

Abstract
There is increasing experimental evidence that ADP-ribosylation of host proteins is an important means to regulate gene expression of bacteriophage T4. Surprisingly, this phage codes for three different ADP-ribosyltransferases, gene products Alt, ModA, and ModB, modifying partially overlapping sets of host proteins. While gene product Alt already has been isolated as a recombinant protein and its action on host RNA polymerases and transcription regulation have been studied, the nucleotide sequences of the two mod genes was published only recently. Their mode of action in the course of the infection cycle and the consequences of the ADP-ribosylations catalyzed by these enzymes remain to be investigated. Here we describe the cloning of the genes, the overexpression, purification, and partial characterization of ADP-ribosyltransferases ModA and ModB. Both proteins seem to act independently, and the ADP-ribosyl moieties are transferred to different sets of host proteins. While gene product ModA, similarly to the Alt protein, acts also on the α-subunit of host RNA polymerase, the ModB activity serves another set of proteins, one of which was identified as the S1 protein associated with the 30S subunit of the E. coli ribosomes.
Keywords: ADP-ribosyltransferase, Lysogeny, ModA, ModB, Alt, Overexpression, Phage T4, Protein purification, Transcription regulation, Translation regulation
SIMILAR to protein phosphorylation, mono-ADP-ribosylation of proteins is an enzyme-catalyzed, posttranslational modification that may be reversed for regulatory purposes by the action of corresponding ADP-ribosylhydrolases. The transfer of the ADP-ribose moiety from the substrate NAD+ to specific amino acid residues of the target protein modulates their activities. Mono-ADP-ribosyltransferases originally had been discovered as the pathogenic principle of diphtheria, cholera, pertussis, and other bacterial toxins (9,11,22). ADP-ribosyltransferases now are known to exist in both procaryotic and eucaryotic systems (13). Interestingly, at least part of the “bacterial” toxins seem to be encoded as pathogenic factors on the genomes of lysogenic phages incorporated into the DNA of their host bacteria (39).
Bacteriophage T4 codes for three ADP-ribosyltransferases, the Alt, the ModA, and the ModB gene products (GenBank accession numbers X15811 and X98695, respectively). Antagonizing ADP-hydrolases have not been detected and might not exist. The Alt protein is also a structural component of the phage head and about 50 copies of this protein enter the host cell in the process of infection together with the phage DNA (10). The Alt polypeptide immediately ADP-ribosylates host RNA polymerase, presumably at only one of the two α-subunits (6,27). One target of this reaction is amino acid ARG265 of the α-subunit (25); however, other arginine residues of the polypeptide as well as other RNA polymerase subunits and proteins may also be ADP-ribosylated by this enzyme (6,7,14). As a consequence of the transfer of the ADP-ribosyl moiety to host RNA polymerase by gene product Alt, the transcription from T4 early promoters is enhanced (40): the Alt-catalyzed ADP-ribosylation of host RNA polymerase leads to an accelerated expression of T4 “early” gene products, thus supporting the phage’s program to gain control over the infected host cell.
Later in the T4 infection cycle ADP-ribosylation of both α-subunits is brought to completion by a second, newly synthesized ADP-ribosyltransferase, the 23-kDa ModA gene product (29). In contrast to gene product Alt, modifying several E. coli proteins (14), ModA reacts more specifically towards the RNA polymerase α-subunit. The biological effects of this ADP-ribosylation on gene expression are not precisely known; however, the association of modified RNA polymerase with phage-encoded proteins, and linked to this process the recognition of middle-mode and late promoters, may be a consequence of the modification (23,24). Surprisingly, a double mutant (alt −, modA −) did not show any significant effect in phage burst or latent period under laboratory conditions (10). In the course of experiments designed to sequence T4 gene modA, a second reading frame, modB, was detected (5,40), and threading experiments predicted that both reading frames code for ADP-ribosyltransferases (2,40). In this article we report the cloning of genes modA and modB, and the overexpression of their gene products. Both recombinant proteins show ADP-ribosylation patterns different from each other and from those of gene product Alt, suggesting that the three ADP-ribosyltransferases act as independent enzymes, and modifying overlapping sets of host proteins. While Alt in vitro targets all subunits of host RNA polymerase (14), ModA transfers the ADP-ribosyl label preferentially to the α-subunit of this enzyme; however, a 26- and a 70-kDa polypeptide are also labeled. ModB transfers the ADP-ribosyl moiety to unknown proteins with approximate molecular masses of 10, 19, 21, 30, 36, and 70 kDa. We shall give evidence that the 70-kDa protein modified by ModB is the SI ribosomal protein of E. coli.
MATERIALS AND METHODS
Materials
DNA-dependent RNA polymerase, alkaline phosphatase, and nucleoside triphosphates were purchased from Boehringer (Mannheim, Germany). [Adenylate-32P]NAD+ was from Hartmann (Braunschweig, Germany). Acrylamid, N,N′-bisacrylamid (Rotiphorese 40%, 29:1) for SDS gels came from Roth (Karlsruhe, Germany) and Coomassie brilliant blue as well as antibiotics from Serva (Heidelberg, Germany). T4 DNA ligase, Taq polymerase, and all restriction enzymes were purchased from Gibco-BRL (Gaithersburg, MD). Minisart filters were obtained by Sartorius (Goettingen, Germany) and agarose from FMC (Rockland, ME). Chromatography Talon™ resin came from Clontech (Palo Alto, CA). Enzymatic DNA manipulations were performed according to Ausubel et al. (1).
