

Abstract
The RET proto-oncogene encodes a receptor tyrosine kinase activated by the binding of factors from the glial cell line-derived neurotrophic factor (GDNF) family to receptor-α components such as GDNF family receptor α-1 (GFRα-1). Mutations within the sequence of the RET proto-oncogene are associated with multiple endocrine neoplasia type 2 (MEN 2), an inherited tumor syndrome characterized by the development of medullary thyroid carcinoma (MTC) and other neuroendocrine tumors. Despite Northern analysis showing that RET is expressed in the majority of MTCs, the factors regulating this expression are poorly understood. To address this issue we examined RET expression in response to glucocorticoids in the TT cell line, derived from a metastatic MTC. The synthetic glucocorticoid dexamethasone was found to reduce RET expression at both mRNA and protein levels. This effect was dose responsive and maximal at 24 h. The reduction in RET mRNA was shown to be specific to glucocorticoids and was also seen in a primary MTC culture. Nuclear run-on studies revealed the reduction in steady-state RNA to be due to a decrease in RET mRNA transcription and the effect was shown to be independent of new protein synthesis or RNA stability. Dexamethasone was also found to exert an inhibitory effect upon cell growth, suggesting a potential use for glucocorticoids in the treatment of medullary carcinoma and MEN 2.
Keywords: RET proto-oncogene, Multiple endocrine neoplasia type 2, Gene expression, Alternative splicing, Dexamethasone
THE RET proto-oncogene encodes a membrane-bound receptor tyrosine kinase (7). Recent work has shown that the RET tyrosine kinase is activated by factors from the glial cell line-derived neurotrophic factor (GDNF) family, consisting of related neurotrophic factors with homology to transforming growth factor-β (27,31). Factors binding to RET from this family include GDNF, neurturin, persephin, and artemin (3,4,21,46). Physiological activation of RET only occurs in the presence of additional extracellular proteins from the GFRα family such as GFRα-1 (7). Expression of RET and its activation by GDNF family members are thought to be especially important in embryogenesis. Embryological studies and RET and GDNF knockout mouse models show a role for RET in renal organogenesis and in the development of central and peripheral nervous systems (33,35,41,43). More specifically, RET is believed to be active in proliferation, differentiation, and migration of cells from the neural crest (19,38). Abnormal expression and activation of RET can also occur within neural crest-derived tissue as a result of mutations in the RET coding sequence. These abnormal events are associated with the inherited disorder multiple endocrine neoplasia type 2 (MEN 2).
MEN 2 is divided into three subgroups, all associated with mutations in the RET proto-oncogene. Mutations altering a cysteine-rich extracellular region within the RET protein, and less commonly in other areas, are linked to familial medullary thyroid carcinoma (FMTC) (20). Mutations within the cysteine-rich region are also associated with MEN 2A, predisposing affected individuals to MTC, pheochromocytoma, and parathyroid hyperplasia. These mutations generally produce a gain of function effect through dimerization and consequent activation of the RET tyrosine kinase in a manner independent of its ligands (1,42). In contrast, a single mutation affecting the methionine at codon 918 within the intracellular tyrosine kinase domain is linked to MEN 2B (20). This is a more aggressive variant of MEN 2 and is associated with a Marfanoid habitus, mucosal neuroma formation, and ganglioneuromatosis in addition to pheochromocytoma and earlier onset MTC. The codon 918 mutation also occurs as a somatic event in up to 80% of sporadic MTCs and has been found to alter the substrate specificity of the RET tyrosine kinase (42,44).
While the mutations described above provide a clear mechanism for tumorigenesis in MEN 2, many events in tumor formation remain unexplained. Importantly, the factors controlling RET expression in both normal and malignant cells remain poorly understood. A single GC-rich promoter region has been identified near the RET exon 1 transcription start site containing putative binding sequences for AP-2, SP-1, epidermal growth factor receptor-specific transcription factor, and GC factor, the latter generally acting to suppress transcription (25). No mutations within this sequence or elsewhere have been published to explain the variation in RET expression seen in different tumors and cell lines. Similarly, current data do not explain how RET mRNA splicing and polyadenylation are regulated. Two protein isoforms can be detected on Western blot arising from this alternative splicing and appear to vary in their transforming ability, especially in the presence of RET mutations (2).
In the TT cell line, derived from a metastatic MTC (30) with a codon 634 mutation characteristic of MEN 2A (8), expression of the RET proto-oncogene can be silenced by activation of the signal transduction factor Raf-I (9). This decrease in RET mRNA levels occurs in association with cell differentiation and a concomitant decrease in tumor cell growth. A similar decrease in growth has been seen when this cell line was treated with the synthetic glucocorticoid dexamethasone (14). We examined the hypothesis that glucocorticoids could similarly inhibit expression of the RET proto-oncogene in MTC cells and here present data to confirm this hypothesis and to show an association between RET expression and cell growth.
MATERIALS AND METHODS
Materials
All cell culture media and antibiotics were obtained from Gibco BRL (Mulgrave, Australia) and fetal calf serum (FCS) from Trace Biosciences (Castle Hill, Australia). Dexamethasone, hydrocortisone, and β2 estradiol were from Sigma Chemical Company (St. Louis, MO), ORG 2058 from Amersham Life Science (Little Chalfont, UK), and dihydrotestosterone from E. Merck (Darmstadt, Germany). Cycloheximide, actinomycin D, and all other reagents, unless otherwise stated, were from Sigma and were of molecular biology grade.
RNA was analyzed with the aid of TRI Reagent (Molecular Research Center, Cincinnati, OH), Genescreen membrane (Dupont NEN Research Products, Boston, MA), and Elutip-D columns (Schleicher and Schuell, Dassel, Germany). Plasmids used in riboprobe synthesis and run-on analysis included pCR-Script (Stratagene, La Jolla, CA) and pGEM-4Z (Promega Corporation, Madison, WI); T3 and T7 polymerases and reagents for riboprobe synthesis were also from Promega. Hybridizations were performed using salmon sperm DNA from Calbiochem (La Jolla, CA) and yeast tRNA from Boehringer Mannheim GmbH (Mannheim, Germany), [α-32P]UTP (800 Ci/mmol, 1480 MBq/ml) was from Dupont NEN Research Products and [methyl-3H]thymidine (60–90 Ci/mmol) from ICN Biochemicals (Costa Mesa, CA).
Cell Culture
TT cells were obtained from the American Type Culture Collection (Rockville, MD) and were grown in RPMI-1640 containing 2 mM glutamine and supplemented with 16% FCS. For steroid specificity studies, phenol red-free RPMI was used because of the estrogen-like activity of phenol red. Penicillin (50 IU/ml) and streptomycin (50 μg/ml) were used in all experiments and cells were routinely tested for Mycoplasma using the Gen-Probe hybridization detection system (Gen-Probe, San Diego, CA). All expression studies were performed at equivalent passage. Before any treatment, cells were plated freshly at 105 cells/cm2 and allowed to settle for 3 days. Cells were then given fresh medium containing either ethanol vehicle at a concentration of 0.1% or dexamethasone in a concentration range of 10−10 to 10−6 M. Following treatment, cells were trypsinized and pelleted for storage at –70°C.
For primary culture, MTC samples were obtained at surgery from a 27-year-old woman with a sporadic tumor. The samples treated were taken from a metastatic deposit within a cervical lymph node. The tissue was finely minced before being placed as ex-plants in RPMI-1640 supplemented with penicillin, streptomycin, and 10% FCS. A number of dispersed, single cells were included from the mincing process. Dexamethasone or vehicle was added in fresh medium after 24 h. Sampling and culture of this tumor tissue was approved by the Royal North Shore Hospital Medical Research Ethics Committee.
