding between FGFR, FGF and HS (Belov and Mohammadi, 2013) led to a biphasic behavior of the dependency of the concentration of receptor-bound FGF on the expression level of Anos1: In the absence or at very low levels of Anos1, FGF was retained strongly by HS in the front of the primordium, leading to very low levels of ligated FGF receptors in the rear (Fig. 8C). When expressed at intermediate levels, Anos1 prevented this overly strong retention of FGF by HS in the front but still allowed FGF to form trimeric complexes with HS and FGFR. When strongly overexpressed, however, Anos1 suppressed most binding of FGF by HS and FGF became almost uniformly distributed without becoming available for the formation of the stable FGFR:FGF:HS complexes. These simulations indicate that Anos1 can, depending on its concentration, play a dual role, enhancing or interfering with FGF signaling in the rear of the primordium.
Discussion
Our analysis shows that Fgf10a secreted from the front will only enrich to detectable levels in microlumina in the rear of the primordium if Anos1 is present. Molecularly, wild-type levels of Anos1 halve the pool of slow moving Fgf10a and thus increase the overall diffusivity of Fgf10. Intriguingly, at least 12 % of Fgf10a also codiffuses with Anos1 in the early embryo, suggesting that Anos1 liberates surface-bound, slow moving Fgf10a from the ECM and preserves it in the freely diffusing Fgf10a pool. In contrast, over-expression of Anos1 does not decrease the pool of slow moving Fgf10a further, suggesting that Anos1 cannot access and liberate all ECM-bound Fgf10. Instead, excess Anos1 reduces the mobility of the pool of freely moving Fgf10, probably by binding to and clustering Fgf10. Similar to the loss of Anos1 activity, this also leads to an overall reduction of Fgf10a diffusivity, although by a different molecular mechanism. Together, these observations suggest that Anos1 liberates Fgf10a from the ECM and shuttles it to increase its signaling range.
Previous studies have shown that Anos1 acts on the FGF receptor (de Castro et al., 2013). At low concentrations, Anos1 enhances FGF-FGF receptor complex formation and thus signaling by either binding to FGF (Hu et al., 2009) or the preformed complex of FGF, FGF receptor and the adhesion molecule L1CAM (Diaz-Balzac et al., 2015; Yamada et al., 2012). Intriguingly, this interaction requires a unique HS sulfation pattern (Diaz-Balzac et al., 2014; Tecle et al., 2013; Tornberg et al., 2011). At high concentrations, Anos1 binds unligated FGF receptor. Although this complex can still bind FGF, it cannot recruit the obligate co-factor HS and thus cannot signal (Hu et al., 2009). Our results indicate an additional function for Anos1 in regulating FGF signaling upstream of FGF ligand-receptor complex formation. We find that Anos1 increases the diffusivity of FGF and thus its signaling range. Importantly, excess levels of Anos1 affect the ability of endogenous FGF to activate FGF receptor signaling but do not block overexpressed FGF from FGF receptor activation. This is probably because over-expressed Anos1 binds and dissipates locally secreted, endogenous FGF but cannot efficiently sequester and dissipate ubiquitously over-expressed FGF to levels below that required for receptor activation.
Our computational modeling suggests that this is a reasonable scenario. If HSbound FGF has a greater affinity for the FGF receptor than free FGF and if Anos1 and HS compete for FGF binding, then increasing Anos1 concentrations yield a biphasic FGF receptor response. At low Anos1 concentrations, FGF binds to HS and does not diffuse far from its source. At intermediate Anos1 concentrations, Anos1 liberates HSbound FGF and FGF diffuses further. At high Anos1 concentrations, most FGF is bound by Anos1, disperses evenly and is not available for signaling. Together with previous reports (Díaz-Balzac et al., 2015; Hu et al., 2009; Yamada et al., 2012), these observations suggest that Anos1 modulates FGF signaling at the receptor and ligand level to ensure proper FGF receptor activation.
After Fgf10a is enriched in patches on the apical side of forming neuromasts, it becomes enclosed in sealed-off, extracellular lumina in the center of maturing neuromasts. While Anos1 facilitates the transport of Fgf10a from the front to the rear in the primordium, several of our observations suggest that it does not trap Fgf10a on the apical side above forming neuromasts. First, although Anos1 concentrates in microlumina together with Fgf10a, it does not localize in patches on the apical side of forming neuromasts like Fgf10a. Second, Anos1 does not decrease but instead increases the mobility of Fgf10a and is itself mostly mobile. Third, locally increased production of Anos1 in the primordium does not retain or enrich Fgf10a (data not shown). Fourth, Anos1-Fgf10a complexes do not diffuse slowly as expected for an Fgf10a trap but instead diffuse rapidly in the early embryo. Thus, another, unknown molecular mechanism traps Fgf10a on the apical side above forming neuromasts. The Fgf10a trap could entail immobile proteins or specifically sulfated HSPGs in the ECM that tether Fgf10a to the apical side of forming neuromasts, a notion that is consistent with the observation of less restricted FGF signaling in the primordium of embryos with defective HS synthesis (Galanternik et al., 2015).
Fgf is secreted from two sites in the primordium, the front and the central cells of the neuromasts (Lecaudey et al., 2008; Nechiporuk and Raible, 2008). Similar to the front cells, the central cells initially secrete Fgf to the apical side of the primordium but later directly pump Fgf into the microlumina. Previous studies have shown that Fgf production is compensatory in the primordium. In the absence of Fgf10a from the central cells or the entire primordium, Fgf3 expression in the central cells increases and compensates for the loss of Fgf10a resulting in normal neuromast development (Lecaudey et al., 2008; Matsuda and Chitnis, 2010; Nechiporuk and Raible, 2008). Our analysis shows that restricting Fgf production in the primordium to only Fgf10a secreted from the front still results in almost normal neuromast development, indicating that Fgf10a secretion from the front alone is sufficient for sensory organ formation. However, when also removing Anos1 activity in this scenario, smaller and fewer neuromasts are deposited. This is presumably because in the absence of Anos1, Fgf10a is less mobile and requires more time to diffuse from the front towards the rear of the primordium, where it takes longer to concentrate to levels that are sufficient to induce neuromast formation. These observations suggest a redundant sensory organ formation mechanism in which Fgf secreted from the front is shuttled by Anos1 towards the rear of the primordium where it becomes locally trapped on the apical side above the site of a future neuromast through an unknown mechanism. Once Fgf accumulation reaches a certain threshold, central cells are induced. The central cells then start locally secreting more Fgf towards the center of the forming neuromast, which induces lumen assembly and enclosure. Once the lumen is sealed off, the central cells secrete directly into it and Fgf becomes progressively more and more concentrated in the lumen of developing neuromasts. Such a feed-forward loop for local Fgf accumulation by the central cells paired with the molecular redundancy of the Fgf ligands provides a robust mechanism for sensory organ formation and may also underlie the robustness of other biological processes.
The affinity of FGF ligands for HS largely determines how far FGF ligands diffuse from their site of production through the ECM (Goetz and Mohammadi, 2013). Ligands with a high affinity for HS diffuse only over short distances and signal locally while ligands with a weak affinity for HS diffuse further and signal over a longer range. The FGF ligand affinity for HS is dictated by its protein sequence and cannot be altered to regulate the diffusion range (Makarenkova et al., 2009). In principle, however, a diffusible FGF-binding protein could compete with HS for FGF ligand binding and shuttle the growth factor through the ECM. Our results suggest that Anos1 is such a FGF shuttle protein. It moves together with Fgf10a through the extracellular space and increases Fgf10a’s diffusivity and signaling range. Besides altering the affinity of HS for FGF through local modifications, this is an elegant way to tailor the FGF signaling range in a context-dependent manner.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Holger Knaut (Holger.Knaut@med.nyu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Zebrafish strains
Zebrafish were raised and housed at 28 °C. 5 day old zebrafsih larvae were raised until 21 dpf on Cylop-eeze (Argent Laboratories, USA). Zebrafish older than 21 dpf were raised and maintained on brine shrimp (Brine Shrimp Direct, USA). For experiments, embryos were collected 15 min past egg laying, raised at 28 °C, unfertilized eggs were discarded and embryos were staged by comparing it to the staging series reporter by Kimmel and colleagues (Kimmel et al., 1995). Embryos were an approximately equal mix of male and female, as there does not exist a good method for distinguishing between the genders at the developmental stages presented in this paper. The fgf3/liat24149 allele contains a missense mutation (E138K) resulting in strong loss of function (Herzog et al., 2004). The fgf10a/daetbvbo allele contains a nonsense mutation that results in a premature stop codon at amino acid position 5 (Norton, 2005).
The fgfr1a/spdt3R705H allele contains a missense mutation that changes arginine 705 in the second kinase domain to a histidine and results in a loss of function (Rohner et al., 2009). The cldnb:lyn2GFP transgenic line contains an 8 kb fragment upstream of the cldnb start codon fused to a membrane-tethered GFP (Haas and Gilmour, 2006). The hsp70:dn-fgfr1 transgenic line contains a 1.5 kb fragment from the zebrafish hsp70 promoter (Halloran et al., 2000) driving the expression of a modified zebrafish Fgfr1 where the tyrosine kinase domain was replaced by EGFP (Lee, 2005). The dusp6:d2EGFP transgenic line contains a 10 kb fragment that includes the 5’ UTR of dusp6 driving the expression of a destabilized EGFP (Molina et al., 2007). The Tcf/LefminiP:dGFP transgenic line contains 6 tandem consensus Tcf/Lef binding sites fused to a minimal promoter derived from pGL4 (Promega Corporation) driving the expression of a destabilized EGFP (Shimizu et al., 2012). The cdh2:cdh2-sfGFP-TagRFP bacterial artificial chromosome (BAC) transgenic line expresses the sfGFP-TagRFP timer from the cdh2 locus (Revenu et al., 2014). The cxcr4b:cxcr4b-Kate2-ires-GFP-CAAX bacterial artificial chromosome (BAC) transgenic line expresses Cxcr4b-Kate2 and membrane tethered GFP from the cxcr4b locus (Venkiteswaran et al., 2013). Other strains used in this manuscript were generated using the methods detailed in the Method Details section below.
