

Abstract
We compared different methods (absorbance, fluorescent dye-binding, and digital PCR) for measuring the concentrations of human genomic DNA from cultured cells and absorbance measurements of a synthetic DNA oligonucleotide. NIST Standard Reference Material (SRM) 2082, a pathlength absorbance standard, was used to benchmark the absorbance measurements done with microvolume spectrophotometers and a microvolume plate reader. Control absorbance values were measured on a high accuracy spectrophotometer and a NIST calibrated pathlength cuvette. Measurements of the human genomic DNA sample were done with several types of fluorescent dye binding assays using different DNA calibrators. The fluorescent dye binding methods gave different results for genomic DNA depending upon the type of DNA calibrator and the fluorescent dye that was used. The human genomic DNA sample was also characterized by using six different droplet digital PCR assays (amplicons located on different chromosomes) to measure the average copy number. Conversion of the digital PCR data to copy numbers was sensitive to the droplet size used for calculations and conversion to mass concentration was dependent upon the molecular weight of the human genome used for the calculations. The results from the different methods were compared and the caveats for each measurement method were discussed.
Keywords: absorbance, digital PCR, fluorescent dye binding, genomic DNA, human, measurements, standard
Accurate and reproducible measurements of the concentrations of human genomic DNA samples are essential to achieve reliable results obtained by PCR and genomic sequencing methods (1). Accurate determination of the concentration of human genomic DNA is an essential task, but to achieve it requires an understanding of the limitations of the methods used and the sample preparation methods needed to achieve reproducible results. This task is complicated by the chemical and physical complexity of genomic DNA and the different methods used to prepare the samples. Storage conditions can change the physical properties of the materials, which can influence how the samples behave with different measurement methods (2).
Nucleic acid concentrations are routinely measured by absorbance, fluorescent dye-binding, and digital PCR methods. The simplest and most rapid method is arguably direct absorbance measurements of nucleic acids at 260 nm using relatively simple instruments (3–5). New spectrophotometers that utilize short pathlengths can measure the absorbance of microliter sized samples to conserve samples. Sensitive assays (compared to absorbance methods) using fluorescent dyes have been developed to specifically measure double-stranded or single-stranded nucleic acids. Digital PCR is rapidly becoming an important method because of its sensitivity, specificity, and dynamic range for a variety of nucleic acid samples (6). Researchers are investigating digital PCR to determine the best ways to reliably quantify the concentration of DNA samples. The accessibility of some human genomic DNA targets can be increased by restriction enzyme fragmentation, but treatments can also reduce the number of targets in some cases (7). NIST has developed methods to more accurately measure the targets per volume (8) and the droplet volumes (9). Control samples and reference materials are necessary to ensure that the sample processing, analytical methods, and instruments are working correctly (7,8,10,11).
DNA derived from human cell lines that have been modified to immortalize the cells are renewable sources of materials. The NIST-led public–private consortium, Genome in a Bottle, is developing human genomic DNA reference materials (derived from cell lines) as well-annotated reference materials for benchmarking DNA sequencing methods (12).
Our goals in this study were to compare the different methods for measuring the concentration of nucleic acids for in-house DNA control materials and to identify the sources of variability in these measurements.
Materials and Methods
Samples
A DNA oligonucleotide (10 micromole scale) was ordered from Eurofins MWG Operon LLC (Louisville, KY) with the sequence of TCCTCAAGGCTAGCACTGTTC (21 bases, 52.4 % GC content) and a molecular weight of 6,357.2. The dried salt-free oligonucleotide was dissolved in 50 ml of a buffer composed of 10 mmol/L TRIS 1 mmol/L EDTA pH 8.0 (TE buffer) to make samples referred to as the high concentration oligo-nucleotide (Oligo High). A portion of the Oligo High sample (10 ml) was diluted to a final volume of 40 ml using TE buffer to produce the solution (Oligo Low). The oligo-nucleotide samples were dispensed into tubes and stored at −20°C. These samples were brought to ambient temperature (approximately 30 min.) and then mixed by vortexing to achieve a uniform solution. The thawed samples can be kept at 4°C for at least 1 month.