To act as a negative control for RET expression, primary fibroblasts were grown from human skin samples within the laboratory of Dr. R. Mason (Sydney University, Australia) and maintained by us for RNA extraction in DMEM supplemented with 10% FCS.
RNA Isolation and Northern Blot Analysis
Total cellular RNA was isolated using TRI Reagent (11). RNA (5 μg) from each treated flask was resolved on a 1% agarose gel containing 4.3 M formaldehyde and transferred to Genescreen membrane by Northern blotting. Membranes were prehybridized and hybridized at 65°C in 50% deionized formamide, 0.8 M NaCl, 1 mM EDTA, 50 mM phosphate buffer, 2% SDS, and 2.5× Denhardt’s solution. Salmon sperm DNA (0.1 mg/ml) and tRNA (0.2 mg/ml) were also added. The RET riboprobe used was constructed by amplifying a 1.5-kb RET fragment encoding exons 11 to 20 from cDNA sequence. This fragment was cloned into pCR-Script and digested for riboprobe synthesis using an Nhe I site introduced in the PCR process. 36B4 was derived from a 220-bp Pst I fragment cloned elsewhere into pGEM-4Z (16). Sequence was transcribed using T3 and T7 polymerases, respectively, in a mixture containing 100 μCi [α-32P]UTP and then purified using an Elutip-D column. Following hybridization, filters were washed twice at 65°C in 0.1 × SSC and 0.1% SDS and analyzed using a PhosphorImager (Molecular Dynamics, Sunnyvale, CA). Bands were quantitated with the aid of ImageQuant software (Molecular Dynamics) and adjusted for loading using 36B4 expression levels.
Western Blot Analysis
TT cell pellets were lysed using TDS lysis buffer [1% Triton X-100 (v/v), 0.5% sodium deoxycholate (w/v), and 0.1% SDS (w/v) in 1× PBS]. Lysates were subjected to SDS-PAGE using 7.5% acrylamide gels (Bio-Rad Laboratories, Hercules, CA) and were transferred to nitrocellulose membranes by Western blotting. Membranes were then blocked in TBS (47 mM Tris, 39 mM glycine, and 20% methanol) containing 1% bovine serum albumin (w/v), 0.02% sodium azide (w/v), and 0.05% Nonidet P-40 (v/v) overnight. Blocked samples were exposed first to rabbit anti-RET antiserum (Santa Cruz Biotechnology, 1:1000) and second to antiserum raised against rabbit and conjugated to horseradish peroxidase (Bio-Rad Laboratories, 1:10000). Bands were detected using SuperSignal chemiluminescent substrate (Pierce, Rockford, IL).
Cycloheximide and Actinomycin D Treatment
Cycloheximide was dissolved in water and added to medium to give a final concentration of 20 μg/ml, a level sufficient to block protein synthesis within 5 min (6). The agent was used to pretreat cells by adding 10 min before dexamethasone.
Actinomycin D was dissolved in DMSO to give a final concentration of 5 μg/ml, a concentration used to examine RET stability in neuroblastoma cell lines (6,45). Actinomycin D and dexamethasone were added concurrently.
Nuclear Run-on Analysis
TT cells subjected to run-on analysis were first treated with l0−6 M dexamethasone or equivalent vehicle for 24 h. Nuclei were isolated by lysis in Nonidet P-40 (23) and incubated with 400 μCi [α-32P]UTP as previously described (22), except that the run-on reaction was performed at 30°C for 45 min (12). Run-on transcripts were purified by extraction in TRI Reagent as for total RNA and equivalent counts used in the subsequent hybridization.
To act as DNA probes, the RET and 36B4 templates used in riboprobe preparation were linearized and immobilized on Genescreen membrane. α-Tubulin was added as an additional control using a 75-bp fragment cloned elsewhere into pBR322 (15). The plasmid pGEM-4Z was also used to control for nonspecific binding. RET (5 μg) and 2.5 μg control DNA were applied via a slot-blot apparatus and cross-linked by baking at 80°C.
Slot-blot membranes were prehybridized at 42°C in 50% deionized formamide, 5× SSPE, 5× Denhardt’s solution, 0.8% SDS, and 100 μg/ml tRNA (36). Hybridization was performed under the same conditions for 36–72 h. Slot-blots were washed three times for 15 min at room temperature in 2× SSC, 0.1% SDS, and 1× Denhardt’s solution without BSA (36) before exposure and PhosphorImager analysis.
Growth Assays
Two assays were used for the analysis of growth. First, the MTS assay was performed using the Cell-Titer 96 AQueous Non-Radioactive Cell Proliferation Assay kit (Promega Corporation). Cells were plated and allowed to settle and grow for 3 days. Each well was then treated with dexamethasone or with an equivalent volume of ethanol vehicle for the times shown. At the commencement of each treatment, the tetrazolium compound MTS was mixed with phenazine methosulphate and added to each well. Plates were incubated for 4 h and their absorbance measured at 490 nm. The background at each time point was also read and subtracted from the readings shown.
[3H]Thymidine uptake assays were performed on cells plated in 24-well plates and allowed to settle and grow for 3 days. Wells were refed with dexamethasone or with an equivalent volume of vehicle for the times shown. At the conclusion of each treatment, 2 μCi/ml [methyl-3H]thymidine was added to each well and cells incubated in this mixture for 4 h. Tritiated material was precipitated with 10% trichloroacetic acid and solubilized in 0.5% SDS. Again, background readings were subtracted from each result before analysis.
Statistical Analysis
RET expression and cell growth were assessed using the Student’s two sample t-test. Equal variance was assumed for each assay and values are presented from both tails. Probability values (p) come from this analysis and were considered significant when p < 0.05. Differences in the RET transcripts were further assessed by two-way analysis of variance using the software package SPSS for Windows, with data tested for significance using multiple group comparisons and with Scheffe adjustment for samples in a normal distribution.
RESULTS
Dexamethasone Suppresses RET Expression Levels
TT cells express the RET proto-oncogene in a characteristic pattern (7), with specific mRNA species of sizes 7.0, 6.0, 4.5, and 3.9 kb seen on Northern blot (Fig. 1A). An additional band noted above the 4.5-kb RET transcript was found to be nonspecific and was detected by our RET riboprobe in all cell types, regardless of their RET expression status (data not shown). Adding dexamethasone to the TT cell line resulted in a decrease in abundance of 7.0-, 6.0-, 4.5-, and 3.9-kb species when compared to vehicle-treated control lanes. This decrease was seen despite loading similar amounts of RNA, as shown by hybridization to 36B4, a probe derived from the gene for human acidic ribosomal phosphoprotein P0 and used extensively as a control for loading (10,28,32).
FIG. 1.
