

Abstract
Background
DNA methylation, an epigenetic modification, can be affected by environmental factors and thus regulate gene expression levels that can lead to alterations of certain phenotypes. Network analysis has been used successfully to discover gene sets that are expressed differently across multiple disease states and suggest possible pathways of disease progression. We applied this framework to compare DNA methylation levels before and after lipid-lowering medication and to identify modules that differ topologically between the two time points, revealing the association between lipid medication and these triglyceride-related methylation sites.
Methods
We performed quality control using beta-mixture quantile normalization on 463,995 cytosine-phosphate-guanine (CpG) sites and deleted problematic sites, resulting in 423,004 probes. We identified 14,850 probes that were nominally associated with triglycerides prior to treatment and performed weighted gene correlation network analysis (WGCNA) to construct pre- and posttreatment methylation networks of these probes. We then applied both WGCNA module preservation and generalized Hamming distance (GHD) to identify modules with topological differences between the pre- and posttreatment. For modules with structural changes between 2 time points, we performed pathway-enrichment analysis to gain further insight into the biological function of the genes from these modules.
Results
Six triglyceride-associated modules were identified using pretreatment methylation probes. The same 3 modules were not preserved in posttreatment data using both the module-preservation and the GHD methods. Top-enriched pathways for the 3 differentially methylated modules are sphingolipid signaling pathway, proteoglycans in cancer, and metabolic pathways (p values < 0.005). One module in particular included an enrichment of lipid-related pathways among the top results.
Conclusions
The same 3 modules, which were differentially methylated between pre- and posttreatment, were identified using both WGCNA module-preservation and GHD methods. Pathway analysis revealed that triglyceride-associated modules contain groups of genes that are involved in lipid signaling and metabolism. These 3 modules may provide insight into the effect of fenofibrate on changes in triglyceride levels and these methylation sites.
Background
Epigenetic changes are biochemical modifications in chromosomes that do not alter the DNA sequence [1]. DNA methylation is one such epigenetic process implicated in human disease. DNA methylation involves the addition of a methyl group to DNA, which usually occurs at cytosine-phosphate-guanine (CpG) dinucleotides in the promoter region or within genes [1]. It is known to regulate gene expression levels by changing the chromatin structure, thereby preventing transcription factors from binding to the gene promoter, which can lead to alterations of phenotypes [2]. DNA methylation can be modulated by external factors, such as smoking or exposure to toxins [3]. As such, epigenetic information is considered to be fundamental in understanding the interaction between the human genome and the environment.
Recent research has unveiled the potential involvement of DNA methylation on the regulation of fasting blood lipids [4–6]. One way to visualize interactions and changes in the DNA methylation profile is to construct methylation networks and compare their topology. Structural changes resulting from external stimuli can be detected with network comparison algorithms that can identify subnetworks that are either preserved or structurally different. Several papers have performed network-based methods to identify trait-related modules [7–9] and examined the preservation of such modules, either between different tissues or different data sets. For example, Horvath et al. [7] conducted weighted gene correlation network analysis (WGCNA) to examine the effect of aging on DNA methylation modules in humans and reported a robustly defined age-related comethylation module that is present in multiple human issues including blood and brain. Rickabaugh et al. [8] also found a preserved methylation module that was associated with age and HIV-1 status in 2 different HIV data sets via WGCNA. However, to our best knowledge, there are no previous studies using network-based approaches to identify triglyceride-associated modules and assess topological differences for each module between 2 time points. We investigated the topological differences of triglyceride-associated methylation networks constructed before and after the administration of fenofibrate.