Human genomic DNA (gDNA) was obtained from Coriell Institute for Medical Research (Camden, NJ). The purified male human DNA (NA24385) was produced from cell line GM24385 by Coriell Institute for Medical Research. The NIST Human Subject Protection Office reviewed and approved the use of this human cell line derived material. NIST reference material (RM) 8391 is produced from the same cell line source, but our samples were obtained directly from Coriell Institute for Medical Research. The concentrated DNA stock solution was diluted tenfold with TE buffer and the resulting solutions (approximately 50 μg/ml) were distributed into individual tubes (0.05 ml) and stored at 4°C. The sample was gently mixed to aid in obtaining uniform measurements.
Reference values of SRM 2082, human genomic DNA, and synthetic oligonucleotides
NIST Standard Reference Material (SRM) 2082 has three solutions: a blank consisting of 10 mmol/L 2-amino-2-hydroxymethyl-propane-1,3 diol, pH 8.0 buffer (TRIS buffer); tryptophan in TRIS buffer; and uracil in TRIS buffer. SRM 2082 was stored at −20°C. It was brought to ambient temperature by placing the vials at room temperature (approximately 30 min) and then suspended by inversion of the vials at least 20 times to ensure a uniform solution. The suspended solutions can be stored at 4°C for at least 3 months. The reference values of the tryptophan and uracil solution of NIST SRM 2082 were measured using NIST calibrated cuvettes (0.1 mm to 2 mm) using a Cary 6000i dual beam spectrophotometer with a spectral bandwidth of 0.8 nm and a temperature of 22°C (13). Samples were scanned from 340 nm to 240 nm (1 nm resolution) and the buffer blanks were subtracted from the samples. The absorbance values for the Oligo Low concentration, the Oligo High concentration, and the human genomic DNA (gDNA) samples were measured using three separate samples in NIST calibrated cuvettes (0.5115 mm) at a temperature of 22°C and spectral band width of 0.8 nm on a Cary 6000i spectrophotometer and a Perkin Elmer Lambda 900 s pectrophotometer.
Microvolume spectrophotometers and cuvettes
The samples were run on different microliter volume (MV) spectrophotometers, including a Nanodrop One C (Thermo Scientific, Willington, DE, USA), UV5Nano (Mettler Toledo, Columbus, OH, USA), and NanoPhometer NP-80 (IMPLEN, Westlake Village, CA, USA). The instruments were operated at ambient temperature (22°C) using the default settings. The MV spectrophotometers were checked to ensure that they were operating according to the specifications of the manufacturer.
A BioTek Synergy MX plate reader with a Take3 microvolume plate was used according to the operations and calibration procedures provide by the manufacturer. A basic system test was run and evaluated to confirm full operation of the reader’s motors, lamp, the PMT, and various subsystems. A calibrated absorbance test plate was used to confirm mechanical alignment, including optical density accuracy, linearity, repeatability, and wavelength accuracy. An absorbance liquid test was performed to confirm repeatability and alignment of the reader when a solution is used in a microplate. Briefly, 2 μl of respective buffer was loaded onto the microspot slide and blanked. Both the microspot slide and top slide were cleaned using a dry laboratory wipe and DI water. Samples of interest were loaded onto the microspot slide and absorbance measured using the recommended wavelengths relative to sample type. All values were imported via the Gen5 software and exported into Excel for analysis. Corrected pathlength and background values were used to calculate normalized absorbance.
Fluorescent dye binding measurements
A compact fluorimeter (Qubit 3.0, Thermo Fisher Scientific) was used with the PicoGreen or proprietary dyes with similar fluorescence properties. The Qubit DNA Broad Range assay (Thermo Fisher #Q32853, 2 ng to 1000 ng) Assay Kit or the Qubit High Sensitivity assay (Thermo Fisher # Q32851, 0.2 ng to 100 ng) was used according to the manufacturer’s protocols. Briefly, 190 μl of a Qubit working solution was added to each 0.5 ml Qubit tube, and then 10 μl of each sample was added and mixed. A blank and a lambda DNA sample in TE buffer were used as the single point standard. A calf thymus DNA standard (10 μl of 10 ng/μl) from the AccuGreen kit (below) was used with the high sensitivity assay for comparison.