METHOD DETAILS
Generation of transgenic strains
For the prim:lyn2mCherry transgene, we cloned the lyn2-mCherry coding sequence into pME and used Gateway recombination to assemble pME-lyn2-mCherry, p5E-sox10(−7.2 kb) and p3E-polyA into pDestTol2pA. We verified the final construct by sequencing and co-injected it along with tol2 transposase mRNA into zebrafish embryos at the one-cell stage. Stable transgenic fish were identified by out-crossing adults injected with the transgene and raising larvae from fish whose offspring expressed Lyn2-mCherry. Note, this transgenic line does not recapitulate the sox10 expression pattern but labels among other structures the posterior lateral line primordium. We termed this transgenic line Tg(prim:lyn2-mCherry).
For the hsp70:anos1b transgene, we used the Tol2 gateway cloning kit generated by the Chien lab (Kwan et al., 2007). We cloned the zebrafish anos1b coding sequence into pME and used Gateway recombination to assemble pME-hsanos1b, p5E-hsp70l and p3E-polyA into pDestTol2pA. We verified the final construct by sequencing and coinjected it along with tol2 transposase mRNA into zebrafish embryos at the one-cell stage. Stable transgenic fish were identified by out-crossing adults injected with the transgene and raising larvae from fish whose offspring were identified to express anos1b upon heat shock as determined by RNA in situ hybridization. Initial founder fish with multiple insertions were outcrossed to obtain single-insertion lines as determined by the fraction of progeny carrying the hsp70:anos1b transgene. The full name of this transgenic line is Tg(hsp70:anos1b)p2.4.17.
For the hsp70:sec-mCherry transgene, the pDestTol2pA-hsp70l-anos1b-sv40pA plasmid used to generate the hsp70:anos1b line (see above) was used as a template. Using Gibson cloning (Gibson et al., 2009), we replaced the anos1b coding sequence with the coding sequence of mCherry fused to the 3’ end of the fgf3 secretion signal (amino acids 1 to 18). We verified the final construct by sequencing and co-injected it along with tol2 transposase mRNA into zebrafish embryos at the one-cell stage. Stable transgenic fish were identified by out-crossing adults injected with the transgene and raising larvae from fish whose offspring were identified to express mCherry upon heat shock as determined by red fluorescence. Founder fish were verified to carry a single copy of the transgene by determining the fraction of progeny carrying the transgene. The full name of this transgenic line is Tg(hsp70:secP-mCherry)p1.
For the hsp70:fgf10a transgene, a genomic fragment spanning 1.5 kb upstream of the hsp70 start codon (Halloran et al., 2000) was fused to the coding sequence of fgf10a followed by the SV40pA signal and cloned into a vector in which the multiple cloning site is flanked by I-SceI sites (Thermes et al., 2002). For transgenesis, 25 ng/μI of this construct was co-injected with the I-SceI enzyme (New England Biolabs, Inc.) into onecell-stage embryos. Stable transgenic fish were identified by out-crossing adults injected with the transgene and raising larvae from fish whose offspring were identified to express fgf10a upon heat shock as determined by in situ hybridization against fgf10a. Founder fish were verified to carry a single copy of the transgene by determining the fraction of progeny carrying the transgene. The full name of this transgenic line is Tg(hsp70:fgf10a).
For the fgf10a:GFP-fgf10a transgene, we used the bacteria artificial chromosome (BAC) clone CH211–262I10. This BAC clone spans 165 kb of genomic zebrafish DNA and encompass the entire fgf10a locus including 55 kb upstream and 75 kb downstream sequence based on the database ensembl and confirmed by PCR amplicons placed every 10 kb. We used recombineering to modify this BAC clone in three ways (Warming et al., 2005). First, we inserted the Tol2(exon4)-FRT-galK-FRT-Tol2(exon1)-alpha- Crystallin-dsRed cassette into the pTARBAC2 backbone (Fuentes et al., 2016) using galK as a selection marker, replacing nucleotides 3008–3052 of the pTARBAC2 backbone. We then removed galK by Flippase-mediated recombination. Second, we inserted a kanamycin resistance cassette to replace the sequence between the pTARBAC backbone and nucleotide 99078 of the genomic insert in the BAC, using kanamycin as a selection marker. This removed 65 kb of sequence that is 10 kb 3’ to the last exon of the Fgf10a gene and reduced the total length of the BAC from 165 kb to 99 kb. The targeting cassette for this recombineering step was assembled by using 38 bp homologous to the 5’ end of the pTARBAC backbone and 50 bp homologous to nucleotides 99029–99078 of the CH211–262I10 BAC. These arms of homology were added to the kanamycin resistance gene in the targeting cassette by PCR using primers with overhangs containing the regions of homology. Third, the EGFP coding sequence was inserted in-between the last codon of the Fgf10a signal peptide (amino acids 31 of Fgf10a) and the first codon of the Fgf10a mature protein using seamless galK-mediate recombineering. 577 bp upstream and 573 downstream of the end of the last codon of the Fgf10a signal peptide were added to the EGFP targeting cassette using Gibson cloning. The final BAC was characterized by restriction digest and PCR amplification followed by sequencing of regions near the sites where the Kanamycin resistance and EGFP were inserted. BAC DNA was then purified with the nucleobond BAC 100 kit (Clonetech) and co-injected with tol2 transposase mRNA into one-cell-stage zebrafish embryos. Stable transgenic larvae were identified by out-crossing adults injected with the transgene and by raising larvae with red fluorescence in the lens of the eye from the transgenesis marker cryaa-dsRed at 4 dpf. Initial founder fish with multiple insertions were outcrossed to obtain single-insertion lines as determined by the fraction of progeny carrying the transgene. The full name of this transgenic line is TgBAC(fgf10a:EGFPfgf10a)p7.2.br.
For the fgf10a:secGFP transgene, we used the same strategy as described above for the fgf10a:GFP-fgf10a transgene, except the EGFP coding sequence followed by a stop codon and the fgf10a 3’UTR was inserted in-between the last codon of the Fgf10a signal peptide and the first codon of the Fgf10a mature protein using seamless galK-mediate recombineering (instead of only the EGFP coding sequence as for the fgf10a:GFPfgf10a transgene). 577 bp upstream and 573 bp downstream of the end of the last codon of the Fgf10a signal peptide along with EGFP, a stop codon, and the fgf10a 3’ UTR were assembled using Gibson cloning to obtain a pUC19 plasmid that harbors the 5’arm- EGFP-STOP-3’UTR-3’arm targeting cassette. The final BAC was characterized by restriction digest and PCR amplification followed by sequencing of the regions across the site where the EGFP-STOP-3’UTR was inserted. The BAC was then purified with the nucleobond BAC 100 kit (Clonetech) and co-injected with tol2 transposase mRNA into one-cell-stage zebrafish embryos. Stable transgenic larvae were identified by outcrossing adults injected with the transgene and raising larvae with red fluorescence in the lens of the eye from the transgenesis marker cryaa-dsRed at 4 dpf. Founder fish were verified to have a single copy of the transgene by determining the fraction of progeny carrying the transgene. The full name of this transgenic line is TgBAC (fgf10a:sec-EGFP- STOP-3’UTR) p2a.
For the anos1a:GFP-anos1a transgene, we used the CH211–267B13 BAC clone. This BAC clone spans 149 kb of genomic zebrafish DNA and encompass the entire anos1a locus including 45 kb upstream and 68 kb downstream of the first and last exon, respectively, based on the ensembl database (http://ensembl.org/index.html) and confirmed by PCR amplicons placed every 10 kb for 40 kb up- and downstream of the first and last exon, respectively. We used recombineering to modify this BAC clone in two ways. First, we inserted the Tol2(exon4)-FRT-galK-FRT-Tol2(exon1)-cryaa-Citrine cassette into the pTARBAC2 backbone of the CH211–267B13 BAC clone using galK as a selection marker, replacing nucleotides 3008–3052 of the pTARBAC2 backbone (Fuentes et al., 2016). We then removed galK by Flippase-mediated recombination. Second, the EGFP coding sequence was inserted in-between the last codon of the anos1a signal peptide (amino acid 34 of Anos1a) and the first codon of the Anos1a mature protein using seamless galK-mediated recombineering. For this, we first inserted galK using 904 bp upstream and 776 bp downstream of the end of the last codon of the Anos1a signal peptide as arms of homology. We then used the same arms of homology flanking the coding sequence of EGFP to replace galK with EGFP. Both targeting cassettes were assembled by stitching PCR. The final BAC was characterized by restriction digest and PCR amplification followed by sequencing of regions near the site where EGFP was inserted. BAC DNA was then purified with the nucleobond BAC 100 kit (Clonetech) and co-injected with tol2 transposase mRNA into one-cell-stage zebrafish embryos. Stable transgenic larvae were identified by out-crossing adults injected with the transgene and by raising larvae with yellow fluorescence in the lens of the eye from the cryaa-Citrine transgenesis marker at 4 dpf. Founder fish were verified to have a single copy of the transgene by determining the fraction of progeny carrying the transgene. The full name of this transgenic line is TgBAC(anos1a:GFP-anos1a)p1.
For the cxcr4b:H2A-mCherry BAC transgene, we used the BAC clone DKEY-169F10. This BAC clone contains the cxcr4b locus, and was modified in two ways by recombineering. First, the Tol2 sites and the cryaa:dsRed transgenesis marker were inserted into the BAC backbone (Fuentes et al., 2016). Second, a cassette consisting of H2A-mCherry-FRT-galK-FRT flanked by 411 bp and 433 bp of homology upstream of cxcr4b exon 2, and downstream of the cxcr4b stop codon, respectively, was inserted to replace the cxcr4b coding sequence in cxcr4b exon 2 (amino acid 6–358, the last amino acid before the stop codon) using galK-mediated recombineering (Warming et al., 2005). The galK cassette was removed by Flippase-mediated recombination. This transgene expresses the first five amino acids from cxcr4b exon 1 fused to H2A-mCherry from the cxcr4b promoter. The final BAC transgene was characterized by EcoRI restriction digestion and sequencing of PCR amplicons of the modified locus. The DKEY-169F10 BAC clone was obtained from ImaGenes GmbH, Germany, (sales@imagenes-bio.de). The BAC was purified with the nucleobond BAC 100 kit (Clontech). We co-injected 1 nl of 50–250 μg/ml BAC transgene DNA, and 40 ng/μl Tol2 mRNA into the lifting cell of the zygote of 0 to 20-minute-old embryos. The Tol2 mRNA was transcribed from pCS2FAtransposase (Kwan et al., 2007) using the mMessage mMachine SP6 Transcription Kit (Thermo Fisher). Stable transgenic larvae were identified by out-crossing adults injected with the cxcr4b:H2A-mCherry BAC transgene, and by raising larvae positive for the red fluorescent transgenesis marker in the lens of the eye at 4 dpf. The full name of this transgenic line is TgBAC(cxcr4b:H2A-mCherry)p1.
