

Abstract
Here we present a mathematical model of movement in an abstract space representing states of cellular differentiation. We motivate this work with recent examples that demonstrate a continuum of cellular differentiation using single cell RNA sequencing data to characterize cellular states in a high-dimensional space, which is then mapped into or with dimension reduction techniques. We represent trajectories in the differentiation space as a graph, and model directed and random movement on the graph with partial differential equations. We hypothesize that flow in this space can be used to model normal and abnormal differentiation processes. We present a mathematical model of hematopoeisis parameterized with publicly available single cell RNA-Seq data and use it to simulate the pathogenesis of acute myeloid leukemia (AML). The model predicts the emergence of cells in novel intermediate states of differentiation consistent with immunophenotypic characterizations of a mouse model of AML.
Keywords: diffusion mapping, hematopoiesis, single cell RNA-Sequencing, developmental trajectories, nonlinear dimension reduction, cellular differentiation, acute myeloid leukemia, AML, differentiation continuum
1. Introduction
The recent advance of single cell RNA sequencing (scRNA-Seq) technologies has enabled a new, high-dimensional definition of cell states. In contrast to conventional gene expression analyses based on measuring the average levels in a tissue or given cell population, single cell analysis captures the cellular heterogeneity and provides resolution at the level of individual cells within the tissue or cell population. This level of resolution coupled with genome wide gene expression provides an unprecedented opportunity to quantitatively probe cellular behavior, cellular variation and dynamics in a wide range of biological contexts.
There are on the order of 20,000 protein encoding genes that compose the transcriptome, which constitute a dimensional space. Therefore, the configuration of the transcriptome at a point in time can be represented as a coordinate vector in space. When a cell expresses genes, it “moves” in this high-dimensional gene expression phenotype space. Over time, the sequence of locations in the space of a given cell creates a trajectory. Dimension reduction techniques are commonly used to map the larger space into a lower dimensional space, for instance, or , at which point the cells are clustered based on a similarity metric and recategorized. This process has revealed a continuum of cell phenotypes, with intermediate states connecting canonical cell states. The most prominent example of this process is in hematopoietic cell differentiation.
Normal hematopoiesis is long thought to occur through stepwise differentiation of hematopoietic stem cells following a tree-like hierarchy of discrete multipotent, oligopo-tent and then unipotent lineage-restricted progenitors (Figure 1A). The classical model of hematopoiesis considers differentiation as a stepwise process of binary branching decisions, famously represented as a potential landscape by Waddington (Waddington 1957). However, this model is based on bulk characterization of prospectively purified immunophenotypic cell populations. Recent advances in scRNA-Seq technologies now allow resolution of single cell heterogeneity and reconstruction of differentiation trajectories which have been applied to a number of different cellular systems, from hematopoiesis to breast endothelial cell differentiation (Hamey et al. 2016; Velten et al. 2017; Bach et al. 2017; Nestorowa et al. 2016a).
Figure 1.
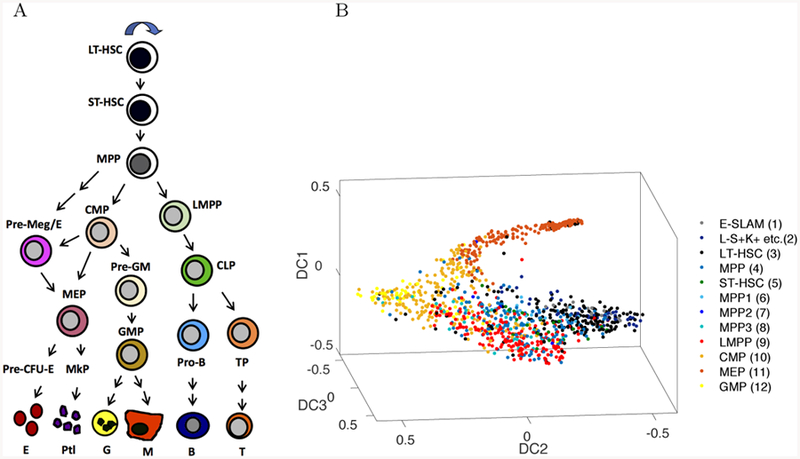
A) Classic representation of a linear hierarchy of discrete cell states, from long-term hematopoietic stem cell (LT-HSC), short-term (ST)-HSC, multipotent progenitor (MPP) to committed common myeloid progenitor (CMP), pre-megakaryocyte/erythrocyte (Pre-Meg/E) and megakaryocyte-erythroid progenitor (MEP),pre-granulocyte/macrophage (Pre-GM), granulocyte-macrophage progenitor (GMP), and lymphoid primed-MPP (LMPP), common lymphoid progenitor (CLP) cells, on down to terminally differentiated cells such as erythrocytes (E) platelets (Plt), granulocytes (G) macrophages (M), B and T cells. B) The classical view is contrasted with a nonlinear continuum representation of hematopoietic cell differentiation states using diffusion map dimension reduction of scRNA-Seq data (figure recreated from data available in Nestorowa et al. (2016a)). Colors representing cell identities in A) and B) are coordinated. Cell types in B) are a subset of cells represented in A).
These efforts have led to the new view that hematopoietic lineage differentiation occurs as a continuous process, which can be mapped into a continuum of cellular and molecular phenotypes (Figure 1B). Hematopoietic malignancies such as acute myeloid leukemia (AML) arise from dysregulated differentiation and proliferation of hematopoietic stem cells and progenitor cells upon accumulation of oncogenic genetic mutations and/or epigenetic alterations. Therefore, characterizing disordered hematopoiesis based on discretely defined phenotypic populations can be challenging. Moreover, “discrete” phenotypic cell populations are in fact highly heterogeneous in terms of functional capacity and gene expression profiles. It is now possible to view pathologic hematopoiesis through a continuum of cellular and molecular phenotypes and capture the heterogeneity, differentiation plasticity and dysregulated gene expression associated with malignant transformation.
This new view of biology forces us to reconsider the mathematical approaches we use to model cell states and behaviors. Instead of building mathematical models which identify discrete cell populations and assign mathematical rules for their evolution and interactions, we may now consider a continuum of cellular states, and model movement between these states in aggregate as a flow of mass on a structured graph. Modeling differentiation in this manner reduces the number of parameters and thus the complexity of the mathematical model by representing many cell populations and states in a single variable. At the same time this increases biological resolution of the system by characterizing an infinite number of sub-states in a continuum representation. Here we consider a model of hematopoietic cell differentiation and associated disorders as a flow and transport process in a continuous differentiation space as a test system for a more general approach of modeling the temporal evolution of a continuum of cell states.
This manuscript is structured as follows: first, we review the state of the art of dimension reduction methods that are used to construct and define hematopoietic differentiation spaces that can be represented as graphs, including a review of Schienbinger et al.’s method for modeling transport on a graph from reduced dimension gene expression data (Schiebinger et al. 2017). Then we introduce our partial differential equation (PDE) model of flow and transport on a graph, and illustrate the model on simple “Y” shaped graph with symmetric and asymmetric differentiation. We then calibrate our model to a graph (Figure 3) constructed from publicly available scRNASeq data of normal hematopoiesis. Finally, we use our model to simulate abnormal hematopoietic cell differentiation processes observed during the pathogenesis of AML, a form of aggressive hematologic malignancy. We conclude with a brief discussion of prior literature on modeling differentiation as a continuum, and the limitations and potential future applications of this modeling approach.
Figure 3.
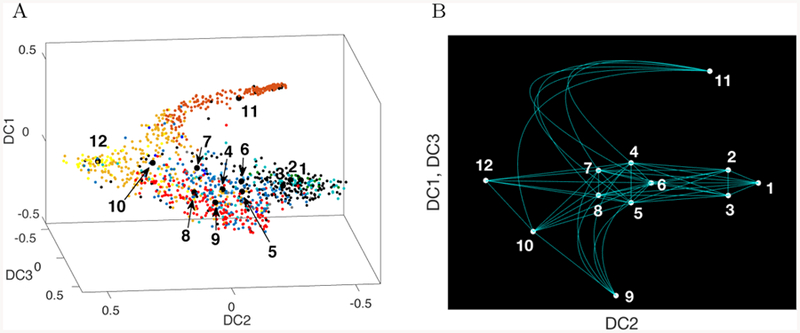
A) For the 12 cell types idenfied in Nestorowa et al. (2016a), the center of mass of each cell type is used to define a node on an abstracted graph B). Edges between nodes are constructed based on inferred trajectories on the graph based on diffusion pseudotimes starting from nodes 1, 2, and 3, to nodes 4–8, then to the progenitor nodes 9–12. The graph represents a continuum of cell states (edges) that includes identification of canonical cell states along the continuum (nodes 1–12) (Table 1).
2. Construction of a differentiation continuum
In order to describe the entire modeling process, in this section we briefly describe methods for reducing the dimension of high dimensional scRNA-data, before reviewing pseudotime reconstruction techniques, and conclude this section by examining a technique from Schiebinger et al. (2017) for construction of a directed graph that represents hematopoietic differentiation space. While the focus of this paper is not dimension reduction techniques or pseudotime reconstruction, we summarize some of these techniques that are most relevant to our modeling approach, without advocating for one over another. We should emphasize that this is a review of already existing algorithms; the novel work begins in Section 3. The relationship between time and pseudotime within a mathematical model of cell differentiation is analogous to the relationship between age structured and stage structured models in ecology. Cell differentiation data yield information about cells at various stages of differentiation, but generally do not provide time-specific data. A pseudotime model is one that considers the differentiation stage of a cell population instead of the time in which a cell is in a certain state.
In Figure 2, we lay out the steps required for going from high dimensional data to construction of the PDE model. Section 2.1 will review various dimension reduction techniques, including a more thorough discussion of the technique used in our application, diffusion mappings. Section 2.2 summarizes techniques such as Wishbone and Wanderlust, that are available for pseudotime reconstruction given dimension reduced data. And finally, Section 2.3 will give an overview of the technique presented in Schiebinger et al. (2017) for construction of a directed graph that indicates how cell populations evolve in pseudotime.
Figure 2.

Flow chart of our modeling process: This chart organizes the steps taken toward constructing the PDE model. First, high-dimensional data such as single cell RNA-Sequencing (scRNA-Seq) are represented in 2- or 3-dimensional space through one of many dimension reduction techniques. Then, temporal events (pseudotime trajectories) are inferred from the dimension reduced reduced data. We then use the reduced dimension representation and pseudotime trajectories to model flow and transport in the reduced space. In Section 2, we summarize dimension reduction techniques and reconstructing pseudotime trajectories. In Section 4 we show the results of our modeling. Data is from Nestorowa et al. (2016a).
2.1. Dimension reduction techniques
A broad range of techniques have been developed to provide insight into interpretation of high dimensional biological data. These techniques provide a first step in our approach to modeling the evolution of cell states in a continuum and play a critical role in characterizing differentiation dynamics. We note that the application of different data reduction techniques, clustering methods, and pseudotime ordering on the same data set will produce different differentiation spaces on which to build a dynamic model. We will use one particular dimension reduction approach as an example, but our framework allows one to select from a variety of approaches. In this section we provide a brief description of a subset of such techniques to give the reader a sense of the field.
