

Abstract
In previous studies we demonstrated that the E1A DNA and proteins of group C adenovirus are present in excess in the lungs of patients with chronic obstructive pulmonary disease (COPD). Because adenovirus E1A gene products are known to regulate the expression of many genes by interacting with cellular transcription factors, we postulated that E1A enhances the production of inflammatory mediators and exacerbates the inflammatory process in smokers’ lungs. We reported that LPS-induced ICAM-1 expression in A549 cells is upregulated by E1A. In the current study we investigated whether this regulation is mediated through the ICAM-1 promoter. A549 cells and primary human bronchial epithelial (HBE) cells were transiently cotransfected with a plasmid containing the ICAM-1 enhancer-promoter linked to the chloramphenicol acetyltransferase (CAT) reporter gene (pBS-CAT-P) and either a plasmid carrying the adenovirus 5 E1A gene (pE1Aneo) or a control plasmid (pneo). To compare the effect of transient versus stable E1A expression on the activity of this promoter, we also transiently transfected stable E1A-expressing A549 cells with pBS-CAT-P. Transient cotransfection of pE1Aneo and pBS-CAT-P had no effect on basal ICAM-1 promoter activity in A549 or HBE cells. After stimulation of A549 cells with TNF-α, IFN-γ, or LPS, promoter activity was increased by two- to threefold in the presence of adenovirus E1A. In HBE cells, on the other hand, E1A repressed the ICAM-1 promoter after stimulation with IFN-yand LPS with little change after TNF-α stimulation. In stable E1A transfectants, ICAM-1 promoter activity was 2 to 2.5 times higher than in control transfectants with or without stimulation with TNF-α or LPS. These findings suggest that E1A can modulate the activity of the ICAM-1 promoter in lung epithelial cells and this modulation is different in cells of alveolar origin compared to bronchial epithelial cells.
Keywords: Adenovirus E1A, Lung epithelial cells, ICAM-1 promoter, Inflammatory stimuli
CIGARETTE smoking is the major risk factor for the development of chronic obstructive pulmonary diseases (COPD), but only 10–20% of smokers actually develop airways obstruction (8). Childhood respiratory infection, predominantly in the first year of life, is another independent risk factor for the development of COPD (10) with adenovirus and respiratory syncytial virus as the major viral agents responsible for these childhood illnesses (3). The group C adeno-viruses are of particular interest in this regard because they persist in tonsils (12), peripheral blood lymphocytes (16), and the lungs of asymptomatic adults (1,26). Studies from our laboratory showed that the ElA region of the adenovirus genome is found in greater copy numbers in lungs of smokers with COPD than in lungs of controls with normal lung function matched for age, gender, and smoking history (26). Subsequent studies established that the E1A proteins could be detected in epithelial cells lining the airways, alveoli, and submucosal glands in human lungs (7). Our working hypothesis is that the adenoviral E1A DNA that persists in the lung following these childhood infections is expressed and its gene products amplify the inflammatory process present in the lungs of smokers. This could help explain why cigarette smoking produces airway inflammation in everyone but only causes airway obstruction in a minority of smokers.
The adenovirus E1A gene is the first viral gene expressed during infection and encodes nuclear phosphoproteins that transactivate a selective set of both viral and cellular genes. This regulation by E1A requires endogenous host transcription factors. Of these, specific members of the family of cellular activating transcription factors interact with E1A through their DNA binding domains and directly mediate E1A-inducible transcriptional activation (23). The activity of other transcription factors such as NF-κB are modulated by E1A but by more indirect mechanisms (9,29). Here, evidence has been presented that the interactions between E1A and the transcription factor are mediated by coactivators, cAMP-responsive element binding (CREB) binding protein (CBP) and the related protein, p300. These findings are in keeping with the current concept that CBP/p300 can interact with a wide variety of nuclear proteins that affect transcription, including DNA binding proteins, the basal transcriptional machinery, as well as viral nuclear proteins such as E1A, and that some of these can bind to each other [reviewed in (17)]. Through similar mechanisms, we postulate that E1A, chronically expressed by latent adenovirus, could alter the activity of specific host genes that control the inflammatory process.
Intercellular adhesion molecule-1 (ICAM-1) is involved in a wide variety of immune and inflammatory interactions [reviewed in (31)]. As a natural ligand for the lymphocyte adhesion molecule LFA-1 and the adhesion molecule Mac-1 found on monocytes and granulocytes, it plays an important role in immune cell trafficking. Reports showing that ICAM-1 expression is related to the pathogenesis of airway diseases include those of increased ICAM-1 expression in basal epithelial cells in COPD patients (6) and of its contribution to the pathogenesis of the asthma (37).
Previously we found that adenovirus 5 E1A increased ICAM-1 and interleukin-8 (IL-8) expression, which were induced by LPS stimulation of A549 lung epithelial cells (19,20). This increased expression correlated with increased levels of the mRNA of these two inflammatory mediators and suggested that the regulation of their expression occurs mainly at the transcriptional level (19,20). This upregulation of inflammatory mediator expression also correlated with increases in LPS-induced NF-κB activity in these E1A-positive cells (21). As an NF-κB binding site is present in the ICAM-1 enhancer-promoter (5), the present study was designed to investigate whether the ICAM-1 promoter activity, as determined by the chloramphenicol acetyltransferase (CAT) reporter gene assay, is affected by E1A proteins in alveolar as well as bronchial epithelial cells.
MATERIALS AND METHODS
Reagents
LPS from E. coli 0111:B4 obtained from Sigma Chemical Co. (St. Louis, MO) was dissolved in sterile distilled water at 10 mg/ml. Recombinant human TNF-α from Calbiochem (LaJolla, CA) and IFN-γ from Gibco BRL (Gaithersburg, MD) were reconstituted in 1% BSA in phosphate-buffered saline (PBS) at 5 × 105 and 7 × 105 U/ml, respectively. N-Butyryl coenzyme A (Sigma) was dissolved in 0.25 M Tris-HCl, pH 8, at 5 mg/ml. These reagents were stored at −70°C and diluted to the appropriate concentration before use.