A SpectraMax Quant AccuBlue HiRange (2 ng to 2000 ng) dsDNA Assay Kit (Cat# R8359, Molecular Devices, Sunnyvale, CA, USA) was used with a BioTek Synergy MX microplate reader with excitation at 350 nm and emission at 460 nm. In brief, 10 μl of each dsDNA standard or unknown DNA sample was added to a well in a black, 96-well microplate, and then 200 μl of the working solution was added and mixed. A set of dilutions of calf thymus dsDNA was used to generate the standard curve, which was used to calculate the concentrations of the unknown DNA samples.
The fluorescent dye binding assay kits were obtained from Biotium, Inc. (Fremont, CA, USA). AccuBlue High Sensitivity Double Stranded DNA kit (Excitation 485 nm and Emission 530 nm, 0.2 ng to 100 ng) and AccuClear Ultra High Sensitivity Double Stranded DNA kit (Excitation 468 nm and Emission 507 nm, 0.03 ng to 250 ng) were used with a BioTek Synergy MX plate reader and 96-well plates. AccuGreen High Sensitivity double-stranded DNA kit (Excitation 502 nm and Emission 523 nm, 0.1 ng to 100 ng) was used with the compact fluorimeter (Qubit 3.0, Thermo Fisher Scientific). The Biotium kits were supplied with calf thymus DNA samples as the standard.
Quantification of RNA
Possible RNA contamination in the purified genomic DNA was assessed using the compact fluorimeter (Qbit 3.0). A Qubit® RNA HS (high sensitivity, 5 ng to 100 ng) Assay Kit (Cat# Q32852, Thermo Fisher Scientific) was used according to the manufacturer’s protocols. Briefly, 190 μl of a Qubit working solution was added to each 0.5 ml Qubit assay tube and then 10 μl of each standard or unknown DNA sample was added and mixed. RNA standards (0 ng and 10 ng, supplied by the kit manufacturer) in TE buffer were used as standards.
Genomic DNA size determination
The size of purified genomic DNA was evaluated using a Fragment Analyzer Automated CE System (AATI, Ankeny, IA, USA) with a High Sensitivity Large Fragment 50 Kb Analysis Kit (Cat# DNF464, AATI). A 2 μl (1 ng/μl) genomic DNA sample was loaded into each well and used to determine the size distribution according to the manufacturer’s software.
Quantification of DNA using droplet dPCR
The sequences of the primers and probes for the digital PCR assays developed at NIST for measuring EGFR, MET, EIF5B, DCK, PMM1, and RPS27A gene copies, and chromosomal locations are shown in Table 1. The amplicon lengths and % GC contents of the amplicons for the gene targets EGFR, MET, EIF5B, DCK, PMM1, and RPS27A are 112 base pairs (40.2%), 91 base pairs (47.3%), 112 base pairs (45.5%), 122 base pairs (43.4%), 78 base pairs (53.8%), and 97 base pairs (43.3%), respectively. A QX200 Droplet Digital PCR system (Bio-Rad, Pleasanton, CA, USA) was used. The TaqMan PCR reaction mixture consists of 1x Droplet dPCR Supermix for probes (no dUTP, Bio-Rad), 900 nmol/L primers, and 250 nmol/L probe (final concentrations), and approximately 20 ng of genomic DNA template or non-template control in a total volume of 25 μl. 20 μl of the 25 μl droplet dPCR reaction mixture was transferred to the droplet generator DG8 cartridge. After droplet generation, 40 μl of the generated droplet emulsion was transferred to a new 96-well PCR plate (Eppendorf). The plate was placed on an Applied Biosystems ProFlex PCR System and amplified using the following thermal cycling conditions: 95°C for 10 min, followed by 40 cycles of 94°C for 30 s and 60°C for 1 min, then 98°C for 10 min, with the temperature ramp rate at 50% (2°C/second). After