Several techniques have been developed to interpret the high-dimensional differentiation space, including principal component analysis (PCA), diffusion maps (DM) and t-distributed stochastic neighbor embedding (t-SNE). Each of these methods map high-dimensional data into a lower dimensional space. As discussed in this section, different techniques produce different shapes and differentiation spaces, and so some techniques are better suited to certain data sets than others. For instance, one commonly used dimension reduction technique is principal component analysis (PCA), a linear projection of the data. While PCA is computationally simple to implement, the limitation of this approach lies in its linearity - the data will always be projected onto a linear subspace of the original measurement space. If the data shows a trend that does not lie in a linear subspace—for instance, if the data lies on an embedding of a lower-dimensional manifold in Euclidean space that is not a linear subspace —then this trend will not be e ciently captured with PCA (Khalid, Khalil, and Nasreen 2014).
In contrast, diffusion mapping (DM) and t-stochastic neighbor embedding (t-SNE), as well as a variant of t-SNE known as hierarchical stochastic neighbor embedding (HSNE), are non-linear dimension reduction techniques. t-SNE, introduced by Maaten and Hinton (2008) is a machine learning dimension reduction technique that is particularly good at mapping high dimensional data into a two or three dimensional space, allowing for the data to be visualized in a scatter plot.
Given a data set in , we can transform the Euclidean distances between two points into a probability distribution. Intuitively, this distribution gives the probability that data point xj is a neighbor of point xi, where the probability of being a neighbor of xi has a Guassian distribution (Maaten and Hinton (2008)):
| (1) |
The t-SNE algorithm aims to find a map from the data set to two or three dimensional Euclidean space that minimizes the Kullback-Leibler divergence between the probability distributions in the original and reduced space. This optimization problem is often solved using gradient descent methods.
In van Unen et al. (2017), a new technique for examining high dimensional mass cytometry data, known as hierarchical stochastic neighbor embedding (HSNE) is presented. Mass cytometry allows for the examination of several cellular markers on samples made up of vast quantities of cells. These data sets are truly “big” in the sense that they are very large (a sample for each cell) as well has high-dimensional. Therefore, pre-existing dimension reduction techniques are not optimal for mass cytometry data. HSNE, as suggested by its name, is hierarchical by nature, allowing for refinement in the level of detail. HSNE ultimately constructs a hierarchy of subsets of the dataset X:
The hierarchy begins with the data set itself . A weighted k-nearest neighbor (kNN) graph is constructed on the data set, and individual points, or “landmarks” are selected from each node on the graph to represent the data set at the next, coarser, level, . This process is repeated as desired. These subsets can each be embedded in lower dimensional space. This hierarchical embedding scheme allows the user to view the data at different resolutions, from a broad overview (level ) to a more refined understanding of cell types associated with markers (intermediate levels). Starting with a certain subset , the user is able to “drill in” to the data by selecting a subset . Thus, HSNE is an approach that is useful for data that requires different levels of detail at different scales. An illuminating graphical representation of the HSNE process can be found in van Unen et al. (2017), Figure 1.
Diffusion maps work by taking advantage of the relationship between heat diffusion and random walk Markov chains. Let X be a data set of size n. The diffusion map algorithm begins by considering a kernel function on pairs of data points; this function must be symmetric and nonnegative. The Gaussian kernel
is one popular choice. Similar to the conditional probability defined in (1), the kernel k(x, y) is used to specify the probability of going from x to y in one step of a random walk on the data, found by normalizing the kernel to ensure the random walk probabilities integrate to 1:
By letting the number of steps in this random walk go to infinity, we can consider the stationary distribution pt of the Markov chain. This stationary distribution is used to formulate a new metric on the data space, known as the diffusion distance:
Intuitively speaking, the diffusion distance between two points will be low if there are many paths in the random walk that connect them, and high if there are few. Because it is computationally expensive to repeatedly compute the diffusion distance between each pair of points, it is easier to map data points to a new Euclidean space using the function defined as:
The Euclidean distance in this space, known as the Diffusion Space, is then equivalent to the Diffusion Distance in the data space. It can be demonstrated that the linearly independent eigenvectors of the diffusion matrix (the transition matrix associated with the aforementioned Markov Chain) form a basis for the diffusion space. Therefore, by opting to keep the k-eigenvectors corresponding to the k largest eigenvalues, we obtain a map from the original data to a k-dimensional subspace of the diffusion space that most e ciently captures the structure of the data; this map is called the diffusion map. A more in depth explanation can be found in Coifman et al. (2005).
Each of these dimension reduction methods has strengths and weaknesses depending on the question(s) being asked of the data. Moreover, each method will produce a distinctly different shape in the lower dimensional representation. Therefore, the choice of dimension reduction technique is a critical step in analyzing any high-dimensional data set. For the purpose of analyzing cell transition probabilities and inferring trajectories within the reduced space, Nestorowa et al. (2016a) and others have chosen to use diffusion mapping to analyze cell differentiation.
2.2. Pseudotime ordering of differentiation states
For data without temporal information, pseudotime methods are available to infer a sequence of biological states from single time point data. Diffusion mapping can be used to infer a “diffusion pseudotime” (Haghverdi et al. 2016; Nestorowa et al. 2016a). In particular, Haghverdi et al. (2016) develops an e cient diffusion pseudotime approach by rescaling the diffusion components by a weighted distance in terms of the eigenvalues, derived by considering a random walk according to a transition matrix that specifies the probability of transitioning from any single cell to another in an infinitesimal amount of time. Alternative pseudotime approaches include Wishbone (Setty et al. 2016) that uses shortest paths in a k-nearest neighbor (kNN) graph constructed in diffusion component space to construct an initial ordering of cells, TASIC (Rashid, Kotton, and Bar-Joseph 2017) that is able to incorporate time information and identify branches and incorporate time information in single cell expression data by considering it as developmental processes emitting expression profiles from a finite number of states, and Monocle (Qiu et al. 2017b,a) that fits a principal graph (Mao et al. 2015) and uses a reversed graph embedding technique which simultaneously learns a low dimensional embedding of the data and a graphical structure spanning the dataset.
Finally, when the data are collected at multiple time points, the transition rates between the nodes can be obtained after partitioning the cell data. For instance, Schiebinger et al. (2017) employs graph clustering (Levine et al. 2015; Shekhar et al. 2016) and optimal transport methods to understand the dynamics in the reduced space of cell data. We describe the optimal transport (OT) method in an effort to provide a clear distinction between the OT algorithm and our modeling approach.
2.3. Optimal transport
Schiebinger et al. (2017) have proposed a model and algorithm for constructing a directed graph oriented in pseudotime given temporal data. The optimal transport algorithm itself is a classical problem studied in the mathematical area of Transportation Theory, which aims to optimally transport and allocated resources given certain cost functions. Schiebinger et al. (2017) apply this theory to a time series of reduced dimension single cell gene expression profiles. The time series is made up of a sequence of samples {S1, …, Sn}, at different times ti for i ∈ {1, …, n}. Suppose that each sample consists of points in . A distribution is defined by each sample Si. For each set :
Where δx represents a Delta Distribution centered at x:
Together, as a sequence, these inferred distributions form what is known as an “empirical developmental process.” The goal is then to determine, as closely as possible, what the true underlying Markov developmental process is by examining what are known as transport maps between pairs and . A transport map for a pair is a distribution π defined on such that and are the two marginal distributions of π. Thus, given a function c(x, y) that represents the cost to transport some unit mass from x to y, the goal is to minimize
subject to
Schienbinger et al. further refine this algorithm by including a growth term in their transport plan to allow for cellular proliferation between time points. This differs from the classical optimal transport algorithm in that the classical optimal transport algorithm is formulated with conservation of mass in mind. Optimal transport can thus be used to estimate the ancestors and descendants of a set of cells. Cells are clustered using the Louvain-Jaccard community detection algorithm on the reduced dimension gene expression data in 20 dimensional space. Schienbinger et al. thus identified 33 cell nodes, which were then used as starting populations from which developmental trajectories could be analyzed. These can be thought of as nodes on a graph visualized with force-directed layout embedding, and edges represent the motion in pseudotime.
In the following section, we assume that the first two steps in Figure 2 have been completed by one of the methods described above. In other words, we start with samples in high-dimensional space, we map the data to a lower dimensional space, and then we produce pseudotime trajectories in this lower dimensional space. In the final step, we model the differentiation process in continuous (pseudo)-time and (reduced-dimensional) space using partial differential equations.
3. Modeling on the differentiation continuum
To illustrate our modeling technique, we assume that we have constructed a simple branched manifold or graph situated in the differentiation space. This graph is not a set of discrete nodes, rather, the graph and its edges represent a continuum of canonical states and intermediate states of differentiation. Assuming that the graph and the temporal evolution on the graph is obtained by any one of the various data analysis techniques summarized in Section 2 including optimal transport (Schiebinger et al. 2017), diffusion pseudotime methods (Haghverdi et al. 2016; Nestorowa et al. 2016a; Haghverdi et al. 2016), Wishbone (Setty et al. 2016), TASIC (Rashid, Kotton, and Bar-Joseph 2017), and Monocle (Qiu et al. 2017b,a), we develop a PDE model that describes the dynamics in this differentiation continuum. Cell differentiation models in the continuous space have been developed in (Gwiazda, Grzegorz, and Marciniakczochra 2012; Doumic et al. 2011) that extends the discrete multi-compartment models (Lander et al. 2009; Lo et al. 2008; Marciniak-Czochra et al. 2009; Stiehl and Marciniak-Czochra 2011).
3.1. PDE model on a graph
Let us define the graph G obtained in the differentiation continuum space. We comment that although we can consider a cell distribution on the actual reduced space, we further reduce our model on a graph that makes it convenient to employ the biological insights from the classical discrete models. The node set of G is denoted as where nv is the total number of nodes, and the edge of the graph connecting in the direction from the i-th to the j-th node as eij. We also introduce an alternate description of the graph with respect to the edge, that is more convenient for describing the PDE model. If the total number of nontrivial edges is ne, we take with the index mapping on the set of nontrivial edges , and the end points in the direction of cell transition as and ,respectively. We remark that .
We denote u(x, t) as the cell distribution on the graph G at the differentiation continuum space location x ∈ G and time (or pseudotime) t. Thus, we follow the dynamics of the cell density at x ∈ G. We annotate the cell distribution on each edge ek as uk(x, t) such that and model the cell density by an advection-diffusion-reaction equation (Evans 2010) as
| (2) |
where x is a one dimensional variable parameterized on each edge ek from ak to bk. The advection coeffcient Vk(x) models the cell differentiation and the transition between the different cell types, that is, the nodes. The transition rate per unit time (e.g., day−1) or pseudotime can be taken as Vk(x) computed using the periods of cell differentiation. For instance, Vk(x) can be computed by smoothly interpolating the speed of cell differentiation from the multi-compartment discrete models as Vk(x) = , where cn is the differentiation rate of cell type vn and is an interpolation function1
Cell proliferation and apoptosis can be modeled by the reaction coeffcient Rk(x). Similar to Vk, if only the proliferation at the discrete cell types are available, we interpolate as , where rn is the growth rate at node vn. In addition to natural proliferation and apoptosis, this term can also model abnormal tumorous cell growth or the effect of targeted therapy by localized Gaussian or Dirac-delta functions centered at the location of the corresponding cell type on the graph.
