

Abstract
Major histocompatibility complex (MHC) class I genes encode highly polymorphic antigens that play an essential role in a number of immunological processes. Their expression is activated in response to a variety of signals and is mediated through several promoter elements among which the enhancer A is one of the key control regions. It contains binding sites for several transcription factors, for example: (i) a well-characterized binding site for rel/NF-κB transcription factors in its 3′-end (the H2TF1 or κB1 element), (ii) a second κB site (the κB2 element), which is located immediately adjacent 5′ to the H2TF1 element and which is recognized by p65/relA in the human HLA system, and (iii) an AP-1/ATF recognition sequence in the 5′ end (EnA-TRE). Here we demonstrate that latter element is bound by at least two distinct heterodimers of the AP-1/ATF transcription factor family, namely c-Jun/ATF-2 and c-Jun/Fra2. Moreover, our data reveal that the enhancer A is simultaneously bound by AP-1/ATF and rel/NF-κB transcription factors and that the cellular coactivator p300, which enhances enhancer A-driven reporter gene expression if cotransfected, is recruited to the enhancer A through this multiprotein complex. In contrast to the complete enhancer A, neither the EnA-TRE nor the H2TF1 element on their own are able to confer activation on a heterologous promoter in response to the phorbol ester tumor promoter TPA or the cytokine TNFα. Moreover, deletion of any one of the enhancer A control elements results in a dramatic loss of its inducibility by TNFα, and point mutations in either the EnA-TRE or the H2TF1 element lead to the loss of AP-1/ATF or NF-κB binding, respectively, and to the loss of enhancer A inducibility. Therefore, we conclude that the enhancer A is synergistically activated through a multiprotein complex containing AP-1/ATF, NF-κB transcription factors as well as the cellular coactivator p300.
Keywords: AP-1/ATF, MHC class I, NF-κB, p300
MAJOR histocompatibility (MHC) class I proteins play a key role in the recognition of virally infected or tumor cells by the host immune system presenting antigenic peptides derived from infectious agents or tumor-specific proteins on the cell surface. These complexes are recognized and bound by cytotoxic T lymphocytes, which results in the lysis of the antigen-carrying cell (40).
MHC class I genes are expressed on nearly all somatic cells of the adult organism although their level of expression varies considerably between different tissues and different cell types (46). The level of MHC class I expression is determined transcriptionally—a process in which various cellular transcription factors are involved. The mouse MHC class I H-2Kb promoter, for instance, possesses several regulatory regions, among them two enhancers: the enhancer A (also known as class I regulatory element, CRE) and the enhancer B (13,51). The latter contains a recognition sequence for the cellular transcription factor AP-l/ATF [nucleotides (nt) −95 to −88 relative to the start site of transcription] (31). Studies of Dey et al. (14) indicate that the enhancer B is responsible for the enhancement of the constitutive MHC class 1 gene transcription in a cell type-specific manner. Moreover, a very recent report demonstrates that the enhancer B is bound by ATF-l/ATF-1, CREB-1/CREB-1, and ATF-l/CREB-1 dimers and that at least ATF-1 is involved in normal regulation of H-2 gene expression and in the stimulation of transcription from the H-2Dd gene after radiation leukemia virus infection of mouse thymocytes (24).
The enhancer A (nt −203 to −159), which partially overlaps with a consensus interferon response sequence (13), is of central importance for the expression of the H-2Kb gene. It contains the H2TF1 element (or κB1 site) in its 3′ end (45), which is a positive regulator of H-2Kb transcription (4,10,13,25). Mutations within this enhancer element abolish basal enhancer activity as well as the ability to respond to various stimuli such as tumor necrosis factor-α (TNFα) or phorbol esters like 12-O-tetradecanoyl phorbol-13-acetate (TPA) (26). Activation through the H2TF1 element is regulated by transcription factors belonging to the re/NF-κB family (27,57), for example, by a homodimer consisting of two 50-kDa subunits (p50-NF-κBl), which are encoded as precursor proteins by the pl05-NF-κBl gene or by the heterodimer NF-κB, which consists of a p50-NF-κBl and a 65-kDa subunit (p65/relA). The latter protein is encoded by the relA gene (5).
A second κB element (κB2) is located next to the H2TF1 element. In the case of the human HLA-A promoter, this site is most probably bound by p65/relA, although its dimerization partner on this specific promoter element is not yet clear (37). In contrast to the H2TF1 element, which is highly conserved among human HLA-A, HLA-B, and mice H-2 promoters, the κB2 site is less conserved (12,30). For instance, the κB2 site from the HLA-A2 promoter (5′-GGGGAGrcCCA-3′) differs in the central two nucleotide positions from the respective site of the H-2Kb promoter (5′-GGGGAAGCCCA-3′). Recent experiments have shown evidence that κB2 is probably as important as κB2 in regulating HLA-A promoter activation (37). This finding was supported through data obtained in the mouse system where either the natural κB,-κB2 element or artifical κB1-κB1 or κB2-κB2 elements can confer activation on a heterologous promoter by signals such as TNFα, whereas a monomer (either κB, or κB2) alone cannot (26).
The regulatory sequences at the 5′ end of the enhancer A are less well characterized. Overlapping binding sites for several transcription factors are located in this region, among them CRE2 (1), members of the superfamily of nuclear hormone receptors (23,34,43), and members of the AP-l/ATF family (9,31,58). Deletion of the AP-l/ATF binding site (henceforth referred to as EnA-TRE) from the H-2Kb promoter results in a greatly diminished promoter activity (9), indicating that this promoter element is of comparable importance for transcriptional activity as the H2TF1 site. On the other hand, a heterologous promoter construct carrying one copy of the AP-1/ATF binding site was not inducible by either TPA or TNFα (26) and a respective reporter carrying three copies of the AP-l/ATF binding site was only slightly inducible by cotransfected c-Jun in transient expression assays (9). These results indicate that factors binding to the AP-l/ATF recognition sequence in the enhancer A have to cooperate with transcription factors bound to the other enhancer A regulatory elements to efficiently activate gene expression driven from the enhancer A.
The composition of the AP-1 transcription factor complex binding to the AP-l/ATF motif of the H-2Kb promoter was not clear (31). In general, AP-1 consists of gene products of the Jun (c-Jun, JunB, JunD) and Fos families (c-Fos, FosB, Fral, Fra2) (28). After forming either homodimers (e.g., Jun/Jun) or heterodimers (e.g., Jun/Fos), these dimers bind preferentially to the typical AP-1 recognition sequence (TGAg/cTCA). Furthermore, c-Jun can heterodimerize with members of the ATF family. Such heterodimers are able to activate target gene expression via ATF/CREB consensus sequences (TGAC GTCA).
Our previous findings indicate that c-Jun is one of the constituents of the AP-l/ATF transcription factor bound to the EnA-TRE (9). Moreover, initial studies of Yamit-Hezi and coworkers (58) suggest that c-Fos might be the dimerization partner of c-Jun on this promoter element. However, DNA sequence analyses demonstrate that the EnA-TRE (TGAGGTCA) resembles much more a typical ATF/CREB binding site than an AP-1 consensus sequence. The aim of this study was therefore (i) to analyze the composition of the AP-l/ATF transcription factor complex bound to the EnA-TRE in detail, and (ii) to investigate its cooperation with factors interacting with the H2TF1 and the κB2 element in the activation process of the enhancer A. We found that the EnA-TRE is bound by at least two distinct heterodimeric AP-1/ATF complexes in vitro, namely c-Jim/Fra2 and c-Jun/ATF-2. Our data suggest that these complexes interact with transcription factors of the rel/NF-κB family (p50-NF-κBl, p65/relA), which bind to the H2TF1 element and/or κB2 site, to synergistically activate H-2Kb gene expression in response to TNFα and TPA. Furthermore, the cellular cofactor p300 is recruited into this multiprotein complex and cotrans-fected p300 enhances enhancer A-driven reporter gene expression in TNFα-treated HeLa-tk− cells. From these results we conclude that the activation of the enhancer A depends on the assembly of a multi-protein complex.
MATERIALS AND METHODS
Plasmids
The (−2000/+12)-H-2Kb-CAT reporter construct [the numbers refer to the position of the respective nt in the H-2Kb promoter as described by Kimura et al. (30)] was a gift of Eric Blair, Department of Biochemistry and Molecular Biology, The University of Leeds, Leeds, UK. To obtain EnA-tk-CAT5 oligonucleotides representing the H-2Kb enhancer A (nt −210 to nt −151) were synthesized on a Gene Assembler Plus (PharmaciaLKB), annealed, and cloned via Bam HI restriction sites, which were synthesized on the 5′ ends of the oligonucleotides, into the respective restriction sites of the basal CAT vector pBLCAT5 (6). H2TFl-tk-CAT5, EnA-TRE-tk-CAT5, κB2/H2TFl-tk-CAT5, TRE/H2TF1-tk-CAT5, and TRE/κB2-tk-CAT5 were cloned in a similar way using oligonucleotides spanning the H2TF1 element (nt −175 to −156), the TRE of the enhancer A (nt −202 to −189), the κB2/H2TF1 region (nt −191 to −159), the EnA-TRE as well as the H2TF1 element (nt −201 to −184 + nt −172 to −159), or the TRE/κB2 region (nt −201 to −173) of the enhancer A. EnA+5-tk-CAT5 and EnA+10-tk-CAT5 were obtained by insertion of 5 (5′-ATATA-3′) or 10 nucleotides (5′-ATATATA TAT-3′) in front of nt −189 between the TRE and κB2 site. Pm-TRE-EnA-tk-CAT5, pm-H2TFl-EnA-tk-CAT5, and pm-TRE/H2TFl-EnA-tk-CAT5 were cloned by inserting a TRE point-mutated enhancer A (5′-GGAGGTTG-3′), a H2TF1 point-mutated enhancer A (5′-GGCGATTCCGG-3′), or a TRE/H2TF1 point-mutated enhancer A into the basal CAT vector pBLCAT5.
The sequences of all H-2Kb promoter mutants were verified by dideoxy sequencing (41) using the automated laser fluorescent DNA sequencer of Pharmacia.
pCMVβ-p300 was kindly provided by D. M. Livingston, Dana-Faber Cancer Institute and Harvard Medical School, Boston, MA.
Cell Culture and CAT Assays
HeLa-tk− cells were grown in DMEM supplemented with 10% FCS in 5% C02. Twenty-four hours after seeding, HeLa-tk− cells were transfected with 0.4 μg of the respective reporter construct using the lipofectamine method (Gibco BRL). For serum starvation HeLa-tk− cells were grown for 48 h in DMEM containing 0.5% serum followed by transfection. In some experiments TPA (50 μg/ml) or TNFα (100 U/ml) was added to the serum-starved cells 8 h after transfection. Sixteen to 24 h later, cells were harvested and lysed by freeze-thaw treatments. The protein concentration of the cellular extracts was measured using the Bradford method (7). Equal amounts of proteins were used to determine CAT activity (22). CAT activity was quantified using the automatic TLC linear analyzer of Berthold (Bad Wild-bad, Germany).
