

Abstract
Cisplatin is an antitumor drug used in the treatment of a wide variety of malignancies. However, its primary dose-limiting side effect is kidney injury, which is a major clinical concern. To help understand mechanisms involved in the development of kidney injury, cisplatin rodent model has been developed. Given the complex pathogenesis of kidney injury, which involves both local events in the kidney and interconnected and interdependent systemic effects in the body, cisplatin rodent model is indispensable in the investigation of underlying mechanisms and potential treatment strategies of both acute and chronic kidney injury. Cisplatin rodent model is well appreciated and widely used model due to its simplicity. It has many similarities to human cisplatin nephrotoxicity, which are mentioned in the paper. In spite of its simplicity and wide applicability, there are also traps that need to be taken into account when using cisplatin model. The present paper is aimed at giving a concise insight into the complex characteristics of cisplatin rodent model and heterogeneity of cisplatin dosage regimens as well as outlining factors that can severely influence the outcome of the model and the study. Challenges for future research are also mentioned.
1. Introduction
Cisplatin is an antitumor drug used in the treatment of a wide variety of malignancies (head and neck, lung, testis, ovary, and breast). It has limited use in clinical practice due to its side effects, particularly nephrotoxicity. Nowadays, 20-30% of patients develop acute kidney injury (AKI) after cisplatin treatment despite improvements in therapy [1]. Patients who develop AKI have an increased risk of mortality and are more likely to develop chronic kidney injury (CKI) [2].
To help understand complex mechanisms involved in the development of kidney injury cisplatin rodent model has been developed and extensively used to investigate cisplatin metabolism and the molecular mechanisms of cisplatin nephrotoxicity [3, 4] and to test new generations of platinum-based chemotherapy drugs or adjunctive therapies [5, 6] or other potential agents [7] or strategies to prevent or treat AKI (for instance, stem cells) [8].
Cisplatin rodent model has been recognized as a simple and reproducible model with high clinical relevance [9]. In the past, it was mainly used to investigate acute nephrotoxicity. The investigation of the development of CKI was not gaining any attention, although it was known that cisplatin can have long term effects on the structure and function of the rat kidney after single [10–12] or multiple [13–15] administration. Due to the recent recognition that repeated cisplatin treatment in humans is often associated with renal interstitial fibrosis, leading to CKI [16], and the fact that CKI may develop without being detected [2, 17], the development of better cisplatin mouse models was proposed with aim of increasing the likelihood of identifying novel therapeutic targets for the treatment of cisplatin-induced kidney injury [18].
Namely, cisplatin rodent model has been a subject of a critique as well. Because it has failed to translate treatment strategies of AKI to humans [19] it was argued that it lacks resemblance with human AKI in some aspects, such as morphological changes in the kidney [20] or cisplatin dosing regimen [19]. Thus, it is important to note that cisplatin model has limitations. The main weakness of the model, particularly mouse cisplatin model, is the fact that it is not standardized. This means that almost every laboratory uses its own protocol. Consequently, the protocols among studies differ significantly (i.e., from low nephrotoxic to extremely high lethal doses of cisplatin). The use of wide variety of protocols, thus, causes difficulties in the comparison of results and in the establishment of valid therapeutic strategies. In addition, it also raises the doubt into the usefulness of a model and serious ethical consideration upon animal research. As demonstrated in the paper, rodents, particularly mice, are too frequently exposed to severe suffering. Namely, depending on the cisplatin dosage regimen (cisplatin protocol) rodents may develop not only different severity of nephrotoxicity but also extrarenal toxicity or even systemic toxicity. Thus, for both ethical and scientific reasons a concise insight into complex characteristics of a model in regard to cisplatin dosing regimen is more than needed. Although many excellent articles about cisplatin rodent model have already been published [9, 18, 21], none of them summarized the complexity of its characteristics or discussed the influence of cisplatin dosage regimen on the results or study outcome.
Thus, the first aim of this paper is to briefly summarize the various cisplatin protocols to demonstrate that variations in cisplatin protocols significantly influence not only the results but also the model itself and in some cases may even hamper the comparison and interpretation of the results. The second aim is to point out factors that can significantly influence the model, the outcome of the study, and consequently the validity of the results. The third aim is to point out ethical and scientific concerns and to expose challenges that need to be addressed in the future research.
2. Cisplatin Nephrotoxicity in Rodents
The experimental cisplatin-induced nephrotoxicity was first reported in 1971 [22]. Since then numerous studies were published. Over the past years researchers have demonstrated that cisplatin nephrotoxicity is dose dependent and cumulative. Nephrotoxicity can be induced by single or multiple applications of cisplatin. Depending on the dosage, frequency of cisplatin injection, and cumulative dose of cisplatin, animals may develop different severity of acute (early) and chronic (advanced) kidney injury. In rodents cisplatin is usually injected intraperitoneally (ip) and less frequently intravenously (iv) or subcutaneously (sc).
To better understand variations among cisplatin protocols (single/multidosage treatment, low/high nephrotoxic dose, and lethal dose) basic knowledge about pharmacokinetics and underlying mechanisms of cisplatin nephrotoxicity is briefly summarized. However, more information about the pathogenesis of cisplatin nephrotoxicity can be found in many excellent papers [23–28].
2.1. The Uptake and Elimination of Cisplatin
Cisplatin is water soluble and low-molecular-weight drug. Following single intraperitoneal administration, cisplatin reaches systemic circulation, where it irreversibly binds to plasma proteins to form inactive complexes, which are considered metabolically inactivated [29]. Unbound cisplatin undergoes distribution to nearly all organs very rapidly. Within 1 hour plasma cisplatin levels decline significantly. Cisplatin is eliminated predominantly by the kidney, much less by biliary (1.2%) [30] and intestinal excretion [31]. The kinetics of cisplatin decay is a biphasic in nature. Cisplatin concentration decreases in the kidney very rapidly after the initial accumulation of the drug (within 15 min) but then again increases and reaches the second peak 48-72 h after a single cisplatin administration [32]. Thereafter it decreases very slowly [33]. Significant levels of the drug were found in the kidney for as long as 1 month [11, 33] or even 3 months after a single nephrotoxic dose of cisplatin [33]. About 43-50% of the drug is eliminated in the urine in the first 24 hours [23, 30], 60-76% in the first 48 hours [23, 34], and about 91% in 72 hours after single cisplatin administration (dose 4-10 mg/kg) [35]. At 72 h after cisplatin administration, the highest concentration of cisplatin was found in the mitochondria (37%), followed by cytosol (27%), nuclei (22%), and microsomes (14%) [32]. Using visualization methodology, it was shown that cisplatin accumulates mostly in the inner cortex and corticomedullary junction of the rat kidney, which is the location of proximal and distal tubules (dose: 5 mg/kg; observation time: day 5). When lethal dose was used (16 mg/kg) cisplatin was found also in renal columns (observation time: day 3), while in the medulla (location of the loop of Henley and collecting tubule) the levels of cisplatin were the lowest, regardless of the dose [36]. In addition, it was found that the intraperitoneal application of cisplatin has a reservoir effect, which prolongs the serum half-life of cisplatin in comparison to the intravenous route [37].
In addition, accumulation and elimination of cisplatin after multidosage treatment has been also examined. It was found that after multiple repeated administration of cisplatin (ip; 5 cycles of 16 mg/m2; the elapsed time between each cycle was 21 days) relative cisplatin concentration in the kidney decreased between the first and the second cycle; from the second to the fifth cycle it remained almost constant, while after the fifth cycle it significantly increased [33]. Similarly, another research group observed that multiple repeated administration of cisplatin resulted in decreased renal clearance and increased accumulation of cisplatin in the kidney by each cycle (iv; 3 cycles of 5 mg/kg, a 21-day washout period between each cycle), suggesting long elimination half-life of cisplatin and a collapse of the elimination/detoxification mechanisms [29].
2.2. The Pathogenesis of Cisplatin Nephrotoxicity
In rodents cisplatin enters the renal epithelial cells from glomerular filtrate mostly by active transport mediated by the copper transporter 1 (Ctr1) [38], by organic cation transporter 2 (OCT 2), [39, 40], the multidrug and toxin extrusion 1 (MATE1) [41], and to a lesser extent by a passive diffusion (pinocytosis) [36]. In rodents Ctr1 is mainly localized at the basolateral side of both proximal and distal tubules [38], OCT 2 is highly expressed at the basolateral membrane of proximal tubules [39, 40], and MATE1 is localized at the brush-border membrane of proximal tubules [41], while a passive diffusion (pinocytosis) takes place through the cellular membrane (lipid rafts) at the apical membrane of epithelial cells of the proximal tubule [36].
In the renal epithelial cell cisplatin then undergoes metabolic activation to highly reactive molecule, which affects cellular antioxidant system (oxidative and nitrosative status) [4] (demonstrated by decreased superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx) activity, and decreased glutathione (GSH), glutathione disulfide (GSSG), and nicotinamide adenine dinucleotide phosphate (NADPH) levels) and interacts with different cellular components and macromolecules causing functional and structural damage of proteins (demonstrated by the formation of carbonyl and decrease of P-SH-sulfhydryl proteins), lipids (increased malondialdehyde (MDA), 4-hydroxynonenal- (4-HNE-) oxidative damage) [42], and cellular organelles such as mitochondria [42–44] and endoplasmatic reticulum. Many molecular pathways are triggered in the tubular epithelial cells. Cisplatin nephrotoxicity results in alteration in the number and size of lysosomes and mitochondria [10], disruption of the cytoskeletal integrity and cell polarity, loss of brush border [10, 44], mislocalisation of membrane proteins such as the sodium/potassium ATPase, decreased number of aquaporin water channels (AQP2 and AQP3 in collecting duct and AQP1 in proximal nephron and renal microvasculature) [45], which are responsible for urinary concentration defect [46]. Depending on the dosage cisplatin may lead to cell injury or cell death, i.e., autophagy, apoptosis, and necrosis [47, 48]. In response to cisplatin a number of cytokines are upregulated; various receptors and variety of leukocyte populations are either increased or activated leading to the inflammation. The inflammation of the renal interstitium additionally contributes to the damage [49]. Locally secreted cytokines attract circulating leukocytes into the renal tissue. Erythrocyte accumulation, leukocyte plugging, increased capillary permeability, and leakage of the plasma water into the renal interstitium affect renal function and result in impaired proximal and distal tubular reabsorptive capacity, reversible changes in the renal blood flow, and increased renal vascular resistance (reduced filtration pressure due to afferent arteriolar vasoconstriction was observed 2-3 days after cisplatin administration in rats, 5 mg/kg ip) [49, 50]. Cisplatin has direct effects on the vasculature and glomeruli in rodents. In the kidney vascular injury was reported 1 day after cisplatin administration [51, 52]. Structural changes of the peritubular microvasculature were seen by electron microscopy as endothelial cell swelling, cytoplasmic vacuolization, nuclear degeneration, and detachment [53]. The damage of proximal and distal tubules reduces reabsorptive capacity of the tubular cells, which result in a reduction in glomerular filtration rate (GFR) [54–56], polyuria (reduced reabsorption in tubules due to decreased expression of water channels along the nephron, i.e., aquaporins) [36, 57, 58], a marked defect in urine concentrating ability, increased excretion of proteins [36, 54, 55, 57, 58], glycosuria (urinary glucose wasting) [55], increased excretion of magnesium (Mg) [55], sodium (Na) [57], reduced creatinine (Cr) clearance [55], increased production of hydrogen peroxide, and reduced antioxidant capacity [54]. Extensive morphological damage and functional impairment ultimately lead to the failure of the kidneys to clear nitrogenous wastes from the blood. As a result, blood urea nitrogen (BUN) and uric acid accumulate in the blood (i.e., azotemia denotes elevated levels of nitrogenous waste products in the blood and hyperuricemia denotes excess uric acid in the blood) [59]. Most common metabolites used as biomarkers to diagnose the nephrotoxicity in rodents are BUN and serum Cr, while GFR measurement is less frequent due to technical reasons. Classical GFR measurements involve repeated blood and/or continuous urine sampling over a prolonged time period (5-24 hours), while a novel GFR method involves implantation of transcutaneous device [60].
2.3. Molecular Mechanisms and the Inflammation
With use of microarray technology [61–63] it was demonstrated that cisplatin affects numerous genes that are involved in various functions in the kidney, such as biochemical pathways related to creatinine biosynthesis, osmoregulation, kinase signaling, cell cycle-related genes, renal transporters, renal injury, regenerative responses, gene expression changes related to drug metabolism, detoxification, and drug resistance. Researchers have also directed their interest to the investigation of time-associated changes in the kidney gene expression patterns and have showed that there is a wide interplay among numerous genes, whose expression not only depends on cisplatin dosage but also greatly varies during the time course of AKI [61, 64].
In the last two decades experimental models have demonstrated that cisplatin nephrotoxicity is associated with the inflammation and oxidative stress. Numerous studies have been performed to evaluate the role of different immune cells and inflammatory molecules/mediators in cisplatin nephrotoxicity. Plethora of information has been obtained and many controversies emerged. Nevertheless, to elucidate the role of inflammatory cells in the pathogenesis of cisplatin nephrotoxicity, various methods were used, i.e., inhibition of particular cell type using inhibitors or antibodies, generation of mutated mice with specific gene deletion, adoptive transfer of particular inflammatory cell type to mutated mice, or combination of all mentioned methods. Studies report that inhibition of macrophages [52], neutrophils [65–67], or nuclear killer cells [68] does not affect cisplatin nephrotoxicity, while inhibition of both neutrophils and nuclear killer cells results in reduced acute nephrotoxicity [67]. Deletion of either total T cells [69, 70] or CD4+ or CD8+ T cells [69] or mast cells (KitW-sh/W-sh mice)[71] attenuates cisplatin nephrotoxicity. The increase in CD4+CD25+ regulatory T cells (Treg cells) has protective effects as well [69, 70, 72], while the deletion of dendritic cells (CD11c-DTRg mice) worsens cisplatin nephrotoxicity [66, 73] (for more information see Table 1).
Table 1.
Genetically altered mice used in the investigation of cisplatin nephrotoxicity.
| age | ||||||||
|---|---|---|---|---|---|---|---|---|
| background | sex | cisplatin | ||||||
| GEM model | (breeder) | N | dose | end | mortality | S | measured parameters | Ref. |
| 25-30g | ||||||||
| 129/SV | male | 10 mg/kg | BUN, Cr, Cr clearance, HO-1, p21mRNA | |||||
| CYP2e1−/− | (NIH) | N=10 | ip | D3 | nr | ↓ | and protein, TUNEL | [74] |
|
| ||||||||
| Oct1−/− | 8-12wk | ns | ||||||
| Oct2−/− | FVB | male | 10 mg/kg | ns | ||||
| Oct1/2/−/− | (Taconic) | N=11 | ip | D3 | nr | ↓ | Histology, Cr, BUN, ALT↑, ALP↓ | [35] |
|
| ||||||||
| 6-10wk | ||||||||
| C57BL/6 | male, female | 15 mg/kg | ||||||
| GstP1/P2−/− | (UK) | N=5-9 | ip | D5 | 0% | ↓ | Cr, ATN, WBC | [75] |
|
| ||||||||
| 11-15wk | D3:BUN, Cr, AST | |||||||
| Mate1−/− | male | 15 mg/kg | mortality | |||||
| (pyrimethamine)∗ | C57BL/6 | N=6 | ip | D6 | 100% | ↑ | [41] | |
|
| ||||||||
| Mdk−/− | 129SV | 8-12wk | 12 mg/kg | D7 | nr | ↓ | ↓BUN, TUNEL, histology, ≈leukocytes | [76] |
| male | ip | infiltration (neutrophil, macrophages, Tcells), | ||||||
| N=6 | Bcl-2, KC protein, MCP-1, MIP-2 | |||||||
|
| ||||||||
| 8-10wk | Survival, histology, BUN, Cr, leukocyte | |||||||
| male | 15 mg/kg | infiltration, TUNEL, TNFα, ICAM1 mRNA, | ||||||
| PI3Kγ−/− | C57BL/6 | N=25 | ip | D5 | 44% | ↑ | proteins, p-Akt, p-Bad | [77] |
|
| ||||||||
| 10-12wk | ||||||||
| B6x129/SF2/J | male | 20 mg/kg | ||||||
| E2F1−/− | (Jax) | N=6 | ip | D3 | nr | ↓ | ↓BUN, Cr, histology, | [78] |
|
| ||||||||
| p53−/− | nr | N=6 | 20 mg/kg | |||||
| (pifithrin-α)∗ | (Jax) | ip | D3 | nr | ↓ | ↓BUN, Cr, histology | [79] | |
|
| ||||||||
| p53−/− | C57BL/6J | 8-10 wk, male | 30 mg/kg | |||||
| (pifithrin-α)∗ | (Jax) | N=22-29 | ip | D3 | nr | ↓ | ↓BUN, Cr, histology | [80] |
|
| ||||||||
| C57BL/6 | 8-10wk, male | 30 mg/kg | ||||||
| Bax−/− | (Jax) | N=3-8 | ip | D3 | nr | ↓ | BUN, Cr, ATN, TUNEL, cytochrome C | [81] |
|
| ||||||||
| Histology, BUN, Cr, neutrophils infiltration, | ||||||||
| C57BL/6 | 8-10wk, male | TUNEL, Casp-1,-3,-8,-9 activity, IL-1β | ||||||
| Casp-1−/− | (Japan) | N=4-15 | 30 mg/kg | D3 | nr | ↓ | [82] | |
|
| ||||||||
| 8-15 wk, male | BUN, histology, TUNEL | |||||||
| p21-/- | nr | N=5-6 | 20 mg/kg | D3 | 30% | ↑ | [83] | |
|
| ||||||||
| HO-1+/- | 12-16wk | 20 mg/kg | ≈ | |||||
| HO-1−/− | nr | N=8 | ip | D3 | ↑ | ↓BW, ↑BUN | [84] | |
|
| ||||||||
| C57BL/10Sn | 6-8 wk | D3 | ||||||
| C5−/− | (Jax) | male | ↓ | BUN, Cr, histology, MDA, MPO, | [85] | |||
| BALB/c | N=5-12 | 20 mg/kg | nitrotyrosine, TNF-α, IL-1β, ICAM-1, casp-3,8,9, STAT3, NF-κB | |||||
| C5aR−/− | (Jax) | SPF | ip | ↓ | ||||
|
| ||||||||
| DBA/1lacJ x | 3-4 months | BW, histology, BUN, Cr, Hct, | ||||||
| mPGES-1−/− | C57BL/6 x | male | 20 mg/kg | TNFα, IL-1β; | ||||
| (celecoxib)∗ | 129/SV | N=6-16 | ip | D3 | nr | ↓ | [86] | |
|
| ||||||||
| TNFα -/- | 8-9 wk | Histology, BUN, leukocyte infiltration | ||||||
| (GM6001, | B6129SF2/J | male | ||||||
| Pentoxifylline)∗ | (Jax) | N=7 | 20 mg/kg | D3 | nr | ↓ | [87] | |
|
| ||||||||
| C57BL/6 | 8-10wk, male | Histology, BUN, Cr, neutrophil infiltration, | ||||||
| TLR4−/− | (Jax) | N=4-7 | 20 mg/kg | D3 | nr | ↓ | cytokines | [88] |
|
| ||||||||
| D1 | Kidney: ↑neutrophils, CXCL1, CXCL2, | |||||||
| 8-10wk | ≈CD4+, ↓CD4+CD25+, Foxp3 | |||||||
| N=6-10 | ||||||||
| TLR9−/− | C57BL/6 | 20 mg/kg | D3 | nr | ↑ | Blood: ↑BUN; Kidney: ↑histology score | [72] | |
|
| ||||||||
| C57BL/6 | 8-10 wks | 15mg/kg | Blood: ≈BUN; Kidney: ≈histology score | |||||
| Tcrd−/− | (Jax) | ♂, N=9 | ip | D4 | nr | ns | [67] | |
|
| ||||||||
| Asc−/− | C57BL/6 | 8-10 wk, male | 15mg/kg | Blood: ↓BUN; | ||||
| TLR2−/− | (Jax) | N=5 | ip | D4 | nr | ↓ | Kidney: ↓histology score, IL-17A, IL-1β | [67] |
|
| ||||||||
| C57BL/6 | 8-10 wk, male | 15mg/kg | Blood: ↓BUN; Kidney: ↓histology score | |||||
| Rorγt-/- | (Jax) | N=9 | ip | D4 | nr | ↓ | [67] | |
|
| ||||||||
| 8-10 wk, | D1 | Kidney:↓neutrophils, ≈CD4+ T cells, CXCL1, | ||||||
| IL-17A−/− | C57BL/6 | male | 15mg/kg | CXCL2, CCL2, CCL5 | ||||
| (anti-IL-17A)∗ | (Jax) | N=8 | ip | D4 | nr | ↓ | Blood: ↓BUN; Kidney: ↓histology score | [67] |
|
| ||||||||
| C57BL/6 | 8-10 wk, male | Histology, BUN, Cr, | ||||||
| IL-6 −/− | (Jax) | N=4-6 | 30 mg/kg | D3 | nr | ns | [65] | |
|
| ||||||||
| 10-12wk, | ↓Mortality, | |||||||
| C57BL/6J | male | ↑BUN | [89, 90] | |||||
| IL-6−/− | (Jax) | N=9-18 | 30 mg/kg | D8 | 47% | ↓∗∗ | ||
|
| ||||||||
| 6-8 wk | Morality, Cr, histology, MPO, | |||||||
| nu/nu B6.Cg-Foxn1nu |
C57BL/6 | male | TNFα, IFN-γ,IL-1β, KC | |||||
| (T-cell transfer)∗ | (Jax) | N=3-14 | 40 mg/kg | D3 | 0% | ↓ | [69] | |
|
| ||||||||
| 6-8wk | D2 | 6% | Mortality, BUN, Cr, histology, macrophage | |||||
| nu/nu (CD4+CD25+Treg |
BALB/c | male | D3 | 31% | infiltration (F4/80 positive cells), TNFα, IL-1β | |||
| transfer) | (South Korea) | N=16 | 40 mg/kg | D4 | 13% | ↓ | [70] | |
|
| ||||||||
| CD4−/− | B6129S2 | 6-8 wk, male | ↓Morality, Cr, histology | |||||
| B6.129S2-Cd4tm1Mak | (Jax) | N=16 | 40 mg/kg | D3 | 38% | ↓ | [69] | |
|
| ||||||||
| 8-10wk | Blood: ↓Cr; Kidney: ↓histology score | |||||||
| CD4−/− CD4 tm1Mak |
C57BL/6 | male | ||||||
| GK1.5 monoclonal AB∗ | (Jax) | N= | 25 mg/kg | D3 | nr | ↓ | [51] | |
|
| ||||||||
| 8-10wk | 10 mg/kg | Blood: ≈BUN, Cr; Kidney: ≈caspase-3, | ||||||
| CD4−/− CD4 tm1Mak |
C57BL/6 | male | per week | TUNEL,≈ IL-1, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12, CXCL1, TNFα, IFN-γ | ||||
| GK1.5 monoclonal AB∗ | (Jax) | N=10 | for 4 wks | 4wks | nr | ns | [91] | |
|
| ||||||||
| CD8−/− | B6129S2 | 6-8 wk, male | Mortality, Cr, histology | |||||
| B6.129S2-Cd8atm1Mak | (Jax) | N=16 | 40 mg/kg | D3 | 56% | ↓ | [69] | |
|
| ||||||||
| C57BL/6 | 8-10wk, male | Blood: ↓Cr; Kidney:↓histology score, | ||||||
| CXCR2−/− | (Jax) | N=4 | 25 mg/kg | D3 | nr | ↓ | apoptosis | [51] |
|
| ||||||||
| C57BL/6 (wt) | 8-10wks | 12 mg/kg | Blood: ↓BUN, serum TNF; Kidney: | |||||
| KitW-sh/W-sh | (Jax) | N=7 | ip | D4 | 0% | ↓ | ↓histology score | [71] |
∗A pharmacological inhibitor of particular gene/protein. ∗∗IL-6 deficiency accelerates the development of nephrotoxicity at the early stage but resists the progress of systemic injury [89].
