

Abstract
Diabetic kidney disease (DKD) is a devastating condition associated with increased morbidity and premature mortality. The etiology of DKD is still largely unknown. However, the risk of DKD development and progression is most likely modulated by a combination of genetic and environmental factors. Patients with autoimmune diseases, like type 1 diabetes, inflammatory bowel disease, and celiac disease, share some genetic background. Furthermore, gastrointestinal disorders are associated with an increased risk of kidney disease, although the true mechanisms have still to be elucidated. Therefore, the principal aim of this review is to evaluate the impact of disturbances in the gastrointestinal tract on the development of renal disorders.
Keywords: type 1 diabetes, diabetic nephropathy, chronic kidney disease, gut permeability, inflammatory bowel disease, gastrointestinal inflammation, contact activation, intestinal alkaline phosphatase
Introduction
Type 1 diabetes is an autoimmune disorder that is primarily associated with elevated blood glucose due to loss of the insulin-producing pancreatic beta-cells. Long disease duration and poor glycemic control increase the risk of micro- and macrovascular complications, and the incidence and severity of these complications is modulated by genetic and environmental factors. Diabetic nephropathy, affecting up to one third of individuals with type 1 diabetes, is a global health problem that is associated with significant and continuously growing health care costs (1, 2). Diabetic nephropathy is not only strongly associated with cardiovascular disease, but is also one of the leading risk factors for premature death in the Western countries (3–5).
The presence and progression of diabetic nephropathy is associated with low-grade chronic inflammation, which could also be seen as an indication of a systemic infection (6). It is evident that hyperglycemia by itself increases the risk of bacterial infections (7, 8), and particularly patients with poor glycemic control are susceptible to respiratory, urinary, skin, and intestinal infections (9–11). Moreover, severe bacterial infections increase the risk of acute kidney injury especially in patients with diabetes, in the elderly, and in those with renal impairment and hypertension (12). Dysbiosis of the gut microbiota has frequently been reported in various autoimmune and metabolic disease conditions such as in allergy, asthma, inflammatory bowel disease, celiac disease, systemic lupus erythematosus, arthritis, chronic kidney disease, diabetes, obesity, and cardiovascular disease (13–18). Based upon the hygiene hypothesis, frequent exposure to infective microbial agents in childhood protects from the development of autoimmune diseases later in life. Apparently, crosstalk between pathogenic and commensal gut bacteria play an important role in this process (19). In support of this hypothesis, certain viruses and bacteria have been linked to the development of autoimmunity (20–22).
It is likely that gastrointestinal disorders associated with dysbiosis may play a significant role in the development of diabetic nephropathy, since disruption of the gastrointestinal wall increases the translocation of microbial compounds, which have been shown to have proinflammatory and nephrotoxic properties. In support of this theory, we and others have shown that elevated systemic levels of bacterial endotoxins are associated with the development and progression of renal disease (23–26). Given the large amount of emerging new data suggesting cross-talk between the gut and the kidneys, the aim of this review is to explore such interconnections and particularly the link between gastrointestinal and renal disorders (Figure 1).
Figure 1.
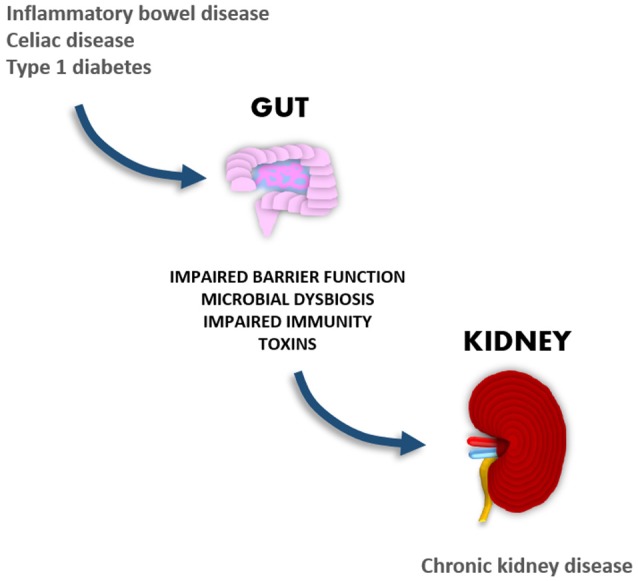
Gastrointestinal manifestations in autoimmune-mediated diseases are associated with the development of chronic kidney disease.
Gastrointestinal manifestations in patients with diabetes
In addition to the classical microvascular (retinopathy, nephropathy, neuropathy) and macrovascular complications (coronary artery disease, stroke, peripheral vascular disease), individuals with type 1 diabetes also display gastrointestinal problems. Although gastrointestinal-related complications are relatively common in individuals with diabetes, their recognition and management are often challenging. The most common gastrointestinal problems are esophageal dysmotility, gastro-esophageal reflux disease, gastroparesis, and enteropathy, and the severity of the problems often correlates with the presence of micro- and macrovascular complications. It is of note that life-style related factors such as composition of diet and exercise as well as medication (e.g., metformin, statins, incretin-based therapies) also have an impact on the gastrointestinal problems (27). Up to 75% of individuals with diabetes may experience gastrointestinal symptoms such as early satiety, postprandial fullness, nausea, abdominal pain, vomiting, bloating, diarrhea and/or constipation affecting their quality of life (28). Although many of these symptoms could be attributed to diabetes-related autonomic neuropathy, the etiology of these gastrointestinal problems often remains unclear. However, in individuals with diabetes, a relationship between cardiovascular autonomic neuropathy and small-bowel bacterial overgrowth has been reported (29, 30).
Exposure to antibiotics may lead to disruption of the normal microbiota, which eventually could lead to colonization of pathogenic microbes (e.g., Helicobacter pylori, Clostridium difficile, Candida etc,). In individuals with type 1 diabetes in the UK, the rate of gastrointestinal infections was ~2-fold higher compared to the general population (8). The frequent use of antibiotics, which often disrupts the balance of the normal intestinal microflora, has also been shown to correlate with the severity of diabetic nephropathy in patients with type 1 diabetes (7). However, chronic gastrointestinal symptoms could also arise from the presence of other systemic diseases such as celiac disease, lactose intolerance, irritable bowel syndrome, and inflammatory bowel disease. Type 1 diabetes and inflammatory bowel disease are multifactorial autoimmune diseases, which share many genetic and immunological aspects (31, 32). Both are chronic diseases that are associated with increased risk of cardiovascular disease and premature mortality (33, 34). Despite the potential overlaps, earlier studies have not been able to demonstrate a higher risk of diabetes in patients with inflammatory bowel disease (35–38). However, in a recent study from the UK, the prevalence of clinical inflammatory bowel disease in adult individuals with type 1 diabetes was ~6-fold higher compared to non-diabetic controls (1.5 vs. 0.3%, respectively) (39). Nevertheless, although gastrointestinal manifestations are frequently reported in individuals with diabetes, the potential impact of gastrointestinal-related disorders on the development and progression of diabetic nephropathy remains to be elucidated (40).
Gastrointestinal manifestations in patients with renal diseases
Chronic kidney disease is a multifactorial disorder primarily associated with the gradual loss of kidney function over time. Based on a large systematic review and meta-analysis comprising nearly seven million adult patients, the global estimate of the prevalence was about 13% (41). Diabetes is one of the leading causes, and the prevalence of chronic kidney disease is about 5 times higher among individuals with diabetes than in the general population (42). Patients with advanced chronic kidney disease carry a significantly higher risk of premature mortality due to increased risk of infection related diseases (43, 44). Consequently, sepsis is a severe life-threatening condition especially in patients with impaired renal function (45, 46).
A key problem in chronic kidney disease is the gut dysbiosis and the accumulation of uremic toxins. These uremic toxins are derived from the diet, the protein metabolism, and the metabolic action of the gut bacteria. The most intensively studied toxins are p-cresylsulphate and indole sulfate. Tyrosine and phenylalanine are the main sources of p-cresol, while indole originates from tryptophan. These intermediate by-products are absorbed from the gut and finally sulfated by the liver. The accumulation of the uremic toxins has many adverse effects, and for instance the accumulation of urea increases the urea influx into the intestinal lumen, where it is hydrolyzed to ammonia by microbial urease. Consequently, ammonium hydroxide, a by-product of ammonia, increases the intestinal pH leading to mucosal irritation and structural damage. The uremic condition in combination with chronic inflammation and impaired renal function may increase the risk of kidney disease progression (47, 48).
Metabolic dysregulation seen in individuals with diabetes is exacerbated by microbial dysbiosis and associated with expansion of anaerobic bacteria, defects in the intestinal barrier function, and increased translocation of microbial compounds, e.g., bacterial endotoxins. Moreover, significant alterations of the gut microbiota have been reported in patients with advanced kidney disease (49, 50). Also, the immunosuppressive treatment in patients with kidney transplants may have adverse effects on the gastrointestinal tract (51). Previous studies in animals and humans have shown that therapies targeting the gut might provide novel tools for the correction of intestinal metabolism and microbial dysbiosis in chronic kidney disease (52). For instance, in collagen type 4α3–deficient mice with progressive chronic kidney disease, the eradication of anaerobic microbiota with antibiotics prevented bacterial translocation, reduced the serum endotoxin levels, and reversed the systemic inflammation to the level of non-uremic controls (53). Although the results from dietary interventions in humans are still somewhat controversial, modulation of the intestinal microbiota by supplementation with probiotics may have beneficial therapeutic effects in individuals with impaired renal function (54).