Methods
Quality control
Data from GAW20 was used and included epigenetic and pharmacogenomics data for 188 unique families [10]. A total of 995 individuals with pretreatment DNA methylation profiles at visit 2 and 530 individuals with posttreatment DNA methylation profiles at visit 4 (with 446 individuals overlapping during the 3-week treatment period) were used to construct, separately, a pre- and posttreatment network. Because of systemic differences in the range of Infinium I and Infinium II probe methylation values, we performed beta-mixture quantile normalization (BMIQ) using the bmiq function from wateRmelon package in R on a total of 463,995 methylation probes to adjust the beta values of Type II probes to align with the distribution of Type I probes [11, 12]. Probes with single nucleotide polymorphisms (SNPs) under the actual CpG sites, SNPs at the nucleotide right next to a CpG site, or cross-reactive probes that have a target sequence similar to another location in the genome are potentially problematic, and were excluded from analysis [13]. A total of 423,004 pretreatment CpG sites were used in the analysis.
Network
We were interested in DNA methylation changes caused by fenofibrate. To build the networks, we limited the methylation probes to those that are nominally associated (p < 0.05) with the log-transformed triglyceride level from visit 2, resulting in 14,850 probes from pretreatment data. If we limited our study to those probes that are significantly associated (false discovery rate [FDR] < 0.05), we might lose many informative probes that only show nominal significance without considering their potential interactions with other probes used in the network constructions.
To isolate the treatment effect from other covariates, we built the networks from pre- and posttreatment residual methylation values. We performed linear mixed models using the lmekin function in the coxme package in R for each of the 14,850 pretreatment CpG sites, adjusting for family relatedness, the first 10 principal components accounting for residual batch effects, and the covariates age, sex, field center, and smoking status [14]. Similarly, we obtained residuals for posttreatment methylation for the same set of CpG sites. Samples with more than half missing residuals and CpG sites with variance smaller than 1 × 10− 10*max (abs [residuals of all probes among all samples]) were removed. We applied hierarchical clustering to samples to detect sample outliers at a branch cut height of 7 using the hclust function from the stats package in R. Overall, 47 probes and 3 samples were excluded in both pre- and posttreatment methylation data.
We used the residuals to construct pretreatment and posttreatment networks using WGCNA, which uses pairwise correlations between the variables to create a weighted network [15]. We chose soft thresholding power β according to the approximate scale-free topology criterion and computed the adjacency matrix by raising co-expression similarities by power β [16]. We chose a power of β = 3 as it was the lowest power at which the scale-free topology had a good fit of R2 greater than 0.9.
To minimize the effects of noise and spurious associations, the adjacency matrix was transformed into the topological overlap matrix, which reflects the relative interconnectedness between a pair of methylation probes, and the corresponding dissimilarity matrix was calculated by subtracting the adjacency matrix from 1. We performed hierarchical clustering of the dissimilarity matrix and a dynamic tree-cut algorithm to identify modules in the pretreatment network, with a minimum module size of 30 to avoid small modules and pass more informative results to the subsequent pathway analysis [17, 18]. Transformation of the adjacency matrix and network construction were performed using the blockwiseModules function from WGCNA package in R [15].
We tested whether pretreatment methylation modules changed after treatment more than we would expect by chance using the module-preservation method and the generalized Hamming distance (GHD) method. The module-preservation statistic is comprised of many factors that describe the density of the network and network connectivity. These statistics are aggregated and were used to calculate the z-score, Zsummary, for each preservation measure [19]. A Zsummary > 10 indicated strong evidence of module preservation, whereas a Zsummary between 2 and 10 indicated weak evidence of preservation. If Zsummary was < 2, we concluded there was no evidence of module preservation. Module changes were also ranked according to a rank statistic medianRank, which is also composed of statistics related to a network’s density and connectivity, but is less sensitive to module size according to Langfelder et al. [19]. We used both statistics to determine whether or not a certain module was preserved between pre- and posttreatment. Module preservation analyses were conducted using the modulePreservation function from the WGCNA package in R [15]. GHD is a distance metric that uses one-step topological overlap as the edge weights to account for its direct neighboring structure around the pair of nodes. Formal hypothesis testing can be done by finding the null distribution using permutation, but it is computationally inefficient. In a scale-free network, GHD permutation distribution is approximately normally distributed, ie, [20]. We further standardized the GHD value so it follows a standard normal distribution and evaluated its significance against the normal quantiles.