Plasmid Used in Transfections
The plasmid, pElAneo, was a generous gift from Dr. F. L. Graham (McMaster University, Hamilton, Ontario). This expression vector carries the promoter and entire coding region of the E1A gene of adenovirus 5 from nucleotides 25 to 1770. A control plasmid, pneo, was generated from pElAneo by deleting the 1.8-kb BamHI-SacI fragment containing the E1A DNA (19). The plasmid, pBS-CAT-P, was a kind gift of Dr. S. W. Caughman (Emory University, Atlanta, GA) (5). This vector carries the 1165 nucleotide fragment of the ICAM-1 5′-flanking region linked to the CAT coding region. The plasmid pCAT-Control (Promega, Madison, WI), which contains the SV40 promoter and enhancer sequences placed 5’ of the CAT gene, was used as a positive control for CAT expression and as a means of monitoring differences in transfection efficiency between cell lines. Plasmid DNAs were prepared using the Plasmid Maxi Kit from Qiagen Inc. (Chatsworth, CA).
Cell Isolation and Culture Conditions
A549 cells from the American Type Culture Collection (Rockville, MD) are a human lung epithelial cell line originally derived from a patient with bronchioloalveolar carcinoma (22). E23 is a clone of the A549 cell stably transfected with pElAneo that is positive for adenovirus E1A expression while C5, a clone of the A549 cell transfected with pneo, is not (19). These cells were grown in Eagle’s minimum essential medium (MEM) from Gibco BRL supplemented with 10% fetal bovine serum (FBS) from Hyclone (Logan, UT). Primary human bronchial epithelial (HBE) cells from bronchial tissue from two patients undergoing resection for a lung tumor at St. Paul’s Hospital, Vancouver, BC, Canada were isolated according to a procedure described previously (13). The clinical data for these patients are presented in Table 1. In brief, 1-cm-long pieces of human bronchial tissue excised from a site remote from the tumor were treated at 4°C for 24 h with 0.1% protease (Type 14, Sigma) solution prepared in MEM. The epithelial cells were then scraped off and washed with bronchial epithelial cell growth medium (BEGM) (Clonetics, San Diego, CA). We also examined HBE cells from Clonetics using the same growth and transfection conditions as those applied to the HBE cells from the local source. HBE cells from the local source were used for transfection from passage 3 to 6 while those from Clonetics, received at passage 2, were used up to passage 4.
TABLE 1.
CLINICAL DATA OF PATIENTS USED FOR HBE CELL SOURCE
| HBE Cell Source | Age | Sex | Smoke* | % DLCO† | % FEV1‡ | % FVC§ | % RV¶ | % TLC# |
|---|---|---|---|---|---|---|---|---|
| Local: patient #1 | 52 | F | 1080 | 85 | 73 | 82 | 152 | 108 |
| Local: patient #2 | 49 | F | 0 | 85.5 | 90 | 91 | 87 | 91 |
| Clonetics | 10 | M | na | na | na | na | na | Na |
na: not available.
Number of cigarettes smoked per day times the years of smoking.
Percent of predicted CO diffusion capacity of the lung.
Percent of predicted forced expiratory volume in 1 s.
Percent of predicted forced vital capacity of the lung.
Percent of predicted residual volume of the lung.
Percent of predicted total lung capacity.
Transient Transfection of E23, C5, A549, and HBE Cells
E23, C5, and A549 cells or HBE cells were seeded on six-well plates in MEM or BEGM, respectively. When 70–80% confluent, approximately 24 h after seeding, the cells were transfected with plasmid DNA using a DEAE-dextran protocol (33), which was optimized for each cell line investigated. The plasmid DNA was suspended in a solution containing DEAE-dextran, 50 mM Tris-HC1, pH 7.1, in MEM (serum free) or BEGM. The concentration of dextran used for E23, C5, and A549 cells was 200 μg/ml; for HBE cells it was 30 μg/ml. E23 and C5 cells were transfected with pBS-CAT-P or pCAT-Control. A549 and HBE cells were cotransfected with pBS-CAT-P and either pElAneo or pneo. The final concentration of each plasmid DNA used for transfection was 1 μg/ml. The cells were exposed to the plasmid DNAs for 6 h then placed in 10% dimethyl sulfoxide in 21 mM HEPES, 135 mM NaCl, 5 mM KCl, 0.8 mM Na2HPO4, pH 7.4, and 5 mM dextrose for 5 min. After washing with PBS, cells were placed in complete medium, either MEM with 10% FBS for A549 cells and their derivatives or BEGM for HBE cells. All cells were transfected under the same conditions except for differences in the DEAE-dextran concentration and culture media specifications already noted. To examine whether differences in the culture conditions might affect how HBE cells respond to adenoviral E1A regulation, HBE cells from patient #2 (Table 1) were also placed in BEGM with 10% FBS or MEM with 10% FBS after transfection and washing with PBS. One day after the transfection all cells either remained unstimulated or were stimulated with LPS (10 μg/ml), TNF-α (100 U/ml), or IFN-γ (100 U/ml) for 24 h. After this period all cells were analyzed by the CAT assay.
To compare the proliferation and viability of the HBE cells under the different culture conditions, passage 6 HBE cells from patient #1 (Table 1) were seeded in BEGM onto three six-well plates and 24 h after seeding the medium was changed in each of the plates with either fresh BEGM, BEGM with 10% FBS, or MEM with 10% FBS. After 6, 24, and 48 h cells were washed in PBS, trypsinized, counted, and then stained with trypan blue to measure viability.
CAT Assay
CAT assays were performed as described previously (11). 14C-Labeled chloramphenicol (CM) was separated from its butyrylated derivatives by ascending thin-layer chromatography using chloroform/ methanol (95:5). After chromatography the plates were used to expose Kodak XAR-5 film for 96 h before the film was developed. The conversion of CM to its butyrylated derivatives was quantified on the ULTROSCAN laser densitometer using the Gelscan analysis software, ver. 2.1 (Pharmacia LKB, Uppsala, Sweden). The CAT activity was expressed as percentage of the sum of the densities of the autoradiographic signal representing the two butyrylated CM divided by the sum of the densities of the signals from the butyrylated CM and the substrate. To take into consideration differences in the transfection efficiency between E23 cells and C5 cells, the ICAM-1 promoter activity was expressed as a ratio of CAT activity as measured above to that found in the same cells transfected with the pCAT-Control plasmid.
