

Abstract
The imaging of real-time fluxes of K+ ions in live cell with high dynamic range (5-150 mM) is of paramount importance for neuroscience and physiology of the gastrointestinal tract, kidney and other tissues. In particular, the research on high-performance deep-red fluorescent nanoparticle-based biosensors is highly anticipated. We found that BODIPY-based FI3 K+-sensitive fluoroionophore encapsulated in cationic polymer RL100 nanoparticles displays unusually strong efficiency in staining of broad spectrum of cell models, such as primary neurons and intestinal organoids. Using comparison of brightness, photostability and fluorescence lifetime imaging microscopy (FLIM) we confirmed that FI3 nanoparticles display distinctively superior intracellular staining compared to the free dye. We evaluated FI3 nanoparticles in real-time live cell imaging and found that it is highly useful for monitoring intra- and extracellular K+ dynamics in cultured neurons. Proof-of-concept in vivo brain imaging confirmed applicability of the biosensor for visualization of epileptic seizures. Collectively, this data makes fluoroionophore FI3 a versatile cross-platform fluorescent biosensor, broadly compatible with diverse experimental models and that crown ether-based polymer nanoparticles can provide a new venue for design of efficient fluorescent probes.
Keywords: Bionanotechnology, Core/Shell Nanoparticles, Live cell imaging, Medical Applications, Sensors/Biosensors
Graphical abstract
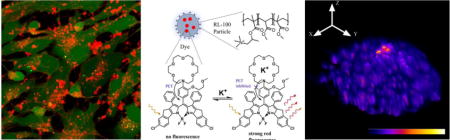
Amphiphilic nanoparticles produced from the positively charged polymer and ion-selective fluoroionophore display remarkable cell staining ability over the broad range of live cell and tissue models: adherent cells, spheroids, organoids and brain tissue. Using BODIPY-based K+-sensitive fluorescent red emitting nanoparticles authors demonstrated their utility in neural cell and functional brain imaging.
1. Introduction
Potassium (K+) is one of the principal ions in living cells and multicellular organisms.[1] Its concentration is dynamically regulated through numerous membrane protein-based channels and transporters, to maintain a gradient of ~ 150 mM inside vs. ~ 5 mM outside the cell. The essential processes of cell growth, acid-base equilibrium, maintenance of electrolyte balance and transmembrane transport are all regulated by potassium. In tissue-specific manner, potassium regulation is crucial for brain (resting potential, firing, interactions between astrocytes and neurons),[2] gut, [3] muscle, [4] kidney[5] and implicated in progression of numerous diseases such as cancer[6] and ischemia.[7] Understanding molecular mechanisms of these processes and monitoring of potassium ion fluxes in different tissues and cellular compartments are important tasks for biochemistry, physiology, pathology and bioanalytical sciences. In particular, development of sensitive, non-invasive and efficient fluorescent chemical sensors for K+ is in high demand. [8] Various optical and fluorescence microscopies allow for high-resolution, fast and quantitative readout, with minimal interferences from electrochemical gradients of other ions present in the cell. Recent developments in high-resolution and super-resolution selective plane illumination, FLIM, confocal and two-photon microscopies demand for bright, highly selective and photostable probes.[9]
Over the last decade the ‘traditional’ method of synthesizing ion-selective dyes is slowly being replaced by their modification with functional and targeting groups and encapsulation in nanoparticles.[10] Nanoparticle technology helps improving the functional properties of fluorophores (brightness, photostability, minimal cross-interferences to other ions and environment) and brings additional features of targeting and biological delivery. For instance, encapsulation in nanoparticles can help solving problems of selectivity of the chemical probe, introduce second (reference) dye and improve its ability to stain cells and tissues.[10b, 11] Various methods such as precipitation of biocompatible polymers allow preparing nanoparticles with virtually any possible specificity and appropriate photophysical properties.[12] However, the control of interaction of nanoparticles with cells and tissues still represents a challenging task:[13] nanoparticles display complex interactions at the nano-bio interface (e.g. salt-induced agglomeration or formation of ‘protein corona’), often resulting in endocytosis-dependent transport of nanoparticles across the cell membrane, and their subsequent entrapment in endosomes, lysosomes and other intracellular organelles.[13a, 14] Compared to small molecule dyes, cell staining with nanoparticles is usually 10-20 times slower, rarely leads to cytoplasmic localization and is often cell-specific. Consequently, staining of more complex cell models such as 3D spheroids or tissues becomes more problematic, limiting the progress in use of fluorescent nanosensors. Significant research is now directed towards modifying the size, charge, shape and surface with functional groups, and understanding the structure-activity relationships.[13a, 15]
The positively charged polymeric nanoparticles based on Eudragit RL100[16] encapsulated with hydrophobic Pt(II)-porphyrins and other fluorescent dyes frequently display cell-specific intracellular staining.[17] Such nanoparticles stain well a number of adherent cell lines (fibroblasts, HeLa, HCT116, HepG2) but display poor staining efficacy of neural cells and multicellular spheroids.[18] While changing to negatively and mixed-charged polymers can address these issues,[11, 19] it is interesting and equally important to understand how the cell staining properties of nanoparticles based on this cationic polymer can be improved.
Here, we report the use of RL100-based nanoparticles, impregnated with a red emitting K+-sensitive fluoroionophore FI3.[8g] As the chemical modification with targeting groups can represent multi-step and laborious procedure,[15b, 20] we examined how the presence of this amphiphilic sensor dye, conjugated with crown ether affects the cell staining properties of RL100-based nanoparticles. Surprisingly, we found that FI3 nanoparticles have improved ability to internalize in a number of ‘hard’ cell types and models, including primary neurons and 3D models in a manner, superior to the free (non-encapsulated) dye and to previously described RL100-based nanosensors. We studied the mechanisms of internalization and demonstrated broad biological application of FI3 in monitoring of K+ fluxes in in vitro and in vivo. This report provides new framework for design of sensing and labeling nanoparticle agents and demonstrates its high practical application potential.
2. Results and discussion
2.1. FI3 nanoparticles display versatile intracellular staining pattern across the range of 2D and 3D tissue models
Recently, we reported a family of new fluoroionophores based on borondipyrromethenes (BODIPY) and aza-crown ether-based sensing unit, emitting in green to near-infrared parts of electromagnetic spectrum. The compound FI3 showed attractive properties with high molar absorption (109,300 M−1 cm−1 at 640 nm), deep red fluorescence (λmax exc./em. 640/660 nm), high quantum yields (47-6.3%, depending on K+ complexation) and selectivity towards K+, independent from pH, within the relevant range [8g] (Figure 1).
Figure 1.
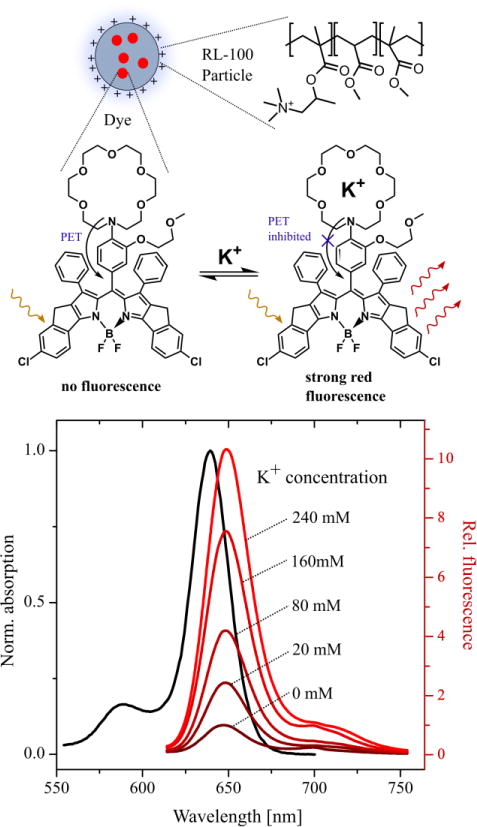
Scheme and spectral properties of K+-sensitive FI3 dye encapsulated in RL100 nanoparticles. Spectra were measured in 20 mM Tris-HCl, pH 7.4 with concentration of nanoparticles adjusted to A630 =0.1.
We found that the dye is relatively easy to impregnate in polymer nanoparticles (1%, by weight of indicator dye, ~780 dye molecules per particle), however their physical properties and interaction with cells were not studied in detail.[8g] The amphipathic nature of fluoroionophore suggests that the structure of nanoparticles can differ from the other RL100-based nanosensors, in which the impregnated material represents highly hydrophobic dyes.[17a, 17b, 21] Using scanning transmission electron microscopy (STEM) we confirmed the nanoscale dimensions of FI3 nanoparticles (30.6±10.6 nm, PDI 0.34) and found them comparable to previously described RL100-based nanosensors[17a, 17b] (Figure S1).