PCR, the 96-well PCR plate was loaded onto the QX200 droplet digital reader. The results are calculated in copies/μl based on a Poisson distribution curve using QuantaSoft software (ver 1.7.4.0917) with a droplet size of 0.85 nl. NIST measurements on the droplet size formed with the BioRad Supermix for probes (no dUTP) had a value of 0.7681 nl (expanded uncertainty 2.3%, k=2) with the method described in a NIST Special Publication 260–184 (9). More recent measurements using the same method and type of master mix but a different lot of master mix had a value 0.7349 nl (expanded uncertainty 2.3%, k = 2) (14). Additional measurements are in progress. A one-way ANOVA was performed on the data to test the hypothesis that the average mean values across the digital PCR assays were equal. A Newman-Keuls multiple comparison test was used to perform pairwise comparisons. Statistics were performed using WINKS SDA Software (Texasoft, Cedar Hill, TX, USA). Statistical decisions were made at p = 0.05. The mean copy numbers and standard deviations for the DCK, EGFR, EIF5B, MET, PMM1, and the RP27A assays were 16,366.8 (SD 783.4, N = 27), 15,606.6 (705.6, N = 27), 16,476.8 (587.9, N = 27), 16,104.2 (825.1, N = 27), 16,849.4 (502.2, N = 27), and 17,062.0 (685.9), respectively.
Table 1.
PCR probes and primers for the six genes using droplet digital PCR assays. Values in parentheses are the percentage of GC content.
| Gene (probe, primers) | Sequence 5’ - 3’ | Human chromosome (location, GRCh38.p7) |
|---|---|---|
| EGFR (probe) | FAM- TGCTCTTAAAGGGATATCCTCTCCTGGT -BHQ-1 (46.4%) | 7 (55109660 to 55109687) |
| MET (probe) | FAM- CCTAGAGTGTGGGTTGGCCTTCCTA-BHQ-1 (60.9%) | 7 (11625065 to 116725087) |
| EIF5 (probe) | FAM-TTCAGCCTTCTCTTCTCATGCAGTTGTCAG-BHQ-1 (46.7%) | 2 (99357710 to 99357739) |
| RPS27A (probe) | FAM-TTTGTCTACCACTTGCAAAGCTGGCCTTT-BHQ-1 (44.8%) | 2(55235204–55235232) |
| DCK (probe) | FAM-CCTTCCAAACATATGCCTGTCTCAGTCGA-BHQ-1 (48.3%) | 4 (71022443 to 71022471) |
| PMM1 (probe) | FAM-CAAATCACCTGAGGTCAAGGCCAGAACA-BHQ-1 (50%) | 22 (41577733 to 41577706) |
| EGFR (forward) | ACCTTTGCAGAGAGGCTTAAT (42.9%) | 7 (55109631 to 55109651) |
| EGFR (reverse) | CCTAGGCCCAAAGGAATGATAG (50%) | 7 (55109742 to 55109721) |
| MET (forward) | TGGGCATGCTCATTCTTCTT (45%) | 7 (11625023 to 116725042) |
| MET (reverse) | CATCATACTTCTTACGTACAGGCA (41.7%) | 7 (11625113 to 116725090) |
| EIF5 (forward) | GGCCGATAAATTTTTGGAAATG (36.4%) | 2 (99357677 to 99357798) |
| EIF5 (reverse) | GGAGTATCCCCAAAGGCATCT (52.4%) | 2 (99357677 to 99357788) |
| DCK (forward) | CTCAGAAAAATGGTGGGAATGTT (39.1%) | 4 (71022380 to 71022402) |
| DCK (reverse) | GCCATTCAGAGAGGCAAGCT (55%) | 4 (71022501 to 71022482) |
| RPS27A (forward) | CGGGTTTGGGTTCAGGTCTT (55%) | 2(55235180–55235199) |
| RPS27A (reverse) | TGCTACAATGAAAACATTCAGAAGTCT (33.3%) | 2 (55235180–55235276) |
| PMM1 (forward) | AGGTCTGGTGGCTTCTCCAAT (52.4%) | 22 (415755 to 41577735) |
| PMM1 (reverse) | CCCCTAAGAGGTCTGTTGTGTTG (52.2%) | 22 (41577678 to 41577700) |
The amount of mitochondrial DNA present in the human genomic DNA sample was determined using droplet digital PCR assays. Two assays for nuclear genomic DNA were NEIF and ND6 (7) and two assays for mitochondrial DNA assays were mtND1 and mtBatz (15,16). The annealing temperature used was 62°C. At this temperature the mitochondrial assays amplify mitochondrial DNA only. The ratios of mitochondrial DNA copies to nuclear DNA copies were calculated from the results of these assays.