Immunocytochemical Detection of ElA Expression
Cells grown on coverslips in six-well plates were fixed in 10% formalin for 5 min then in acetone for 30 s. Immunocytochemistry was performed on the fixed cells by the alkaline phosphatase antialkaline phosphatase (APAAP) method previously described (7). In brief, the fixed cells were equilibrated in 0.15 M NaCl, 0.05 M Tris-HCl, pH 7.6 (TBS), and prein-cubated in 5% normal rabbit serum in TBS for 20 min. The cells were then incubated with anti-ElA monoclonal antibody M73 (15) (Oncogene Sciences, Uniondale, NY) or control mouse IgG2a, and then with rabbit anti-mouse immunoglobulins (Dako, Glostrup, Denmark) followed by the APAAP complex (Dako). After each incubation the cells were rinsed with TBS three times. The substrate, naphthol AS-BI phosphate (Sigma) with new fuchsin (Merck, Rahway, NJ), was applied to the preparations for color development. Endogenous alkaline phosphatase activity was blocked by adding 1 mM levamisole (Sigma) to the substrate.
Statistical Analysis
Data were analyzed by the analysis of variance (ANOVA) and are expressed as the mean ± SEM. A value of p< 0.05 was considered significant.
RESULTS
Effect of ElA on ICAM-1 Promoter Activity in A549 Cells Stably Transfected With E1A
To measure differences in transfection efficiency between the stable E1A transfectant, E23, and control cells, C5, these cells were transiently transfected with pCAT-Control. CAT activity was higher in E1A-ex-pressing cells than in control cells, indicating a higher efficiency of transfection of the stable E1A transfec-tants (Fig. 1a). To examine the effect of E1A on the ICAM-1 promoter activity in stable E1A transfec-tants, pBS-CAT-P was transiently transfected into E1A-expressing cells, E23, and control cells, C5, and the ICAM-1 promoter activity was measured (Fig. 1a) and expressed as a ratio of CAT activity found in these cells compared to that found in the same cells transfected with pCAT-Control (Fig. 1b). After this correction for differences in transfection efficiency was made, ICAM-1 promoter activity in control C5 cells, in the presence or absence of stimulation, was found to be low. Overall, this activity was much higher in ElA-expressing E23 compared to C5 and this increased activity was statistically significant in unstimulated E23 cells as well as in these cells stimulated with TNF-α and LPS (Fig. 1b).
FIG. 1.
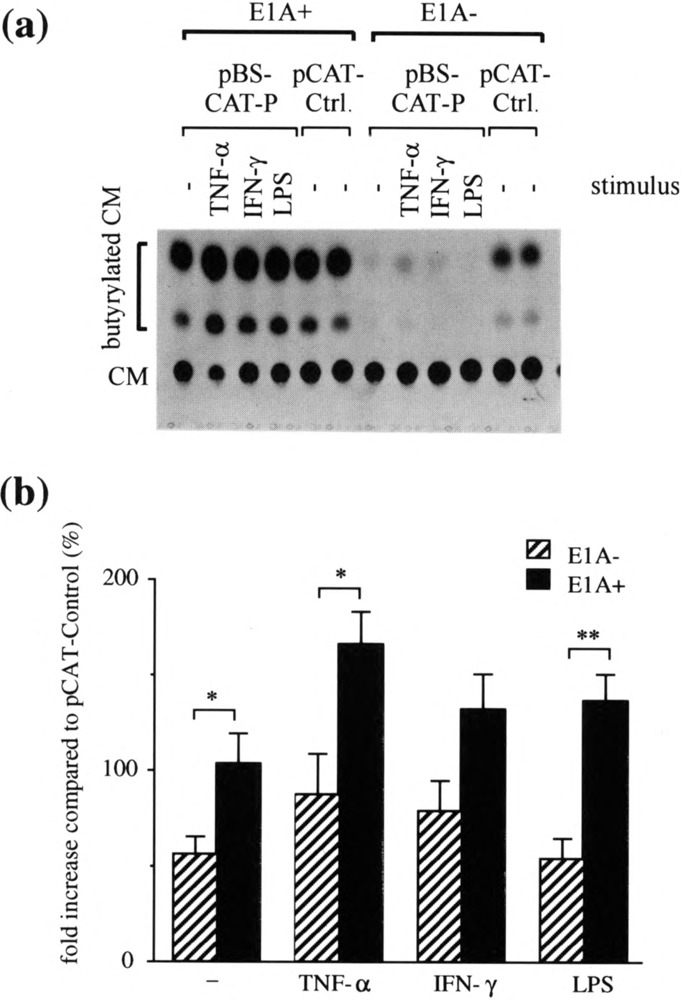
The effect of stably transfected E1A on ICAM-1 promoter activity in A549 cells. E23, a stable E1A transfectant (E1A+) and C5, a control transfectant (E1A−) (19) were transfected with pBS-CAT-P (1 μg/ml) or pCAT-Control (pCAT-Ctrl.) (1 Ug/ml). The cells were left unstimulated (−) or stimulated with TNF-α (100 U/ml), IFN-γ (100 U/ml), or LPS (10 μg/ml) for 24 h and then analyzed by the CAT assay, (a) A representative autoradiogram of a chromatogram from one experiment, (b) CAT activity in cells transfected with pBS-CAT-P was quantitated by densitometric scanning of an autoradiogram such as that shown in (a) and normalized to the CAT activity found in the same El A+ or E1A- cells transfected with pCAT-Control. Data are expressed as means ± SEM of six independent experiments. *p < 0.05, **p < 0.001.
Effect of E1A on ICAM-1 Promoter Activity After Transient Transfection of A549 Cells
Because adenovirus E1A is known to repress the activity of the SV40 enhancer-promoter (35), the pCAT-Control used to monitor differences in transfection efficiency between E23 and C5 could also reflect differences due to the regulation of the SV40 enhancer-promoter by E1A. To avoid both this problem and that of transfection differences between these cells, parent A549 cells were cotransfected with pElAneo and pBS-CAT-P or, as control, with pneo and pBS-CAT-P. After pE1Aneo transfection, E1A protein was detected in 4.5 ± 0.8% of the A549 cells (n = 4) with staining found in the nuclei of cells (Fig. 2a). No staining was seen when these cells were stained with an isotypic control antibody (Fig. 2b). CAT activity was measured 48 h after transfection, which included 24 h in the presence or absence of various inflammatory stimuli (Fig. 3). In the absence of adenovirus E1A little or no ICAM-1 promoter activity was detected in unstimulated A549 cells. TNF-α induced weak ICAM-1 promoter activity in these cells while IFN-γ and LPS had little or no effect. Transient transfection of the ElA gene had no effect on the ICAM-1 promoter activity in unstimulated A549 cells. On the other hand, stimulation with TNF-α, IFN-γ, or LPS in the presence of the viral ElA gene enhanced promoter activity two- to threefold over that found in the absence of ElA.