For assessment of cell staining efficacy, we chose a number of previously tested adherent cell lines, including human colon cancer HCT116 cells, mouse embryonic fibroblasts (MEF), neuronal origin cancer cells (PC12, SHSY5Y) and mixed culture of rat primary neural cells (Figure 2). When analyzed by confocal fluorescence microscopy, we found efficient and bright staining of cells, including culture of primary neural cells. Bright intracellular fluorescence was observed for both differentiated (not shown) and non-differentiated SHSY5Y neuroblastoma cells and for two principal types of neural cells present in primary culture, astrocytes and neurons. With mixed culture of primary neurons we also observed some extracellular staining, which can be caused by non-specific aggregation of nanoparticles or their interaction with dead cells or cell debris; however, at the same time, the bright cytoplasmic staining of both cell bodies and neuritis was evident, proving efficient intracellular accumulation in neural cells (Figure 2).
Figure 2. FI3 nanoparticles provide efficient staining of various 2D and 3D tissue models.
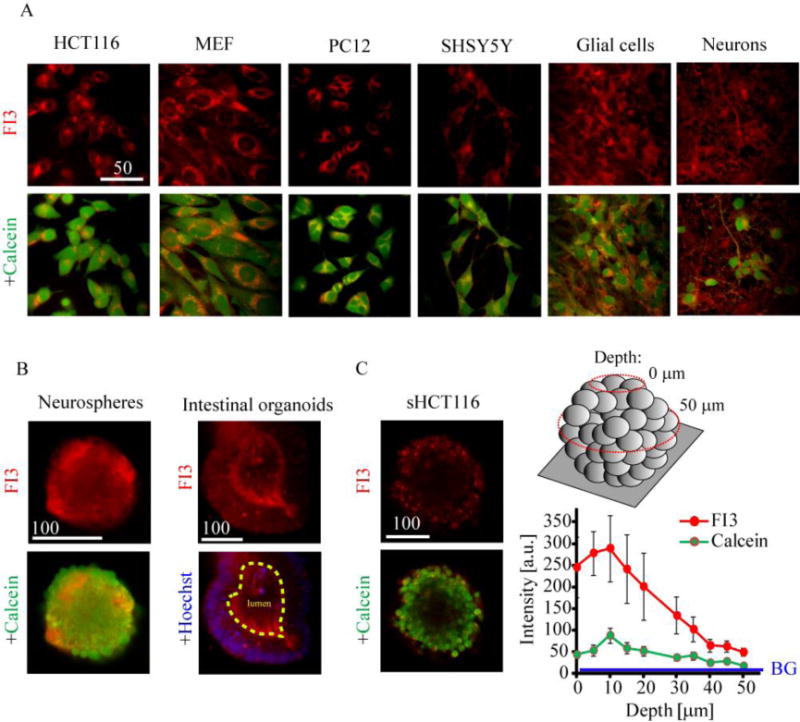
A: adherent cell models of neuronal and non-neuronal origin. Live cells were incubated with FI3 nanoparticles (10 μg/ml, 16 h), washed and counter-stained with Calcein Green AM (1 μM, 0.5 h) and imaged. B: staining of rat primary neurospheres and mouse intestinal organoids. Live neurospheres and organoids were incubated with FI3 nanoparticles (10 μg/ml) for 16 h and 3 h, respectively, counter-stained with Calcein Green or Hoechst 33342 and imaged. C: staining of tumor spheroids from HCT116 cells. Formed tumor spheroids were incubated with FI3 nanoparticles (3 h) and counter-stained with Calcein Green. The image represents cross-section at depth 50 μm, as indicated. Right graph shows distribution of fluorescence intensity in the center of spheroids at depths 0-50 μm. BG indicates background intensity. Scale bar is in μm.
This is in contrast with the poor staining efficacy anticipated from positively charged RL100-based nanoparticles: for control, we stained primary neurons with RL100-nanoparticles impregnated with hydrophobic metalloporphyrin PdTFPP (Figure S2A). In this case we observed mostly extracellular aggregates, in agreement with our previous data.[19]
The unusually efficient staining of neural cells with FI3 nanoparticles prompted us to test them with 3D tissue models. We produced neurospheres from rat primary neurons (E18) and observed efficient in-depth staining achieved in 6-16 h incubation time (Figure 2B). With more complex mouse intestinal organoid model [22] we found comparably efficient staining of epithelium and lumen and some degree of aggregation (Figure 2). In comparison, two other types of nanoparticle biosensors, based on anionic polymer (PA1)[19] and RL100 polymers (NanO2)[17a] lacked the ability to stain intestinal organoids under similar conditions (Figure S2B): instead, only strong autofluorescence of lumen compartment was observed. Since FI3, PA1 and NanO2 nanosensors are all emitting in the same spectral window, the luminal fluorescence can be related to strong red autofluorescence, which we reported before.[23] FI3 showed rather homogeneous staining of epithelium with enrichment in certain cell types (showing a 5-6 times higher fluorescence), which can be explained by the cell-specific difference in staining efficiency (Figure S2B).
We also evaluated relative staining efficacy of FI3 nanoparticles with culture of tumor spheroids from HCT116 cells. Using optical sectioning we found strong fluorescent signals at 40-50 μm depths, indicating similar or higher staining efficacy than with conventional Calcein Green dye (Figure 2C).
Thus, we observed highly efficient and unique, in comparison to previously reported RL100-based probes, intracellular accumulation of FI3 nanoparticles with all tested cell models: from ‘simple’ adherent to multicellular aggregates and intestinal organoids, containing multiple differentiated cell types.
2.2. Intracellular staining differs between FI3 nanoparticles and non-encapsulated dye
The amphipathic nature of FI3 dye can potentially lead to its leakage from nanoparticles upon or prior to the cell internalization and thus explain the observed ‘efficient’ intracellular staining pattern. To reveal if this is the case, we performed a number of additional experiments. First, we compared the staining of neural cells with the free dye and encapsulated in RL100 nanoparticles (Figure 3): free dye displayed intracellular staining only of certain cells in culture and with lower intensity. The comparison of average brightness inside the cells showed that the dye impregnated in nanoparticles displayed 3-5 times higher brightness (0.1 μM of free dye corresponds to 10 μg/ml concentration of nanoparticles) suggesting its retention within them after cell internalization.
Figure 3. FI3 dye remains encapsulated in the nanoparticles after cell internalization.
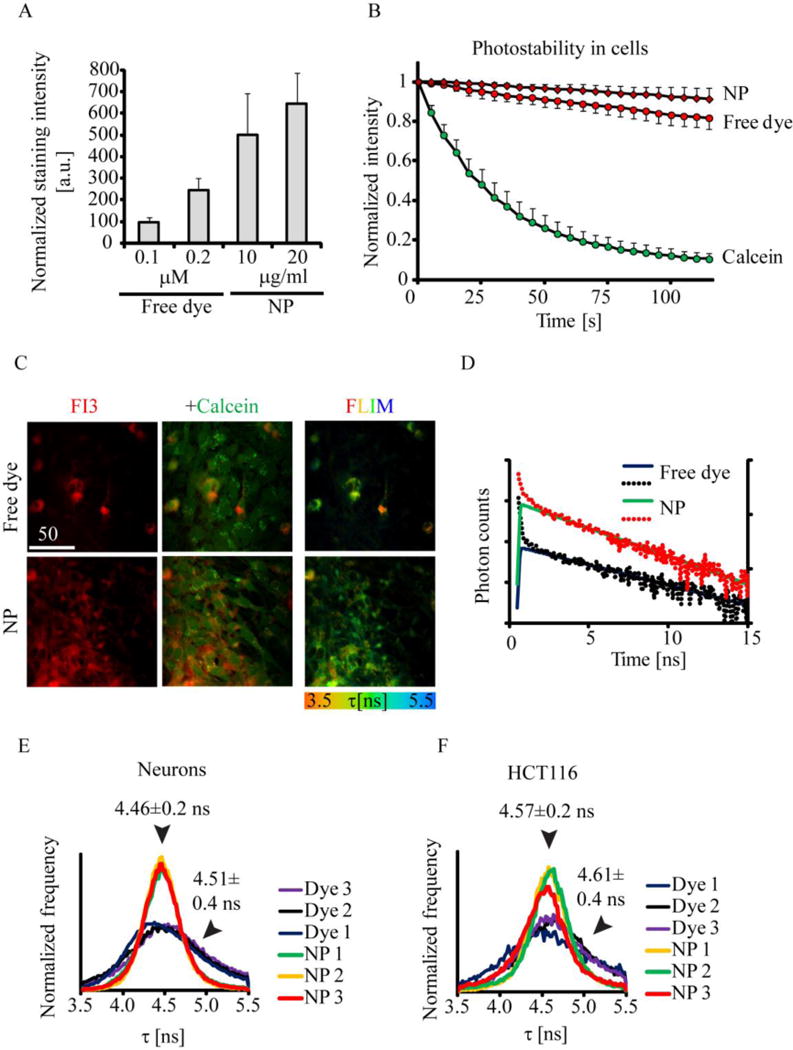
A-B: comparison of staining efficiency of live primary neurons with FI3 as free dye and encapsulated in nanoparticles (NP). The dye concentration of 0.1 μM equals to 10 μg/ml of nanoparticles. Cells were incubated with dye and dye/NP at indicated concentrations (16 h), washed and imaged. A: Comparison of fluorescence intensity in cells for the free dye and NP at different concentrations. B: Comparison of photostability between free dye, NP and Calcein Green. N=8. C-F: The differences in fluorescence lifetimes observed in cells stained with free dye and NP, measured by FLIM. C: characteristic images of mixed culture of astro-glial cells and neurons stained with FI3. Intensity (red fluorescence), co-staining with Calcein Green and FLIM images are shown. D: typical fluorescence lifetime decay curves (dots) and the respective fits (lines), shown in logarithmic scale. E, F: distribution histograms for free dye and NP, observed for primary neurons and HCT116 cells. N = 3. Scale bar is in μm.