Pathway analysis
After we compared the pre−/posttreatment module pairs, we performed pathway-enrichment analysis for module pairs that had topological change between the 2 time points to determine whether the genes from the modules are associated with a certain biological function. Pathway analysis was done using the gometh function in missMethyl library in R, which conducts hypergeometric tests to determine if the gene sets from the modules contain more genes than expected in a particular pathway [21]. We used IlluminaHumanMethylation450kanno.ilmn12.hg19 to get annotations and tested 319 pathways in the Kyoto Encyclopedia of Genes and Genomes [22].
Results
Module assignment
Six subnetworks, whose module sizes are 210, 177, 123, 112, 91, and 44, were identified in the pretreatment network, while 14,049 probes remained outside of these modules. Each module was assigned a color: pink, blue, brown, yellow, green, and red that are in descending order of module size.
Module preservation analysis
In Fig. 1, methylation probes assigned to blue, pink, and yellow modules clearly grouped together in the posttreatment network, indicating fairly good preservation for these 3 modules. As seen in Fig. 2, pink, blue, and yellow modules also had a Zsummary > 10 and were ranked at first, second, and third, respectively, when applying medianRank statistic, revealing their good preservation in the posttreatment network. Green and brown modules were tied for fifth place, and their Zsummary statistic values were < 10, so it is likely these 2 modules were not preserved between pre- and posttreatment. The red module had a Zsummary of 1.234, which was < 2, and was ranked fourth in terms of medianRank statistic.
Fig. 1.

Clustering dendrogram of pre- and posttreatment methylation probe network, together with assigned module colors (pink, blue, yellow, red, brown, and green)
Fig. 2.

The medianRank and Zsummary statistics of module preservation of pretreatment modules in posttreatment modules (y-axis) versus module size (x-axis)
GHD
Unlike WGCNA, the GHD method requires a (0,1)-adjacency matrix. We obtained such a matrix by thresholding the absolute value of the correlation matrix obtained from WGCNA. We used a threshold of 0.2, which results in a nearly scale-free network. It is thought that biological networks are scale-free, which are characterized by a small number of highly connected nodes known as hubs that hold the network together [16]. Under the null hypothesis, pretreatment and posttreatment modules are independent, which means that there is topological difference between the two modules. For each pre–post module pair, we calculated the GHD statistic, the mean, and the variance, and computed the corresponding Z score and the p value.
We used Bonferroni-corrected p values to account for multiple testing, ie, a significance threshold of 0.05/6 = 0.00833. Table 1 shows that the pink, blue, and yellow module pairs were not independent, which implies that the pre- and posttreatment modules did not have topological differences. Conversely, the green, brown, and red modules show a differential topological structure between pre- and posttreatment networks. Of note, the red module has a borderline p value of 0.009, which is very close to the Bonferroni corrected alpha of 0.00833, suggesting that it is nearly significant in having no topological difference between pre- and posttreatment.
Table 1.
Results of independence test of each module pair between pre- and posttreatment networks
| Modules | GHD | Z score | p Value |
|---|---|---|---|
| Pink | 0.025 | − 11.774 | 5.307 × 10−32 |
| Blue | 0.092 | −9.384 | 6.350 × 10−21 |
| Yellow | 0.059 | −8.106 | 5.225 × 10−16 |
| Red | 0.016 | 2.609 | 0.009 |
| Brown | 0.041 | 0.953 | 0.340 |
| Green | 0.006 | 0.462 | 0.643 |
Null: pre- and posttreatment modules are dependent (there are structural changes); alternative: pre- and posttreatment modules are independent (no structural changes)
We found that rankings of 6 modules from GHD and medianRank statistic were similar (ranked first to sixth for module-preservation level: pink, blue, yellow, red, brown, and green) by comparing the results from module-preservation and GHD methods. Although its Zsummary showed the weakest preservation, the red module had a borderline p value of showing no topological difference and was ranked fourth; further investigation may be necessary to evaluate module preservation of red module.