Generation of mutant strains
We used the plasmid set described by Dahlem et al. to generate transcription activatorlike effector nucleases (TALENs) targeting the codons for cysteine 163 in anos1a and cysteine 155 in anos1b (Dahlem et al., 2012). These cysteines are homologous to cysteine 172 in human ANOS1, which forms a crucial disulfide bond. In humans the C172R mutation leads to the loss of ANOS1 function, resulting in Kallmann Syndrome. TALEN sequences were designed using TALEN Targeter (https://tale-nt.cac.cornell.edu). The plasmids encoding the TALEN proteins were constructed by golden gate cloning and verified by sequencing. We used pTal3DD and pTal3RR instead of pTal3 as the destination vector. These plasmid variants encode a more efficient FokI endonuclease that is also engineered to be a constitutive heterodimer to minimize off-target effects. To generate mutant lines, 400 pg of in vitro transcribed mRNAs encoding the TALENs targeting anos1a and anos1b were injected into separate clutches of zebrafish embryos at the one-cell stage. Stable mutant lines were identified by outcrossing adults injected with TALEN mRNA and raising fish whose offspring had mutations in anos1a or anos1b as determined by PCR followed by restriction enzyme digest. Alleles in stable mutant lines were characterized by sequencing. For anos1a, we recovered two alleles: a 5 nucleotide (nt) deletion and a 7 nt deletion (Fig. S3B). For anos1b, we recovered four alleles: a 5 nt deletion (d5), a 79 nt deletion (d79), a 6 nt deletion (d6), and a 15 nt deletion (Fig. S3B). Anos1a d7/d7; anos1b d5/d5 and anos1a d5/d5; anos1b d79/d79 were generated by first crossing anos1a+/− to anos1b+/− fish and then genotyping and in-crossing the anos1a+/−; anos1b+/− fish. All anos1a and anos1b alleles were outcrossed at least three times before generating anos1a−/−; anos1b−/− double mutants. All anos1a−/−; anos1b−/− fish described in the experiments are anos1a d5/d7; anos1b d5/d79 double compound heterozygotes in order to minimize any potential off-target effects from the TALEN mutagenesis. Note that all these alleles delete a critical cysteine in the whey-acidic protein (WAP) domain and result in a frame shift and premature stop codon (Fig. S3D). Anos1a and anos1b single mutants and anos1a; anos1b double mutants are viable.
Genotyping primers and restriction enzymes
The following PCR primers and restriction enzymes were used to genotype the corresponding alleles. For a list of primers see also Table S1. Anos1a gene
Outer PCR primers: 5’-AGCTGTGTGCAAAGTGTTGTAG-3’/ 5’-CATATAGTTTGATAGAGCGCTTGGAC-3’
Inner PCR primers: 5’-AGATGAGAGTGTGTGTACTATGGC-3’/5’-CCCAGAAGCCACTTTGTGTG-3’
Restriction enzyme digest with BsrI
Anos1b gene
Outer PCR primers (all alleles): 5’-GTGTGACGAGCGCTGAGTTCCTG-3’/5’-ATTTCACCTGTTTCTTTTAGTATG-3’
Inner PCR primers (5 nt deletion allele): 5’-TGTGTGGAGAGCTGTGCGCGGGACCGCGAG-3’/5’-TAAATACCTTTATTGAAATTCCCCAGTATCATCCAGCCGTGCTCTCCAGTGTG-3’ (these primers contain several mismatches to remove the additional Hpych4V sites surrounding the mutation)
Restriction enzyme digest with Hpych4v
Inner PCR primers (6, 15 and 79 nt deletion allele): 5’-TCGCTACGTGTGCAGAAGCAGGG-3’/5’-TAAATACCTTTATTGAAATTCC-3’
fgf3 gene
Outer PCR primers: 5’-ATCCCGCCATGCCACAAT-3’/5’- TCTCGTACCCCACATAAAACTGAC-3’
Inner PCR primers: 5’-CTGCTCTTGTTGTTACTGAGC-3’/5’- CTCAAATATCAAACGGTTTACTCAC-3’
Restriction enzyme digest with ApoI
fgf10a gene
Outer PCR primers: 5’-TGCATCACCCTTTCTCCCATCCAG-3’/5’- TCGTCCTTGCTTTTGGTGCCATTG-3’
Inner PCR primers: 5’-GCTCTTCCCAGTTTTCCGAGCTCCAGGACAATGTGCAAATCG-3’/5’- TCCGTTCTTATCGATCCTGAG-3’ (forward primer is dCAPS primer) Restriction enzyme digest with TaqαI
fgfr1a gene
PCR primers: 5’- TTTGCCGGTGAAATGGATGGCTCC-3’/5’- AGTCTTACAGCTCATGTGTGCATG-3’
Restriction enzyme digest with AciI
hsp70:anos1b transgene
Outer PCR primers: 5’-TGAGCATAATAACCATAAATACTA- 3/5’TCAGGCAGCGGGACACG-3’
Inner PCR primers: 5’-AGCAAATGTCCTAAATGAAT-3’/5’- CGCAACGCTCACCTCAAAC-3’
hsp70:fgf10a transgene
Outer PCR primers: 5’-TGAGCATAATAACCATAAATACTA-3’/5’-TCGTCCTTGCTTTTGGTGCCATTG-3’
Inner PCR primers: 5’-AGCAAATGTCCTAAATGAAT-3’/5’-TCCGTTCTTATCGATCCTGAG-3’
Mosaic analysis
At the 1,000-cell to sphere stage, 50 cells were transplanted from a cldnb:lyn2GFP; prim:lyn2mCherry or cldnb:lyn2GFP; prim: lyn2mCherry; fgf10a:GFP-fgf10a donor embryo into a cxcr4b:H2A-mCherry recipient embryo of an equivalent stage. Embryos were incubated at 28 degree C until 36 hpf, sorted for embryos with a mosaic primordium based on the membrane GFP expression from the cldnb:lyn2GFP transgene and imaged on a Leica SP5 II confocal microscope using a 40× NA 1.1 objective. Forming and deposited neuromasts were analyzed from mosaic primordia that contained donorderived cells in the front of the primordium and no donor-derived cells in the analyzed neuromasts to exclude the possibility of direct Fgf10a-GFP secretion from donor-cells into the microlumen.
Heat shock
For confocal imaging experiments, hsp70:anos1b, hsp70:fgf10a and hsp70:anos1b; hsp70:fgf10a embryos were heat shocked twice at 39.5 degrees C for 1 hour at the 27–28 hpf and the 31–32 hpf stage in a water bath and imaged at 36–38 hpf. hsp70:anos1b; cdh2-GFP embryos were heat shocked at 39.5 degrees C for 1 hour at the 31–32 hpf stage and imaged starting at 34 hpf. hsp70:dn-fgfr1 embryos were heat shocked at 39.5 degrees C for 20 minutes starting at the 31 hpf stage because longer heat shocks resulted in lethality.
For overview images, hsp70:anos1b and hsp70:fgf10a embryos were heat-shocked at 39.5 degrees C for 1 hour and hsp70:dn-fgfr1 embryos were heat-shocked for 20 minutes at 28 hpf for overview images of the phenotype shown in Fig. 1B and S7J. Images were taken at 50–52 hpf.
Morpholino injections
1 nl of 0.05 mM antisense morpholino targeting atoh1a with the sequence 5’- ATCCATTCTGTTGGTTTGTGCTTTT-3’ (Gene Tools, (Millimaki et al., 2007)) was injected into embryos at the one-cell stage.
Brefeldin treatment
Embryos were incubated with 28 μΜ brefeldin (Bfa, Sigma) dissolved in fish water (0.3g/l Instant Ocean Aquarium Sea Salt Mixture, Instant Ocean) and mounted in 0.5% low melt agar also containing 28 μΜ Bfa. Z-stacks were taken every 30 minutes for a total duration of 3 hours using a Leica SP5 II confocal equipped with hybrid detectors in photon counting mode. We used a 40× NA 1.1 objective (HC PL APO 40×/1.10 W CORR CS2) with the pinhole set to Airy 3. The 488 nm laser was set to 150 μW and the 561 nm laser was set to 57 μW.
Whole-mount in situ hybridization
Preparation of RNA probes and whole mount in situ hybridization were performed as previously described (Thisse and Thisse, 2008). Briefly, we cloned DNA templates with the primers listed below using a zebrafish 36 hpf cDNA library generated from polyA RNA extracted with Trizol (Invitrogen) and reverse transcribed with ThermoScript (Invitrogen). Probes were in vitro transcribed with digoxigenin-labelled nucleotides (DIG, Roche). They were detected with anti-DIG-AP antibody (1:2000, Roche) and NBT/BCIP (1:1000 each, Roche).
Embryos were fixed in 4% PFA (Sigma Aldrich) overnight at room temperature. Following fixation, they were permeabilized in methanol (Fisher Chemical) at −20°C, rehydrated, permeabilized with 10 μg/ml proteinase K (Sigma Aldrich) in PBST for 8 minutes at room temperature, and post-fixed in 4% PFA for 20 minutes at room temperature. Blocking and probe hybridization were performed at 68°C overnight. Following probe hybridization and washes, embryos were blocked in 2% BSA (Sigma Aldrich) in PBST and incubated with anti-DIG-AP antibody overnight at 4°C. Embryos were washed and developed in NBT/BCIP solution overnight at room temperature. For assessing nonsense-mediated decay of mRNA transcripts for anos1a and anos1b mutant alleles, anos1a−/−; anos1b−/− double mutant embryos and wild-type embryos were stained and developed in the same tube. Wild-type embryos were marked by removing the tips of their tails after fixation and before staining.