The diffusion term represents the instability on the phenotypic landscape of the cells that should be taken account into when modeling the macroscopic cell density. In particular, we consider the diffusion term in Eq. (2) in such form that is appropriate to model the dynamics on a graph that is reduced from a higher-dimensional narrow domain. It involves two parameters Dk(x) and wk(x) describing the magnitude of cell fluctuation and the width of the narrow domain around the edge, respectively. Considering the phenotypic fluctuation of the cell density as a random process subject to Brownian motion with magnitude σ, the diffusion term becomes Dk = σ2 and wk = 1 (Evans 2010). In addition, the width or the area of the cross section of the narrow domain that is vertical to the projecting edge can be taken as wk(x), which is called Fick-Jacobs equation considering deterministic PDEs (Zwanzig 1992; Valero Valdes and Herrera Guzman 2014) and can be similarly derived for stochastic PDEs (Cerrai and Freidlin 2017; Freidlin and Hu 2013). wk(x) can be measured as the length of maximal fluctuation in the vertical direction along the graph.
In addition to the governing equation on the edges, the boundary condition at the nodes are critical when describing the dynamics on the graph. The boundary condition on the cell fate PDE model can be classified into three types, the initial nodes that do not have inflow ,e.g., stem cells, the final nodes without outflow ,e.g., most differentiated cells, and the intermediate nodes,
On the intermediate nodes vn ∈ NT , mixed boundary conditions can be imposed to balance the cell inflow and outflow as
| (3) |
where and is the right end point of the edge between nodes i and n. Similarly, is the left end point of the edge between nodes n and j. In addition, continuity conditions are taken as Dirichlet boundary conditions as
for a fixed n. The cell outflow boundary conditions on the final nodes vn ∈ NF are imposed as reflecting boundary conditions
and u(bI[i,n]) = u(bI[j,n]) for all (i, n) and (j, n) in . Similarly this can be imposed on the initial nodes vn ∈ NI as or depending on whether the model describes the cell inflow flux or a prescribed density.In particular, the prescribed value when v(aI[n,j]) represents the density of stem cell, one can model the discrete stem cell state as a separate ODE and impose its solution as the boundary condition at aI[n,j] (Doumic et al. 2011; Gwiazda, Grzegorz, and Marciniak-czochra 2012). This approach makes it possible to distinguish the stem cell proliferation into the division that remains as stem cell and the one that differentiates to a matured cell.
3.1.1. Example on a Y shaped graph
To illustrate our approach, we apply the PDE model given in (Eq. 2) to a simple Y shaped graph. This example is motivated by cell differentiation data that reveals multiple branching procedures in the continuous space (Haghverdi, Buettner, and Theis 2015; Velten et al. 2017; Moris, Pina, and Arias 2016; Rizvi et al. 2017), therefore we assume the simplest case that the differentiated cells have two different cell fates with one branching. For instance, assume that the cell data projected onto the first two diffusion components, DC1 and DC2, are as in Figure 4A and the temporal direction of cell differentiation is from left to right, as indicated by the arrows in the figure. We define the Y shaped graph with four nodes v1 = (–1, 0), v2 = (0, 0), v3 = (1, 1), and v4 =(1, –1), and three edges , and . This corresponds to the set of nontrivial edges and index mapping I on as I(1, 2) = 1, I(2, 3) = 2, and I(2, 4) = 3, that yields the end points of the edges ak and bk as v1 = a1, v2 = b1 = a2 = a3, v3 = b2, and v4 = b3. For simplicity, we assume that the edges are straight lines and parametrize the variables on each edge as e1(x) = (x 1, 0), e2(x) = (x, x), and e3(x) = (x, −x), so that x ∈ [0, 1]. When there is possibility for confusion, we use subscripts on the x-variables to specify which edge is parametrized. So, for example, x2 parametrizes e2. Then, the PDE model on each parametrized edge ek can be written as
| (4) |
Figure 4.

We use a simple “Y” shaped graph to illustrate our model. A) The graph is defined by four nodes and three edges e1 = eI(1,2), e2 = eI(2,3), and e3 = eI(2,4) within two components of a diffusion map (DC1, DC2). The transfer rate from v2 to v3 and v4 is taken to be proportional to 1 − p and p, respectively. B) The evolution of the cell density solution from the initial condition (t = 0) concentrated at the left end, DC1 = −1, to a density concentrated at the right ends, DC1 = 1, at t = 15. In the symmetric case, p = 0.5, the two branches evolve in the same way; C) in the asymmetric case, p = 0.25, the cell density is larger at t = 15 on the upper branch, shown in blue, compared to the lower branch, shown in red.
We consider the case that the cells transfer from v1 to v2 in nT = 5 unit time, differentiate into each cell type with proportion p and 1–p, and accumulate at DC1 = 1, where the cells are fully differentiated2. Here, we simplify the differentiation rate to be constants assuming that the single branching Y graph lies locally and close enough in the differentiation space that the differentiation rate does not change. Then,
| (5) |
where V2 and V3 reflect the accumulation at cell types v3 and v4 (x = 1). Also, we assume that the differentiation process is subject to fluctuations such as trans-differentiation (cross-lineage) and de-differentiation (stem state reversion) that is modeled as Brownian motion with a constant variance σ so that Dk = σ2 = 0.02. Also, the maximal fluctuation in the vertical direction of the edge is assumed to be a constant that is independent to x and w1 = 2w2 = 2w3 so that the fluctuation in the vertical direction reduces by half in e2 and e3. wk cancels out in the diffusion term in (Eq. 4). Figure 4 plots the two examples of symmetric differentiation p = 0.5 and asymmetric differentiation p = 0.25.
In this example, to demonstrate our model focusing on the cell differentiation and branching, we assume that the proliferation is zero as Rk = 0 (see Appendix A for the detail of modeling Rk). The boundary type of the nodes are classified, according to our description above, as NI = {v1}, NI = {v2} and NI = {v3, v4} Thus, we impose the gluing condition as in Eq. (3) at v2, as
with continuity conditions u1(b1) = u2(a2) = u3(a3). In addition, an inflow boundary condition is imposed at v1, and reflecting boundary conditions at the end nodes v3 and v4 as ,,and The Dirichlet condition of u1(a1, t) is given to resemble the transition of a certain cell state to fully differentiated cells from the initial distribution
Simulations of this simple model are shown in Figure 4, where densities on edges e2 and e3 are plotted in different colors. We see that an initial cell distribution concentrated near the cell state v1 moves to the right as the cells differentiate, branches at v2, and becomes absorbed at the fully differentiated cell states v3 and v4. In the symmetric case, when p = 0.5, the density is the same on each of the two branches to the right of v2, so that the two curves are plotted on top of each other. When p = 0.25, the density profile is not symmetric: more cells move along the upper branch than on the lower branch. This provides a simple illustration of the mathematical details of our modeling framework, which we apply on more complicated graph structured derived from data as follows.
4. Simulation results
In this section, we employ the framework developed in section 3.1 to the mouse hematopoietic stem and progenitor cell data in Nestorowa et al. (2016a). See Appendix A for details, including the model parameters and simulation codes.
4.1. Model of normal adult hematopoiesis
To calibrate our model, we first apply it to normal hematopoietic cell differentiation trajectories identified in Nestorowa et al. (2016a). Nestorowa et al. characterize early stages in hematopoiesis with twelve cell types, shown in Table 1 and Figure 3, including E-SLAM (CD48-CD150+CD45+EPCR+), long-term HSCs (LT-HSCs), short-term HSCs (ST-HSCs), lymphoid-primed multipotent progenitors (LMPPs), multipo-tent progenitors (MPPs), and megakaryocyte-erythroid progenitors (MEPs), common myeloid progenitors (CMPs), and granulocyte-macrophage progenitors (GMPs). We consider these twelve cell types as the twelve nodes, vk, in our graph, and add 51 edges to model the hematopoietic cell hierarchy (see Figure 1A) and pseudotime computed in Nestorowa et al. (2016a) (see Figure 5A). This graph represents a continuum of canonical and intermediate states of hematopoietic differentiation with nodes and edges, respectively. The spatial variable in our PDE model represents the differentiation state of the cell.
Table 1.
Index of cell identities and labels. Long- and short-term hematopoietic stem cells (LT-HSC, ST-HSC); multipoent potent progenitors (MPP), lymphoid-primed multipotent progenitors; common myeloid progenitors (CMP); megakaryocyte-erythroid progenitors (MEP); granulocyte-macrophage progenitors (GMP).
| Cell identities and labels | |||
|---|---|---|---|
| ID | Cell type | ID | Cell type |
| 1 | E-SLAM | 7 | MPP2 |
| 2 | L-S+K+ CD34- Flk2+ CD48-CD150+ |
8 | MPP3 |
| 3 | LT-HSC | 9 | LMPP |
| 4 | MPP | 10 | CMP |
| 5 | ST-HSC | 11 | MEP |
| 6 | MPP1 | 12 | GMP |
Figure 5.
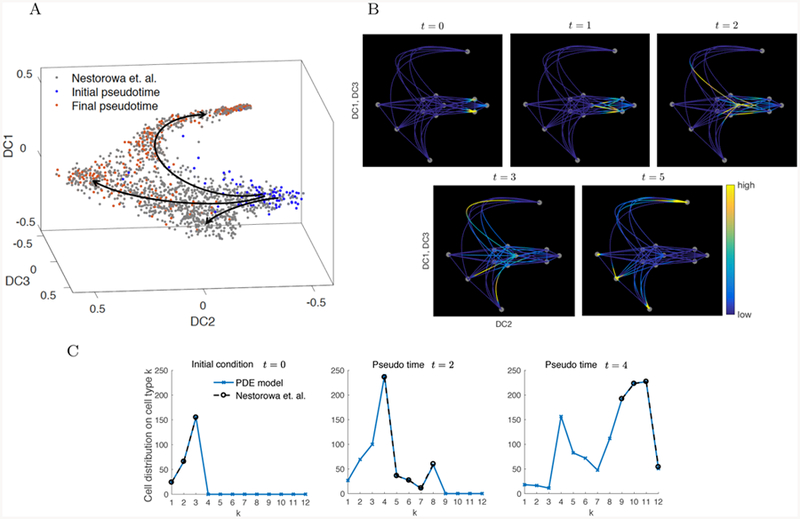
A) The cell data colored by pseudotime analysis produced by the Wanderlust algorithm applied to data mapped to diffusion space in Nestorowa et al. (2016a). The initial point in pseudotime is taken from the HSC cells and the final pseudotime from the progenitor cells. B) Cell distribution computed by the PDE model describing normal conditions on the graph from t = 0 to t = 5. The cells flow from E-SLAM and LT-HSC nodes on the right to the LMPP, CMP, MEP, and GMP nodes on the other three ends (bottom, top, and left), following the pseudotime trajectories identified in A). C) Comparison of the cell type distribution computed by the PDE model described in Eq. (2) and the reference data from Nestorowa et al. (2016a). The reference distribution (Nestorowa et al.) is computed by clustering the initial, middle and final pseudotime cells from A) into 12 cell nodes. By considering t = 4 as the final pseudotime in the PDE model, the values of the solution at the nodes agree well with the reference data.