N: number; S: susceptibility to cisplatin toxicity; ip: intraperitoneally; D: day; ≈: ns; ↓: decrease; ↑: increase; BUN: blood urea nitrogen; Cr: serum creatinine; GFR: glomerular filtration rate; ATN: acute tubular necrosis; BW: body weight; WBC: white blood cells; Hct: hematocrit; TNFα: tumor necrosis factor alpha; IL: interleukin; IFNγ: interferon gamma; MCP-1: monocyte chemoattractant protein-1; HO-1: heme oxygenase-1; ICAM-1: intracellular adhesion molecule-1; TLR4: toll like receptor 4; COX: cyclooxygenase; Casp3: caspase 3; KC: keratinocyte-derived chemokine; MPO: myeloperoxidase; SOD: superoxide dismutase; CAT: catalase; GSH: glutathione; GPx: glutathione peroxidase; GR: glutathione reductase; GST: glutathione S transferase; MDA: malondialdehyde; ALT: alanine transaminase; AST: aspartate transaminase; ALP: alkaline phosphatase; Cas3: caspase 3; Cas9: caspase 9; CXCL: chemokine (C-X-C motif) ligand; CCL: chemokine (C-C motif) ligand.
In response to renal injury, various inflammatory proteins such as cytokines and intracellular adhesion molecules are produced. Increased expression of various inflammatory proteins (tumor necrosis factor alpha (TNFα), intracellular adhesion molecule- (ICAM-) 1, monocyte chemoattractant protein- (MCP-) 1, etc.) or their receptors (TNFR1, TNFR2) was found in the kidney as early as the first day after cisplatin administration [46]. Studies show that the expression of inflammatory mediators differs in a time dependent manner. For instance, interleukin- (IL-) 17A (proinflammatory cytokine) and chemokine (C-X-C motif) ligand- (CXCL-) 1 (neutrophil chemoattractant) peaked at 24 h after cisplatin administration, but 96 h after cisplatin they both returned to the baseline levels. On the other hand, chemokine (C-C motif) ligand- (CCL-) 20 (chemokine for CD4+) and CXCL2 (neutrophil chemoattractant) significantly increased not earlier than 96h after cisplatin administration [67]. Renal, circulatory, and urinary levels of various cytokines in acute phase of cisplatin nephrotoxicity are demonstrated in Table 2. Results show that the expression of cytokines differs in accordance with the cisplatin dosage. For instance, nonlethal nephrotoxic dose (10 mg/kg in mice) did not exert significant changes in measured inflammatory proteins in serum or urine (TNFα was not increased within first 3 days) [92], while lethal dose (20 mg/kg) exerted significant changes already 24 hours after cisplatin administration (see Table 2).
Table 2.
Inflammatory, apoptotic, and oxidative factors in the acute cisplatin nephrotoxicity in rodents.
|
tissue/ method |
analytes measured | D1-D2 | D3 | strain | Ref. |
|---|---|---|---|---|---|
| dose | |||||
| Serum, urine |
ELISA: TNFα | ns ns |
ns ns |
C57BL/6, | [92] |
| Urine/MCA | IL-1β, IL-2, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12(p70), IL-17, IFN-γ, IP-10, G-CSF, MIP-1α, MCP-1, RANTES | ns | n.a. | 10wk, male 25-30g 10 mg/kg |
|
| Kidney/RT-PCR | IL-1β, TNFα, HO-1, IL-18, MCP-1, ICAM-1, TLR4, HMG1 | n.a. | ns | ||
| Kidney/ELISA | TNFα, MCP-1, MIP-2 | n.a. | ↑MCP-1, MIP-2 | ||
|
| |||||
| kidney/RPA | TNF-α, TGF-β, RANTES, MIP-2, IP-10, MCP-1, TCA3, IL-5, IL-9, IL-15, IFN-γ | TNF-α, TGF-β, RANTES, MIP-2, IP-10, MCP-1, TCA3 | TNF-α, TGF-β, RANTES, MIP-2, IP-10, MCP-1, TCA3 | Swiss Webster |
[87] |
| kidney/RT-PCR | TNF-α, TGF-β, RANTES, MIP-2, MCP-1,ICAM-1, IL-1ɑ, IL-1β | n.a. | TNF-α, TGF-β, RANTES, MIP-2, MCP-1, ICAM-1, IL-1β | 20 mg/kg | |
|
| |||||
| Kidney/RT-PCR | TNFα, MCP-1, IL-10 | ↑ TNFα, MCP-1 | ↑TNFα, MCP-1, IL-10 | CD-1, Crl Male |
[93] |
| ELISA | ↑MCP-1 | ↑MCP-1 | 25-35g | ||
| Plasma/ELISA | TNFα, MCP-1, IL-10 | ↑MCP-1 | ↑ TNFα, MCP-1 | 30 mg/kg | |
|
| |||||
| kidney/RT-PCR | TNF-α, IL-6, IL-11, LIF, SOCS3, gp130, OSM, CNTF, CT-1, c-fos, HO-1, Bax, Bcl-xl, Bcl-2, Nrf2, MT-1, MT-2, SOD1, SOD2, COX-2, Casp3 | ↑TNF-α, IL-6, IL-11, LIF, SOCS3, OSM, CNTF, c-fos, HO-1, Bax, Bcl-xl, Bcl-2, Nrf2, MT-1, MT-2 |
↑TNF-α, IL-6, IL-11, LIF, SOCS3, gp130, OSM, CNTF, c-fos, HO-1, Bax, Bcl-xl, Bcl-2, Nrf2, MT-1, MT-2, COX-2, ↓SOD1 | ||
| C57BL/6 Jax 30 mg/kg |
[89, 90] | ||||
| serum/ELISA | IL-6 | ↑IL-6 | ↑IL-6 | ||
|
| |||||
| analytes measured | D3 | ||||
|
| |||||
| kidney/RT-PCR | TNFα, IL-1β, ICAM-1, MCP-1, TGF-β1, HO-1 | ↑TNF-α, MCP-1, ICAM-1 | C57BL/6 20 mg/kg |
[94] | |
| kidney/ELISA | TNFα | ↑TNFα | 10-12 wk, male |
||
|
| |||||
| serum/ELISA | TNF-α | ↑TNF-α | C57BL/6 male, Jax |
||
| kidney/RT-PCR | TNF-α, TNFR1, TNFR2, MCP-1,ICAM-1, HO-1 | ↑TNF-α, TNFR1, TNFR2, MCP-1,ICAM-1, HO-1 | 20 mg/kg N=4-8; ip |
[95] | |
|
| |||||
| serum/ | TNF-α, IL-1β, IL-2, IL-6, IL-10, IP-10, MCP-1, RANTES, KC | ↑TNF-α, IL-1β, IL-6, IL-10, IP-10, ↓IL-2 | [88] | ||
| kidney/ | ↑TNF-α, IL-6, MCP-1, KC, IP-10 | C57BL/6 Jax 20 mg/kg |
|||
| urine/MCA | ↑TNF-α, IL-6, IL-2, RANTES, MCP-1, KC, IP-10 | ||||
| kidney/RT-PCR | TNF-α, IL-1β, IL-2, IL-6, IL-18, RANTES, MCP-1, ICAM-1, KC, IP-10, TLR4, iNOS, IFN-ɑ, IFN-β | ↑TNF-α, IL-1β, IL-6, IL-18, MCP-1, ICAM-1, KC, TLR4, iNOS | |||
| kidney/activity | p38MAPK activity, p-JNK phosphorylation - Western | ↑p38MAPK, p-JNK | |||
|
| |||||
| urine/MCA | IL-1β, IL-2, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 (p70), IL-17, IFN-γ, IP-10, G-CSF, TNF-ɑ, MIP-1ɑ, MCP-1, RANTES | ↑TNF-α, IL-6, IL-2, RANTES, IP-10 | C57BL/6 Jax |
[96] | |
| kidney/RT-PCR | HO-1, IL-1β, IL-10, TNF-ɑ, TNFR1, TNFR2 | ↑HO-1, IL-1β, TNF-ɑ, TNFR1, TNFR2 | 20 mg/kg | ||
| serum/ELISA | TNF-α | ↑TNF-α | |||
|
| |||||
| kidney/MCA | TNF-α, IL-1β, KC, IFNγ | ↑TNF-α, IL-1β, KC; ns (24h) | C57BL/6J 40 mg/kg |
[69] | |
|
| |||||
| kidney/RT-PCR | IL-1β, TNFɑ, COX-2, mPGES-1, mPGES-2, cPGES, gp91phox, p47phox, NOX-1, NOX-3, SOD1, SOD2, SOD3, Bak, Bax, Bcl-2, | ↑IL-1β, TNFɑ, COX-2, mPGES-1, ↑gp91phox, p47phox, ↓SOD2, SOD3, ↑Bax, Bak, | DBA/1lacJx C57BL/6 x 129/SV 20 mg/kg |
||
| [86] | |||||
| ELISA/serum | TNF-α | ↑TNF-α | |||
| Kidney | PGE2, TNF-α | ↑ PGE2, TNF-α | |||
D: day; n.a.: not analyzed; MCA: multiplexed cytokine assay; RPA: ribonuclease protection assay; TNFα: tumor necrosis factor alpha; IL: interleukin; IFNγ: interferon gamma; MCP-1: monocyte chemoattractant protein-1; HO-1: heme oxygenase-1; ICAM-1: intracellular adhesion molecule-1; TLR4: toll like receptor 4; TGFβ: tumor growth factor; LIF: leukemia inhibitory factor; CNTF: ciliary neurotrophic factor; gp130: glycoprotein 130; Nrf2: nuclear factor erythroid 2-related factor; OSM: oncostatin M; MT: metallothioneins; SOD: superoxide dismutase; COX: cyclooxygenase; Casp3: caspase 3; iNOS: inducible nitric oxide synthase; KC: keratinocyte-derived chemokine; TNFR1: TNF receptor 1; TNFR2: TNF receptor 2; cPGES: cytosolic prostaglandin E synthase; mPGES: microsomal prostaglandin E synthase; CT-1: cardiotrophin-1; gp91phox and p47phox: two major subunits of NADPH oxidase; MIF: macrophage migration inhibitory factor.
2.4. Histological Characteristics
Cisplatin nephropathy in rodents is histologically characterized by degenerative changes in the proximal tubules that consist of hydropic degeneration, pycnotic nuclei, increased cytoplasmic vesicles, cytoplasmic vacuolization, evident loss of the brush border, necrosis and apoptosis of tubular cells, and desquamation of necrotic epithelial cells filling the tubular lumens and forming hyaline casts (see Figure 1) [10].
Figure 1.

Proximal tubules of BALB/c male mice 4 days after a single cisplatin treatment (17 mg/kg ip): (a,b) hydropic degeneration, pycnotic nuclei, increased cytoplasmic vesicles, cytoplasmic vacuolization, apoptosis, necrosis, loss of brush border, and small hyaline droplets in the cytoplasm (PAS, 400x). (c) Evident loss of brush border, necrosis, desquamation and hyaline/proteinaceous casts, hyaline droplets, and dilated tubules (PAS, 200x). (d) Hyaline casts, hydropic degeneration, and necrosis in distal and collecting tubules (PAS, 200x).
These morphological changes are more pronounced in the proximal tubules but are also induced in distal and collecting tubules. Studies report wide variability in severity of histological changes ranging from no damage [63] to moderate or severe damage of the kidney [97], as well as reports describing different sites of the damage, i.e., only S3 segments or only S3 segments and distal tubules or throughout the cortex and outer medulla [59] or in the medulla (S3 segments of PT, loops of Henle, and collecting ducts) [4, 36, 98–100]. Mild interstitial inflammation and edema in renal cortex and outer medulla together with some glomeruli exhibiting thickening of the basement membrane were also reported [99]. Nevertheless, studies show that depending on the cisplatin dosage and the time of observation (sampling), histological alterations of AKI can range from no damage to severe damage limited to S3 segment or extended to distal tubules or collecting ducts (see Tables 3 and 4). In contrast, in case of chronic kidney disease repeated administration of low cisplatin dose induced firstly minimal morphological signs of AKI (such as tubules with dispersed heterochromatin and segregated nuclei in epithelial cells) followed by flattened epithelial cells with elongated nuclei. At this time small amount of collagen fibers with mononuclear cell infiltrate can be observed. With repeated administration progressive alterations are induced, such as occasional dilated tubules with mononuclear cells and desquamated epithelial cells. In the recovery period, the size of dilated tubules (lined by flattened and polygonal epithelial cells) gradually diminishes, and atrophic tubules (lined by regenerative cuboidal or cylindrical epithelial cells) appear. Around the affected tubules gradual development of fibrotic areas can be observed. Fibrotic areas are accompanied by infiltration of inflammatory cells such as macrophages and lymphocytes [13, 14]. It was found that the most abundant population of resident leukocytes in the healthy kidney is CD11c+MHCII+ dendritic cells (renal dendritic cells are CD4− and CD8−) [73]. First inflammatory cells that appear in the kidney after cisplatin administration (40 mg/kg) are T cells (CD3+), which infiltrate kidney parenchyma within 1 h, reach the peak at 12 h, and decline back on the baseline level by 24 and 72 h. The macrophage and neutrophil infiltration follows later [69]. The reports about the time of increased macrophage or neutrophil infiltration in the kidney vary between 24 h [46, 66], 48 h [52, 70], and 72 h after cisplatin injection [82].
Table 3.