The staining with free dye did not reach saturation at 0.1-0.2 μM, implying at difference in cell uptake mechanisms between the free dye and RL100 nanoparticles (Figure 3A). Similarly, we found lower staining efficacy for the dye with HCT116 and MEF cells (Figure S3A). Next, we found that FI3 encapsulated in nanoparticles displayed higher photostability than free dye and Calcein Green dye (Figure 3B). While testing other cell lines (HCT116, MEF, PC12) we saw comparable efficacy of staining between nanoparticles and free dye (not shown), however with mixed culture of neural cells we observed the differences in cell specificity (Figure 3C). We found that free dye stained predominantly certain neuron-like cells, while nanoparticles provided efficient staining of all the cells in population (both neurons and astrocytes). We attempted to confirm the cell specificity of free dye using immunofluorescent staining of fixed cells with respective antibodies but found that dye and nanoparticles did not remain visible after this procedure (not shown).
Further, we reasoned that after internalization into the cell different environments of the free dye (directly exposed to proteins, lipids or other biomolecules) and protected by nanoparticle shell can affect the fluorescence lifetime of FI3. Using fluorescence lifetime imaging microscopy (FLIM) as a method to study the intracellular degradation of nanoparticles[24], we found that both free dye and RL100 nanoparticles displayed fluorescence decays, which could be fit with mono-exponential function (Figure 3C-F). The dye encapsulated in nanoparticles showed consistently narrower distributions (±0.2 ns) of fluorescence lifetimes across the imaged areas, than the free dye (twice broader, ±0.4 ns) in mixed culture of neurons (Figure 3E) and in other cell types, such as HCT116 (Figure 3F).
This data suggests that: (1) the fluorescence lifetime of FI3 is environment-sensitive; (2) encapsulation in RL100 nanoparticles helps minimizing the effect of biological environment on the fluorescence lifetime; (3) the dye does not leak from the nanoparticles after cell internalization. Collectively, these results demonstrate the improvement of photophysical properties of FI3 encapsulated in nanoparticles, in the staining of neural cells.
2.3. Mechanism of intracellular accumulation of FI3 nanoparticles
With culture of primary neural cells, FI3 nanoparticles displayed relatively fast intracellular accumulation different from the free dye (Figure 4A): the dye-stained cells appeared already after 1 h incubation but they represented only minor fraction of culture; in contrast, FI3 nanoparticles stained all cells in the culture progressively accumulating the signals over 3-6 h staining time.
Figure 4. Mechanism of cell entry for FI3 nanoparticles.
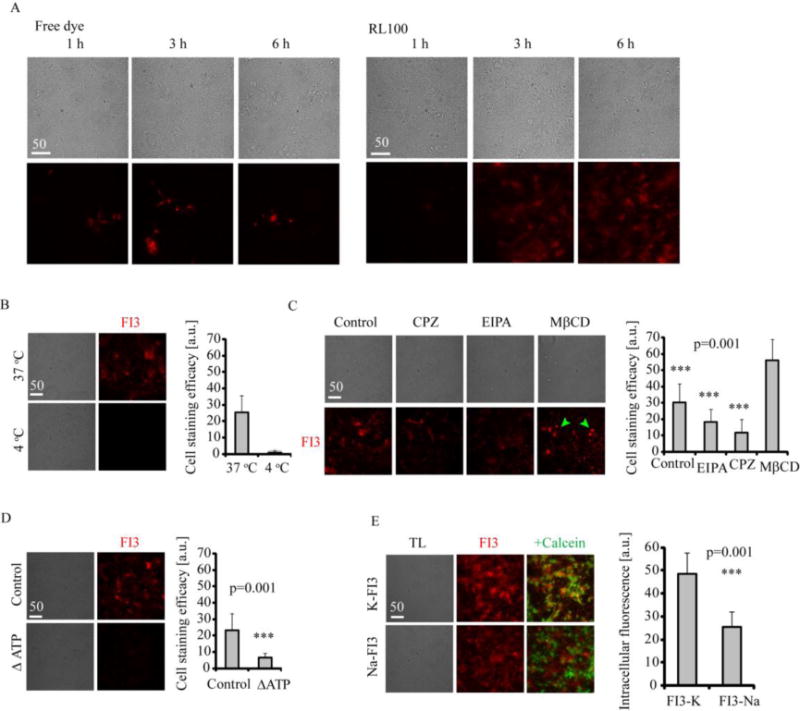
A: Staining kinetics for FI3 (0.1 μM free dye or 10 μg/ml RL100 nanoparticles) in live rat primary neural cells. Cells were incubated with FI3 for indicated time intervals, washed and immediately imaged. B-D: Staining of primary neural cells with FI3 nanoparticles (10 μg/ml, 3 h). B: Effect of temperature on staining efficiency. C: Effects of various endocytosis inhibitors on cell staining with FI3. Cells were pre-treated with inhibitors (50 μM EIPA, 10 μg/ml CPZ and 5 mg/ml MβCD) for 30 min, followed by staining procedure. MβCD-treated cells displayed round morphology, indicated with green arrows. N= 17. D: Effect of ATP depletion on cell staining efficiency. For ATP depletion, cells were pre-incubated in no-glucose medium and treated with oligomycin (10 μM), followed by staining procedure. N=20. E: Cell staining efficacy for K-FI3 and Na-FI3. Cells were incubated with nanoparticles (10 μg/ml, 16 h) washed, counter-stained with Calcein Green and imaged. Scale bar is in μm.
Although this was still slower than with regular small molecule dyes such as Calcein Green, TMRM and fluorescent conjugates of transferrin or cholera toxin, staining with FI3 nanoparticles was comparable or better than loading with previously reported nanoparticle-based biosensors,[19, 25] allowing shortening the incubation time with cells to as little as 3 h. Staining time of 3 h allowed us to study the mechanism of intracellular accumulation of FI3 nanoparticles using the culture of primary neural cells (Figure 4B-D). As expected, we observed that intracellular staining with FI3 was nearly completely inhibited by decreasing temperature to 4 °C (Figure 4B) and was mainly energy-dependent (Figure 4D), suggesting endocytosis as its cell entry mechanism. The incomplete blocking of uptake can be explained by partial depletion of cell energy sources, when both glycolytic and oxidative phosphorylation ATP production pathways were blocked. Staining of cells with FI3 nanoparticles in presence of endocytosis inhibitors[26] revealed reliance of cell uptake on macropinocytosis (EIPA) and largely on clathrin-mediated endocytosis (chlorpromazine, CPZ) (Figure 4C). The treatment with methyl-β-D-cyclodextrin (MβCD) targeting cholesterol-rich plasma membrane regions, resulted in extensive cell condensation (round phenotype) and apparent increase of brightness, allowed to conclude that there was no effect of lipid raft-mediated endocytosis on cell uptake. This data shows with culture of primary neurons FI3 nanoparticles utilize mostly clathrin-dependent endocytosis (main energy dependent delivery route for small molecules), with lower degree of macropinocytosis. Thus, with culture of primary neural cells, FI3 nanoparticles employ different cell entry pathway than negatively charged polymeric nanoparticles.[19] The molecular weight of RL100 polymer (Mw = 150,000 g/mol) can be also the factor allowing efficient endocytic cell entry.[26–27]
We wondered if the K+-sensitive crown ether sensor groups exposed at the surface of nanoparticles could participate in observed improvement of cellular uptake. To test this, we stained neural cells with Na+-sensitive FI3 analog (Na-FI3)[28] under the same conditions and analyzed them by microscopy (Figures 4E, S3B). We found that both K+ and Na+-sensitive nanoparticles displayed comparable ability to stain live cells suggesting the potential role of crown ether in improving the cell staining efficiency of RL100-based nanoparticles. With Na-FI3 we observed expected ~50% ‘lower’ signal intensities inside the cells, which can be explained by much lower concentration of Na+ in the cells. However, comparing quantum yield efficiency of the Na-FI3 (0.7%) and K-FI3 (6.3% at physiological pH) implied at higher staining efficacy for Na-FI3.