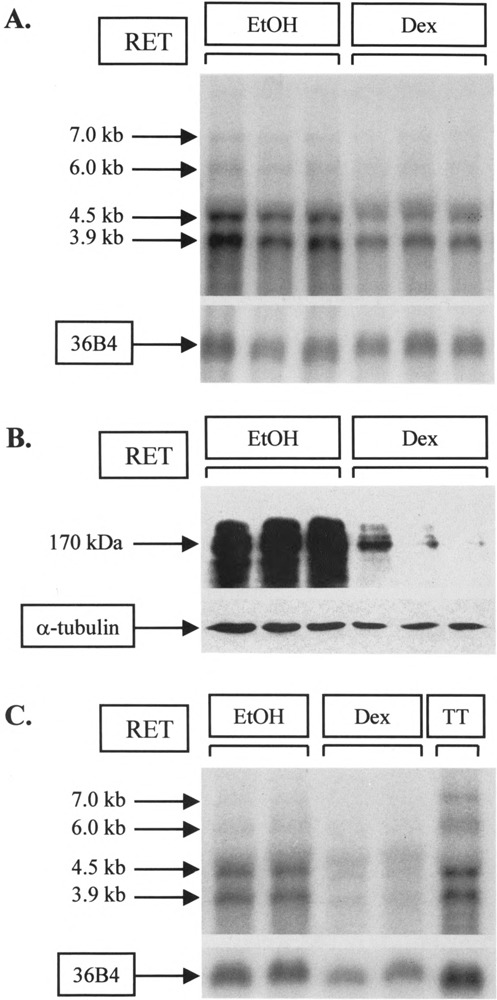
Effect of dexamethasone on RET expression. (A) Assessing RNA expression using the TT cell line. Flasks were treated in triplicate with 10−6 M dexamethasone (Dex) or an equivalent volume of ethanol (EtOH) for 24 h. Total RNA (5 μg) was subjected to electrophoresis and Northern blotting. The resulting filter was hybridized to RET and 36B4 riboprobes concurrently. (B) Assessing protein expression using the TT cell line. Cells were treated in triplicate with 10−6 M dexamethasone or ethanol for 24 h as in (A) and 50 μg protein used for SDS-PAGE and Western blotting. Membranes were hybridized to RET and α-tubulin antibodies sequentially. (C) Assessing RNA expression using primary medullary carcinoma cells. Explant cultures from a 27-year-old woman with metastatic MTC were treated 1 day after establishment with vehicle (EtOH) or 10−6 M dexamethasone (Dex) for 24 h. The duplicate lanes shown after Northern blotting and hybridization were derived from single treatment samples; less was loaded in dexamethasone-treated lanes. A single lane of TT RNA was included for comparison with (A).
To determine whether both RET RNA and protein levels are affected by dexamethasone, this experiment was repeated using cells that were lysed for total protein and exposed to a RET polyclonal antibody. Treatment with 10−6 M dexamethasone for 24 h resulted in a clear decrease in RET protein levels (Fig. 1B) as well as in RET mRNA (Fig. 1A) when compared to vehicle-treated control lanes. Hybridization to α-tubulin antiserum as a control for loading shows that this decrease was not caused by a variation in the total protein present.
We finally explored the applicability of this finding beyond the TT cell line by establishing primary cultures from a 27-year-old female with a sporadic MTC and treating these cultures with dexamethasone (Fig. 1C). The tumor cells expressed all four transcripts, although 7.0- and 6.0-kb species were present at relatively low level compared to TT. Dexamethasone had little impact upon 7.0-kb transcripts but clearly reduced steady-state levels of 6.0-, 4.5-, and 3.9-kb species, as with the TT cell line. Taken together, these results are consistent with dexamethasone exerting an effect on both RET mRNA and protein in medullary thyroid carcinoma cells in vitro.
The Effect of Dexamethasone Is Dose and Time Dependent
The effect of dexamethasone on RET mRNA expression was further characterized in TT cells by varying the amount given over 24 h (Fig. 2). Increasing the concentration of dexamethasone from 10−10 to 10−6 M yielded an initial increase in all mRNA species relative to control, followed by a dose-dependent decrease (Fig. 2A). Of these two changes, the decrease seen at higher doses, was more obvious and was found to be significant for all transcripts (Fig. 2B). This decrease was also found to be transcript specific, with levels of 7.0- and 6.0-kb mRNAs declining to a greater degree than 4.5- and 3.9-kb species (Fig. 2B). Analysis of variance showed that these transcript-specific changes were significant for this experiment (data not shown).
FIG. 2.
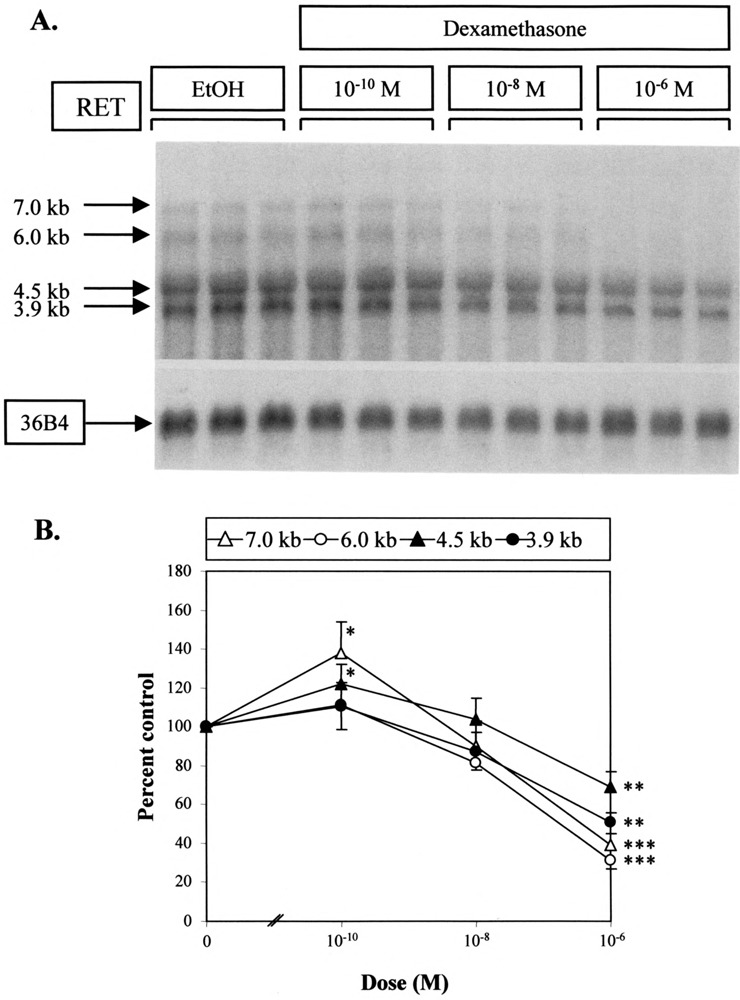
Dose-dependent effect of dexamethasone on RET mRNA. (A) Northern analysis. TT flasks were treated in triplicate with vehicle (EtOH) or with dexamethasone at the doses shown for 24 h. Lanes were hybridized to RET and 36B4 riboprobes sequentially. (B) Quantification of the results obtained by Northern analysis. RET mRNA species were quantitated and adjusted using hybridization to 36B4 sequence as a control for loading. The mean and SD of each triplicate were then expressed as a percentage of vehicle-treated control samples. Significant differences when compared to control results are shown: *p < 0.05, **p < 0.01, ***p < 0.005.
To further investigate the changes seen in RET expression, TT cells were exposed to 10−6 M dexamethasone for up to 72 h (Fig. 3). All RET mRNA levels declined significantly in response to treatment, with the greatest reduction at 24 h followed by at least partial recovery for all species. If dexamethasone was removed after 24-h treatment and the effect observed, a rapid increase in RET mRNA abundance resulted to greater than control levels over a further 24 h (data not shown). These results would indicate that the effect of dexamethasone on the TT cell line is both dose and time responsive.
FIG. 3.