IgA nephropathy is the most common form of primary glomerulonephritis worldwide. The renal injury is mediated by deposition of IgA antibodies in the glomerular mesangium leading to increased local inflammation and renal dysfunction. Increased reactivity to dietary antigens has been associated with mucosal inflammation. Interestingly, a recent genome-wide association study on individuals with IgA nephropathy of European and East Asian ancestry identified several risk loci, which have previously been associated with the risk of inflammatory bowel disease, intestinal epithelial barrier function, and immune response to mucosal pathogens (55). Furthermore, increased intestinal permeability, typically seen in inflammatory bowel disease and celiac disease, has also been reported in patients with IgA nephropathy (56–58).
Kidney manifestations in patients with gastrointestinal disease
The inflammatory bowel disease is a multifactorial disease primarily associated with microbial dysbiosis, intestinal epithelial damage, immune dysregulation, and inflammation of the gut. Despite the growing knowledge, the etiology and pathogenesis of this complex disease is not yet fully understood. The incidence of extra-intestinal manifestations (e.g., in the eye, skin, bone, liver, pancreas, kidney, lung, and the heart) and other autoimmune related diseases (e.g., celiac disease, type 1 diabetes, sarcoidosis, asthma, psoriasis, and rheumatoid arthritis) seem to be significantly higher in patients with inflammatory bowel disease compared to the general population (59, 60). It has been estimated that 4–23% of patients with inflammatory bowel disease suffer from reno-ureteral complications (61), of which the most common are nephrolithiasis, tubulointerstitial nephritis, glomerulonephritis, and amyloidosis. A significant proportion of these renal complications are, however, associated with the use of medication (e.g., aminosalisalicytes, cyclosporine, azathioprine, and tumor necrosis factor alpha inhibitors) (62). Based on the histopathologic examination of kidney biopsies, IgA nephropathy and tubulointerstitial nephritis were the most common findings observed in patients with inflammatory bowel disease with subsequent kidney disease (63). Notably, inflammatory bowel disease patients with active disease have shown higher urinary albumin levels compared to those in the remission phase (64, 65).
Celiac disease is an autoimmune disease mainly associated with defects in the small intestine. Patients with celiac disease suffer from diarrhea, malabsorption, weight loss, iron deficiency, and anemia. Celiac disease is primarily caused by an aberrant autoimmune reaction toward gluten, which is an abundant protein present in various grains e.g., wheat, oats, rye, and barley. The diagnosis of celiac disease requires specific serologic blood tests (tissue transglutaminase IgA, deaminated gliadin peptide IgA/IgG, endomysial IgA) and small intestinal biopsies. Abnormal villous structures, villous atrophy, crypt hyperplasia, and increased number of intraepithelial lymphocytes are common endoscopic findings in patients with active disease. The primary treatment of celiac disease is gluten-free diet, which results in improvements in many cases (66). In Europe, the prevalence of celiac disease in individuals with type 1 diabetes is about 6–8 times higher compared to the general population (67). Despite the increased risk of celiac disease, many patients with type 1 diabetes and celiac disease do not display gastrointestinal symptoms (68, 69). It has become evident that patients with type 1 diabetes and celiac disease share many genetic (e.g., HLA and non-HLA genes) and environmental (e.g., dietary antigens, viral infections) risk factors. Based on previous clinical surveys, celiac disease was also associated with other extra-intestinal manifestations such as endocrine, connective tissue, and pulmonary disorders (70, 71). Compared to the general population, patients with celiac disease carry a higher risk of renal diseases e.g., IgA nephropathy (72–75). Based on eight Northern European studies, the relative risk of kidney disease was ~2-fold higher in patients with celiac disease compared to those without. Subsequent subgroup analyses in patients with celiac disease showed ~1.5-fold and ~2.6-fold increased risks of diabetic nephropathy and IgA nephropathy, respectively (76).
Potential novel therapeutic tools
Probiotics
Probiotics are living non-pathogenic micro-organisms, the ingestion of which should provide health benefits to the host. Overall, probiotics are thought to have anti-inflammatory, anti-oxidative, and many other favorable gut-modulating properties (77). Especially bacterial species belonging to the genus of Bifidobacterium and Lactobacillus, support the humoral immune responses against environmental toxins and antigens. Over the past decade, the impact of supplementation with probiotics has been intensively studied in individuals with mild or severe gastrointestinal symptoms e.g., diarrhea, irritable bowel syndrome, and inflammatory bowel disease (78, 79). Probiotic treatment with butyrate-producing bacteria (e.g., Faecalibacterium prausnitzii, Butyricicoccus pullicaecorum) may improve epithelial barrier integrity and suppression of intestinal inflammation (80). Among the newly discovered gut residing bacteria, Akkermansia muciniphila has shown promise for the treatment of metabolic disorders (81). In patients with kidney disease, probiotic supplementation has also shown some beneficial effects on the glycemic control, uremic toxins, blood urea nitrogen, oxidative stress, and markers of inflammation (82–87). Similar trends have been observed in clinical trials using synbiotics, a combination of probiotics and prebiotics, which support the growth and activity of the beneficial micro-organisms in the gut (54, 88). In a mouse model of acute kidney injury, short-term probiotic pretreatment decreased the inflammation and protected the mice from the development of severe kidney damage (89).
Resistant starch
Starches are considered prebiotics, which support the health and growth of the gastrointestinal microflora. Natural plant starches with high amylose content are resistant to digestion by intestinal amylases. Enzyme-resistant starches pass the upper intestinal tract and are finally fermented by the gut bacteria in the large intestine leading to increased production of beneficial metabolites. Many of the propitious effects (e.g., increased number of butyrate-producing bacteria, reduced cell proliferation, decreased inflammation, and cellular apoptosis) are thought to be mediated by an increased production of short-chain fatty acids including acetate, propionate, and butyrate. Clinical trials with resistant starches have shown some promising effects in the management of metabolic diseases including colon cancer, inflammatory bowel disease, obesity and diabetes (90). In a recent study in non-obese diabetic mice, resistant starch diet decreased the number of autoreactive T-cells and protected the mice against type 1 diabetes (91). In addition to improved glycemic control and insulin sensitivity (92), resistant starch containing diet could also exert renoprotective effects e.g., via the vitamin D metabolism (93). Dietary supplementation of digestion resistant starches has been shown to ameliorate the progression of kidney disease in rodents (94, 95). In hemodialysis patients, consumption of resistant starch for 6 weeks reduced the plasma levels of uremic toxins (96). Despite these promising effects, more studies are needed to evaluate the impact of dietary factors on the gut and the kidney function in patients with chronic kidney disease (97).
Fecal transplantation
The basic principle of fecal microbiota transplantation is to transfer the normal microbiome from a healthy donor to individuals suffering from microbial dysbiosis. Isolated stool preparations can be transferred to the recipient with the aid of colonoscopy, an orogastric tube, enema, or by orally given capsules. The fecal microbiota transplantation strategy has successfully been applied in the treatment of infections caused by Clostridium difficile and especially in those patients that do not respond to the antibiotic treatment. It is of note that chronic Clostridium difficile infections increase the risk of colon-related complications such as megacolon and pseudomembraneous colitis. Alternative treatment options have become increasingly important as the prevalence of antibiotic resistant bacterial strains have exploded all around the world. The risk of Clostridium difficile infections is particularly high among the patients with inflammatory bowel disease—up to eight times higher risk than in matched controls (98). Also patients receiving immunosuppressive medication, e.g., patients with a kidney transplant, have a higher risk of colonization of pathogenic bacteria. In addition to Clostridium difficile infections, the fecal microbiota transplantation treatment has also been shown to be an effective tool for the eradication of gut residing Salmonella and ESBL-producing E. coli bacterial strains (99, 100). Besides chronic gastrointestinal infections, the fecal microbiota transplantation therapy may also offer novel treatment strategies for various metabolic diseases such as diabetes, obesity, non-alcoholic fatty liver disease, and inflammatory bowel disease (101, 102).
Potential novel therapeutic targets
In relation to the manifestations of the gastrointestinal diseases, there are potential novel biological mechanisms associated with the activity and permeability of the microbial toxins in the gut. New evidence supports the hypothesis that intestinal alkaline phosphatase (IAP) and components of the kallikrein-kinin system play an important role in the detoxification and the transport of bacterial endotoxins (Figure 2).
Figure 2.

Putative interconnections between LPS-detoxification and intestinal barrier function. Intestinal alkaline phosphatase (IAP) detoxifies bacterial lipopolysaccharides (LPS; endotoxins) by dephosphorylation. Butyrate, a short-chain fatty acid produced as a result of bacterial fermentation, stimulates IAP gene expression. IAP may also modulate composition of microbial community via regulation of secretory IgA release. Fecal calprotectin is a commonly used marker for increased neutrophil (NEU) migration and local inflammation in patients with gastrointestinal diseases. Lower luminal IAP-activity may lead to increased production of toxic LPS-molecules, which in turn may stimulate Factor XII regulated contact activation pathway in the gut. Subsequent activation of kallikrein-kinin system by endotoxins may potentiate leakiness of the intestinal wall. Modified from Lassenius et al. (103).