The colored and labeled clustered cell data and the corresponding graph are shown in Figure 3. The location of the nodes on the graph is not chosen to be identical to the data, but for an illustrative purpose to represent DC2 and DC1/DC3. The edges are chosen according to the pseudotime progression from the E-SLAM and HSCs (nodes 1–3) to the progenitor cells (nodes 9–12).
The parameters of the PDE model of cell differentiation under normal conditions are chosen to reproduce the distribution of cell types from Nestorowa et al. (2016a) at the initial and final pseudotime (Figure 5C). Considering the data in Nestorowa et al. (2016a) grouped by sorting gate of LT-HSC, hematopoietic stem and progenitor cell (HSPC), and progenitor cells, we denote the subsets of nodes that are representative of each group as for HSC, for HSPC, and, for progenitor cell, where we also take , and The reference distribution is computed by counting the relative number of cells in each cluster at the initial and final pseudotime. The initial and final cell distribution is concentrated on nodes 1–3 of and 9–12 of , respectively.
The distribution of cells in the remaining states, represented by nodes 4–8 of , goes from 0 at time t = 0 to positive at time t = 2, and reduces at t = 4. We remark that the ratios of the number of cells in each node are used to compute the advection coeffcients Vk in (Eq. A3), where we take the drift VI[i,j] from cell type i to another other cell types j to be proportional to the ratio plotted in Figure 5C. For instance, the outflow from v5 to nodes 9–12 is taken to be proportional to the reference distribution at pseudotime t = 4. With the ratios fixed, we assume a constant parameter that represents the differentiation rate on each node, and find the values that reproduce the given cell data by simple root finding algorithms such as secant method. The range of the values are 0 ≤ Vk ≤ 3. The detailed procedure is explained in Appendix A.
The diffusion coeffcient is taken as Dk = DI(i,j) = 10−2 within the either subsets of nodes or , and Dk = 10−3 on the other edges. The magnitude Dk = 10−2 corresponds to the phenotypic fluctuation of 2.5456 × 10−2 in the diffusion space and Dk = 10−3 takes into account of the increased average distance between the nodes that yields smaller diffusion coeffcient due to relatively smaller fluctuation. We assume that the proliferation of the progenitor cell nodes are a constant as rn = 1.3648 at t ≤ 2 and rn = 0.4 at t > 2 for , where the proliferation rate reflects the increment of cell number from HSC to progenitor cells in the data. Also, the proliferation at the HSC nodes are assumed to be negligible compared to progenitor cells as (Passegué et al. 2005). See Appendix A for the model parameters and detailed discussion.
For the implementation, we discretize the system using a fourth-order finite difference method and 100 grid points on each edge, and a third-order Runge-Kutta method in time with time step 10−4. Figure 5C compares the solution to the PDE in the normal condition to the reference distribution. The initial condition of the PDE is taken as the initial reference distribution, and we compute the solution up to time t = 5. The solution at t = 4 is similar to the reference distribution at final pseudotime. Also, the solution at t = 2 is close to the distribution of the remaining cells excluding the initial and final cells. Figure 5B shows the cell distribution on the graph from time t = 0 to t = 5. We observe that the support of the cell density shifts from the initial nodes 1–3 representing HSCs, to nodes 9–12 representing progenitor cells.
4.2. Acute myeloid leukemia (AML)
AML results from aberrant differentiation and proliferation of transformed leukemia-initiating cells and abnormal progenitor cells. Parallel to the hierarchy of normal hematopoiesis, malignant hematopoiesis has also been considered to follow a hierarchy of cells at various differentiation states although with certain levels of plasticity (see Figure 6). Given the aberrant differentiation and plasticity associated with the pathology of AML, modeling in a continuous differentiation space o ers the advantage over discrete models that all pathological and intermediate cell states can be captured. With our model calibrated to data obtained from normal hematopoietic differentiation trajectories, we now model the progression of AML using a genetic knock-in mouse model that recapitulates somatic acquisition of a chromosomal rearrangement, inv(16)(p13q22)(Liu et al. 1993, 1996), commonly found in approximately 12 percent of AML cases. Inv(16) rearrangement results in expression of a leukemogenic fusion protein CBFβ-SMMHC, which impairs differentiation of multiple hematopoietic lineages at various stages (Castilla et al. 1999; Kuo et al. 2006; Kuo, Gerstein, and Castilla 2008).
Figure 6.
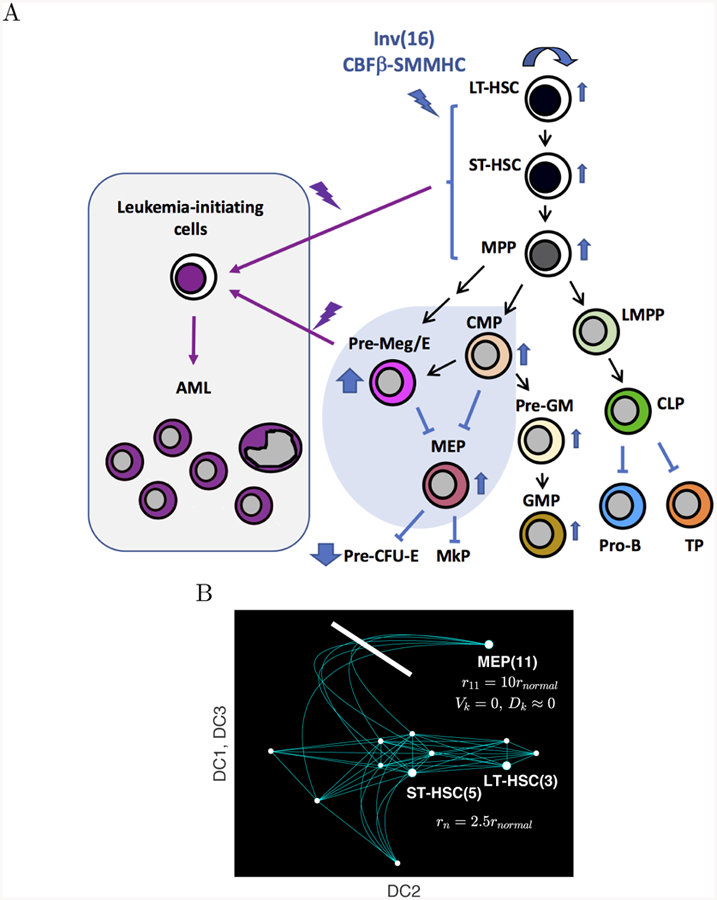
A) Acute myeloid leukemia (AML) is a cancer of aberrant differentiation and proliferation of hematopoietic progenitor cells. Previous studies demonstrated that expression of inv(16) leukemogenic fusion protein CBFβ-SMMHC results differentiation block at multiple hematopoetic stages along with expansion of preleukemic stem/progenitor cells and abnormal myeloid progenitors, including CMP, Pre-Meg/E and MEP. These preleukemic stem/progenitor cells and abnormal myeloid progenitors are susceptible to malignant transformation into leukemia-initiating cells that drive and sustain AML pathogenesis. B) Schematic illustration of AML pathogenesis in the differentiation continuum. To simulate inv(16) driven AML, the proliferation Rk(x) connecting the nodes 3, 5, and 11 is increased and the flow toward the node 11, Vk(x) and Dk(x) for k = I(i, 11) is blocked.
Our previous studies using the inv(16) AML mouse model demonstrate that expression of CBFβ-SMMHC leukemogenic fusion protein results in expansion of preleukemic hematopoietic stem and progenitor populations susceptible to transformation into leukemia-initiating cells which can initiate and propagate AML. Most notable was the increased in abnormal myeloid progenitors, which had an MEP-like immunopheno-type and a CMP-like differentiation potential (Kuo et al. 2006). Further separation of myeloid-erythroid progenitors with additional phenotypic markers (Pronk et al. 2007) show a predominant increase in pre-megakaryocyte/erythroid (Pre-Meg/E) population (ranging from 5 to 12 fold) accompanied by impaired erythroid lineage differentiation (Figure 6A)(Cai et al. 2016). This refined phenotypic Pre-Meg/E population consists partly of the CMP and MEP populations using conventional markers (Akashi et al. 2000)(Nestorowa et al. 2016a).
The simulation of inv(16) initiated AML pathogenesis is motivated by our previous observations that AML is preceded by expansion of preleukemic myeloid progenitor cells, particularly the Pre-Meg/E and MEP-like populations with impaired differentiation. These abnormal progenitors are predisposed to subsequent cooperating events necessary to transform to overt AML (Kuo et al. 2006; Cai et al. 2016; Castilla et al. 1999). To simulate AML pathogenesis, we increase the proliferation rate of MEP (node 11) by 10 times, that is, rI[i,11] = 10rnormal, to reflect the abnormal expansion of MEP-like cells (ranging from 5 to 12 fold based on previous data) (Kuo et al. 2006; Cai et al. 2016). Here, rnormal is the value that is used in the normal condition in section 3.1. Thus, the proliferation is assumed to be maximal at the MEP node, Rk(v11) and the proliferation of intermediate cells that are phenotypically close to MEP, that is, RI[i,11](x) near x = v11, also increase. Also, the flow to the MEP is blocked by taking zero advection coeffcient on the edge that is connected to v11, i.e., VI(i,11)(x) = 0. We also lower diffusion by ten as DI(i,11)(x) = Dnormal/10 to model the phenotypic fluctuations and imperfect differentiation block involved in AML pathogenesis. The differentiation block is imperfect because there is a continuum of leukemic cell pheno-types (states).
In addition, the proliferation rate of LT-HSC and ST-HSC (nodes 3 and 5), that is, r3 and r5, is increased by 2.5 times as 2.5rnormal (Figure 6B). We model the induction of the leukemogenic fusion protein CBFβ-SMMHC resulting from the chromosome inversion inv(16) (p13q22) as the “start of AML.” In this murine model of AML, inv(16) is the initial founder event that results in differentiation block and expansion of abnormal progenitors, which are predisposed to subsequent cooperating events necessary to transform to overt AML (Kuo et al. 2006; Cai et al. 2016; Castilla et al. 1999). The approach used here directly models the sequence of events observed during leukemia initiation. Finally, we denote tAML as the effective time that the aforementioned tenfold proliferation change in MEP and other abnormal differentiation and proliferations due to AML are observed. The other parameters except the ones described in this section follows the ones from section 4.1.
Figure 7 shows the total number of cells in each cell type in normal condition and AML condition starting at t = 4. In normal condition, the CMP, MEP, and LMPP cells dominates the population after t ≥ 4. However, in the AML case, the MEP cells increases up to 100 times of the normal condition after a single psuedotime and dominates the population. Figure 7C plots the number of cells in each cell type separately, where we can observe the increasing number of cells not only in MEP, but also the intermediate cell types, 4–8. Figure 8 compares the cell distribution on the graph between the normal and AML case at time t = 7. In the AML case, the peak is shown on the edges near MEP cells.
Figure 7.