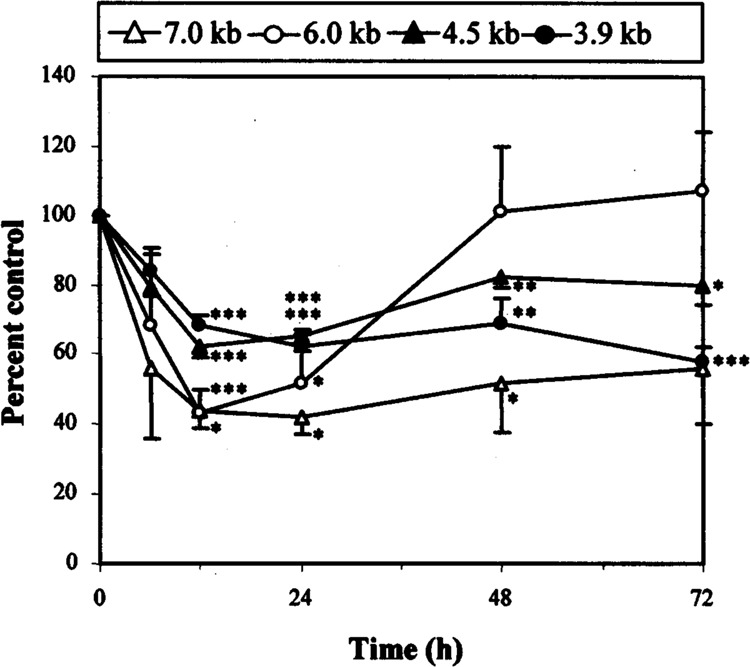
Time-dependent effect of dexamethasone on RET mRNA. Quantification of results obtained by Northern blot analysis (not shown). TT cells were treated in triplicate with 10−6 M dexamethasone for 6, 12, 24, 48, and 72 h. RET mRNAs were again quantitated, adjusted against 36B4 species for loading, and expressed as a percentage of control lanes. Cells at time zero were used as a control, because RET expression had not been found to alter significantly over 72 h in previous experiments (data not shown). Significant differences when compared to control results are shown: *p < 0.05, **p < 0.01, ***p < 0.005.
The Effect of Dexamethasone Is Steroid Specific
Having characterized the effect of dexamethasone, a panel of steroids was used to treat TT cells and determine the specificity of the effect seen on RET expression levels (Fig. 4A). Dexamethasone and hydrocortisone were found to reduce RET mRNA levels while β2 estradiol, dihydrotestosterone, and the bioactive progesterone analogue, ORG 2058, had no net effect (Fig. 4B). Considering the similar effect of hydrocortisone and dexamethasone, we would suggest that the inhibition of RET expression seen occurs via the glucocorticoid receptor. This receptor was found to be expressed in the TT cell line by hybridizing TT RNA to a glucocorticoid receptor riboprobe, supporting this hypothesis (data not shown).
FIG. 4.
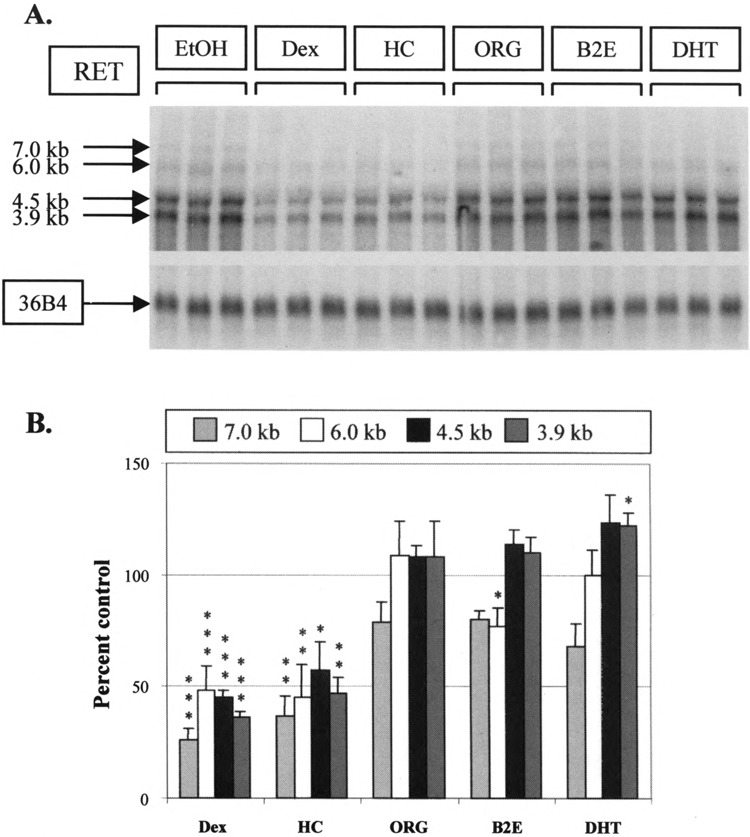
Steroid specificity and RET mRNA expression. (A) Northern analysis. TT cells were treated in triplicate with the steroids shown, all at 10−6 M, for 24 h. ORG 2058 was used as an active analogue of progesterone. EtOH, ethanol vehicle; Dex, dexamethasone; HC, hydrocortisone; ORG, ORG 2058; β2E, β2 estradiol; DHT, dihydrotestosterone. (B) Quantification of results obtained from Northern analysis. Band intensities were again corrected for loading with 36B4 and expressed as a percentage of the mean of vehicle-treated control lanes. Significant differences when compared to control results are shown: *p < 0.05, **p < 0.01, ***p < 0.005.
Dexamethasone Suppresses RET Expression Through Directly Altering RNA Transcription
The 12-h delay between the addition of dexamethasone and an effect on RET expression suggested that this effect could be mediated via a separate, newly synthesized protein. In order to investigate this possibility, TT cells were treated with cycloheximide to eliminate all new protein synthesis (Fig. 5A). Cycloheximide alone (C) produced an increase in RET mRNA levels compared to control lanes (Et). Adding dexamethasone in conjunction with cycloheximide (D/C) resulted in a decrease in RET mRNA levels relative to cycloheximide alone (C) at 12 h, a time at which suppression of these levels by dexamethasone could be seen (Fig. 5B). The fact that this decrease could still be seen even after adding cycloheximide would indicate that the effect of dexamethasone is not dependent on new protein synthesis.
FIG. 5.
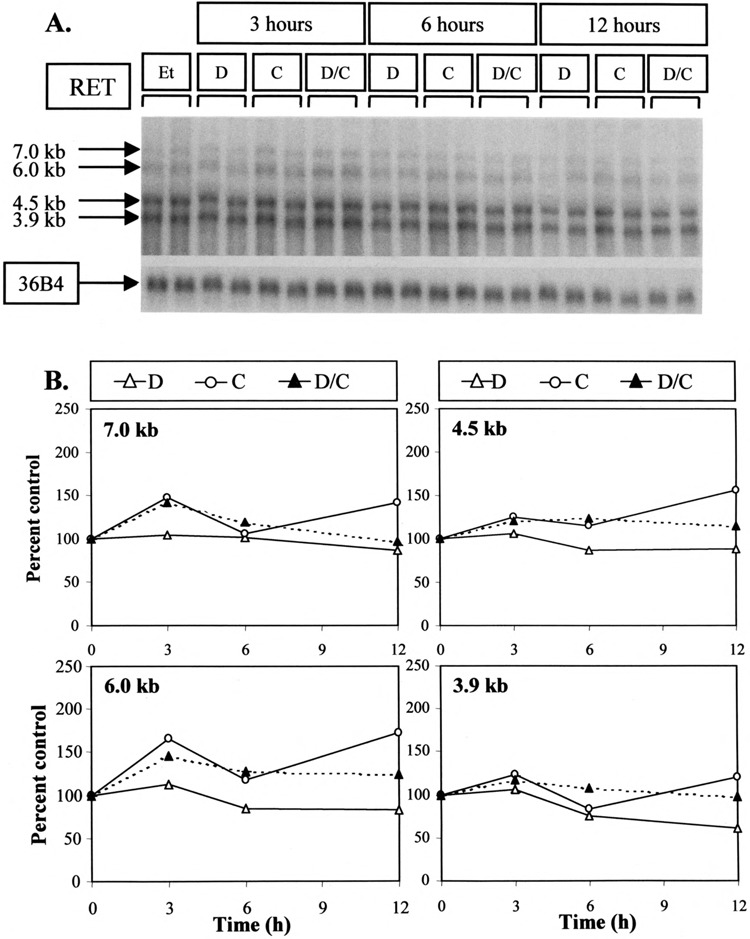
Effect of cycloheximide on RET mRNA levels and their dexamethasone-induced inhibition. (A) Northern analysis. TT cells were treated for the time shown with 10-8 M dexamethasone (D), 20 μg/ml cycloheximide (C), or both substances (D/C) as described in Materials and Methods. Representative results from duplicate samples are shown. (B) Quantification of results obtained from Northern analysis. Each of the 7.0-, 6.0-, 4.5-, and 3.9-kb RET species are assessed in separate panels here. The effects of dexamethasone alone (D), cycloheximide (C), or both substances (D/C) are expressed as a percentage of the mean vehicle-treated control lanes (Et) after correction for loading with 36B4.