Intestinal alkaline phosphatase
The human gut contains about 1.0–1.5 kg of bacteria. Notably, the brush-border enzyme intestinal alkaline phosphatase (IAP) plays an important role in the gut mucosal defense by suppressing inflammatory mediators, including microbial compounds (endotoxins, polyphosphates), and luminal adenosine triphosphate by dephosphorylation. IAP has also been shown to be involved in the regulation of duodenal bicarbonate secretion and fatty acid absorption. A decrease in the luminal IAP activity could increase the risk of gastrointestinal disease through changes in the microbial composition, intestinal inflammation, and the gut permeability (104, 105). Exogenous administration of IAP has on the other hand been shown to have potential therapeutic effects. In animal models of intestinal damage, oral supplementation of IAP reduced the gut epithelial damage and inflammation (106). Moreover, administration of exogenous IAP to mice suppressed the Salmonella and Clostridium associated disease activity and improved survival (107, 108). Interestingly, in IAP-knockout mice, the development of the metabolic syndrome was also prevented by oral IAP supplementation (109, 110). Both recombinant and bovine IAP has been utilized in human clinical trials. Promising results has so far been obtained from the treatment of patients with rheumatoid arthritis, heart surgery, sepsis, acute kidney injury, and inflammatory bowel disease (110–112). The histone deacetylase inhibitor butyrate, produced by intestinal bacteria through carbohydrate fermentation, is a positive activator of IAP gene expression. Oral butyrate supplementation has also shown beneficial effects in individuals with various gastrointestinal problems such as irritable bowel syndrome, inflammatory bowel disease, diverticulitis, and traveler's diarrhea (113). Butyrate has also been shown to alleviate renal fibrosis, inflammation, and kidney damage in rodent animal models (114–116).
Importantly, we recently observed in individuals with type 1 diabetes that decreased fecal IAP activity was accompanied by lower fecal butyrate, and IgA antibody concentrations. Of note, individuals with uncomplicated type 1 diabetes also exhibited higher fecal calprotectin concentrations compared to the non-diabetic controls. It was also shown that oral IAP supplementation increased the mucosal IgA secretion, which modulate immunity and host-microbe interactions in the gut (103, 117). Reduced fecal IAP levels have been reported in individuals with inflammatory bowel disease, celiac disease, and type 2 diabetes (118–120). Hypothetically, low intestinal IAP-activity could result in increased translocation of cytotoxic bacterial compounds, higher systemic endotoxin activity, and could thereby increase the risk of kidney or other organ injuries. In IAP-knockout mice, the dextran sodium sulfate-induced colitis phenotype was significantly aggravated compared to dextran sodium sulfate-treated wild-type mice. Oral administration of calf IAP attenuated the intestinal inflammation and normalized the mucosal architecture in dextran sodium sulfate-treated IAP-knockout mice (121). The first compound heterozygous mutations in the human ALPI gene leading to IAP deficiency have recently been described. Two index cases with different IAP mutations exhibited gastrointestinal manifestations similar to inflammatory bowel disease (122). A novel deletion-insertion mutation in the C-terminal end of the hALPI gene, was recently associated with familial hyperphosphatemia in Japan (123).
The effects of exogenous IAP administration have been evaluated in recent human clinical trials involving patients with sepsis, heart surgery, and rheumatoid arthritis. Of note, intravenously administrated bovine IAP has been shown to have promising renoprotective effects in sepsis-induced acute kidney injury (124–126). In animal models of intestinal damage, oral administration of IAP reduced gut epithelial damage, and inflammation (106, 127, 128). Despite these promising effects in animals, human trials with oral IAP supplementation are currently at the very early stage (111). However, endogenous IAP expression could potentially be enhanced via modulation of the gut microbiome (diet, probiotics), dietary supplements (e.g., resistant starch, fiber, butyrate) or with specific drug-therapy (e.g., Rifampicin, Zanamivir) (108, 129, 130).
Contact activation pathway
Patients with impaired intestinal barrier function also display anomalous changes in the factor XII (FXII; Hageman factor) regulated contact activation pathway. Upon contact with negatively charged surfaces and anionic compounds (e.g., glass, heparin, polyphosphates, dextran sulfate, endotoxins) FXII is autoactivated, which eventually leads to sequential proteolytic cleavage of the down-stream targets. Cleavage of high-molecular-weight kininogen leads to subsequent production of bradykinin, a potent vasodilator of blood vessels (131). The question arises whether bradykinin release could also be linked to the increased gut permeability e.g., bacterial endotoxins, which may in turn boost the activation of FXII in the submucosal tissue. The increased gut permeability and translocation of bacterial components may also aggravate systemic inflammation and renal damage.
Hereditary angioedema is a rare genetic defect, primarily caused by mutations in the SERPING1 gene, associated with complement 1 inhibitor deficiency (132). Gene mutations associated with hereditary angioedema have also been found in the Factor XII and plasminogen genes (133–136). Moreover, the non-genetic and non-allergic form of angioedema has frequently been seen in subjects using angiotensin-converting enzyme inhibitors (132). Angioedema is commonly characterized by short-lived episodes of serious edema involving the lungs, the skin, and the gastrointestinal tract (137, 138). Over 90% of hereditary angioedema patients have experienced episodic or chronic abdominal pain, which is accompanied by other gastrointestinal symptoms such as diarrhea, nausea or vomiting (139). Similar complications have been reported in individuals with angiotensin-converting enzyme inhibitor-induced angioedema (140). Based on the surveillance of antihypertensive medication (angiotensin-converting enzyme inhibitors/angiotensin receptor blockers) in the UK between 2007 and 2014, gastroenteritis was shown to be a significant risk factor for acute kidney injury (141). In addition to the gastrointestinal problems, kidney related disorders have also been documented in patients with hereditary angioedema (142–148). Cutaneous and systemic lupus erythematosus are the most often described autoimmune conditions associated with hereditary angioedema. It has been estimated that ~20–25% of hereditary angioedema patients with a subsequent lupus diagnosis exhibit renal complications (149).
Defects in the contact activation pathway have also been reported in patients with inflammatory bowel disease. Zeitlin et al. reported significantly higher kinin-forming activity in the colonic muscle tissue isolated from patients with ulcerative colitis (150). In patients with inflammatory bowel disease, increased interstitial kallikrein activity has also been shown to correlate with the inflammation in the isolated tissue samples (151, 152). In an animal model of colitis, elevated levels of portal endotoxins were found in mice treated with dextran sodium sulfate (153). However, in wild-type mice with dextran sodium sulfate induced colitis, complement 1 inhibitor treatment reduced the severity of the disease (154). Furthermore, the use of bradykinin receptor blockers have been shown to improve the general health condition of mice with chemically induced colitis (155–157). Moreover, inhibition of the kallikrein-kinin system reduced peptidoglycan-polysaccharide induced enterocolitis also in rats (158–160). In support of these earlier findings, Wang et al. has recently demonstrated that mice deficient in kallikrein, kininogen, or bradykinin receptors are protected from dextran sodium sulfate-induced mucosal damage and inflammation (161). In addition to the dextran sodium sulfate-induced gut related phenotype, the presence of other extra-intestinal manifestations should also be considered. Noteworthy, dextran sodium sulfate-induced colitis has also been associated with the development of acute kidney injury in mice (162, 163). There is a substantial number of studies supporting the impact of the kallikrein-kinin system on the development of microvascular complications such as diabetic retinopathy, macular edema, and diabetic nephropathy in patients with diabetes (164, 165). Apparently, more studies are needed to elucidate whether the contact activation pathway play a significant role in the pathogenesis of gastrointestinal and kidney related disorders.
Conclusion
Based on the present overview, interconnections between gastrointestinal and renal disorders exist. One of the future challenges is to find diagnostic tools to determine the incidence of gut related diseases especially among patients with type 1 diabetes. Early identification and effective management of gut related disorders may slow down or even prevent the development of secondary complications e.g., renal and vascular injuries. In the future novel treatment strategies could potentially be adapted once the pathophysiological mechanisms behind the gut-kidney axis has been clarified.
Author contributions
ML reviewed the literature and wrote manuscript. P-HG reviewed and edited the manuscript. Both authors made intellectual contribution to the present work and approved the final version of the manuscript for publication.
Conflict of interest statement
P-HG has received research grants from Eli Lilly and Roche, is an advisory board member for AbbVie, Astra Zeneca, Boehringer-Ingelheim, Cebix, Eli Lilly, Janssen, MSD, Medscape, Novartis, and Sanofi. He has received lecture fees from Astra Zeneca, Boehringer-Ingelheim, Eli Lilly, Elo Water, Genzyme, Medscape, MSD, Novartis, Novo Nordisk, and Sanofi. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by Folkhälsan Research Foundation, Wilhelm and Else Stockmann Foundation, Novo Nordisk Foundation (#NNF14SA0003), and Diabetes Research Foundation.