Clinical alterations and the toxicity of cisplatin in mice.
|
strain (breeder) |
age sex, N |
R | cisplatin dose | TP |
BUN mg/dl |
Cr mg/dl |
end | M | histology and other comments | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| BALB/c | MTD | iv | D2-4 | ns | ns | D10 | 0% | Histology: proximal tubular epithelial necrosis observed not earlier than D6 - D10; D4: ↓GFR; Urine: ↑proteins, glycosuria, |
[55] | |
| female | 7 mg/kg | D6 | ns | ns | ||||||
| N=3-8 | D10 | ns | ns | |||||||
|
| ||||||||||
| BALB/c | female | ip | 8 mg/kg | D4 | ns | ns | D4 | 0% | Histology: not evaluated D4:↓GFR |
[56] |
|
| ||||||||||
| C57BL/6J | 20-25g | ip | 10 mg/kg | D1 | 24 ±17 ns | 1.4 ± 0.5 ns | D4 | 0% | Histology: D2 rare changes, D3-D4 evident loss of brush border, pyknotic nuclei, vesicles, Ultrastructure: changes in mitochondria from D1 | [44] |
| Jax | male | D2 | 44 ±10 ns | 1.9 ± 0.9 ns | ||||||
| N=6 | D3 | 105 ±9 | 12.7 ±1.2 | |||||||
| D4 | 228 ± 18 | 22.4 ±1.8 | ||||||||
|
| ||||||||||
| C57BL/6 Harlan, UK |
male N=6 |
ip | 7.5 mg/kg | D4 | 1.6x ns | 1.2x ns | D4 | 0% | ↓BW (4.4±5%); histology: not evaluated | [103] |
| 10 mg/kg | D4 | ↑4x | ↑3.1x | D4 | 0% | ↓BW (15.3±11%), histology: moderate ATN: degradation of the brush border, loss of cell-cell adhesion, cellular vacuolation and sloughing of cellular material into lumen | ||||
| 12.5mg/kg | D4 | ↑5x | ↑3.6x | D4 | HEP | BW (↓27±1%)∗, systemic toxicity | ||||
|
| ||||||||||
| C57BL/6 | 8-12 wk | ip | 15 mg/kg | D5 | n.a. | 4.7 ± 0.5 | D5 | 12% | Histology: all survived mice (7/8) had severe ATN although one had normal Cr levels (6/7); survived mice: ↓BW (19.5%), lack of grooming, lethargy, high morbidity, ↓WBC |
[114] |
| USA | female | (6/7) | (1/8) | |||||||
| N=8 | ||||||||||
|
| ||||||||||
| C57BL/6 | male | ip | 15 mg/kg | D5 | ↑4x | n.a. | D5 | 55% | ↓BW (24%), histology: severe ATN: extensive tubular necrosis, loss of the epithelial brush- border, presence of protein casts in the lumen mice began dying on day 3 |
[115] |
| Harlan | 6-8 wk | (6/11) | ||||||||
| N=11 | ||||||||||
|
| ||||||||||
| C57BL/6 | 11-15wk | ip | 15 mg/kg | D2 | ns | n.a. | D10 | 100% | Histology: not evaluated D4:↓BW (20-30%); mice began dying on day 5 |
[41] |
| Japan | male | D3 | 4x | 2x | ||||||
| N=5-6 | ||||||||||
|
| ||||||||||
| Swiss | Male | ip | D3 | ↑3x | ↑10x | D3 | 0% | Histology: not evaluated Kidney:↓SOD, CAT, GPx, ↓GSH, ↑MDA |
[116] | |
| albino | 6-7wks | 10 mg/kg | ||||||||
| (India) | N=6 | |||||||||
|
| ||||||||||
| Swiss | female | ip | 12 mg/kg | D3 | ↑4x | ↑4x | D3 | 0% | Histology: severe ATN; marked necrosis in proximal tubules Kidney:↓SOD, CAT, GPx; ↓GSH, ↑MDA |
[117] |
| albino | 6wks | |||||||||
| (India) | N=6 | |||||||||
|
| ||||||||||
| ICR Thailand |
25-30g male N=8 |
ip | 7.5 mg/kg | D3-5 | ↑ns | ns | 0 | Preliminary experiment to determine cisplatin dose | [118] | |
| 10 mg/kg | D3 D5 |
31.7±2.9 ns 53.8 ± 4.2 |
0.88±0.02 ns 1.74 ± 0.43 ns |
D5 | 0 | dose- and time-dependent elevation of BUN and Cr | ||||
| 12.5mg/kg | D3 D4 D5 |
52.2 ± 8.3 88.9 ± 18.8 131.8 ± 22 |
ns ns 2.01 ±0.53 |
D5 | 0 | 15 mg/kg resulted in marked BW loss at d5; | ||||
| 15 mg/kg | D3 D4 D5 |
63.8 ± 17.2 158 ± 77 225 ± 41 |
ns 2.32 ± 0.59 2.49 ± 0.59 |
D5 | HEP | 12.5 mg/kg was chosen. Histology: severe ATN: focal necrosis of the proximal tubules throughout the cortex, hyaline casts in tubular lumen, desquamation, and parenchyma degeneration of the tubular epithelium cells | ||||
|
| ||||||||||
| FVB/n | N=10 | ip | 4 x 7 mg/kg | D25 | ns | ns | D25 | 0% | Histology: evident loss of brush borders, tubular dilation, less tubular necrosis, evident inflammatory cells and interstitial fibrosis (red Sirius) Urine: ≈KIM-1, ↑NGAL |
[119] |
| Jax | per week | |||||||||
| 4 x 9 mg/kg | D25 | ns | ns | D25 | 90% | |||||
| per week | ||||||||||
|
| ||||||||||
| High lethal dose – severe systemic toxicity | ||||||||||
|
| ||||||||||
| BALB/c | 6-8wk | ip | 25 mg/kg | D1 | ns | ns | D3 | 0% | [70] | |
| South | male | D2 | ↑2x | ↑2x | D4 | 80% | ||||
| Korea | N=11 | D3 | ↑4x | ↑4x | D5 | 100% | ||||
|
| ||||||||||
| BALB/c | 6-8wk | ip | 40 mg/kg | D1 | 185 ± 22 | 2.4 ±0.48 | D1 | 20% | [70] | |
| South | male | D2 | 60% | |||||||
| Korea | N=21 | D3 | 100% | |||||||
|
| ||||||||||
| Nephrotoxicity and antitumor activity in mice with cancer | ||||||||||
|
| ||||||||||
| BALB/c | 8 wk | ip | 11.5 mg/kg | D4 | 70±42 | 1.6 ± 0.6 | D7 | 0-12% | Histology: ATN (dose dependent) nephrotoxicity and antitumor activity |
[120] |
| (Harlan) | female | 13 mg/kg | D4 | 130 ± 38 | 4.5 ± 1.0 | D7 | 0-50% | |||
| N=8 | 14.5 mg/kg | D4 | 175±52 | 5.8 ± 0.7 | D7 | 100% | ||||
| 16 mg/kg | D4 | 232 ± 40 | 6.4 ± 1.0 | D7 | nr | |||||
| 19 mg/kg | D4 | 275 ± 26 | 6.9 ± 0.2 | D7 | nr | |||||
|
| ||||||||||
| C57BL/6 | female | iv | 7x 1mg/kg every other day |
D18 | n.a. | n.a. | D18 | 0/8 | Low-dose cisplatin no changes in the BW Antitumor activity |
[121] |
| N=8 | ||||||||||
|
| ||||||||||
| C57BL/6 | female N=8 |
iv | 7x 5mg/kg every other day | D18 | n.a. | n.a. | D18 | 4/8 | High-dose cisplatin 15-30% BW loss, antitumor activity (experiment was stopped when mice started dying) |
[121] |
N: number; M: mortality; R: route of injection; TP: time point; n.a.: not analyzed; ip: intraperitoneally; iv: intravenously; sc: subcutaneously; D: day; ≈: ns; ↓: decrease; ↑: increase; BUN: blood urea nitrogen; Cr: serum creatinine; GFR: glomerular filtration rate; ATN: acute tubular necrosis; HEP: humane endpoint (animals were humanely euthanized due to severe illness). ∗Animals receiving high dose of cisplatin exhibited systemic toxicity as demonstrated by significant weight loss in excess of 25% of their starting bodyweight, requiring termination of the experiment on day 4 [103].
Table 4.
Clinical alterations and the toxicity of cisplatin in rats.
|
strain (breeder) |
age | R |
cisplatin dose |
TP |
BUN mg/dl |
Cr mg/dl |
end | M | histology and other comments | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| sex | ||||||||||
| N | ||||||||||
| Han-Wistar (Harlan) |
7wks female N=10 |
ip | 2.5 mg/kg | D5 D8 D22 |
Ns Ns ns |
Ns Ns ns |
D22 | 0% | Various doses (0.1; 1; 2.5mg/kg): the effects appeared in a temporal and dose-dependent manner; Histology: D5: mild ATN, D8: minimal ATN, D22: regeneration; majority of alteration seen in the S3 segment of proximal tubules, rarely in distal tubules and collecting ducts | [122, 123] |
|
| ||||||||||
| Han-Wistar (Crl) |
7wks Male N=10 |
ip | 3 mg/kg | D2 D3 D5 |
Ns Ns ↑3.5x |
Ns Ns ↑2.5x |
D5 | 0% | Histology: D2,D3,D5: moderate ATN, Urine: ↑leukocytes, ↑ total protein (5.6x) |
[104] |
|
| ||||||||||
| Sprague-Dawley (Crl) |
6wks Male N=10 |
ip | 3 mg/kg | D2 D3 D5 |
Ns ↑1.37x ↑2.2x |
Ns ↑1.25x ↑2.4x |
D5 | 0% | Histology: D3-D5: minimal to moderate degeneration/necrosis of the S3 segment of proximal tubules in less than half of rats Urine: ↑leukocytes, ↑ total protein (4.6x) |
[104] |
|
| ||||||||||
| Wistar WKY (Spain) |
7wks female N=4 |
ip | 5 mg/kg | D5 | ↑2x | ↑3.5x | D5 | 0% | Histology: tubule swelling, loss of brush border membrane, epithelial vacuolization, hyaline casts and cell debris detachment in proximal tubular cells; ↓GFR 3.3x Urine:↑volume (1.9x), proteins (1.9x) |
[36, 124] |
|
| ||||||||||
| Wistar ( Spain) |
7wks male N=8 |
ip | 5mg/kg | D5 | ↑2.4x | ↑3.3x | D5 | 0% | Histology: tubular necrosis, swelling and tubular dilatation, extensive epithelial vacuolization, hyaline casts in tubules; ↓BW (8.5%); ↓GFR (4.1x) Urine: ↑Na+, ↑volume (2x), ↑proteins (1.7x), ↑urine hydrogen peroxide production, ↓antioxidant capacity in urine |
[54] |
|
| ||||||||||
| F344 (NIH) |
Male N=7-45 |
ip | 6 mg/kg | D1 D2 D3 D4 D5 D7 |
Ns ↑1.5x ↑4x ↑3.5x ↑5.3x ↑3.2x |
Ns Ns ↑3.7x ↑4.7x ↑4.7x ↑2.6x |
D7 | 0% | ↓5 g of BW per day; Histology: D3: focal area of mild ATN in proximal tubules: loss of the brush border, necrotic cells sloughing into the tubular lumen, condensed cytoplasm of many cells, increased number of cytoplasmic vesicles in injured cells; D5: widespread moderate ATN in proximal as and distal tubules: desquamation of necrotic cells, patches of denuded basement membrane, necrotic debris filling the tubular lumens and hyaline casts. Some cells show similar changes as those at D3, lower in height, brush border loss, increased vesicles and swelling; D7: first signs of regeneration, necrotic cells and necrotic debris are still present |
[10, 32] |
|
| ||||||||||
| Sprague Dawley (Turkey) |
8wk, Male N=6 |
ip | 7 mg/kg | D5 | 107±5.1 | 1.4±0.16 | D5 | 0% | Histology: necrosis in proximal tubules, karyomegaly, hyaline casts, desquamation and parenchyma degeneration of tubular epithelial cells, interstitial nephritis; ↓BW (9%); ≈plasma Na+ and K+ Kidney: ↓GSH, GPx, CAT, ↑MDA |
[125] |
|
| ||||||||||
| Wistar (Turkey) |
200-250g female N=10 |
ip | 7 mg/kg | D5 | ↑8x | ↑4.5x | D5 | 0% | Histology: D5: extensive epithelial cell vacuolization, swelling and desquamation Kidney: ↓ SOD, CAT, GPx,↑ MDA, MPO |
[126] |
|
| ||||||||||
| Wistar Hanover (Harlan) |
male N=5 |
iv | 8 mg/kg | D3 D5 |
↑2x ↑5.5x |
↑1.3x ↑6.5x |
D5 | 0% | Histology: D5: widespread tubular degeneration and regeneration, tubules contained granular and proteinaceous casts, mineralization (5/5) D3: ↑AST (1.2x), urine proteins, glycosuria D5:↓AST, TBIL, GGT, ALP, CI; ↑urine proteins, glycosuria |
[64] |
|
| ||||||||||
| Wistar Hanover (Harlan) |
male N=5 |
iv | 4x 1 mg/kg | D3 D5 |
ns ns |
ns ns |
D5 | 0% | Histology: D5: similar to 8 mg/kg but less extensive (5/5) | [64] |
|
| ||||||||||
| Nephrotoxicity and antitumor activity in rats with cancer | ||||||||||
|
| ||||||||||
| rats | N=3-5 | iv | 5mg/kg | D3 D6 |
51±10 (↑2x) 56±20 (↑2x) / |
D6 | 0% | ↓5% BW | [127] | |
|
| ||||||||||
| rats | N=3-5 | ip | 3 x 2.5 mg/kg daily | D3 D6 |
34±4 (↑0.5x) 73±28 (↑3x)) / |
D6 | 0% | ↓17% BW | [127] | |
|
| ||||||||||
| rats | N=3-5 | ip | 7.5 mg/kg | D3 D6 |
60±10(↑0.2x) 216±82 (↑8x) / |
D6 | 0% | ↓22% BW | [127] | |
N: number; M: mortality; R: route of injection; TP: time point; ip: intraperitoneally; iv: intravenously; sc: subcutaneously; D: day; ≈: ns; ↓: decrease; ↑: increase; BUN: blood urea nitrogen; Cr: serum creatinine; GFR: glomerular filtration rate; ATN: acute tubular necrosis; HEP: humane endpoint (animals were humanely euthanized due to severe illness).
3. The Heterogeneity of Cisplatin Protocols
The literature search (PubMed; key words “cisplatin, nephrotoxicity, mice, rats”) has shown a wide variation in doses used for cisplatin-induced nephrotoxicity in both single and multiple dosage protocols. In rats, cisplatin is mostly injected in dose ranging inside nonlethal nephrotoxic range (i.e., between 1 and 8 mg/kg), while in mice a wide variation in cisplatin dose has been used, from low nephrotoxic to highly lethal dose (i.e., between 5 mg/kg and 40 mg/kg).
We have categorized cisplatin protocols according to the extent and severity of cisplatin nephrotoxicity to demonstrate that both the selection of the cisplatin dosage and the time of observation are very important variables that can significantly affect measured parameters and the model. Namely, depending on the cisplatin dosage and time of observation rodents develop acute (early) or chronic (advanced) kidney injury, extrarenal toxicity, or even systemic toxicity. The heterogeneity of protocols and their effects are summarized in Tables 3 and 4.
3.1. Acute Nephrotoxicity
According to the definition, AKI is a life-threatening disease that occurs over a period of hours or days as a consequence of septic, ischemic, or toxic insults [102]. However, it is important to note that in case of cisplatin AKI occurs over a period of days in both rodents and humans.
In rodents, acute nephrotoxicity is most frequently induced by a single intraperitoneal injection of cisplatin followed by euthanasia few days later. It can also be induced by multiple applications of low doses of cisplatin for several consecutive days. However, in case of multiple applications, the clinical and histological changes in the kidney develop more slowly than in a single dose treatment.
A single low nephrotoxic dose of cisplatin (for instance, 5-8 mg/kg in mice and 1-3 mg/kg in rats) causes mild kidney injuries that can be seen 4-5 days after cisplatin administration histologically (as nuclear pleomorphism (karyomegaly), as mild basophilia and presence of a few necrotic cells), or sometimes as changes in some urine and blood markers (glycosuria, decreased GFR). Importantly, in mice low nephrotoxic dose of cisplatin does not affect serum BUN or Cr levels [44, 55, 56, 103, 104].
Repeated administration of cisplatin results in time related increase of many parameters. However, the time course of the disease depends on the dosage, frequency of cisplatin injection, and cumulative dose of cisplatin. For instance, cisplatin treatment in rats (1mg/kg daily for 14 days) resulted in functional renal damage from day 5 onwards. Creatinine increased 2-3-fold from day 5 on, while BUN showed 3-fold on day 5 up to 6-fold by day 14 gradual increase. Glucose was detected in urine from day 5 onwards (150-fold increase on day 5, 18-fold increase on day 7, and 5-fold increase on day 14), without any alterations in serum glucose. Cr clearance decreased to 24-40% on day 5, indicating decreased renal functionality. Histological examination revealed that the incidence and severity of morphological changes increased over time. Minimal severity, such as tubular basophilia with apoptosis in the par recta of proximal tubules (S3), was seen after 3 days of treatment. With increased dosing duration, other degenerative changes were noted, firstly tubular giant cells, crowded basophilic nuclei with prominent nuclei and karyomegaly, and thickened basement membrane in basophilic/degenerative tubules followed by tubular necrosis, hyaline casts and cell debris/exfoliation in the lumen, and tubular dilatation. The distribution of the changes extended from the outer stripe of the outer medulla to the medullary rays (straight tubules, then upper collecting ducts), the cortex (proximal, S2 tubules), and the papilla (loop of Henle, lower collecting ducts) [105].
When using a single high nephrotoxic dose (for instance, 10-13 mg/kg in mice and 3-8 mg/kg in rats) 1-2 days after administration only few minimal changes (such as decrease in mitochondria, focal loss of the microvillus brush border, pycnotic nuclei, and increased cytoplasmic vesicles) can be found [10, 44], while morphological changes (such as loss of the brush border, necrotic cells sloughing into the tubular lumen) are usually seen not earlier than 3-4 days after cisplatin administration [10, 44]. Increased BUN/Cr levels are usually observed 3-7 days after cisplatin administration [106–109] and then return to the baseline levels within 14 days [110]. First signs of structural regeneration were observed 7 days after cisplatin injection [10, 111]. However, in case of lethal dose death may occur within 10 days [112].
3.2. Chronic Nephrotoxicity
It has been shown that a single nephrotoxic dose of cisplatin not only exerts acute nephrotoxicity but also can have long term effects on the structure and function of the rat kidney [11, 12, 15, 111]. Twenty days after cisplatin injection (5 mg/kg) histological features of chronic nephropathy such as interstitial fibrosis, tubular atrophy, and dilation were found [12]. Gradually developing fibrosis was observed around the affected tubules 14 and 28 days after a single dose of cisplatin (6 mg/kg). Infiltration of macrophages into the injured kidney reached a peak on day 7 and was accompanied by an increase in muscle actin-positive myofibroblasts. On days 14 and 28, the number of macrophages declined, while the number of muscle actin-positive myofibroblasts in the fibrotic area was still high. Cytoplasmic myofilaments were observed in myofibroblasts by electron microscopy [111]. Fifteen months after single cisplatin injection (6 mg/kg) rats had significantly reduced GFR and urinary osmolality and increased number of abnormal proximal tubules (atrophic or hyperplastic), such as presence of glomerular sclerosis and interstitial fibrosis and dilated tubules filled with hyaline casts and lined by simple squamous cells [10, 11]. This indicates that the nephrotoxic effects of cisplatin are long-lasting in rats, like in humans [18]. Unfortunately, we were unable to find any study investigating long term effects of single injection of cisplatin on the structure and function of the mouse kidney.
Nevertheless, chronic nephrotoxicity is usually induced by multiple applications of low doses of cisplatin once a week for a few weeks or once in three-week interval. Few decades ago, Yamate et al. [13] established renal interstitial fibrosis model by administering multiple doses of cisplatin (2 mg/kg once weekly ip for 7 weeks). First mild histological alterations (dispersed heterochromatin and segregated nucleoli) in epithelial cells of the proximal tubules were observed 4 weeks after first injection of cisplatin (at that time BUN and Cr levels were normal), while necrosis or desquamation of renal epithelial cells was seen not earlier than 7 weeks after first injection. At this time BUN and Cr levels increased, tubules were markedly dilated, and regenerative process was observed as well as fibrotic area that developed around affected tubules, accompanied by infiltration of inflammatory cells (macrophages and lymphocytes). Fibrosis was present until the end of the study, i.e., 19 weeks after the last injection of cisplatin, when BUN and Cr finally reached control levels [14].