Preparation of Nuclear Extracts and Gel Retardation Assays
Nuclear extracts of HeLa-tk− cells were prepared as described by Shapiro et al. (44). The extracts were stored at −80°C until usage.
For gel retardation assays (18,19) the following synthetic oligonucleotids were prepared on a Gene Assembler Plus (Pharmacia):
EnA-oligonucleotide [nt −204 to −154 of the mouse H-2Kb promoter (30)]: 5′-(G)GGCAGTGAGGTCAGGGGTGGGGAAGCCCAGGGCTGGGGATTCCCCATCTCC-3′
EnA-TRE-oligo [nt −207 to −186 of the mouse H-2Kb promoter (30)]: 5′-(GGG)CCAGGCAGTGAGGTCAGGGGTG-3′
H2TFl-oligo [nt −175 to −156 of the mouse H-2Kb promoter (30)]: 5′-GGGCTGGGGATTCCCCATCT-3′
colTRE-oligo [nt −79 to −59 of the human collagenase promoter (2)]: 5′-(GGG)TAAAGCATGAGTCAGACACCT-3′
c-jun2TRE-oligo [nt −194 to −178 of the c-jun promoter (53)]: 5′-(GGG)AGCATTACCTCATCCCG-3′
T/G-EnA-TRE-oligo [nt −207 to −186 of the mouse H-2Kb promoter (30); this mutant oligonucleotide contains a T to G transversion mutation at position nt −199; (9)]: 5′-(GGG)CCAGGCAGGGAGGTCAGGGGTG-3′
pm-EnA-TRE-oligo [nt −207 to −186 of the mouse H-2Kb promoter (30); this mutant oligonucleotide contains three point mutations in the enhancer A TRE; compare to oligonucleotide 2, EnA-TRE-oligo]: 5′-(GGG)CCAGGCAGGGAGGTGGGGTG-3′
pm-H2TFl-oligo [nt −175 to −156 of the mouse H-2Kb promoter (30); this mutant oligonucleotide contains three point mutations in the H2TF1-element; compare to oligonucleotide 3, H2TFl-oligo]: 5′-GGGCTGGCGATTCCGGATCT-3′
A complementary oligonucleotide was used in each case to generate double-stranded DNA fragments. To allow labeling with [32P]dCTP and the Klenow fragment of E. coli DNA polymerase (36) either three guanosines were synthesized at the 5′ ends of the oligonucleotides or the oligonucleotides were digested with the appropriate restriction endonuclease. All binding assays (except the gel retardation assays using the NF-κB oligonucleotide) were performed as described (3,8). In case of the NF-κB band shift assays, binding reactions were performed in 10 mM Tris/HCl, pH 7.5, 50 mM NaCl, 1 mM DTT, 1 mM EDTA, 5% glycerol, 0.075 μg BSA/μl. Up to 5 μg of nuclear proteins was incubated with the radioactively labeled oligonucleotide. To compare the band shift pattern of nuclear extracts prepared from TNFα-treated HeLa-tk− cells with the band shift pattern of nuclear extracts prepared from serum-starved HeLa-tk− cells, we performed gel retardation assays in which we used nuclear extracts of a constant amount of cells instead of a constant protein concentration.
The following antibodies were used for antibody perturbation assays (Santa Cruz Biotechnology): anti-c-Jun (KM-1), anti-c-Jun/AP-1 (D), anti-c-Fos (K-25), anti-c-Fos (4), anti-Fral (R20), anti-Fra2 (Q-20), anti-ATF-2 (F2BR-1), anti-ATF3 (C-19), anti-NF-κB-p50 (NLS), anti-NF-κB-p65 (C20), anti-DP-1 (K-20), and anti-p300 (C-20). Antiserum (2 μl) was added to the incubation mixture (either overnight at 4°C or for 15 min at room temperature) prior to the addition of the labeled probe.
GST Pull-Down Assays Followed by Western Blot Analyses
The GST-235R fusion protein as well as the GST-235R deletion mutants were obtained by cloning the respective coding region of the adenovirus serotype 12 ElA wild-type/mutant 235R protein, obtained via PCR, into the Bam HI cloning site of the bacterial expression vector pGEX-2T (Pharmacia). GST-235RΔN lacks aa 1–29 of the 235R protein, GST-235RΔCR1 lacks the conserved region 1 (corresponding to aa 39–79 of the 235R protein), and GST-235RΔ1-79 lacks aa 1–79.
Whole-cell extract was prepared as described by Wang et al. (55) except that 150 mM NaCl was used instead of 250 mM NaCl. GST pull-down assays were performed as described (33). GST-E1A fusion protein (50 μg) was incubated with 4 mg cellular protein for protein-protein interaction assays. Interacting proteins were analyzed on 6% SDS/polyacrylamide gels (32) followed by Western blotting using the anti-p300 (C-20) antibody (Santa Cruz Biotechnology) as primary antibody. p300 was detected using the Super-Signal ULTRA system from Pierce (Rockford, IL), in combination with the horseradish peroxidase-conjugated anti-rabbit Ig from Amersham Life Science.
RESULTS
The EnA-TRE as well as the H2TF1 Element Is Necessary for an Efficient Activation of the H-2Kb Enhancer A
The enhancer A is one key element for the activation of the H-2Kb promoter (13). It contains several transcription factor binding sites, among them an AP-1/ATF recognition sequence (EnA-TRE) and a well-characterized NF-κB binding site (H2TF1 element). To determine whether both sites function independently or whether they cooperate in enhancer A activation, we have cloned three heterologous reporter constructs (Fig. 1A) and compared their activity in transient expression assays in HeLa-tk− cells: (i) EnA-tk-CAT5 containing the complete enhancer A in front of the herpes simplex virus (HSV) thymidine kinase (tk) promoter (nt −105 to +51) in the basal CAT reporter construct pBLCAT5 (6), (ii) H2TF1-tk-CAT5, which contains the enhancer A H2TF1 element, and (iii) EnA-TRE-tk-CAT5 carrying the AP-1/ATF binding site of the enhancer A. Heterologous reporter constructs were chosen instead of mutants of the natural H-2Kb promoter to prevent a possible interference with one or more of the other MHC class I promoter elements in our analyses.
FIG. 1.
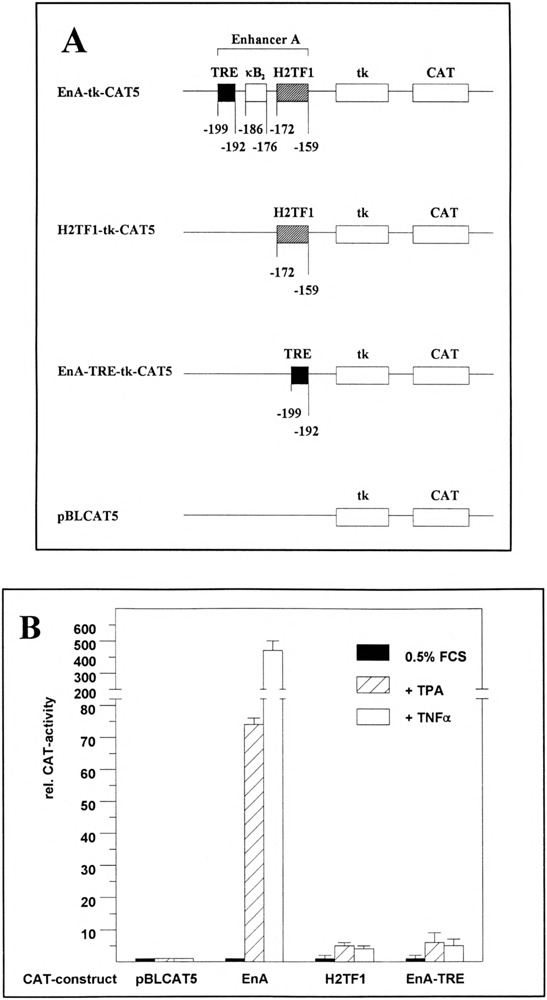
Neither the TRE on its own nor the H2TF1 element on its own of the H-2Kb enhancer A is able to mediate efficient reporter gene activation in response to TPA or TNFα. (A) Reporter constructs. In the reporter construct EnA-tk-CAT5 the expression of the CAT gene is driven by the enhancer A (nt −210 to −151) of the H-2Kb promoter and the HSV tk promoter (nt −105 to +51). The EnA-TRE, the κB2 site, and the H2TF1 element are indicated. The numbers refer to the positions of nt in the H-2Kb promoter relative to the CAP site as described by Kimura et al. (30). H2TFl-tk-CAT5 contains the H2TF1 element (nt −172 to −159), EnA-TRE-tk-CAT5 contains the TRE (nt −199 to −192) of the enhancer A. The basal reporter construct pBLCAT5 contains the HSV tk promoter and the CAT gene (6). CAT, chloramphenicol acetyltransferase gene. (B) Determination of CAT enzyme activity in cellular extracts obtained from HeLa-tk− cells. The respective reporter constructs (0.4 μg) [pBLCAT5, EnA-tk-CAT5 (EnA), H2TFl-tk-CAT5 (H2TF1), or EnA-TRE-tk-CAT5 (EnA-TRE)] were transfected in serum-starved (0.5% FCS) HeLa-tk− cells (black columns). TPA-treated (50 μg/ml) HeLa-tk− cells are represented by hatched columns; TNF-α treated (100 U/ml) HeLa-tk− cells are represented by empty columns. The quantification is the average of three independent CAT assays. EnA-tk-CAT5 promoter activity in serum-starved HeLa-tk− cells was set at 1.
EnA-tk-CAT5 displayed a very low CAT activity in HeLa-tk− cells kept under low serum condition (0.5%), which was set at 1 in Fig. 1B. However, CAT activity was strongly induced by TPA (75-fold; Fig. 1B) as well as TNFα (440-fold; Fig. 1B). Both substances have been shown to be very efficient activators of the cellular transcription factors AP-l/ATF and NF-κB (17,28,54). Contrary to EnA-tk-CAT5, TPA and TNFα were only slightly able to induce reporter gene expression from H2TFl-tk-CAT5 (sixfold or fourfold, respectively, Fig. 1B) or EnA-TRE-tk-CAT5 (sevenfold or sixfold, respectively; Fig. 1B). Both findings are consistent with published data of Israel et al. (26) and our own results showing that a reporter construct carrying three copies of the EnA-TRE is only poorly activated by cotransfecting a c-Jun expression plasmid in F9 cells (9). As expected, neither TPA nor TNFα was able to activate reporter gene expression from the basal reporter construct pBLCAT5 (Fig.1B). From these results we draw the conclusion that neither the EnA-TRE nor the H2TF1 element on their own can confer efficient promoter activation in response to TPA or TNFα.