References
- 1.Lithovius R, Harjutsalo V, Saraheimo M, Forsblom C, Groop PH; FinnDiane Study Group. Longitudinal trends in use and costs of prescription medication in patients with type 1 diabetes: the impact of renal disease. Ann Med. (2013) 45:112–9. 10.3109/07853890.2012.671535 [DOI] [PubMed] [Google Scholar]
- 2.Tuttle KR, Bakris GL, Bilous RW, Chiang JL, de Boer IH, Goldstein-Fuchs J, et al. Diabetic kidney disease: a report from an ADA Consensus Conference. Am J Kidney Dis. (2014) 64:510–33. 10.1053/j.ajkd.2014.08.001 [DOI] [PubMed] [Google Scholar]
- 3.Rossing P, Hougaard P, Borch-Johnsen K, Parving HH. Predictors of mortality in insulin dependent diabetes: 10 year observational follow up study. BMJ (1996) 313:779–84. 10.1136/bmj.313.7060.779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Groop PH, Thomas MC, Moran JL, Wadèn J, Thorn LM, Mäkinen VP, et al. The presence and severity of chronic kidney disease predicts all-cause mortality in type 1 diabetes. Diabetes (2009) 58:1651–8. 10.2337/db08-1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groop PH, Thomas M, Feodoroff M, Forsblom C, Harjutsalo V. FinnDiane Study Group. Excess mortality in patients with type 1 diabetes without albuminuria-separating the contribution of early and late risks. Diabetes Care (2018) 41:748–54. 10.2337/dc17-1618 [DOI] [PubMed] [Google Scholar]
- 6.Saraheimo M1, Teppo AM, Forsblom C, Fagerudd J, Groop PH. Diabetic nephropathy is associated with low-grade inflammation in Type 1 diabetic patients. Diabetologia (2003) 46:1402–7. 10.1007/s00125-003-1194-5 [DOI] [PubMed] [Google Scholar]
- 7.Simonsen JR, Harjutsalo V, Järvinen A, Kirveskari J, Forsblom C, Groop PH, et al. Bacterial infections in patients with type 1 diabetes: a 14-year follow-up study. BMJ Open Diabetes Res Care (2015) 3:e000067. 10.1136/bmjdrc-2014-000067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carey IM, Critchley JA, DeWilde S, Harris T, Hosking FJ, Cook DG. Risk of infection in type 1 and type 2 diabetes compared with the general population: a matched cohort study. Diabetes Care (2018) 41:513–21. 10.2337/dc17-2131 [DOI] [PubMed] [Google Scholar]
- 9.Benfield T, Jensen JS, Nordestgaard BG. Influence of diabetes and hyperglycaemia on infectious disease hospitalisation and outcome. Diabetologia (2007) 50:549–54. 10.1007/s00125-006-0570-3 [DOI] [PubMed] [Google Scholar]
- 10.Gornik I, Gornik O, Gasparović V. HbA1c is outcome predictor in diabetic patients with sepsis. Diabetes Res Clin Pract. (2007) 77:120–5. 10.1016/j.diabres.2006.10.017 [DOI] [PubMed] [Google Scholar]
- 11.Kornum JB, Thomsen RW, Riis A, Lervang HH, Schønheyder HC, Sørensen HT. Diabetes, glycemic control, and risk of hospitalization with pneumonia: a population-based case-control study. Diabetes Care (2008) 31:1541–5. 10.2337/dc08-0138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cartin-Ceba R, Kashiouris M, Plataki M, Kor DJ, Gajic O, Casey ET. Risk factors for development of acute kidney injury in critically ill patients: a systematic review and meta-analysis of observational studies. Crit Care Res Pract. (2012) 2012:691013. 10.1155/2012/691013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis. (2015) 26:26191. 10.3402/mehd.v26.26191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hevia A, Milani C, López P, Cuervo A, Arboleya S, Duranti S, et al. Intestinal dysbiosis associated with systemic lupus erythematosus. MBio (2014) 5:e01548–14. 10.1128/mBio.01548-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Battson ML, Lee DM, Weir TL, Gentile CL. The gut microbiota as a novel regulator of cardiovascular function and disease. J Nutr Biochem. (2017) 56:1–15. 10.1016/j.jnutbio.2017.12.010 [DOI] [PubMed] [Google Scholar]
- 16.Lau WL, Vaziri ND. The leaky gut and altered microbiome in chronic kidney disease. J Ren Nutr. (2017) 27:458–61. 10.1053/j.jrn.2017.02.010 [DOI] [PubMed] [Google Scholar]
- 17.Li X, Watanabe K, Kimura I. Gut microbiota dysbiosis drives and implies novel therapeutic strategies for diabetes mellitus and related metabolic diseases. Front Immunol. (2017) 8:1882. 10.3389/fimmu.2017.01882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Picchianti-Diamanti A, Rosado MM, D'Amelio R. Infectious agents and inflammation: the role of microbiota in autoimmune arthritis. Front Microbiol. (2018) 8:2696. 10.3389/fmicb.2017.02696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bach JF. The hygiene hypothesis in autoimmunity: the role of pathogens and commensals. Nat Rev Immunol. (2018) 18:105–20. 10.1038/nri.2017.111 [DOI] [PubMed] [Google Scholar]
- 20.Vatanen T, Kostic AD, d'Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell (2016) 165:842–53. 10.1016/j.cell.2016.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Honkanen H, Oikarinen S, Nurminen N, Laitinen OH, Huhtala H, Lehtonen J, et al. Detection of enteroviruses in stools precedes islet autoimmunity by several months: possible evidence for slowly operating mechanisms in virus-induced autoimmunity. Diabetologia (2017) 60:424–31. 10.1007/s00125-016-4177-z [DOI] [PubMed] [Google Scholar]
- 22.Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science (2018) 359:1156–61. 10.1126/science.aar7201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nymark M, Pussinen PJ, Tuomainen AM, Forsblom C, Groop PH, Lehto M. Serum lipopolysaccharide activity is associated with the progression of kidney disease in finnish patients with type 1 diabetes. Diabetes Care (2009) 32:1689–93. 10.2337/dc09-0467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J Clin Invest. (2009) 119:2868–78. 10.1172/JCI39421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lassenius MI, Pietiläinen KH, Kaartinen K, Pussinen PJ, Syrjänen J, Forsblom C, et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care (2011) 34:1809–15. 10.2337/dc10-2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doi K. Role of kidney injury in sepsis. J Intensive Care (2016) 4:17. 10.1186/s40560-016-0146-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krishnan B1, Babu S, Walker J, Walker AB, Pappachan JM. Gastrointestinal complications of diabetes mellitus. World J Diabetes (2013) 4:51–63. 10.4239/wjd.v4.i3.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bytzer P, Talley NJ, Leemon M, Young LJ, Jones MP, Horowitz M. Prevalence of gastrointestinal symptoms associated with diabetes mellitus: a population-based survey of 15,000 adults. Arch Intern Med. (2001) 161:1989–96. 10.1001/archinte.161.16.1989 [DOI] [PubMed] [Google Scholar]
- 29.Zietz B, Lock G, Straub RH, Braun B, Schölmerich J, Palitzsch KD. Small-bowel bacterial overgrowth in diabetic subjects is associated with cardiovascular autonomic neuropathy. Diabetes Care (2000) 23:1200–1. 10.2337/diacare.23.8.1200 [DOI] [PubMed] [Google Scholar]
- 30.Ojetti V, Pitocco D, Scarpellini E, Zaccardi F, Scaldaferri F, Gigante G, et al. Small bowel bacterial overgrowth and type 1 diabetes. Eur Rev Med Pharmacol Sci. (2009) 13:419–23. [PubMed] [Google Scholar]
- 31.Claesson MH, Nicoletti F, Stosic-Grujicic S, Doria A, Zampieri S. Interactions between infections and immune-inflammatory cells in type 1 diabetes mellitus and inflammatory bowel diseases: evidences from animal models. Clin Exp Rheumatol. (2008) 26 (1 Suppl. 48):S8–11. [PubMed] [Google Scholar]
- 32.Gjymishka A, Coman RM, Brusko TM, Glover SC. Influence of host immunoregulatory genes, ER stress and gut microbiota on the shared pathogenesis of inflammatory bowel disease and Type 1 diabetes. Immunotherapy (2013) 5:1357–66. 10.2217/imt.13.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gandhi S, Narula N, Marshall JK, Farkouh M. Are patients with inflammatory bowel disease at increased risk of coronary artery disease? Am J Med. (2012) 125:956–62. 10.1016/j.amjmed.2012.03.015 [DOI] [PubMed] [Google Scholar]
- 34.Lind M, Svensson AM, Kosiborod M, Gudbjörnsdottir S, Pivodic A, Wedel H, et al. Glycemic control and excess mortality in type 1 diabetes. N Engl J Med. (2014) 371:1972–82. 10.1056/NEJMoa1408214 [DOI] [PubMed] [Google Scholar]
- 35.Weng X, Liu L, Barcellos LF, Allison JE, Herriton LJ. Clustering of inflammatory bowel disease with immune mediated diseases among members of a Northern-California-Managed Care organization. Am J Gastroenterol. (2007) 102:1429–35. 10.1111/j.1572-0241.2007.01215.x [DOI] [PubMed] [Google Scholar]