Results and Discussion
Absorbance values of the samples
The control absorbance values for the human genomic DNA, the Oligo Low concentration, and the Oligo High concentration samples were measured using high accuracy dual-beam spectrophotometers, a 0.5115 mm cuvette, and normalized to 1 cm (Table 2). The values for the tryptophan and uracil solutions (SRM 2082) are the NIST-certified values normalized to a 1 cm pathlength (13). The ratio of absorbance at 260 nm to 280 nm provides a measure of purity for genomic DNA samples (17). The value for the 260 nm to 280 nm ratio for the human genomic DNA sample was in the recommended range (Table 2). The absor bance spectrum of the uracil component of SRM 2082, the human genomic DNA sample, and the synthetic DNA oligonucleotide sample are compared in Figure 1.
Table 2.
Absorbance control values of the samples at 1 cm pathlength measured using a high accuracy dual beam spectrophometer at 22°C and 0.8 nm spectral bandwidth. The values in the parentheses are CV (%) of the mean from N = 9 (genomic DNA) and N = 4 (oligoncleotide samples), except for the *SRM 2082 components that are the standard uncertainties calculated as described in the NIST certificate of analysis (https://www.nist.gov/srm) calcuated using extensive measurements.
| Test sample | Abs. 260 nm | Abs. 280 nm | Ratio 260 nm/280 nm |
|---|---|---|---|
| Human genomic DNA | 1.029 (5.0%, N = 9) | 0.553 (6.5%, N = 9) | 1.85 |
| Oligonucleotide low | 6.247 (0.2%, N = 4) | 3.755 (0.1%, N = 4) | 1.66 |
| Oligonucleotide high | 24.943 (0.1%, N = 4) | 14.972 (0.1%, N = 4) | 1.67 |
| SRM 2082 Uracil | 7.990 (0.003)* | ||
| SRM 2082 Tryptophan | 8.350 (0.003)* |
Figure 1. UV absorbance spectra of uracil, synthetic oligonucleotide, and human genomic DNA.
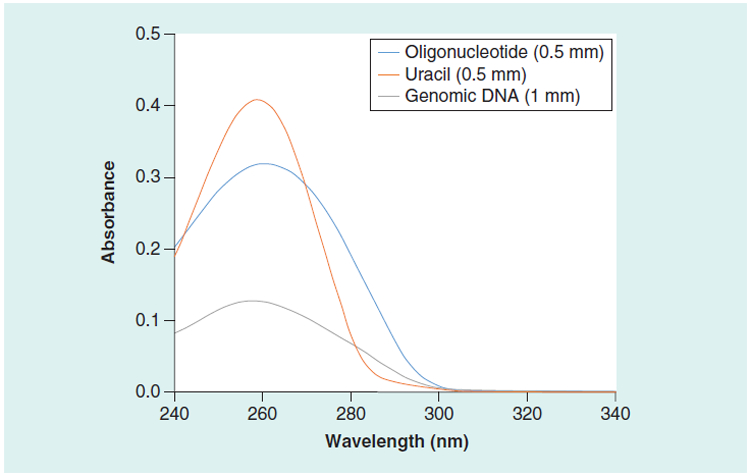
The spectra were measured at 22°C with a spectral band width of 0.8 nm.