To determine the role of the κB2 site in the activation process of the enhancer A we cloned three enhancer A deletion mutants (Fig. 2A): (i) κB2/H2TF1-tk-CAT5, lacking the EnA-TRE; (ii) TRE/H2TFl-tk-CAT5 in which the κB2 site is deleted; and (iii) TRE/κB2-tk-CAT5, lacking the H2TF1 element. After transfection in HeLa-tk− cells and treatment with TNFα, CAT activity was determined. The activity of EnA-tk-CAT5 was set at 100% (Fig. 2B). In contrast to the complete enhancer A, none of the three deletion mutants was able to confer a high reporter gene activation in response to TNFα (Fig. 2B). These results clearly show that the κB2 site is not able to substitute for either the EnA-TRE or the H2TF1 element in transcriptional activation. Moreover, they demonstrate that all three m-acting elements are necessary to confer efficient reporter gene activation and indicate that a correct spacing between the EnA-TRE and the H2TF1 element might be essential for activation. However, we cannot exclude that factors bound to the κB2 site are directly involved in the transcription activation process.
FIG. 2.
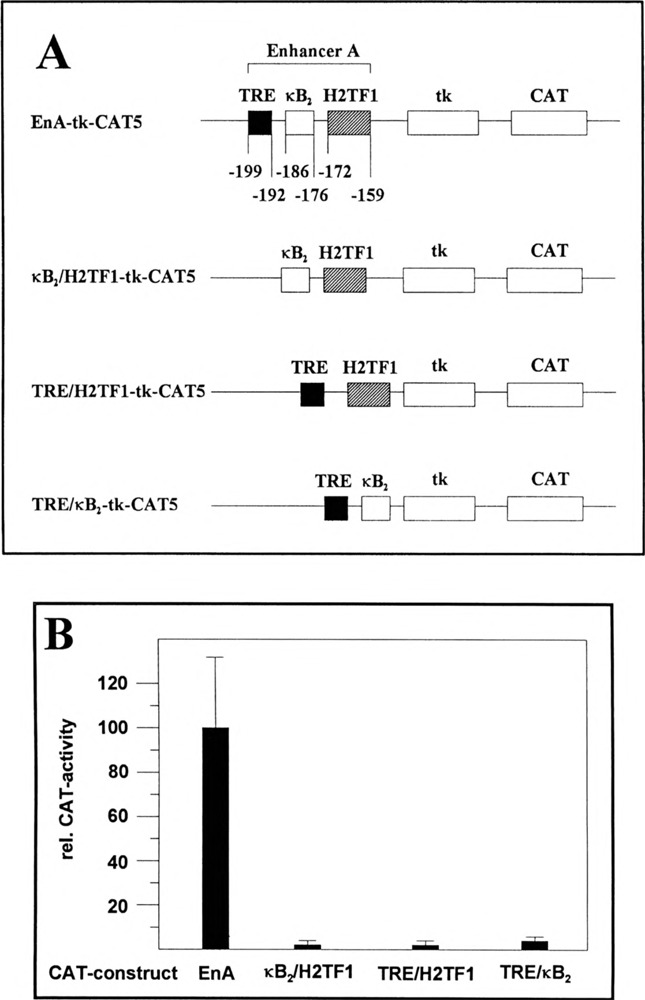
The κB2 site is indispensable for H-2Kb enhancer A activity. (A) Reporter constructs. In the reporter construct EnA-tk-CAT5 the expression of the CAT gene is driven by the enhancer A (nt −210 to −151) of the H-2Kb promoter and the HSV tk promoter (nt −105 to +51). κB2/H2TFl-tk-CAT5 lacks the EnA-TRE, TRE/H2TFl-tk-CAT5 lacks the κB2 site, and TRE/κB2-tk-CAT5 lacks the H2TF1 element. The numbers refer to the positions of nt in the H-2Kb promoter relative to the CAP site as described by Kimura et al. (30). (B) Determination of CAT enzyme activity in cellular extracts obtained from TNFα-treated HeLa-tk− cells. The respective reporter constructs (0.4 μg) were transfected. The quantification is the average of three independent CAT assays. EnA-tk-CAT5 promoter activity was set at 100%.
A Correct Spacing Between the EnA-TRE and the κB2/H2TF1 cis Elements Is Necessary for the Activation of the Enhancer A
Two enhancer A insertion mutants were constructed to analyze whether a correct spacing between the EnA-TRE and the other transcription factor binding sites is one prerequisite for the activation of the enhancer A: (i) EnA+5-tk-CAT5 containing five additional nucleotides between the EnA-TRE and the κB2 site (Fig. 3A) and (ii) EnA+10-tk-CAT5 containing 10 additional nucleotides between the EnA-TRE and the κB2-site (Fig. 3A). The activity of both promoter mutants was compared to the EnA-tk-CAT5 wild-type reporter construct in transient expression assays in TNFα-treated HeLa-tk-cells. These CAT-assays demonstrated that both insertion mutants were unable to confer CAT gene expression in response to TNFα (Fig. 3B), underscoring our assumption that a correct spacing of the cis-acting elements in the enhancer A is necessary for its activation.
FIG. 3.
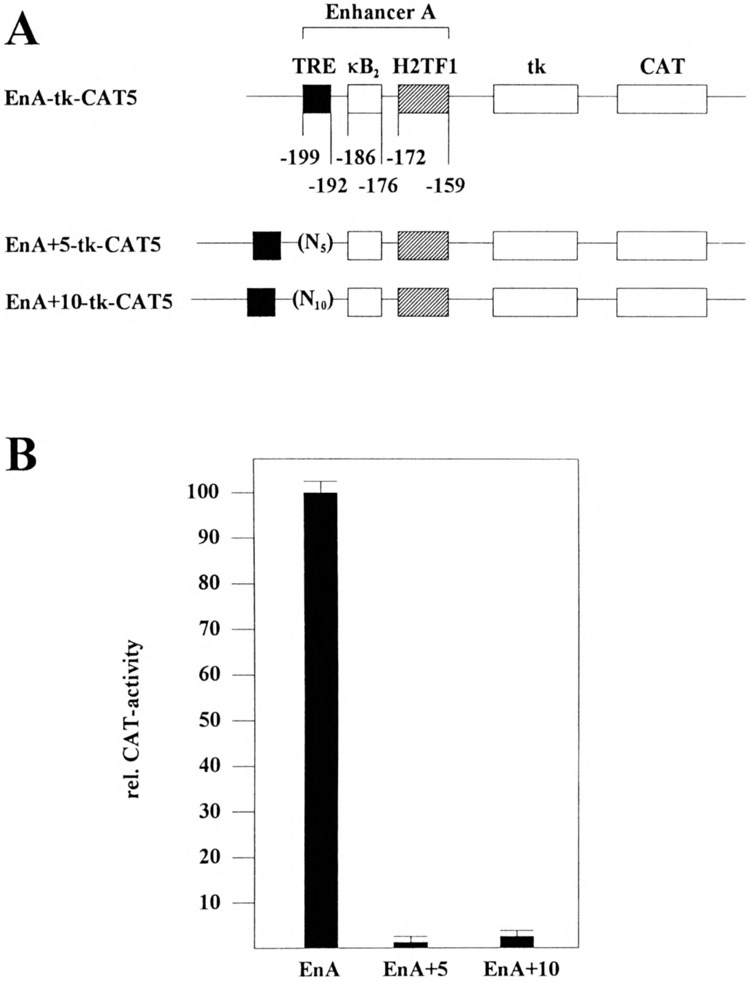
H-2Kb enhancer A activation depends on a correct spacing between the EnA-TRE and the κB2 site and H2TF1 element. (A) Reporter constructs. In the reporter construct EnA-tk-CAT5 (EnA) the expression of the CAT gene is driven by the enhancer A (nt −210 to −151) of the H-2Kb promoter and the HSV tk promoter (nt −105 to +51). EnA+5-tk-CAT5 (EnA+5) and EnA+10-tk-CAT5 (EnA+10) contain an insertion of 5 or 10 nucleotides, respectively, in front of nt −189 between the EnA-TRE and κB2 site. The numbers refer to the positions of nt in the H-2Kb promoter relative to the CAP site as described by Kimura et al. (30). Transcription factor binding sites are indicated. (B) Determination of CAT enzyme activity in cellular extracts obtained from TNFα-treated HeLa-tk− cells. The respective reporter constructs (0.4 ug) were transfected. The quantification is the average of three independent CAT assays. EnA-tk-CAT5 promoter activity was set at 100%.
The EnA-TRE Is Bound by at Least Two Heterodimers of the AP-l/ATF Transcription Factor Family
We have shown previously that the c-Jun transcription factor binds to the EnA-TRE (9). To identify its dimerization partner on this specific promoter element we performed gel retardation assays using a radioactively labeled EnA-TRE oligonucleotide in combination with nuclear extract prepared from either serum-starved or TNFα-treated HeLa-tk− cells and a panel of antibodies directed against distinct members of the AP-l/ATF transcription factor family.
Incubation of the EnA-TRE oligonucleotide with nuclear extract prepared either from TNFα-treated or from serum-starved HeLa-tk− cells gave rise to two complexes, marked 1 and 2 in Fig. 4A. However, in contrast to the band shift pattern obtained from TNFα-treated cells, the intensity of bands was reduced using nuclear extracts from serum-starved cells (data not shown), reflecting a lower concentration of these factors in serum-starved cells. This result is in agreement with published data demonstrating that c-Jun is expressed in many cell types at low levels and that the expression of members of the AP-l/ATF transcription factor family can be greatly induced by TNFα (28). Moreover, ATF-2 is constitutively expressed and preexisting ATF-2 is activated, too, through TNFα (28). Preincubation of the nuclear extract prepared from TNFα-treated HeLa-tk− cells with an anti-c-Jun mouse monoclonal antibody reduced the amount of both complexes (Fig. 4A, lanes 3 and 11). In addition, a more slowly migrating complex was generated (Fig. 4A, lanes 3 and 11). The formation of both complexes was also prevented using an independent goat polyclonal anti-c-Jun antiserum (Fig. 4A, lanes 4 and 12). However, no supershifted complex could be detected, which might be due to the fact that this anti-c-Jun antiserum recognizes the DNA binding domain of the transcription factor, thereby preventing its interaction with DNA. These data confirm our previous findings showing that c-Jun is one of the constituents of the transcription factor complex bound to the EnA-TRE of the H-2Kb promoter (9).
FIG. 4.