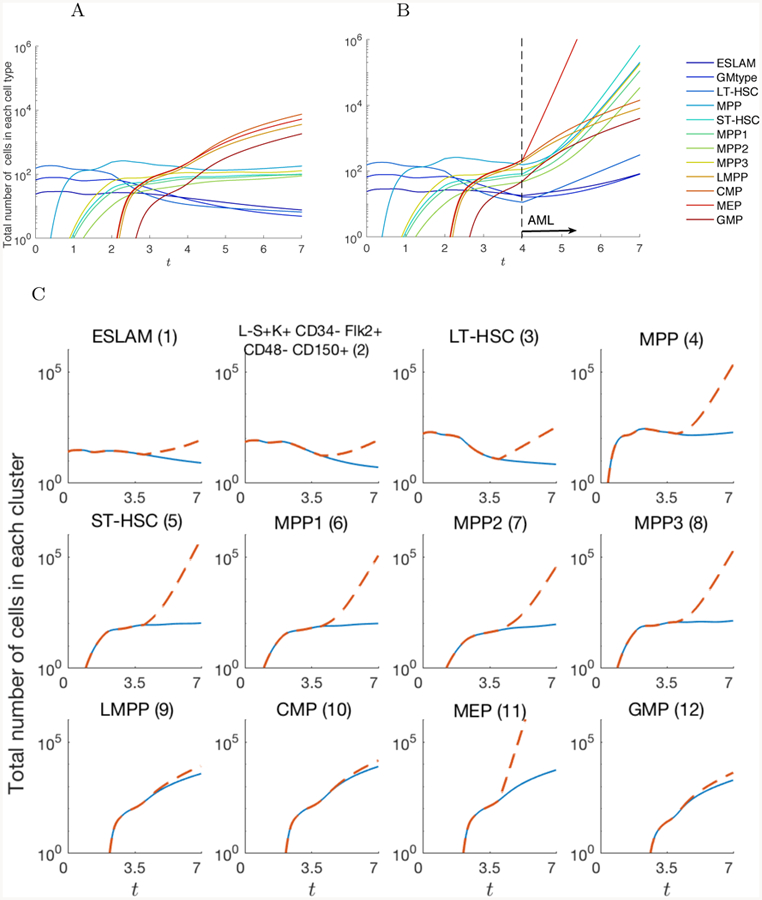
Total number of cells in each node up to t = 7 in A) normal condition and B) AML pathogenesis. The AML simulation is started at t = 4. Compared to the normal case, cells in MEP, LT-HSC, and ST-HSC increase as well as other cell types. Figure C) compares the number of cells between the normal and AML case for each cell type individually.
Figure 8.
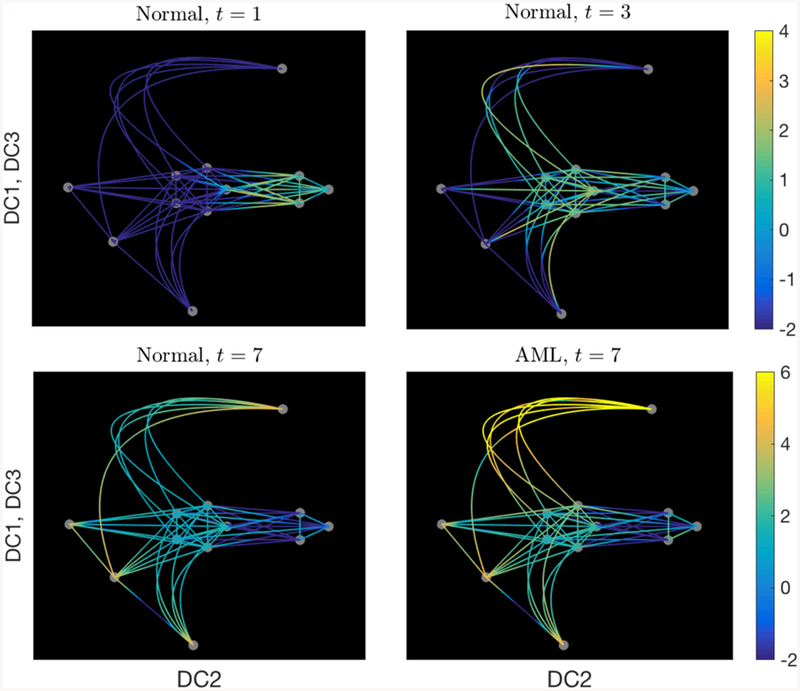
The cell distribution on the graph in a log10 scale, comparing the normal and AML conditions at t = 7. The AML condition shows increased density on the edges near the MEP state (node 11) at t = 7.
The continuum of intermediate cell types, represented as numbers of cells along the edges of the graph are plotted in Figure 9. The cell distribution in the normal case at t = 1 and t = 3 shows the cell population moving on the edges from HSCs to progenitors states. Under normal hematopoeisis, we observe the flow of cells along the continuum from a stem cell like state to a progenitor state, with an even distribution of all types of progenitor cells. However in the AML case, we predict the emergence of novel intermediate cell types, including a mixed CMP-MPP3 and CMP-MEP cell type. These indeterminate cells may exhibit phenotypic and/or functional properties of both cell types on either side of the edge (node i and/or node j). This cell state may be unstable, phenotypically plastic, may be in an abnormal state or process of differentiation, or perhaps even undergoing a selection pressure to induce transformation. Of note, this prediction of a mixed CMP-MEP cell type echoes the biological observation that abnormal myeloid progenitors seen during AML progression exhibit an MEP-like immunophenotype with a CMP-like functional readout (Kuo et al. 2006). This mixed identity/functionality coincides with a strong differentiation block towards erythrocyte and megakaryocytes (Cai et al. 2016).
Figure 9.
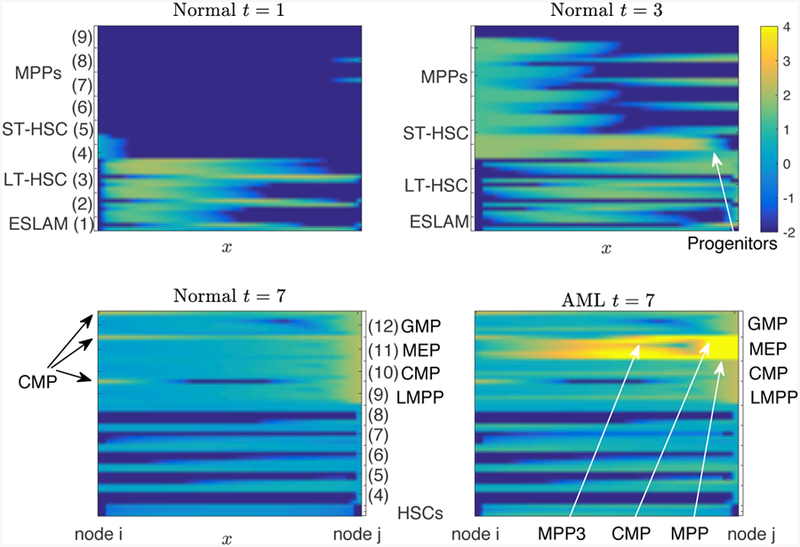
The continuum of cell states can be visualized as the density of cells along the 51 edges of the graph (rows) connecting node i (left) to node j (right) for all nodes i, j. Cell distribution (log10 scale) on the edge comparing the normal condition and AML. In addition to an accumulation of MEP cells, novel intermediate cell states emerge resulting from the differentiation block and increased proliferation rate resulting from AML. These novel cell states are indicated with white arrows and generally fall between the CMP, MPP, and MEP canonical cell states. The presented edges in the first row (t < 4) are lexicographically ordered with respect to the left end (an) to visualize which nodes are the differentiating cells departing from and with respect to the right end (bn) in the second row (t > 4) to visualize which nodes are the arriving cells differentiated into.
This highlights the advantage of modeling pathologic conditions in a continuum of cell states as the phenotypic properties and the differentiation processes are often abnormal during pathogenesis. This approach also circumvents the limitations of varying phenotypic definitions used in different studies in the literature (e.g., MEP vs. Pre-Meg/E) and the varying degree of heterogeneity within phenotypically defined cell populations in health and in disease.
We also simulated AML starting at different time points from t = 1 to t = 6. Since our initial condition assumes that the cells have not yet developed to MEP, the total number of cells is maximized when the AML occurs after a critical amount of cells have differentiated into an MEP state. Figure 10 shows the results of model simulations, where we observe that the number of cells are maximal at later times when AML is started at t = 3. From these simulations, we infer that the short and long term evolution of AML may depend on the state and composition of the hematopoietic landscape at the time of AML initiation.
Figure 10.
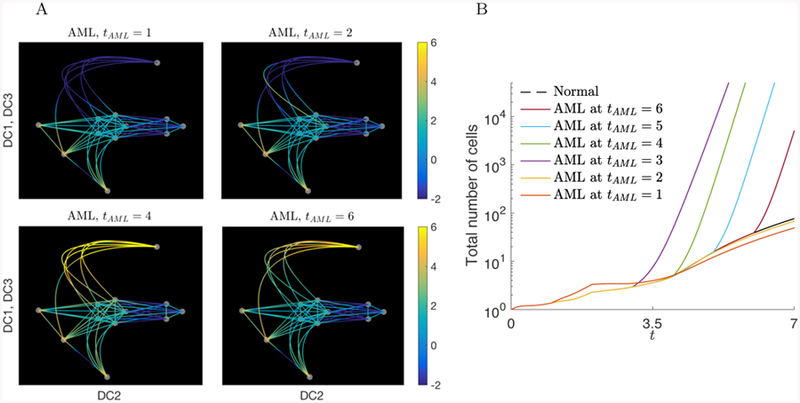
A) Cell distribution on the graph at t = 7 for AML occurring at different times, tAML = 1, 2, 4, and 6. MEP (11) blows up when AML occurs after t ≥ 2. The dominating intermediate cells are also distinct. B) Relative total number of cells when AML occurs at tAML = 1 to tAML = 6 compared to the normal case (dashed line) up to time t = 7. The total number of cells is maximized when AML occurs at tAML = 3.
5. Discussion
We present a mathematical model of movement in an abstract space representing states of cellular differentiation. We represent trajectories in the differentiation space as a graph and model the directed and random movement on the graph with partial differential equations. We demonstrate our modeling approach on a simple graph, and then apply our model to hematopoiesis with publicly available scRNA-seq data. We calibrate the PDE model to pseudotime trajectories in the diffusion map space and use the model to predict the early stages of pathogenesis of acute myeloid leukemia.
A more traditional approach for modeling the process of cell differentiation is to use a discrete collection of ordinary differential equations (ODEs) that describe dynamics of cells at n different maturation stages and the transition between those stages, c.f.,Lander et al. (2009); Lo et al. (2008); Marciniak-Czochra et al. (2009); Stiehl and Marciniak-Czochra (2011). These discrete models are also referred to as “multicompartmental models,” and are based on the biological assumption that in each lineage of cell precursors there are discrete steps in the maturation process that are followed sequentially, c.f., Lord (1997); Uchida et al. (1993).
This view of the differentiation process being discrete does not capture biological observations that indicate that cell differentiation is more likely a continuous process, and that maturation may, in fact, even be decoupled from cell division, c.f., Doumic et al. (2011); Dontu et al. (2003). A number of mathematical models have been created that aim to capture the continuous process of cell differentiation (Adimy, Crauste, and Ruan 2005; Pujo-Menjouet, Crauste, and Adimy 2004; Alarcon et al. 2011; Bélair, Mackey, and Maha y 1995; Colijn and Mackey 2005; Doumic-Jau ret, Kim, and Perthame 2010; Doumic et al. 2011; Gwiazda, Grzegorz, and Marciniak-czochra 2012).