Immunostaining was performed after stopping NBT/BCIP development with PBST supplemented with 1mM EDTA. Embryos were blocked in 2 % BSA in PBST and incubated with affinity-purified goat anti-GFP (1:100, (Venkiteswaran et al., 2013)) overnight at 4 degree C, followed by washes, incubation with donkey anti-goat-Alexa488 (1:1000, Invitrogen) overnight at 4 degree C, and additional washes.
Primers for cloning RNA in situ probe templates
The following primers were used to clone probe templates into the pCRII vector using TOPO TA cloning (Invitrogen).
anos1a: 5’-GGTGGCACGGACATCAGAAGAACG-3’, 5’-GAAGACCGGGAAGGCTGGCAAAAT-3’
anos1b: 5’- TCCCGCTGAAGCCAAGAAGAGAC −3’, 5’-CAGCAGGCCGGTAATCACAAAATG-3’
fgf3: 5’-ATGGTTATAATTCTGCTCTT-3’, 5’-TTAAATGTCAGCCCTTCTGT-3’
fgfr1a: 5’-AGGCGATGGGGATGGATAAAGAAA-3’, 5’- TCCGGCGTCATGAGAAAACACT-3’
fgfr1b: 5’-CTGGCGGAGTGATTTGTTTTGATT-3’, 5’-ATGCTCCCGTATTCGTTCTCCACA-3’
The following primer pairs were used to amplify probe templates. The reverse primers contain the T7 promoter sequence, and purified PCR product was used directly as a template for the in vitro RNA transcription reaction using reagents from Roche. fgf10a: 5’-CTACAACCCCACCAAAGGGAAC-3’, 5’-GAAATTAATACGACTCACTATAGGCATGTGTAACCGATAGAATAGC-3’
EGFP: 5’-GTGAGCAAGGGCGAGGAGCTG-3’, 5’-GAAATTAATACGACTCACTATAGGGCTTGTACAGCTCGTCCATGCC-3’
Whole mount immunostaining
Immunostaining was performed as previously described (Lewellis et al., 2013). Briefly, embryos were fixed in 4% PFA for two hours at room temperature. Following fixation, they were permeabilized in methanol at −20°C, rehydrated, permeabilized with 10 μg/ml proteinase K in PBST for 8 minutes at room temperature, and post-fixed in 4% PFA for 20 minutes at room temperature. Embryos were blocked in 2% BSA in PBST for 1 hour and incubated in primary antibody at 4 degree C overnight. Following PBST washes, they were incubated in secondary antibody overnight at 4 degree C. Rabbit anti-GFP (1:1000, Invitrogen), affinity-purified goat anti-GFP (1:100, (Venkiteswaran et al., 2013)), affinity-purified sheep anti-mCherry (1:1000, custom-made antibody generated against bacterially-produced, recombinant full-length mCherry protein by Covance), zns-2 (1:50, Developmental Studies Hybridoma Bank) and mouse anti-ZO-1 (1:500, ThermoFisher) were detected with goat anti-rabbit-Alexa488 (1:1000, Invitrogen), donkey anti-rabbit- Cy3 (1:1000, Jackson ImmunoResearch), donkey anti-goat-Alexa488 (1:1000, Invitrogen), donkey anti-goat-Cy3 (1:1000, Jackson ImmunoResearch), donkey anti-sheep-Alexa647 (1:1000, Jackson ImmunoResearch), donkey anti-mouse-Cy3 (1:1000, Jackson ImmunoResearch) and donkey anti-mouse Alexa647 (1:1000, Jackson ImmunoResearch) secondary antibodies.
Wide field imaging
GFP from the cldnb:lyn2GFP transgene:
Embryos were imaged using a Leica M165 FC stereo microscope (fluorescent dissecting scope) with a 1× objective at 3.2× zoom. One-second exposures were taken using a GFP filter or two-second exposures were taken using a GFP/mCherry dual color filter. Images of the posterior lateral line in hsp70:dn-fgfr1 embryos were taken with a Texas red filter with a two-second exposure to block out GFP signal from the hsp70:dn-fgfr1 transgene.
Whole mount RNA in situ hybridization
Intact embryos were mounted in 3 % methylcellulose in PBS and imaged on a Zeiss Discovery V8 stereo microscope using a 0.67× objective at 8× zoom.
Flat mount RNA in situ hybridization
Embryo tails were dissected and mounted in 50 % glycerol on slides with thin wells made from a single reinforcement label (Avery). They were covered with a No. 1.5 coverslip and imaged using a Zeiss Axioplan equipped with a 20× NA 0.75 objective or Zeiss Apotome equipped with a 20× NA 0.8 objective.
Reflection imaging
Reflection imaging of in situ hybridizations stained with NBT/BCIP was performed for anos1a and anos1b mRNA stainings. Tails were dissected and mounted in 50 % glycerol on slides with thin wells made from a single reinforcement label (Avery, Staples). Z-stacks of the primordium were taken on a Leica SP5 II confocal with a 40× NA 1.1 water immersion objective (HC PL APO 40×/1.10 W CORR CS2). The NBT/BCIP signal was captured with 633 nm laser reflection. The collection wavelength was set to 630–640 nm while illuminating with the 633 nm laser line. The GFP signal was collected simultaneously with a second detector, exciting with the 488 nm laser line and collecting signal in a 495–550 nm window.
Confocal imaging
Still images
All embryos were mounted in 0.5 % low melt agar dissolved in fish water and supplemented with 0.4 mg/ml MS-222 anesthetic.
The Tcf/Lef-miniP:dGFP Wnt reporter line was imaged using the Leica SP5 II confocal microscope with hybrid detectors set to photon counting mode and a Leica 40× NA 0.8 water dipping objective (HCX APO L 40×/0.80 W U-V-I). The pinhole was set to 1 airy unit. The 488 nm laser was set to 300 μW and the 561 nm laser was set to 57 μW.
The dusp6:GFP, fgf10a:fgf10a-GFP, fgf10a:secGFP, and anos1a:anos1a-GFP transgenic lines were imaged using the Leica SP5 II confocal microscope with hybrid detectors set to photon counting mode and a 40× NA 1.1 water immersion objective (HC PL APO 40×/1.10 W CORR CS2). The pinhole was set to 2 airy units (150 μm). The 488 nm laser was set to 300 μW and the 561 nm laser to 57 μW.
The laser power was calibrated before each imaging session using an X-Cite Power Meter Model XR2100 (Lumen Dynamics), which measures the power of laser light emitted from the objective onto the stage.
Movies
Embryos were mounted in 0.5 % low melt agar dissolved in fish water and anesthesized with MS-222 (0.4mg/ml). The embryos were imaged on a Leica SP5 II laser scanning confocal microscope equipped with two HyD hybrid detectors. For time-lapse imaging, we used a Leica 20× NA 0.5 objective (HCX APO L 20×/0.50 W U-V-I) for all movies except the cdh2:cdh2-sfGFP; prim:iyn2mCherry transgenic embryos, which were imaged with a Leica 20× NA 0.75 objective (HC PL APO 20×/.75 CS2). We acquired Z stacks every 10 or 15 minutes. Embryos were maintained at 28 degree C for the duration of imaging using a heated stage (Warner Instruments). For experiments requiring a heat shock, embryos were first mounted and then heat shocked immediately before imaging. The first image was collected 30–45 minutes after the end of the heat shock.
Light sheet imaging
Anos1a:anos1a-GFP; prim:iyn2mCherry and fgf10a:fgf10a-GFP; prim:lyn2mCherry embryos were mounted in 1% low melt agarose dissolved in fish water with 0.4 mg/ml MS-222 and imaged on the Zeiss Lightsheet Z. 1 using a W Plan Apochromat 20× NA 1.0 water immersion detection objective and two PCO.Edge sCMOS detectors. Samples were illuminated from the dorsal side using a LSFM 10× NA 0.2 objective with pivot scan enabled. We used a 405/488/561 nm laser blocking filter combined with a LP 560 beam splitter to capture the red and green channels simultaneously. For anos1a:anos1a-GFP, prim:lyn2mCherry embryos the 488 nm laser power was set to 5% and the 561 nm laser power was set to 2%, with exposure times of 400 ms for each plane and an optical zoom of 2.5. For fgf10a:fgf10a-GFP; prim:lyn2mCherry embryos the 488 nm laser power was set to 5% and the 561 nm laser power was set to 2%, with exposure times of 800 ms for each plane and an optical zoom of 1.5.
mRNA constructs
cDNA encoding fgf10a-GFP, anos1a-mCherry (N-terminus tag), sec-mCherry, secGFP-linker-mCherry and iyniyn-mCeruiean were assembled into the pDest-hsp70-SV40pAtol2 plasmid backbone using Gibson cloning. For Fgf10a-GFP, the GFP was inserted between the secretion signal and mature Fgf10a protein. For Anos1a-mCherry, the mCherry was inserted after the Fgf3 secretion signal and immediately upstream of the mature Anos1a protein. Sec-mCherry and secGFP-linker-mCherry both consist of the respective fluorescent proteins with an upstream Fgf3 secretion signal. For secGFP-linker-mCherry, the two fluorescent proteins are separated by a 46 amino acid glycinealanine flexible linker. For lynlyn-mCerulean, mCerulean was cloned directly downstream of two tandem repeats of lyn transmembrane domain sequence. Templates for in vitro transcription were generated by PCR using the plasmid constructs as a template. The forward primer included a SP6 transcription start site, and the reverse primer primed at the end of the SV40pA site. To generate mRNA, we transcribed these templates using the mMessage mMachine SP6 Kit (Thermo Fisher) and purified the mRNA with phenol-chloroform extraction followed by isopropanol precipitation.
Dual color FCCS sample preparation
Wild-type zebrafish embryos were injected at the one-cell stage with 400 pg each of mRNAs encoding fgf10a-GFP, anos1a-mCherry, sec-mCherry and/or secGFP-linkermCherry and 100 pg of mRNA encoding lynlyn-mCerulean to mark the cell membranes with CFP.