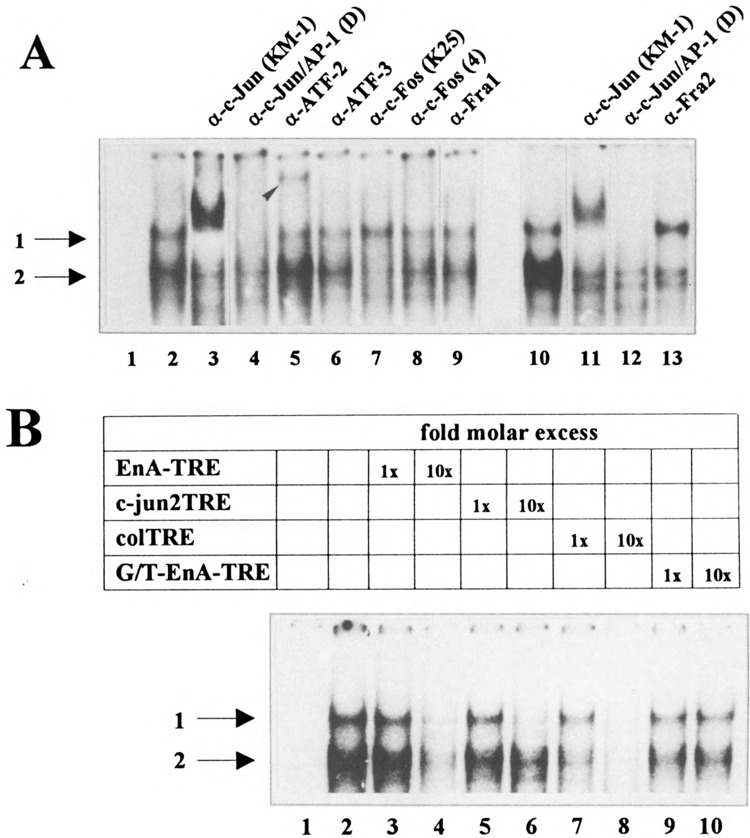
The EnA-TRE is bound by at least two distinct AP-l/ATF transcription factor complexes in vitro. (A) For antibody perturbation assays nuclear extracts prepared from TNFα-treated HeLa-tk− cells were incubated with a radioactively labeled synthetic EnA-TRE-oligo representing nucleotides −207 to −186 of the H-2Kb promoter in the absence (lanes 2 and 10) or the presence of two different anti-c-Jun antibodies (lanes 3, 4, 11, and 12), an anti-ATF-2 mouse monoclonal antibody (lane 5), an anti-ATF-3 rabbit polyclonal antiserum (lane 6), two different anti-c-Fos goat polyclonal antisera (lanes 7 and 8), an anti-Fral rabbit polyclonal antiserum (lane 9), or an anti-Fra2 rabbit polyclonal antiserum (lane 13). Lane 1 shows the labeled oligo without extract. A complex supershifted by the anti-ATF-2 antibody is indicated by an arrow (lane 5). Protein-DNA complexes were resolved on 5% nondenaturing polyacrylamide gels. The positions of the retarded complexes are indicated. (B) Competition analyses using nuclear extracts of TNFα-treated HeLa-tk− cells and a radioactively labeled synthetic EnA-TRE oligonucleotide in combination with a onefold or 10-fold molar excess of unlabeled EnA-TRE-oligo (lanes 3 and 4), of unlabeled c-jun2TRE-oligo (lanes 5 and 6), of unlabeled colTRE-oligo (lanes 7 and 8), or of the unlabeled mutant G/T-EnA-TRE (lanes 9 and 10). This mutant oligonucleotide contains a T to G transversion mutation at position nt −199 (9). Lane 2 shows the band shift pattern without competitor, lane 1 the EnA-TRE oligo-without nuclear extract.
Preincubation of the nuclear extract with an anti-ATF-2 mouse monoclonal antibody led to the reduction of the amount of complex 1 and to a supershifted complex (Fig. 4A, lane 5; the supershifted complex is marked by an arrow), indicating that complex 1 also contains the ATF-2 protein, most probably as c-Jun/ATF-2 heterodimer. Nevertheless, we cannot rule out the presence of a comigrating ATF-2/ATF-2 homodimer. In contrast to the ATF-2 antibody, the usage of a rabbit polyclonal anti-ATF-3 antiserum only slightly interfered with the generation of complex 1 and complex 2 (Fig. 4A, lane 6). As we obtained no supershifted complex, we suppose that this slight decrease in the amount of complex 1 and complex 2 is rather due to a weak cross-reactivity of the anti-ATF-3 polyclonal antiserum with other members of the AP-l/ATF transcription factor family included in complex 1 and 2 than to a specific interaction. Therefore, ATF-3 is most probably not included in one of the protein complexes bound to the EnA-TRE in this cell system.
Interestingly, preincubation with an anti-c-Fos goat polyclonal antiserum, which is broadly reactive with c-Fos, FosB, Fral, and Fra2, completely inhibits the formation of complex 2 (Fig. 4A, lane 7). A second anti-c-Fos-specific goat polyclonal antiserum [anti-c-Fos(4); Fig. 4A, lane 8] or an anti-Fral rabbit polyclonal antiserum (Fig. 4A, lane 9) reduces modestly the amount of complex 1 and complex 2. In contrast, the formation of complex 2 was nearly completely inhibited using an anti-Fra2-specific rabbit polyclonal antiserum (Fig. 4A, lane 13). As Fra2 is able to heterodimerize with c-Jun (48), we conclude that complex 2 consists, at least in part, of a c-Jun/Fra2 heterodimer. However, due to the weak interaction of the anti-c-Fos(4) and the anti-Fral antiserum with complex 2, we cannot exclude that this complex contains minor amounts of c-Fos and/or Fral.
It was described that the EnA-TRE overlaps with a retionoid X receptor p recognition sequence in the enhancer A (43). Therefore, we included an anti-retinoid X receptor β rabbit polyclonal antiserum in our analyses. However, the usage of this antiserum did not interfere with the generation of complex 1 or complex 2 (data not shown).
To confirm our findings that the EnA-TRE can be bound by at least two different AP-l/ATF transcription factor complexes, we performed competition experiments. The radioactively labeled EnA-TRE oligonucleotide was incubated with nuclear extract prepared from TNFα-treated HeLa-tk− cells in the presence of a 1- or 10-fold molar excess of an unlabeled c-jun2TRE or colTRE oligonucleotide. The c-jun2TRE oligonucleotide [TTACCTCA; nt −190 to −183 of the c-jun promoter (53)] was shown to bind predominantly c-Jun/ATF-2 heterodimers (53), whereas the colTRE [TGAGTCA; nt −72 to −65 of the human collagenase promoter (2)] represents the classical c-Jun/c-Fos recognition sequence (28), which can be bound by c-Jun/Fra2 and c-Jun/c-Fos with a similar affinity (48). Therefore, the colTRE should compete efficiently complex 2 if c-Jun/Fra2 and/or c-Jun/c-Fos is present. A 10-fold molar excess of the jun2TRE oligonucleotide completely inhibits the formation of complex 1 whereas complex 2 was slightly affected (Fig. 4B, lane 6). On the other hand, in the presence of the same molar concentration of unlabeled colTRE oligocucleotide complex 2 was greatly diminished (Fig. 4B, lane 7), whereas a 10-fold molar excess inhibits the formation of complex 1 as well as of complex 2 (Fig. 4B, lane 8). These data are consistent with our findings obtained with the antibody perturbation assays, indicating that complex 1 contains predominantly c-Jun/ATF-2 heterodimers whereas complex 2 contains at least c-Jun/Fra2 heterodimers.
To demonstrate the specificity of our competition experiments, we synthesized an EnA-TRE oligonucleotide point mutant in which the thymine was replaced by a guanine [TGAGGTCA → GGAGGTCA (9)]. A comparable mutation was shown to greatly inhibit the transactivation of the human collagenase promoter by c-Jun/c-Fos (2), indicating that this nucleotide is essential for a functional interaction between the promoter element and the transcription factor. Most interestingly, neither the formation of complex 1 nor the formation of complex 2 was completely competed by a 10-fold molar excess of the mutant EnA-TRE oligonucleotide (Fig. 4B, lane 10). Together with the competition experiments using the unlabeled wild-type EnA-TRE oligonucleotide (Fig. 4B, lanes 3 and 4), these results demonstrate the specificity of the protein-DNA interaction. Moreover, they indicate that the first T of the binding site octamer seems to be of importance for the interaction of c-Jun/ATF-2 and c-Jun/Fra2 with the EnA-TRE.
The Coactivator p300 Is Recruited Through NF-κB to the H2TF1 Element
It is well documented that the H2TF1 element is bound by NF-κB as well as by a homodimer consisting of the NF-κB p50 subunit (27,57). Furthermore, very recent results demonstrate that p300 functions as a transcriptional coactivator for the p65 subunit of NF-κB on E-selectin- and VCAM-l-CAT-reporter constructs (20). To check whether p300 is also involved in the transcriptional activation of the enhancer A of the H-2Kb promoter, we performed gel retardation and transient expression assays.
For gel retardation assays an oligonucleotide (H2TF1 oligonucleotide) spanning nt −175 to −156 of the H-2Kb promoter and nuclear extract prepared from either serum-starved or TNFα-treated HeLa-tk− cells was used. Protein-DNA complexes were resolved on 5% nondenaturing polyacrylamide gels and detected by autoradiography.
Incubation of the H2TF1 oligonucleotide with nuclear extracts obtained from TNFα-treated HeLa-tk− cells gave rise to two complexes (complex 1 and complex 2; Fig. 5A, B, lanes 2), which is in agreement with data published by Schouten et al. (42) and Liu et al. (35) using protein extracts of other cells. The formation of both complexes was efficiently prevented by an anti-p50-specific goat polyclonal antiserum. In addition, a supershifted complex was generated (Fig. 5A, B, lanes 3). The formation of complex 1 was also completely inhibited by an anti-p65-specific goat polyclonal antiserum whereas complex 2 was not affected (Fig. 5A, B, lanes 4). In this case two more slowly migrating complexes were generated. The reason why two slowly migrating complexes are formed in the presence of the anti-p65-specific goat polyclonal antiserum is not yet clear, but might be due to the presence of two or more antibody species recognizing different p65 epitopes resulting in two immunocomplexes with different migration properties. From these results we conclude (i) that the protein-DNA complex 2 includes the p50-NF-κB1 protein probably as p50/p50 homodimer, and (ii) that the protein-DNA complex 1 contains p50-NF-κBl as well as p65/relA. The latter result was confirmed using a second independent goat polyclonal anti-p65-specific antiserum (data not shown).
FIG. 5.
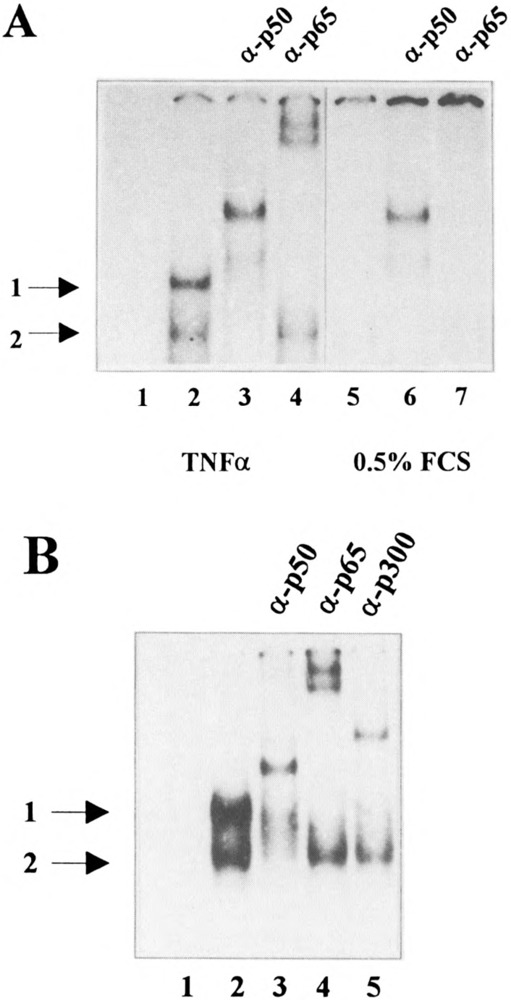
The cellular coactivator p300 is recruited to the H2TF1 element through NF-κB. Nuclear extracts prepared from TNFα-treated HeLa-tk− cells (A, lanes 1–4; B) or from serum-starved HeLa-tk− cells (A, lanes 5–7) were incubated with a radioactively labeled synthetic H2TFl-oligo representing nucleotides −175 to −156 of the H-2Kb promoter in the presence of an anti-p50 goat polyclonal antiserum (A, lanes 3 and 6; B, lane 3), in the presence of an anti-p65 goat polyclonal antiserum (A, lanes 4 and 7; B, lane 4) or in the presence of an anti-p300 rabbit polyclonal antiserum (B, lane 5). Lane 1 shows the labeled oligo without extract, lanes 2 (and 5 in A) show the labeled oligo with extract. Protein-DNA complexes were resolved on 5% nondenaturing polyacrylamide gels. The positions of the retarded complexes are indicated.