For example, in Doumic et al. (2011), the authors present a model of cell differentiation that assumes that the dynamics of differentiated precursors can be approximated by a continuous maturation model. The model is created by extending the multicompartment discrete system of Marciniak-Czochra et al. (2009). The authors provide a careful comparison that shows that the continuous structured population model is not a mathematical limit of the discrete multicompartment model. In particular, the dynamics of the continuous model allow commitment and maturation of cell progenitors to be a continuous process that can take place between cell divisions. They do show, however, that there is overlap in model dynamics with a particular choice of maturation rate. In Gwiazda, Grzegorz, and Marciniak-czochra (2012), the authors subsequently developed a continuous model that can be viewed as a generalization that admits both the continuous model of Doumic et al. (2011) and the discrete model of Marciniak-Czochra et al. (2009) as special cases. In Prokharau, Vermolen, and Garca-Aznar (2014), the authors develop a PDE-based continuous model of cell differentiation that allows cells to differentiate into an arbitrary number of cell types. A particular differentiation trajectory can be determined by any number of parameters, such as biochemical factors, the current differentiation state, or just by a random variable, so their approach allows differentiation to be either a deterministic or a stochastic process.
The modeling approach we present differs from previous cell differentiation models in that it is centered on capturing cell differentiation dynamics that take place within a space that has been created via a dimension reduction transformation of high dimensional data. Within that reduced space, our model assumes that maturation and differentiation take place along a continuous trajectory. (The dimension reduction outcomes on the data sets we tested indicate that the trajectory will, in fact, be continuous.) Cells can differentiate along an arbitrary number of paths with an arbitrary number of end states, all of which are determined by the data set and dimension reduction technique employed. Thus, the reduced differentiation space is not predetermined, but is generated as a function of the dimension reduction technique and the data set of interest.
Although methods exist to characterize differentiation trajectories, such as optimal transport (Schiebinger et al. 2017) and diffusion pseudotime methods (Haghverdi et al. 2016), an advantage of our approach is the ability to use a mathematical model to predict the outcomes of abnormal trajectories and to perturb the system mathematically with the model. We use this advantage of the mathematical model to simulate and explore AML pathogenesis based on immunophenotypic characterization of a mouse model for inv(16) AML. Our simulation results are consistent with the evolution of inv(16) driven AML, and predicts dynamics in canonical cell populations as well as cells in novel, intermediate states of differentiation. The intermediate cell states such as CMP-MEP seen in our simulation is consistent with previous observations that CBFβ-SMMHC expressing phenotypic MEP cells confer CMP-like progenitor cell activity (Kuo et al. 2006). Given the phenotypic plasticity and aberrant differentiation occurring during leukemia evolution, it is particularly informative to model cell dynamics in a continuum of differentiation space.
The novelty and power of this modeling approach is the ability to capture and predict dynamics of many interconnected cell types. We now consider a continuum of cellular states, and model movement between these states in aggregate by representing many cell populations and states in a single variable. This approach increases biological resolution of the system by characterizing an infinite number of sub-states in a continuum representation and allows us to make predictions with one equation and very few model parameters, which can be directly calibrated to experimental data, for example with time-series cell differentiation experiments. These data could be used in place of the inferred pseudotime methods to construct more realistic differentiation trajectories, as well as estimate parameters such as the transport rates between locations in the differentiation space. We note that this is not equivalent to rates of cellular differentiation, since this allows inference of transition between intermediate states of differentiation which may not be directly calculated from differentiation assays which rely on specific lineage markers.
A limitation of our approach is that it does not include physical properties of the living biological system, such as the cellular microenvironment, which is known to play a critical role in the transformation of cell state and function. Furthermore, we recognize and acknowledge that cellular state transition dynamics as represented as a projection in a low dimensional space is an approximation of the dynamics in the original high dimensional space. Moreover, the dynamics observed and predicted in the lower dimensional space critically depend on the method of dimension reduction. This logic motivates our use of diffusion maps as the method to construct the differentiation space.
In addition, our current model assumes that the cell properties of the intermediate cell types change linearly between the node cell types. Although it is reasonable to assume that the overall cell properties in the macro scale changes linearly depending on the distance in the phenotypic space when no other information in between is given, our future work involves using the expression levels of the intermediate cells that are related to cell dynamics, e.g., cell cycle, differentiation, and proliferation, to develop more appropriate models for the intermediate cells. A limitation of the Nestorowa et al. (2016a) data set is that it includes only stem and committed progenitor cells, and lacks a population of fully differentiated cells (e.g. erythrocytes, platelets, B-cells, T-cells, etc.), which yields an incomplete differentiation trajectory. Although we note that the stem and progenitor cell populations are the leukemia-initiating cell populations most immediately relevant to the pathogenesis of inv(16) driven AML Cai et al. (2016). Data sets covering the full spectrum of differentiation trajectory during normal and abnormal (AML) hematopoeisis will enable modeling of differentiation blocks occurring at later stages of differentiation.
However, despite these limitations, we contend that this kind of analysis is a critical and valuable first step towards understanding the evolution of the higher dimensional system, and that low dimensional approximations have value, particularly when predictions in the lower dimensional space can be experimentally validated. We postulate that when dynamics in low dimensional representations are su ciently characterized, they may eventually be used as a surrogate for high dimensional data, thus reverting the trend of “big data” back down to more informative “small data.”
We note that our modeling approach can be applied to any data set or manifold shape. As more normal and abnormal cellular state transitions are characterized at single cell resolution, we may apply similar computational and modeling methods to those systems. We emphasize our modeling approach is general and is not tailored or adapted to hematopoiesis in particular. Future applications of this approach may be useful to model the effects of therapies which target specific states of differentiation or the differentiation process itself, including other hematologic malignancies.
Acknowledgements
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572 and R01CA178387. The content is solely the responsibility of the authors and does not necessarily represent the o cial views of the National Institutes of Health.
Appendix A. Model and parameters
Here we present the PDE model and parameter calculations used to produce the results presented in Section 4. MATLAB files used to generate the figures and results are included as supplemental files and can be downloaded from the journal website. The cell distribution u(x, t) is computed on the graph G as
| (A1) |
where uk is the solution projected on the edge ek as and are the 51 edges connecting the twelve nodes as in Figure 3B. We assume that the edges are unit length as and find the coeffcients in (Eq. A1) that are scaled to the unit length edge.
The total number of cells can be computed as , and we compute the number of cells in the n-th cluster as
| (A2) |
Alternatively, since the boundary of the cell types are not distinctive, one can compute it as a weighted sum along the edges adjacent to the node n with linear weight functions such as and along the entire edge.
To obtain the transfer rate between the cell nodes, we assume three discrete psuedotimes at those three sorted groups starting from LT-HSC to HSPC, and finally to progenitor cells. As remarked in section 4.1, we consider subsets of nodes I1 = {1, 2, 3} as HSC, I2 = {4, …, 8} as HSPC, I3= {9, …, 12} as progenitor cell group. This follows the cell data in Nestorowa et al. (2016b) that is classified with ComBat from the SVA package using the sorting gate of LT-HSC, HSPC, and progenitor, and then processed with diffusion mapping initialized from a subpopulation of LT-HSC to the progenitor cells of different lineage of Erythroid, Granulocyte-macrophage, and Lymphoid. Accordingly, we consider three discrete psuedotimes considering LT-HSC (t0), HSPC (t1), and progenitor (t2) and compute the number of cells in each node that is summarized in Table A1. We comment that diffusion pseudotime is not a physical time unit (i.e. days) and that the differentiation process is modeled based on the inferred pseudotime trajectories. with the following mapping of pseudotimes t0 = 0, t1 = 2, and t2 = 4. The time mapping procedure can be refined with time series differentiation assay data. The transfer rates between the nodes are taken from the ratios at each psuedotime.
Table A1.
Number of cells in each node (ID) at three distinct psuedotimes to, ti, and t2. We notate it as pjD (t). The cell numbers are plotted in Figure 5C comparing to our simulation.
| ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| t0 | 24 | 66 | 155 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| t1 | 0 | 0 | 0 | 236 | 36 | 27 | 11 | 60 | 0 | 0 | 0 | 0 |
| t2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 192 | 223 | 227 | 54 |
We compute ratio as time independent within the subsets as
that is, γ1 = 24/245, γ2 = 66/245, γ3 = 155/245 for I1, γ 4 = 236/370, γ 5 = 36/370, γ 6 = 27/370, γ 7 = 11/370, γ 8 = 60/370 for I2, and γ9 = 192/696, γ10 = 223/696, γ11 = 227/696, γ12 = 54/696 for I3. We remark that the transfer rates can be time dependent as γn(t) if the data is collected at sequential timepoints, which is one way that the model could be parameterized.
We take these values as the in and out transfer rate imposed in the advection coeffcient. For each node, we assume a constant parameter cn ≠ 0 that determines the magnitude of the advection coeffcient, that is, the speed of the cell differentiation.We take the transfer in rate at the node , as , for and transfer out rate as , for . Using the fixed transfer rates at the nodes, the advection coeffcient is linearly interpolated as , that is,
| (A3) |
In addition, we apply the weight (1 − x2) to model the accumulation of cells at the progenitor nodes ,
and take , for other pairs of nodes. For instance, ,for within the same hierarchy of cells, and the transition between these nodes are only governed by diffusion. The constant parameter at each node cn are taken to reproduce the cell distribution as in Figure 5 as following,
The values are computed by a simple root finding algorithm such as secant method.
The diffusion coeffcients on the edges are taken as Dk(x) = DI(i,j)(x) = 10−2 between the nodes that are within and , assuming that the perturbation of the cells are in unit psuedotime in the rescaled edges is in the order of , where L is the average length of the edges within and . Considering that the average length of the other combinations of (i, j) are increased by threefold, therefore we take DI(i,j)(x) = 10−3.
The proliferation rate is also obtained by the secant method to match the given data in Table A1 at t1 and t2. The computed values are rn = 1.3648, t < t1 and rn = 0.4, t ≥ t1 for the HSPC and progenitor cells . In addition, the fact that LT-HSC cells proliferate relatively less than the progenitor cells (Passegué et al. 2005) are imposed as for . The intermediate level of proliferation is linearly interpolated as
| (A4) |
assuming that the overall proliferation of intermediate cell states change gradually. If the time variable is taken as the actual time, the rate in each node can be computed considering the proportion of proliferating stem cells (5–10%) and cell cycle (36–145 days) (Hao, Chen, and Cheng 2016; Pietras, Warr, and Passegué 2011). Moreover, the abnormal proliferation of cancerous cells with cell cycle λ and apoptosis of the differentiated cells with rate d at expression level x* can be modeled with a localized Gaussian function with variance ∈ as , and , respectively. The choice of localized Gaussian function assumes that the center x* is location in the diffusion space that most closely resembles the “prototypical,” or “ideal” cell type identity.
The described parameters are summarized in Table A2.
Table A2.