FIG. 2.

The expression of E1A proteins in A549 and HBE cells transiently transfected with pE1Aneo. Cells grown on coverslips were stained with the E1 A monoclonal antibody M73. (a) A549 cells transfected with pElAneo and pBS-CAT-P. The staining (black nuclei) shows that the ElA protein is found in the nuclei of these cells, (b) The same cells as in (a) stained with an isotypic control antibody. No staining is seen herein, (c) HBE cells from a surgical specimen from patient #2 transfected with pElAneo and pBS-CAT-P. The staining (black nuclei) shows that the E1A protein is located in the nuclei of these cells, (d) The same cells as in (c) stained with an isotypic control antibody. No staining is seen herein. Bar = 10 μm.
FIG. 3.
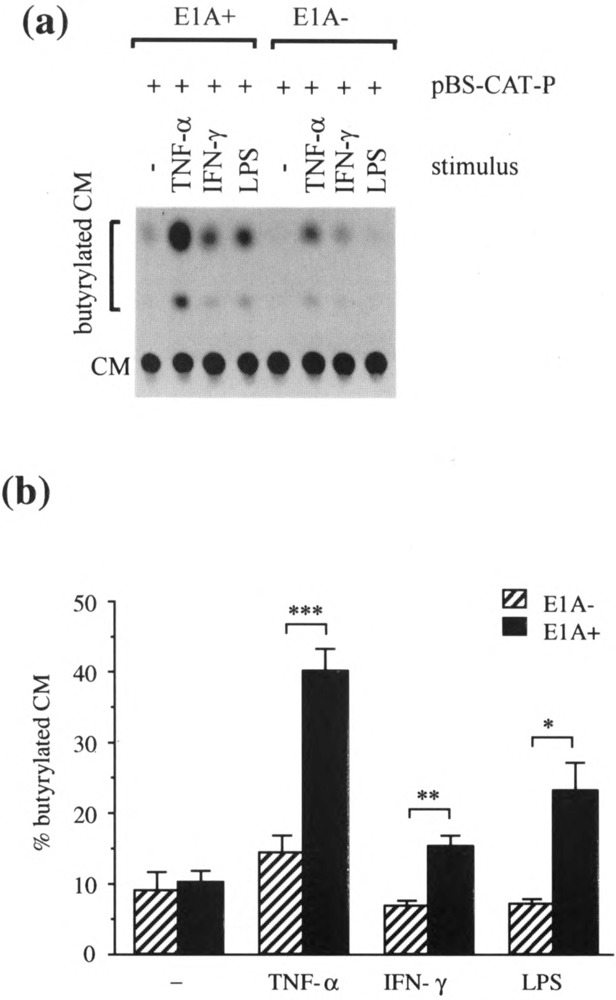
The effect of transient ElA transfection on ICAM-1 promoter activity in A549 cells. A549 cells were cotransfected with pE1Aneo (1 μg/ml) (E1A+) or control plasmid pneo (1 μg/ml) (E1A−) and with pBS-CAT-P (1 μg/ml). Twenty-four hours after transfection cells were left unstimulated (−) or were stimulated with TNF-α (100 U/ml), IFN-γ (100 U/ml), or LPS (10 ug/ml) for another 24 h then analyzed by the CAT assay, (a) A representative autoradiogram of a chromatogram from one experiment, (b) CAT activity was quantitated by densitometric scanning of an autoradiogram such as that shown in (a) and expressed as the percent conversion of CM to butyrylated CM. Data are expressed as means ±SEM of four independent experiments. *p<0.01, **p< 0.005, ***p< 0.001.
Effect of the ElA Gene on ICAM-1 Promoter Activity in HBE Cells After Transient Transfection With pE1Aneo
To examine the effect of ElA on the ICAM-1 promoter activity in HBE cells, primary bronchial epithelial cells from surgically resected lungs were cultured and cotransfected with pBS-CAT-P and pElAneo or pneo. The ElA protein was detected in 1.8 ±0.4% of HBE cells transfected with pElAneo (n = 4) (Fig. 2c). Consistent with results found for A549 cells transiently transfected with pElAeno, the staining showed that the ElA protein is found in the nuclei of HBE cells. No staining was seen when these cells were stained with an isotypic control antibody (Fig. 2d).
As with A549 cells, ICAM-1 promoter activity in HBE cells was very weak in the absence of additional stimuli (Fig. 4). LPS and IFN-γ stimulation of these cells increased promoter activity with IFN- γ as the strongest inducer (Fig. 4b). TNF-α had a minimal effect. ElA did not affect the basal level of the ICAM-1 promoter activity in HBE cells; however, it drastically repressed the effect of IFN-γ on this promoter. ElA also reduced promoter activity significantly after LPS stimulation (Fig. 4b).
FIG. 4.
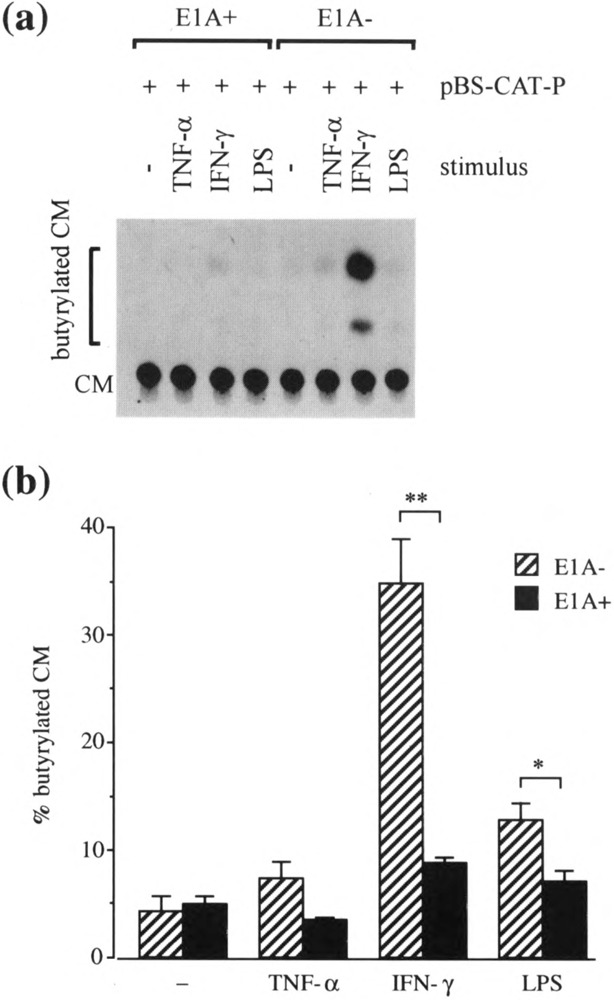
The effect of transient adenovirus ElA transfection on ICAM-1 promoter activity in HBE cells from resected lung specimens. HBE cells were cotransfected with pElAneo (1 μg/ml) (E1A+) or control plasmid, pneo (1 μg/ml) (El A−), and with pBS-CAT-P (1 μg/ml). Twenty-four hours after transfection cells were left unstimulated (−) or were stimulated with TNF-a (100 U/ml), IFN-γ (100 U/ml), or LPS (10 μg/ml) for another 24 h then analyzed by the CAT assay, (a) A representative autoradiogram of a chromatogram from one experiment, (b) CAT activity was quantitated by densitometric scanning of an autoradiogram such as that shown in (a) and expressed as the percent conversion of CM to butyrylated CM. Data are expressed as means ± SEM of four separate experiments performed on two batches of cells from each of the two patients. *p < 0.05, **p < 0.001.