The samples were measured using the microvolume (MV) spectrophotometers and a MV 16-well plate in a plate reader. The absorbance values of the samples were calculated as percentages of the control values (Table 2) to facilitate comparison of the different samples (Figure 2). The average absorbance values and coefficients of variation (CV, percentage) for the MV spectrophotometers and the MV plate reader for the three days of measurements (shown in Figure 2) are summarized in Table 3. The MV spectrophotometers and the MV plate reader gave coefficients of variation in the range of 0.5% to 2% for the tryptophan, uracil, and oligonucleotide samples, and agreement with the control or reference values had a similar range of 1% to 2%. The results with the high accuracy dual-beam spectrophotometers had coefficients of variation at 0.2% or lower for the components of SRM 2082 and the oligo-nucleotide samples.
Figure 2. Typical results for three different microvolume spectrophotometers.
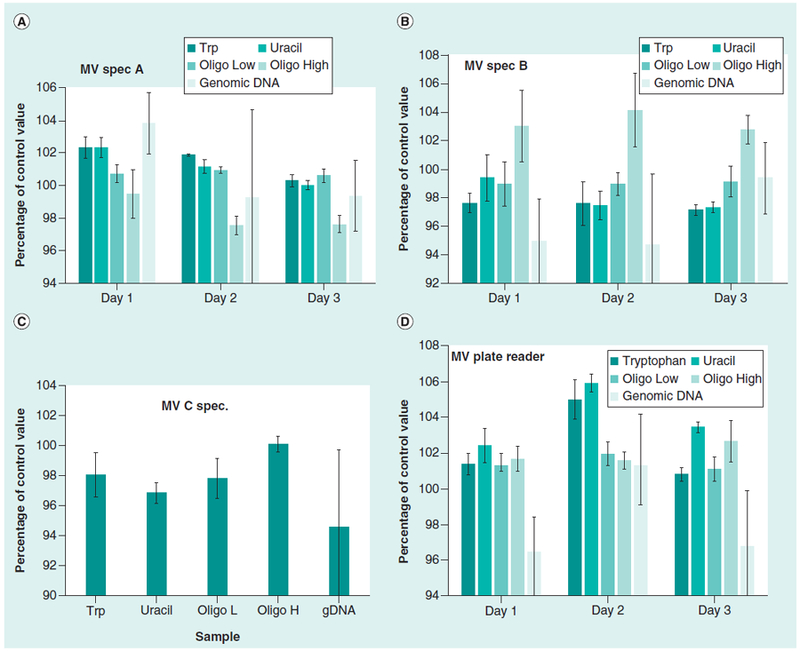
Results from MV Spec A is shown in (A), MV spectrophotometer B is shown in (B), MV spectrophotometer C is shown (C), and the MV plate reader is shown in (D). The error bars are 1 CV (%). The data for these measurements are summarized in Table 3.
Table 3.
Percentage of control values and coefficients of variation (% CV) using microvolume spectrophometers (MV Spec), and a microvolume (MV) plate reader. The values are the average of data acquired over three days as shown in Figure 2, with the exception of MV Spec C, where the data were acquired in one day. The values in the parentheses are 1 CV (%) and the number (N) of measurements.