- 36.Cohen R, Robinson D, Jr, Paramore C, Fraeman K, Renahan K, Bala M. Autoimmune disease concomitance among inflammatory bowel disease patients in the United States, 2001-2002. Inflamm Bowel Dis. (2008) 14:738–43. 10.1002/ibd.20406 [DOI] [PubMed] [Google Scholar]
- 37.Virta LJ, Kolho KL. The risk of contracting pediatric inflammatory bowel disease in children with celiac disease, epilepsy, juvenile arthritis and type 1 diabetes–a nationwide study. J Crohns Colitis (2013) 7:53–7. 10.1016/j.crohns.2012.02.021 [DOI] [PubMed] [Google Scholar]
- 38.Bähler C, Schoepfer AM, Vavricka SR, Brüngger B, Reich O. Chronic comorbidities associated with inflammatory bowel disease: prevalence and impact on healthcare costs in Switzerland. Eur J Gastroenterol Hepatol. (2017) 29:916–25. 10.1097/MEG.0000000000000891 [DOI] [PubMed] [Google Scholar]
- 39.Penny HA, Leeds JS, Kurien M, Averginos A, Hopper AD, Hadjivassiliou M, et al. The relationship between inflammatory bowel disease and type 1 diabetes mellitus: a study of relative prevalence in comparison with population controls. J Gastrointestin Liver Dis. (2015) 24:125-6. [PubMed] [Google Scholar]
- 40.Chen Z, Zhu S, Xu G. Targeting gut microbiota: a potential promising therapy for diabetic kidney disease. Am J Transl Res. (2016) 8:4009–16. [PMC free article] [PubMed] [Google Scholar]
- 41.Hill NR, Fatoba ST, Oke JL, Hirst JA, O'Callaghan CA, Lasserson DS, et al. Global Prevalence of Chronic Kidney Disease - A Systematic Review and Meta-Analysis. PLoS ONE (2016) 11:e0158765. 10.1371/journal.pone.0158765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zelnick LR, Weiss NS, Kestenbaum BR, Robinson-Cohen C, Heagerty PJ, Tuttle K, et al. Diabetes and CKD in the United States Population, 2009-2014. Clin J Am Soc Nephrol. (2017) 12:1984–90. 10.2215/CJN.03700417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheikh Hassan HI, Tang M, Djurdjev O, Langsford D, Sood MM, Levin A. Infection in advanced chronic kidney disease leads to increased risk of cardiovascular events, end-stage kidney disease and mortality. Kidney Int. (2016) 90:897–904. 10.1016/j.kint.2016.07.013 [DOI] [PubMed] [Google Scholar]
- 44.Xu H, Gasparini A, Ishigami J, Mzayen K, Su G, Barany P, et al. eGFR and the risk of community-acquired infections. Clin J Am Soc Nephrol. (2017) 12:1399–408. 10.2215/CJN.00250117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishigami J, Grams ME, Chang AR, Carrero JJ, Coresh J, Matsushita K. CKD and Risk for Hospitalization With Infection: The Atherosclerosis Risk in Communities (ARIC) Study. Am J Kidney Dis. (2017) 69:752–61. 10.1053/j.ajkd.2016.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chua HR, Wong WK, Ong VH, Agrawal D, Vathsala A, Tay HM, et al. Extended mortality and chronic kidney disease after septic acute kidney injury. J Intensive Care Med. (2018) 1:885066618764617 10.1177/0885066618764617 [DOI] [PubMed] [Google Scholar]
- 47.Barrios C, Beaumont M, Pallister T, Villar J, Goodrich JK, Clark A, et al. Gut-microbiota-metabolite axis in early renal function decline. PLoS ONE (2015) 10:e0134311. 10.1371/journal.pone.0134311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Armani RG, Ramezani A, Yasir A, Sharama S, Canziani MEF, Raj DS. Gut microbiome in chronic kidney disease. Curr Hypertens Rep. (2017) 19:29. 10.1007/s11906-017-0727-0 [DOI] [PubMed] [Google Scholar]
- 49.Lee JR, Muthukumar T, Dadhania D, Toussaint NC, Ling L, Pamer E, et al. Gut microbial community structure and complications after kidney transplantation: a pilot study. Transplantation (2014) 98:697–705. 10.1097/TP.0000000000000370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stadlbauer V, Horvath A, Ribitsch W, Schmerböck B, Schilcher G, Lemesch S, et al. Structural and functional differences in gut microbiome composition in patients undergoing haemodialysis or peritoneal dialysis. Sci Rep. (2017) 7:15601 10.1038/s41598-017-15650-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dobies A, Renke M, Wołyniec W, Palenicek L, Januszczyk J, Król E, et al. Gastrointestinal pathologies in patients after successful renal transplantation-A pilot study. Transplant Proc. (2016) 48:1566–9. 10.1016/j.transproceed.2016.02.060 [DOI] [PubMed] [Google Scholar]
- 52.Al Khodor S, Shatat IF. Gut microbiome and kidney disease: a bidirectional relationship. Pediatr Nephrol. (2017) 32:921–31. 10.1007/s00467-016-3392-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andersen K, Kesper MS, Marschner JA, Konrad L, Ryu M, Kumar Vr S, et al. Intestinal dysbiosis, barrier dysfunction, and bacterial translocation account for CKD-related systemic inflammation. J Am Soc Nephrol. (2017) 28:76–83. 10.1681/ASN.2015111285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rossi M, Johnson DW, Morrison M, Pascoe EM, Coombes JS, Forbes JM, et al. Synbiotics easing renal failure by improving gut microbiology (SYNERGY): a randomized trial. Clin J Am Soc Nephrol. (2016) 11:223–31. 10.2215/CJN.05240515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. (2014) 46:1187–96. 10.1038/ng.3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davin JC, Forget P, Mahieu PR. Increased intestinal permeability to (51 Cr) EDTA is correlated with IgA immune complex-plasma levels in children with IgA-associated nephropathies. Acta Paediatr Scand. (1988) 77:118–24. 10.1111/j.1651-2227.1988.tb10609.x [DOI] [PubMed] [Google Scholar]
- 57.Rostoker G, Terzidis H, Petit-Phar M, Meillet D, Lang P, Dubert JM, Lagrue G, Weil B. Secretory IgA are elevated in both saliva and serum of patients with various types of primary glomerulonephritis. Clin Exp Immunol. (1992) 90:305–11. 10.1111/j.1365-2249.1992.tb07947.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kovács T, Kun L, Schmelczer M, Wagner L, Davin JC, Nagy J. Do intestinal hyperpermeability and the related food antigens play a role in the progression of IgA nephropathy? I. Study of intestinal permeability. Am J Nephrol. (1996) 16:500–5. 10.1159/000169050 [DOI] [PubMed] [Google Scholar]
- 59.Rothfuss KS, Stange EF, Herrlinger KR. Extraintestinal manifestations and complications in inflammatory bowel diseases. World J Gastroenterol. (2006) 12:4819–31. 10.3748/wjg.v12.i30.4819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Halling ML, Kjeldsen J, Knudsen T, Nielsen J, Hansen LK. Patients with inflammatory bowel disease have increased risk of autoimmune and inflammatory diseases. World J Gastroenterol. (2017) 23:6137–46. 10.3748/wjg.v23.i33.6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katsanos KH, Tsianos EV. The kidneys in inflammatory bowel disease. Ann Gastroenterol. (2002) 15:41–52. [Google Scholar]
- 62.Corica D, Romano C. Renal involvement in inflammatory bowel diseases. J Crohn's Colitis (2016) 10:226–35. 10.1093/ecco-jcc/jjv138 [DOI] [PubMed] [Google Scholar]
- 63.Ambrutzs JM, Walker PD, Larsen CP. The histopathologic spectrum of kidney biopsies in patients with inflammatory bowel disease. Clin J Am Soc Nephrol. (2014) 9:265–70. 10.2215/CJN.04660513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mahmud N, Stinson J, O'Connell MA, Mantle TJ, Keeling PW, Feely J, et al. Microalbuminuria in inflammatory bowel disease. Gut (1994) 35:1599–604. 10.1136/gut.35.11.1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mahmud N, O'Connell MA, Stinson J, Goggins MG, Weir DG, Kelleher D. Tumour necrosis factor-alpha and microalbuminuria in patients with inflammatory bowel disease. Eur J Gastroenterol Hepatol. (1995) 7:215–9. [PubMed] [Google Scholar]
- 66.Cohn A, Sofia AM, Kupfer SS. Type 1 diabetes and celiac disease: clinical overlap and new insights into disease pathogenesis. Curr Diab Rep. (2014) 14:517. 10.1007/s11892-014-0517-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elfström P, Sundström J, Ludvigsson JF. Systematic review with meta-analysis: associations between coeliac disease and type 1 diabetes. Aliment Pharmacol Ther. (2014) 40:1123–32. 10.1111/apt.12973 [DOI] [PubMed] [Google Scholar]