To confirm the results from HBE cells cultured from specimens resected at our institution, we also analyzed HBE cells purchased from a commercial source (Clonetics) in the same manner (Fig. 5). Compared to the very low ICAM-1 promoter activity found in these cells in the control state, all three stimuli (IFN-γ, TNF-α, and LPS) induced promoter activity. As with HBE cells obtained locally, IFN-γ was the strongest inducer in the commercially obtained HBE cells. Also, consistent with results from HBE cells obtained locally, adenovirus E1A significantly repressed the ICAM-1 promoter activity induced by IFN-γ and LPS in the cells obtained commercially. In addition, a significant repression by ElA of the TNF-α-induced promoter activity was observed.
FIG. 5.
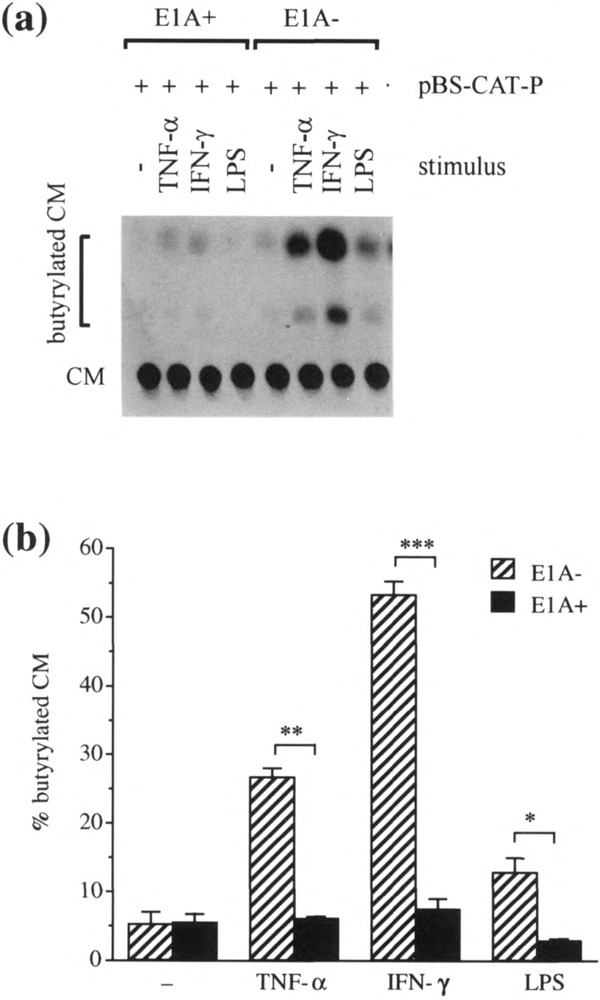
The effect of transient adenovirus E1A transfection on ICAM-1 promoter activity in HBE cells from a commercial source. HBE cells were cotransfected with pElAneo (1 μg/ml) (E1A+) or control plasmid, pneo (1 pg/ml) (E1A-), and with pBS-CAT-P (1 μg/ml). Twenty-four hours after transfection cells were left unstimulated (−) or were stimulated with TNF-a (100 U/ml), IFN-γ (100 U/ml), or LPS (10 μg/ml) for another 24 h then analyzed by the CAT assay, (a) A representative autoradiogram of a chromatogram from one experiment, (b) CAT activity was quantitated by densito-metric scanning of an autoradiogram such as that shown in (a) and expressed as the percent conversion of CM to butyrylated CM. Data are expressed as means ± SEM of three separate experiments. *p < 0.01, **p < 0.0005, ***p < 0.0001.
Culturing HBE cells in BEGM or MEM supplemented with 10% FBS for 48 h did not alter the ICAM-1 promoter activity observed when cells were grown in BEGM alone, either under basal conditions or after stimulation with TNF-α, IFN-γ, or LPS (data not shown). Even in the presence of adenovirus E1A, the ICAM-1 promoter activity, either at the basal level or after treatment with the three stimuli, was not affected by these changes in culture conditions (data not shown).
Incubation of HBE cells in BEGM or MEM with 10% FBS for 6 and 24 h did not affect their growth (Fig. 6) or viability (data not shown) compared to cells grown in BEGM alone. After 48-h incubation, although the viability of the adherent cells grown with FBS supplement was more than 90% of those grown in BEGM alone, total cell counts in MEM and BEGM supplemented with FBS were reduced to 50% and 54%, respectively, of those in BEGM alone (Fig. 6).
FIG. 6.
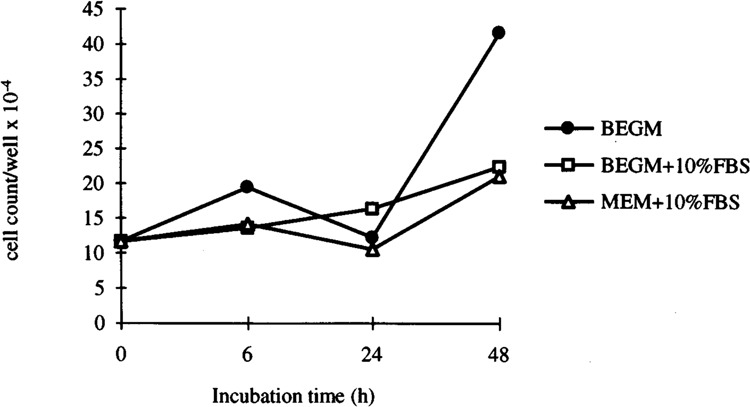
HBE cell proliferation under different medium conditions. The HBE cells were seeded onto six-well plates in BEGM and after 24 h the medium was changed to BEGM with or without 10% FBS or MEM with 10% FBS. After 6, 24, and 48 h proliferation under these conditions was determined by counting the number of cells (n = 2).