Visualization
As mentioned previously, the red module required further investigation to determine if there is any topological difference between pre- and posttreatment. We examined the structural changes for the red module by making network plots. We used the same threshold of 0.2 as in the first step of the GHD method, and converted each cell of the adjacency matrix from absolute value of the correlation coefficient into a value of 0 or 1. If the dichotomous correlation coefficient between 2 probes was 1, we drew an edge between them.
In the 2 network plots in Fig. 3, there was an apparent difference of network structure between pre- and posttreatment of the red module. The strong relationships between most of the methylation probes in the red module disappeared in the posttreatment network. The network plots of the red module were similar to those of the green and brown modules, indicating that the red module was also differentially methylated between pre- and posttreatment. Further investigation into the probes is needed to determine if the changes were the result of a drug effect. One such potential investigation would be to perform a pathway-enrichment analysis with these modules to determine if methylation probes can be linked to genes related to lipid signaling.
Fig. 3.
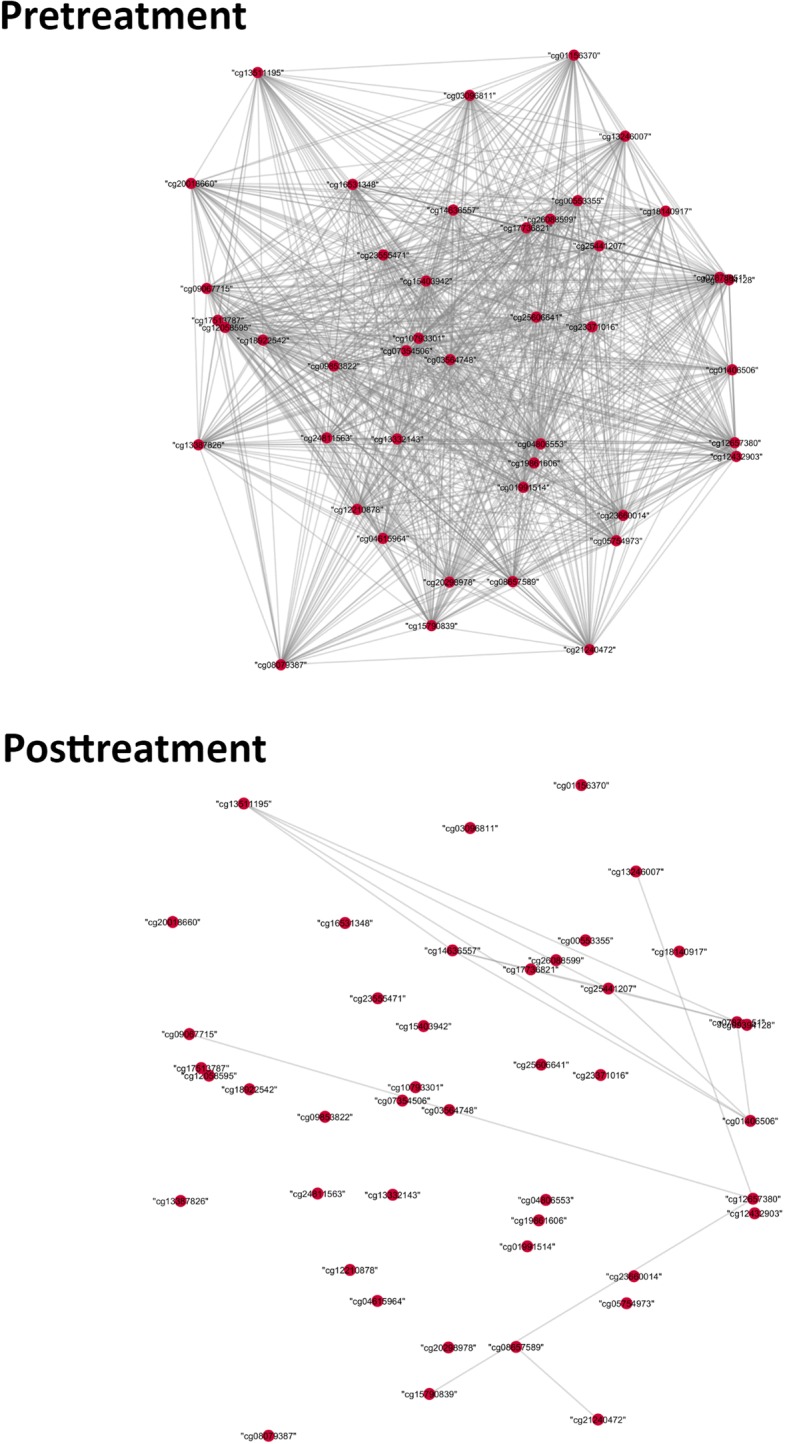
Pretreatment and posttreatment network plots of red module
Pathway analysis
For the 3 differentially methylated modules (green, brown, red), we performed pathway-enrichment analysis to test for overrepresentation. Table 2 shows the results of the pathway analysis for the red, green, and brown modules. Compared to the brown and green modules, the red module is made up of more genes that are involved in lipid signaling and metabolism, which points to overrepresentation in the gene list in the red module. For example, LPCA1 and AGPAT3 are known to be involved in encoding enzymes that play a role in phospholipid metabolism [23]. This implies that fenofibrate had a bigger effect on triglyceride levels through methylation probes in the red module, leading to a striking structural change between the 2 time points as is shown in Fig. 3.