A large inter- and intraindividual variation was reported in repeated multidose treatment [29, 110]. When cisplatin (4 mg/kg) was repeatedly injected four times at intervals of three weeks (ip, 4 cycles of 4 mg/kg with 21 days of washout period), a decrease in the levels of BUN and Cr was observed after the 2nd injection, but after the 3rd and 4th injection the levels of Cr and BUN increased in an accumulative manner [110]. Finally, it was demonstrated that animals that are recovering from a single injection of cisplatin are less susceptible to a subsequent insult with cisplatin. Ming et al. showed that both the increase in Cr and tubular damage were significantly lower in rats which had received cisplatin (3 mg/kg, ip) 14 days prior to the rechallenge with cisplatin (5 mg/kg, ip) compared with the previously untreated rats. However, attenuation of nephrotoxicity was more obvious in the histological index than in the increase in Cr concentration. Increase in Cr concentration did not correlate with tubular necrosis [113].
3.3. Systemic-Extrarenal Toxicity
As demonstrated in Table 3, the lethal dose of cisplatin (exceeding LD50) markedly decreases the survival time of animals [108]. For instance, cisplatin in a dose of 20 mg/kg causes severe morphological injuries in mouse kidney and increased BUN levels already 3 days after single intraperitoneal injection, leading to death within 5 days after the injection [84, 128]. Cisplatin in a dose 40 mg/kg causes systemic toxicity within 1-2 days and death within 4 days after single cisplatin injection [44].
In contrast to the nephrotoxic dosage of cisplatin (single or cumulative), where primary injury is located in the kidney, the lethal dosage (single or cumulative) causes systemic toxicity, i.e., injuries in various organs and tissues.
The fact that cisplatin can cause numerous extrarenal injuries is very important issue although it is rarely mentioned in the cisplatin nephrotoxicity research literature. It is important to keep in mind that cisplatin is an antitumor drug that exerts no specific selectivity to certain cell type. Consequently, cisplatin damages not only the dividing cancer cells but also other fast-dividing cells in the body, thus affecting function of many different tissues. Gastrointestinal toxicity, myelosuppression, ototoxicity, neuropathy, nephrotoxicity, and vascular injury (i.e., thrombotic microangiopathy) [129] due to cisplatin treatment are well known side effects in humans [130]. Severe nephrotoxicity, myelosuppression, nausea, and vomiting are particularly dose related effects of cisplatin and occur in up to 30% of patients treated with recommended dosage protocols (with cumulative 50-100 mg cisplatin/m2 exposure per cycle) [131].
Extensive signs of cisplatin toxicity can be found in rodents as well [106, 132], like decrease in white blood cells in bone marrow and circulating peripheral blood [106], injuries in gastrointestinal tract [133], testis, lymph tissue, and heart [134], disruption in spermatogenesis [135], systemic endothelial cell injury [52], etc. (for more details see Table 5). It is important to take into consideration the fact that cisplatin causes injuries in various organs in a dose and time dependent manner [106, 132]. Minor injuries were found already after application of nonlethal nephrotoxic dosage of cisplatin. However, when high nephrotoxic dosage is used (which in general severely exceeds LD10 that is by convention the maximal dose used in phase I human studies)[131] toxic effects of cisplatin are more pronounced and when lethal dosage is used (exceeding LD50) systemic injury with multiorgan involvement appears. It is important to note that severe systemic toxicity is accompanied by a generalized host inflammatory response known as the systemic inflammatory response syndrome (SIRS), characterized by intense proinflammatory reaction and release of a cascade of potent inflammatory mediators into the systemic circulation, including TNF-α, IL-1β, and IL-6 [136], which were regularly observed in the serum and urine of mice treated with lethal dose of cisplatin but not observed when nephrotoxic dose was used (10 mg/kg) (see Table 1). Thus, when using cisplatin animal model, cisplatin dosage and its side effects should be properly included and discussed in the study (i.e., nephrotoxicity versus systemic toxicity).
Table 5.
Cisplatin causes injuries also in other organs and tissues in the body.
| Strain (breeder), | cisplatin | Other | Ref. | |
|---|---|---|---|---|
| sex, age, N | dose | Time | comment | |
| Myelotoxicity | ||||
|
| ||||
| CBAxC57BLF1, female, 10-12wk, 20-25g N=3 | 12 mg/kg ip |
D1-23 |
gastroenteritis and body weight loss; Femoral bone marrow, blood (WBC, RBC) more toxic for earlier haemopoietic progenitor cells than for the mature cells. Reduction isseen after 1 day: CFU-S number dropped to 5%, and CFU-C to 6%, BFU-E to0.85%, CFU-E to 60% of the control values on D1; WBC decreased to 53% and MNC to65 % of the control values on D3. |
[101] |
|
| ||||
| Gastrointestinal toxicity | ||||
|
| ||||
| Wistar Male, 200-300g |
6 mg/kg ip |
D0-2 | Day 1: reduction in food and water intake Day 2: ↓food intake (86%),↓water intake(78%), ↑weight of gastric contents (10x) |
[137] |
|
| ||||
| C57BL/6 Male, 8-10wk |
6 mg/kg ip |
D0-2 | Day 1: reduction in food and water intake Day 2: ↓food intake (68%),↓water intake(45%), ↑weight of gastric contents (3x) |
[137] |
|
| ||||
| B6D2F1 Adult, male 26-30g |
8 mg/kg 10 mg/kg 12 mg/kg 14 mg/kg ip |
D1,3,6,10,14 |
Weight loss was dose dependent, maximal on day 6 (11- 26%); reticulocytopenia was dose dependent with the lowest one on day 6; necrosis of renal epithelium (dose dependent) was present by day 10 posttreatment and was still apparent 21-22 days after treatment azotemia BUN (dose dependent); dose-dependent lesions in intestinal epithelium appeared on days 1-6 (necrosis, hypertrophy, hyperplasia, and macronucleus); lesions in thecolon appeared later and were less severe. Mucosal recovery was evident by day 10 posttreatment |
[132] |
|
| ||||
| C57BL/6 8-12 wk, 20-25g N=6 |
27 mg/kg ip |
D1-3 | Villi in the small intestine shortened, reduced number of goblet cells, with increase of apoptosis 3h after cisplatin there were many apoptotic cells in the lamina propria of villi and massive apoptosis of vascular endothelial cells in the lamina propria (microvascular endothelial apoptosis) which preceded apoptosis in the epithelium (columnar epithelial cells) Similar results were observed in the thymus: massive apoptosis of endothelial cells 3-12h after cisplatin, followed by massive apoptosis of thymocytes 36-72h after cisplatin |
[138] |
|
| ||||
| ICR Male, Crl N=5 |
45 μmol/kg sc |
D4 | Decrease of total leucocytes, diarrhea observed in all animals | [108] |
|
| ||||
| CBA, UK Male, 6wk N=15 |
10 mg/kg ip |
D10 1,3,5,7,10 |
Alteration in the kinetics, morphology, and function of the mouse small intestinal mucosa.Severe depression in crypt cell production, evident by 2h and maximal between 12 and 24 h, with only a slow recovery on day 7 and marked depletion in both maltase and sucrose activity in jejunum on days 3 and 5. | [139] |
|
| ||||
| CBA Male, adult N=4 |
10 mg/kg ip |
D1,3,5,7,10 | Reduced height of villi and increased height of crypts in small intestine, temporary ablation of crypts, diminished mucosal function (↓disaccharidase activity) observed at D3-10 | [140] |
|
| ||||
| Wistar, Adult male N=15 |
10 mg/kg ip |
h1 | Mucosal damage, reduced jejunal net fluid and electrolyte absorption (sodium, potassium, chloride) | [141] |
|
| ||||
| Wistar Adult male |
6 mg/kg ip |
D3 | decreased activities of the brush border membrane enzymes (alkaline phosphatase, leucine aminopeptidase, γ-glutamyltransferase) in the lining the epithelial cells of intestine, increased production of ROS and alteration in the activities of several antioxidant enzymes (↓SOD, CAT, GPx, GR,G6PD, ↑GST, TR, MDA) | [142] |
|
| ||||
| Wistar Male 7 months |
7 mg/kg ip |
D5 | Changed structure of crypts and villi. Enlarged crypts, reduced length of the villi and epithelium denuded, infiltration of inflammatory cells. In the colon the epithelial tissues and subepithelial layer were affected the most due to cell degeneration, laminin immunopositivity | [143] |
|
| ||||
| Hepatotoxicity | ||||
|
| ||||
| Wistar female adult | 5 mg/kg ip |
D5 | Histological abnormalities: dispersed area of necrotic hepatocytes, inflammatory cellular infiltration, vacuolation and degeneration of hepatocytes, ↑ ALT, AST, γGT, ALP,↑ total bilirubin in serum; ↓SOD, GSH, GPx, GST, GR,↑MDA and NO, ↑expression of Cas3, Cas9, Bax in liver | [144] |
|
| ||||
| Wistar Male N=15 |
12 mg/kg ip |
D30 | ↑ ALT, AST, ALP, LDH, γGGT, albumin,↑ total bilirubin in serum ↓SOD, CAT, GSH, GPx, GST, GR,↑MDA, NO, TNF-α in liver |
[145] |
|
| ||||
| Testicular toxicity | ||||
|
| ||||
| Sprague-Dawley, male 8wk, 190-250g N=6 | 7 mg/kg ip |
D5 D50 |
Decreased weight of testes, epididymis, accessory glands (seminal vesicles, prostate), lower sperm concentration, ↓sperm motility, head abnormalities, ↑MDA, GPx, ↓GSH activity in testes | [146, 147] |
|
| ||||
| C57BL/6J Male, 6-16wk, Jax |
10 mg/kg ip |
D5-31 | Changes most evident at D5: majority of tubules were devoid of the late stages of spermatogenesis. Sperm production improved by D12-18 but the decreased number of spermatogenic cells was still evident; spermatocytes and spermatid were TUNEL positive at D14-18. Reduced number of sperm/ml on D31; decreased testis size and weight. | [135] |
|
| ||||
| Cardiotoxicity | ||||
|
| ||||
| Wistar Male, adult |
7 mg/kg ip |
D6 | ↑LDH, CK, TBARS, NO, ↓GSH, ATP degenerative changes and vacuolated cytoplasm of many muscle cells |
[134] |
|
| ||||
| Ototoxicity | ||||
|
| ||||
| CBA/J, female Jax4-8 wk, N=5 | 14 mg/kg ip |
D8 | Hearing loss; click evoked auditory brainstem responses threshold elevation at 12± 7 dB | [148, 149] |
|
| ||||
| Wistar Fisher 344 |
16 mg/kg 3x5 mg/kg ip |
D5 | Hearing loss, loss of cochlear outer hair cells, sensory and motor nerves conduction velocities alteration; threshold shift of more than 30 dB (at 14kHz measured by auditory evoked brain stem response); 20 dB (at 16 and 24 kHz) | [150] |
N: number; D: day; h: hour; ip: intraperitoneally; sc: subcutaneously; WBC: white blood cells; RBC: red blood cells; MNC: mononuclear cells; ↓: decrease; ↑: increase; BUN: blood urea nitrogen; ROS: reactive oxygen species; SOD: superoxide dismutase; CAT: catalase; GSH: glutathione; GPx: glutathione peroxidase; GR: glutathione reductase; GST: glutathione S transferase; G6PD: glucose 6-phosphate dehydrogenase; TR: thioredoxin reductase; MDA: malondialdehyde; TNFα: tumor necrosis factor alpha; ALT: alanine transaminase; AST: aspartate transaminase; ALP: alkaline phosphatase; γGT: gamma glutamyl transpeptidase; NO: nitric oxide; Cas3: caspase 3; Cas9: caspase 9; LDH: lactate dehydrogenase; CK: creatine kinase; TBARS: thiobarbituric acid reactive substances; ATP: adenosine triphosphate.
4. Factors Modifying Cisplatin Nephrotoxicity
Various factors can influence the susceptibility, onset, severity, and responsiveness to cisplatin-induced AKI. It is mostly accepted that the onset, severity, and mortality rate of cisplatin nephrotoxicity depend on the cisplatin dosage. However, much less attention is given to other factors such as microbiological state, genetic background, and physical conditions of animals.
4.1. Variability among Studies
Susceptibility to cisplatin nephrotoxicity is species specific. Rats are more susceptible to cisplatin toxicity than mice [151]. The rat kidney is also more sensitive to the effects of cisplatin than human kidney [64]. In addition, differences in the susceptibility between strains were also reported (shown in Table 6).
Table 6.
The acute lethal dose of cisplatin varies among strains of mice and rats.
| Strain (origin), | LD100 | End | Ref. |
|---|---|---|---|
| sex, age | |||
| BALB/c (Harlan) | 14.5 mg/kg; ip | D7 | [109, 120] |
| female, N=8 | |||
|
| |||
| C57BL/6, Japan | 15 mg/kg; ip | D10 | [41] |
| Male, 11-15wk; N=5-6 | |||
|
| |||
| CBA; female, | 16 mg/kg; ip | D7 | [149] |
| 24 months, N=3-7 | |||
|
| |||
| Wistar rats, | 8 mg/kg; ip | D11 | [110] |
| Male, N=17 | |||
|
| |||
| LD90 | |||
|
| |||
| B6D2F1 | 14 mg/kg; ip | D8 | [106] |
| Male, N=10 | |||
|
| |||
| DBA2 | 16 mg/kg; ip | D10 | [112] |
| Female; N=10 | |||
|
| |||
| Swiss Webster | 19.5 ± 0.8; iv | D10 | [106] |
| Male, N=10 | |||
|
| |||
| LD50 | |||
|
| |||
| DBA2 | 10.7 mg/kg; ip | D10 | [112] |
| Female; N=10 | (9.6-11.9mg/kg) | ||
|
| |||
| Swiss Webster | 16.0 ± 0.8; iv | D10 | [106] |
| Male, N=10 | |||
|
| |||
| NMRI | 17.0 mg/kg; ip | D10 | [112] |
| Female; N=10 | (14.9-19.7mg/kg) | ||
|
| |||
| Wistar rats | 10.8 mg/kg; ip | D10 | [112] |
| Female; N=10 | (9.1-12.8 mg/kg) | ||
|
| |||
| Fischer 344 rats | 11 mg/kg; ip | D6 | [133, 152] |
| female, 8wks | |||
N: number; ip: intraperitoneally; iv: intravenously; sc: subcutaneously; D: day; LD: lethal dose; LD100: dose of cisplatin that results in 100% mortality; LD50: dose of cisplatin that results in 50% mortality.
Another factor that can significantly affect cisplatin nephrotoxicity is age. Cisplatin nephrotoxicity was found to be less pronounced in 2-3-week-old unweaned rats compared to 7-8-week-old rats [98, 168, 169]. Younger rats were found to accumulate less cisplatin in their kidneys than older ones [169]. For instance, 6 days after single cisplatin injection (6 mg/kg) 3- and 7-week-old rats had 50 and 30%, respectively, lower concentrations of cisplatin in the kidney than 24-week-old rats [58]. In addition, nephrotoxicity occurred faster in 1-2-week-old rats (6 h after cisplatin) than in 7-week-old rats (3 days after cisplatin). By the time that older rats developed nephrotoxicity, damage in young rats was nearly completely repaired [169, 170]. In addition, differences between neonatal and adult rats were also found in the development of renal interstitial fibrosis. Adult rats developed extensive interstitial fibrosis [111], while in neonatal rats the formation of fibrotic lesions was delayed, and the lesions were limited to the area around the affected nephrons [15]. The reason for differences in cisplatin nephrotoxicity between young and adult rats may be in the stage of kidney development. In contrast with humans and mice, rats nephrogenesis completes 4 to 6 weeks after birth (see Table 7) [153].
Table 7.
Comparison of the kidney development among species.
| Glomerular nephrogenesis | GFR ∗ | Concentrating ability ∗ | ||
|---|---|---|---|---|
| species | onset | completion | age | age |
| Human | GD 35-37 | 35 weeks of gestation | 1-2 years | 1 year |
|
| ||||
| Rat | GD12.5 | Postnatal weeks 4-6 | 6 wk | 6 wk |
|
| ||||
| Mouse | GD11 | Before birth | nr | nr |
∗time when adult levels are reached. GD: gestation day; GFR: glomerular filtration rate. Data from Zoetis et al. [153].
In addition, nutritional status of the mother may significantly affect cisplatin nephrotoxicity as well. Offspring of mother rats fed low-protein diets during gestation have lower numbers of nephrons and low renal size until 19 weeks of their age [153].
Similarly to humans [18], aging animals are more susceptible to cisplatin toxicity. Single cisplatin injection (1 mg/kg) resulted in significantly increased BUN and Cr levels and structural degeneration of the proximal tubules in aging rats (52-week-old), beginning 3 days after injection, while in young rats (9-week-old) no significant changes were found (observed period 10 days) [155]. Single dose of cisplatin (16 mg/kg) in aging and young mice resulted in 100% mortality of 24-month-old female mice [149] and 40% mortality of 4-8-week-old mice within 7 days after administration [148, 149]. Increased susceptibility to cisplatin toxicity in aging animals can be ascribed to age-related changes in various tissues, including kidney. Thus, to avoid unwanted toxicity and death in aging animals lower dose of cisplatin should be used.
On the other hand, the influence of sex on cisplatin susceptibility in rodents is less conclusive. Some researchers reported that male Wistar rats have higher susceptibility to cisplatin nephrotoxicity than females [122, 171, 172], while others found no difference [36, 54, 124, 168]. Interestingly, in ovariectomized Wistar rats estrogen showed no effects on cisplatin nephrotoxicity [173], while in castrated Wistar rats testosterone showed protective effects at low dose but harmful effects at high dose [174]. Contradicting results have been reported in mice as well. One study found that male C57BL/6 mice are more susceptible to cisplatin nephrotoxicity than the females [75], while another one found female mice (C57BL/6J and 129Sv) more susceptible [59].
4.2. Variability within the Study
Although cisplatin model is reported as reproducible [9], animals do not always respond equally to the same cisplatin protocol (identical dose and administration regimens), even within the same group.
Animals may show enhanced susceptibility [69] or no response [63, 104] to cisplatin toxicity. For instance, in one study 3 of 5 rats developed moderate kidney injury and elevated BUN and Cr levels, while two rats (2/5) had no elevation in the BUN and Cr. Histological assessment revealed mild kidney injury in one rat but no histological alteration in another [63]. According to our experience lethal dose of cisplatin (17 mg/kg) may result in variable response as well. In our case 9/10 mice developed severe AKI, while 1 mouse remained healthy without any increase in BUN values or any clinical signs of illness. Mouse (1/10) experienced only slight drop of body weight (less than 4 %) and remained active, curious, and in good health.