Another possible mechanism for a decline in RET mRNA levels could involve a change in RNA stability. Such a possibility was tested by treating TT cells with actinomycin D to eliminate all new RNA synthesis (Fig. 6A). Treatment with actinomycin D (A) for 24 h resulted in a decline in 7.0- and 6.0-kb species to approximately 50% and in 4.5- and 3.9-kb transcripts to 70% of control (Et) levels. Adding dexamethasone as well as actinomycin D (A/D) did not provoke a more rapid decrease in any of these four transcripts relative to actinomycin D alone (Fig. 6B). This was true even at 24 h, the time at which dexamethasone was found to exert its greatest effect on RET expression (Fig. 3). The fact that adding dexamethasone did not induce a more rapid decline in RNA expression than actinomycin D alone would imply that its effect on RET mRNAs was not caused by a change in RNA stability.
FIG. 6.
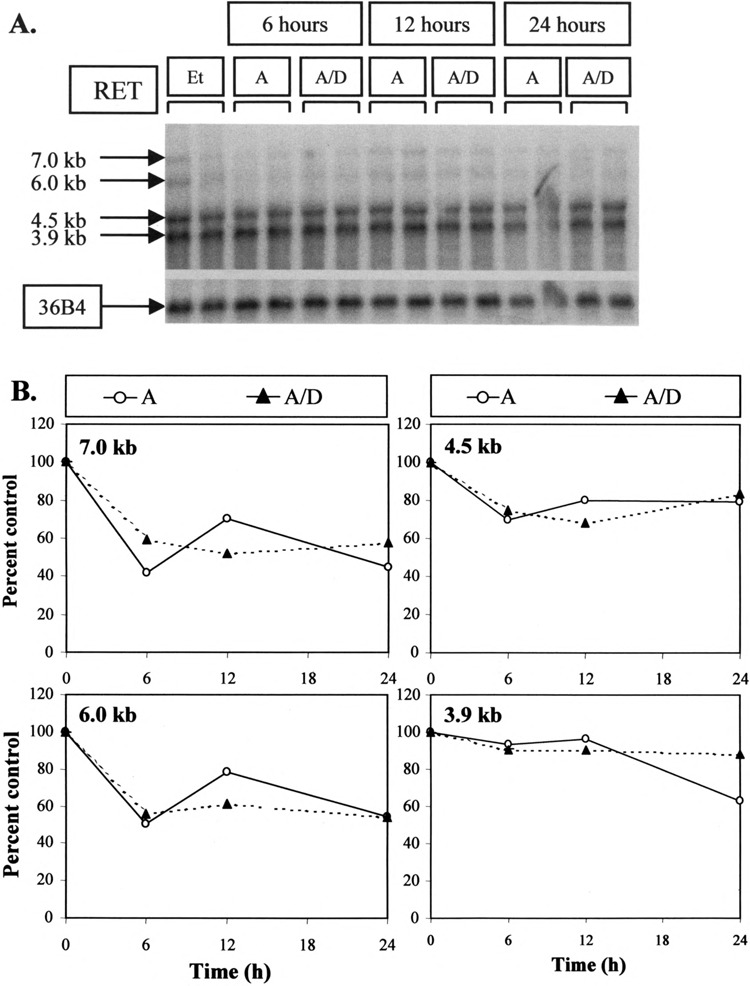
Effect of actinomycin D on RET mRNA levels with and without dexamethasone. (A) Northern analysis. TT cells were treated with 5 μg/ml actinomycin D alone (A) or with the addition of 10−8 M dexamethasone (A/D) for the times shown. Again, representative results from duplicate samples are shown. (B) Quantification of results obtained from Northern analysis. Each of the 7.0-, 6.0-, 4.5-, and 3.9-kb RET species are again assessed in separate panels. The effects of actinomycin D alone (A) or in combination with dexamethasone (A/D) are expressed as a percentage of the mean vehicle-treated control lanes (Et) after correction for loading with 36B4.
To finally investigate the effect of dexamethasone on overall RET transcription, nuclear run-on transcription studies were performed to compare cells treated with dexamethasone to others treated with vehicle alone. Nuclear RNA transcripts from treated cells were extended and labeled in run-on assays before their hybridization to slot-blots containing RET sequence encoding transmembrane and tyrosine kinase domains (Fig. 7). Slot-blots also included 36B4, α-tubulin, and plasmid pGEM-4Z sequence as controls. Treatment with dexamethasone resulted in a specific decrease in RET hybridization, indicating that its inhibitory effect on RET expression was due to a direct effect on the transcription rate.
FIG. 7.
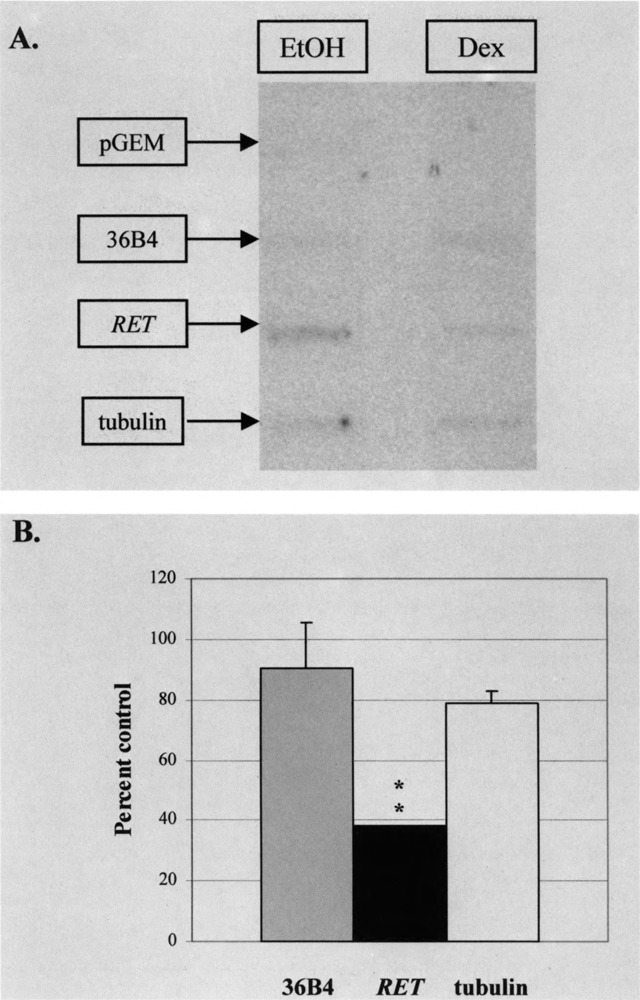
Effect of dexamethasone on RET transcription. TT cells were treated with 10−6 M dexamethasone (Dex) or equivalent vehicle (EtOH) for 24 h and cells harvested for nuclear extraction. Relative nuclear transcription rates were determined by run-on assay, allowing the elongation of nascent RNA transcripts in the presence of [α-32P]UTP as described in Materials and Methods. Transcripts were hybridized to identical slot-blots containing RET transmembrane and tyrosine kinase sequence. As controls for the action of dexamethasone on other transcripts, 36B4 and a-tubulin constructs were included on these slot-blots. To further control for hybridization specificity, the plasmid pGEM-4Z was also added. Results were reproducible on two occasions. Significant differences when compared to control results are shown: **p< 0.01.