Bacteria and Plasmids
T4dC DNA was prepared by growing T4 mutant alc536 [42− (amC87), 56− (amE51), denB– (amS19), ale (unG9)] in E. coli B834 [gal− (U56), (met-, gal−(U), su−)]. For the cloning experiments we used plasmid vectors pET-11d and pET-16b, which were grown in E. coli BL21 [F−, ompT, hsdSB (r− B m− B) gal dcm] (Novagen, Madison, WI).
PCR Amplification of DNA
The following PCR protocol was used to amplify the modA and modB genes from XhoI-hydrolyzed T4dC DNA as a template (25 ng), with primer concentrations of 50 pmol. PCR was performed with 35 cycles and the following program: DNA denaturation for 1 min at 95°C, primer annealing for 2 min at 52°C, DNA extension for 1 min at 72°C, and a final extension for 10 min at 72°C. Samples of PCR-amplified DNAs were checked by gel electrophoresis in TBE buffer, pH 8.0 (89 mM Trisborate, 89 mM boric acid, 2 mM EDTA-Na2). The gels were stained with ethidium bromide (0.4 mg/1) and photographed on a 312-nm UV light source.
The primers used for the amplification of modA were TGAGGTAGTTCCATGGAATACTCA and TGGTATAATGGATCCAAGTCCTTC for the cloning into vector pET-11d, or TTGAATGAAGGATCCAGTAATGCA and TGGTATAATGGATCCAAGTCCTTC for pET-16b. Correspondingly, for the amplification of modB the primers were ACGGAGGCTACCATGGTTATTAAT, and GCACCGATGGATCCATTGATTTTA (pET-11d), or ACGGAGGCTCATATGATTATTAAT and GCACCGATGGATCCATTGATTTTA (pET-16b). The restriction sites introduced with the primer sequence (NcoI, BamHI, and NdeI, respectively) are underlined.
Overexpression of the T4 ADP-Ribosyltranferases ModA and ModB
Overexpression of the ModA and ModB proteins was performed by cloning the PCR products of genes modA and modB into the restriction sites NcoI-BamHI of pET-11d. To obtain both proteins as Histagged polypeptides modA was cloned into the BamHI-BamHI and modB into the NdeI-BamHI restriction sites of pET-16b. The ligation mixture was transformed into E. coli BL21 by electroporation. For the overexpression, a single recombinant colony was resuspended in 2 ml of LB medium [1% (w/v) Bacto tryptone, 0.5% (w/v) yeast extract, 1% (w/v) NaCl] supplemented with 0.2% of maltose and 200 μg/ml of ampicillin. Cells were grown at 37°C to an OD590 of 0.6–1.0. Bacteria then were centrifuged for 5 min at 5000 × g, resuspended in 10 ml of fresh medium (as above), and grown to the same OD590. Cells again were centrifuged and resuspended in 10 ml of fresh medium and the culture was induced by the addition of IPTG and MgSO4 to final concentrations of 1 and 10 mM, respectively. In addition, the culture was infected with 4 × 109 pfu/ml of λCE6, resulting in a multiplicity of infection of 5–10. The culture was allowed to grow for 3 h at 37°C. Cells were harvested by centrifugation and kept at −20°C until further use. Overexpression of the Mod proteins was monitored on 13% SDS-polyacrylamide gels (17).
Purification of ADP-Ribosyltransferases ModA andModB
For the purification of the ModA and ModB proteins the above procedure was scaled up to a volume of 250 ml. After sonic oscillation of the cells and centrifugation at 3000 × g the overexpressed proteins were found in the pellet. It was concluded that most of the recombinant proteins were tied down as inclusion bodies. The inclusion bodies were resuspended in 5 ml of a lysis buffer containing 50 mM NaH2PO4, 10 mM Tris-HCl, pH 8.0, 7 M guanidinium chloride. Centrifugation for 15 min at 20,000 × gat room temperature removed insoluble debris. The supernatant then was filtered through Minisart filters (pore size 0.45 μm). The flow-through contained approximately 90 mg of protein in a total volume of 5 ml. The sample was applied to 2 ml of Talon™ resin equilibrated with lysis buffer. The resin was treated according to the batch/gravity flow procedure (for further details see Talon™ user manual), poured onto a small column, and washed. The proteins bound were renatured on the column by developing the column at room temperature with 4 volumes of a buffer containing 6 M urea, 50 mM NaH2PO4, 10 mM Tris-HCl, pH 8.0, followed by 4 volumes of the same buffer containing 3 M urea. In a final step, which was performed at 4°C, urea was entirely omitted from the last 5 volumes of renaturation buffer. The proteins then were eluted with 100 mM imidazole, 20 mM Tris-HCl, pH 8.0. Fractions of 500 μl were collected and the ModA or ModB proteins were monitored on 13% SDS-polyacrylamide gels. Fractions containing ModA or ModB were pooled and dialyzed against 50% glycerol in TE buffer (10 mM Tris-HCl, pH 8.0, 0.5 mM EDTA-Na2).