- 68.Goh C, Banerjee K. Prevalence of coeliac disease in children and adolescents with type 1 diabetes mellitus in a clinic based population. Postgrad Med J. (2007) 83:132–6. 10.1136/pgmj.2006.049189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scaramuzza AE1, Mantegazza C, Bosetti A, Zuccotti GV. Type 1 diabetes and celiac disease: The effects of gluten free diet on metabolic control. World J Diabetes (2013) 4:130–4. 10.4239/wjd.v4.i4.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Collin P, Reunala T, Pukkala E, Laippala P, Keyriläinen O, Pasternack A. Coeliac disease–associated disorders and survival. Gut (1994) 35:1215–8. 10.1136/gut.35.9.1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rodrigo L. Celiac disease. World J Gastroenterol. (2006) 12:6585–93. 10.3748/wjg.v12.i41.6585 [DOI] [PubMed] [Google Scholar]
- 72.Collin P, Syrjänen J, Partanen J, Pasternack A, Kaukinen K, Mustonen J. Celiac disease and HLA DQ in patients with IgA nephropathy. Am J Gastroenterol. (2002) 97:2572–6. 10.1111/j.1572-0241.2002.06025.x [DOI] [PubMed] [Google Scholar]
- 73.Ludvigsson JF, Montgomery SM, Olén O, Ekbom A, Ludvigsson J, Fored M. Coeliac disease and risk of renal disease-a general population cohort study. Nephrol Dial Transplant. (2006) 21:1809–15. 10.1093/ndt/gfl117 [DOI] [PubMed] [Google Scholar]
- 74.Welander A, Sundelin B, Fored M, Ludvigsson JF. Increased risk of IgA nephropathy among individuals with celiac disease. J Clin Gastroenterol. (2013) 47:678–83. 10.1097/MCG.0b013e318284792e [DOI] [PubMed] [Google Scholar]
- 75.Nurmi R, Metso M, Pörsti I, Niemelä O, Huhtala H, Mustonen J, et al. Celiac disease or positive tissue transglutaminase antibodies in patients undergoing renal biopsies. Dig Liver Dis. (2018) 50:27–31. 10.1016/j.dld.2017.09.131 [DOI] [PubMed] [Google Scholar]
- 76.Wijarnpreecha K, Thongprayoon C, Panjawatanan P, Thamcharoen N, Pachariyanon P, Nakkala K, et al. Celiac disease and the risk of kidney diseases: a systematic review and meta-analysis. Dig Liver Dis. (2016) 48:1418–24. 10.1016/j.dld.2016.08.115 [DOI] [PubMed] [Google Scholar]
- 77.Cavalcanti Neto MP, Aquino JS, Romão da Silva LF, de Oliveira Silva R, Guimarães KSL, de Oliveira Y, et al. Gut microbiota and probiotics intervention: a potential therapeutic target for management of cardiometabolic disorders and chronic kidney disease? Pharmacol Res. (2018) 130:152–63. 10.1016/j.phrs.2018.01.020 [DOI] [PubMed] [Google Scholar]
- 78.Verna EC, Lucak S. Use of probiotics in gastrointestinal disorders: what to recommend? Therap Adv Gastroenterol. (2010) 3:307–19. 10.1177/1756283X10373814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, et al. Expert consensus document. The international scientific association for probiotics and prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. (2014) 11:506–14. 10.1038/nrgastro.2014.66 [DOI] [PubMed] [Google Scholar]
- 80.Van Immerseel F, Ducatelle R, De Vos M, Boon N, Van De Wiele T, Verbeke K, et al. Butyric acid-producing anaerobic bacteria as a novel probiotic treatment approach for inflammatory bowel disease. J Med Microbiol. (2010) 59:141–3. 10.1099/jmm.0.017541-0 [DOI] [PubMed] [Google Scholar]
- 81.Cani PD, de Vos WM. Next-generation beneficial microbes: the case of akkermansia muciniphila. Front Microbiol. (2017) 8:1765. 10.3389/fmicb.2017.01765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Simenhoff ML, Dunn SR, Zollner GP, Fitzpatrick ME, Emery SM, Sandine WE, et al. Biomodulation of the toxic and nutritional effects of small bowel bacterial overgrowth in end-stage kidney disease using freeze-dried Lactobacillus acidophilus. Miner Electrolyte Metab (1996) 22:92–6. [PubMed] [Google Scholar]
- 83.Ranganathan N1, Friedman EA, Tam P, Rao V, Ranganathan P, Dheer R. Probiotic dietary supplementation in patients with stage 3 and 4 chronic kidney disease: a 6-month pilot scale trial in Canada. Curr Med Res Opin. (2009) 25:1919–30. 10.1185/03007990903069249 [DOI] [PubMed] [Google Scholar]
- 84.Ranganathan N, Ranganathan P, Friedman EA, Joseph A, Delano B, Goldfarb DS, et al. Pilot study of probiotic dietary supplementation for promoting healthy kidney function in patients with chronic kidney disease. Adv Ther. (2010) 27:634–47. 10.1007/s12325-010-0059-9 [DOI] [PubMed] [Google Scholar]
- 85.Vaziri ND, Yuan J, Norris K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am J Nephrol. (2013) 37:1–6. 10.1159/000345969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Di Cerbo A, Pezzuto F, Palmieri L, Rottigni V, Iannitti T, Palmieri B. Clinical and experimental use of probiotic formulations for management of end-stage renal disease: an update. Int Urol Nephrol. (2013) 45:1569–76. 10.1007/s11255-012-0335-5 [DOI] [PubMed] [Google Scholar]
- 87.Soleimani A, Zarrati Mojarrad M, Bahmani F, Taghizadeh M, Ramezani M, et al. Probiotic supplementation in diabetic hemodialysis patients has beneficial metabolic effects. Kidney Int. (2017) 91:435–42. 10.1016/j.kint.2016.09.040 [DOI] [PubMed] [Google Scholar]
- 88.Guida B, Germanò R, Trio R, Russo D, Memoli B, Grumetto L, et al. Effect of short-term synbiotic treatment on plasma p-cresol levels in patients with chronic renal failure: a randomized clinical trial. Nutr Metab Cardiovasc Dis. (2014) 24:1043–9. 10.1016/j.numecd.2014.04.007 [DOI] [PubMed] [Google Scholar]
- 89.Andrade-Oliveira V, Amano MT, Correa-Costa M, Castoldi A, Felizardo RJ, de Almeida DC, et al. Gut bacteria products prevent AKI induced by ischemia-reperfusion. J Am Soc Nephrol. (2015) 26:1877–88. 10.1681/ASN.2014030288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Birt DF, Boylston T, Hendrich S, Jane JL, Hollis J, Li L, et al. Resistant starch: promise for improving human health. Adv Nutr. (2013) 4:587–601. 10.3945/an.113.004325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol. (2017) 18:552–62. 10.1038/ni.3713 [DOI] [PubMed] [Google Scholar]
- 92.Shen L, Keenan MJ, Raggio A, Williams C, Martin RJ. Dietary-resistant starch improves maternal glycemic control in Goto-Kakizaki rat. Mol Nutr Food Res. (2011) 55:1499–508. 10.1002/mnfr.201000605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smazal AL, Borcherding NC, Anderegg AS, Schalinske KL, Whitley EM, Rowling MJ. Dietary resistant starch prevents urinary excretion of 25-hydroxycholecalciferol and vitamin D-binding protein in type 1 diabetic rats. J Nutr. (2013) 143:1123–8. 10.3945/jn.112.173278 [DOI] [PubMed] [Google Scholar]
- 94.Vaziri ND, Liu SM, Lau WL, Khazaeli M, Nazertehrani S, Farzaneh SH, et al. High amylose resistant starch diet ameliorates oxidative stress, inflammation, and progression of chronic kidney disease. PLoS ONE (2014) 9:e114881. 10.1371/journal.pone.0114881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kieffer DA, Piccolo BD, Vaziri ND, Liu S, Lau WL, Khazaeli M, et al. Resistant starch alters gut microbiome and metabolomic profiles concurrent with amelioration of chronic kidney disease in rats. Am J Physiol Renal Physiol. (2016) 310:F857–71. 10.1152/ajprenal.00513.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sirich TL, Plummer NS, Gardner CD, Hostetter TH, Meyer TW. Effect of increasing dietary fiber on plasma levels of colon-derived solutes in hemodialysis patients. Clin J Am Soc Nephrol. (2014) 9:1603–10. 10.2215/CJN.00490114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moraes C, Borges NA, Mafra D. Resistant starch for modulation of gut microbiota: Promising adjuvant therapy for chronic kidney disease patients? Eur J Nutr. (2016) 55:1813–21. 10.1007/s00394-015-1138-0 [DOI] [PubMed] [Google Scholar]
- 98.Allegretti J1, Eysenbach LM, El-Nachef N, Fischer M, Kelly C, Kassam Z. The current landscape and lessons from fecal microbiota transplantation for inflammatory bowel disease: past, present, and future. Inflamm Bowel Dis. (2017) 23:1710–7. 10.1097/MIB.0000000000001247 [DOI] [PubMed] [Google Scholar]
- 99.Singh R, van Nood E, Nieuwdorp M, van Dam B, ten Berge IJ, Geerlings SE, et al. Donor feces infusion for eradication of extended spectrum beta-lactamase producing Escherichia coli in a patient with end stage renal disease. Clin Microbiol Infect. (2014) 20:O977–8. 10.1111/1469-0691.12683 [DOI] [PubMed] [Google Scholar]
- 100.Lahtinen P, Mattila E, Anttila VJ, Tillonen J, Teittinen M, Nevalainen P, et al. Faecal microbiota transplantation in patients with Clostridium difficile and significant comorbidities as well as in patients with new indications: a case series. World J Gastroenterol. (2017) 23:7174–84. 10.3748/wjg.v23.i39.7174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Han JL, Lin HL. Intestinal microbiota and type 2 diabetes: from mechanism insights to therapeutic perspective. World J Gastroenterol. (2014) 20:17737–45. 10.3748/wjg.v20.i47.17737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Khajah MA. The potential role of fecal microbiota transplantation in the treatment of inflammatory Bowel disease. Scand J Gastroenterol. (2017) 52:1172–84. 10.1080/00365521.2017.1347812 [DOI] [PubMed] [Google Scholar]