Embryos were mounted in 0.5 % low melt agar with the animal pole directly against the coverslip at the 2000-cell stage. Dual color FCS measurements were performed with the laser positioned in the extracellular space of the cell layer directly under the enveloping layer at the animal pole between the oblong and dome stage.
Fluorescence correlation spectroscopy (FCS) and fluorescence cross correlation spectroscopy (FCCS) imaging
FCS measurements were taken using a Zeiss 880 laser scanning confocal microscope equipped with a C-Apochromat 40× NA=1.2 water-immersion objective, a spectral detector (32 channel GaAsP photomultiplier tube array), and an incubation chamber set to 28 degrees C. For single-color measurements, the laser beam (488 nm from a 25 mW argon laser with attenuators set to 1.5 μW (0.2 %) for Rhodamine 6G and 0.08 μW (0.01 %) for GFP) was coupled into the objective via a 488/561/633 nm main beam splitter, and the emitted fluorescence between 490–552 nm was recorded on the spectral detector for GFP and Rhodamine 6G. For dual-color measurements with high laser intensities, the 488 nm 25 mW laser was set to 3.86 μW (0.5 %) for Rhodamine 6G and GFP and the 561 nm 20mW laser set to 63 μW (2 %) for mCherry and Alexa Fluor 594, with the same optical setup as the single-color measurements.
For dual-color measurements with low laser intensities, the 488 nm 25 mW laser was set to 1.9 μW (0.25 %) for Rhodamine 6G and GFP and the 561 nm 20 mW laser set to 3.4 μW (0.1 %) for mCherry and Alexa Fluor 594, with the same optical setup as the singlecolor measurements. We note that the dual-color measurements with high laser intensities (488 nm at 3.86 μW, 561 nm at 63 μW), yielded better signal-to-noise ratios but caused bleaching of the fluorescent proteins during measuring. This resulted in an overestimation of the diffusion coefficients and an underestimation of the number of codiffusing particles. Dual-color measurements with low laser intensities (488 nm at 1.9 μW, 561 nm at 3.4 μW), did not result in significant fluorescent protein bleaching but in decreased signal-to-noise ratios. Emission wavelengths 499–552 nm were collected for GFP and Rhodamine 6G and 606–695 nm were collected for mCherry and Alexa Fluor 594. All samples were kept at 28 degree C during the measurements. The pinhole was set to 1 airy unit (31.8 μm in radius for single-color and 34.2 μm for dual-color measurements) and the confocal volumes of the two laser lines were calibrated using 10 nM Rhodamine 6G (R6G, ACROS Organics) and 5 nM Alexa Fluor 594 (A594, Thermo Fisher), respectively, in deionized water. All correlations of the FCS measurements were normalized to G(τ =1.6 μs) except for the measurements of Anos1a-GFP which were normalized to G(τ=10 μs)
Modeling description
The spatial modeling platform Simmune builds computational representations of (multi-) molecular complexes and their reaction-diffusion dynamics based on the specification of pairwise interactions between molecular binding sites (Angermann et al., 2012; Cheng et al., 2014; Zhang et al., 2013). These specifications can be entered into the software through a visual interface that uses iconographic symbols for molecules, their subdomains and binding sites. All model assumptions about the biochemical properties of the interacting molecular species are accessible through this interface. There are thus no ‘hidden’ assumptions in the mathematical representation of the model.
To perform simulated experiments with Simmune, one needs to specify the molecular interactions of the simulated model. This information is stored in the model file. The supplementary model file (Data S1) can be loaded into the Simmune Modeler for facile inspection (and, if desired, modification) of model parameters and molecular interactions. In addition to the model file describing the biochemistry, one needs to define the geometry (for instance a single cell with a particular cell morphology or an arrangement of multiple cells). This spatial information is stored in a geometry file that can be created and modified with the Simmune Cell Designer. Simulations are run by loading the simulation description file (Data S1) into the Simulator. The simulation description file contains the name of the model file and the geometry file (Data S1) and the initial molecular concentrations in the volume elements of the simulation. Thus, in order to rerun the simulations the results of which are reported here, it suffices to run the Simmune simulator and load the description file FGF_Anos1_HS.sdc. As long as the name and location of the model file (FGF_Anos1_HS.dbf) and the geometry file (FGF_Anos1_HS.geo) are not changed (the model description file assumes that all three files are in one directory/folder) the model file can be modified to alter, for instance, Anos1 production parameters. This is described in the model file annotation as well.
Model details
For the simulations of the reaction-diffusion system consisting of FGF, Anos1 and HS, we modeled the primordium as a 3D rectangular structure consisting of 126 individual cells arranged in a 3×3×14 shape (Fig. 8A). Each cell is represented as a cube of 7 microns side length, giving the primordium in our model a length (x-axis) of 98 microns and a width (y- and z-axis) of 21 microns. To have more control over the localized secretion of FGF and Anos1 towards the top of the primordium, the cells were divided into three cells along the vertical axis (as along the depth, the y-axis) even though the actual primordium consists of only one layer of cells vertically. Three cells on top of the primordium produce FGF while Anos1 is being released by a stripe of cells (Fig. S8B, right panel). All cells express HS on their surfaces while only the back half of the simulated primordium expresses the FGF receptor (Fig. 8A, S8B). The extracellular space is 140 microns long and 35 microns wide. To account for loss of FGF into the surrounding space, FGF was assigned a tag that represents availability of FGF and switches from on to off with a transition time of 1 second for freely diffusing FGF while receptor bound FGF will remain available for 1000 seconds (16.7 minutes) reflecting internalization and eventual diffusional loss. During the simulation, intra- and extracellular concentrations can be assessed through a visual user interface (Fig. 8A, S8B). All molecular reactions and rates for associations, dissociations and productions are shown in Fig. S8C.
QUANTIFICATION AND STATISTICAL ANALYSIS Image quantification
Wnt signaling reporter
We generated a mask of the primordium by thresholding the prim:lyn2mCherry channel and filling the holes to encompass the cytoplasm in addition to the membranes. We multiplied the GFP channel to this mask, removed the zeroes, and plotted the average GFP intensity at each anterior-posterior position against the distance from the front of the primordium. This procedure was automated using a custom-written imageJ script (Data S1).
FGF signaling reporter
We cropped a single Z-slice from the Z-stack that did not contain GFP signal from glial cells medial to the primordium and generated a mask of the primordium by thresholding the prim:lyn2mCherry channel and filling the holes to encompass the cytoplasm in addition to the membrane. We multiplied the GFP channel by this mask, removed the zeroes, and plotted the average GFP intensity at each anterior-posterior position against the distance from the front of the primordium. This procedure was automated using a custom-written imageJ script (Data S1). We defined the rear of the primordium to begin at the inflection point of the plots of the average GFP intensity at each anterior-posterior position. We used the position of the inflection point to calculate the average GFP intensity in the rear of each primordium.
Fgf10a-GFP and secGFP
To account for changes in microlumen intensity as proneuromasts matured, GFP intensity measurements were taken from the microlumen at the center of the fourth apical constriction from the front of the primordium. Confocal stacks were cropped in the axial dimension to only include planes within the boundary of each microlumen. These stacks were then sum projected, and an ROI for each microlumen was manually defined. Total GFP intensities were determined by measuring the integrated density (pixel area times mean pixel intensity) within each ROI.
Microlumina Volume Calculation
Embryos were fixed, stained with GFP and ZO-1 antibodies, and imaged using the Leica SP5 II confocal microscope with hybrid detectors set to standard mode and a Leica 40× NA 0.8 water-dipping objective (HCX APO L 40×/0.80 W U-V-I). Confocal stacks were cropped in the axial dimension to only include planes within the boundary of each microlumen. We generated a mask of the microlumen by thresholding the GFP channel and filling the holes. We multiplied the ZO-1 channel to this mask, removed zeroes, and sum projected the stacks. This procedure was automated using a custom-written imageJ script (Data S1). An ROI for each microlumen position was manually defined. Total volumes were determined by measuring the integrated density (pixel area times mean pixel intensity) within each ROI.
Neuromast size measurement
For the calculation of neuromast size, the region of the neuromast was manually defined by cropping. The GFP channel was thresholded to generate a mask that encompasses the neuromast. Then, the volume of the mask was determined by summing up the voxels in the mask. This procedure was automated using a custom-written ImageJ script (Data S1).
Fgf10a-GFP production and degradation rate measurement
To quantify the degradation of Fgf10a-GFP intensity in the microlumen, confocal stacks from each time point in embryos treated with Brefeldin were cropped in the axial dimension to only include planes within the boundary of each microlumen. These stacks were then sum projected, and a region of interest (ROI) for each microlumen was manually defined. Total GFP intensities were determined by measuring the integrated density (pixel area x mean pixel intensity) within each ROI. All these measurements were performed using ImageJ (NIH). The degradation of Fgf10a-GFP with time was fitted to a one phase decay model (y=(y(0) - a)*exp(−b*x) + a) using Prism 7 (GraphPad). To quantify Fgf10a-GFP production, confocal stacks from each time point were cropped in the axial dimension to only include planes with intracellular GFP signal in the central cells. These were sum projected, an ROI was manually defined, and total GFP intensity measured as integrated density. All these measurements were performed using ImageJ (NIH). The production of Fgf10a-GFP with time was fitted to a linear model (y=a* x + b) using Prism 7 (GraphPad).
P value calculation
All statistical analysis for image quantification were performed with Graphpad Prism 7 software. P-values reported for T-tests were calculated using unpaired one-tailed twosample t-tests assuming unequal sample variance (Welch’s t-test). For experiments involving multiple testing, samples were first compared using ordinary one-way ANOVA. If this test was significant, then the relevant pairwise comparisons were made. Reported P-values were calculated using Sidak’s multiple comparisons test with a single pooled variance.