ADP-Ribosyltransferase Assay
The activities of the overexpressed transferases were assayed by radioactive labeling of the proteins in vitro, according to Rohrer et al. (27), followed by gel electrophoresis and autoradiography. Because the proteins of a cell lysate prepared from Mod-overexpressing cells might already be ADP-ribosylated with residual cellular NAD+, for testing we always added an equal amount of cell lysate of cells not having been in contact with the Mod genes. Hence, incubation mixtures contained, in a final volume of 0.1 ml, 35 μg of externally added RNA polymerase holoenzyme (optional), 30 μg E. coli crude cell lysate, 30 Hg E. coli crude cell lysate of the overexpressing bacteria, or, alternatively, 4 μg of the purified ADP-ribosyltransferases, 1 μCi [32P]NAD+ (specific activity: 800 Ci/mmol) in transferase buffer [0.05 M Tris-acetate, pH 7.5, 0.01 M Mg(OAc)2, 22 mM NH4C1, 1 mM EDTA-Na2, 0.01 M 2-mercaptoethanol, 10% glycerol]. The reaction mixture was incubated for 30 min at 15°C and the reaction was stopped by precipitation with 10% trichloroacetic acid. To reduce residual radioactive NAD+, the precipitate was centrifuged, the sediment was washed with 70% acetone/ water (v/v), dried, dissolved in 80 μ1 of electrophoresis sample buffer, and incubated at 95°C for 5 min. Aliquots of 15 μ1 were applied per channel and separated on a 13% SDS-polyacrylamide gel (17). Gels were stained with Coomassie brilliant blue, dried, and autoradiographed on Fuji-RX films, usually with exposure times of 24 h.
Blotting of the ADP-Ribosylated Proteins
The plasmid p11modB was transformed into E. coli BL21 and transformed cells were grown as described above. After induction and 3 h of continued growth, cells were harvested and opened by sonic oscillation. An aliquot of the soluble fraction was submitted to an ADP-ribosyltransferase assay in the presence of [32P]NAD+. To achieve an efficient separation of the 70-kDa polypeptide from the bulk of accompanying proteins the samples were separated on 8% SDS-PAGE and blotted onto a PVDF membrane (Immobilon™-P, Millipore, Bedford). This membrane was stained with Coomassie brilliant blue to monitor the protein transfer, air dried, and autoradiographed. Comparison of the electrophoretical pattern with the autoradiography shows a labeled protein of approximately 70 kDa. The labeled protein was excised from the membrane and directly subjected to amino acid sequencing.
Amino Acid Sequencing
N-terminal protein sequence analysis was executed with a 473A protein sequencer (Perkin Elmer, Applied Biosystems), according to Edman (4). The phenylthiohydrated amino acids were identified by HPLC on a Brewrilee™ column.
Isolation of 70S Ribosomes From E. coli BL2J Cells
The purification of the 70S ribosomes followed the procedure of Jelenc (3).
Cloning and Overexpression of the S1 Gene
For the amplification of the rspA gene we followed the PCR protocol as outlined above, with the following exceptions. The primers were annealed at 42°C; the primers .5′-TGAAGATTAACCATGGCTGAA-3′ and 5′-TCGGAATAAGAATTCCGAAGA 3′ were derived from the rspA-flanking regions as published (28). DNA isolated from E. coliBL21 served as template. The amplified rspA gene was cloned into the Ncol and BamHl restriction sites (as underlined in the primer sequences) of vector pBAD (Invitrogen, Carlsbad, USA). The vector was transformed into E. coli LMG194 and the cells were grown to an OD590 of 0.5. The expression of gpRspA was induced by adding 0.2% of arabinose. The culture was kept for 4 h at 37°C prior to cell disruption and the consecutive ADP-ribosylation test.
RESULTS
Cloning, Overexpression, and Purification of ModA and ModB
Starting material for the cloning of the genes modA and modB was T4 dCDNA as obtained by growing T4 strain alc536 in E. coli B834. This T4 mutant replaces the 5-hydroxymethyl cytosine bases usually found in T4 wild-type DNA by cytosine. T4 dCDNA is accessible to restriction enzyme digestion while the T4 wild-type DNA is refractory to an attack of most of these enzymes. To obtain smaller DNA fragments and to isolate the restriction fragment carrying both genes, total T4 dCDNA was digested with nuclease XhoI, resulting in 17 DNA fragments (8). Genes modA and modB reside on a 19.459-kbp XhoI fragment in an approximately central position, between map positions 13.135 and 11.914 kbp on the genomic map of bacteriophage T4 (16). The corresponding fragment can be prepared from agarose gels. The nucleotide sequence of this region revealed that both genes are arranged in one transcription unit, served by T4 early promoter PI3.149 (designated PI2.8 in publications prior to the sequencing and editing of the total T4 sequence) (18). Their reading frames are linked by an ATGA sequence, with the start codon ATG of the downstream gene overlapping the stop codon TGA of the upstream gene by one base pair. For an overview of this genomic region see Fig. 1.
FIG. 1.

The arrangement of the reading frames modB and modA between the early promoters P13.149 and P11.815. The positions in the T4 genome and the sizes of the corresponding gene products are indicated. The positions of PCR primers for DNA amplification with restriction sites introduced to facilitate cloning are also shown.