Summary of the required data and corresponding parameters. In our simulation, Vk and Rk are estimated from in Table A1.
| biological meaning and parameters | |
|---|---|
| Vk (x) | cell differentiation rate ck, branching ratio |
| Rk (x) | growth rate rk |
| Dk (x) | phenotypic fluctuation , wk |
The initial condition is taken by considering the cell data at pseudotime t0 with ratios , , , for k = 4,…, 12. We remark that this is shown in Figure 5C. Accordingly, the initial distribution is taken as
With this choice, the total number of cells in each node ρn(t0) computed as in Eq. (A2) is similar to the given ratios . The boundary condition defined as in Eq. (3) around the node vn, that is, at x = bI(i,n) and x = aI(n,j) becomes
| (A5) |
with continuity boundary conditions for fixed n. The condition (A5) reduces to in our model since
Sensitivity of model parameters
We test the sensitivity of the results with respect to the parameters in the diffusion, advection, and reaction coeffcient. The values of Dk, Vk, and Rk are varied by −10%, −1%, 1%, and 10%, and Figure A1 presents the difference in the total number of cells ρ(t) in percentage. While it is expected that the total number of cells are sensitive to the reaction coeffcient, since it governs the proliferation rate, it also strongly depends on the advection coeffcient as well, particularly in AML condition. On the other hand, the results are less dependent on the diffusion coeffcient. The number of intermediate cells while varying the coeffcients are plotted in Figure A2. In particular, we present the dynamics of LT-HSC(3)-STHSC(5), ST-HSC(5)-MPP1(6), MPP(4)-LMPP(9), and CMP(10)-MEP(11) cells in normal and AML condition. We observe similar results as in the total number of cells, however, the overall trend of the dynamics is independent to the variation in the coeffcients.
Figure A1.
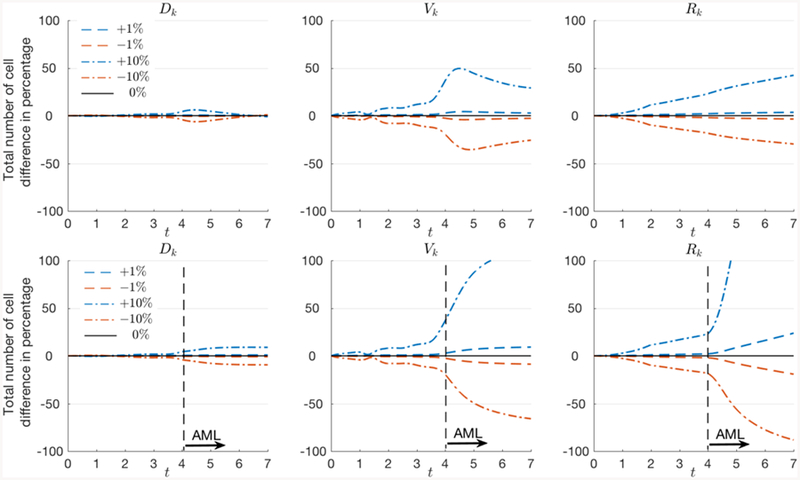
Change in the total number of cells ρ(t) in percentage with respect to the model parameters, diffusion Dk, advection Vk, and reaction Rk. We test the cases where the coeffcients change their values by − 10%, −1%, 1%, and 10%. The results are sensitive to the reaction and advection coeffcients particularly in AML condition. On the other hand, the results are less dependent on the di usion coeffcient.
Figure A2.
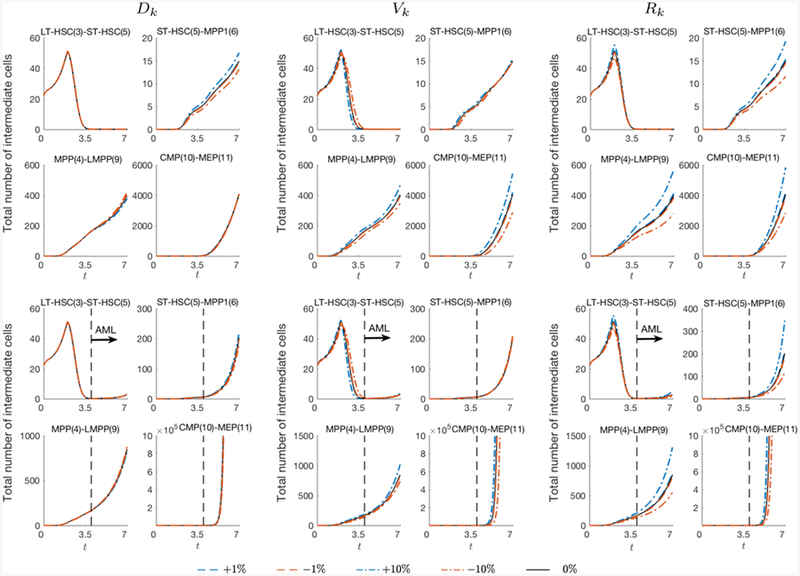
Number of intermediate cells with respect to the model parameters, diffusion Dk, advection Vk, and reaction Rk. The results are computed by varying the coeffcients by −10%, −1%, 1%, and 10%. Although the result varies from the reference case (0%), the overall trend of the cell−dynamics are observed to be similar.
Appendix B. Supplementary figures
Figure B1.
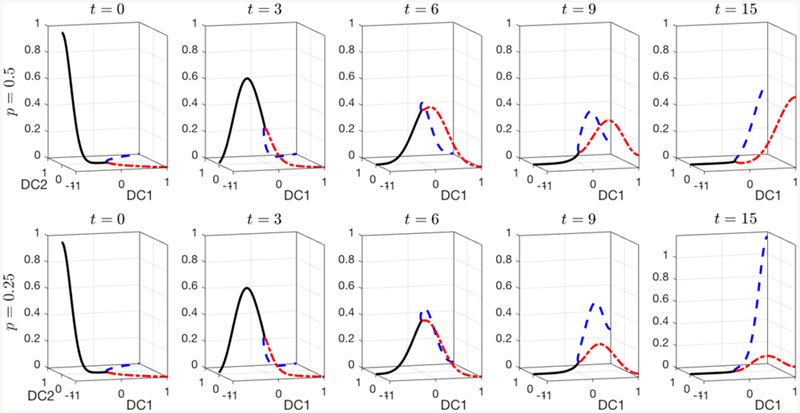
Solutions of the PDE model on the Y shaped graph from the initial condition centered at the left end DC1 = –1 (black line in (a-b)) with diffusion D = 10−2 and drift c = –0:2 for the symmetric (top row) and asymmetric cases (bottom row).
Figure B2.
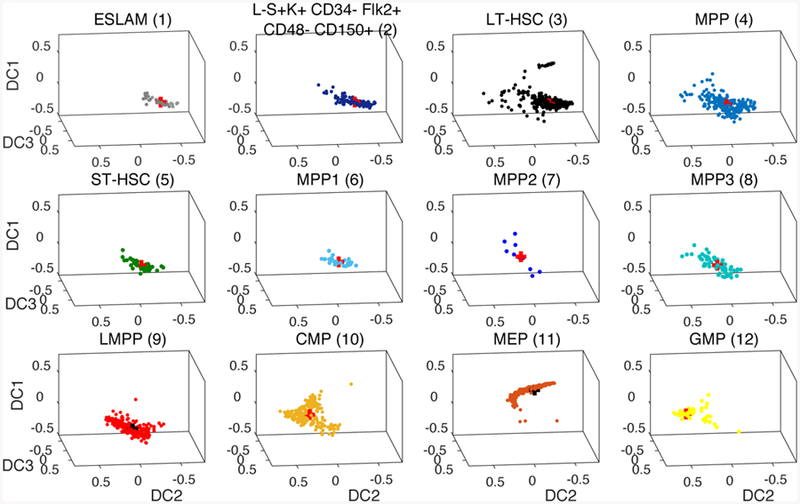
The cell data from (Nestorowa et al. 2016a) is grouped into 12 cell nodes according to 12 commonly sorted HSPC phenotypes including LT-HSC, ST-HSC, and MEP. The center of mass of each cluster is marked as a red cross and used to establish nodes and edges on the graph which is then used as a computational domain for our simulations.
Footnotes
The interpolation function can be taken, for instance, as a linear function as , where k = I(i, j). This assumes that the cell property changes linearly in terms of thedistance in the diffusion component space (Doumic et al. 2011; Gwiazda, Grzegorz, and Marciniak-czochra 2012). In addition, the values of VI(n,j)(x) near x = vn will take into account of the ratio of cells that branch out to different cell types vj, while the values of VI(i,n)(x) consider the ratio of cells that are flowing in from different cell types vi.
Using the notation in Appendix A, γ3 = p and γ4 = 1 − p.
Disclosure statement
No potential conflicts of interest are disclosed by the authors.