Table 2.
Result of pathway analysis for the red, green, and brown modules
| Pathway | # DE | p Value |
|---|---|---|
| Red module | ||
| Sphingolipid signaling pathway | 5 | 1.81 × 10−32 |
| Glycerophospholipid metabolism | 4 | 0.00016 |
| Phospholipase D signaling pathway | 4 | 0.0008 |
| Cyclic adenosine monophosphate signaling pathway | 4 | 0.00149 |
| Choline metabolism in cancer | 3 | 0.00561 |
| Ether lipid metabolism | 2 | 0.01523 |
| Glycerolipid metabolism | 2 | 0.03437 |
| Green module | ||
| Metabolic pathway | 6 | 0.00471 |
| Epstein-Barr virus infection | 3 | 0.1168 |
| Notch signaling pathway | 2 | 0.1168 |
| Pyrimidine metabolism | 2 | 0.1492 |
| Basal cell carcinoma | 2 | 0.1492 |
| Brown module | ||
| Proteoglycans in cancer | 6 | 0.00208 |
| Salmonella infection | 4 | 0.00208 |
| Terpenoid backbone biosynthesis | 3 | 0.00208 |
| Pathways in cancer | 7 | 0.00239 |
| Gonadotropin-releasing hormone signaling pathway | 4 | 0.00239 |
Pathway indicates the KEGG pathway being tested; # DE is the number of genes that are differentially expressed in our module; and the false discovery rate is the p value for overrepresentation
Discussion
By performing network analyses on triglyceride-associated DNA methylation, we found 2 modules (green and brown) that are differentially methylated between pre- and posttreatment based on the results of both 2 statistics (Zsummary and medianRank) in the WGCNA module-preservation method and the GHD method. As the red module has a borderline p value of showing no topological difference and was ranked fourth, we created network plots for pre- and posttreatment, separately, and observed that it appeared to be differentially methylated. Therefore, 3 modules (red, green, and brown) were found to have topological difference between pre- and posttreatment.