DISCUSSION
The results of this study show that adenovirus ElA can modulate the activity of the promoter in the isolated 5′ regulatory region of the human ICAM-1 gene in human lung epithelial cells and this modulation depends on various factors, among them the inducing stimulus and whether the expression of ElA in these cells is stable or transient, but the main modifier is the cell type, whether they are transformed alveolar carcinoma cells or primary bronchial epithelial cells. In A549 cells where this promoter is minimally responsive to stimulation by TNF-α, LPS, and IFN-γ, the adenovirus El A, present either permanently or transiently for the duration of the experiment, reverses the response of the promoter to one of increased activity. On the other hand, in HBE cells where the promoter is readily activated by these same stimuli, the effect of ElA is to reduce this activity. However, as in A549 cells, the overall effect of ElA is again a reversal of the response found in the absence of this viral nucleoprotein.
The interpretation of the results from the stable E1A transfectants of A549 cells relies on the ability to measure differences in transfection efficiency between the E1A-positive and control cells. It could be argued that the pCAT-Control used for this purpose could underestimate the transfection efficiency of E1A-expressing cells because the activity of the SV40 promoter driving the CAT gene is known to be repressed by adenovirus E1A (35). Consequently, the ratio of CAT activity from pBS-CAT-P to that from pCAT-Control would be overestimated in El A-positive cells. However, as results of an enhancement of ICAM-1 promoter activity by E1A in transiently cotransfected A549 cells, where correction for transfection efficiency using pCAT-Control was not necessary, support our findings in A549 cells stably expressing El A, this repression of the SV40 promoter in pCAT-Control is considered minimal.
The effect of ElA to enhance the activity of the promoter in the isolated 5’ regulatory region of the ICAM-1 gene in A549 cells in response to LPS stimulation is consistent with our findings of increased ICAM-1 mRNA and protein expression in response to this inflammatory stimulus in stable ElA transfec-tants of A549 cells (19). The mechanism by which adenovirus El A, a nuclear protein, brings about this increased expression in the absence of the LPS receptor, CD 14, has not been elucidated (19). In initial experiments we have shown, however, that the transcription factor NF-κB is induced by LPS in these cells if ElA is present (21). As the 1165-bp 5′ regulatory sequence of the ICAM-1 gene used in this study includes the binding site for this transcription factor (5,32), a similar induction of NF-κB in response to LPS is expected to take place to activate this promoter in A549 cells when adenovirus ElA is present, either as a result of stable or transient transfection.
The well-documented repression of the interferon signal transduction pathway by ElA proteins (14,24, 30,34) supports our finding of a reduction in IFN-γ -induced ICAM-1 promoter activity in HBE cells by El A. Our results are similar to those from Hela cells where adenovirus ElA prevented the IFN-γ induction of CAT activity driven by a transcriptional regulatory region of an IFN- γ responsive gene (18). ElA gene products are known to block the expression of IFN-inducible genes at the level of transcription, possibly by decreasing the amount of the signal transducer and activator of transcription family of proteins (34). Contrary to these findings, an IFN- γ response by the ICAM-1 promoter was very weak in our A549 cells and, furthermore, this response was enhanced in the presence of adenovirus El A. This result contrasts the endogenous ICAM-1 expression in A549 cells which, in keeping with the established ElA repression, showed that both mRNA and protein expression induced by IFN-γ were significantly reduced in the presence of ElA (19). One explanation for the difference between the effect of ElA on the response to IFN-γ as monitored by the expression of the reporter gene driven by the isolated ICAM-1 promoter-enhancer reported here and by the expression of the endogenous ICAM-1 mRNA reported earlier (19) is the IFN-γ stabilization of an otherwise labile ICAM-1 mRNA, labile because of destabilizing sequences in its coding region (28). However, because of the overwhelming evidence in favor of a strong activation of the ICAM-1 promoter by IFN-γ, we consider mRNA stability a minor player in accounting for differences between results of the isolated promoter and the endogenous one. Furthermore, it is interesting to note that Voraberger and coworkers (36) reported that in A549 cells, while the 1.3-kb ICAM-1 upstream region mediates responsiveness to IL-1, it did not to IFN-γ. When a more extensive 5-kb flanking region was included, reporter activity was increased twofold after IFN-γ stimulation. These authors suggest that regulatory elements mediating IFN-γ responsiveness in A549 cells are located further upstream of the 1.3-kb fragment. Because the 1.165-kb ICAM-1 promoter used in our studies is similar to the 1.3-kb fragment used by Voraberger and coworkers, our result of a lack of IFN-γ responsiveness could be related to the missing response element. Taken together, our results suggest that in HBE cells ElA is very effective in suppressing the 1.16-kb ICAM-1 promoter tested here while in A549 cells additional sequences more distal than those included here may be required to reveal the full regulatory control by IFN-γ and its subsequent repression by E1A.
The basal ICAM-1 promoter activity was generally higher in stable E1A transfectants of A549 cells than in the corresponding controls. On the other hand, A549 cells transiently transfected with E1A, like their controls, showed little or no basal promoter activity. Two possibilities could account for this difference. One is the duration of E1A activity in the cell. In the stable ElA transfectant this viral gene is expressed in the cell continually while in transient transfectants E1A is expressed at most only for the 24 h prior to the application of stimuli. The 24-h period of ElA expression may be too short to reproduce the changes that are manifested in the modulation of the activity of the ICAM-1 promoter in the stable transfectants. Another difference is the state of the E1A gene in these cells. In stable transfectants the ElA gene may be integrated into the genomic DNA of the cell, while in the transient transfectants it most likely resides as plasmid DNA. Therefore, the regulation of the expression of the E1A gene itself and hence ElA’s ability to alter the basal level of expression of the reporter gene may be affected. Contrary to this notion, ElA’s ability to modify the host response to inflammatory stimuli was not altered by the stability of the ElA transfection because the increased activity of the ICAM-1 promoter in transiently transfected A549 cells appeared to equal that in stable transfectants.