References
- Adimy Mostafa, Crauste Fabien, and Ruan Shigui. 2005. “A Mathematical Study of the Hematopoiesis Process with Applications to Chronic Myelogenous Leukemia.” SIAM Journal on Applied Mathematics 65 (4): 13281352. [Google Scholar]
- Akashi K, Traver D, Miyamoto T, and Weissman IL. 2000. “A clonogenic common myeloid progenitor that gives rise to all myeloid lineages.” Nature 404 (6774): 193–7. [DOI] [PubMed] [Google Scholar]
- Alarcon Tomas, Getto Philipp, Marciniak-Czochra Anna, and Vivanco D. 2011. “A model for stem cell population dynamics with regulated maturation delay.” Discrete Contin Dyn Syst 32–43. [Google Scholar]
- Bach Karsten, Pensa Sara, Grzelak Marta, Hadfield James, Adams David J., Marioni John C., and Khaled Walid T.. 2017. “Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing.” Nature Communications 8 (1): 2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélair Jacques, Mackey Michael C, and Mahaffy Joseph M. 1995. “Age-structured and two-delay models for erythropoiesis.” Mathematical biosciences 128 (1–2): 317–346. [DOI] [PubMed] [Google Scholar]
- Cai Qi, Jeannet Robin, Hua Wei-Kai K., Cook Guerry J., Zhang Bin, Qi Jing, Liu Hongjun, et al. 2016. “CBFβ-SMMHC creates aberrant megakaryocyte-erythroid progenitors prone to leukemia initiation in mice.” Blood 128 (11): 1503–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla LH, Garrett L, Adya N, Orlic D, Dutra A, Anderson S, Owens J, Eckhaus M, Bodine D, and Liu PP. 1999. “The fusion gene Cbfb-MYH11 blocks myeloid differentiation and predisposes mice to acute myelomonocytic leukaemia.” Nat Genet 23 (2): 144–6. [DOI] [PubMed] [Google Scholar]
- Cerrai S, and Freidlin M. 2017. “SPDEs on narrow domains and on graphs: an asymptotic approach.” Annales de lInstitut Henri Poincar 53: 865–899. [Google Scholar]
- Coifman RR, Lafon S, Lee AB, Maggioni M, Nadler B, Warner F, and Zucker SW. 2005. “Geometric diffusions as a tool for harmonic analysis and structure definition of data: Diffusion maps.” Proceedings of the National Academy of Sciences 102 (21): 7426–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colijn Caroline, and Mackey Michael C.. 2005. “A mathematical model of hematopoiesisI. Periodic chronic myelogenous leukemia.” Journal of Theoretical Biology 237 (2): 117 – 132. [DOI] [PubMed] [Google Scholar]
- Dontu Gabriela, Muhammad Al-Hajj Wissam M. Abdallah, Clarke Michael F., and Wicha Max S.. 2003. “Stem cells in normal breast development and breast cancer.” Cell Proliferation 36: 59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doumic Marie, Anna Marciniak-Czochra Benoit Perthame, and Zubelli Jorge P.. 2011. “A structured population model of cell differentiation.” SIAM J. Appl. Math 71 (6): 1918–1940. [Google Scholar]
- Doumic-Jauffret Marie, Kim Peter S., and Perthame Benot. 2010. “Stability Analysis of a Simplified Yet Complete Model for Chronic Myelogenous Leukemia.” Bulletin of Mathematical Biology 72 (7): 17321759. [DOI] [PubMed] [Google Scholar]
- Evans Lawrence C. 2010. Partial Differential Equations. 2nd ed American Mathematical Society. [Google Scholar]
- Freidlin M, and Hu W. 2013. “On diffusion in narrow random channels.” J. Stat. Phys 152: 136–158. [Google Scholar]
- Gwiazda Piotr, Grzegorz Jamroz, and Marciniak-czochra Anna. 2012. “Models of discrete and continuous cell differentiation in the framework of transport equation.” SIAM J. Math. Anal 44 (2): 1103–1133. [Google Scholar]
- Haghverdi L, Büttner M, Wolf FA, Buettner F, and Theis FJ. 2016. “Diffusion pseudotime robustly reconstructs lineage branching.” Nat. Methods 13 (10): 845–848. [DOI] [PubMed] [Google Scholar]
- Haghverdi Laleh, Buettner Florian, and Theis Fabian J. 2015. “Diffusion maps for high-dimensional single-cell analysis of differentiation data.” Bioinformatics 31 (18): 2989–2998. [DOI] [PubMed] [Google Scholar]
- Hamey Fiona K., Nestorowa Sonia, Wilson Nicola K., and Göttgens Berthold. 2016. “Advancing haematopoietic stem and progenitor cell biology through single-cell profiling.” FEBS Lett 590 (22): 4052–4067. [DOI] [PubMed] [Google Scholar]
- Hao Sha, Chen Chen, and Cheng Tao. 2016. “Cell cycle regulation of hematopoietic stem or progenitor cells.” International Journal of Hematology 103 (5): 487–497. [DOI] [PubMed] [Google Scholar]
- Khalid Samina, Khalil Tehmina, and Nasreen Shamila. 2014. “A survey of feature selection and feature extraction techniques in machine learning” In Science and Information Conference (SAI) 2014, August. IEEE. [Google Scholar]
- Kuo Ya-Huei H., Gerstein Rachel M., and Castilla Lucio H.. 2008. “Cbfβ-SMMHC impairs differentiation of common lymphoid progenitors and reveals an essential role for RUNX in early B-cell development.” Blood 111 (3): 1543–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Ya-Huei H., Landrette Sean F., Heilman Susan A., Perrat Paola N., Garrett Lisa, Liu Pu P., Beau Michelle M. Le, Kogan Scott C., and Castilla Lucio H.. 2006. “Cbfβ-SMMHC induces distinct abnormal myeloid progenitors able to develop acute myeloid leukemia.” Cancer Cell 9 (1): 57–68. [DOI] [PubMed] [Google Scholar]
- Lander Arthur D, Gokoffski Kimberly K, Wan Frederic Y. M, Nie Qing, and Calof Anne L. 2009. “Cell Lineages and the Logic of Proliferative Control.” PLOS Biology 7 (1): 1–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine Jacob H, Simonds Erin F, Bendall Sean C, Davis Kara L, Amir El-ad D, Tadmor Michelle, Litvin Oren, et al. 2015. “Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis.” Cell 162 (1): 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Tarlé SA, Hajra A, Claxton DF, Marlton P, Freedman M, Siciliano MJ, and Collins FS. 1993. “Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia.” Science 261 (5124): 1041–4. [DOI] [PubMed] [Google Scholar]
- Liu PP, Wijmenga C, Hajra A, Blake TB, Kelley CA, Adelstein RS, Bagg A, et al. 1996. “Identification of the chimeric protein product of the CBFB-MYH11 fusion gene in inv(16) leukemia cells.” Genes Chromosomes Cancer 16 (2): 77–87. [DOI] [PubMed] [Google Scholar]
- Lo Wing-Cheong, Chou Ching-Shan, Gokoffski Kimberly, Wan Frederic, Lander Arthur, Calof Anne, and Nie Qing. 2008. “Feedback regulation in multistage cell lineages.” Mathematical Biosciences and Engineering 6 (1): 5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord Brian I. 1997. Stem Cells, Chap. Growth factors and the regulation of haemopoietic stem cells, 401–422. Academic Press, Cambridge, UK. [Google Scholar]
- Maaten Laurens van der, and Hinton Geoffrey. 2008. “Visualizing data using t-SNE.” Journal of machine learning research 9 (Nov): 2579–2605. [Google Scholar]
- Mao Qi, Wang Li, Goodison Steve, and Sun Yijun. 2015. “Dimensionality Reduction Via Graph Structure Learning” In Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD ‘15, New York, NY, USA, 765–774. ACM. [Google Scholar]
- Marciniak-Czochra Anna, Stiehl Thomas, Ho Anthony D., Jger Willi, and Wagner Wolfgang. 2009. “Modeling of Asymmetric Cell Division in Hematopoietic Stem CellsRegulation of Self-Renewal Is Essential for Effcient Repopulation.” Stem Cells and Development 18 (3): 377386. [DOI] [PubMed] [Google Scholar]
- Moris Naomi, Pina Cristina, and Alfonso Martinez Arias. 2016. “Transition states and cell fate decisions in epigenetic landscapes.” Nature Reviews Genetics 17 (11): 693–703. [DOI] [PubMed] [Google Scholar]
- Nestorowa Sonia, Hamey Fiona K, Sala B. Pijuan, Diamanti Evangelia, Shepherd Mairi, Laurenti Elisa, Wilson Nicola K., et al. 2016a. “A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation.” Blood 128 (8): 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestorowa Sonia, Hamey Fiona K., Blanca Pijuan Sala Evangelia Diamanti, Shepherd Mairi, Laurenti Elisa, Wilson Nicola K., Kent David G., and Göttgens Berthold. 2016b. “A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation.” Blood 128 (8): e20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegué Emmanuelle, Wagers Amy J., Giuriato Sylvie, Anderson Wade C., and Weissman Irving L.. 2005. “Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates.” The Journal of Experimental Medicine 202 (11): 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras Eric M., Warr Matthew R., and Passegué Emmanuelle. 2011. “Cell cycle regulation in hematopoietic stem cells.” Journal of Cell Biology 195 (5): 709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokharau Pavel A., Vermolen Fred J., and Garca-Aznar Jos Manuel. 2014. “A mathematical model for cell differentiation, as an evolutionary and regulated process.” Computer Methods in Biomechanics and Biomedical Engineering 17 (10): 1051–1070. [DOI] [PubMed] [Google Scholar]
- Pronk Cornelis J. H., Rossi Derrick J., Mansson Robert, Attema Joanne L., Gudmundur Logi Norddahl Charles Kwok Fai Chan, Sigvardsson Mikael, Weissman Irving L., and Bryder David. 2007. “Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy.” Cell Stem Cell 1 (4): 428–42. [DOI] [PubMed] [Google Scholar]
- Pujo-Menjouet Laurent, Crauste Fabien, and Adimy Mostafa. 2004. “On the stability of a nonlinear maturity structured model of cellular proliferation.” Discrete and Continuous Dynamical Systems 12 (3): 501522. [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C. 2017a. “Reversed graph embedding resolves complex single-cell trajectories.” Nat. Methods 14 (10): 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Xiaojie, Hill Andrew, Packer Jonathan, Lin Dejun, Trapnell Cole, and Cellular Biology Program. 2017b. “Single-cell mRNA quantification and differential analysis with Census.” Nat. Methods 14 (3): 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid S, Kotton DN, and Bar-Joseph Z. 2017. “TASIC: determining branching models from time series single cell data.” Bioinformatics 33 (16): 2504–2512. [DOI] [PubMed] [Google Scholar]
- Rizvi Abbas H., Camara Pablo G., Kandror Elena K., Roberts Thomas J., Schieren Ira, Maniatis Tom, and Rabadan Raul. 2017. “Single-cell topological RNA-seq analysis reveals insights into cellular differentiation and development.” Nature Biotechnology 35 (6): 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebinger Geoffrey, Shu Jian, Tabaka Marcin, Cleary Brian, Subramanian Vidya, Solomon Aryeh, Liu Siyan, et al. 2017. “Reconstruction of developmental landscapes by optimal-transport analysis of single-cell gene expression sheds light on cellular reprogramming.” bioRxiv. [Google Scholar]
- Setty Manu, Michelle D Tadmor Shlomit Reich-Zeliger, Angel Omer, Tomer Meir Salame Pooja Kathail, Choi Kristy, Bendall Sean, Friedman Nir, and Pe’er Dana. 2016. “Wishbone identifies bifurcating developmental trajectories from single-cell data.” Nature Biotechnology 34: 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar Karthik, Lapan Sylvain W, Whitney Irene E, Tran Nicholas M, Evan Z, Kowalczyk Monika, Adiconis Xian, et al. 2016. “Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics.” Cell 166 (5): 1308–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiehl Thomas, and Marciniak-Czochra Anna. 2011. “Characterization of stem cells using mathematical models of multistage cell lineages.” Mathematical and Computer Modelling 53 (7–8): 1505–1517. [Google Scholar]
- Uchida Nobuko, Fleming William H., Alpern Eytan J., and Weissman Irving L.. 1993. “Heterogeneity of hematopoietic stem cells.” Current Opinion in Immunology 5 (2): 177–184. [DOI] [PubMed] [Google Scholar]
- Carlos Valero Valdes, and Herrera Guzman Rafael. 2014. “Fick-Jacobs equation for channels over three-dimensional curves.” Physical Review E 90 (5): 1–11. [DOI] [PubMed] [Google Scholar]
- van Unen Vincent, Thomas Höllt Nicola Pezzotti, Li Na, Reinders Marcel J. T., Eisemann Elmar, Koning Frits, Vilanova Anna, and Lelieveldt Boudewijn P. F.. 2017. “Visual analysis of mass cytometry data by hierarchical stochastic neighbour embedding reveals rare cell types.” Nature Communications 8 (1): 1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velten Lars, Haas Simon F., Raffel Simon, Blaszkiewicz Sandra, Islam Saiful, Hennig Bianca P., Hirche Christoph, et al. 2017. “Human haematopoietic stem cell lineage commitment is a continuous process.” Nat Cell Biol 19 (4): 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH 1957. The strategy of the genes a discussion of some aspects of theoretical biology London: George Allen; & Unwin, Ltd. [Google Scholar]
- Zwanzig Robert. 1992. “Diffusion past an entropy barrier.” Journal of Physical Chemistry 96 (10): 3926–3930. [Google Scholar]