Dexamethasone Alters Cell Proliferation in Association With its Effect on RET Expression
The functional significance of dexamethasone was last assessed by measuring cell growth in two different assay systems. Treatment with 10−6 M dexamethasone for up to 6 days resulted in a decrease in cell number when compared using the MTS assay to vehicle-treated control samples (Fig. 8A). This decrease became apparent after only 1 day but did not become significant until the second day of treatment. Similarly, treatment with dexamethasone for 24 h resulted in a decrease in cell growth when assessed by [3H]thymidine uptake assay (Fig. 8B). This decrease became significant after treating cells with more than 10−8 M dexamethasone. The results of both assays would indicate that the decrease in RET expression seen with dexamethasone is associated with a similar decrease in cell proliferation.
FIG. 8.
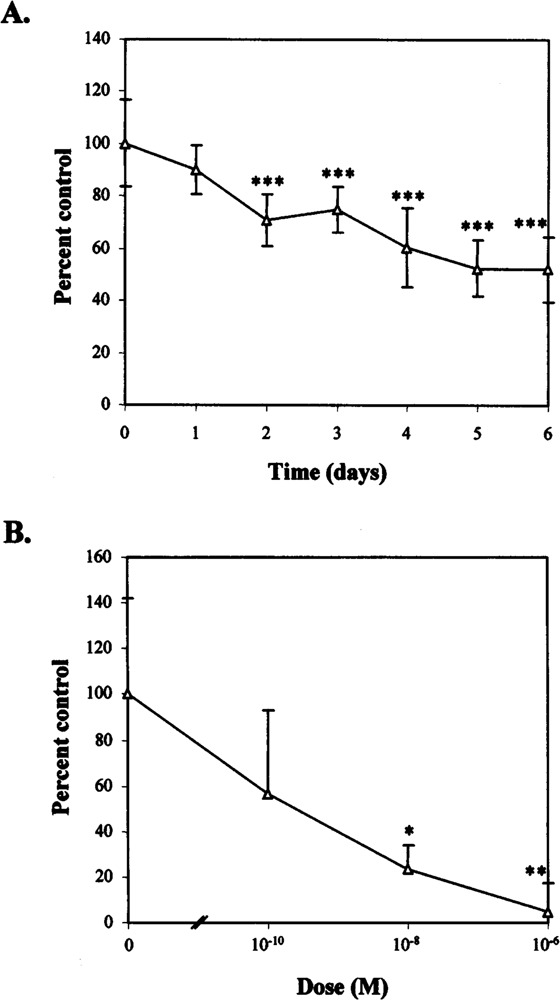
Growth in response to dexamethasone. (A) Using the MTS assay. TT cells were treated with 10−6 M dexamethasone or with an equivalent amount of ethanol for up to 6 days. MTS assays were performed daily and background readings subtracted from each day’s result. Treatment results are presented here as a percentage of vehicle-treated control lanes ± 1 SD (n = 8). Significant differences when compared to control samples are shown: *p < 0.05, **p < 0.01, ***p < 0.005. (B) Using the [3H]thymidine uptake assay. TT cells were treated with either 10−6 M dexamethasone or vehicle for 24 h. [3H]Thymidine was then added for the final 4 h of this incubation and the uptake assay performed. Treatment results are presented, minus background readings, as a percentage of vehicle-treated control lanes ± 1 SD (n = 3). Significant differences are shown as in (A).
DISCUSSION
Expression of the RET proto-oncogene and its activation by the mutations of MEN 2 are important events for tumorigenesis in certain neuroendocrine tissues. Silencing of RET expression has been found to be associated with endocrine differentiation and a decrease in cell growth (9,40), a finding of potential interest for tumor treatment. Because dexamethasone has previously been found to have a similar effect on cell growth, an investigation of its effect on RET expression was warranted.
Treatment of the TT cell line, an accepted in vitro model for medullary thyroid carcinoma, with dexamethasone resulted in a clear decrease in both RET mRNA and protein levels (Fig. 1). The decrease in RNA expression seen with dexamethasone was evident with concentrations at or above 10−8 M (Fig. 2) and appeared at higher concentrations after only 12 h of treatment (Fig. 3). This effect was found to be specific to glucocorticoids (Fig. 4). While neither β2 estradiol nor dihydrotestosterone reduced RET mRNA levels, both hydrocortisone and dexamethasone produced a significant decrease in RET expression for all transcripts. Interestingly, the progesterone analogue ORG 2058 did not produce a similar decrease. This lack of activity is surprising, because progesterone is known to bind competitively to the glucocorticoid receptor (24), but may be explained by the existence of a separate progesterone receptor in the TT cell line (13). The fact that dexamethasone reduced RET mRNA levels in both TT cells and in primary culture (Fig. 1C) would suggest that this finding is applicable to more than a single cell line.
The various methods used to study the effects of glucocorticoids on RET expression suggested a direct effect on RET transcription. First, cycloheximide was used to eliminate protein synthesis and assess the importance of new protein (Fig. 5). Cycloheximide alone was found to yield a net increase in RET mRNA levels, implying that protein was normally synthesized to suppress RET expression. A similar effect has been noted elsewhere in SK-N-BE cells (6). Adding dexamethasone resulted in a decline in RET mRNA levels relative to cycloheximide alone, suggesting that the steroid exerted its effect directly without new protein mediators. Second, actinomycin D was employed to abolish new RNA synthesis and assess the importance of changes in RNA stability (Fig. 6). Adding actinomycin D showed that existing RET mRNA species degraded slowly, with half-lives of greater than 24 h. Similar half-life observations have previously been made in neuroblastoma cell lines (6,45). It could be seen that 7.0- and 6.0-kb species declined more rapidly with actinomycin D alone, suggesting that these longer species are more susceptible to degradation. Adding dexamethasone, however, did not materially affect the degradation of any of the four RET species when compared to treatment with actinomycin D alone. Finally, transcription run-on assays supported this result by showing that treatment with dexamethasone resulted in a decrease in overall RET mRNA synthesis (Fig. 7).
The mechanism for such a decrease in RET transcription remains unclear after analysis of known regulatory sequence. Binding of a glucocorticoid–receptor complex to specific regulatory elements is likely, but neither a full glucocorticoid regulatory element (GRE) nor the rarer negative GRE (18) have been identified within the RET promoter region [(25), L. Mulligan, personal communication]. Additional regulatory sequence may lie further upstream. Further examination of the published promoter and intronic sequence (25), however, has identified two glucocorticoid receptor half-sites. The first lies 382 bp upstream from the transcription start site, while the second is located 947 bp downstream of exon 19 (data, not shown). Both half-sites are identical to that implicated in repression of calcitonin/calcitonin gene-related peptide expression by glucocorticoids (47). The importance of either glucocorticoid receptor half-site for RET expression is currently unknown. Alternately, it remains possible that glucocorticoids exert their effect through interaction with other transcription factors in the absence of DNA binding. Such a mechanism frequently results in suppression of gene transcription (5) and may involve transcription factors such as AP-1 (26,48) or NF-κB (37). Neither of these factors has yet been implicated in binding to the RET promoter and additional promoter sequence is required to investigate these mechanisms.
The functional significance of glucocorticoids is confirmed by studying TT cell growth. As previously documented by other groups, treatment with dexamethasone resulted in a significant growth inhibition using two assay methods. This inhibition was dose responsive and became more pronounced with time, as seen in both TT and CA77 cell lines (14,29,39). It should be noted that a similar effect on cell growth and RET expression has also been noted after activation of Raf-1 (9) and after treatment with phorbol esters and sodium butyrate [(17,34), A. Capes-Davis, submitted]. Considering these results, we suggest that the decrease seen in RET mRNA levels may be responsible for the changes in cell behavior seen with these agents.