| Test sample | MV Spec A | MV Spec B | MV Spec C | MV plate reader |
|---|---|---|---|---|
| Uracil | 101.4 (0.4%, N = 18) | 97.5 (0.9%, N = 21) | 96.8 (0.7%, N = 6) | 102.4 (0.7%, N = 144) |
| Trptophan | 101.2 (0.4%, N = 18) | 98.1 (1.0%, N = 15) | 98.1 (1.5%, N = 6) | 103.4 (0.6%, N = 144) |
| Oligo Low | 100.8 (0.4%, N = 9) | 99.0 (1.1%, N = 12) | 97.8 (1.3%, N = 6) | 101.5 (0.6%, N = 144) |
| Oligo High | 98.2 (0.9%, N = 9) | 103.3 (2.0%, N = 12) | 100.1 (0.5%, N = 6) | 102.0 (0.8%, N = 144) |
| Human genomic DNA | 100.8 (3.2%, N = 9) | 96.3 (3.5%, N = 30) | 94.6 (5.2%, N = 6) | 98.2 (2.6%, N = 144) |
However, the human genomic DNA sample had CVs in the range of 2% to 5% for both the MV instruments and the dual-beam spectrophotometer (Tables 2 and 3). One reason for this higher CV is that the absorbance of the human genomic DNA sample (0.05 at 0.5 mm pathlength) is below the optimal range for absorbance measurements. The complexity of the high molecular weight human genomic DNA samples makes quantitation difficult. The size of the human genomic DNA sample had a mean value of approximately 43,000 base pairs (data not shown). Human DNA molecules are highly complex with regions that are highly repetitive or enriched in GC, resulting in a complex heterogenous analytical target. The absorbance values for the different instruments and the resulting mass concentrations are summarized in Table 4.
Table 4.
Mass concentration values of human genomic DNA samples obtained using the different methods. Absorbance values and derived mass values of the human genomic DNA samples from a double-beam (DB) spectrophotometer, microliter volume (MV) spectrophotometers, microvolume (MV) plate reader, fluorescent dye binding, and digital PCR assays.
| Test method | Absorbance | Mass (ng/μl) |
|---|---|---|
| Absorbance DB Spec. | 1.029 (.051, N = 9) | 51.5 (2.6)* |
| Absorbance MV Spec. A | 1.04 (0.05, N = 29) | 52.0 (2.5)* |
| Absorbance MV Spec. A (0.2 M NaOH) | 0.71 (0.01, N = 6) | 52.4 (0.4)** |
| Absorbance MV Spec. B | 1.012 (0.086, N = 30) | 50.6 (4.3)* |
| Absorbance MV Spec. C | 0.973 (0.050, N = 6) | 48.7 (2.5)* |
| Absrobance MV Plate | 1.011 (0.036, N = 26) | 50.6 (1.8)* |
| Compact Flurorimeter (Broad Range Lambda DNA manufacturer’s standard) | 38.0 (0.3, N = 6) | |
| Compact Flurorimeter (High Sensitivity Lambda DNA manufacturer’s standard) | 46.5 (0.5, N = 4) | |
| Compact Flurorimeter (High Sensitivity Calf Thymus DNA standard) | 46.9 (0.7, N = 4) | |
| Compact Flurorimeter (AccuGreen, Calf Thymus DNA manufacturer’s standard) | 52.5 (0.7, N = 36) | |
| Microplate Fluorimeter (AccuBlue Dye A, Calf Thymus manufacturer’s standard) | 54.6 (1.8, N = 3) | |
| Microplate Fluorimeter (AccuBlue Dye B, Calf Thymus manufacturer’s standard) | 49.9 (1.0, N = 22) | |
| Microplate Fluorimeter (AccuClear Dye, Calf Thymus manufacturer’s standard) | 47.6 (0.8, N = 18) | |
| Droplet Digital PCR Assays*** | 53.9 (2.5, N = 81) |
The mass concentrations were calculated using the assumption that 1 Absorbance Unit at 260 nm and 1 cm pathlength equals 50 ng/μl. The values in the parentheses are 1 standard deviation.
The DNA sample was denatured by adding an equal volume of 0.4 mol/L NaOH and converted to mass by multiplying a dilution factor of 2 with the assumption that 1 Absorbance Unit at 260 nm and 1 cm pathlength equals 37 ng/μl.
Calculated using the assumptions shown in Table 5.