Both genes were amplified separately by PCR with the corresponding XhoI restriction fragment as a template. Restriction sites suitable to clone both fragments in proper orientation into multiple cloning sites of commercial expression vectors were introduced by the synthesis of eight PCR primers, each of 24 nucleotides length, carrying the sequences of the particular restriction sites (see Materials and Methods). For the overexpression of the two Mod polypeptides we took advantage of the T7 polymerase-based pET system (35). The vectors pi1modA, pi1modB, pl6modA, and pl6modB were transformed into E. coli BL21 and grown as described in Materials and Methods. Expression was induced by infection with bacteriophage λCE6 carrying the gene for T7 RNA polymerase. Concomitantly IPTG was added to open transcription of the cloned mod genes. The synthesis of gpModA (23 kDa), gpModB (24 kDa), gpHis-ModA (26 kDa), and gpHis-ModB (27 kDa) was monitored on SDS-polyacrylamide gels.
After cell disruption, soluble and insoluble portions of the preparation were separated by centrifugation. Essential amounts of the overexpressed proteins remained with the insoluble fraction of the cell debris. It was concluded that the ModA and ModB proteins aggregated in inclusion bodies. Separation of the inclusion bodies by centrifugation at 3000 × g immediately resulted in preparations of the proteins that were pure to about 90% (Fig. 2). Inclusion bodies were dissolved in lysis buffer containing 7 M guanidinium chloride and the preparation was applied to a Talon™ column. Affinity chromatography of the His-Mod proteins and their renaturation (for details see Materials and Methods) yielded preparations close to 100% pure as judged from silver-stained polyacrylamide gels (Fig. 3). Though hydrophobicity plots of the two enzymes (data not shown) do not reveal extended hydrophobic or charged regions, ModA and ModB are endowed with a remarkable tendency to aggregate and precipitate at concentrations above 1 mg/ml.
FIG. 2.
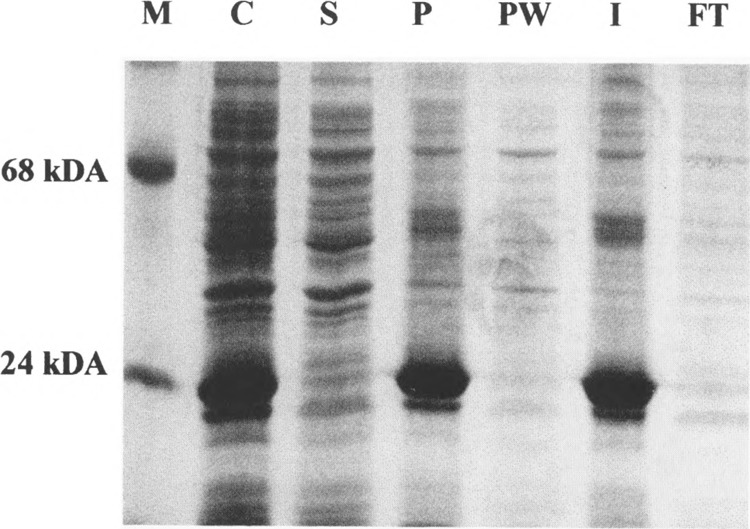
Solubility and prepurification of ModA. Polypeptides were separated on a 13% SDS-polyacrylamide gel and stained with Coomassie brilliant blue. M: marker proteins; C: total protein fraction of a ModA-expressing culture; S: soluble and P: insoluble protein fractions of this culture; PW: inclusion body wash; I: solubilized inclusion bodies; FT: column flow-through. For further information see text.
FIG. 3.

Purified ModA, 1 and 3 μg protein per lane, respectively, separated on a 13% SDS-polyacrylamide gel. Proteins were detected by silver staining.
Determination of the ADP-Ribosyltransferase Activity of the Overexpressed Enzyme
The ADP-ribosyltransferase activity of the overexpressed and purified Mod proteins was determined following the protocol outlined in Materials and Methods. In these experiments no differences were observed among the ribosylation reactions performed by the untagged proteins or their His-tagged counterparts. Because the His-tagged proteins usually are faster to purify, most of the experiments shown here were executed with gpHis-ModA and gpHis-ModB. The ADP-ribosylation products resulting from these tests were analyzed by SDS-polyacrylamide gel electrophoresis and subsequent autoradiography. As can be seen from Fig. 4, the ribosylation patterns of His-ModA and His-ModB are different. While His-ModA ADP-ribosylates the α-subunit of the RNA polymerase as well as a 26- and a 70-kDa protein, His-ModB modifies proteins with apparent sizes of 10, 19, 21, 30, 36, and 70 kDa. ModA and ModB, like the Alt enzyme (15), perform an autoribosylation reaction.
FIG. 4.

Autoradiograph of soluble E. coli proteins incubated in the presence of T4 ADP-ribosyltransferases ModA and ModB, respectively. The crude cell extract was supplemented with 20 μg of RNAP. The in vitro labeling reaction followed the protocol as given in Materials and Methods. The radioactive substrate was [32P]NAD+. The reaction products were separated by electrophoresis on a 13% SDS-polyacrylamide gel and stained with Coomassie brilliant blue (A). (B) The autoradiograph of (A). As a molecular weight standard a 10-kDa ladder was employed.