FCS and FCCS quantification
Auto-Correlation Functions (ACF) were fitted with a model accounting for one diffusive particle with possible photophysical fluctuations:
| (1) |
Where Napp is the average number of fluorophores detected in the confocal volume, τp is the photophysics relaxation time, Fp is the fraction of the particles undergoing photophysical fluctuations, where ωxy and ωz are the 1/e2 intensity radii of the confocal volume in lateral and axial direction, respectively, and τD is the characteristic time that the observed molecules require to diffuse through the confocal volume. was calibrated using the known Rhodamine 6G diffusion coefficient D(301K) = 447μm2s_1(Müller et al., 2008), and the known Alexa A594 diffusion coefficient D(301K) = 374μm2s−1 (Nitsche et al., 2004). The apparent size of the overlapping volume between the two laser lines was calculated as:
where and ωz,cross are the apparent sizes of the overlapping volume in the lateral and axial directions, respectively; ωxy,G/R and ωz,G/R are the sizes of the illumination volume of the 488 nm/561 nm laser, in the lateral and axial directions, respectively (Schwille et al., 1997). The diffusion coefficient of the Cross-Correlation
Function (CCF) was calculated following:ACF measurements in fgf10a:fgf10a-GFP, fgf10a:fgf10a-GFP; anos1a−/−; anos1b−/−, fgf10a:fgf10a-GFP; hs:anos1b, fgf10a:fgf10a-GFP; fgfr1a−/−, fgf10a:sec-GFP, fgf10a:sec-GFP; anos1a−/− and anos1b−/−, anos1a:anos1a-GFP embryos and anos1a:anos1a-GFP embryos; hs:fgf10a, and CCF measurements in embryos injected with mRNA coding for fgf10a-GFP+anos1a-mCherry; fgf10a-GFP+sec-mCherry and sec-GFP-linker-mCherry were performed by positioning the illumination volume at the center of the microlumen of the first neuromast trailing the primordium (for FCS) or in the extracellular space directly underneath the enveloping layer of 3.5 to 4.5 hpf embryos (oblong to dome stage, for FCCS), and the emitted fluorescence was recorded for 10 seconds and its ACF and CCF were calculated in situ using an online auto-correlator (Zeiss) for every 10 second interval. In general, 10 such 10 second-ACFs and 5 such 10 second-CCFs were collected and averaged for each measurement. However, measurements with evident photo-bleaching and/or stage-drifting or sample movement as judged by ACF/CCF curves with non-zero convergence values were discarded before averaging. All measurements were performed at 28 degree C.
The ACF in the GFP channel in both ACF and CCF measurements, and the ACF in the mCherry channel in CCF measurements was fitted using a custom-written MATLAB script (The MathWorks Inc.) (Data S1). The ACF measurements in the early embryo were fitted to Equation (1) (Fig. S6). The ACF measurements in the mircolumen were fitted to Equation (1) (Fig. S5A-C) and to a 3D free-diffusion model according to Equation (2).
| (2) |
| (3) |
Where fi is the fraction of particles with their characteristic diffusion time τDi. The fit to Equation (1) included short time-lags (starting at 1.6 μs). The fit to Equation (2) excluded short time-lags (starting at 50 μs) to avoid the relatively lower signal-to-noise ratio in short time-lags. For the fitting to Equation (2), each ACF was fitted to both single-particle ( N = 1 ) and dual-particle ( N = 2 ) models, and we performed an F-test to determine if the latter was a significantly better fit against the single-particle model (with the p-value < 0.05). Note that the fast and slow diffusing components were assumed of similar brightness (see below), otherwise the and should be weighed as where Qi is the brightness of the ith component and needs to be carefully calibrated (Thompson, 2002). We note that there were unreasonable regression results for the dual-particle model (e.g. fi < 1 or τDi < 10 μs) in 8/61 (8 out of 61 measurements) for fgf10a:fgf10a-GFP, 6/83 for fgf10a:fgf10a-GFP; anos1a−/−; anos1b−/−, 5/53 for fgf10a:fgf10a-GFP; heat-shock control, 5/42 for fgf10a:fgf10a-GFP; heat-shocked hsp70:anos1b and 9/79 for anos1a:anos1a-GFP. We considered these dual-particle fits as failed fits despite the F-test indicating that the dualparticle model was a better fit than the single-particle model for these ACFs.
For the fit to Equation (2) we note that there were unreasonable regression results (e.g. Fp < 0 or τD < 10μs) in 30/61 (30 out of 61 measurements) for fgf10a:fgf10a-GFP, 38/83 for fgf10a:fgf10a-GFP; anos1a−/−; anos1b−/−, 16/53 for fgf10a:fgf10a-GFP; heatshock control, 21/42 for fgf10a:fgf10a-GFP; heat-shocked hsp70:anos1b and 30/79 for anos1a:anos1a-GFP. We considered these fits as failed fits due to the poor signal-tonoise ratio in the time window shorter than ~ 50 μs.
The CCF measurements in the early embryo were fitted using a one-diffusive particle model (Fig. S6A-C):
| (4) |
The diffusion coefficient was then calibrated according to with known diffusion coefficient of Rhodamine 6G or Alexa A594, or the calibrated overlapping confocal volume.
The CCF measurements using higher laser intensities did not yield unreasonable convergence during fitting (Fig. 6B, C). The CCF measurements using lower laser intensities yield unreasonable convergence during fitting for the ACF of mCherry and CCF of 1 out of 4 measurements of Fgf10-GFP and Anos1a-mCherrry and for the ACF of mCherry and CCF of 2 out of 7 measurements of Fgf10-GFP and sec-mCherrry (Fig. 6D, E).
The q-value was calculated as:
| (5) |
where the Gcross (0), GG (0), and Gr (0) are the fitted value at τ = 0 of CCF, ACF in GFP channel, and ACF in mCherry channel, respectively.
The concentration of GFP tagged protein was estimated via parameters fitted from the ACF in GFP channel:
| (6) |
where Fmean is the average fluorescence intensity, and β is the fluorescence background; NA is the Avogadro constant, and is the observation volume (Schwille et al., 1997).
In Fig. 5B and5C, we show the average of the ACFs for each genotype and the dualparticle fit of these averaged ACFs for the microlumen FCS measurements. Residuals for the dual-particle fit of these averaged ACFs are plotted in the graph below the ACFs. Individual ACFs for each genotype are plotted in Fig. S5A-S5C and overlaid with the dual particle fit of the averaged ACFs. Residuals for two-particle fits or one-particle fits of individual ACFs and the averaged ACFs are plotted below the ACFs in Fig. S5A-S5C.
In Fig. 5F, we show the averaged results for dual-particle fits of individual ACFs for Fgf10a-GFP and Anos1a-GFP and the averaged results for single-particle fits of individual ACFs for secreted GFP expressed from fgf10a:secGFP for the microlumen FCS measurements. Other FGF ligands have been shown to diffuse as two populations (Yu et al., 2009), and a significant portion of Anos1a ACFs were better fit by the dualparticle model than the single-particle model. For dual-particle fits, we only averaged fits where the F-test indicated that the dual-particle model fitted better than the singleparticle model and where the dual-particle model did not produce unreasonable fits (see above). We assume that signal-to-noise limitations prevented us from detecting the second population in the subset of ACFs where the dual-particle model did not fit the data significantly better than the single-particle model. Likewise, for single-particle fits, we only averaged fits where the F-test indicated that the dual-particle model did not fit better than the single-particle model. The excluded single-particle fits for Fgf10a-GFP and Anos1a-GFP and dual-particle fits for secGFP are displayed in Fig. S5D. In Fig. S5E we show the single-particle fits for the FCS measurements in the microlumen when including the short time-lags.
We note that the FCS measurements in the microlumen are only best described by a two-component model when we exclude the noisier short time-lags (Fig. S5D). When including shorter time-lags, the FCS measurements are best described by a onecomponent fit (Fig. S5E). However, in both cases the overall diffusivity of Fgf10a decreases in the absence of Anos1 activity.
To determine the low intensities for the 488 nm laser line, we calibrated the laser intensities given in percentage to actual laser intensity measurements on the microscope stage (X-Cite Power Meter, Lumen Dynamics Group Inc.) and fitted these to a linear model (y = a*x). The 0.01 % and 0.25 % laser intensities for the 488 nm laser line are extrapolated values.
Photon counting histogram analysis
The open source Fiji plugin-”analysis PCH” by Jay Unruh1 (Stowers Institute, http://research.stowers.org/imagejplugins/zipped_plugins.html) was used to read the Carl Zeiss confocor3 raw data (*.RAW files), to calculate the photon counting histogram and to fit the photon counting histogram to a 3D Gaussian point spread function model. A bin time of 17 μs was used to calculate the PCH from acquisitions each lasting 100 s.
DATA AND SOFTWARE AVAILABILITY
The Simmune software we used for modeling can be downloaded (without the need to register) from https://www.niaid.nih.gov/research/simmune-project. The downloaded Simmune software package contains documentation and a tutorial which illustrate the process of model creation and simulation.
Data S1. ImageJ and MATLAB scripts and Simmune modeling files, Related to Figures 3 and 5 to 8.
(File 1) ImageJ script used for Wnt signaling reporter analysis, related to Figures 3 and 7. (File 2) ImageJ script used for Fgf signaling reporter analysis, related to Figures 3 and 7. (File 3) ImageJ script used for ZO-1 volume analysis, related to Figures 3 and 7.
(File 4) ImageJ script used for neuromast volume analysis, related to Figure 3.
(File 5) MATLAB script used for fitting FCS auto- and cross-correlation curves, related to Figures 5 and 6.
(File 6 to 8) Files for Simmune model, related to Figure 8.
Supplementary Material
Data S1. ImageJ and MATLAB scripts and Simmune modeling files, Related to Figures 3 and 5 to 8.
(File 1) ImageJ script used for Wnt signaling reporter analysis, related to Figures 3 and 7.
(File 2) ImageJ script used for Fgf signaling reporter analysis, related to Figures 3 and 7.
(File 3) ImageJ script used for ZO-1 volume analysis, related to Figures 3 and 7.
(File 4) ImageJ script used for neuromast volume analysis, related to Figure 3.
(File 5) MATLAB script used for fitting FCS auto- and cross-correlation curves, related to Figures 5 and 6.