Variability in cisplatin toxicity within the study [69, 98, 104] shows, that there are also other nongenetic factors that play important role in the susceptibility to cisplatin nephrotoxicity or even mortality. Such factors are dietary Mg-depletion [158], reduced intestinal Mg absorption [159], decreased dietary level of selenium [160], hydration status of animals, repeated blood collections during experiment, application of substances, etc. To reduce variability some investigators withhold food and water for few hours prior to cisplatin injection [65]. In addition, circadian timing of cisplatin administration was also shown to have significant effect on BUN, urine volume, urinary concentration of cisplatin, and morphology [133, 161, 175] as well as preventive effect of hydration on cisplatin nephrotoxicity and survival rate when given in lethal dose [152]. Kidney exhibits circadian rhythm of its function in both rodents and humans. The excretion of water and electrolyte and clearance of BUN and Cr from the blood are highly rhythmically regulated within the circadian time scale [176, 177]. In addition, it was shown that IL-6 production is rhythmically regulated, which suggests that cisplatin nephrotoxicity might also depend on kidney sensitivity to diurnal variation in inflammatory reaction without direct cisplatin toxicity [175].
Importantly, it was found that the presence of endotoxin (LPS) increases susceptibility to cisplatin nephrotoxicity, suggesting that coexisting infections might result in synergistic effects and influence cisplatin susceptibility [92, 93]. Since TLR4 receptor was found to be responsible for synergistic effects it was also suggested that acquired or genetic differences in TLR4 signaling or downstream pathways, such as NF-κB activation, might also influence the risk of cisplatin nephrotoxicity [93]. All these results show that microbiological state (latent infection) and genetic background of animals are important factors in cisplatin nephrotoxicity. Unfortunately, both microbiological status and genetic background of the animals are rarely properly reported, which may additionally contribute to confusion and discrepancy of the results.
5. Challenges of Cisplatin Rodent Model
The results demonstrate that cisplatin rodent model has many similarities to human nephrotoxicity (Table 8), offering an insight into underlying mechanisms under standardized and controlled conditions in time and dose dependent manner. However, there are also challenges that need to be addressed in the future studies.
Table 8.
Examples of the risk factors associated with cisplatin nephrotoxicity in humans and rodents.
| Risk factors | rodents | humans |
|---|---|---|
| Race, strain | Some strains are more susceptible than others (see Tables 3 and 4) |
African-Americans have high risk than Caucasians [154] |
|
| ||
| Age | Aging rats and mice are more susceptible [149, 155] | Incidence increases with age [18, 156] |
|
| ||
| Hydration | Hydration reduces nephrotoxicity and mortality [133, 152] |
Hydration is used to prevent cisplatin nephrotoxicity [157] |
|
| ||
| Mg supplementation | Increased risk in case of dietary Mg-depletion [158], or reduced intestinal Mg absorption [159], or decreased dietary level of Selenium [160] | Magnesium supplementation is used to prevent cisplatin nephrotoxicity [157] |
|
| ||
| Circadian rhythms | Reduced risk when injected in the middle of the dark period (when the urinary volume is maximal). Difference in survival can be 8-fold and in BUN levels 1.6-fold [152, 161] | ? |
|
| ||
| Dose | High doses of cisplatin increase the risk (see Tables 3 and 4) | High doses of cisplatin (↑50 mg/m2) increase the risk [28] |
|
| ||
| Frequency | Renal injury is more likely when cisplatin is administered at repetitively close time intervals (daily vs weekly vs 3-week interval). | Renal injury is much more likely when cisplatin is administered at repetitively close time intervals [28] |
|
| ||
| Long-term administration | Nephrotoxicity worsens with the time and repeated long-term treatment [29, 110] | Nephrotoxicity worsens with the time and repeated long-term treatment [18] |
|
| ||
| BUN, Cr, GFR | Unspecific and insensitive A need for better markers |
Unspecific and insensitive [17] A need for better markers |
|
| ||
| Extra-renal toxicity | Similar to humans (see Table 5) | Gastrointestinal toxicity, myelosuppression, ototoxicity, neuropathy, nephrotoxicity, vascular injury [130] |
5.1. A Need for Better Markers
In clinical practice, the diagnosis of kidney injury is mostly based on clinical markers such as BUN and serum Cr, supplemented with data on GFR estimated with different equations, which include additional variables like age, gender, and race [178]. In rodents, the diagnosis of nephrotoxicity is based on determination of BUN and/or Cr in the serum and histological assessment (see Tables 1, 3, and 4). However, the BUN and Cr are not ideal markers and have important deficiencies that need to be taken into consideration. They are unspecific and insensitive in both rodents and humans. The levels of BUN and Cr can be influenced by many other physiological events such as changes in protein synthesis, degradation due to starvation or loss of body weight, gastric or intestinal bleeding, and dehydration [56, 98, 104], all of which are usually seen in cisplatin model (see Table 4) and may contribute to potential underestimation of the actual degree of renal damage. In addition, BUN and Cr lack sensitivity in detecting early stages of kidney injury. As demonstrated in the paper, structural damage within the kidney can be present before BUN or Cr increases. In case of mild form of AKI, BUN and Cr are usually in normal levels. Increase in the BUN and Cr is usually not seen until more than 50 % of the nephrons are functionally damaged in rats and humans, while in mice a rise in the BUN and Cr occurs following the loss of 70-75% of the nephrons, which usually denotes severe nephrotoxicity [55, 56, 104]. Due to the drawbacks of current renal functional parameters, histological assessment is currently the most reliable method to determine the degree of nephrotoxicity in animal research, particularly in mice with mild to moderate AKI.
Nevertheless, since histological evaluation involves invasive procedure and requires experienced pathologist, there is intensive search for more reliable and sensitive biomarkers of nephrotoxicity in both rodents and man [179, 180]. Biomarkers should be noninvasive, indicative of kidney damage, and with ability to detect renal injury before development of marked histological or functional changes [104]. In rodents, collecting blood (for biomarker measurement) during acute phase of kidney injury is dissuaded because it worsens the course of AKI and may result in death of animals. Several candidate biomarkers of AKI in rats have been identified, such as kidney injury molecule-1 (KIM-1), clusterin, and osteopontin [105]. Some of proteins, detected in either urine or blood, were approved for nonclinical drug development. They can be species specific; thus, it is important to stress that they were evaluated only on rats. More information can be found elsewhere [100, 122, 123, 181–183]. Recently, also urinary miRNA as noninvasive biomarker of AKI in humans [184] and rodents [185] was proposed.
5.2. The Resemblance to Human Kidney Injury
One oftentimes criticized issue concerning the cisplatin model, particularly mice model, is that morphological changes in the rodents are not equivalent to those observed in human biopsy [20]. It was argued that an acute tubular necrosis in human renal biopsy “does not accurately reflect the morphological changes in this condition. In essence, ATN (acute tubular necrosis) is the situation in which there is adequate renal perfusion such that there is sufficient blood flow to largely maintain tubular integrity, but not to sustain glomerular filtration” [20]. Studies report that patients that are suffering from severe AKI in clinical settings [186] show almost normal histological picture or only sporadic mild lesions consisting of degeneration, necrosis, and regenerative changes in the proximal tubule, distal tubule, and collecting ducts [187]. Importantly, in clinical practice, diagnosis of AKI is mostly based on the rise of clinical markers such as BUN and serum Cr and/or the fall in urine output [2]. The renal biopsies are rarely performed in critically ill patients, mainly due to the perceived risk of bleeding complications and general lack of therapeutic consequences. The current knowledge is consequently limited. Animal studies have shown that the morphological changes in the kidney are focally distributed. In human renal biopsy only a small amount of the kidney is captured and thus the histological picture may not be representative. In animals usually the whole size of the kidney is morphologically examined which enables more representative results.
As demonstrated in the paper, depending on the cisplatin protocol rodents may develop various forms of kidney injury, from mild to severe. However, the evaluation of nephrotoxicity with current functional markers is limited particularly in mice, because in mice the BUN and Cr are elevated only when the kidney is already severely damaged. Thus, the critique refers more likely to the use of unspecific markers of nephrotoxicity and to the selection of lethal cisplatin protocols. Therefore, use of better cisplatin rodent models (i.e., use of appropriate cisplatin protocols) is needed to more accurately define the progression of structural and functional changes in the kidney (i.e., from AKI to CKI).
5.3. The Use of the Lethal Dose of Cisplatin
Appropriate dosing for cytotoxic anticancer agents in humans has been largely determined upon animal studies in which the goal was to maximize the efficacy and minimize the toxicity, whereas the occurrence of hematologic toxicity (i.e., anemia, leukopenia) usually correlated with proper dose to achieve optimal anticarcinogenic effect [188, 189]. In the 1980s numerous studies on cisplatin acute toxicity alone or in combination with various preventive agents have been reported. It was found that when cisplatin doses were increased above therapeutic doses (the maximum tolerated dose for rats was reported to be 6 mg/kg) the therapeutic effect was reduced due to the toxic side effects of cisplatin [112]. Recently, it was reported that the deletion of CD4+ T cell in mice not only did not protect against the kidney injury, but also had harmful effect on the cancer [91]. Thus, to use appropriate cisplatin protocol it is important to consider all side effects of cisplatin (Table 4), including immunosuppressive and carcinogenic ones. These results additionally show that the use of extremely high cisplatin dose (i.e., lethal dose) to produce model to investigate human situation is not scientifically justified and valid.
In addition, since lethal dose induces systemic toxicity, characterized by multiorgan-injuries, the use of lethal dose also raises a question on ethical justification of such experimental protocols. According to severity classification of EU Directive 2010/63, death of animal due to induced illness denotes severe suffering. In accordance with a good laboratory practice, EU legislation and a good science severe suffering of animals in experiments should be avoided. Thus, in case of cisplatin rodent model it is advised to use nonlethal doses of cisplatin or to intervene before severe suffering of animals is manifested.
5.4. Clinical Manifestation of The Toxicity
Monitoring and reporting the clinical signs of laboratory animals in experiments are necessary for many reasons such as “the assessment of animal welfare, compliance with the principle of refinement (e.g., humane endpoints), regulatory compliance (e.g., reporting severity) and, importantly, as a scientific outcome, e.g., in animal models of diseases or safety studies” [190]. In contrast, we noticed that clinical signs or mortality of animals in cisplatin nephrotoxicity studies are rarely reported. Moreover, we even found statement that “renal failure per se is not painful” [191].
However, in cisplatin rodent model clinical signs develop with a delay of a few days and are progressive in nature and dose related. After cisplatin administration, rodents show progressive dehydration and loss of body weight, anemia, and reduced activity. Although rats and mice cannot vomit (nausea and vomiting are major adverse effects of cisplatin therapy in humans) they severely suffer from gastrointestinal malaise already 2 days after low nephrotoxic dose of cisplatin in both mice and rats (6 mg/kg). Gastrointestinal malaise reflects as reduced food and water intake and impaired gastric function (demonstrated by enormously increased gastric content) [137].
In case of the lethal dose of cisplatin (17 mg/kg) clinical signs can be observed not earlier than 2 days after cisplatin injection, when mice show a slight drop of body weight and slight dehydration without other clinical signs of illness. Obvious clinical signs develop progressively on day 3 (obvious dehydration, ruffled hair, and reduced activity), reaching the peak on days 4-5, when gross pancytopenia and significant increase of BUN and Cr in the serum are detected. Mice suffer from hemorrhagic diarrhea and show clinical signs of severe pain (hunched posture, lethargy, orbital tightening, nose bulge, cheek bulge, and changed ear and whisker). Death occurs 4-7 days after cisplatin administration. Before death ataxia with loss of coordination, tremor and rotating body movements after upholding mice by their tails are regularly observed. Systemic injury with multiorgan involvement is reflected by systemic side effects, such as body weight loss, diarrhea, and mortality. At autopsy thymus and splenic involution are found, markedly enlarged stomach filled with food but empty small intestine, both with areas of hemorrhage, similar to what was described by Aggarwal et al. in rats [192]. Thus, to prevent severe suffering of animals it is important to intervene when animals show first signs of severe suffering (see coding of facial expression of pain [193]), are lethargic and moribund, or lose more that 20-25% of body weight.
5.5. Challenges in the Investigation of Inflammation
Although numerous studies investigated the role of inflammatory cells in cisplatin nephrotoxicity and demonstrated that various inflammatory cells are implicated in the pathogenesis of cisplatin nephrotoxicity (see Section 2.3), currently the role of inflammatory cells (dendritic cells, T cells, B cells, macrophages, neutrophils, Treg cells, and NK cells) is still difficult to interpret. Lack of surface markers unique to individual type of inflammatory cell population hampers the investigation and limits the interpretation of the results. In addition, the use of surface markers and inhibitors is not always properly reported. Immunology is complex and constantly developing field; thus, results should rely on surface markers and not cell types (i.e., CD11c+ instead of dendritic cell). Table 9 lists recently recognized surface markers expressed on particular inflammatory cells. Additional limitation represents the time and the duration of investigation of inflammatory process in animal studies. The role of inflammatory cells was investigated only in acute state, which is usually in the first three days after cisplatin administration. Since it is known that the role of inflammatory cells in cisplatin nephrotoxicity may differ in response to the phase and severity of the disease, this issue needs to be properly addressed in the future research. For instance, the deletion of CD4+ T cells reduced cisplatin nephrotoxicity in acute phase [51, 69], but it had no beneficial effect during disease progression 4 weeks later [91]. Finally, the results are obtained using variety of methods with different degree of sensitivity (morphology, immunohistochemistry, and flow cytometry), which needs to be taken into consideration.
Table 9.
Surface markers of inflammatory cells in cisplatin rodent model.
| Surface marker | Inflammatory cells that express surface marker | Ref. |
|---|---|---|
| CD45+ | pan-leukocyte marker | [162] |
|
| ||
| CD19+ (CD79α) | marker of B cells | [163] |
|
| ||
| CD3+ | marker of T cells | [163] |
|
| ||
| F4/80+ | canonical marker of macrophages, monocytes | [73] |
|
| ||
| CD11c+ | canonical marker of DC, recently also macrophages, activated CD8+ T cells, plasma B cell blasts, NK | [66, 73, 164] |
|
| ||
| CD14+ | monocytes, macrophages, dendritic cells, neutrophils | [165] |
|
| ||
| CD11b+ | macrophages, monocytes, neutrophils, dendritic cells | [73] |
|
| ||
| CD11b+CD27+ | NK (immature, mature) | [166] |
|
| ||
| CD49b+ | pan-NK marker, small subset of CD8+ T cells, platelets |
[68] |
|
| ||
| F4/80+CD11c−CD206+ | macrophages M2 | [167] |
|
| ||
| F4/80+CD11c+ | macrophages M1 | [167] |
|
| ||
| F4/80−CD11c+ | dendritic cells | [167] |
|
| ||
| GR-1+ | neutrophils, monocytes | [73] |
|
| ||
| GR-1+ CD11b+ CD11c+ | monocyte-derived proinflammatory DC | [73] |
|
| ||
| GR-1+ CD11b+ CD11c− | neutrophils, monocytes | [73] |
|
| ||
| 7/4+ | monocytes, neutrophils | [73] |
|
| ||
| Ly6G+ | neutrophils | [73] |
|
| ||
| 7/4+Ly6G− | monocytes | [73] |
DC: dendritic cells; NK: natural killer cells
The most important factor that can influence the validity of the results is the fact that depending on the dose cisplatin may exert immunosuppressive effects (Figure 2) [101, 106] or cause systemic toxicity. Therefore, to study inflammatory process in acute or chronic nephrotoxicity the use of appropriate cisplatin dosage regimen reflecting those used in cancer patients is necessary. Otherwise investigation may provide contradictory results, such as in case of anti-inflammatory drugs, where one study reported beneficial effect of COX-2 inhibitor on cisplatin toxicity (cisplatin: 20mg/kg, high lethal dose) [86], while in another study no effect was found (rats (6mg/kg), Wistar Kyoto rats (5 mg/kg, nephrotoxic dose)) [194].
Figure 2.
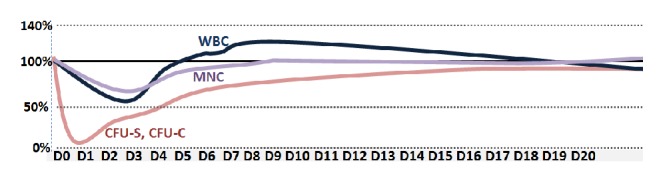
Cisplatin has immunosuppressive effects and exhibits cytotoxicity to spleen (CFU-S), granulocyte-macrophage (CFU-C) colony-forming units, and mononuclear cells (MNC) in bone marrow and white blood cells (WBC) in F1 CBAxC57BL female mice (cisplatin: 12 mg/kg, single ip), adapted and modified from Nowrousian et al. [101].
6. Conclusions
Cisplatin nephrotoxicity is very complex disease, which involves both complex local events in the kidney and complex interconnected and interdependent systemic effects in the body. Therefore, animal models are indispensable in the investigation of both acute and chronic kidney injury. Although cisplatin rodent model is simple to induce, is not expensive, and has many similarities with human cisplatin nephrotoxicity, it is important to take into consideration that the selection of the cisplatin dosage regimen (cisplatin protocol) significantly affects the characteristics of the model and the outcome of the study. The heterogeneity of cisplatin protocols may contribute to improved knowledge and insight into the development and progression of cisplatin kidney injury, but on the other hand, when used inappropriately, it may hamper the interpretation and usefulness of the results, for instance, when cisplatin dosage results in systemic toxicity instead of nephrotoxicity or when the time frame or method to diagnose mild kidney injury is not optimally selected. Therefore, it is important to recognize that each cisplatin protocol has its own advantages and limitations. The choice of the protocol should thus be based on the scope and aims of a particular study and the characteristics and limitation of a particular cisplatin protocol. However, use of cisplatin protocols that cause acute systemic toxicity should be avoided due to ethical and scientific reasons.
In addition, studies have shown that many factors can affect susceptibility to cisplatin toxicity in rodents. To avoid misinterpretation of the published results, research on animal models should be properly reported in accordance with the ARRIVE guidelines [195] or gold standard publication checklist [196], FELASA recommendations [197, 198], and standardized genetic nomenclature of rodents (http://www.informatics.jax.org/nomen/strains.shtml).
Acknowledgments
This work was in part supported by ARRS (Slovenian Research Agency, Programs P3-054 and P3-0323).