Collectively, this data confirms that FI3 nanoparticles show different kinetics of staining than free dye, utilizing endocytic, energy-dependent cell entry pathway. The charge of nanoparticles and their complexation with K+ unlikely plays significant role in their cell entry but the presence of crown ether is seemingly beneficial.
2.4. Evaluation of FI3 nanoparticles as K+ nanosensor with primary neural cells
The previously unseen ability of RL100-based FI3 nanoparticles to stain neural cells prompted us to evaluate it as a functional tool in monitoring of intra- and extracellular K+ dynamics in mixed culture of primary neural cells. First, we tested whether the successful staining of cells with this fluoroionophore has any effects on cell viability. Using analysis of total cellular ATP, primary marker of cell viability, we found minor or no toxicity of nanoparticles at up to 20 μg/ml concentrations (6 and 25 h staining times) (Figure S4A). Similarly, we did not see toxic effects of FI3 nanoparticles when analyzed the membrane integrity (not shown). However, prolonged staining time and increase of concentration resulted in some toxic effects, more profound than for free dye. We also further tested if the toxicity increases at higher cell density (100%) and upon metabolic stimulation (cells exposed to glucose-free galactose medium or treated with mitochondrial uncoupler FCCP), when the effects of FI3 ionophore activity on cell function can become more profound: in this case we observed strong toxicity (25 h incubation time), reflected in loss of cell mass due to cell detachment, and overall inhibition of mitochondrial function (Figure S4B). The potential effect on cell energy metabolism at high doses of FI3 nanoparticles (20 μg/ml, 25 h incubation time) was also confirmed with analysis of glycolytic flux via analysis of extracellular acidification rate (Figure S4C). While our data showed minimal toxicity upon short incubation time and small concentrations of FI3, it should be kept in mind that use of FI3 (and any other fluoroionophore) at high concentrations can have adverse effects on cell function. However, the high brightness, quantum yield and cell staining efficiency with FI3 allow for use of minimal doses of these nanoparticles for cell-based measurements.
Next, we evaluated the performance of FI3 in monitoring of K+ dynamics with mixed culture of neural cells. Using confocal fluorescence microscopy, we observed nearly equal staining efficacy of both astro-glial cells and neurons in mixed culture (Figure S5). Astrocytes showed stronger punctate staining of cytoplasm with FI3 than neurons (diffuse cytoplasmic staining), which can be attributed to their higher capacity[2a] in storing intracellular K+. To demonstrate the responses of FI3 nanoparticles in kinetic measurements, we treated neural cells with KCl (increases extracellular K+) and then with ionophore valinomycin (increases membrane permeability and equalizes extra- and intracellular K+) (Figure 5A-C). We found that application of KCl led to cell contraction, changing cell shape and size. In order to better interpret the differences between extra- and intracellular (visualized by co-staining with Calcein Green) pools we used line profiles across optical sections and integrated peak areas (grey boxes indicate extracellular signals, see Methods for details and Figure 5B). We found that line profiles are more appropriate for quantitative measurements compared to selecting individual ROIs: the cells frequently change shape and contract during fluxes of intra- and extracellular K+ leading to inaccurate quantification or underestimation of drug effects (Figures S6, S7). As expected, we observed overall increase in extracellular K+ after addition of 40 mM KCl and subsequent decrease after applying valinomycin (Figure 5C). Upon such treatment, application of external KCl resulted in minor decrease of intracellular pool of K+, potentially due to partial depolarization of cell membrane, while adding valinomycin helped to slightly ‘restore’ it. In another experiment, we reversed the order of these treatments and monitored only intracellular K+: we added first valinomycin and observed ~40% decrease in K+, and again slightly ‘restored’ it by applying KCl (Figure 5D, S7, S8). We also looked at the effects of treatment with nigericin (H+/K+ ionophore) and ouabain (inhibitor of Na+/K+-ATPase).[5] Treatment with these drugs led to decrease of intracellular K+ (Figure S9).
Figure 5. Monitoring of extra- and intracellular K+ in neural cells with FI3 nanoparticles.
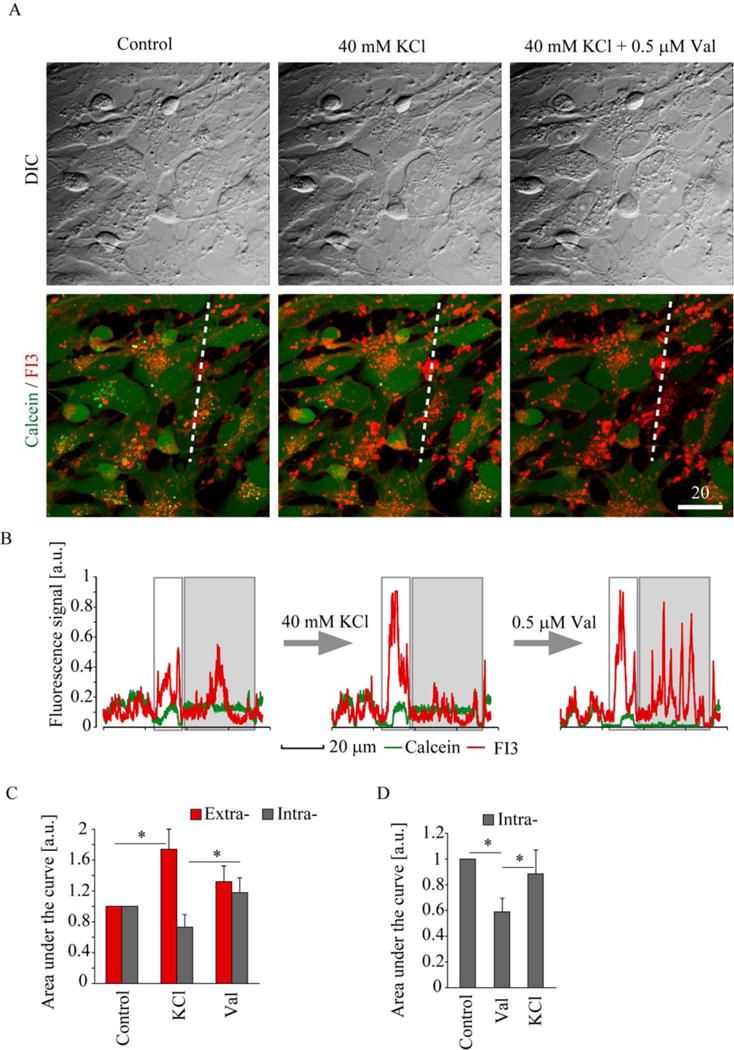
The live rat primary neural cells were stained with nanoparticles (10 μg/ml, 16 h), counter-stained with Calcein Green and treated with KCl and valinomycin (Val). A: Representative transmission light (DIC) and superimposed confocal fluorescence images of FI3 and Calcein before and after treatment with KCl (40 mM) and valinomycin (Val, 0.5 μM). B-C: Line profile analysis of the changes in FI3 and Calcein fluorescence signals, induced by treatments in (A). The areas of intra- and extracellular FI3 pools are highlighted in (B) in grey and white, respectively. D. Results of line profile analysis of the changes in intracellular FI3 signal, induced by the sequential addition of Val and KCl. Images (A) represent stacks of 2 confocal planes taken with 0.5 μm step. Error bars indicate SEM. Asterisks indicate significant difference (p < 0.01, U test). N = 4 (D). Scale bar is in μm.