In contrast to the nuclear extract prepared from TNFα-treated HeLa-tk− cells, nuclear extract obtained from serum-starved HeLa-tk− cells gave rise to only a residual amount of the p50/p50 homodimer (Fig. 5A, lane 5). Interestingly, the anti-p50-specific goat polyclonal antiserum stabilizes the interaction between the p5G7p50 homodimer and the H2TF1 element (Fig. 5A, lane 6). A p50/p65 NF-κB hetero-dimer could not be detected (Fig. 5A, lanes 5 and 7), which can be explained by published data showing that TNFα treatment of cells results in the translocation of the p50/p65 NF-κB heterodimer into the nucleus due to the degradation of IκB via the ubiquitination-proteosomes pathway and in the processing of the p50 precursor p105 (54).
To analyze whether p300 is included in the protein-DNA complex 1 containing NF-κB, we performed gel retardation assays in the presence of an anti-p300 rabbit polyclonal antiserum. Most interestingly, this antiserum prevents the formation of complex 1 whereas complex 2 was only slightly affected (Fig. 5B, lane 5). Moreover, a more slowly migrating protein-DNA complex was formed (Fig. 5B, lane 5). This result suggests that complex 1 might also contain the coactivator p300.
To confirm that the anti-p300 rabbit polyclonal antiserum used in gel retardation assays is highly specific for p300, we performed GST pull-down assays followed by Western blot analyses in which we took advantage of the finding that the 235R ElA protein of adenovirus type 12 interacts with the cellular co-activator p300 in an N-terminus- and CR1-dependent manner [(56); K. S. Lipinsκi, H. Esche, and D. Brockmann, unpublished data]. This interaction is necessary for the viral regulation of expression of specific target genes as well as for the adenovirus-mediated cell transformation (39).
Whole-cell extract was prepared from HeLa-tk− cells and incubated with the GST leader sequence (GST) or a fusion protein consisting of the GST leader sequence and the 235R ElA protein of adenovirus type 12 (GST-235R). GST-235R-interacting proteins were resolved on a 6% SDS/polyacrylamide gel followed by Western blotting using the same anti-p300 rabbit polyclonal antiserum as for the gel retardation assays shown in Fig. 5B. This antiserum recognizes a cellular protein in the size of 300 kDa in Western blot analyses (Fig. 6, lane 1; indicated by an arrow). A cellular protein of the same size interacts with GST-235R (Fig. 6, lane 3) but not with the GST leader sequence (Fig. 6, lane 2). Removal of the conserved region 1 greatly diminishes the capacity of the resultant mutant to bind to p300 (Fig. 6, lane 5), whereas deletion of aa 1-29 (GST-235RΔN; Fig. 6, lane 4) or deletion of aa 1-79 (GST-235RΔN/CR1; Fig. 6, lane 6) completely prevents their interaction. These data are consistent with our finding that in case of the Ad12 ElA 235R protein aa 1–29 are sufficient for an interaction with p300 (K. S. Lipinski, H. Esche, and D. Brockmann, unpublished data). To exclude that the difference in the binding capacity of the 235R mutants is due to different concentrations of the respective mutants used in GST pull-down assays, conventional SDS/polyacrylamide gel electrophoresis was performed with a constant aliquot of the glutathione sepharose beads coupled GST fusion proteins used in the pull-down assays. Coomassie blue staining showed the comparable concentration of fusion proteins in these assays (data not shown). These results strongly indicate that the anti-p300 antiserum recognizes specifically p300 and therefore support our presumption that the cellular coactivator p300 and NF-κB are able to form a complex on the H2TF1 element of the H-2Kb promoter.
FIG. 6.
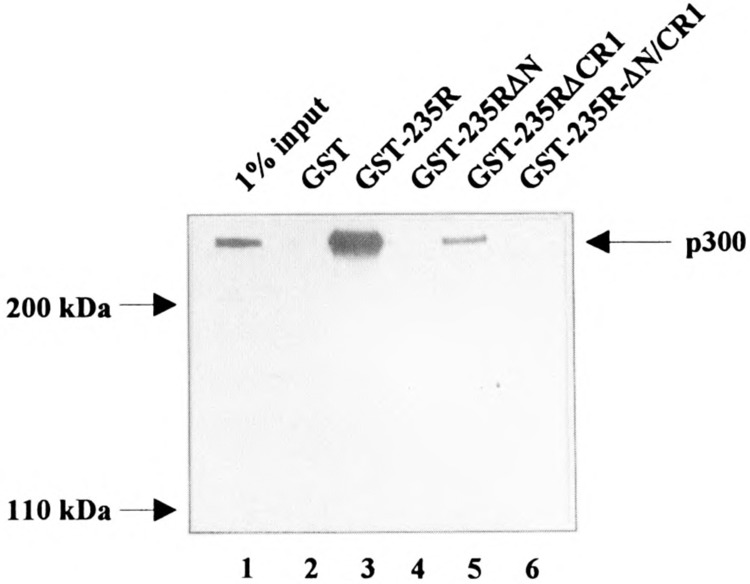
The cellular coactivator p300 interacts with aa 1–79 of the 235R ElA protein of adenovirus type 12. Whole-cell extract was prepared from HeLa-tk− cells and incubated with the GST leader sequence (GST, lane 2), a fusion protein consisting of the GST leader sequence and the 235R ElA protein of adenovirus type 12 (GST-235R, lane 3), or fusion proteins containing 235R mutants (lanes 4–6). After extensive washing to remove unspecifically bound cellular proteins, GST-235R-interacting proteins were resolved on a 6% SDS/polyacrylamide gel followed by Western blotting using the same anti-p300 rabbit polyclonal antiserum as in the experiment described in Fig. 5. GST-235RAN lacks aa 1–29 of the 235R protein (lane 4), GST-235ACR1 lacks aa 39–79 (lane 5), and GST-235RAN/CR1 lacks aa 1–79 (lane 6). Lane 1 represents 1% of the cellular extract input. The position of the p300 protein as well as of the marker proteins is indicated.
p300 Functions as a Coactivator for Enhancer A-Driven Gene Expression
If p300 functions as a coactivator in the activation mechanism of the enhancer A its cotransfection should increase CAT gene expression from EnA-tk-CAT5. This hypothesis was tested in transient expression assays. Rising amounts of the p300 expression vector pCMVβ-p300 were cotransfected with EnA-tk-CAT5 in HeLa-tk− cells. After treatment with TNFα, cells were harvested and CAT activity determined. The activity of EnA-tk-CAT5 in the absence of pCMVβ-p300 was set at 1 (Fig. 7, empty box). Cotransfection of 0.2 or 0.35 μg pCMVβ-p300 results in a 2.4-fold activation of CAT gene expression (Fig. 7, black boxes) compared to cotransfection experiments using the empty expression vector. Cotransfection of 0.05 μg pCMVβ-p300 has no effect on the enhancer A-driven reporter gene expression whereas a pCMVβ-p300 concentration above 0.5 μg per dish represses reporter gene expression, most probably due to a squelching mechanism (data not shown).
FIG. 7.
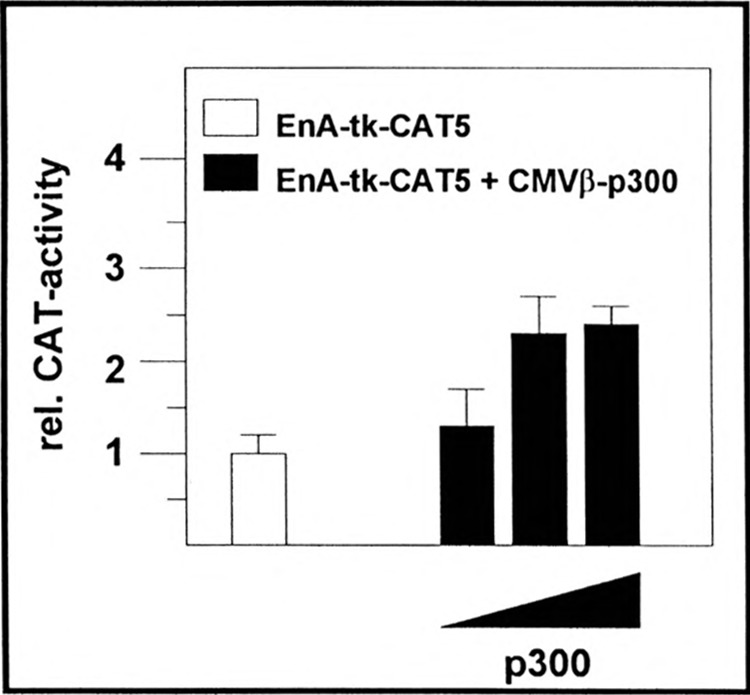
Cotransfected p300 enhances enhancer A-driven gene expression. EnA-tk-CAT5 (0.4 μg) was cotransfected with pCMVβ (empty box) or rising amounts of the p300 expression vector pCMVβ-p300 (0.05–0.35 μg; black columns) in HeLa-tk− cells. After treatment with TNFα, cells were harvested and CAT activity determined. Empty expression vector pCMVβ was always cotransfected to keep the DNA concentration constant. EnA-tk-CAT5 in the absence of pCMVβ-p300 was set at 1.
To make sure that p300 activates CAT gene expression through factors bound to the enhancer A and not through the tk promoter, we performed transient expression assays using the mutants κB2/H2TFl-tk-CAT5, TRE/H2TF1-tk-CAT5, and TRE/κB2-tk-CAT5 described in Fig. 2A. None of these enhancer A mutants was inducible by cotransfected p300 in the presence of TNFα (data not shown). Taken together these data demonstrate that p300 functions as a coactivator for enhancer A-driven gene expression.