Although both are lung epithelial cells, modulation of the ICAM-1 promoter activity by adenovirus E1A in HBE cells was opposite to that found in A549 cells. In A549 cells this promoter was minimally responsive to TNF-α, LPS, or IFN-γ stimulation while the presence of the adenovirus E1A markedly enhanced this response. In contrast, in HBE cells this promoter was more responsive to these stimuli and the presence of E1A repressed this activation. This difference is most likely due to inherent properties of the distinct cell types where A549 cells are from a pulmonary carcinoma and have features of type II alveolar cells while the HBE cells are primary cells from large airways. Although differences in cell type are a major consideration, differences in culture conditions must also be considered. A549 cells were cultured in medium with added serum, while HBE cells were cultured in serum-free medium with the addition of retinoic acid and corticosteroid, which are known to regulate ICAM-1 expression in other types of cells (2,4). To examine the effect of serum, we performed the same CAT assay on HBE cells grown in BEGM plus FBS or in MEM plus FBS. Although the rate of proliferation of the cells was reduced with the addition of serum, cell viability was not altered and the results from the CAT assay were not different from that found in serum-free medium. Because long-term incubation of HBE cells in serum causes terminal differentiation, most likely due to the presence of TGF-β1, causing squamous cell differentiation of these cells (27), we were limited in the length of time these cells could be analyzed under these conditions. Therefore, we cannot rule out the possibility that long-term effects of the serum could alter the response of HBE cells to inflammatory stimuli.
Even in the absence of ElA the response of HBE cells to inflammatory stimuli differed from that of A549 cells. In HBE cells the ICAM-1 promoter appeared to be more responsive to IFN-γ than TNF-α, while in A549 cells it was more responsive to TNF-α than IFN-γ. The results from the HBE cells are consistent with those from tracheal bronchial epithelial and BEAS-2B cells where a marked increase in ICAM-1 mRNA and protein was observed after IFN-γ but not with TNF-α stimulation (25). Similarly, the results from the A549 cells are supported by results from our previous study in A549 cells showing that ICAM-1 expression as measured by ELISA was higher after TNF stimulation compared to IFN-γ (19). Again, cell type-specific attributes such as differences in the expression of the receptors for these two ligands in the two cell types might offer a possible explanation for these differences.
In conclusion, we have demonstrated that adenovirus E1A can modulate the activity of the ICAM-1 promoter in both alveolar and bronchial epithelial cells. The viral protein upregulates ICAM-1 promoter activity in response to an inflammatory stimulus in A549 cells and downregulates this activity in HBE cells. The mechanisms(s) by which ElA alters the activity of the ICAM-1 promoter in lung epithelial cells remains to be determined.
ACKNOWLEDGMENTS
We thank Dr. W. S. Caughman and Dr. F. Graham for their generous gifts of plasmid DNAs, namely, pBS-CAT-P and pE1Aneo, respectively. N. Keicho was a recipient of the Canadian Cystic Fibrosis Foundation fellowship and Y. Higashimoto a recipient of a MRC/PMAC Health Program-Astra Pharma Respiratory Medicine fellowship. This work was supported by the National Centres of Excellence for Respiratory Health and by the B.C. Lung Association.
REFERENCES
- 1. Bateman E. D.; Hayashi S.; Kuwano K.; Wilke T. A.; Hogg J. C. Latent adenoviral infection in follicular bronchiectasis. Am. J. Respir. Crit. Care Med. 151:170–176; 1995. [DOI] [PubMed] [Google Scholar]
- 2. Bouillon M.; Audette M. Retinoic acid-stimulated intercellular adhesion molecule-1 expression on SK-N-SH cells: Calcium/calmodulin-dependent pathway. Cancer Res. 54:4144–4149; 1994. [PubMed] [Google Scholar]
- 3. Brandt C. D.; Kim H. W.; Vargosko A. J.; Jeffries B. C.; Arrobio J. O.; Rindge B.; Parrott R. H.; Chanock R. M. Infections in 18,000 infants and children in a controlled study of respiratory tract disease. I. Adenovirus pathogenicity in relation to serologic type and illness syndrome. Am. J. Epidemiol. 90:484–500; 1969. [DOI] [PubMed] [Google Scholar]
- 4. Cronstein B. N.; Kimmel S. C.; Levin R. I.; Martiniuk F.; Weissmann G. A mechanism for the anti-inflammatory effects of corticosteroids: The glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA 89:9991–9995; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Degitz K.; Lian-Jie L.; Caughman S. W. Cloning and characterization of the human intercellular adhesion molecule-1 gene. J. Biol. Chem. 266:14024–14030; 1991. [PubMed] [Google Scholar]
- 6. Di Stefano A.; Maestrelli P.; Roggeri A.; Turato G.; Calabro S.; Potena A.; Mapp C. E.; Ciaccia A.; Covacev L.; Fabbri L. M.; Saetta M. Upregulation of adhesion molecules in the bronchial mucosa of subjects with chronic obstructive bronchitis. Am. J. Respir. Crit. Care Med. 149:803–810; 1994. [DOI] [PubMed] [Google Scholar]
- 7. Elliott W. M.; Hayashi S.; Hogg J. C. Immunodetection of adenoviral ElA proteins in human lung tissue. Am. J. Respir. Cell Mol. Biol. 12:642–648; 1995. [DOI] [PubMed] [Google Scholar]
- 8. Fletcher C.; Peto R. The natural history of chronic airflow obstruction. Br. Med. J. 1:1645–1648; 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gerritsen M. E.; Williams A. J.; Neish A. S.; Moore S.; Shi Y.; Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc. Natl. Acad. Sci. USA 94:2927–2932; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gold D. R.; Tager I. B.; Weiss S. T.; Tosteson T. D.; Speizer F. E. Acute lower respiratory illness in childhood as predictor of lung function and chronic respiratory symptoms. Am. Rev. Respir. Dis. 140:877–884; 1989. [DOI] [PubMed] [Google Scholar]