These results demonstrate that FI3 can be successfully used for monitoring of intracellular K+ fluxes in live cells, by high-resolution live cell confocal fluorescence microscopy. The counter-staining with markers of cytoplasm and simultaneous monitoring of FI3 can be also used for analysis of extracellular K+. The quick response time and the molecular structure of BODIPY dye[29] constituting FI3 nanoparticles allow for advanced two-photon excited imaging applications and multi-color imaging.[30]
2.5. Application of FI3 nanoparticles to brain imaging
To further evaluate the application potential of FI3 we tested more complex biological models, such as live brain slices and intact brain. Using confocal fluorescence microscopy of live organotypic rat brain slices (ex vivo), we observed efficient staining of cortex, different from distribution of cholera toxin-positive cells (Figure 6A-B). This confirmed that FI3 can be used for staining of intact live tissue and prompted us to apply FI3 nanoparticles in functional brain imaging.
Figure 6. Application of FI3 nanoparticles to ex vivo and in vivo brain imaging.
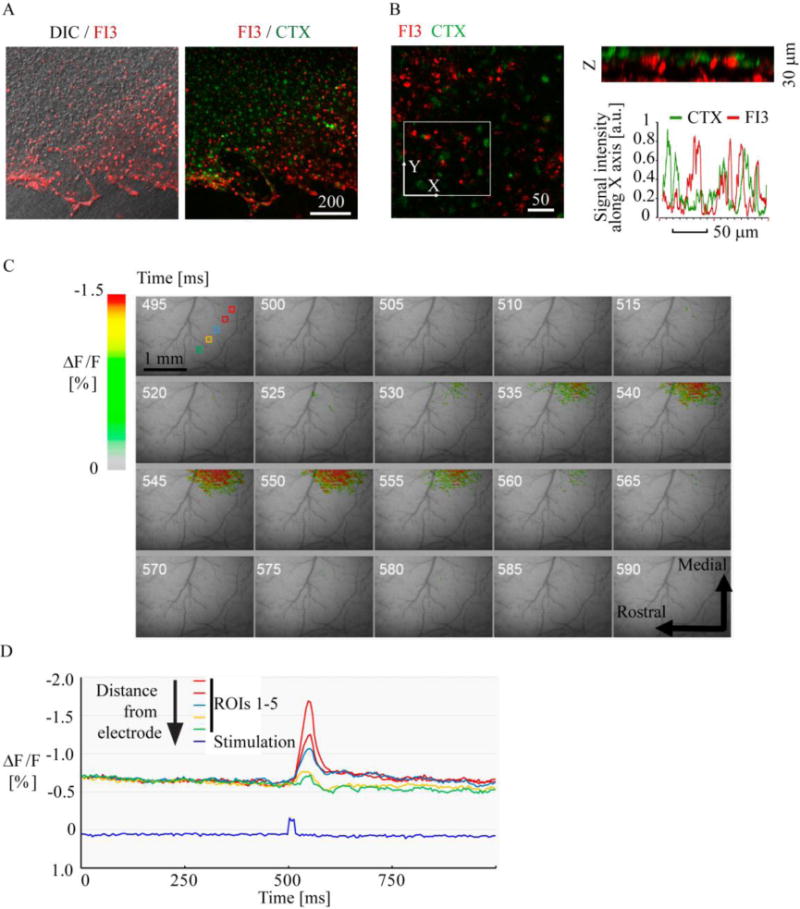
A-B: staining efficiency and distribution of FI3 (10 μg/ml, 6 h) in live organotypic rat brain slices. Cortex region is shown. Staining with FI3 (red) and cholera toxin-Alexa Fluor 488 conjugate (green) is shown. B: 3D reconstruction (the views of XY and Z projections) of FI3 and CTX localization in the tissue, and corresponding line profile analysis. Images represent stacks of 21 (A) and 16 (B) confocal planes taken with 2 μm step. C-D: In vivo imaging of stained mouse brain. C: Pseudocolor images of the cortex before and after stimulus onset. Time (ms) after beginning of the trial is indicated by numbers. D: Time-course of recorded response at the locations (depicted in C) having different distances from the electrode.
Using previously described in vivo imaging setup[31] we applied FI3 nanoparticles to live mouse brains (intracortical administration) and recorded the responses to electrical stimulation. Functional pseudocolor maps images of recorded optic signal were constructed using ‘the first frame analysis’ (Figure 6C). Due to short incubation time, we expected that FI3 nanoparticles partially localized inside the cells and predominantly in the brain parenchyma. The changes of FI3 fluorescence were observed near the electrode tip immediately following the stimulation onset, reaching a maximum at 40–50 ms after stimulation onset and then decaying during the subsequent frames. This response occurred simultaneously but its intensity was proportional to the distance from the electrode tip (Figure 6C-D). In another experiment, we looked if the drug-induced epileptic seizures can be monitored with FI3 in intact mouse brain. To do this, we applied 4-aminopyridine (4-AP) and monitored fluorescence changes together with EEG (Figure S10): thus, we found periodic responses lasting for 2 – 3 h (30 – 200 s with few min intervals). Figure S10 shows pattern of the EEG typically observed during epileptic seizures. Epileptic seizure on the EEG signal (30 seconds window) and profound changes in fluorescence were well correlated with each other.
Thus, we demonstrated for the first time that K+-sensitive FI3 nanoparticles are useful for in vivo mapping of epileptic seizures with high spatial and temporal resolution in animal model. This suggests that FI3 can become a new tool for mapping neural activity in various areas of brain imaging.
The resolution of the used optical system is about 50 μm per pixel, and the obtained data represents 2D projection of the 3D object. Therefore, each pixel reflects averaged fluctuations of intra- and extracellular K+ in the particular volume of the brain tissue. It is well known that electrical stimulation of the cortical tissue causes an increase in extracellular K+ concentration[32] and thus positive and negative fluorescence changes in particular pixels can be observed on a micro scale. In our method, measured relative changes in the optical signals inform on the fluxes of intra- and extracellular [K+]. This is relatively complicated process, which proceeds on the scale of tens of milliseconds and is related to the evoked neural activity. The biphasic character of the observed signal reflects the processes of [K+] release and transport through the cellular membranes. Overall, the brain activity patterns obtained in our experiments are very similar to voltage-sensitive dye optical imaging (VSDi).[33] Altogether, measured relative changes in FI3 fluorescence displayed the responses, concomitant with the evoked neural activity, within the useful scale of milliseconds. FI3 showed good sensitivity, reproducibility of signals, selectivity and thus can be applied for functional brain imaging and such applications as development of better therapy against epileptic seizures.
3. Conclusion
We discovered that polymeric Eudragit RL100 nanoparticles drastically improve their cell permeability, when combined with crown ether-based BODIPY FI3 dyes. For the first time, we show that such RL100-based nanoparticles can provide efficient staining of primary neural cells (astrocytes and neurons) and 3D tissue cancer and stem cell-based models (spheroids, organoids and ex vivo tissue sections). The encapsulation of FI3 in cationic RL100 polymer facilitated brighter cell staining, with no observable cell specificity, via energy-dependent clathrin-mediated and macropinocytosis endocytosis mechanisms. These features and relatively short cell staining time (3 h) demonstrate that FI3 nanoparticles are as easy to use as small molecule fluorescent dyes. We also performed detailed evaluation of FI3 nanoparticles in quantitative live cell imaging and in vivo brain imaging applications. The sensitivity, brightness and versatility of the described nanoparticles make them a promising tool both as a versatile K+-sensitive biosensor and as a prototype for advanced design of polymer-based nanoparticles and imaging agents, helping to solve the problem of their efficient biodistribution.
4. Methods
4.1. Materials
CellTox Green™ and CellTiter-Glo™ viability assay kits were from Promega (MyBio, Ireland). Lipidure™ 96-well plates were from Amsbio (UK). Palladium (II) meso-tetra(pentafluorophenyl)porphine (PdTFPP) was from Frontier Scientific (Inochem Ltd, Lancashire, UK). IntestiCult organoid growth medium (mouse) kit and Gentle dissociation reagent were from StemCell technologies (UK). Matrigel™ matrix with reduced growth factors was from Corning (USA). Eudragit RL-100 copolymer (poly-(ethylacrylate-co-methylmethacrylate-co-trimethyl-aminoethyl methacrylate), Mw ≈ 150,000 g/mol, 8.8 - 12% of quaternary ammonium groups) was from Degussa, Germany (www.evonik.com). MitoImage-NanO2™ and pH-Xtra™ probes were from Luxcel Biosciences (Cork, Ireland). PdTFPP/RL100 nanoparticles and PA1 O2-sensitive nanoparticles were prepared as described previously.[19] FI3 free dye and RL100-based nanoparticles were prepared as described previously.[8g] Na-FI3 was synthesized as described before.[28] Cholera toxin, subunit B, Alexa Fluor 488 conjugate, CoroNa Green-AM and B-27 serum-free media supplement were from ThermoFisher (BioSciences, Dublin, Ireland). Fibroblast growth factor (FGF), Epidermal growth factor (EGF), R-spondin and MilliCell membranes were from Millipore (Cork, Ireland). Galactose, valinomycin, oligomycin, Calcein Green AM, bis-benzimide Hoechst 33342, chlorpromazine, 5-(N-ethyl-N-isopropyl)amiloride, methyl-β-cyclodextrin and all the other reagents were from Sigma-Aldrich (Dublin, Ireland). Standard cell culture grade plasticware was from Sarstedt (Wexford, Ireland), glass bottom mini-dishes were from MatTek (Ashland, USA) and Ibidi μ-slide 8/12-well chambers were from Ibidi (Martinsried, Germany).