Protein Sequencing
As a first step to identify the target proteins of ModA and ModB, other than the subunits of RNA polymerase, we purified the 70-kDa protein, apparently targeted by both proteins, but seemingly preferred by ModB (Fig. 4). Taking advantage of the transfer reaction as catalyzed by ModB, the E. coli proteins sensitive to ADP-ribosylation by this enzyme were radioactively labeled, as described in Materials and Methods. The proteins were separated on an 8% SDS-polyacrylamide gel and blotted onto a PVDF membrane. The membrane was autoradiographed, the labeled 70-kDa protein excised and submitted to NH2-terminal amino acid sequencing. The comparison of these sequences with the E. coli database is shown in Fig. 5. The only homology found was that to the ribosomal protein SI of E. coli. To further substantiate this finding we isolated the 70S ribosome fraction of E. coli BL21 cells. The purified ribosomes were also subjected to ADP-ribosylation by ModB and the incubation mixture then was separated on a 13% SDS-polyacrylamide gel; the gel was dried and the proteins autoradiographed (Fig. 6). Again, we found a 70-kDa protein carrying the ADP-ribosyl label. Control experiments in which ModB was omitted from the reaction mix did not show the 70-kDa band radioactively labeled. It is well documented that the actual molecular weight of the SI protein is 61.159 kDa, while the apparent molecular weight as determined on polyacrylamide gels is 70 kDa (12). In a second approach we overexpressed the S1 protein (gpRspA) as outlined in Materials and Methods, and again found this protein labeled in a cell-free extract by the transfer reaction as catalyzed by ModB (Fig. 6).
FIG. 5.

The amino acid sequences of two independent isolates of the ADP-ribosylated 70-kDa protein, aligned with the sequence of the E. coli ribosomal protein S1.
FIG. 6.
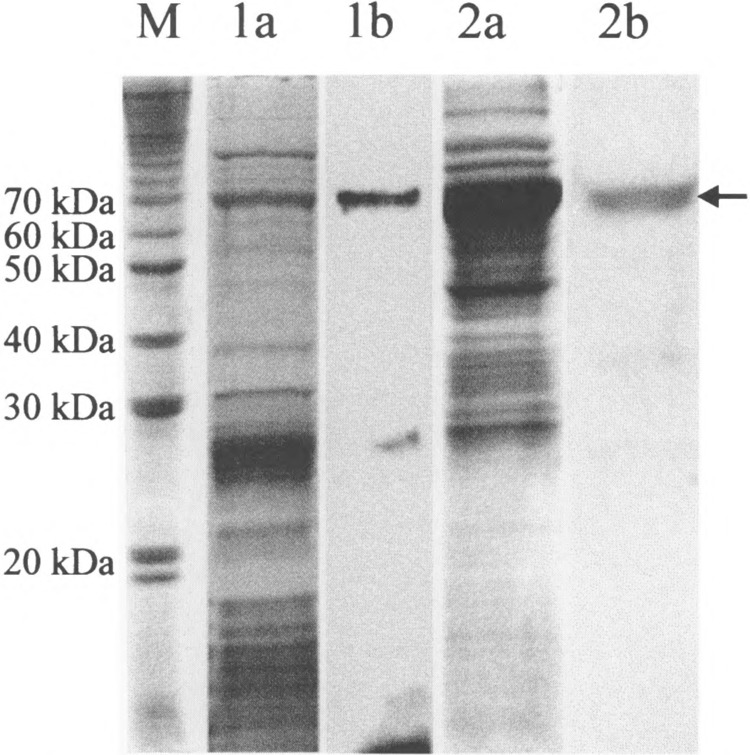
ADP-ribosylation of purified 70S ribosomes and of the overexpressed S1 protein. The in vitro labeling reaction with [32P]NAD+ as a substrate followed the protocol given in Material and Methods. After ADP-ribosylation the products were seperated by 13% SDS-PAGE and stained with Coomassie brilliant blue. The band representing the S1 protein is marked by an arrow. M: 10-kDa protein ladder; 1a: purified 70S ribosomes; 1b: autoradiography of la; 2a: E. coli crude extract with the overexpressed SI protein; 2b: autoradiography of 2a.
DISCUSSION
In this article we describe the cloning and the overexpression of phage T4 genes modA and modB, as well as the purification and partial characterization of their gene products, ADP-ribosyltransferases ModA and ModB. Gene modA already had been linked to T4 transcription regulation by biochemical and genetic studies (7,27,32), and it had been shown that this gene product ADP-ribosylates the α-subunit of E. coli RNA polymerase. However, the isolation of the protein from phage-infected E. coli cells had been difficult and the limited yield of active enzyme then did not allow extended biochemical experimentation.
Gene modB even escaped attention until recently, when it was discovered in the course of sequencing experiments targeting gene modA(5,40). Both reading frames reside next to each other on the T4 genome, in the same transcription unit, overlapping by one base pair, and they code for polypeptides of very similar M r. This bicistronic transcription unit is controlled by the strong T4 early promoter P13.149 (N. Sommer et al., in preparation), possibly reflecting the importance of these two gene products to the T4 replication cycle. Surprisingly, threading experiments predicted secondary structures for both gene products to fit the structural characteristics of ADP-ribosyltransferases, and amino acids constituting putative active sites were found to be conserved in both polypeptides (2,40). With the aim of confirming the results of the threading experiments and of gaining knowledge on the ribosylation characteristics of these enzymes, we isolated both as recombinant proteins.