Abbreviations
- 4-HNE:
4-Hydroxynonenal
- AKI:
Acute kidney injury
- ALP:
Alkaline phosphatase
- ALT:
Alanine transaminase
- AST:
Aspartate transaminase
- ATN:
Acute tubular necrosis
- ATP:
Adenosine triphosphate
- BUN:
Blood urea nitrogen
- Casp:
Caspase
- CAT:
Catalase
- CFU-C:
Granulocyte-macrophage colony-forming unit
- CFU-S:
Spleen colony-forming unit
- CK:
Creatine kinase
- CKI:
Chronic kidney disease
- CNTF:
Ciliary neurotrophic factor
- COX:
Cyclooxygenase
- cPGES:
Cytosolic prostaglandin E synthase
- Cr:
Creatinine
- CT-1:
Cardiotrophin-1
- Ctr 1:
Copper transporter 1
- G6PD:
Glucose 6-phosphate dehydrogenase
- GFR:
Glomerular filtration rate
- gp130:
Glycoprotein 130
- GPx:
Glutathione peroxidase
- GR:
Glutathione reductase
- GSH:
Glutathione
- GSSG:
Glutathione disulfide
- GST:
Glutathione S transferase
- HEP:
Humane endpoint
- HO-1:
Heme oxygenase-1
- ICAM-1:
Intracellular adhesion molecule-1
- IFNγ:
Interferon gamma
- IL:
Interleukin
- iNOS:
Inducible nitric oxide synthase
- ip:
Intraperitoneally
- iv:
Intravenously
- KC:
Keratinocyte-derived chemokine
- KIM-1:
Kidney injury molecule-1
- LDH:
Lactate dehydrogenase
- LIF:
Leukemia inhibitory factor
- MATE1:
Multidrug and toxin extrusion 1
- MCA:
Multiplexed cytokine assay
- MCP-1:
Monocyte chemoattractant protein-1
- MDA:
Malondialdehyde
- MIF:
Macrophage migration inhibitory factor
- MNC:
Mononuclear cells
- mPGES:
Microsomal prostaglandin E synthase
- MPO:
Myeloperoxidase
- MT:
Metallothioneins
- NADPH:
Nicotinamide adenine dinucleotide phosphate
- NO:
Nitric oxide
- Nrf2:
Nuclear factor erythroid 2-related factor
- OCT 2:
Organic cation transporter 2
- OSM:
Oncostatin M
- PGE:
Prostaglandin E
- ROS:
Reactive oxygen species
- RPA:
Ribonuclease protection assay
- sc:
Subcutaneously
- SOD:
Superoxide dismutase
- TBARS:
Thiobarbituric acid reactive substances
- TGFβ:
Tumor growth factor
- TLR4:
Toll like receptor 4
- TNFR1:
TNF receptor 1
- TNFR2:
TNF receptor 2
- TNFα:
Tumor necrosis factor alpha
- TR:
Thioredoxin reductase
- WBC:
White blood cells
- γGT:
Gamma glutamyl transpeptidase.
Conflicts of Interest
The authors declare that no financial interest or conflicts of interest exist.
References
- 1.Uccelli A., Moretta L., Pistoia V. Mesenchymal stem cells in health and disease. Nature Reviews Immunology. 2008;8(9):726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 2.Chawla L. S., Eggers P. W., Star R. A., Kimmel P. L. Acute kidney injury and chronic kidney disease as interconnected syndromes. The New England Journal of Medicine. 2014;371(1):58–66. doi: 10.1056/NEJMra1214243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozkok A., Edelstein C. L. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Research International. 2014 doi: 10.1155/2014/967826.967826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J., Goering P. L., Espandiari P., et al. Differences in immunolocalization of Kim-1, RPA-1, and RPA-2 in kidneys of gentamicin-, cisplatin-, and valproic acid-treated rats: potential role of iNOS and nitrotyrosine. Toxicologic Pathology. 2009;37(5):629–643. doi: 10.1177/0192623309339605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cepeda V., Fuertes M. A., Castilla J., Alonso C., Quevedo C., Pérez J. M. Biochemical mechanisms of cisplatin cytotoxicity. Anti-Cancer Agents in Medicinal Chemistry. 2007;7(1):3–18. doi: 10.2174/187152007779314044. [DOI] [PubMed] [Google Scholar]
- 6.Zhu S., Pabla N., Tang C., He L., Dong Z. DNA damage response in cisplatin-induced nephrotoxicity. Archives of Toxicology. 2015;89(12):2197–2205. doi: 10.1007/s00204-015-1633-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ali B. H., Al Moundhri M. S. Agents ameliorating or augmenting the nephrotoxicity of cisplatin and other platinum compounds: a review of some recent research. Food and Chemical Toxicology. 2006;44(8):1173–1183. doi: 10.1016/j.fct.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 8.Večerić-Haler Ž., Cerar A., Perše M. (Mesenchymal) Stem cell-based therapy in cisplatin-induced acute kidney injury animal model: risk of immunogenicity and tumorigenicity. Stem Cells International. 2017:17. doi: 10.1155/2017/7304643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heyman S. N., Rosen S., Rosenberger C. Animal models of renal dysfunction: acute kidney injury. Expert Opinion on Drug Discovery. 2009;4(6):629–641. doi: 10.1517/17460440902946389. [DOI] [PubMed] [Google Scholar]
- 10.Dobyan D. C., Levi J., Jacobs C., Kosek J., Weiner M. W. Mechanism of cis-platinum nephrotoxicity: II. Morphologic observations. The Journal of Pharmacology and Experimental Therapeutics. 1980;213(3):551–556. [PubMed] [Google Scholar]
- 11.Dobyan D. C. Long-term consequences of cis-platinum-induced renal injury: a structural and functional study. Anatomical Record. 1985;212(3):239–245. doi: 10.1002/ar.1092120304. [DOI] [PubMed] [Google Scholar]
- 12.Sendao M. C., Francescato H. D., Antunes L. M., Costa R. S., Bianchi Mde L. Comparative study of multiple dosage of quercetin against cisplatin-induced nephrotoxicity and oxidative stress in rat kidneys. Pharmacological Reports. 2006;58(4):526–532. [PubMed] [Google Scholar]
- 13.Amate J. Y., Ishida A., Tsujino K., et al. Immunohistochemical study of rat renal interstitial fibrosis induced by repeated injection of cisplatin, with special reference to the kinetics of macrophages and myofibroblasts. Toxicologic Pathology. 1996;24(2):199–206. doi: 10.1177/019262339602400208. [DOI] [PubMed] [Google Scholar]
- 14.Yamate J., Sato K., Ide M., et al. Participation of different macrophage populations and myofibroblastic cells in chronically developed renal interstitial fibrosis after cisplatin-induced renal injury in rats. Veterinary Pathology. 2002;39(3):322–333. doi: 10.1354/vp.39-3-322. [DOI] [PubMed] [Google Scholar]
- 15.Yamate J., Machida Y., Ide M., et al. Cisplatin-Induced Renal Interstitial Fibrosis in Neonatal Rats, Developing as Solitary Nephron Unit Lesions. Toxicologic Pathology. 2005;33(2):207–217. doi: 10.1080/01926230490523978. [DOI] [PubMed] [Google Scholar]
- 16.Palant C. E., Amdur R. L., Chawla L. S. The acute kidney injury to chronic kidney disease transition: A potential opportunity to improve care in acute kidney injury. Contributions to Nephrology. 2016;187:55–72. doi: 10.1159/000442365. [DOI] [PubMed] [Google Scholar]
- 17.Palant C. E., Chawla L. S., Faselis C., et al. High serum creatinine nonlinearity: A renal vital sign? American Journal of Physiology-Renal Physiology. 2016;311(2):F305–F309. doi: 10.1152/ajprenal.00025.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharp C. N., Siskind L. J. Developing better mouse models to study cisplatin-induced kidney injury. American Journal of Physiology-Renal Physiology. 2017;313(4):F835–F841. doi: 10.1152/ajprenal.00285.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skrypnyk N. I., Siskind L. J., Faube S., de Caestecker M. P. Bridging translation for acute kidney injury with better preclinical modeling of human disease. American Journal of Physiology-Renal Physiology. 2016;310(10):F972–F984. doi: 10.1152/ajprenal.00552.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosen S., Heyman S. N. Difficulties in understanding human "acute tubular necrosis": Limited data and flawed animal models. Kidney International. 2001;60(4):1220–1224. doi: 10.1046/j.1523-1755.2001.00930.x. [DOI] [PubMed] [Google Scholar]
- 21.Heyman S. N., Rosenberger C., Rosen S. Acute kidney injury: lessons from experimental models. Contributions to Nephrology. 2011;169:286–296. doi: 10.1159/000313957. [DOI] [PubMed] [Google Scholar]
- 22.Kociba R. J., Sleight S. D. Acute toxicologic and pathologic effects of cis-diamminedichloroplatinum (NSC-119875) in the male rat. Cancer Chemotherapy Reports. 1971;55(1):1–8. [PubMed] [Google Scholar]
- 23.Fillastre J. P., Raguenez-Viotte G. Cisplatin nephrotoxicity. Toxicology Letters. 1989;46(1-3):163–175. doi: 10.1016/0378-4274(89)90125-2. [DOI] [PubMed] [Google Scholar]
- 24.Yao X., Panichpisal K., Kurtzman N., Nugent K. Cisplatin nephrotoxicity: a review. The American Journal of the Medical Sciences. 2007;334(2):115–124. doi: 10.1097/MAJ.0b013e31812dfe1e. [DOI] [PubMed] [Google Scholar]
- 25.Dasari S., Tchounwou P. B. Cisplatin in cancer therapy: molecular mechanisms of action. European Journal of Pharmacology. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.dos Santos N. A. G., Rodrigues M. A. C., Martins N. M., dos Santos A. C. Cisplatin-induced nephrotoxicity and targets of nephroprotection: an update. Archives of Toxicology. 2012;86(8):1233–1250. doi: 10.1007/s00204-012-0821-7. [DOI] [PubMed] [Google Scholar]
- 27.Miller R. P., Tadagavadi R. K., Ramesh G., Reeves W. B. Mechanisms of cisplatin nephrotoxicity. Toxins (Basel) 2010;2(11):2490–2518. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sánchez-González PD, López-Hernández F. J., López-Novoa J. M., Morales A. I. An integrative view of the pathophysiological events leading to cisplatin nephrotoxicity. Critical Reviews in Toxicology. 2011;41(10):803–821. doi: 10.3109/10408444.2011.602662. [DOI] [PubMed] [Google Scholar]
- 29.Okada A., Fukushima K., Fujita M., et al. Alterations in cisplatin pharmacokinetics and its acute/sub-chronic kidney injury over multiple cycles of cisplatin treatment in rats. Biological & Pharmaceutical Bulletin. 2017;40(11):1948–1955. doi: 10.1248/bpb.b17-00499. [DOI] [PubMed] [Google Scholar]
- 30.Siddik Z. H., Newell D. R., Boxall F. E., Harrap K. R. The comparative pharmacokinetics of carboplatin and cisplatin in mice and rats. Biochemical Pharmacology. 1987;36(12):1925–1932. doi: 10.1016/0006-2952(87)90490-4. [DOI] [PubMed] [Google Scholar]
- 31.Litterst C. L. Alterations in the toxicity of cis-Dichlorodiammineplatinum-II and in tissue localization of platinum as a function of NaCl concentration in the vehicle of administration. Toxicology and Applied Pharmacology. 1981;61(1):99–108. doi: 10.1016/0041-008X(81)90011-9. [DOI] [PubMed] [Google Scholar]
- 32.Levi J., Jacobs C., Kalman S. M., McTigue M., Weiner M. W. Mechanism of cis-platinum nephrotoxicity: I. Effects of sulfhydryl groups in rat kidneys. The Journal of Pharmacology and Experimental Therapeutics. 1980;213(3):545–550. [PubMed] [Google Scholar]
- 33.Esteban-Fernández D., Verdaguer J. M., Ramírez-Camacho R., Palacios M. A., Gómez-Gómez M. M. Accumulation, fractionation, and analysis of platinum in toxicologically affected tissues after cisplatin, oxaliplatin, and carboplatin administration. Journal of Analytical Toxicology. 2008;32(2):140–146. doi: 10.1093/jat/32.2.140. [DOI] [PubMed] [Google Scholar]
- 34.Jongejan H. T. M., Provoost A. P., Wolff E. D., Molenaar J. C. Nephrotoxicity of Cis-Platin comparing young and adult rats. Pediatric Research. 1986;20(1):9–14. doi: 10.1203/00006450-198601000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Filipski K. K., Mathijssen R. H., Mikkelsen T. S., Schinkel A. H., Sparreboom A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clinical Pharmacology & Therapeutics. 2009;86(4):396–402. doi: 10.1038/clpt.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moreno-Gordaliza E., Giesen C., Lázaro A., et al. Elemental bioimaging in kidney by LA-ICP-MS as a tool to study nephrotoxicity and renal protective strategies in cisplatin therapies. Analytical Chemistry. 2011;83(20):7933–7940. doi: 10.1021/ac201933x. [DOI] [PubMed] [Google Scholar]
- 37.Sekiya S., Iwasawa H., Takamizawa H. Comparison of the intraperitoneal and intravenous routes of cisplatin administration in an advanced ovarian cancer model of the rat. American Journal of Obstetrics & Gynecology. 1985;153(1):106–111. doi: 10.1016/0002-9378(85)90605-2. [DOI] [PubMed] [Google Scholar]
- 38.Pabla N., Murphy R. F., Liu K., Dong Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. American Journal of Physiology-Renal Physiology. 2009;296(3):F505–F511. doi: 10.1152/ajprenal.90545.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jonker J. W., Schinkel A. H. Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1-3) The Journal of Pharmacology and Experimental Therapeutics. 2004;308(1):2–9. doi: 10.1124/jpet.103.053298. [DOI] [PubMed] [Google Scholar]
- 40.Yonezawa A., Masuda S., Nishihara K., Yano I., Katsura T., Inui K.-I. Association between tubular toxicity of cisplatin and expression of organic cation transporter rOCT2 (Slc22a2) in the rat. Biochemical Pharmacology. 2005;70(12):1823–1831. doi: 10.1016/j.bcp.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 41.Nakamura T., Yonezawa A., Hashimoto S., Katsura T., Inui K.-I. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochemical Pharmacology. 2010;80(11):1762–1767. doi: 10.1016/j.bcp.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 42.Santos N. A. G., Catão C. S., Martins N. M., Curti C., Bianchi M. L. P., Santos A. C. Cisplatin-induced nephrotoxicity is associated with oxidative stress, redox state unbalance, impairment of energetic metabolism and apoptosis in rat kidney mitochondria. Archives of Toxicology. 2007;81(7):495–504. doi: 10.1007/s00204-006-0173-2. [DOI] [PubMed] [Google Scholar]
- 43.Gordon J. A., Gattone V. H. Mitochondrial alterations in cisplatin-induced acute renal failure. The American Journal of Physiology. 1986;250((6 Pt 2)):F991–F998. doi: 10.1152/ajprenal.1986.250.6.F991. [DOI] [PubMed] [Google Scholar]
- 44.Singh G. A possible cellular mechanism of cisplatin-induced nephrotoxicity. Toxicology. 1989;58(1):71–80. doi: 10.1016/0300-483X(89)90105-4. [DOI] [PubMed] [Google Scholar]
- 45.Kishore B. K., Krane C. M., Di Iulio D., Menon A. G., Cacini W. Expression of renal aquaporins 1, 2, and 3 in a rat model of cisplatin-induced polyuria. Kidney International. 2000;58(2):701–711. doi: 10.1046/j.1523-1755.2000.00216.x. [DOI] [PubMed] [Google Scholar]
- 46.Lee S., Kim W., Moon S., et al. Rosiglitazone ameliorates cisplatin-induced renal injury in mice. Nephrology Dialysis Transplantation . 2006;21(8):2096–2105. doi: 10.1093/ndt/gfl194. [DOI] [PubMed] [Google Scholar]
- 47.Yang C., Kaushal V., Shah S. V., Kaushal G. P. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. American Journal of Physiology-Renal Physiology. 2008;294(4):F777–F787. doi: 10.1152/ajprenal.00590.2007. [DOI] [PubMed] [Google Scholar]
- 48.Price P. M., Safirstein R. L., Megyesi J. The cell cycle and acute kidney injury. Kidney International. 2009;76(6):604–613. doi: 10.1038/ki.2009.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daugaard G., Abildgaard U., Larsen S., et al. Functional and histopathological changes in dog kidneys after administration of cisplatin. Kidney and Blood Pressure Research. 1987;10(1):54–64. doi: 10.1159/000173114. [DOI] [PubMed] [Google Scholar]
- 50.Winston J. A., Safirstein R. Reduced renal blood flow in early cisplatin-induced acute renal failure in the rat. American Journal of Physiology-Endocrinology and Metabolism. 1985;249(4):F490–496. doi: 10.1152/ajpcell.1985.249.5.C490. [DOI] [PubMed] [Google Scholar]
- 51.Akcay A., Nguyen Q., He Z., et al. IL-33 exacerbates acute kidney injury. Journal of the American Society of Nephrology. 2011;22(11):2057–2067. doi: 10.1681/ASN.2010091011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu L. H., Oh D. J., Dursun B., et al. Increased macrophage infiltration and fractalkine expression in cisplatin-induced acute renal failure in mice. The Journal of Pharmacology and Experimental Therapeutics. 2008;324(1):111–117. doi: 10.1124/jpet.107.130161. [DOI] [PubMed] [Google Scholar]
- 53.Morigi M., Rota C., Montemurro T., et al. Life-sparing effect of human cord blood-mesenchymal stem cells in experimental acute kidney injury. Stem Cells. 2010;28(3):513–522. doi: 10.1002/stem.293. [DOI] [PubMed] [Google Scholar]
- 54.Humanes B., Lazaro A., Camano S., et al. Cilastatin protects against cisplatin-induced nephrotoxicity without compromising its anticancer efficiency in rats. Kidney International. 2012;82(6):652–663. doi: 10.1038/ki.2012.199. [DOI] [PubMed] [Google Scholar]
- 55.McKeage M. J., Morgan S. E., Boxall F. E., Murrer B. A., Hard G. C., Harrap K. R. Lack of nephrotoxicity of oral ammine/amine platinum (IV) dicarboxylate complexes in rodents. British Journal of Cancer. 1993;67(5):996–1000. doi: 10.1038/bjc.1993.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jodrell D. I., Morgan S. E., Clinton S., et al. The renal effects of N10-propargyl-5,8-dideazafolic acid (CB3717) and a non-nephrotoxic analogue ICI D1694, in mice. British Journal of Cancer. 1991;64(5):833–838. doi: 10.1038/bjc.1991.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Braunlich H., Kuo C. H., Slocombe R., Hook J. B. cis-Dichlorodiammineplatinum nephrotoxicity in rats of different ages. Research Communications in Chemical Pathology and Pharmacology. 1984;44(2):279–291. [PubMed] [Google Scholar]
- 58.Ali B. H., Al-Moundhri M., Tageldin M., et al. Ontogenic aspects of cisplatin-induced nephrotoxicity in rats. Food and Chemical Toxicology. 2008;46(11):3355–3359. doi: 10.1016/j.fct.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 59.Wei Q., Wang M. H., Dong Z. Differential gender differences in ischemic and nephrotoxic acute renal failure. The American Journal of Nephrology. 2005;25(5):491–499. doi: 10.1159/000088171. [DOI] [PubMed] [Google Scholar]
- 60.Santeramo I., Perez Z. H., Illera A., et al. Human kidney-derived cells ameliorate acute kidney injury without engrafting into renal tissue. Stem Cells Translational Medicine. 2017;6(5):1373–1384. doi: 10.1002/sctm.16-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang Q., Dunn R. T., II, Jayadev S., et al. Assessment of cisplatin-induced nephrotoxicity by microarray technology. Toxicological Sciences. 2001;63(2):196–207. doi: 10.1093/toxsci/63.2.196. [DOI] [PubMed] [Google Scholar]