4.2. Preparation of nanoparticles
100 mg of Eudragit RL100 was dissolved in 50 ml acetone, followed up addition of 1 mg of indicator dye K-FI3 (or Na-FI3). To start precipitation, 250 ml water was added quickly under vigorous stirring (within 3 s); acetone was removed using rotary evaporator and the particle dispersion was further concentrated to a volume of 10 ml.
4.3. Cells, tissues and organoids
HCT116, PC12, MEF and SH-SY5Y cells (ATCC) were handled and differentiated as described before.[11, 34] Typically, cells cultured at 30-50% confluence were incubated with dyes and nanoparticles in regular growth medium (McCoy 5A supplemented with 10% heat-inactivated fetal bovine serum, penicillin-streptomycin, 10 mM HEPES-Na, pH 7.2 for HCT116 cells; RPMI 1640 supplemented with 1% heat inactivated horse serum, penicillin-streptomycin, 10 mM HEPES-Na, pH 7.2 for differentiated PC12 cells; high-glucose DMEM supplemented with 10% heat-inactivated fetal bovine serum, penicillin-streptomycin, 10 mM HEPES-Na, pH 7.2 for MEF and SHSY5Y cells) at indicated concentrations and staining time, washed three times with growth medium and immediately proceeded to live cell imaging. In studies of cell entry mechanism, cells were pre-incubated with respective drugs for 30 min (or 1.5 h in glucose-free DMEM supplemented with 10 mM galactose, followed by 15 min pre-incubation with 10 μM oligomycin, for ATP depletion), followed by incubation in the same medium with FI3 (10 μg/ml, 3 h), washing and analysis. Tumor spheroids were produced by seeding in Lipidure™ plates (300 cells, 3 days growth per spheroid) essentially as described previously.[21]
All the procedures with animals were performed under the license issued by the Department of Health and Children (Ireland) and in accordance with the Directive 2010/63/EU adopted by the European Parliament and the Council of the European Union. Cultures of embryonic rat (E18) neurons, neurospheres and organotypic brain slices were produced and handled as described previously.[18b, 19] Mixed culture of primary neurons and astro-glial cells (primary neural cells) were allowed to differentiate in DMEM F12 Ham medium supplemented with 1% heat-inactivated fetal bovie serum, 2% B-27 and penicillin-streptomycin for 4-8 days, before experiments. To obtain the culture enriched in astrocytes, cells were first grown in neurospheres, then trypsinized and seeded for additional 5 days differentiation.[18b] Neurospheres were typically grown in suspension state for 5-7 days before staining and analysis. For staining and microscopy, they were collected, washed in Hanks’ balanced salt solution and allowed to attach under differentiation conditions (1% FBS, 2% B-27) for 3 h on poly-D-lysine pre-coated glass or plasticware as described previously.[25] Organotypic brain slices were cut at 300 μm thickness on a vibratome and cultured on MilliCell (0.4 μm) membranes for 7-9 days prior the staining and analysis, essentially as described previously.[25]
Mouse intestinal organoids were produced, grown in Matrigel and handled as described before.[23a] Organoids were incubated with FI3 nanoparticles (10 μg/ml, 16 h), washed and imaged directly in Matrigel™ in growth medium.
4.4. Microscopy
Cell staining kinetics, comparison of efficiency of staining, photostability and mechanism of cell internalization were studied on widefield live fluorescence microscope Axiovert 200 (Carl Zeiss)[18b] equipped with 40×/1.3 Plan Neofluar oil-immersion objective, integrated T/CO2 climate control (PeCon), pulsed LED excitation module (390, 470, 590 nm), gated CCD camera, respective green (FITC) and red (635LP) emission filters and image acquisition and processing ImSpector software (LaVision Bio-Tec).
Comparative analysis of staining of different cell models (including neurospheres, tumor spheroids and organoids) and FLIM were performed on an upright laser scanning Axio Examiner Z1 (Carl Zeiss) microscope[11] equipped with 20×/1.0 and 63×/1.0 W-Plan Apochromat dipping objectives, integrated T and Z-axis control, ps diode BDL-SMC 405 nm (Becker & Hickl GmbH) and ps supercontinuum (450-700 nm) SC400-4 (Fianium, UK) lasers, DCS-120 confocal TCSPC scanner, photon counting detector and dedicated SPCM and SPCImage software (Becker & Hickl GmbH). Calcein Green dye was excited using 488 nm laser (emission collected at 512-536 nm), Hoechst 33342 was excited using 405 nm laser (emission collected at 428-468 nm), FI3 was excited using either 405 nm or 632 nm lasers (emission collected at 635-675 nm). FLIM measurements of FI3 (excited at 632 nm) were performed essentially as described previously,[21] with application of mono-exponential decay (longer component) fitting function.
Analysis of brain slices and monitoring of kinetic responses of primary neural cells to drug stimulation were performed on an inverted Olympus FV1000 confocal laser scanning microscope[35] with controlled temperature, CO2 and humidity. Calcein Green and CTX were excited at 488 nm (2-8% of laser power) with emission collected at 500-560 nm. FI3 was excited at 633 nm (5-20% of laser power); emission was collected at 560-600 nm. Acquisition of each spectral signal was done using UPLSAPO 20×/0.75 and UPLSAPO 60×/1.35 oil immersion Super Apochromat objectives in sequential laser mode to avoid spectral overlap. In all experiments differential interference contrast (DIC) images were collected to complement fluorescence images. In kinetic experiments, images (8-12 focal planes) were collected with approximately 3 min intervals before and after stimulations. The changes in FI3 intensity signals (Figure 5C-D and S5) were quantified as areas under the curve (from line profiles), calculated according to the equation S (the area) = Σ[(X(n+1)-Xn)×(Yn+Y(n+1))/2], where Xn is the position on the analyzed line (μm) and Yn is the value of corresponding fluorescence signal.
Analysis of cellular responses to ouabain (50 μM) and nigericin (10 μM in the presence of 20 mM KCl) was performed in a kinetic mode, with 3 focal planes imaged repeatedly with 30 s intervals before and after addition of drugs. The resulting single plane and z-stacked images were analysed using FV1000 Viewer software (Olympus) and assembled using Adobe Photoshop and Illustrator software.
Scanning transmission electron microscopy (STEM) was performed on a FEI Talos F200X at the Scope M facility (Zürich, Switzerland). The nanoparticle solution (0.6 mg ml−1) was diluted (1:2) with distilled water, then 1 μL of solution was drop-casted on a carbon support film copper grid Cu 400 and used for measurements.
4.5. Assessment of toxicity
The effects of cell staining with FI3 (free dye and nanoparticles) were evaluated by using analysis of membrane integrity (staining of cells with 0.2% CellTox Green), total cellular ATP (CellTiter-Glo assay) and extracellular acidification (unsealed system), as described before.[11] Briefly, primary neurons grown on poly-D-lysine pre-coated microplates were incubated with FI3 at indicated concentration and time, washed and either measured in presence of pH-Xtra probe on Victor2 time-resolved fluorescence microplate reader (PerkinElmer) over 1 h at 37 °C or lysed using CellTiter-Glo reagent and measured for total luminescence. For normalization, cells were lysed with buffer containing 50 mM HEPES-Na, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Igepal CA630, protease inhibitor cocktail (Sigma P2714) and total protein concentration was determined using BCA protein assay kit (Pierce, Thermofisher, Ireland).[36]
4.6. In vivo imaging
All the procedures with animals were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) revised in 1996 and a protocol approved by the University of Maryland School of Medicine Institutional Animal Use and Care Committee. The in vivo imaging experiments were performed on C57BL/6N (Charles River Laboratory, MD) mice (weight 25–35 g, 3–6 months old, N=4) using a MiCAM-02 optical imaging system (SciMedia Ltd). Animals were anesthetized with urethane (IP injection, 1.15 g/kg), fixed on a stereotaxic frame and proceeded to surgical removal of the skin of the dorsal part of the head. The exposed area was washed with a hemostatic sponge with the artificial cerebrospinal fluid (ACSF). A cranial opening (~4–5 mm2) was made using a dental drill and the exposed dura mater surface was cleaned with ACSF. After each experiment, the animal was euthanized. After craniotomy, FI3 nanoparticles (1 μl of 2 mg/ml solution) were injected into 3-5 sites in the cortex. For electrical stimulation experiments a bipolar tungsten electrode MX216TW(VT1) (FHC Inc.) was introduced into the cortical tissue at a 0.3–0.4 mm depth using XYZ manipulator. A 100 μA stimulus was generated by A-M Systems stimulator (Model 2100), controlled by computer and consisted of a 10 ms train of 0.2 ms 300 Hz. For the epileptic seizures experiment, 4-aminopyridine (4-AP, 0.3 μL of 30 mM solution in ACSF) was injected into cortical layer III, using an injector mounted on a micromanipulator (Nanojet II) with a 20–30 μm diameter glass microcapillary as described previously.[37] A single-channel electroencephalogram (EEG) was recorded through a screw-type electrode placed in the operated hemisphere in the bone, then amplified by the differential amplifier (A-M Systems, Model 3000), digitized at 5000 Hz, and recorded using custom made computer software. The EEG data acquisition was initiated manually.