Comparison between module-preservation and GHD methods should also be considered. Rankings of 6 modules from GHD and medianRank statistic were similar (ranked first to sixth for module preservation level: pink, blue, yellow, red, brown, and green). However, the Zsummary statistic showed slightly different results, especially for the red module, meaning the red module has the weakest preservation in terms of Zsummary, but it was at the middle level when applying the GHD and medianRank statistic. This difference between results of Zsummary and medianRank statistic may be a result of Zsummary’s sensitivity to module size. Because the red module has a relatively small module size of 44, its lack of preservation based on low Zsummary value may not be as credible as other large modules [19]. Thus, further investigation using network visualization was necessary to evaluate the preservation level of red module.
Some limitations of WGCNA and 2 network comparison methods surfaced during the analysis. Edge direction was not considered in any of the statistics, possibly resulting in loss of information regarding relationships between methylation probes. It would be more telling if we could use a two-step or even more general topological overlap rather than one-step topological overlap to include more information about the neighboring structure around a pair of methylation probes to define the GHD statistic, although it may not be straightforward to prove normality of the sampling distribution under the null.
Conclusions
We explored the relationship between triglyceride-associated DNA methylation and fenofibrate using WGCNA to construct pre- and posttreatment co-methylation networks and detect topological differences between the 2 networks. Using both module-preservation and GHD methods, we found 3 modules that were differentially methylated in posttreatment. Enrichment analysis of these 3 modules revealed that some of these modules are made up of genes that are involved in phospholipid metabolism, which may provide insight into the effect of fenofibrate on changes in methylation and triglyceride levels. Although we cannot conclude that fenofibrate induced the epigenetic alterations, our network-based approach seems promising for detecting treatment-induced changes in comethylation.
Acknowledgments
Funding
Publication of this article was supported by NIH R01 GM031575. Research reported in this publication was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant R01 DK089256.
Availability of data and materials
The data that support the findings of this study are available from the Genetic Analysis Workshop (GAW) but restrictions apply to the availability of these data, which were used under license for the current study. Qualified researchers may request these data directly from GAW.
About this supplement
This article has been published as part of BMC Proceedings Volume 12 Supplement 9, 2018: Genetic Analysis Workshop 20: envisioning the future of statistical genetics by exploring methods for epigenetic and pharmacogenomic data. The full contents of the supplement are available online at https://bmcproc.biomedcentral.com/articles/supplements/volume-12-supplement-9.
Authors’ contributions
All authors contributed to the design of the overall study. HX, EL, and PW conducted statistical analyses and drafted the manuscript. All other authors provided advice and critically revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Elise Lim, Email: elise625@bu.edu.
Hanfei Xu, Email: hfxu@bu.edu.
Peitao Wu, Email: peitaowu@bu.edu.
Daniel Posner, Email: dcposner@bu.edu.
Jiayi Wu, Email: jiayiwu@bu.edu.
Gina M. Peloso, Email: gpeloso@bu.edu
Achilleas N. Pitsillides, Email: anp4r@bu.edu
Anita L. DeStefano, Email: adestef@bu.edu
L. Adrienne Cupples, Email: adrienne@bu.edu
Ching-Ti Liu, Email: ctliu@bu.edu.