- 103.Lassenius MI, Fogarty CL, Blaut M, Haimila K, Riittinen L, Paju A, et al. Intestinal alkaline phosphatase at the crossroad of intestinal health and disease - a putative role in type 1 diabetes. J Intern Med. (2017) 281:586–600. 10.1111/joim.12607 [DOI] [PubMed] [Google Scholar]
- 104.Lorenz B, Schröder HC. Mammalian intestinal alkaline phosphatase acts as highly active exopolyphosphatase. Biochim Biophys Acta (2001) 1547:254–61. 10.1016/S0167-4838(01)00193-5 [DOI] [PubMed] [Google Scholar]
- 105.Bilski J, Mazur-Bialy A, Wojcik D, Zahradnik-Bilska J, Brzozowski B, Magierowski M, et al. The role of intestinal alkaline phosphatase in inflammatory disorders of gastrointestinal tract. Mediators Inflamm. (2017) 2017:9074601. 10.1155/2017/9074601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bol-Schoenmakers M, Fiechter D, Raaben W, Hassing I, Bleumink R, Kruijswijk D, et al. Intestinal alkaline phosphatase contributes to the reduction of severe intestinal epithelial damage. Eur J Pharmacol. (2010) 633:71–7. 10.1016/j.ejphar.2010.01.023 [DOI] [PubMed] [Google Scholar]
- 107.Alam SN, Yammine H, Moaven O, Ahmed R, Moss AK, Biswas B, et al. Intestinal alkaline phosphatase prevents antibiotic-induced susceptibility to enteric pathogens. Ann Surg. (2014) 259:715–22. 10.1097/SLA.0b013e31828fae14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang WH, Heithoff DM, Aziz PV, Sperandio M, Nizet V, Mahan MJ, et al. Recurrent infection progressively disables host protection against intestinal inflammation. Science (2017) 358:eaao5610. 10.1126/science.aao5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kaliannan K, Hamarneh SR, Economopoulos KP, Nasrin Alam S, Moaven O, Patel P, et al. Intestinal alkaline phosphatase prevents metabolic syndrome in mice. Proc Natl Acad Sci USA. (2013) 110:7003–8. 10.1073/pnas.1220180110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Estaki M, DeCoffe D, Gibson DL. Interplay between intestinal alkaline phosphatase, diet, gut microbes and immunity. World J Gastroenterol. (2014) 20:15650–6. 10.3748/wjg.v20.i42.15650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lukas M, Drastich P, Konecny M, Gionchetti P, Urban O, Cantoni F, et al. Exogenous alkaline phosphatase for the treatment of patients with moderate to severe ulcerative colitis. Inflamm Bowel Dis. (2010) 16:1180–6. 10.1002/ibd.21161 [DOI] [PubMed] [Google Scholar]
- 112.Peters E, Heemskerk S, Masereeuw R, Pickkers P. Alkaline phosphatase: a possible treatment for sepsis-associated acute kidney injury in critically ill patients. Am J Kidney Dis. (2014) 63:1038–48. 10.1053/j.ajkd.2013.11.027 [DOI] [PubMed] [Google Scholar]
- 113.Borycka-Kiciak K, Banasiewicz T, Rydzewska G. Butyric acid - a well-known molecule revisited. Prz Gastroenterol. (2017) 12:83–9. 10.5114/pg.2017.68342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Machado RA, Constantino Lde S, Tomasi CD, Rojas HA, Vuolo FS, Vitto MF, et al. Sodium butyrate decreases the activation of NF-κB reducing inflammation and oxidative damage in the kidney of rats subjected to contrast-induced nephropathy. Nephrol Dial Transplant. (2012) 27:3136–40. 10.1093/ndt/gfr807 [DOI] [PubMed] [Google Scholar]
- 115.Khan S, Jena G. Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-β1-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem Toxicol. (2014) 73:127–39. 10.1016/j.fct.2014.08.010 [DOI] [PubMed] [Google Scholar]
- 116.Huang W, Zhou L, Guo H, Xu Y, Xu Y. The role of short-chain fatty acids in kidney injury induced by gut-derived inflammatory response. Metabolism (2017) 68:20–30. 10.1016/j.metabol.2016.11.006 [DOI] [PubMed] [Google Scholar]
- 117.Gutzeit C, Magri G, Cerutti A. Intestinal IgA production and its role in host-microbe interaction. Immunol Rev. (2014) 260:76–85. 10.1111/imr.12189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Malo MS. A high level of intestinal alkaline phosphatase is protective against type 2 diabetes mellitus irrespective of obesity. EBioMedicine (2015) 2:2016–23. 10.1016/j.ebiom.2015.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Molnár K, Vannay A, Sziksz E, Bánki NF, Gyorffy H, Arató A, et al. Decreased mucosal expression of intestinal alkaline phosphatase in children with coeliac disease. Virchows Arch. (2012) 460:157–61. 10.1007/s00428-011-1188-5 [DOI] [PubMed] [Google Scholar]
- 120.Molnár K, Vannay A, Szebeni B, Bánki NF, Sziksz E, Cseh A, et al. Intestinal alkaline phosphatase in the colonic mucosa of children with inflammatory bowel disease. World J Gastroenterol. (2012) 18:3254–9. 10.3748/wjg.v18.i25.3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ramasamy S, Nguyen DD, Eston MA, Alam SN, Moss AK, Ebrahimi F, et al. Intestinal alkaline phosphatase has beneficial effects in mouse models of chronic colitis. Inflamm Bowel Dis. (2011) 17:532–42. 10.1002/ibd.21377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Parlato M, Charbit-Henrion F, Pan J, Romano C, Duclaux-Loras R, Le Du MH, et al. Human ALPI deficiency causes inflammatory bowel disease and highlights a key mechanism of gut homeostasis. EMBO Mol Med. (2018) 10:e8483. 10.15252/emmm.201708483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ishige T, Itoga S, Utsuno E, Nishimura M, Yoshikawa M, Kato N, et al. Variant in C-terminal region of intestinal alkaline phosphatase associated with benign familial hyperphosphatasaemia. J Med Genet. (2018). 10.1136/jmedgenet-2017-104964. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 124.Pickkers P, Heemskerk S, Schouten J, Laterre PF, Vincent JL, Beishuizen A, et al. Alkaline phosphatase for treatment of sepsis-induced acute kidney injury: a prospective randomized double-blind placebo-controlled trial. Crit Care (2012) 16:R14. 10.1186/cc11159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Peters E, Masereeuw R, Pickkers P. The potential of alkaline phosphatase as a treatment for sepsis-associated acute kidney injury. Nephron Clin Pract. (2014) 127:144–8. 10.1159/000363256 [DOI] [PubMed] [Google Scholar]
- 126.Peters E, Ergin B, Kandil A, Gurel-Gurevin E, van Elsas A, Masereeuw R, et al. Effects of a human recombinant alkaline phosphatase on renal hemodynamics, oxygenation and inflammation in two models of acute kidney injury. Toxicol Appl Pharmacol. (2016) 313:88–96. 10.1016/j.taap.2016.10.015 [DOI] [PubMed] [Google Scholar]
- 127.Lee C, Chun J, Hwang SW, Kang SJ, Im JP, Kim JS. The effect of intestinal alkaline phosphatase on intestinal epithelial cells, macrophages and chronic colitis in mice. Life Sci. (2014) 100:118–24. 10.1016/j.lfs.2014.02.003 [DOI] [PubMed] [Google Scholar]
- 128.Hwang SW, Kim JH, Lee C, Im JP, Kim JS. Intestinal alkaline phosphatase ameliorates experimental colitis via toll-like receptor 4-dependent pathway. Eur J Pharmacol. (2018) 820:156–66. 10.1016/j.ejphar.2017.12.026 [DOI] [PubMed] [Google Scholar]
- 129.Oscarson M, Burk O, Winter S, Schwab M, Wolbold R, Dippon J, et al. Effects of rifampicin on global gene expression in human small intestine. Pharmacogenet Genomics (2007) 17:907–18. 10.1097/FPC.0b013e3280143dfc [DOI] [PubMed] [Google Scholar]
- 130.Melo AD, Silveira H, Bortoluzzi C, Lara LJ, Garbossa CA, Preis G, et al. Intestinal alkaline phosphatase and sodium butyrate may be beneficial in attenuating LPS-induced intestinal inflammation. Genet Mol Res. (2016) 15:gmr15048875. 10.4238/gmr15048875 [DOI] [PubMed] [Google Scholar]
- 131.Nickel KF, Long AT, Fuchs TA, Butler LM, Renne T. Factor XII as a therapeutic target in thromboembolic and inflammatory diseases. Arterioscler Thromb Vasc Biol. (2017) 37:13–20. 10.1161/ATVBAHA.116.308595 [DOI] [PubMed] [Google Scholar]
- 132.Maurer M, Magerl M, Ansotegui I, Aygören-Pürsün E, Betschel S, Bork K, et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2017 revision and update. Allergy (2018) 73:1575–96. 10.1111/all.13384 [DOI] [PubMed] [Google Scholar]