Initial attempts to clone the mod genes into a number of vectors, like pBluescript (30), pT7-5 (38), or pGEX-3X (33), were without success. However, the tightly controlled pET expression vectors proved to be useful for the cloning and overproducing the ADP-ribosyltransferases. The difficulties encountered in cloning the mod genes into expression vectors other than pET are most probably due to their toxicity for the host cells: few copies of both gene products as synthesized (e.g., by the leakiness of a vector system) might prevent E. coli from growing. Moreover, transformed lysogenic strains, recommended for efficient expression of the genes cloned on the vectors [e.g., E. coli BL21 (DE3) or BL21 (DE3) pLysS] lyse rather quickly and produce phage progeny. Therefore, ADP-ribosylation catalyzed by either one of these proteins may interfere with lysogeny in these bacterial strains.
For a fast and one-step purification of the Mod gene products, His-tags were linked to the amino-termini, allowing to purify the enzymes by metal ion affinity chromatography close to 100%. All recombinant enzymes, his-tagged or not, showed transferase activity with radiolabeled NAD+ as a substrate. In crude extracts of transformed and induced E. coli cells, the ModA protein modified only two polypeptides (i.e., the α-subunit of the RNA polymerase and a 70-kDa protein), while ModB catalyzed the ADP-ribosylation of at least six different polypeptide chains. Upon exposure of the corresponding ADP-ribosylated proteins for more than 1 week, a number of additional fainter bands appear on the autoradiographs, and several polypeptides then seem to comigrate in the ModA and ModB channels. Whether these bands represent specific ADP-ribosylations of minor proteins, with relevance to the T4 replication cycle, or whether they reflect proteins labeled with less specificity, remains to be investigated. ADP ribo-syltransferases have already been reported to react rather unspecifically (19,21,34). However, the ModA and ModB proteins are good examples to show that preferences exist. This preference for certain target proteins may possibly be linked with the tendency of these enzymes to aggregate: for example, small clusters of hydrophobic and/or charged amino acid side chains might direct the enzymes to corresponding contact sites at specific target proteins from where “docked” enzyme molecules react with the arginine residue in reach. This mechanism would also give clues as to the preference for Arg265 on the carboxy-terminus of the α-subunit of RNA polymerase, to the autoribosylation reaction, and to a partially unspecific reaction of these enzymes.
Polyacrylamide gels, separating the E. coli proteins that had been ADP-ribosylated by ModA or ModB, show a 70-kDa polypeptide comigrating on both gels. Because ModB exhibits a preference to react with this protein we have recovered the protein after labeling and submitted it to amino-terminal sequencing. Comparison of the amino acid sequence obtained to those deposited in the EMBL database (BlastP 2.0) revealed a high homology with the amino-terminal sequence of the SI protein (Fig. 5). The ribosomal protein SI is the largest out of the E. coli ribosomal 30S subunit proteins. It is composed of 557 amino acid residues, has an M r of 61.159 kDa, and migrates as a 70-kDa protein on SDS-polyacrylamide gels (28). The SI protein plays an essential role in the binding of mRNA to ribosomes (37). It had been reported to have a two-domain structure, with one domain identified to be responsible for the binding to the ribosome and the other for mRNA binding. The S1 protein also proved to be essential for initiation of the translation and for the Qβ replicase function (36). Ribosylation of the SI protein by phage T4 gene products may interfere with the translation of E. coli mRNA or, alternatively, as a consequence of a T4-specific mRNA recognition by the ribosylated S1 protein, may enhance the translation of T4 mRNA. Areas of high sequence homologies, as reported for T4 early promoters between positions −10 and +1 and further downstream (8), might be a basis for a modified mRNA recognition scheme and specific interactions. Though modifications of E. coli ribosomal proteins after T4 infection to date have not been documented, the ADP-ribosylation of the E. coli S1 protein may be a first hint towards a general mechanism affecting T4-specific translation or its regulation (20). Global effects on the E. coli translation apparatus as a consequence of phage infection already had been observed with phage T7, where gene product 0.7, a protein kinase, phosphorylates among others the ribosomal protein SI, supporting viral reproduction under suboptimal growth conditions (26).
While Skorko and Kur (31) only found the α-subunit of RNA polymerase as well as a 20-kDa protein modified by the action of Mod (these authors might have been unable to separate ModA and ModB), the recombinant enzymes ADP-ribosylate a larger number of proteins in vitro. At present it is not known whether these ADP-ribosylations are of relevance to the T4 replication cycle, or whether these ribosylations occur mainly at random (e.g., at arginine residues easily accessible to the ModA and ModB proteins). The high toxicity of the Mod proteins directed towards E. coli seems to underline the significance of these reactions. The recombinant Mod proteins in the future will be valuable tools to investigate the intricate interactions of phage and host proteins to regulate and optimize the phage T4 replication cycle.