References
- 1.Zaina S, Lund G. Epigenetics: a tool to understand diet-related cardiovascular risk? J Nutrigenet Nutrigenomics. 2012;4(5):261–274. doi: 10.1159/000334584. [DOI] [PubMed] [Google Scholar]
- 2.Lim DHK, Maher ER. DNA methylation: a form of epigenetic control of gene expression. The Obstetrician & Gynaecologist. 2010;12(1):37–42. doi: 10.1576/toag.12.1.037.27556. [DOI] [Google Scholar]
- 3.Pacchierotti F, Spanò M. Environmental impact on DNA methylation in the germline: state of the art and gaps of knowledge. Biomed Res Int. 2015;2015:123484. doi: 10.1155/2015/123484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irvin MR, Zhi D, Joehanes R, Mendelson M, Aslibekyan S, Claas SA, Thibeault KS, Patel N, Day K, Jones LW, et al. Epigenome-wide association study of fasting blood lipids in the genetics of lipid-lowering drugs and diet network study. Circulation. 2014;130(7):565–572. doi: 10.1161/CIRCULATIONAHA.114.009158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfeiffer L, Wahl S, Pilling LC, Reischl E, Sandling JK, Kunze S, Holdt LM, Kretschmer A, Schramm K, Adamski J, et al. DNA methylation of lipid-related genes affects blood lipid levels. Circ Cardiovasc Genet. 2015;8(2):334–342. doi: 10.1161/CIRCGENETICS.114.000804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kriebel J, Herder C, Rathmann W, Wahl S, Kunze S, Molnos S, Volkova N, Schramm K, Carstensen-Kirberg M, Waldenberger M, et al. Association between DNA methylation in whole blood and measures of glucose metabolism: KORA F4 study. PLoS One. 2016;11(3):e0152314. doi: 10.1371/journal.pone.0152314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, van den Berg LH, Ophoff RA. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13(10):R97. doi: 10.1186/gb-2012-13-10-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rickabaugh TM, Baxter RM, Sehl M, Sinsheimer JS, Hultin PM, Hultin LE, Quach A, Martínez-Maza O, Horvath S, Vilain E, et al. Acceleration of age-associated methylation patterns in HIV-1-infected adults. PLoS One. 2015;10(3):e0119201. doi: 10.1371/journal.pone.0119201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West J, Beck S, Wang X, Teschendorff AE. An integrative network algorithm identifies age-associated differential methylation interactome hotspots targeting stem-cell differentiation pathways. Sci Rep. 2013;3(1):1630. doi: 10.1038/srep01630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aslibekyan S, Irvin MR, Hidalgo BA, Perry RT, Jeyarajah EJ, Garcia E, Shalaurova I, Hopkins PN, Province MA, Tiwari HK, et al. Genome- and CD4+ T-cell methylome-wide association study of circulating trimethylamine-N-oxide in the genetics of lipid lowering drugs and diet network (GOLDN) J Nutr Intermed Metab. 2017;8:1–7. doi: 10.1016/j.jnim.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pidsley R, CC WY, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Terry M. Therneau (2015). Coxme: mixed effects cox models. R package version 2.2–5. https://CRAN.R-project.org/package=coxme
- 15.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9(1):559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barabási A, Bonabeau E. Scale-free networks. Sci Am. 2003;3(1):50–59. doi: 10.1038/scientificamerican0503-60. [DOI] [PubMed] [Google Scholar]
- 17.Xu X, Zhou Y, Miao R, Chen W, Qu K, Pang Q, Liu C. Transcriptional modules related to hepatocellular carcinoma survival: coexpression network analysis. Front Med. 2016;10(2):183–190. doi: 10.1007/s11684-016-0440-4. [DOI] [PubMed] [Google Scholar]
- 18.Liu R, Guo C-X, Zhou H-H. Network-based approach to identify prognostic biomarkers for estrogen receptor–positive breast cancer treatment with tamoxifen. Cancer Biol Ther. 2015;16(2):317–324. doi: 10.1080/15384047.2014.1002360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7(1):e1001057. doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruan D, Young A, Montana G. Differential analysis of biological networks. BMC Bioinformatics. 2015;16(1):327. doi: 10.1186/s12859-015-0735-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phipson B, Maksimovic J and Oshlack A. missMethyl: an R package for analysing methylation data from Illuminas HumanMethylation450 platform. Bioinformatics. 2015;32(2):btv560. [DOI] [PubMed]
- 22.Hansen KD. IlluminaHumanMethylation450kanno.ilmn12.hg19: Annotation for Illumina's 450k methylation arrays. R package version 0.6.0; 2016.
- 23.Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H, et al. GeneCards Version 3: the human gene integrator. Database (Oxford) 2010;2010:baq020. doi: 10.1093/database/baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the Genetic Analysis Workshop (GAW) but restrictions apply to the availability of these data, which were used under license for the current study. Qualified researchers may request these data directly from GAW.