(File 6 to 8) Files for Simmune model, related to Figure 8.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-ZO-1 | Thermo Fisher | Cat No. 33–9100 |
| Mouse monoclonal anti-zns-2 | DSHB | AB 531907 |
| Rabbit polyclonal anti-GFP | Thermo Fisher | A-11122 |
| Goat polyclonal anti-GFP | (Venkiteswaran et al., 2013) | |
| Sheep polyclonal anti-mCherry | Covance, this paper | |
| Donkey anti-Goat 488 | Thermo Fisher | A-11055 |
| Goat anti-Rabbit 488 | Thermo Fisher | A-11034 |
| Donkey anti-Rabbit Cy3 | Jackson Immunoresearch | Cat No. 711–166-152 |
| Donkey anti-Goat Cy3 | Jackson Immunoresearch | Cat No. 705–166-147 |
| Donkey anti-Sheep 647 | Jackson Immunoresearch | Cat No. 713–606-147 |
| Donkey anti-Mouse Cy3 | Jackson Immunoresearch | Cat No. 715–166-150 |
| Donkey anti-Mouse 647 | Jackson Immunoresearch | Cat No. 715–606-150 |
| Sheep anti-DIG AP | Sigma Aldrich | SKU 11093274910 |
| Bacterial and Virus Strains | ||
| NEB 5-alpha | New England Biolabs | Cat No C2987H |
| sw105 | (Warming et al., 2005) | |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| NBT | Sigma Aldrich | SKU 11383213001 |
| BCIP | Sigma Aldrich | SKU 11383221001 |
| Digoxigenin-labeled nucleotides | Sigma Aldrich | SKU 11277073910 |
| Trizol | Thermo Fisher | Cat No 15596026 |
| Paraformaldehyde | Sigma Aldrich | SKU P6148–500G |
| Proteinase K | Sigma Aldrich | SKU P6556–1G |
| Bovine Serum Albumin | Sigma Aldrich | SKU A8022–50g |
| Methycellulose | Sigma Aldrich | SKU 1424506–1G |
| Glycerol | Sigma Aldrich | SKU G7893–1L |
| Tricaine (Ethyl 3-aminobenzoate methanesulfonate) | Sigma Aldrich | SKU E10521– 50G |
| Low melt agarose (AquaPor HR) | National Diagnostics | Cat No EC-205 (100g) |
| Rhodamine 6G | Fischer Scientific | Cat No AC419010050 |
| Alexa Fluor 594 Carboxylic Acid, tris(triethylammonium) salt | Thermo Fisher | A33082 |
| BsrI | New England Biolabs | R0527S |
| Hpych4v | New England Biolabs | R0620L |
| AciI | New England Biolabs | R0551S |
| ApoI | New England Biolabs | R0566L |
| Taqαi | New England Biolabs | R0149L |
| Brefeldin A | Sigma Aldrich | B7651–5MG |
| Critical Commercial Assays | ||
| Thermoscript Reverse Transcriptase | Thermofisher | 11146–024 |
| TOPO TA cloning | Thermofisher | Cat No K461020 |
| mMessage mMachine T7 kit | Thermofisher | AM1344M |
| mMessage mMachine SP6 kit | Thermofisher | AM 1340 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| fgf3/liat24149 | (Herzog et al., 2004) | |
| fgf10a/daetbvbo° | (Norton, 2005) | |
| cldnb:lyn2GFP | (Haas and Gilmour, 2006) | |
| hsp70:dn-fgfr1 | (Halloran et al., 2000) | |
| dusp6:d2EGFP | (Molina et al., 2007) | |
| Tcf/Lef-miniP:dGFP | (Shimizu et al., 2012) | |
| cdh2:cdh2-sfGFP-TagRFP | (Revenu et al., 2014) | |
| cxcr4b:cxcr4b-Kate2-ires-GFP-CAAX | (Venkiteswaran et al., 2013) | |
| fgfr1a/spdt3R705H | (Rohner et al., 2009) | |
| prim:lyn2mCherry | this paper | |
| hsp70:anos1b | this paper | |
| hsp70:sec-mCherry | this paper | |
| hsp70:fgf10a | this paper | |
| fgf10a: GFP-fgf10a | this paper | |
| fgf10a:secGFP | this paper | |
| anos1a:GFP-anos1a | this paper | |
| cxcr4b:H2A-mCherry | this paper | |
| anos1ad5 | this paper | |
| anos1ad7 | this paper | |
| anos1bd5 | this paper | |
| Anos1bd79 | this paper | |
| Oligonucleotides | ||
| For DNA oligonucleotides, see Table S1 | ||
| atoh1a morpholino: 5’- ATCCATTCTGTTGGTTT GTGCTTTT-3’ | Gene Tools | Custom synthesis |
| Recombinant DNA | ||
| pCRII | Thermo Fisher | Cat No K461020 |
| pDest-hsp70-SV40pA-tol2 | (Kwan et al., 2007) | |
| CH211–262I10 (fgf10a BAC) | BACPAC Resources Center | |
| CH211–267B13 (anos1a BAC) | BACPAC Resources Center | |
| DKEY-169F10 (cxcr4b BAC) | Source Bioscience | |
| Golden Gate tAlEN and tAl Effector Kit 2.0 | Addgene | ID 1000000024 |
| Software and Algorithms | ||
| Simmune | https://www.niaid.nih.gov/research/simmune-project | |
| Prism 7 | Graphpad | |
| FIJI (ImageJ v1.48) | https://fiji.sc/ | |
| Other | ||
Highlights.
-
-
Fgf10 diffuses from front to back of migrating primordium to induce sensory organs
-
-
Anosmin1 is required for front-to-back diffusion of Fgf10
-
-
Anosmin1 increases the fraction of fast diffusing Fgf10 molecules
-
-
Anosmin1 co-diffuses with Fgf10 molecules in a complex
Acknowledgement
We thank J. Torres-Vazquez, T. Colak-Champollion and N. Yamaguchi for critical comments; D. Grunwald, D. Gilmour, A. Nechiporuk, M. Harris, M. Tsang and T. Ishitani for reagents and F. Fuentes and T. Gerson for excellent fish care. The zns-2 monoclonal antibody developed by Bill Trevarrow was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242. This work was supported by NIH grants NS069839 (H.K.), DC016073 (H.K.), HD079229 (J.W.), GM007308 (J.W.), in part by the intramural program of the National Institute of Allergy and Infectious Diseases (NIAID), the Singapore Ministry of Education grant MOE2016-T3–1-005 (T.W. and J.S.) and the American Cancer Society grant 130304-RSG-16–241-01-DMC (E.R.).
Footnotes
Declaration of interest
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aman A and Piotrowski T (2008). Wnt/beta-catenin and Fgf signaling control collective cell migration by restricting chemokine receptor expression. Dev. Cell 15, 749–761. [DOI] [PubMed] [Google Scholar]
- Angermann BR, Klauschen F, Garcia AD, Prustel T, Zhang F, Germain RN and Meier-Schellersheim M (2012). Computational modeling of cellular signaling processes embedded into dynamic spatial contexts. Nat. Meth 9, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardouin O, Legouis R, Fasano L, David-Watine B, Korn H, Hardelin J and Petit C (2000). Characterization of the two zebrafish orthologues of the KAL-1 gene underlying X chromosome-linked Kallmann syndrome. Mech. Dev 90, 89–94. [DOI] [PubMed] [Google Scholar]
- Bacia K, Kim SA and Schwille P (2006). Fluorescence cross-correlation spectroscopy in living cells. Nat. Meth 3, 83–89. [DOI] [PubMed] [Google Scholar]
- Belov AA and Mohammadi M (2013). Molecular Mechanisms of Fibroblast Growth Factor Signaling in Physiology and Pathology. Cold Spring Harbor Perspectives in Biology 5, a015958–a015958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JR, Schuksz M and Esko JD (2007). Heparan sulphate proteoglycans finetune mammalian physiology. Nature Cell Biology 446, 1030–1037. [DOI] [PubMed] [Google Scholar]
- Cheng H-C, Angermann BR, Zhang F and Meier-Schellersheim M (2014). NetworkViewer: visualizing biochemical reaction networks with embedded rendering of molecular interaction rules. BMC Syst. Biol 8, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlem TJ, Hoshijima K, Jurynec MJ, Gunther D, Starker CG, Locke AS, Weis AM, Voytas DF and Grunwald DJ (2012). Simple Methods for Generating and Detecting Locus-Specific Mutations Induced with TALENs in the Zebrafish Genome. PLoS Genet. 8, e1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayel MJ, Hom EFY and Verkman AS (1999). Diffusion of Green Fluorescent Protein in the Aqueous-Phase Lumen of Endoplasmic Reticulum. Biophys. J 76, 2843–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro F, Esteban PF, Bribián A, Murcia-Belmonte V, García-González D and Clemente D (2013). The Adhesion Molecule Anosmin-1 in Neurology: Kallmann Syndrome and Beyond In Advances in Neurobiology, Berezin V and Walmod PS, eds. (New York, Springer; New York, USA: ), pp. 273–292. [DOI] [PubMed] [Google Scholar]
- Díaz-Balzac CA, Lázaro-Peña MI, Ramos-Ortiz GA and Bulow HE (2015). The Adhesion Molecule KAL-1/anosmin-1 Regulates Neurite Branching through a SAX- 7/L1CAM-EGL-15/ FGFR Receptor Complex. Cell Reports 11, 1377–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Balzac CA, Lázaro-Peña MI, Tecle E, Gomez N and Bülow HE (2014). Complex cooperative functions of heparan sulfate proteoglycans shape nervous system development in Caenorhabditis elegans. G3; Genes|Genomes|Genetics 4, 1859–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durdu S, Iskar M, Revenu C, Schieber N, Kunze A, Bork P, Schwab Y and Gilmour D (2014). Luminal signalling links cell communication to tissue architecture during organogenesis. Nature 515, 120–124. [DOI] [PubMed] [Google Scholar]
- Foo YH, Naredi-Rainer N, Lamb DC, Ahmed S and Wohland T (2012). Factors Affecting the Quantification of Biomolecular Interactions by Fluorescence Cross-Correlation Spectroscopy. Biophys. J 102, 1174–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes F, Reynolds E, Lewellis SW, Venkiteswaran G and Knaut H (2016). A Plasmid Set for Efficient Bacterial Artificial Chromosome (BAC) Transgenesis in Zebrafish. G3: Genes|Genomes|Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanternik MV, Kramer KL and Piotrowski T (2015). Heparan Sulfate Proteoglycans Regulate Fgf Signaling and Cell Polarity during Collective Cell Migration. Cell Reports 10, 414–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA and Smith HO, (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Meth 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Goetz R and Mohammadi M (2013). Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol 14, 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas P and Gilmour D (2006). Chemokine signaling mediates self-organizing tissue migration in the zebrafish lateral line. Dev Cell 10, 673–680. [DOI] [PubMed] [Google Scholar]