Loss of Binding of AP-l/ATF or NF-κB to Their Respective Promoter Elements in the Enhancer A Correlates With Loss of Enhancer A Inducibility
To analyze whether the binding of AP-l/ATF and NF-κB transcription factors to their respective promoter elements in the enhancer A correlates with the activation of the enhancer A, we constructed several point mutants: (i) pm-TRE-EnA-tk-CAT5 containing a TRE point-mutated enhancer A, (ii) pm-H2TFl-EnA-tk-CAT5 containing a H2TF1 point-mutated enhancer A, and (iii) pm-TRE/H2TF1-EnA-tk-CAT5 containing a TRE/H2TF1 point-mutated enhancer A. These point mutations lead to a complete loss of the binding of AP-l/ATF to the EnA-TRE (Fig. 8A, compare lanes 2–4 with lanes 6–7) and of NF-κB to the H2TF1 element (Fig. 8B, compare lanes 2–4 with lanes 6–8) as demonstrated by gel retardation assays using nuclear extract prepared from TNFα-treated HeLa-tk− cells. To determine their promoter activity, pm-TRE-EnA-tk-CAT5, pm-H2TF1-EnA-tk-CAT5, and pm-TRE/H2TF1-EnA-tk-CAT5 were transfected in HeLa-tk− cells. After treatment with TNFα, cells were harvested and CAT activity was compared to the reporter gene activity driven by the wild-type enhancer A. Figure 8C demonstrates that point mutations in either the EnA-TRE or in the H2TF1 element prevented enhancer A activation through TNFα in transient expression assays. Taken together these results show a strong correlation between the loss of AP-l/ATF or NF-κB binding to their respective cis elements in the enhancer A and loss of the inducibility of the enhancer A.
FIG. 8.
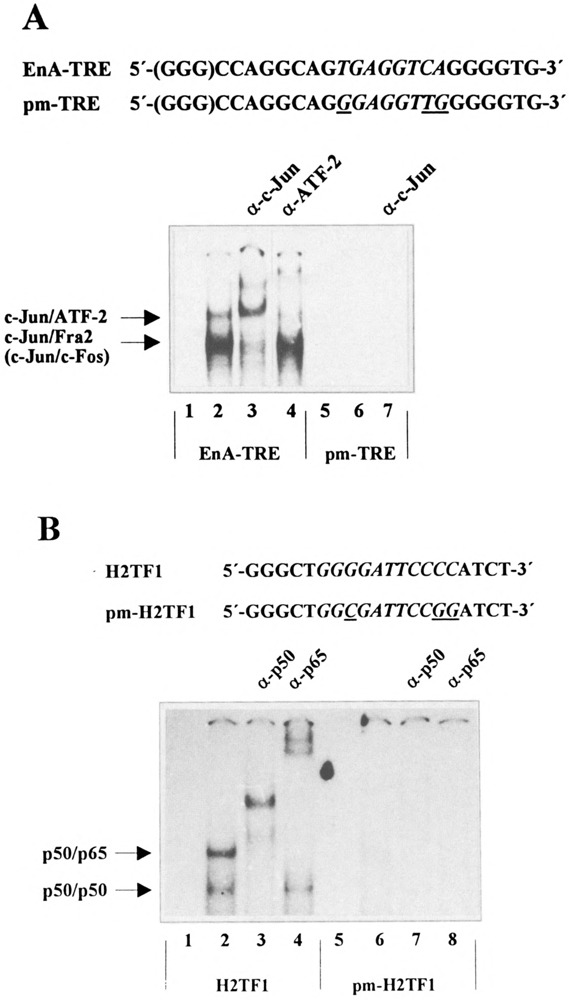
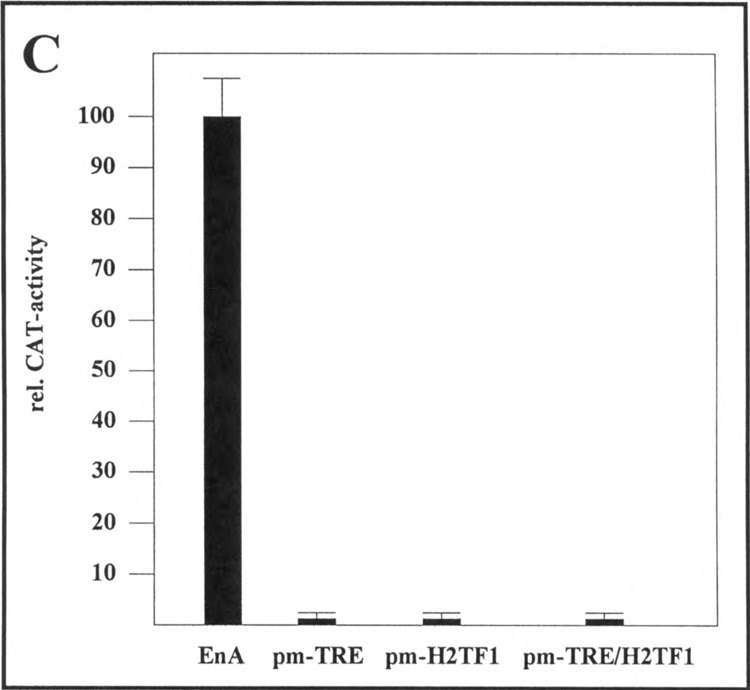
Loss of binding of AP- l/ATF or NK-κB to their respective binding sites in the enhancer A correlates with the loss of the inducibility of the enhancer A. (A) For antibody perturbation assays nuclear extracts prepared from TNFα-treated HeLa-tk− cells were incubated with either a radioactively labeled synthetic EnA-TRE-oligo representing nucleotides −207 to −186 of the H-2Kb promoter (lanes 2–4) or a radioactively labeled point-mutated EnA-TRE-oligo (lanes 6 and 7) as indicated on the top of the figure. Lanes 3 and 7, gel retardation assays in the presence of an anti-c-Jun antibody; lane 4, gel retardation assay in the presence of an anti-ATF-2 antibody; lanes 2 and 6, gel retardation assays in the absence of antisera. Lanes 1 and 5 show the labeled oligos without extract. Protein-DNA complexes were resolved on 5% nondenaturing polyacrylamide gels. The positions of the retarded complexes are indicated. (B) Nuclear extracts prepared from TNFα-treated HeLa-tk− cells were incubated with a radioactively labeled synthetic H2TFl-oligo representing nucleotides −175 to −156 of the H-2Kb promoter (lanes 2–4) or a radioactively labeled point-mutated H2TFl-oligo (lanes 6–8) as indicated on the top of the figure. Lanes 3 and 7, gel retardation assays in the presence of an anti-p50 goat polyclonal antiserum; lanes 4 and 8, gel retardation assays in the presence of an anti-p65 goat polyclonal antiserum. Lanes 2 and 6, gel retardation assays in the absence of antisera; lanes 1 and 5 show the labeled oligos without extract. The positions of the retarded complexes are indicated. (C) 0.4 μg of either EnA-tk-CAT5 (EnA) or the point mutants pm-TRE-EnA-tk-CAT5 (pm-TRE), pm-H2TFl-EnA-tk-CAT5 (pm-H2TFl), pm-TRE/H2TF1-EnA-tk-CAT5 (pm-TRE/H2TFl) was transfected in HeLa-tk− cells. After treatment with TNFα, cells were harvested and CAT activity determined. The quantification is the average of three independent CAT assays. EnA-tk-CAT5 promoter activity was set at 100%.
The Enhancer A Can Be Bound Simultaneously by AP-l/ATF as well as NF-κB Transcription Factors
If the enhancer A is synergistically activated by AP-l/ATF and NF-κB, proteins of both transcription factor families should be present in the same retarded complex in band shift analyses using the enhancer A as oligonucleotide. To proof this assumption, a radio-actively labeled oligonucleotide spanning the entire enhancer A (EnA oligonucleotide; nt −204 to −154) of the H-2Kb promoter was incubated with nuclear extract prepared from TNFα-treated HeLa-tk− cells in the presence of different specific antibodies.
Incubation of the EnA oligonucleotide with nuclear extract prepared from TNFα-treated HeLa-tk− cells gives rise to several retarded complexes, termed A, B, and C (Fig. 9, lane 2). The formation of complex A was prevented through two different anti-c-Jun antisera (Fig. 9, lanes 3 and 4) and through an anti-p65 goat polyclonal antiserum (Fig. 9, lane 7). Moreover, its amount decreased in the presence of an anti-ATF-2 antibody (Fig. 9, lane 5). As an unrelated antiserum directed against the DP-1 protein does not prevent the formation of complex A (and complexes B and C; Fig. 9, lane 8), we conclude that complex A contains c-Jun and ATF-2 (most probably as c-Jun/ATF-2 heterodimer) as well as p65. However, the dimerization partner of p65 is not yet clear due to the fact that we cannot differentiate between the two possibilities that (i) the anti-50 antiserum stabilizes complex A or (ii) a complex shifted by the anti-p50 antiserum comigrates with complex A (Fig. 9, lane 6).
FIG. 9.
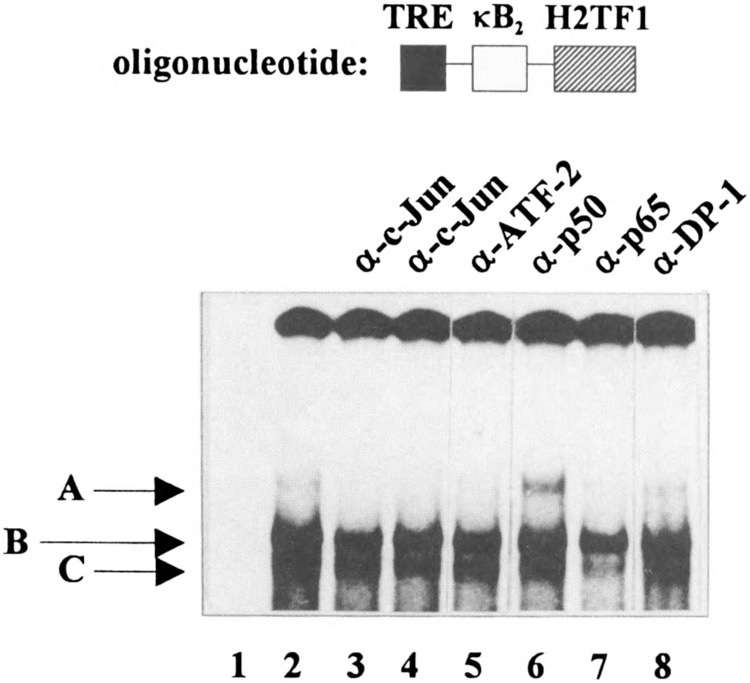
The enhancer A can be bound simultaneously by AP-1/ATF and NK-κB transcription factors in vitro. Nuclear extracts prepared from TNFα-treated HeLa-tk− cells were incubated with a radioactively labeled synthetic enhancer A oligonucleotide representing nucleotides −204 to −154 of the mouse H-2Kb promoter in the presence or absence of specific antibodies as indicated. Protein-DNA complexes were resolved on 5% nondenaturing polyacrylamide gels and retarded complexes were visualized by autoradiography.