4.7. Data assessment
Plate reader data represent averaged values (obtained from >4 replicates) with standard deviation shown as error bars. For microscopy, the experiments were performed in triplicate. For quantification, images were processed in Fiji (ImageJ 1.49k) or FV1000 (Olympus) software, using analysis of independently taken ROIs (N indicated in figure legends). Where appropriate (normal distribution), an independent t-test was performed in Origin 6.0 software, with confidence level P=0.001 accepted as significant. FLIM data were processed using SPCImage and Microsoft Excel software, with mono-exponential tail-enhanced fitting function (χ2 < 1.5) and binning factor as appropriate.
Distribution of fluorescent signals was evaluated using a cumulative histogram approach, as described before.[38] Where appropriate, an independent t-test and Mann-Whitney U-test were performed in Origin 6.0 (Microcal Software) and SPSS software, with confidence level P=0.05 or 0.01 deemed significant.
Supplementary Material
Acknowledgments
This work was supported by Science Foundation Ireland (SFI) 13/SIRG/2144 (RID), NSF CBET-1254743 ‘Career award’ (YC) and National Institutes of Health (NIH) NS084818 (RSE) grants. We thank Dr. C. Waeber and E. Keomani (School of Pharmacy, University College Cork) for help with brain slice culture, and B. Okura, T. Sakuraba and K. Tsubokura (Sci-Media Company) for help with MiCAM-02 data analysis and Evgenia Dmitrieva-Okkelman for inspiration.
Abbreviations
- FI3
BODIPY-based K+-selective fluoroionophore
- FLIM
fluorescence lifetime imaging microscopy
- EEG
electroencephalogram
- NP
nanoparticle
- ROI
region of interest
Footnotes
Supporting Information
Supporting information is available from the Wiley Online Library of from the author.
Competing financial interests
None declared.
References
- 1.a) Burnell JM, Villamil MF, Uyeno BT, Scribner BH. Journal of Clinical Investigation. 1956;35:935. doi: 10.1172/JCI103352. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Post R, Merritt C, Kinsolving CR, Albright C. Journal of Biological Chemistry. 1960;235:1796. [PubMed] [Google Scholar]
- 2.a) Bellot-Saez A, Kékesi O, Morley JW, Buskila Y. Neuroscience & Biobehavioral Reviews. 2017;77:87. doi: 10.1016/j.neubiorev.2017.03.002. [DOI] [PubMed] [Google Scholar]; b) Shieh CC, Coghlan M, Sullivan JP, Gopalakrishnan M. Pharmacological reviews. 2000;52:557. [PubMed] [Google Scholar]
- 3.Agarwal R, Afzalpurkar R, Fordtran JS. Gastroenterology. 1994;107:548. doi: 10.1016/0016-5085(94)90184-8. [DOI] [PubMed] [Google Scholar]
- 4.Lindinger MI. Journal of molecular and cellular cardiology. 1995;27:1011. doi: 10.1016/0022-2828(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 5.Jørgensen P. Physiological Reviews. 1980;60:864. doi: 10.1152/physrev.1980.60.3.864. [DOI] [PubMed] [Google Scholar]
- 6.Comes N, Serrano-Albarrás A, Capera J, Serrano-Novillo C, Condom E, Cajal SRy, Ferreres JC, Felipe A. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2015;1848:2477. doi: 10.1016/j.bbamem.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 7.Kitazono T, Faraci FM, Taguchi H, Heistad DD. Stroke. 1995;26:1713. doi: 10.1161/01.str.26.9.1713. [DOI] [PubMed] [Google Scholar]
- 8.a) Zhou X, Su F, Tian Y, Youngbull C, Johnson RH, Meldrum DR. Journal of the American Chemical Society. 2011;133:18530. doi: 10.1021/ja207345s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang Y, Huang J, Yang X, Quan K, Xie N, Ou M, Tang J, Wang K. Chemical Communications. 2016;52:11386. doi: 10.1039/c6cc05379c. [DOI] [PubMed] [Google Scholar]; c) Padmawar P, Yao X, Bloch O, Manley GT, Verkman A. Nature Methods. 2005;2:825. doi: 10.1038/nmeth801. [DOI] [PubMed] [Google Scholar]; d) Yaron J, Gangaraju S, Rao M, Kong X, Zhang L, Su F, Tian Y, Glenn H, Meldrum D. Cell death & disease. 2015;6:e1954. doi: 10.1038/cddis.2015.277. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yaron JR, Rao MY, Gangaraju S, Zhang L, Kong X, Su F, Tian Y, Glenn HL, Meldrum DR. Biochemical and biophysical research communications. 2016;472:545. doi: 10.1016/j.bbrc.2016.03.021. [DOI] [PubMed] [Google Scholar]; f) Depauw A, Dossi E, Kumar N, Fiorini-Debuisschert C, Huberfeld G, Ha-Thi MH, Rouach N, Leray I. Chemistry-A European Journal. 2016;22:14902. doi: 10.1002/chem.201602209. [DOI] [PubMed] [Google Scholar]; g) Müller BJ, Borisov SM, Klimant I. Advanced Functional Materials. 2016;26:7697. [Google Scholar]; h) Song G, Sun R, Du J, Chen M, Tian Y. Chemical Communications. 2017;53:5602. doi: 10.1039/c7cc00598a. [DOI] [PubMed] [Google Scholar]
- 9.a) Specht EA, Braselmann E, Palmer AE. Annual Review of Physiology. 2017;79:93. doi: 10.1146/annurev-physiol-022516-034055. [DOI] [PubMed] [Google Scholar]; b) Cella Zanacchi F, Lavagnino Z, Perrone Donnorso M, Del Bue A, Furia L, Faretta M, Diaspro A. Nat Meth. 2011;8:1047. doi: 10.1038/nmeth.1744. [DOI] [PubMed] [Google Scholar]; c) Sarder P, Maji D, Achilefu S. Bioconjugate chemistry. 2015;26:963. doi: 10.1021/acs.bioconjchem.5b00167. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Conway JRW, Warren SC, Timpson P. Methods. 2017;128:78. doi: 10.1016/j.ymeth.2017.04.014. [DOI] [PubMed] [Google Scholar]
- 10.a) Wolfbeis OS. Angewandte Chemie International Edition. 2013;52:9864. doi: 10.1002/anie.201305915. [DOI] [PubMed] [Google Scholar]; b) Wolfbeis OS. Chemical Society Reviews. 2015;44:4743. doi: 10.1039/c4cs00392f. [DOI] [PubMed] [Google Scholar]
- 11.Dmitriev RI, Borisov SM, Düssmann H, Sun S, Müller BJ, Prehn J, Baklaushev VP, Klimant I, Papkovsky DB. ACS Nano. 2015;9:5275. doi: 10.1021/acsnano.5b00771. [DOI] [PubMed] [Google Scholar]
- 12.Borisov SM, Mayr T, Mistlberger G, Waich K, Koren K, Chojnacki P, Klimant I. Talanta. 2009;79:1322. doi: 10.1016/j.talanta.2009.05.041. [DOI] [PubMed] [Google Scholar]
- 13.a) Nazarenus M, Zhang Q, Soliman MG, del Pino P, Pelaz B, Carregal-Romero S, Rejman J, Rothen-Rutishauser B, Clift MJD, Zellner R, Nienhaus GU, Delehanty JB, Medintz IL, Parak WJ. Beilstein Journal of Nanotechnology. 2014;5:1477. doi: 10.3762/bjnano.5.161. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pelaz B, Jaber S, de Aberasturi DJ, Wulf V, Aida T, de la Fuente JM, Feldmann J, Gaub HE, Josephson L, Kagan CR, Kotov NA, Liz-Marzán LM, Mattoussi H, Mulvaney P, Murray CB, Rogach AL, Weiss PS, Willner I, Parak WJ. ACS Nano. 2012;6:8468. doi: 10.1021/nn303929a. [DOI] [PubMed] [Google Scholar]