- 11. Gorman C. M.; Moffat L. F.; Howard B. H. Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol. Cell. Biol. 2:1044–1051; 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Green M.; Wold W. S. M.; Mackey J. K.; Rigden P. Analysis of human tonsil and cancer DNAs and RNAs for DNA sequences of group C (serotypes 1, 2, 5 and 6) human adenoviruses. Proc. Natl. Acad. Sci. USA 76:6606–6610; 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gruenert D. C.; Basbaum C. B.; Widdicombe J. H. Long-term culture of normal and cystic fibrosis epithelial cells grown under serum-free conditions. In Vitro Cell. Dev. Biol. 26:411–418; 1990. [DOI] [PubMed] [Google Scholar]
- 14. Gutch M. J.; Reich N. C. Repression of the interferon signal transduction pathway by the adenovirus E1A oncogene. Proc. Natl. Acad. Sci. USA 88:7913–7917; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harlow E.; Franza B. R. Jr.; Schley C. Monoclonal antibodies specific for adenovirus early region 1A proteins: Extensive heterogeneity in early region 1A products. J. Virol. 55:533–546; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horvath J.; Palkonyay L.; Weber J. Group C adenovirus DNA sequences in human lymphoid cells. J. Virol. 59:189–192; 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Janknecht R.; Hunter T. A growing coactivator network. Nature 383:22–23; 1996. [DOI] [PubMed] [Google Scholar]
- 18. Kalvakolau D. V. R.; Bandyopadhyay S. K.; Harter M. L.; Sen G. C. Inhibition of interferon-inducible gene expression by adenovirus ElA proteins: Block in transcriptional complex formation. Proc. Natl. Acad. Sci. USA 88:7459–7463; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Keicho N.; Elliott W. M.; Hogg J. C.; Hayashi S. Adenovirus ElA gene dysregulates ICAM-1 expression in pulmonary epithelial cells. Am. J. Respir. Cell Mol. Biol. 16:23–30; 1997. [DOI] [PubMed] [Google Scholar]
- 20. Keicho N.; Elliott W. M.; Hogg J. C.; Hayashi S. Adenovirus ElA upregulates interleukin-8 expression induced by endotoxin in pulmonary epithelial cells. Am. J. Physiol. 272(Lung Cell. Mol. Physiol. 16): L1046–L1052; 1997. [DOI] [PubMed] [Google Scholar]
- 21. Keicho N.; Higashimoto Y.; Bondy G. P.; Elliott W. M.; Hogg J. C.; Hayashi S. Endotoxin-specific NF-κB activation in pulmonary epithelial cells harbouring adenovirus E1A. Am. J. Physiol. 277(Lung Cell. Mol. Physiol. 21):L523–L532; 1999. [DOI] [PubMed] [Google Scholar]
- 22. Lieber M.; Smith B.; Szakal A.; Nelson-Rees W.; Todaro G. A continuous tumor cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int. J. Cancer 17:62–70; 1976. [DOI] [PubMed] [Google Scholar]
- 23. Liu F.; Green M. R. Promoter targeting by adenovirus ElA through interaction with different cellular DNA-binding domains. Nature 368:520–525; 1994. [DOI] [PubMed] [Google Scholar]
- 24. Look D. C.; Pelletier M. R.; Taguchi M.; Holtzman M. Adenoviral mechanisms that inhibit intercellular adhesion molecule-1 gene expression in airway epithelial cells. Am. J. Respir. Crit. Care. Med. 155:A460; 1997. [Google Scholar]
- 25. Look D. C.; Rapp S. R.; Keller B. T.; Holtzman M. J. Selective induction of intercellular adhesion molecule-1 by interferon-γ in human airway epithelial cells. Am. J. Physiol. 263:L79–L87; 1992. [DOI] [PubMed] [Google Scholar]
- 26. Matsuse T.; Hayashi S.; Kuwano K.; Keunecke H.; Jefferies W. A.; Hogg J. C. Latent adenoviral infection in the pathogenesis of chronic airways obstruction. Am. Rev. Respir. Dis. 146:177–184; 1992. [DOI] [PubMed] [Google Scholar]
- 27. Masui T.; Wakefiled L. M.; Lechner J. F.; LaVeck M. A.; Sporn M. B.; Harris C.C. Type beta transforming growth factor is the primary differentiation inducing serum factor for normal human bronchial epithelial cells. Proc. Natl. Acad. Sci. USA 83:2438–2442; 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ohh M.; Smith C. A.; Carpenito C.; Takei F. Regulation of intercellular adhesion molecule-1 gene expression involves multiple mRNA stabilization mechanisms: Effects of interferon-γ and phorbol myristate acetate. Blood 84:2632–2639; 1994. [PubMed] [Google Scholar]
- 29. Parker S. F.; Felzien L. K.; Perkins N. D.; Imperiale M. J.; Nabel G. J. Distinct domains of adenovirus E1A. interact with specific cellular factors to differentially modulate human immunodeficiency virus transcription. J. Virol. 71:2004–2012; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pilewski J. M.; Sott D. J.; Wilson J. M.; Albelda S. M. ICAM-1 expression on bronchial epithelium after recombinant adenovirus infection. Am. J. Respir. Cell. Mol. Biol. 12:142–148; 1995. [DOI] [PubMed] [Google Scholar]
- 31. Springer T. A. Adhesion receptors of the immune system. Nature 346:425–434; 1990. [DOI] [PubMed] [Google Scholar]
- 32. Stratowa C.; Audette M. Transcriptional regulation of the human intercellular adhesion molecule-1 gene: A short overview. Immunobiology 193:293–304; 1995. [DOI] [PubMed] [Google Scholar]
- 33. Sussman D. J.; Milman G. Short-term, high-efficiency expression of transfected DNA. Mol. Cell. Biol. 4:1641–1643; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takeda T.; Nakajima K.; Kojima H.; Hirano T. ElA repression of IL-6-induced gene activation by blocking the assemby of IL-6 response element binding complexes. J. Immunol. 153:4573–4582; 1994. [PubMed] [Google Scholar]
- 35. Velcich A.; Ziff E. Adenovirus Ela proteins repress transcription from the SV40 early promoter. Cell 40:705–716; 1985. [DOI] [PubMed] [Google Scholar]
- 36. Voraberger G.; Schäfer R.; Stratowa C. Cloning of the human gene for intercellular adhesion molecule 1 and analysis of its 5′-regulatory region. J. Immunol. 147:2777–2786; 1991. [PubMed] [Google Scholar]
- 37. Wegner C. D.; Gundel R. H.; Reilly P.; Haynes N.; Letts L. G.; Rothlein R. Intercellular adhesion molecule-1 (ICAM-1) in the pathogenesis of asthma. Science 247:456–459; 1990. [DOI] [PubMed] [Google Scholar]