ACKNOWLEDGMENTS
The authors thank Karl-Heinz Wüster (MPI, Dortmund) for performing the protein sequencing reaction. W.R. gratefully acknowledges the financial support of the DFG (Ru 123/22-2).
REFERENCES
- 1. Ausubel F. M.; Brent R.; Kingston R. E.; Moore D. D.; Seidman J. G.; Smith J. A.; Struhl K. Current protocols in molecular biology. In: Anonymous current protocols in molecular biology. New York: Greene Publishing and Wiley-Interscience; 1993. [Google Scholar]
- 2. Bazan J. F.; Koch-Nolte F. Sequence and structural links between distant ADP-ribosyltransferase families. Adv. Exp. Med. Biol. 419:99–107; 1997. [DOI] [PubMed] [Google Scholar]
- 3. Dagert M.; Ehrlich S. D. Prolonged incubation in calcium chloride improves the competence of Escherichia coli cells. Gene 6:23–28; 1979. [DOI] [PubMed] [Google Scholar]
- 4. Edman P. Sequence determination. Mol. Biol. Biochem. Biophys. 8:211–255; 1970. [DOI] [PubMed] [Google Scholar]
- 5. Frazier M. W.; Mosig G. The bacteriophage T4 gene mrh whose product inhibits late T4 gene expression in an Escherichia coli rpoH (sigma 32) mutant. Gene 88: 7–14; 1990. [DOI] [PubMed] [Google Scholar]
- 6. Goff C. G. Chemical structure of a modification of the Escherichia coli ribonucleic acid polymerase alpha polypeptides induced by bacteriophage T4 infection. J. Biol. Chem. 249:6181–6190; 1974. [PubMed] [Google Scholar]
- 7. Goff C. G. Coliphage-induced ADP-ribosylation of Escherichia coli RNA polymerase. Methods Enzymol. 106:418–129; 1984. [DOI] [PubMed] [Google Scholar]
- 8. Gram H.; Liebig H. D.; Hack A.; Niggemann E.; Rüger W. A physical map of bacteriophage T4 including the positions of strong promoters and terminators recognized in vitro. Mol. Gen. Genet. 194:232–240; 1984. [DOI] [PubMed] [Google Scholar]
- 9. Honjo T.; Nishizuka Y.; Hayaishi O. Diphtheria toxin-dependent adenosine diphosphate ribosylation of aminoacyl transferase II and inhibition of protein synthesis. J. Biol. Chem. 243:3553–3555; 1968. [PubMed] [Google Scholar]
- 10. Horvitz H. R. Bacteriophage T4 mutants deficient in alteration and modification of the Escherichia coli RNA polymerase. J. Mol. Biol. 90:739–750; 1974. [DOI] [PubMed] [Google Scholar]
- 11. Katada T.; Ui M. ADP ribosylation of the specific membrane protein of C6 cells by islet-activating protein associated with modification of adenylate cyclase activity. J. Biol. Chem. 257:7210–7216; 1982. [PubMed] [Google Scholar]
- 12. Kimura M.; Foulaki K.; Subramanian A. R.; Witt-mann-Liebold B. Primary structure of Escherichia coli ribosomal protein S1 and features of its functional domains. Eur. J. Biochem. 123:37–53; 1982. [DOI] [PubMed] [Google Scholar]
- 13. Koch-Nolte F.; Haag F. Mono(ADP-ribosyl) transfer-ases and related enzymes in animal tissues. Emerging gene families. Adv. Exp. Med. Biol. 419:1–13; 1997. [DOI] [PubMed] [Google Scholar]
- 14. Koch T.; Raudonikiene A.; Wilkens K.; Rüger W. Overexpression, purification, and characterization of the ADP-ribosyltransferase (gpAlt) of bacteriophage T4: ADP-ribosylation of E. coli RNA polymerase modulates T4 “early” transcription. Gene Expr. 4:253–264; 1995. [PMC free article] [PubMed] [Google Scholar]
- 15. Koch T.; Rüger W. The ADP-ribosyltransferases (gpAlt) of bacteriophages T2, T4, and T6: Sequencing of the genes and comparison of their products. Virology 203:294–298; 1994. [DOI] [PubMed] [Google Scholar]
- 16. Kutter E.; Stidham T.; Guttman B.; Batts D.; Peterson S.; Djavakhishvili T.; Arisaka F.; Mesyanzhinov V.; Rüger W.; Mosig G. Genomic map of bacteriophage T4. In: Karam J. D.; Drake J. W.; Kreuzer K. N.; Mosig G.; Hall D. H. , eds. Molecular biology of bacteriophage T4. Washington: ASM Press; 1994:491–519. [Google Scholar]
- 17. Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685; 1970. [DOI] [PubMed] [Google Scholar]
- 18. Liebig H. D.; Rüger W. Bacteriophage T4 early promoter regions. Consensus sequences of promoters and ribosome-binding sites. J. Mol. Biol. 208:517–536; 1989. [DOI] [PubMed] [Google Scholar]
- 19. Mekalanos J. J.; Collier R. J.; Romig W. R. Enzymic activity of cholera toxin. I. New method of assay and the mechanism of ADP-ribosyl transfer. J. Biol. Chem. 254:5849–5854; 1979. [PubMed] [Google Scholar]