- 14.Nel AE, Mädler L, Velegol D, Xia T, Hoek EM, Somasundaran P, Klaessig F, Castranova V, Thompson M. Nature materials. 2009;8:543. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 15.a) Sapsford KE, Algar WR, Berti L, Gemmill KB, Casey BJ, Oh E, Stewart MH, Medintz IL. Chemical reviews. 2013;113:1904. doi: 10.1021/cr300143v. [DOI] [PubMed] [Google Scholar]; b) Algar WR, Prasuhn DE, Stewart MH, Jennings TL, Blanco-Canosa JB, Dawson PE, Medintz IL. Bioconjugate Chemistry. 2011;22:825. doi: 10.1021/bc200065z. [DOI] [PubMed] [Google Scholar]
- 16.Pignatello R, Ferro M, De Guidi G, Salemi G, Vandelli MA, Guccione S, Geppi M, Forte C, Puglisi G. International Journal of Pharmaceutics. 2001;218:27. doi: 10.1016/s0378-5173(01)00597-x. [DOI] [PubMed] [Google Scholar]
- 17.a) Fercher A, Borisov SM, Zhdanov AV, Klimant I, Papkovsky DB. Acs Nano. 2011;5:5499. doi: 10.1021/nn200807g. [DOI] [PubMed] [Google Scholar]; b) Kondrashina AV, Dmitriev RI, Borisov SM, Klimant I, O’Brien I, Nolan YM, Zhdanov AV, Papkovsky DB. Advanced Functional Materials. 2012;22:4931. [Google Scholar]; c) Aigner D, Dmitriev RI, Borisov SM, Papkovsky DB, Klimant I. Journal of Materials Chemistry B. 2014;2:6792. doi: 10.1039/c4tb01006j. [DOI] [PubMed] [Google Scholar]; d) Dmitriev RI, Papkovsky DB. Methods and Applications in Fluorescence. 2015;3:034001. doi: 10.1088/2050-6120/3/3/034001. [DOI] [PubMed] [Google Scholar]
- 18.a) Dmitriev RI, Kondrashina AV, Koren K, Klimant I, Zhdanov AV, Pakan JMP, McDermott KW, Papkovsky DB. Biomaterials Science. 2014;2:853. doi: 10.1039/c3bm60272a. [DOI] [PubMed] [Google Scholar]; b) Dmitriev RI, Zhdanov AV, Nolan YM, Papkovsky DB. Biomaterials. 2013;34:9307. doi: 10.1016/j.biomaterials.2013.08.065. [DOI] [PubMed] [Google Scholar]
- 19.Dmitriev R, Borisov S, Kondrashina A, Pakan JP, Anilkumar U, Prehn JM, Zhdanov A, McDermott K, Klimant I, Papkovsky D. Cell Mol Life Sci. 2015;72:367. doi: 10.1007/s00018-014-1673-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Napp J, Behnke T, Fischer L, Würth C, Wottawa M, Katschinski DrM, Alves F, Resch-Genger U, Schäferling M. Analytical chemistry. 2011;83:9039. doi: 10.1021/ac201870b. [DOI] [PubMed] [Google Scholar]; b) Karamchand L, Kim G, Wang S, Hah HJ, Ray A, Jiddou R, Koo Lee Y-E, Philbert MA, Kopelman R. Nanoscale. 2013;5:10327. doi: 10.1039/c3nr00908d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Koo Lee Y-E, Ulbrich EE, Kim G, Hah H, Strollo C, Fan W, Gurjar R, Koo S, Kopelman R. Analytical chemistry. 2010;82:8446. doi: 10.1021/ac1015358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenkins J, Borisov SM, Papkovsky DB, Dmitriev RI. Analytical Chemistry. 2016;88:10566. doi: 10.1021/acs.analchem.6b02675. [DOI] [PubMed] [Google Scholar]
- 22.Mahe MM, Aihara E, Schumacher MA, Zavros Y, Montrose MH, Helmrath MA, Sato T, Shroyer NF. Current protocols in mouse biology. 2013;217 doi: 10.1002/9780470942390.mo130179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Okkelman IA, Dmitriev RI, Foley T, Papkovsky DB. PLOS ONE. 2016;11:e0167385. doi: 10.1371/journal.pone.0167385. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Okkelman IA, Foley T, Papkovsky DB, Dmitriev RI. Biomaterials. 2017;146:86. doi: 10.1016/j.biomaterials.2017.08.043. [DOI] [PubMed] [Google Scholar]; c) Okkelman IA, Foley T, Papkovsky DB, Dmitriev RI. Multi-Parametric Live Cell Microscopy of 3D Tissue Models. Springer; 2017. p. 85. [Google Scholar]
- 24.a) Romero G, Qiu Y, Murray RA, Moya SE. Macromolecular bioscience. 2013;13:234. doi: 10.1002/mabi.201200235. [DOI] [PubMed] [Google Scholar]; b) Sulheim E, Baghirov H, Haartman E, Bøe A, Åslund AK, Mørch Y, de Lange Davies C. Journal of nanobiotechnology. 2016;14:1. doi: 10.1186/s12951-015-0156-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dmitriev R, Papkovsky D. In: Neuronal Cell Death. Lossi L, Merighi A, editors. Vol. 1254. Springer; New York: 2015. p. 55. Ch. 5. [Google Scholar]
- 26.Ivanov AI. Exocytosis and Endocytosis. Springer; 2008. p. 15. [Google Scholar]
- 27.Ringsdorf H. Presented at Journal of Polymer Science: Polymer Symposia. 1975;135 [Google Scholar]
- 28.Müller BJ, Rappitsch T, Staudinger C, Rüschitz C, Borisov SM, Klimant I. Analytical Chemistry. 2017;89:7195. doi: 10.1021/acs.analchem.7b01373. [DOI] [PubMed] [Google Scholar]
- 29.Bestvater F, Spiess E, Stobrawa G, Hacker M, Feurer T, Porwol T, Berchner-Pfannschmidt U, Wotzlaw C, Acker H. Journal of microscopy. 2002;208:108. doi: 10.1046/j.1365-2818.2002.01074.x. [DOI] [PubMed] [Google Scholar]
- 30.Pawlicki M, Collins HA, Denning RG, Anderson HL. Angewandte Chemie International Edition. 2009;48:3244. doi: 10.1002/anie.200805257. [DOI] [PubMed] [Google Scholar]
- 31.Tsytsarev V, Arakawa H, Borisov S, Pumbo E, Erzurumlu RS, Papkovsky DB. Journal of neuroscience methods. 2013;216:146. doi: 10.1016/j.jneumeth.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kocsis J, Malenka R, Waxman S. The Journal of Physiology. 1983;334:225. doi: 10.1113/jphysiol.1983.sp014491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grinvald A, Hildesheim R. Nature Reviews Neuroscience. 2004;5:874. doi: 10.1038/nrn1536. [DOI] [PubMed] [Google Scholar]
- 34.Dmitriev RI, Ropiak HM, Yashunsky DV, Ponomarev GV, Zhdanov AV, Papkovsky DB. FEBS Journal. 2010;277:4651. doi: 10.1111/j.1742-4658.2010.07872.x. [DOI] [PubMed] [Google Scholar]
- 35.Zhdanov A, Dmitriev R, Papkovsky D. Cell Mol Life Sci. 2011;68:903. doi: 10.1007/s00018-010-0502-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dmitriev RI, Papkovsky DB. FEBS Letters. 2015;589:138. doi: 10.1016/j.febslet.2014.11.038. [DOI] [PubMed] [Google Scholar]
- 37.Tsytsarev V, Maslov KI, Yao J, Parameswar AR, Demchenko AV, Wang LV. Journal of neuroscience methods. 2012;203:136. doi: 10.1016/j.jneumeth.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhdanov AV, Okkelman IA, Golubeva AV, Doerr B, Hyland NP, Melgar S, Shanahan F, Cryan JF, Papkovsky DB. Cell Mol Life Sci. 2017;74:141. doi: 10.1007/s00018-016-2323-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.