- 133.Cichon S, Martin L, Hennies HC, Müller F, Van Driessche K, Karpushova A, et al. Increased activity of coagulation factor XII (Hageman factor) causes hereditary angioedema type III. Am J Hum Genet. (2006) 79:1098–104. 10.1086/509899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Deroux A, Boccon-Gibod I, Fain O, Pralong P, Ollivier Y, Pagnier A, et al. Hereditary angioedema with normal C1 inhibitor and factor XII mutation: a series of 57 patients from the French National Center of Reference for Angioedema. Clin Exp Immunol. (2016) 185:332–7. 10.1111/cei.12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dewald G. A missense mutation in the plasminogen gene, within the plasminogen kringle 3 domain, in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. (2018) 498:193–8. 10.1016/j.bbrc.2017.12.060 [DOI] [PubMed] [Google Scholar]
- 136.Bork K, Wulff K, Steinmüller-Magin L, Braenne I, Staubach-Renz P, Witzke G, et al. Hereditary angioedema with a mutation in the plasminogen gene. Allergy (2018) 73:442–50. 10.1111/all.13270 [DOI] [PubMed] [Google Scholar]
- 137.Bork K, Staubach P, Eckardt AJ, Hardt J. Symptoms, course, and complications of abdominal attacks in hereditary angioedema due to C1 inhibitor deficiency. Am J Gastroenterol. (2006) 101:619–27. 10.1111/j.1572-0241.2006.00492.x [DOI] [PubMed] [Google Scholar]
- 138.Craig TJ, Bernstein JA, Farkas H, Bouillet L, Boccon-Gibod I. Diagnosis and treatment of bradykinin-mediated angioedema: outcomes from an angioedema expert consensus meeting. Int Arch Allergy Immunol. (2014) 165:119–27. 10.1159/000368404 [DOI] [PubMed] [Google Scholar]
- 139.Ali MA, Borum ML. Hereditary angioedema: what the gastroenterologist needs to know. Clin Exp Gastroenterol. (2014) 7:435–45. 10.2147/CEG.S50465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wilin KL, Czupryn MJ, Mui R, Renno A, Murphy JA. ACE inhibitor-induced angioedema of the small bowel: a case report and review of the literature. J Pharm Pract. (2018) 31:99–103. 10.1177/0897190017690641 [DOI] [PubMed] [Google Scholar]
- 141.Mansfield KE, Douglas IJ, Nitsch D, Thomas SL, Smeeth L, Tomlinson LA. Acute kidney injury and infections in patients taking antihypertensive drugs: a self-controlled case series analysis. Clin Epidemiol. (2018) 10:187–202. 10.2147/CLEP.S146757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Plaza J, Malasit P, Kerr DN. Hereditary angio-oedema with mesangiocapillary glomerulonephritis. Postgrad Med J. (1977) 53:627–30. 10.1136/pgmj.53.624.627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hory B, Haultier JJ. Glomerulonephritis and hereditary angioedema: report of 2 cases. Clin Nephrol. (1989) 31:259–63. [PubMed] [Google Scholar]
- 144.Nomura H, Tsugawa Y, Koni I, Tofuku Y, Mabuchi H, Takeda R, et al. Hereditary angioedema complicated with chronic renal failure: report of sibling cases. Intern Med. (1992) 31:94–7. 10.2169/internalmedicine.31.94 [DOI] [PubMed] [Google Scholar]
- 145.Srinivasan J, Beck P. IgA nephropathy in hereditary angioedema. Postgrad Med J. (1993) 69:95–9. 10.1136/pgmj.69.808.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Van Bommel EF, Ouwehand AJ, Mulder AH, Weimar W. Mesangiocapillary glomerulonephritis associated with hereditary angioedema. Nephron (1995) 69:178–9. 10.1159/000188441 [DOI] [PubMed] [Google Scholar]
- 147.Ohsawa I, Satomura A, Fuke Y, Hidaka M, Endo M, Fujita T, et al. Worsening fluid retention in a patient with hereditary angioedema and end-stage renal disease. Intern Med. (2004) 43:708–12. 10.2169/internalmedicine.43.708 [DOI] [PubMed] [Google Scholar]
- 148.Majoni SW, Smith SR. Membranous nephropathy in a patient with hereditary angioedema: a case report. J Med Case Rep. (2008) 2:328. 10.1186/1752-1947-2-328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gallais Sérézal I, Bouillet L, Dhôte R, Gayet S, Jeandel PY, Blanchard-Delaunay C, et al. Hereditary angioedema and lupus: a French retrospective study and literature review. Autoimmun Rev. (2015) 14:564–8. 10.1016/j.autrev.2015.02.001 [DOI] [PubMed] [Google Scholar]
- 150.Zeitlin IJ, Smith AN. Mobilization of tissue kallikrein in inflammatory disease of colon. Gut (1973) 14:133–8. 10.1136/gut.14.2.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Devani M, Cugno M, Vecchi M, Ferrero S, Di Berardino F, Avesani EC, et al. Kallikrein-kinin system activation in Crohn's disease: Differences in intestinal and systemic markers. Am J Gastroenterol. (2002) 97:2026–32. 10.1111/j.1572-0241.2002.05919.x [DOI] [PubMed] [Google Scholar]
- 152.Devani M, Vecchi M, Ferrero S, Avesani EC, Arizzi C, Chao L, et al. Kallikrein-kinin system in inflammatory bowel diseases: intestinal involment and correlation with degree of tissue inflammation. Dig Liv Dis. (2005) 37:665–73. 10.1016/j.dld.2005.01.021 [DOI] [PubMed] [Google Scholar]
- 153.Gäbele E, Dostert K, Hofmann C, Wiest R, Schölmerich J, Hellerbrand C, et al. DSS induced colitis inreases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J Hepatol. (2011) 55:1391–99. 10.1016/j.jhep.2011.02.035 [DOI] [PubMed] [Google Scholar]
- 154.Lu F, Fernandes SM, Davis AE. The role of the complement and contact systems in the dextran sulfate sodium-induced colitis model: the effect of C1 inhibitor in inflammatory bowel disease. Am J Gastrointest Liver Physiol. (2010) 298:G878–83. 10.1152/ajpgi.00400.2009 [DOI] [PubMed] [Google Scholar]
- 155.Arai Y, Takanashi H, Kitagawa H, Wirth KJ, Okayasu I. Effect of icatibant, a bradykinin B2 receptor antagonist, on the development of experimental ulcerative colitis in mice. Dig Dis Sci. (1999) 44:845–51. 10.1023/A:1026694732602 [DOI] [PubMed] [Google Scholar]
- 156.Hara DB, Leite DF, Fernandes ES, Passos GF, Guimarães AO, Pesquero JB, et al. The relevance of kinin B1 receptor upregulation in a mouse model of colitis. Br J Pharmacol. (2008) 154:1276–86. 10.1038/bjp.2008.212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Marcon R, Claudino RF, Dutra RC, Bento AF, Schmidt EC, Bouzon ZL, et al. Exacerbation of DSS-induced colitis in mice lacking B1 receptors through compensatory up-regulation of kinin B2 reseptors: the role of tight juctions and intestinal homeostasis. Br J Pharmacol. (2013) 168:389–402. 10.1111/j.1476-5381.2012.02136.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Stadnicki A, DeLa Cadena RA, Sartor RB, Bender D, Kettner CA, Rath HC, et al. Selective plasma kallikrein inhibitor attenuates acute intestinal inflammation in Lewis rat. Dig Dis Sci. (1996) 41:912–20. 10.1007/BF02091530 [DOI] [PubMed] [Google Scholar]
- 159.Stadnicki A, Sartor RB, Janardham R, Majluf-Cruz A, Kettner CA, Adam AA, et al. Specific inhibition of plasma kallikrein modulates chronic granulomatous intestinal and systemic inflammation in genetically susceptible rats. FASEB J. (1998) 12:325–33. 10.1096/fasebj.12.3.325 [DOI] [PubMed] [Google Scholar]
- 160.Isordia-Salas I, Pixley RA, Li F, Sainz I, Sartor RB, Adam A, et al. Kininogen deficiency modulates chronic intestinal inflammation in genetically susceptible rats. Am J Physiol Gastrointest Liver Physiol. (2002) 283:G180–6. 10.1152/ajpgi.00514.2001 [DOI] [PubMed] [Google Scholar]
- 161.Wang B, Yang A, Zhao Z, He C, Liu Y, Colman RW, et al. The plasma kallikrein-kininogen pathway is critical in the pathogenesis of colitis in mice. Front Immunol. (2018) 9:21. 10.3389/fimmu.2018.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ranganathan P, Jayakumar C, Santhakumar M, Ramesh G. Netrin-1 regulates colon-kidney cross talk through suppression of IL-6 function in a mouse model of DSS-colitis. Am J Physiol Renal Physiol. (2013) 304:F1187–97. 10.1152/ajprenal.00702.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ranganathan P, Jayakumar C, Manicassamy S, Ramesh G. CXCR2 knockout mice are protected against DSS-colitis-induced acute kidney injury and inflammation. Am J Physiol Renal Physiol. (2013) 305:F1422–7. 10.1152/ajprenal.00319.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tomita H, Sanford RB, Smithies O, Kakoki M. The kallikrein-kinin system in diabetic nephropathy. Kidney Int. (2012) 81:733–44. 10.1038/ki.2011.499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Abdulaal M, Haddad NM, Sun JK, Silva PS. The role of plasma kallikrein-kinin pathway in the development of diabetic retinopathy: pathophysiology and therapeutic approaches. Semin Ophthalmol. (2016) 31:19–24. 10.3109/08820538.2015.1114829 [DOI] [PubMed] [Google Scholar]