- 20. Miller E. S.; Karam J. D.; Spicer E. Control of translation initiation: mRNA structure and protein repressors. In: Karam J. D.; Drake J. W.; Kreuzer K. N.; Mosig G.; Hall D. H., eds. Molecular biology of bacteriophage T4. Washington: ASM Press; 1994:193–205. [Google Scholar]
- 21. Moss J.; Manganiello V. C.; Vaughan M. Hydrolysis of nicotinamide adenine dinucleotide by choleragen and its A protomer: Possible role in the activation of adenylate cyclase. Proc. Natl. Acad. Sci. USA 73:4424–1427; 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moss J.; Vaughan M. Mechanism of action of choleragen. Evidence for ADP-ribosyltransferase activity with arginine as an acceptor. J. Biol. Chem. 252:2455–2457; 1977. [PubMed] [Google Scholar]
- 23. Ouhammouch M.; Adelman K.; Harvey S. R.; Orsini G.; Brody E. N. Bacteriophage T4 MotA and AsiA proteins suffice to direct Escherichia coli RNA polymerase to initiate transcription at T4 middle promoters. Proc. Natl. Acad. Sci. USA 92:1451–1455; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ouhammouch M.; Orsini G.; Brody E. N. The asiA gene product of bacteriophage T4 is required for middle mode RNA synthesis. J. Bacteriol. 176:3956–3965; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ovchinnikov Y. A.; Lipkin V. M.; Modyanov N. N.; Chertov O. Y.; Smirnov Y. V. Primary structure of α-subunit of DNA-dependent RNA polymerase from Escherichia coli . FEBS Lett. 76:108–111; 1977. [DOI] [PubMed] [Google Scholar]
- 26. Robertson E. S.; Nicholson A. W. Phosphorylation of Escherichia coli translation initiation factors by the bacteriophage T7 protein kinase. Biochemistry 31:4822–4827; 1992. [DOI] [PubMed] [Google Scholar]
- 27. Rohrer H.; Zillig W.; Mailhammer R. ADP-ribosylation of DNA-dependent RNA polymerase of Escherichia coli by an NAD+: protein ADP-ribosyltransferase from bacteriophage T4. Eur. J. Biochem. 60:227–238; 1975. [DOI] [PubMed] [Google Scholar]
- 28. Schnier J.; Kimura M.; Foulaki K.; Subramanian A. R.; Isono K.; Wittmann-Liebold B. Primary structure of Escherichia coli ribosomal protein S1 and of its gene rpsA. Proc. Nad. Acad. Sci. USA 79:1008–1011; 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seifert W.; Qasba P.; Walter G.; Palm P.; Schachner M.; Zillig W. Kinetics of the alteration and modification of DNA-dependent RNA-polymerase in T4-infected E. coli cells. Eur. J. Biochem. 9:319–324; 1969. [DOI] [PubMed] [Google Scholar]
- 30. Short J. M.; Fernandez J. M.; Sorge J. A.; Huse W. D. Lambda ZAP: A bacteriophage lambda expression vector with in vivo excision properties. Nucleic Acids Res. 16:7583–7600; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Skorko R.; Kur J. ADP-ribosylation of proteins in non-infected Escherichia coli cells. Eur. J. Biochem. 116:317–322; 1981. [DOI] [PubMed] [Google Scholar]
- 32. Skorko R.; Zillig W.; Rohrer H.; Fujiki H.; Mail-hammer R. Purification and properties of the NAD+: Protein ADP-ribosyltransferase responsible for the T4-phage-induced modification of the alpha subunit of DNA-dependent RNA polymerase of Escherichia coli . Eur. J. Biochem. 79:55–66; 1977. [DOI] [PubMed] [Google Scholar]
- 33. Smith D. B.; Johnson K. S. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67:31–10; 1988. [DOI] [PubMed] [Google Scholar]
- 34. Soman G.; Tomer K. B.; Graves D. J. Assay of mono ADP-ribosyltransferase activity by using guanylhydra-zones. Anal. Biochem. 134:101–110; 1983. [DOI] [PubMed] [Google Scholar]
- 35. Studier F. W.; Moffatt B. A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130; 1986. [DOI] [PubMed] [Google Scholar]
- 36. Subramanian A. R. Structure and functions of ribosomal protein SI. Prog. Nucleic Acid Res. Mol. Biol. 28:101–142; 1983. [DOI] [PubMed] [Google Scholar]
- 37. Suryanarayana T.; Subramanian A. R. An essential function of ribosomal protein SI in messenger ribo-nucleic acid translation. Biochemistry 22:2715–2719; 1983. [DOI] [PubMed] [Google Scholar]
- 38. Tabor S.; Richardson C. C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. USA 82:1074–1078; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Uchida T.; Gill D. M.; Pappenheimer A. M. J. Mutation in the structural gene for diphtheria toxin carried by temperate phage . Nat. New Biol. 233:8–11; 1971. [DOI] [PubMed] [Google Scholar]
- 40. Wilkens K.; Tiemann B.; Bazan F.; Rüger W. ADP-ribosylation and early transcription regulation by bacteriophage T4. Adv. Exp. Med. Biol. 419:71–82; 1997. [DOI] [PubMed] [Google Scholar]