The effects of glucocorticoids on RET expression and cell behavior are likely to have clinical significance for the treatment of MTC. Dexamethasone has been shown here to have significant effects on a cell line that is held to be an in vitro model for medullary carcinoma, with a particular impact on the expression of a proto-oncogene known to cause this tumor. Although the adverse effects of glucocorticoids such as dexamethasone can be significant, these steroids are currently available for clinical use and may be helpful for treatment or palliation of patients with end-stage tumors. In the future, with development of synthetic glucocorticoids varying in their metabolic action, further agents may become available minimizing these adverse reactions. The in vitro work described here thus forms a rational basis for animal studies and may lead to subsequent human trials for the use of glucocorticoids in MTC.
ACKNOWLEDGMENTS
The authors are indebted to Dindy Benn for help with the MTS assay and to Ana Guinea for statistical analysis. Christine Clarke is thanked for the generous gifts of 36B4 and α-tubulin DNA, and Rebecca Mason for primary fibroblast cultures. Helpful discussion and the screening for glucocorticoid regulatory elements by Lois Mulligan are gratefully acknowledged. Financial support was provided by the Sydney University Cancer Research Fund, by National Health and Medical Research Council Medical Scholarships (A.G.C, ST., D.L.L.), and by the Westpac Scholarship (A.G.C.) and Mary Jo Reeve Scholarship (D.L.L.).
REFERENCES
- 1. Asai N.; Iwashita T.; Matsuyama M.; Takahashi M. Mechanism of activation of the ret proto-oncogene by multiple endocrine neoplasia 2A mutations. Mol. Cell. Biol. 15:1613–1619; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Asai N.; Murakami H.; Iwashita T.; Takahashi M. A mutation at tyrosine 1062 in MEN2A-Ret and MEN2B-Ret impairs their transforming activity and association with She adaptor proteins. J. Biol. Chem. 271:17644–17649; 1996. [DOI] [PubMed] [Google Scholar]
- 3. Baloh R. H.; Tansey M. G.; Golden J. P.; Creedon D. J.; Heuckeroth R. O.; Keck C. L.; Zimonjic D. B.; Popescu N. C.; Johnson E. M. Jr.; Milbrandt J. TrnR2, a novel receptor that mediates neurturin and GDNF signaling through Ret. Neuron 18:793–802; 1997. [DOI] [PubMed] [Google Scholar]
- 4. Baloh R. H.; Tansey M. G.; Lampe P. A.; Fahrner T. J.; Enomoto H.; Simburger K. S.; Leitner M. L.; Araki T.; Johnson E. M. Jr.; Milbrandt J. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRalpha3-RET receptor complex. Neuron 21:1291–1302; 1998. [DOI] [PubMed] [Google Scholar]
- 5. Bamberger C. M.; Schulte H. M.; Chrousos G. P. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr. Rev. 17:245–261; 1996. [DOI] [PubMed] [Google Scholar]
- 6. Bunone G.; Borrello M. G.; Picetti R.; Bongarzone I.; Peverali F. A.; de Franciscis V.; Della Valle G.; Pierotti M. A. Induction of RET proto-oncogene expression in neuroblastoma cells precedes neuronal differentiation and is not mediated by protein synthesis. Exp. Cell Res. 217:92–99; 1995. [DOI] [PubMed] [Google Scholar]
- 7. Capes-Davis A.; Robinson B. G. Return of the native: Deducing the normal function of the RET proto-oncogene. Curr. Opin. Endocrinol. Diabet. 6:61–69; 1999. [Google Scholar]
- 8. Carlomagno F.; Salvatore D.; Santoro M.; de Franciscis V.; Quadro L.; Panariello L.; Colantuoni V.; Fusco A. Point mutation of the RET proto-oncogene in the TT human medullary thyroid carcinoma cell line. Biochem. Biophys. Res. Commun. 207:1022–1028; 1995. [DOI] [PubMed] [Google Scholar]
- 9. Carson E. B.; McMahon M.; Baylin S. B.; Nelkin B. D. Ret gene silencing is associated with Raf-1-induced medullary thyroid carcinoma cell differentiation. Cancer Res. 55:2048–2052; 1995. [PubMed] [Google Scholar]
- 10. Chambon P.; Dierich A.; Gaub M. P.; Jakowlev S.; Jongstra J.; Krust A.; LePennec J. P.; Oudet P.; Reudelhuber T. Promoter elements of genes coding for proteins and modulation of transcription by estrogens and progesterone. Recent Prog. Horm. Res. 40:1–42; 1984. [DOI] [PubMed] [Google Scholar]
- 11. Chomczynski P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 15:532–537; 1993. [PubMed] [Google Scholar]
- 12. Clarke C. L.; Graham J.; Roman S. D.; Sutherland R. L. Direct transcriptional regulation of the progesterone receptor by retinoic acid diminishes progestin responsiveness in the breast cancer cell line T-47D. J. Biol. Chem. 266:18969–18975; 1991. [PubMed] [Google Scholar]
- 13. Colomer A.; Martinez-Mas J. V.; Matias-Guiu X.; Llorens A.; Cabezas R.; Prat J.; Garcia-Ameijeiras A. Sex-steroid hormone receptors in human medullary thyroid carcinoma. Mod. Pathol. 9:68–72; 1996. [PubMed] [Google Scholar]
- 14. Cote G. J.; Gagel R. F. Dexamethasone differentially affects the levels of calcitonin and calcitonin gene-related peptide mRNAs expressed in a human medullary thyroid carcinoma cell line. J. Biol. Chem. 261:15524–15528; 1986. [PubMed] [Google Scholar]
- 15. Cowan N. J.; Dobner P. R.; Fuchs E. V.; Cleveland D. W. Expression of human alpha-tubulin genes: Interspecies conservation of 3′ untranslated regions. Mol. Cell. Biol. 3:1738–1745; 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Daly R. J.; Darbre P. D. Cellular and molecular events in loss of estrogen sensitivity in ZR-75-1 and T-47-D human breast cancer cells. Cancer Res. 50:5868–5875; 1990. [PubMed] [Google Scholar]
- 17. de Bustros A.; Baylin S. B.; Berger C. L.; Roos B. A.; Leong S. S.; Nelkin B. D. Phorbol esters increase calcitonin gene transcription and decrease c-myc mRNA levels in cultured human medullary thyroid carcinoma. J. Biol. Chem. 260:98–104; 1985. [PubMed] [Google Scholar]
- 18. Drouin J.; Trifiro M. A.; Plante R. K.; Nemer M.; Eriksson P.; Wrange O. Glucocorticoid receptor binding to a specific DNA sequence is required for hormone-dependent repression of pro-opiomelanocortin gene transcription. Mol. Cell. Biol. 9:5305–5314; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Durbec P. L.; Larsson-Blomberg L.; Schuchardt A.; Costantini F.; Pachnis V. Common origin and developmental dependence on c-ret of subsets of enteric and sympathetic neuroblasts. Development 122:349–358; 1996. [DOI] [PubMed] [Google Scholar]
- 20. Eng C. Seminars in medicine of the Beth Israel Hospital, Boston. The RET proto-oncogene in multiple endocrine neoplasia type 2 and Hirschsprung’s disease. N. Engl. J. Med. 335:943–951; 1996. [DOI] [PubMed] [Google Scholar]
- 21. Enokido Y.; de Sauvage F.; Hongo J. A.; Ninkina N.; Rosenthal A.; Buchman V. L.; Davies A. M. GFRalpha-4 and the tyrosine kinase Ret form a functional receptor complex for persephin. Curr. Biol. 8:1019–1022; 1998. [DOI] [PubMed] [Google Scholar]