- 62.Wang E.-J., Snyder R. D., Fielden M. R., Smith R. J., Gu Y.-Z. Validation of putative genomic biomarkers of nephrotoxicity in rats. Toxicology. 2008;246(2-3):91–100. doi: 10.1016/j.tox.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 63.Thompson K. L., Afshari C. A., Amin R. P., et al. Identification of platform-independent gene expression markers of cisplatin nephrotoxicity. Environmental Health Perspectives. 2004;112(4):488–494. doi: 10.1289/ehp.6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vickers A. E. M., Rose K., Fisher R., Saulnier M., Sahota P., Bentley P. Kidney slices of human and rat to characterize cisplatin-induced injury on cellular pathways and morphology. Toxicologic Pathology. 2004;32(5):577–590. doi: 10.1080/01926230490508821. [DOI] [PubMed] [Google Scholar]
- 65.Faubel S., Lewis E. C., Reznikov L., et al. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1β, IL-18, IL-6, and neutrophil infiltration in the kidney. The Journal of Pharmacology and Experimental Therapeutics. 2007;322(1):8–15. doi: 10.1124/jpet.107.119792. [DOI] [PubMed] [Google Scholar]
- 66.Tadagavadi R. K., Gao G., Wang W. W., Gonzalez M. R., Reeves W. B. Dendritic cell protection from cisplatin nephrotoxicity is independent of neutrophils. Toxins. 2015;7(8):3245–3256. doi: 10.3390/toxins7083245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chan A. J., Alikhan M. A., Odobasic D., et al. Innate IL-17A-producing leukocytes promote acute kidney injury via inflammasome and toll-like receptor activation. The American Journal of Pathology. 2014;184(5):1411–1418. doi: 10.1016/j.ajpath.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 68.Kim H.-R., Lee M.-K., Park A.-J., et al. Reduction of natural killer and natural killer T cells is not protective in cisplatin-induced acute renal failure in mice. Nephrology. 2011;16(6):545–551. doi: 10.1111/j.1440-1797.2011.01473.x. [DOI] [PubMed] [Google Scholar]
- 69.Liu M., Chien C., Burne-Taney M., et al. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. Journal of the American Society of Nephrology. 2006;17(3):765–774. doi: 10.1681/ASN.2005010102. [DOI] [PubMed] [Google Scholar]
- 70.Lee H., Nho D., Chung H., et al. CD4+ CD25+ regulatory T cells attenuate cisplatin-induced nephrotoxicity in mice. Kidney International. 2010;78(11):1100–1109. doi: 10.1038/ki.2010.139. [DOI] [PubMed] [Google Scholar]
- 71.Summers S. A., Chan J., Gan P., et al. Mast cells mediate acute kidney injury through the production of TNF. Journal of the American Society of Nephrology. 2011;22(12):2226–2236. doi: 10.1681/ASN.2011020182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alikhan M. A., Summers S. A., Gan P. Y., et al. Endogenous toll-like receptor 9 regulates aki by promoting regulatory T cell recruitment. Journal of the American Society of Nephrology. 2016;27(3):706–714. doi: 10.1681/ASN.2014090927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tadagavadi R. K., Reeves W. B. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. Journal of the Americal Society of Nephrology. 2010;21(1):53–63. doi: 10.1681/ASN.2009040407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu H., Baliga R. Cytochrome P450 2E1 null mice provide novel protection against cisplatin-induced nephrotoxicity and apoptosis. Kidney International. 2003;63(5):1687–1696. doi: 10.1046/j.1523-1755.2003.00908.x. [DOI] [PubMed] [Google Scholar]
- 75.Townsend D. M., Tew K. D., He L., King J. B., Hanigan M. H. Role of glutathione S-transferase Pi in cisplatin-induced nephrotoxicity. Biomedicine & Pharmacotherapy. 2009;63(2):79–85. doi: 10.1016/j.biopha.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kawai H., Sato W., Yuzawa Y., et al. Lack of the growth factor midkine enhances survival against cisplatin-induced renal damage. The American Journal of Pathology. 2004;165(5):1603–1612. doi: 10.1016/S0002-9440(10)63417-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuwana H., Terada Y., Kobayashi T., et al. The phosphoinositide-3 kinase γ-Akt pathway mediates renal tubular injury in cisplatin nephrotoxicity. Kidney International. 2008;73(4):430–445. doi: 10.1038/sj.ki.5002702. [DOI] [PubMed] [Google Scholar]
- 78.Yu F., Megyesi J., Safirstein R. L., Price P. M. Involvement of the CDK2-E2F1 pathway in cisplatin cytotoxicity in vitro and in vivo. American Journal of Physiology-Renal Physiology. 2007;293(1):F52–F59. doi: 10.1152/ajprenal.00119.2007. [DOI] [PubMed] [Google Scholar]
- 79.Yang C., Kaushal V., Haun R. S., Seth R., Shah S. V., Kaushal G. P. Transcriptional activation of caspase-6 and -7 genes by cisplatin-induced p53 and its functional significance in cisplatin nephrotoxicity. Cell Death & Differentiation. 2008;15(3):530–544. doi: 10.1038/sj.cdd.4402287. [DOI] [PubMed] [Google Scholar]
- 80.Wei Q., Dong G., Yang T., Megyesi J., Price P. M., Dong Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. American Journal of Physiology-Endocrinology and Metabolism. 2007;293(4):F1282–F1291. doi: 10.1152/ajprenal.00230.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wei Q., Dong G., Franklin J., Dong Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney International. 2007;72(1):53–62. doi: 10.1038/sj.ki.5002256. [DOI] [PubMed] [Google Scholar]
- 82.Faubel S., Ljubanovic D., Reznikov L., Somerset H., Dinarello C. A., Edelstein C. L. Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney International. 2004;66(6):2202–2213. doi: 10.1111/j.1523-1755.2004.66010.x. [DOI] [PubMed] [Google Scholar]
- 83.Megyesi J., Safirstein R. L., Price P. M. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. The Journal of Clinical Investigation. 1998;101(4):777–782. doi: 10.1172/JCI1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shiraishi F., Curtis L. M., Truong L., et al. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. American Journal of Physiology-Renal Physiology. 2000;278(5):F726–F736. doi: 10.1152/ajprenal.2000.278.5.F726. [DOI] [PubMed] [Google Scholar]
- 85.Pan H., Shen Z., Mukhopadhyay P., et al. Anaphylatoxin C5a contributes to the pathogenesis of cisplatin-induced nephrotoxicity. American Journal of Physiology-Renal Physiology. 2009;296(3):F496–F504. doi: 10.1152/ajprenal.90443.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jia Z., Wang N., Aoyagi T., Wang H., Liu H., Yang T. Amelioration of cisplatin nephrotoxicity by genetic or pharmacologic blockade of prostaglandin synthesis. Kidney International. 2011;79(1):77–88. doi: 10.1038/ki.2010.331. [DOI] [PubMed] [Google Scholar]
- 87.Ramesh G., Brian Reeves W. TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. The Journal of Clinical Investigation. 2002;110(6):835–842. doi: 10.1172/JCI200215606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang B., Ramesh G., Uematsu S., Akira S., Reeves W. B. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. Journal of the American Society of Nephrology. 2008;19(5):923–932. doi: 10.1681/ASN.2007090982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mitazaki S., Kato N., Suto M., Hiraiwa K., Abe S. Interleukin-6 deficiency accelerates cisplatin-induced acute renal failure but not systemic injury. Toxicology. 2009;265(3):115–121. doi: 10.1016/j.tox.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 90.Mitazaki S., Hashimoto M., Matsuhashi Y., et al. Interleukin-6 modulates oxidative stress produced during the development of cisplatin nephrotoxicity. Life Sciences. 2013;92(12):694–700. doi: 10.1016/j.lfs.2013.01.026. [DOI] [PubMed] [Google Scholar]
- 91.Ravichandran K., Wang Q., Ozkok A., et al. CD4 T cell knockout does not protect against kidney injury and worsens cancer. Journal of Molecular Medicine. 2016;94(4):443–455. doi: 10.1007/s00109-015-1366-z. [DOI] [PubMed] [Google Scholar]
- 92.Ramesh G., Zhang B., Uematsu S., Akira S., Reeves W. B. Endotoxin and cisplatin synergistically induce renal dysfunction and cytokine production in mice. American Journal of Physiology-Renal Physiology. 2007;293(1):F325–F332. doi: 10.1152/ajprenal.00158.2007. [DOI] [PubMed] [Google Scholar]
- 93.Zager R. A., Johnson A. C. M., Hanson S. Y., Lund S. Acute nephrotoxic and obstructive injury primes the kidney to endotoxin-driven cytokine/chemokine production. Kidney International. 2006;69(7):1181–1188. doi: 10.1038/sj.ki.5000022. [DOI] [PubMed] [Google Scholar]
- 94.Ramesh G., Reeves W. B. p38 MAP kinase inhibition ameliorates cisplatin nephrotoxicity in mice. American Journal of Physiology-Renal Physiology. 2005;289(1):F166–F174. doi: 10.1152/ajprenal.00401.2004. [DOI] [PubMed] [Google Scholar]
- 95.Ramesh G., Reeves W. B. Salicylate reduces cisplatin nephrotoxicity by inhibition of tumor necrosis factor-alpha. Kidney International. 2004;65(2):490–499. doi: 10.1111/j.1523-1755.2004.00413.x. [DOI] [PubMed] [Google Scholar]
- 96.Zhang B., Ramesh G., Norbury C. C., Reeves W. B. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-α produced by renal parenchymal cells. Kidney International. 2007;72(1):37–44. doi: 10.1038/sj.ki.5002242. [DOI] [PubMed] [Google Scholar]
- 97.Jones T. W., Chorpa S., Kaufman J. S., Flamenbaum W., Trump B. F. Cis-diamminedichloroplatinum (II)-induced acute renal failure in the rat: Enzyme histochemical studies. Toxicologic Pathology. 1985;13(4):296–305. doi: 10.1177/019262338501300406. [DOI] [PubMed] [Google Scholar]
- 98.Espandiari P., Rosenzweig B., Zhang J., et al. Age-related differences in susceptibility to cisplatin-induced renal toxicity. Journal of Applied Toxicology. 2010;30(2):172–182. doi: 10.1002/jat.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Amin R. P., Vickers A. E., Sistare F., et al. Identification of putative gene-based markers of renal toxicity. Environmental Health Perspectives. 2004;112(4):465–479. doi: 10.1289/ehp.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harpur E., Ennulat D., Hoffman D., et al. Biological qualification of biomarkers of chemical-induced renal toxicity in two strains of male rat. Toxicological Sciences. 2011;122(2):235–252. doi: 10.1093/toxsci/kfr112. [DOI] [PubMed] [Google Scholar]
- 101.Nowrousian M. R., Schmidt C. G. Effects of cisplatin on different haemopoietic progenitor cells in mice. British Journal of Cancer. 1982;46(3):397–402. doi: 10.1038/bjc.1982.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schrier R. W., Wang W., Poole B., Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. The Journal of Clinical Investigation. 2004;114(1):5–14. doi: 10.1172/JCI22353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wainford R. D., Weaver R. J., Stewart K. N., Brown P., Hawksworth G. M. Cisplatin nephrotoxicity is mediated by gamma glutamyltranspeptidase, not via a C-S lyase governed biotransformation pathway. Toxicology. 2008;249(2-3):184–193. doi: 10.1016/j.tox.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 104.Gautier J.-C., Riefke B., Walter J., et al. Evaluation of novel biomarkers of nephrotoxicity in two strains of rat treated with cisplatin. Toxicologic Pathology. 2010;38(6):943–956. doi: 10.1177/0192623310379139. [DOI] [PubMed] [Google Scholar]
- 105.Vinken P., Starckx S., Barale-Thomas E., et al. Tissue Kim-1 and urinary clusterin as early indicators of cisplatin-induced acute kidney injury in rats. Toxicologic Pathology. 2012;40(7):1049–1062. doi: 10.1177/0192623312444765. [DOI] [PubMed] [Google Scholar]
- 106.Bodenner D. L., Dedon P. C., Katz J., Borch R. F. Selective protection against cis-diamminedichloroplatinum(II)–induced toxicity in kidney, gut, and bone marrow by diethyldithiocarbamate. Cancer Research. 1986;46(6):2751–2755. [PubMed] [Google Scholar]
- 107.Kramer R. A. Protection against cisplatin nephrotoxicity by prochlorperazine. Cancer Chemotherapy and Pharmacology. 1989;25(3):156–160. doi: 10.1007/BF00689575. [DOI] [PubMed] [Google Scholar]
- 108.Naganuma A., Satoh M., Imura N. Prevention of lethal and renal toxicity of cis-diamminedichloroplatinum(II) by induction of metallothionein synthesis without compromising its antitumor activity in mice. Cancer Research. 1987;47(4):983–987. [PubMed] [Google Scholar]
- 109.van den Hamer C. J., Los G., de Goeij J. J., McVie J. G. Selenium-induced protection against cis-diamminedichloroplatinum(II) nephrotoxicity in mice and rats. Cancer Research. 1989;49(11):3020–3023. [PubMed] [Google Scholar]
- 110.Mizushima Y., Nagahama H., Yokoyama A., Morikage T., Yano S. Studies on nephrotoxicity following a single and repeated administration of cis-diamminedichloroplatinum (CDDP) in rats. The Tohoku Journal of Experimental Medicine. 1987;151(2):129–135. doi: 10.1620/tjem.151.129. [DOI] [PubMed] [Google Scholar]
- 111.Yamate J., Tatsumi M., Nakatsuji S., Kuwamura M., Kotani T., Sakuma S. Immunohistochemical observations on the kinetics of macrophages and myofibroblasts in rat renal interstitial fibrosis induced by cis-diamminedichloroplatinum. Journal of Comparative Pathology. 1995;112(1):27–39. doi: 10.1016/S0021-9975(05)80087-8. [DOI] [PubMed] [Google Scholar]
- 112.Wagner T., Kreft B., Bohlmann G., Schwieder G. Effects of fosfomycin, mesna, and sodium thiosulfate on the toxicity and antitumor activity of cisplatin. Journal of Cancer Research and Clinical Oncology. 1988;114(5):497–501. doi: 10.1007/BF00391499. [DOI] [PubMed] [Google Scholar]
- 113.Ming C. J., Ohishi K., Yonemura K., Hishida A. Acquired resistance to acute renal failure in cisplatin-induced renal failure of rats. The Japanese Journal of Nephrology. 1997;39(7):728–733. [PubMed] [Google Scholar]
- 114.Jenderny S., Lin H., Garrett T., Tew K. D., Townsend D. M. Protective effects of a glutathione disulfide mimetic (NOV-002) against cisplatin induced kidney toxicity. Biomedicine & Pharmacotherapy. 2010;64(1):73–76. doi: 10.1016/j.biopha.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Townsend D. M., Hanigan M. H. Inhibition of gamma-glutamyl transpeptidase or cysteine S-conjugate beta-lyase activity blocks the nephrotoxicity of cisplatin in mice. The Journal of Pharmacology and Experimental Therapeutics. 2002;300(1):142–148. doi: 10.1124/jpet.300.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ajith T. A., Usha S., Nivitha V. Ascorbic acid and alpha-tocopherol protect anticancer drug cisplatin induced nephrotoxicity in mice: a comparative study. Clinica Chimica Acta; International Journal of Clinical Chemistry. 2007;375(1-2):82–86. doi: 10.1016/j.cca.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 117.Nitha B., Janardhanan K. K. Aqueous-ethanolic extract of morel mushroom mycelium Morchella esculenta, protects cisplatin and gentamicin induced nephrotoxicity in mice. Food and Chemical Toxicology. 2008;46(9):3193–3199. doi: 10.1016/j.fct.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 118.Jariyawat S., Kigpituck P., Suksen K., Chuncharunee A., Chaovanalikit A., Piyachaturawat P. Protection against cisplatin-induced nephrotoxicity in mice by Curcuma comosa Roxb. ethanol extract. Journal of Natural Medicines. 2009;63(4):430–436. doi: 10.1007/s11418-009-0345-5. [DOI] [PubMed] [Google Scholar]
- 119.Sharp C. N., Doll M. A., Dupre T. V., et al. Repeated administration of low-dose cisplatin in mice induces fibrosis. American Journal of Physiology-Renal Physiology. 2016;310(6):F560–F568. doi: 10.1152/ajprenal.00512.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Baldew G. S., McVie J. G., van der Valk M. A., Los G., de Goeij J. J. M., Vermeulen N. P. E. Selective Reduction of ci5-Diamminedichloroplatinum(II) Nephrotoxicity by Ebselen. Cancer Research. 1990;50(21):7031–7036. [PubMed] [Google Scholar]
- 121.Mannel R. S., Stratton J. A., Moran G., Rettenmaier M. A., Liao S. Y., DiSaia P. J. Intraperitoneal cisplatin: Comparison of antitumor activity and toxicity as a function of solvent saline concentration. Gynecologic Oncology. 1989;34(1):50–53. doi: 10.1016/0090-8258(89)90105-4. [DOI] [PubMed] [Google Scholar]
- 122.Pinches M., Betts C., Bickerton S., et al. Evaluation of novel renal biomarkers with a cisplatin model of kidney injury: Gender and dosage differences. Toxicologic Pathology. 2012;40(3):522–533. doi: 10.1177/0192623311432438. [DOI] [PubMed] [Google Scholar]
- 123.Wadey R. M., Pinches M. G., Jones H. B., Riccardi D., Price S. A. Tissue expression and correlation of a panel of urinary biomarkers following cisplatin-induced kidney injury. Toxicologic Pathology. 2014;42(3):591–602. doi: 10.1177/0192623313492044. [DOI] [PubMed] [Google Scholar]
- 124.Lazaro A., Humanes B., Jado J. C., Tejedor A. The authors reply. Kidney International. 2013;83(6):1201–1202. doi: 10.1038/ki.2013.42. [DOI] [PubMed] [Google Scholar]
- 125.Atessahin A., Yilmaz S., Karahan I., Ceribasi A. O., Karaoglu A. Effects of lycopene against cisplatin-induced nephrotoxicity and oxidative stress in rats. Toxicology. 2005;212(2-3):116–123. doi: 10.1016/j.tox.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 126.Özyurt H., Yildirim Z., Kotuk M., et al. Cisplatin-induced acute renal failure is ameliorated by erdosteine in a dose-dependent manner. Journal of Applied Toxicology. 2004;24(4):269–275. doi: 10.1002/jat.983. [DOI] [PubMed] [Google Scholar]
- 127.Mayer R. D., Lee K. E., Cockett A. T. Inhibition of cisplatin-induced nephrotoxicity in rats by buthionine sulfoximine, a glutathione synthesis inhibitor. Cancer Chemotherapy and Pharmacology. 1987;20(3):207–210. doi: 10.1007/BF00570486. [DOI] [PubMed] [Google Scholar]
- 128.Ma S.-F., Nishikawa M., Hyoudou K., et al. Combining cisplatin with cationized catalase decreases nephrotoxicity while improving antitumor activity. Kidney International. 2007;72(12):1474–1482. doi: 10.1038/sj.ki.5002556. [DOI] [PubMed] [Google Scholar]
- 129.Fields S. M., Lindley C. M. Thrombotic microangiopathy associated with chemotherapy: Case report and review of the literature. DICP: The Annals of Pharmacotherapy. 1989;23(7-8):582–588. doi: 10.1177/1060028089023007-810. [DOI] [PubMed] [Google Scholar]
- 130.Astolfi L., Ghiselli S., Guaran V., et al. Correlation of adverse effects of cisplatin administration in patients affected by solid tumours: A retrospective evaluation. Oncology Reports. 2013;29(4):1285–1292. doi: 10.3892/or.2013.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Eaton K., Lyman G. Dosing of anticancer agents in adults. UpToDate; 2017. [Google Scholar]