The composition of complexes B and C is not clear. Complex C contains at least the p65 protein (Fig. 9, lane 7). Of note, neither the anti-c-Fos antiserum, which is broadly reactive with c-Fos, FosB, Fral, and Fra2, nor the anti-Fra2 antiserum nor the anti-p300 antiserum had a clear-cut effect on complexes A to C, although the anti-c-Fos antiserum gave rise to a supershifted complex (data not shown). Taken together these data show that the enhancer A can be bound simultaneously by AP-l/ATF and NF-κB transcription factors.
DISCUSSION
The aim of this study was to analyze the factors bound to the TRE of the H-2Kb enhancer A and to clarify their interrelationship in the process of transcriptional activation with transcription factors associated with other cis-acting elements of this enhancer. We identified two distinct AP-l/ATF complexes able to bind to the EnA-TRE: c-Jun/Fra2 and c-Jun/ATF-2. Most interestingly, the EnA-TRE on its own is unable to confer efficient activation on a heterologous promoter construct. AP-l/ATF transcription factors have to cooperate with NF-κB, bound to the H2TF1-element, to highly induce reporter gene expression. In addition, our findings indicate that NF-κB recruits p300 as coactivator to the enhancer A as cotransfected p300 augmented transcriptional activation.
Our data suggest that all three cis elements are involved in mediating transcriptional activation of the enhancer A in response to TNFα and TPA. However, the function of the second NF-κB binding site, κB2, is less clear. Own unpublished antibody perturbation assays show that κB2 can be bound by NF-κB recruiting p300 to the κB2 site (data not shown). On the other hand, they also suggest that the EnA-TRE and the κB2 sites cannot be bound simultaneously by AP-l/ATF transcription factors and NF-κB, respectively (data not shown), and that a mutated enhancer lacking the H2TF1 element is unable to confer activation of a heterologous reporter construct. As the enhancer mutant TRE/H2TF1 lacking 11 nucleotides of the κB2 site is also not functional, one might conclude that a correct spatial distance between the EnA-TRE and the H2TF1 binding site is necessary for a high transcriptional activation. This conclusion is supported by our findings that enhancer A mutants, in which 5 or 10 nucleotides are inserted between the EnA-TRE and the κB2 site, are also unable to mediate reporter gene activation in response to TNFα. These data are partly in agreement with results published by Mansky et al. (37). Using mutants of the HLA-A2 promoter these authors have shown that the complete enhancer A gives rise to the highest CAT activity in transient expression assays compared to H2TF1 or κB2 mutants, which is consistent with findings of Israel et al. (26) using the H-2Kb promoter in TNFα-treated HeLa cells. Interestingly, point substitutions in H2TF1 (bound either by a BI protein complex containing the p50 subunit of NF-κB or by Bill containing p65) as well as in κB2 (not bound by BI) interferes equally with promoter activation. Therefore, Mansky et al. (37) suggest that occupancy of both sites is required for enhancer activation, although a multiprotein complex containing BI as well as Bill was not detected in gel retardation assays. Our gel retardation assays indicate that a simultaneous occupation of the κB2 site and the EnA-TRE is not possible (data not shown), underscoring our interpretation that the DNA sequence itself as spacer is important for activation, most probably to guarantee a correct spacing between both transcription factor binding sites. The different results might be explained by the fact that the κB2 site of the H-2Kb and HLA-A2 promoters is not conserved (12,30), allowing a different kind of regulation.
A very interesting finding of our studies is that at least two distinct AP-l/ATF complexes (c-Jun/ATF-2, c-Jun/Fra2) are able to bind to the EnA-TRE. These results extend published data concerning the composition of the complex bound to the EnA-TRE (9,31,58). For example, using DNAse I protection analyses Korber and coworkers (31) have shown that crude nuclear extracts prepared from cells of the mouse myeloma cell line MPC 11 gave rise to the same protected region in the enhancer A (spanning the EnA-TRE)-like AP-1 complexes purified from HeLa cells. However, the composition of the purified AP-1 complex was not clarified in these studies. In a second investigation (58) it was shown that the incubation of an enhancer A oligonucleotide with nuclear extracts prepared from D122 cells (D122 is a high metastatic cell clone, derived from Lewis lung carcinoma cells obtained from C57BL/6 mice) gave rise to five complexes. Preliminary gel shift assays revealed that the formation of two of these complexes is disturbed in the presence of an anti-c-Fos antibody (58).
c-Jun/ATF-2 is a well-known activator of gene expression via ATF/CREB binding sites [see, e.g., (53)]. c-Jun/Fra2 was shown to bind AP-1 as well as ATF recognition sequences (48). Moreover, as heterodimer with c-Jun, Fra2 was described to have a suppressive function on the colTRE (48), which is in contrast to our expectations that c-Jun/Fra2 should function as an activator for the H-2Kb enhancer A. There are several explanations for this discrepancy, (i) The results obtained in the gel retardation assays do not reflect the in vivo situation during MHC class I gene activation. In vitro c-Jun/Fra2 binds to the EnA-TRE but under physiological conditions this heterodimer might not be involved in the activation of the enhancer A because the EnA-TRE is occupied by a c-Jun/ATF-2 heterodimer, (ii) c-Jun/Fra2 can have opposite effects on gene expression depending on the cell type and/or promoter/enhancer context. In this context it is interesting that a Fra2-containing complex called Mod-1 was shown to bind to a specific sequence in the enhancer of the swine class I (PD1) gene (21), although the sequence under study does not contain an obvious AP-l/ATF recognition sequence (16). In addition to Fra2, Mod-1 contains the p50 but most probably not the p65 subunit of NF-κB and it functions as a transcriptional activator of the swine PD1 gene in response to IFN-α or IFN-γ. However, it is not yet clear whether Fra2 fulfills its transcriptional activity by contacting the Mod-1 recognition sequence directly or by targeting p50. For instance, the latter mechanism was shown for c-Fos, which binds to the p65 subunit of NF-κB, thereby enhancing the transcriptional activity of NF-κB on the κB-dependent 5′ long terminal repeat of the human immunodeficiency virus type 1 (47). The exact role of Fra2 in the process of H-2Kb enhancer A activation will be enlightened in transient expression assays using the F9 teratocarcinoma cell system.
Taken together our data might suggest that the activity of the H-2Kb enhancer A is regulated by a multiprotein complex that might assemble to an en-hanceosome. In enhanceosomes transcription factors synergize with each other as well as the basal transcriptional machinery, which results in a dramatic increase in gene transcription (11). One of the best studied examples for transcriptional synergism is the virus-inducible enhancer of the human interferon-P gene (52). The enhanceosome bound to this enhancer consists of the transcriptional activators NF-κB, IRF1, and the heterodimer c-Jun/ATF-2. Moreover, it contains the architectural protein HMG I(Y), which is required for the cooperative assembly, for the stability of the enhanceosome, and for maximal levels of transcriptional synergy. The activation domains of the transcription factors interact with factors of the basal transcription machinery like TFIID and with specific components of the RNA II holoenzyme, resulting in the recruitment of the transcriptional apparatus to the promoter and in the formation of a stable preinitiation complex (29). Very recent results have shown that the recruitment of the coactivator p300/CBP through the p65 subunit of NF-κB is also required for transcriptional synergy (38). The assembly as well as the transcriptional activity requires a precise helical relationship between individual transcription factor binding sites (50).
There are some striking similarities between the virus-inducible enhancer of the human interferon-P gene and the enhancer A of the H-2Kb gene, (i) A single copy of any regulatory element of both enhancers does not function on its own and inactivation of one of the cis-regulatory elements in the respective enhancer leads to a dramatic decrease of its inducibility (15,49); this study], (ii) The relative position of the individual cis-regulatory elements in the respective enhancer plays a decisive role for transcriptional synergy, (iii) The cellular coactivator p300 is recruited to both enhancers, most probably through the p65 subunit of the NF-κB transcription factor. However, there are also some differences. Most important, we were unable to detect the architectural HMG I(Y) protein in the multiprotein complex bound to the H-2Kb enhancer A up to now, although it contains one putative HMG I(Y) binding site located in the center of the H2TF1 element. Studies are under way to analyze the network of protein-protein interactions of the individual transcription factors with the basal transcriptional machinery to get further insight in the transcriptional activation of the enhancer A of the H-2Kb gene.
ACKNOWLEDGMENTS
We thank Daniela Schäfer for excellent technical assistance and Peter Fax for critical reading of the manuscript. This work was supported by the Deut-sche Forschungsgemeinschaft (DFG) through BR 1150/4–1.