- 22. Fei H.; Drake T. A. A rapid nuclear runoff transcription assay. Biotechniques 15:838; 1993. [PubMed] [Google Scholar]
- 23. Greenberg M. E. Identification of newly transcribed RNA: Nuclear runoff transcription in mammalian cells. In: Ausubel F. M.; Brent R.; Kingston R. E.; Moore D. D.; Seidman J. G.; Smith J. A.; Struhl K., eds. Current protocols in molecular biology. New York: John Wiley & Sons; 1989:4.10.1–4.10.8. [Google Scholar]
- 24. Hollenberg S. M.; Weinberger C.; Ong E. S.; Cerelli G.; Oro A.; Lebo R.; Thompson E. B.; Rosenfeld M. G.; Evans R. M. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 318:635–641; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Itoh F.; Ishizaka Y.; Tahira T.; Yamamoto M.; Miya A.; Imai K.; Yachi A.; Takai S.; Sugimura T.; Nagao M. Identification and analysis of the ret proto-oncogene promoter region in neuroblastoma cell lines and medullary thyroid carcinomas from MEN2A patients. Oncogene 7:1201–1206; 1992. [PubMed] [Google Scholar]
- 26. Jonat C.; Rahmsdorf H. J.; Park K.-K.; Cato A. C. B.; Gebel S.; Ponta H.; Herrlich P. Antitumor promotion and antiinflammation: Down-modulation of AP-1 (fos/jun) activity by glucocorticoid hormone. Cell 62:1189–1204; 1990. [DOI] [PubMed] [Google Scholar]
- 27. Kotzbauer P. T.; Lampe P. A.; Heuckeroth R. O.; Golden J. P.; Creedon D. J.; Johnson E. M. Jr.; Milbrandt J. Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature 384:467–470; 1996. [DOI] [PubMed] [Google Scholar]
- 28. Laborda J. 36B4 cDNA used as an estradiol-independent mRNA control is the cDNA for human acidic ribosomal phosphoprotein P0. Nucleic Acids Res. 19:3998; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lamari Y.; Tahri E. H.; Collignon H.; Garel J. M. Steroid hormones and retinoic acid interact in the regulation of calcitonin and calcitonin gene-related peptide secretion and messenger ribonucleic acid levels in CA-77 C cells. Cell. Mol. Biol. 40:541–550; 1994. [PubMed] [Google Scholar]
- 30. Leong S. S.; Horoszewicz J. S.; Shimaoka K.; Friedman M.; Kawinski E.; Song M. J.; Zeigel R.; Chu T. M.; Baylin S.; Mirand E. A. A new cell line for study of human medullary thyroid carcinoma. In: Andreoli M.; Monaco F.; Robbins J., eds. Advances in thyroid neoplasia. Rome: Field Educational Italia; 1981:95–108. [Google Scholar]
- 31. Lin L. F.; Doherty D. H.; Lile J. D.; Bektesh S.; Collins F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260:1130–1132; 1993. [DOI] [PubMed] [Google Scholar]
- 32. Masiakowski P.; Breathnach R.; Bloch J.; Gannon F.; Krust A.; Chambon P. Cloning of cDNA sequences of hormone-regulated genes from the MCF-7 human breast cancer cell line. Nucleic Acids Res. 10:7895–7903; 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moore M. W.; Klein R. D.; Farinas I.; Sauer H.; Armanini M.; Phillips H.; Reichardt L. F.; Ryan A. M.; Carver-Moore K.; Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature 382:76–79; 1996. [DOI] [PubMed] [Google Scholar]
- 34. Nakagawa T.; Nelkin B. D.; Baylin S. B.; de Bustros A. Transcriptional and posttranscriptional modulation of calcitonin gene expression by sodium n-butyrate in cultured human medullary thyroid carcinoma. Cancer Res. 48:2096–2100; 1988. [PubMed] [Google Scholar]
- 35. Pichel J. G.; Shen L.; Sheng H. Z.; Granholm A. C.; Drago J.; Grinberg A.; Lee E. J.; Huang S. P.; Saarma M.; Hoffer B. J.; Sariola H.; Westphal H. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature 382:73–76; 1996. [DOI] [PubMed] [Google Scholar]
- 36. Prins G. S.; Woodham C. Autologous regulation of androgen receptor messenger ribonucleic acid in the separate lobes of the rat prostate gland. Biol. Reprod. 53:609–619; 1996. [DOI] [PubMed] [Google Scholar]
- 37. Ray A.; Prefontaine K. E. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kB and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 91:752–756; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robertson K.; Mason I. Expression of ret in the chicken embryo suggests roles in regionalisation of the vagal neural tube and somites and in development of multiple neural crest and placodal lineages. Mech. Dev. 53:329–344; 1995. [DOI] [PubMed] [Google Scholar]
- 39. Russo A. F.; Nelson C.; Roos B. A.; Rosenfeld M. G. Differential regulation of the coexpressed calcitonin/alpha-CGRP and beta-CGRP neuroendocrine genes. J. Biol. Chem. 263:5–8; 1988. [PubMed] [Google Scholar]
- 40. Russo A. F.; Clark M. S.; Durham P. L. Thyroid parafollicular cells: An accessible model for the study of serotonergic neurons. Mol. Neurobiol. 13:257–275; 1996. [DOI] [PubMed] [Google Scholar]
- 41. Sanchez M. P.; Silos Santiago I.; Frisen J.; He B.; Lira S. A.; Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature 382:70–73; 1996. [DOI] [PubMed] [Google Scholar]
- 42. Santoro M.; Carlomagno F.; Romano A.; Boltaro D. P.; Dathan N. A.; Grieco M.; Fusco A.; Vecchio G.; Matoskova B.; Kraus M. H.; Di Fiore P. P. Activation of RET as a dominant transforming gene by germ-line mutations of MEN2A and MEN2B. Science 267:381–383; 1995. [DOI] [PubMed] [Google Scholar]
- 43. Schuchardt A.; D’Agati V.; Larsson-Blomberg L.; Costantini F.; Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 367:380–383; 1994. [DOI] [PubMed] [Google Scholar]
- 44. Songyang Z.; Canaway K. L.; Eck M. J.; Harrison S. C.; Feldman R. A.; Mohammadi M.; Schlessinger J.; Hubbard S. R.; Smith D. P.; Eng C.; Lorenzo M. J.; Ponder B. A.; Mayer B. J.; Cantley L. C. Catalytic specificity of protein tyrosine kinases is critical for selective signalling. Nature 373:536–539; 1995. [DOI] [PubMed] [Google Scholar]
- 45. Tahira T.; Ishizaka Y.; Itoh F.; Nakayasu M.; Sugimura T.; Nagao M. Expression of the ret proto-oncogene in human neuroblastoma cell lines and its increase during neuronal differentiation induced by retinoic acid. Oncogene 6:2333–2338; 1991. [PubMed] [Google Scholar]
- 46. Trupp M.; Arenas E.; Fainzilber M.; Nilsson A.-S.; Sieber B.-A.; Grigoriou M.; Kilkenny C.; Salazar-Grueso E.; Pachnis V.; Arumae U.; Sariola H.; Saarma M.; Ibanez C. F. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature 381:785–793; 1996. [DOI] [PubMed] [Google Scholar]
- 47. Tverberg L. A.; Russo A. F. Cell-specific glucocorticoid repression of calcitonin/calcitonin gene-related peptide transcription. J. Biol. Chem. 267:17567–17573; 1992. [PubMed] [Google Scholar]
- 48. Yang-Yen H. F.; Chambard J. C.; Sun Y. L.; Smeal T.; Schmidt T. J.; Drouin J.; Karin M. Transcriptional interference between c-jun and the glucocorticoid receptor: Mutual inhibition of DNA binding due to direct protein-protein interaction. Cell 62:1205–1215; 1990. [DOI] [PubMed] [Google Scholar]