- 132.Harrison S. D. Toxicologic evaluation of cis-diamminedichloroplatinum II in B6D2F1 mice. Toxicological Sciences. 1981;1(5):382–385. doi: 10.1093/toxsci/1.5.382. [DOI] [PubMed] [Google Scholar]
- 133.Levi F. A., Hrushesky W. J. M., Halberg F., Langevin T. R., Haus E., Kennedy B. J. Lethal nephrotoxicity and hematologic toxicity of cis-diamminedichloroplatinum ameliorated by optimal circadian timing and hydration. European Journal of Cancer and Clinical Oncology. 1982;18(5):471–477. doi: 10.1016/0277-5379(82)90116-X. [DOI] [PubMed] [Google Scholar]
- 134.Al-Majed A. A., Sayed-Ahmed M. M., Al-Yahya A. A., Aleisa A. M., Al-Rejaie S. S., Al-Shabanah O. A. Propionyl-L-carnitine prevents the progression of cisplatin-induced cardiomyopathy in a carnitine-depleted rat model. Pharmacological Research. 2006;53(3):278–286. doi: 10.1016/j.phrs.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 135.Cherry S. M., Hunt P. A., Hassold T. J. Cisplatin disrupts mammalian spermatogenesis, but does not affect recombination or chromosome segregation. Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 2004;564(2):115–128. doi: 10.1016/j.mrgentox.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 136.Simmons E. M., Himmelfarb J., Tugrul Sezer M., et al. Plasma cytokine levels predict mortality in patients with acute renal failure. Kidney International. 2004;65(4):1357–1365. doi: 10.1111/j.1523-1755.2004.00512.x. [DOI] [PubMed] [Google Scholar]
- 137.Liu Y. L., Malik N., Sanger G. J., Friedman M. I., Andrews P. L. Pica—a model of nausea? Species differences in response to cisplatin. Physiology & Behavior. 2005;85(3):271–277. doi: 10.1016/j.physbeh.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 138.Rebillard A., Rioux-Leclercq N., Muller C., et al. Acid sphingomyelinase deficiency protects from cisplatin-induced gastrointestinal damage. Oncogene. 2008;27(51):6590–6595. doi: 10.1038/onc.2008.257. [DOI] [PubMed] [Google Scholar]
- 139.Allan S. G., Smyth J. F., Hay F. G., Leonard R. C., Wolf C. R. Protective Effect of Sodium-2-mercaptoethanesulfonate on the Gastrointestinal Toxicity and Lethality of cis-Diamminedichloroplatinum. Cancer Research. 1986;46(7):3569–3573. [PubMed] [Google Scholar]
- 140.Allan S. G., Smyth J. F. Small intestinal mucosal toxicity of cis – platinum comparison of toxicity with platinum analogues and dexamethasone. British Journal of Cancer. 1986;53(3):355–360. doi: 10.1038/bjc.1986.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bearcroft C. P., Domizio P., Mourad F. H., André E. A., Farthing M. J. G. Cisplatin impairs fluid and electrolyte absorption in rat small intestine: A role for 5-hydroxytryptamine. Gut. 1999;44(2):174–179. doi: 10.1136/gut.44.2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Arivarasu N. A., Priyamvada S., Mahmood R. Oral administration of caffeic acid ameliorates the effect of cisplatin on brush border membrane enzymes and antioxidant system in rat intestine. Experimental and Toxicologic Pathology. 2013;65((1-2)):21–25. doi: 10.1016/j.etp.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 143.Kalkan Y., Ozer E., Yucel A. F., et al. The protective effect of infliximab on cisplatin-induced intestinal tissue toxicity. European Review for Medical and Pharmacological Sciences. 2014;18(14):2076–2083. [PubMed] [Google Scholar]
- 144.Al-Quraishy S., Aref A. M., Othman M. S., El-Deib K. M., Abdel Moneim A. E. The potential role of Azadirachta indica treatment on cisplatin-induced hepatotoxicity and oxidative stress in female rats. Oxidative Medicine and Cellular Longevity. 2013 doi: 10.1155/2013/741817.741817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Al-Malki A. L., Sayed A. A. Thymoquinone attenuates cisplatin-induced hepatotoxicity via nuclear factor kappa-β. BMC Complementary and Alternative Medicine. 2014;14, article 282 doi: 10.1186/1472-6882-14-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ateşşahin A., Karahan I., Türk G., Gür S., Yilmaz S., Çeribaşi A. O. Protective role of lycopene on cisplatin-induced changes in sperm characteristics, testicular damage and oxidative stress in rats. Reproductive Toxicology. 2006;21(1):42–47. doi: 10.1016/j.reprotox.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 147.Ateşşahin A., Sahna E., Türk G. Chemoprotective effect of melatonin against cisplatin-induced testicular toxicity in rats. Journal of Pineal Research. 2006;41(1):21–27. doi: 10.1111/j.1600-079X.2006.00327.x. [DOI] [PubMed] [Google Scholar]
- 148.Hill G. W., Morest D. K., Parham K. Cisplatin-induced ototoxicity: effect of intratympanic dexamethasone injections. Otology & Neurotology. 2008;29(7):1005–1011. doi: 10.1097/MAO.0b013e31818599d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Parham K. Can intratympanic dexamethasone protect against cisplatin ototoxicity in mice with age-related hearing loss? Otolaryngology—Head and Neck Surgery. 2011;145(4):635–640. doi: 10.1177/0194599811409304. [DOI] [PubMed] [Google Scholar]
- 150.Li G., Sha S.-H., Zotova E., Arezzo J., Van De Water T. R., Schacht J. Salicylate protects hearing and kidney function from cisplatin toxicity without compromising its oncolytic action. Laboratory Investigation. 2002;82(5):585–596. doi: 10.1038/labinvest.3780453. [DOI] [PubMed] [Google Scholar]
- 151.Katayama R., Nagata S., Iida H., Yamagishi N., Yamashita T., Furuhama K. Possible role of cysteine-S-conjugate β-lyase in species differences in cisplatin nephrotoxicity. Food and Chemical Toxicology. 2011;49(9):2053–2059. doi: 10.1016/j.fct.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 152.Hrushesky W. J., Levi F. A., Halberg F., Kennedy B. J. Circadian Stage Dependence of Cis-diamminedichloroplatinum Lethal Toxicity in Rats. Cancer Research. 1982;42(3):945–949. [PubMed] [Google Scholar]
- 153.Zoetis T., Hurtt M. E. Species comparison of anatomical and functional renal development. Birth Defects Research. Part B, Developmental and Reproductive Toxicology. 2003;68(2):111–120. doi: 10.1002/bdrb.10013. [DOI] [PubMed] [Google Scholar]
- 154.Shord S. S., Thompson D. M., Krempl G. A., Hanigan M. H. Effect of concurrent medications on cisplatin-induced nephrotoxicity in patients with head and neck cancer. Anti-Cancer Drugs. 2006;17(2):207–215. doi: 10.1097/00001813-200602000-00013. [DOI] [PubMed] [Google Scholar]
- 155.Namikawa K., Kinsoku A., Minami T., et al. Relationship between age and nephrotoxicity following single low-dose cisplatin (CDDP) injection in rats. Biological & Pharmaceutical Bulletin. 1995;18(7):957–962. doi: 10.1248/bpb.18.957. [DOI] [PubMed] [Google Scholar]
- 156.Launay-Vacher V., Isnard-Bagnis C., Janus N., Karie S., Deray G. Chemotherapy and renal toxicity. Bulletin du Cancer. 2008;95(8):F96–F103. doi: 10.1684/bdc.2007.0544. [DOI] [PubMed] [Google Scholar]
- 157.Crona D. J., Faso A., Nishijima T. F., McGraw K. A., Galsky M. D., Milowsky M. I. A systematic review of strategies to prevent cisplatin-induced nephrotoxicity. The Oncologist. 2017;22(5):609–619. doi: 10.1634/theoncologist.2016-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lajer H., Kristensen M., Hansen H. H., et al. Magnesium depletion enhances cisplatin-induced nephrotoxicity. Cancer Chemotherapy and Pharmacology. 2005;56(5):535–542. doi: 10.1007/s00280-005-1010-7. [DOI] [PubMed] [Google Scholar]
- 159.Lajer H., Kristensen M., Hansen H. H., Christensen S., Jonassen T., Daugaard G. Magnesium and potassium homeostasis during cisplatin treatment. Cancer Chemotherapy and Pharmacology. 2005;55(3):231–236. doi: 10.1007/s00280-004-0899-6. [DOI] [PubMed] [Google Scholar]
- 160.Masahiko S., Akira N., Nobumasa I. Deficiency of selenium intake enhances manifestation of renal toxicity of cis-diamminedichloroplatinum in mice. Toxicology Letters. 1987;38(1-2):155–160. doi: 10.1016/0378-4274(87)90123-8. [DOI] [PubMed] [Google Scholar]
- 161.Levi F. A., Hrushesky W. J. M., Halberg F., Kennedy B. J. Reduction of cis-diamminedichloroplatinum nephrotoxicity in rats by optimal circadian drug timing. Cancer Research. 1982;42(3):950–955. [PubMed] [Google Scholar]
- 162.Saint-Paul L., Nguyen C.-H., Buffière A., et al. CD45 phosphatase is crucial for human and murine acute myeloid leukemia maintenance through its localization in lipid rafts. Oncotarget . 2016;7(40):64785–64797. doi: 10.18632/oncotarget.11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jang H. R., Kim D. H., Kwon G. Y., et al. Early, but not late treatment with human umbilical cord blood-derived mesenchymal stem cells attenuates cisplatin nephrotoxicity through immunomodulation. Americal Journal of Physiology. Renal Physiology. 2017 doi: 10.1152/ajprenal.00097.2016. [DOI] [PubMed] [Google Scholar]
- 164.Bennett C. L., Clausen B. E. DC ablation in mice: promises, pitfalls, and challenges. Trends in Immunology. 2007;28(12):525–531. doi: 10.1016/j.it.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 165.Buchheister S., Buettner M., Basic M. CD14 plays a protective role in experimental inflammatory bowel disease by enhancing intestinal barrier function. The American Journal of Pathology. 2017;187(5):1106–1120. doi: 10.1016/j.ajpath.2017.01.012. [DOI] [PubMed] [Google Scholar]
- 166.Harms R. Z., Creer A. J., Lorenzo-Arteaga K. M., Ostlund K. R., Sarvetnick N. E. Interleukin (IL)-18 binding protein deficiency disrupts natural killer cell maturation and diminishes circulating IL-18. Frontiers in Immunology. 2017;8:1020. doi: 10.3389/fimmu.2017.01020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Burks S. R., Nguyen B. A., Tebebi P. A., et al. Pulsed focused ultrasound pretreatment improves mesenchymal stromal cell efficacy in preventing and rescuing established acute kidney injury in mice. Stem Cells. 2015;33(4):1241–1253. doi: 10.1002/stem.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Appenroth D., Bräunlich H. Age differences in cisplatinum nephrotoxicity. Toxicology. 1984;32(4):343–353. doi: 10.1016/0300-483X(84)90086-6. [DOI] [PubMed] [Google Scholar]
- 169.Appenroth D., Winnefeld K., Bräunlich H. Nephrotoxicity and pharmacokinetics of cisplatinum in young and adult rats. Biomedica Biochimica Acta. 1988;47(8):791–797. [PubMed] [Google Scholar]
- 170.Appenroth D., Gambaryan S., Gerhardt S., Kersten L., Bräunlich H. Age dependent differences in the functional and morphological impairment of kidney following cisplatin administration. Experimental Pathology. 1990;38(4):231–239. doi: 10.1016/S0232-1513(11)80232-X. [DOI] [PubMed] [Google Scholar]
- 171.Nematbakhsh M., Ebrahimian S., Tooyserkani M., Eshraghi-Jazi F., Talebi A., Ashrafi F. Gender difference in cisplatin-induced nephrotoxicity in a rat model: greater intensity of damage in male than female. Nephro-Urology Monthly. 2013;5(3):818–821. doi: 10.5812/numonthly.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Nematbakhsh M., Pezeshki Z. Sex-related difference in nitric oxide metabolites levels after nephroprotectant supplementation administration against cisplatin-induced nephrotoxicity in wistar rat model: the role of vitamin E, erythropoietin, or N-acetylcysteine. ISRN Nephrology. 2013;2013:5. doi: 10.5402/2013/612675.612675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Pezeshki Z., Nematbakhsh M., Nasri H., et al. Evidence against protective role of sex hormone estrogen in cisplatin-induced nephrotoxicity in ovarectomized rat model. Toxicology International. 2013;20(1):43–47. doi: 10.4103/0971-6580.111568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rostami B., Nematbakhsh M., Pezeshki Z., et al. Effect of testosterone on Cisplatin-induced nephrotoxicity in surgically castrated rats. Nephro-Urology Monthly. 2014;6(5) doi: 10.5812/numonthly.21546.e21546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.To H., Kikuchi A., Tsuruoka S., et al. Time-dependent nephrotoxicity associated with daily administration of cisplatin in mice. Journal of Pharmacy and Pharmacology. 2000;52(12):1499–1504. doi: 10.1211/0022357001777711. [DOI] [PubMed] [Google Scholar]
- 176.Richards J., Gumz M. L. Advances in understanding the peripheral circadian clocks. The FASEB Journal. 2012;26(9):3602–3613. doi: 10.1096/fj.12-203554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gumz M. L. Tick tock: Time to recognize the kidney clock. Journal of the American Society of Nephrology. 2014;25(7):1369–1371. doi: 10.1681/ASN.2014020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Levey A. S., Inker L. A. Assessment of glomerular filtration rate in health and disease: a state of the art review. Clinical Pharmacology and Therapeutics. 2017;102(3):405–419. doi: 10.1002/cpt.729. [DOI] [PubMed] [Google Scholar]
- 179.Ferguson M. A., Vaidya V. S., Bonventre J. V. Biomarkers of nephrotoxic acute kidney injury. Toxicology. 2008;245(3):182–193. doi: 10.1016/j.tox.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mårtensson J., Martling C.-R., Bell M. Novel biomarkers of acute kidney injury and failure: clinical applicability. British Journal of Anaesthesia. 2012;109(6):843–850. doi: 10.1093/bja/aes357. [DOI] [PubMed] [Google Scholar]
- 181.Reeves W. B., Kwon O., Ramesh G. Netrin-1 and kidney injury. II. Netrin-1 is an early biomarker of acute kidney injury. American Journal of Physiology-Renal Physiology. 2008;294(4):F731–F738. doi: 10.1152/ajprenal.00507.2007. [DOI] [PubMed] [Google Scholar]
- 182.Brott D. A., Adler S. H., Arani R., Lovick S. C., Pinches M., Furlong S. T. Characterization of renal biomarkers for use in clinical trials: Biomarker evaluation in healthy volunteers. Drug Design, Development and Therapy. 2014;8:227–237. doi: 10.2147/DDDT.S54956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Pinches M. D., Betts C. J., Bickerton S. J., et al. Evaluation of Novel Urinary Renal Biomarkers with a Cisplatin Model of Kidney Injury: Effects of Collection Period. Toxicologic Pathology. 2012;40(3):534–540. doi: 10.1177/0192623311432437. [DOI] [PubMed] [Google Scholar]
- 184.Wei Q., Mi Q.-S., Dong Z. The regulation and function of micrornas in kidney diseases. IUBMB Life. 2013;65(7):602–614. doi: 10.1002/iub.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kanki M., Moriguchi A., Sasaki D., et al. Identification of urinary miRNA biomarkers for detecting cisplatin-induced proximal tubular injury in rats. Toxicology. 2014;324(1):158–168. doi: 10.1016/j.tox.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 186.Ostermann M., Joannidis M. Acute kidney injury 2016: Diagnosis and diagnostic workup. Critical Care. 2016;20(1):299. doi: 10.1186/s13054-016-1478-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tanaka H., Ishikawa E., Teshima S., Shimizu E. Histopathological study of human cisplatin nephropathy. Toxicologic Pathology. 1986;14(2):247–257. doi: 10.1177/019262338601400215. [DOI] [PubMed] [Google Scholar]
- 188.Di Maio M., Gridelli C., Gallo C., et al. Chemotherapy-induced neutropenia and treatment efficacy in advanced non-small-cell lung cancer: A pooled analysis of three randomised trials. The Lancet Oncology. 2005;6(9):669–677. doi: 10.1016/S1470-2045(05)70255-2. [DOI] [PubMed] [Google Scholar]
- 189.Rankin E. M., Mill L., Kaye S. B., et al. A randomised study comparing standard dose carboplatin with chlorambucil and carboplatin in advanced ovarian cancer. British Journal of Cancer. 1992;65(2):275–281. doi: 10.1038/bjc.1992.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Fentener van Vlissingen J. M., Borrens M., Girod A., Lelovas P., Morrison F., Torres Y. S. The reporting of clinical signs in laboratory animals: FELASA working group report. Laboratory Animals. 2015;49(4):267–283. doi: 10.1177/0023677215584249. [DOI] [PubMed] [Google Scholar]
- 191.Bataille A., Galichon P., Wetzstein M., et al. Evaluation of the ability of bone marrow derived cells to engraft the kidney and promote renal tubular regeneration in mice following exposure to cisplatin. Renal Failure. 2016;38(4):521–529. doi: 10.3109/0886022X.2016.1145521. [DOI] [PubMed] [Google Scholar]
- 192.Aggarwal S. K., Broomhead J. A., Fairlie D. P., Whitehouse M. W. Platinum drugs: combined anti-lymphoproliferative and nephrotoxicity assay in rats. Cancer Chemotherapy and Pharmacology. 1980;4(4):249–258. doi: 10.1007/BF00255269. [DOI] [PubMed] [Google Scholar]
- 193.Langford D. J., Bailey A. L., Chanda M. L., et al. Coding of facial expressions of pain in the laboratory mouse. Nature Methods. 2010;7(6):447–449. doi: 10.1038/nmeth.1455. [DOI] [PubMed] [Google Scholar]
- 194.Al Suleimani Y. M., Abdelrahman A. M., AlMahruqi A. S., et al. Interaction of nimesulide, a cyclooxygenase-2 inhibitor, with cisplatin in normotensive and spontaneously hypertensive rats. Food and Chemical Toxicology. 2010;48(1):139–144. doi: 10.1016/j.fct.2009.09.030. [DOI] [PubMed] [Google Scholar]
- 195.Kilkenny C., Browne W. J., Cuthill I. C., Emerson M., Altman D. G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biology. 2010;8(6) doi: 10.1371/journal.pbio.1000412.e1000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hooijmans C. R., Leenaars M., Ritskes-Hoitinga M. A gold standard publication checklist to improve the quality of animal studies, to fully integrate the three Rs, and to make systematic reviews more feasible. Alternatives to Laboratory Animals. 2010;38(2):167–182. doi: 10.1177/026119291003800208. [DOI] [PubMed] [Google Scholar]
- 197.Mähler M., Berar M., Feinstein R., et al. FELASA recommendations for the health monitoring of mouse, rat, hamster, guinea pig and rabbit colonies in breeding and experimental units. Laboratory Animals. 2014;48(3):178–192. doi: 10.1177/0023677213516312. [DOI] [PubMed] [Google Scholar]
- 198.Rülicke T., Montagutelli X., Pintado B., Thon R., Hedrich H. J. FELASA guidelines for the production and nomenclature of transgenic rodents. Laboratory Animals. 2007;41(3):301–311. doi: 10.1258/002367707781282758. [DOI] [PubMed] [Google Scholar]