REFERENCES
- 1. Ackrill A. M.; Blair G. E. Nuclear proteins binding to an enhancer element of the major histocompatibility class I promoter: Differences between highly oncogenic and nononcogenic adenovirus-transformed rat cells. Virology 172:643–646; 1989. [DOI] [PubMed] [Google Scholar]
- 2. Angel P.; Imagawa M.; Chiu R.; Stein B.; Imbra R. J.; Rahmsdorf H. J.; Jonat C.; Herrlich P.; Karin M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell 49:729–739; 1987. [DOI] [PubMed] [Google Scholar]
- 3. Auwerx J.; Sassone-Corsi P. IP-1: A dominant inhibitor of Fos/Jun whose activity is modulated by phosphorylation. Cell 64:983–993; 1991. [DOI] [PubMed] [Google Scholar]
- 4. Baldwin A. S.; Sharp P. A. Binding of a nuclear factor to a regulatory sequence in the promoter of the mouse H-2Kb class I major histocompatibility gene. Mol. Cell. Biol. 7:305–313; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baldwin A. S. The NF-κB and IκB proteins: New discoveries and insights. Annu. Rev. Immunol. 14:649–681; 1996. [DOI] [PubMed] [Google Scholar]
- 6. Boshart M.; Klüppel M.; Schmidt A.; Schütz G.; Luckow B. Reporter constructs with low background activity utilizing the CAT gene. Gene 110:129–130; 1992. [DOI] [PubMed] [Google Scholar]
- 7. Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254; 1976. [DOI] [PubMed] [Google Scholar]
- 8. Brockmann D.; Feng L.; Kröner G.; Tries B.; Esche H. Adenovirus type 12 early region 1A expresses a 52R protein repressing the trans-activating activity of transcription factor c-Jun/AP-1. Virology 198:717–723; 1994. [DOI] [PubMed] [Google Scholar]
- 9. Brockmann D.; Schäfer D.; Kirch H.-C.; Esche H. Repression of c-Jun-induced mouse major histocompatibility class I promoter (H-2Kb) activity by the adenovirus type 12-unique 52R ElA protein. Oncogene 12:1715–1725; 1996. [PubMed] [Google Scholar]
- 10. Burke P. A.; Hirschfeld S.; Shirayoshi Y.; Kasik J. W.; Hamada K.; Apella E.; Ozato K. Developmental and tissue-specific expression of nuclear proteins that bind the regulatory element of the major histocompatibility complex class I gene. J. Exp. Med. 169:1309–1321; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carey M. The enhanceosome and transcriptional synergy. Cell 92:5–8; 1998. [DOI] [PubMed] [Google Scholar]
- 12. Cereb N.; Yang S. Y. The regulatory complex of HLA class I promoters exhibits locus-specific conservation with limited allelic variation. J. Immunol. 152:3873–3883; 1994. [PubMed] [Google Scholar]
- 13. David-Watine B.; Israël A.; Kourilsky P. The regulation and expression of MHC class I genes. Immunol. Today 11:286–292; 1990. [DOI] [PubMed] [Google Scholar]
- 14. Dey A.; Thornton A. M.; Lonergan M.; Weissman S. M.; Chamberlain J. W.; Ozato K. Occupancy of upstream regulatory sites in vivo coincides with major histocompatibility complex class I gene expression in mouse tissues. Mol. Cell. Biol. 12:3590–3599; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Du W.; Thanos D.; Maniatis T. Mechanism of transcriptional synergism between distinct virus-inducible enhancer elements. Cell 74:887–898; 1993. [DOI] [PubMed] [Google Scholar]
- 16. Ehrlich R.; Maguire J. E.; Singer D. S. Identification of negative and positive regulatory elements associated with a class I major histocompatibility complex gene. Mol. Cell. Biol. 8:695–703; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Foletta V. Transcription factor AP-1, and the role of Fra-2. Immunol. Cell Biol. 74:121–133; 1996. [DOI] [PubMed] [Google Scholar]
- 18. Fried M.; Crothers D. Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res. 9:6505–6525; 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garner M.; Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: Application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 9:3047–3060; 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gerritsen M. E.; Williams A. J.; Neish A. S.; Moore S.; Shi Y.; Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc. Natl. Acad. Sci. USA 94:2929–2932; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giuliani, C; Saji M.; Napolitano G.; Palmer L. A.; Taniguchi S.-L.; Shong M.; Singer D. S.; Kohn L. D. Hormonal modulation of major histocompatibility complex class I gene expression involves an enhancer A-binding complex consisting of Fra-2 and the p50 subunit of NF-κB. J. Biol. Chem. 270:11453–11462; 1995. [DOI] [PubMed] [Google Scholar]
- 22. Gorman C. M.; Moffat L. F.; Howard B. H. Recombinant genomes which express chloramphenicol acetyl-transferase in mammalian cells. Mol. Cell. Biol. 2:1044–1051; 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hamada K.; Gleason S. L.; Levi B.-Z.; Hirschfeld S.; Appella E.; Ozato K. H-2RIIBP, a member of the nuclear hormone receptor superfamily that binds to both the regulatory element of major histocompatibility class I genes and the estrogen response element. Proc. Natl. Acad. Sci. USA 86:8289–8293; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ishiguro N.; Brown G. D.; Meruelo D. Activation transcription factor 1 involvment in the regulation of murine H-2Dd expression. J. Biol. Chem. 272:15993–16001; 1997. [DOI] [PubMed] [Google Scholar]
- 25. Israël A.; Kimura A.; Kieran M.; Yano O.; Kanello-poulos J.; Le Bail O.; Kourilsky P. A common positive trans-acting factor binds to enhancer sequences in the promoters of mouse H-2 and β2-microglobulin. Proc. Natl. Acad. Sci. USA 84:2653–2657; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Israël A.; Le Bail O.; Hatat D.; Piette J.; Kieran M.; Logeat F.; Wallach D.; Fellous M.; Kourilsky P. TNF stimulates expression of mouse MHC class I genes by inducing an NF-κB-like enhancer binding activity which displaces constitutive factors. EMBO J. 8:3793–3800; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Israël A.; Yano O.; Logeat F.; Kieran M.; Kourilsky P. Two purified factors bind to the same sequence in the enhancer of mouse MHC class I genes: One of them is a positive regulator induced upon differentiation of teratocarcinoma cells. Nucleic Acids Res. 17:5245–5257; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karin M.; Liu Z.-G.; Zandi E. AP-1 function and regulation. Curr. Opin. Cell Biol. 9:240–246; 1997. [DOI] [PubMed] [Google Scholar]
- 29. Kim T. K.; Maniatis T. The mechanism of transcriptional synergy of an in vitro assembled interferon-β enhanceosome. Mol. Cell 1:119–129; 1997. [DOI] [PubMed] [Google Scholar]
- 30. Kimura A.; Israël A.; Le Bail O.; Kourilsky P. Detailed analysis of the mouse H-2Kb promoter: Enhancer-like sequences and their role in the regulation of class I gene expression. Cell 44:261–272; 1986. [DOI] [PubMed] [Google Scholar]
- 31. Korber B.; Mermod N.; Hood L.; Stroynowski I. Regulation of gene expression by interferons: Control of H-2 promoter responses. Science 239:1302–1306; 1988. [DOI] [PubMed] [Google Scholar]
- 32. Laemmli E. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685; 1970. [DOI] [PubMed] [Google Scholar]
- 33. Lipinski K. S.; Esche H.; Brockmann D. Amino acids 1–29 of the adenovirus serotypes 12 and 2 ElA proteins interact with rap30 (TFIIF) and TBP in vitro. Virus Res. 54:99–106; 1998. [DOI] [PubMed] [Google Scholar]
- 34. Liu X.; Ge R.; Westmoreland S.; Cooney A. J.; Tsai S. Y.; Tsai M.-J.; Ricciardi R. P. Negative regulation by the R2 element of the MHC class I enhancer in adenovirus-12 transformed cells correlates with high levels of COUP-TF binding. Oncogene 9:2183–2190; 1994. [PubMed] [Google Scholar]
- 35. Liu X.; Ge R.; Ricciardi R. P. Evidence for the involvement of a nuclear NF-κB inhibitor in global down-regulation of the major histocompatibility complex class I enhancer in adenovirus type 12-transformed cells. Mol. Cell. Biol. 16:398–104; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maniatis T.; Sambrook I.; Fritsch E. F. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 37. Mansky P.; Brown W. M.; Park J.-H.; Choi J. W.; Yang S. Y. The second κB element, κB2, of the HLA-A class I regulatory complex is an essential part of the promoter. J. Immunol. 153:5082–5090; 1994. [PubMed] [Google Scholar]
- 38. Merika M.; Williams A. J.; Chen G.; Collins T.; Thanos D. Recruitment of CBP/p300 by the IFNβ enhanceosome is required for synergistic activation of transcription. Mol. Cell 1:277–289; 1998. [DOI] [PubMed] [Google Scholar]
- 39. Moran E. DNA tumor virus transforming proteins and the cell cycle. Curr. Opin. Genet. Dev. 3:63–70; 1993. [DOI] [PubMed] [Google Scholar]
- 40. Nabholz M.; MacDonald H. R. Cytolytic T lymphocytes. Annu. Rev. Immunol. 1:273–306; 1983. [DOI] [PubMed] [Google Scholar]
- 41. Sanger F.; Nicklen S.; Coulson A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463–5467; 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schouten G. J.; Van der Eb A. J.; Zantema A. Down-regulation of MHC class I expression due to interference with pl05-NFκBl processing by Adl2ElA. EMBO J. 14:1498–1507; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Segars J. H.; Nagata T.; Bours V.; Medin J. A.; Franzoso G.; Blanco J. C. G.; Drew P. D.; Becker K. G.; An J.; Tang T.; Stephany D. A.; Neel B.; Siebenlist U.; Ozato K. Retinoic acid induction of major histocompatibility complex class I genes in NT-era-2 embryonal carcinoma cells involves induction of NF-κB (p50-p65) and retinoic acid receptor β-retinoid X receptor β heterodimers. Mol. Cell. Biol. 13:6157–6169; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shapiro D. L.; Sharp P. A.; Wahli W. W.; Keller M. J. A high-efficient HeLa cell nuclear transcription extract. DNA 7:47–55; 1988. [DOI] [PubMed] [Google Scholar]
- 45. Shirayoshi Y.; Miyazaki J.; Burke P. A.; Hamada K.; Appella E.; Ozato K. Binding of multiple factors to the 5′upstream regulatory element of the murine major histocompatibility class I gene. Mol. Cell. Biol. 7:4542–4548; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Singer D. S.; Maguire J. E. Regulation of the expression of class I MHC genes. Crit. Rev. Immunol. 10:235–257; 1990. [PubMed] [Google Scholar]
- 47. Stein B.; Baldwin A. S.; Ballard D. W.; Greene W. C.; Angel P.; Herrlich P. Cross-coupling of the NF-κB p65 and Fos/Jun transcription factors produces potentiated biological function. EMBO J. 12:3879–3891; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Suzuki T.; Okuno H.; Yoshida T.; Endo T.; Nishina H.; Iba H. Difference in transcriptional regulatory function between c-Fos and Fra-2. Nucleic Acids Res. 19:5537–5542; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thanos D.; Maniatis T. The high mobility group protein HMG I (Y) is required for NF-κB-dependent virus induction of the human IFN-β gene. Cell 71:777–789; 1992. [DOI] [PubMed] [Google Scholar]
- 50. Thanos D.; Maniatis T. Virus induction of human IFNβ gene expression requires the assembly of an enhanceosome. Cell 83:1091–1100; 1995. [DOI] [PubMed] [Google Scholar]
- 51. Ting J. P.-Y.; Baldwin A. S. Regulation of MHC gene expression. Curr. Opin. Immunol. 5:8–16; 1993. [DOI] [PubMed] [Google Scholar]
- 52. Tjian R.; Maniatis T. Transcriptional activation: A complex puzzle with few easy pieces. Cell 77:5–8; 1994. [DOI] [PubMed] [Google Scholar]
- 53. Van Dam H.; Duyndam M.; Rottier R.; Bosch A.; de Vries-Smits L.; Herrlich P.; Zantema A.; Angel P.; van der Eb A. J. Heterodimer formation of c-Jun and ATF-2 is responsible for induction of c-jun by the 243 amino acid adenovirus ElA protein. EMBO J. 12:479–487; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Verma. I. M.; Stevenson J. K.; Schwarz E. M.; Van Antwerp D.; Miyamoto S. Rel/NF-κB/IκB family: Intimate tales of association and dissociation. Genes Dev. 9:2723–2735; 1995. [DOI] [PubMed] [Google Scholar]
- 55. Wang H.-G. H.; Draetta G.; Moran E. E1A induces phosphorylation of the retinoblastoma protein independently of direct physical association between the ElA and retinoblastoma products. Mol. Cell. Biol. 11:4253–4265; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang H.-G. H.; Yaciuk P.; Ricciardi R. P.; Green M.; Yokoyama K.; Moran E. The E1A products of oncogenic adenovirus serotype 12 include amino-terminally modified forms able to bind the retinoblastoma protein but not p300. J. Virol. 67:4804–4813; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yano O.; Kanellopoulos J.; Kieran M.; Le Bail O.; Israël A.; Kourilsky P. Purification of KBF1, a common factor binding to both H-2 and β2-microglobulin enhancers. EMBO J. 6:3317–3324; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yamit-Hezi A.; Plaksin D.; Eisenbach L. c-fos and c-jun overexpression in malignant cells reduces their tumorigenic and metastatic potential, and affects their MHC class I gene expression. Oncogene 9:1065–1079; 1994. [PubMed] [Google Scholar]