- Halloran MC, Sato-Maeda M, Warren JT, Su F, Lele Z, Krone PH, Kuwada JY and Shoji W (2000). Laser-induced gene expression in specific cells of transgenic zebrafish. Development 127, 1953–1960. [DOI] [PubMed] [Google Scholar]
- Hardelin JP and Dode C (2008). The Complex Genetics of Kallmann Syndrome: KAL1, FGFR1, FGF8, PROKR2, PROK2, et al. Sex Dev 2, 181–193. [DOI] [PubMed] [Google Scholar]
- Herzog W, Sonntag C, Hardt, von der S, Roehl HH, Varga ZM and Hammerschmidt M (2004). Fgf3 signaling from the ventral diencephalon is required for early specification and subsequent survival of the zebrafish adenohypophysis. Development 131, 3681–3692. [DOI] [PubMed] [Google Scholar]
- Hu Y, Guimond SE, Travers P, Cadman S, Hohenester E, Turnbull JE, Kim SH and Bouloux P-M (2009). Novel mechanisms of fibroblast growth factor receptor 1 regulation by extracellular matrix protein anosmin-1. Journal of Biological Chemistry 284, 29905–29920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecker C and Lumsden A (2012). The Role of Organizers in Patterning the Nervous System. Annu. Rev. Neurosci 35, 347–367. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B and Schilling TF (1995). Stages of embryonic development of the zebrafish. Dev. Dyn 203, 253–310. [DOI] [PubMed] [Google Scholar]
- Kok FO, Shin M, Ni C-W, Gupta A, Grosse AS, van Impel A, Kirchmaier BC, Peterson-Maduro J, Kourkoulis G, Male I et al. (2015). Reverse Genetic Screening Reveals Poor Correlation between Morpholino-Induced and Mutant Phenotypes in Zebrafish. Dev. Cell 32, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP and Chien C-B (2007). The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn 236, 3088–3099. [DOI] [PubMed] [Google Scholar]
- Lecaudey V, Cakan-Akdogan G, Norton WHJ and Gilmour D (2008). Dynamic Fgf signaling couples morphogenesis and migration in the zebrafish lateral line primordium. Development 135, 2695–2705. [DOI] [PubMed] [Google Scholar]
- Lee Y (2005). Fgf signaling instructs position-dependent growth rate during zebrafish fin regeneration. Development 132, 5173–5183. [DOI] [PubMed] [Google Scholar]
- Lewellis SW, Nagelberg D, Subedi A, Staton A, LeBlanc M, Giraldez A and Knaut H (2013). Precise SDF1-mediated cell guidance is achieved through ligand clearance and microRNA-mediated decay. The Journal of Cell Biology 200, 337–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sun C, Yates EA, Jiang C, Wilkinson MC and Fernig DG (2016). Heparin binding preference and structures in the fibroblast growth factor family parallel their evolutionary diversification. Open Biol. 6, 150275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarenkova HP, Hoffman MP, Beenken A, Eliseenkova AV, Meech R, Tsau C, Patel VN, Lang RA and Mohammadi M (2009). Differential interactions of FGFs with heparan sulfate control gradient formation and branching morphogenesis. Science Signaling 2, ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda M and Chitnis AB (2010). Atoh1a expression must be restricted by Notch signaling for effective morphogenesis of the posterior lateral line primordium in zebrafish. Development 137, 3477–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millimaki BB, Sweet EM, Dhason MS and Riley BB (2007). Zebrafish atoh1 genes: classic proneural activity in the inner ear and regulation by Fgf and Notch. Development 134, 295–305. [DOI] [PubMed] [Google Scholar]
- Molina GA, Watkins SC and Tsang M (2007). Generation of FGF reporter transgenic zebrafish and their utility in chemical screens. BMC Dev. Biol 7, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller CB, Loman A, Pacheco V, Koberling F, Willbold D, Richtering W and Enderlein J (2008). Precise measurement of diffusion by multi-color dual-focus fluorescence correlation spectroscopy. Europhys. Lett 83, 46001–6. [Google Scholar]
- Nechiporuk A and Raible DW (2008). FGF-dependent mechanosensory organ patterning in zebrafish. Science 320, 1774–1777. [DOI] [PubMed] [Google Scholar]
- Nitsche JM, Chang H-C, Weber PA and Nicholson BJ (2004). A Transient Diffusion Model Yields Unitary Gap Junctional Permeabilities from Images of Cell-to- Cell Fluorescent Dye Transfer Between Xenopus Oocytes. Biophys. J 86, 2058–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogare DD and Chitnis AB (2017). A framework for understanding morphogenesis and migration of the zebrafish posterior Lateral Line primordium. Mech. Dev 148, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R, Kondev J, Theriot J and Garcia H (2012). Physical Biology of the Cell, 2nd ed. (New York: Garland Science; ). [Google Scholar]
- Rohner N, BercsEnyi M, OrbAn L, Kolanczyk ME, Linke D, Brand M, Nüsslein- Volhard C and Harris MP (2009). Duplication of fgfr1 Permits Fgf Signaling to Serve as a Target for Selection during Domestication. Curr. Biol 19, 1642–1647. [DOI] [PubMed] [Google Scholar]
- Rossi A, Kontarakis Z, Gerri C, Nolte H, Hölper S, Krüger M and Stainier DYR, (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233. [DOI] [PubMed] [Google Scholar]
- Rugarli EI, Di Schiavi E, Hilliard MA, Arbucci S, Ghezzi C, Facciolli A, Coppola G, Ballabio A and Bazzicalupo P (2002). The Kallmann syndrome gene homolog in C. elegans is involved in epidermal morphogenesis and neurite branching. Development 129, 1283–1294. [DOI] [PubMed] [Google Scholar]
- Schwanzel-Fukuda M, Bick D and Pfaff DW (1989). Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Brain Res. Mol. Brain Res 6, 311–326. [DOI] [PubMed] [Google Scholar]
- Schwille P, Meyer-Almes FJ and Rigler R (1997). Fluorescence. Biophys. J 72, 1878–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty S, 2015. Olfactory Agenesis in Kallmann Syndrome (KS). JCDR 1–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Kawakami K and Ishitani T (2012). Visualization and exploration of Tcf/Lef function using a highly responsive Wnt/b-catenin signaling-reporter transgenic zebrafish. Dev. Biol 370, 71–85. [DOI] [PubMed] [Google Scholar]
- Shimizu N, Kawakami K and Ishitani T (2012). Visualization and exploration of Tcf/Lef function using a highly responsive Wnt/b-catenin signaling-reporter transgenic zebrafish. Dev. Biol 370, 71–85. [DOI] [PubMed] [Google Scholar]
- Sudhaharan T, Liu P, Foo YH, Bu W, Lim KB, Wohland T and Ahmed S (2009). Determination of in Vivo Dissociation Constant, K D, of Cdc42-Effector Complexes in Live Mammalian Cells Using Single Wavelength Fluorescence Cross-correlation Spectroscopy. Journal of Biological Chemistry 284, 13602–13609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecle E, Diaz-Balzac CA and Bulow HE (2013). Distinct 3-O-Sulfated Heparan Sulfate Modification Patterns Are Required for kal-1-Dependent Neurite Branching in a Context-Dependent Manner in Caenorhabditis elegans. G3: Genes|Genomes|Genetics 3, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermes V, Grabher C, Ristoratore F, Bourrat F, Choulika A, Wittbrodt J and Joly J-S (2002). I-SceI meganuclease mediates highly efficient transgenesis in fish. Mech. Dev 118, 91–98. [DOI] [PubMed] [Google Scholar]
- Thisse C and Thisse B (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nature Protocols 3, 59–69. [DOI] [PubMed] [Google Scholar]
- Thompson NL (2002). Fluorescence Correlation Spectroscopy In Topics in Fluorescence Spectroscopy, Lakowicz JR, ed. (Springer, Germany: ), pp. 337–378. [Google Scholar]
- Tornberg J, Sykiotis GP, Keefe K, Plummer L, Hoang X, Hall JE, Quinton R, Seminara SB, Hughes V, Van Vliet G, et al. (2011). Heparan sulfate 6-O- sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proceedings of the National Academy of Sciences 108, 11524–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N and Grose R (2010). Fibroblast growth factor signalling: from development to cancer. Nat. Rev. Cancer 10, 116–129. [DOI] [PubMed] [Google Scholar]
- Venkiteswaran G, Lewellis SW, Wang J, Reynolds E, Nicholson C and Knaut H (2013). Generation and Dynamics of an Endogenous, Self-Generated Signaling Gradient across a Migrating Tissue. Cell 155, 674–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA and Copeland NG (2005). Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Research 33, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada YEHI-EK, Ishiwata-Endo H and Yamada KM (2012). Extracellular Matrix Protein Anosmin Promotes Neural Crest Formation and Regulates FGF, BMP, and WNT Activities. Dev. Cell 23, 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanicostas C, Ernest S, Dayraud C, Petit C and Soussiyanicostas N (2008). Essential requirement for zebrafish anosmin-1a in the migration of the posterior lateral line primordium. Dev. Biol 320, 469–479. [DOI] [PubMed] [Google Scholar]
- Yu SR, Burkhardt M, Nowak M, Ries J, Petrášek Z, Scholpp S, Schwille P and Brand M (2009). Fgf8 morphogen gradient forms by a source-sink mechanism with freely diffusing molecules. Nature 461, 533–536. [DOI] [PubMed] [Google Scholar]
- Zhang F, Angermann BR, and Meier-Schellersheim M (2013). The Simmune Modeler visual interface for creating signaling networks based on bi-molecular interactions. Bioinformatics 29, 1229–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.