SRM 2082 is a standard designed to measure short pathlengths for cuvettes and spectrophotometer instruments. We did an additional set of experiments using the tryptophan and uracil components of SRM 2082 to determine the pathlengths of the 16-well microvolume plate (approximately 0.5 mm) using a plate reader instrument. Three sets of tryptophan and uracil sample measurements were done on three separate days and used to calculate the pathlengths of each of the 16 wells in the MV plate. The pathlengths calculated using the tryptophan, uracil, and the average of the two values were compared to the default (factory) measured pathlength values for measurements of the oligonucleotide samples. Figure 3 shows the values for the low and high concentration oligo-nucleotide samples that were calculated using the different pathlength values. The day to day average of the oligonucleotide measurements (each day N = 48) and the CVs for each day are in the range of 0.8% to 3%. The calibrations using the tryptophan values are similar to the factory values and the uracil values were lower. The calibrations obtained using the components of SRM 2082 yielded comparable values to the default (factory) values, providing confidence in the data from the plate reader.
Figure 3. Comparison of the absorbance values of the Oligo Low and Oligo High concentration samples using the default pathlength calibration and SRM 2082 to calculate pathlengths for the microvolume plate reader.
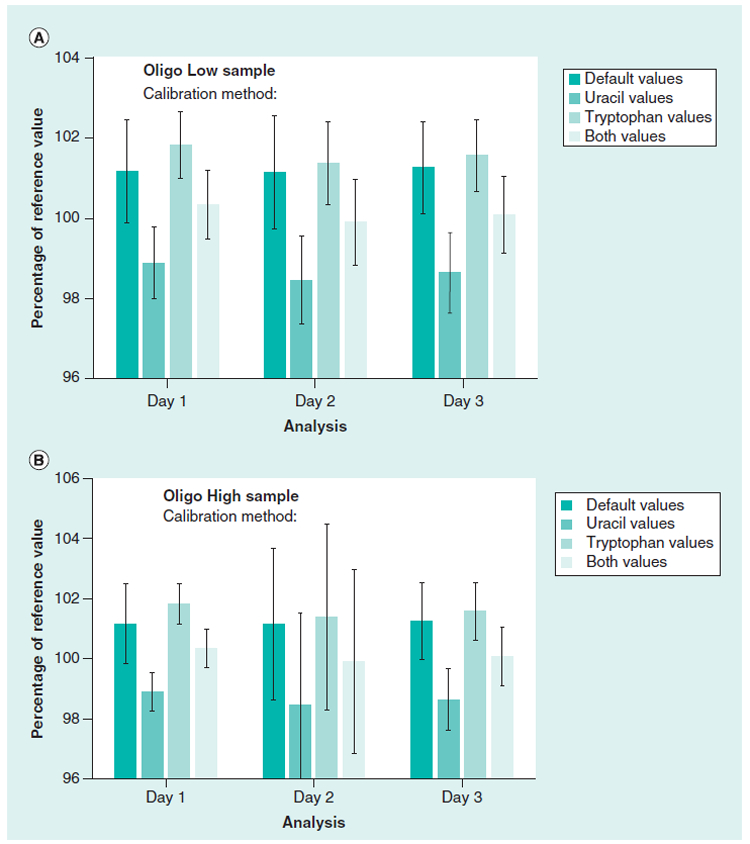
The path length values for the 16-well plate were calculated using either uracil, tryptophan, or the average of both to calculate the absorbance values for the oligonucleotide low and high concentration samples. Values were compared to the default instrument (factory) values. Values were from three different days using three measurments per sample in the 16 wells. The error bars are 1 CV (%).
Fluorescence dye binding assays
Fluorescent dye binding methods for measuring nucleic acid concentrations can increase sensitivity and specificity compared to absorbance measurements if care is taken in the measurements (18,19). PicoGreen assays have been used for many applications in a microplate format for PCR products (20) to quantitate human genomic DNA for large-scale genotyping (1). We used PicoGreen-type assays with a compact tabletop fluorimeter designed specifically for DNA measurements. The broad range Qubit assay (using the supplied lambda DNA standard) returned a mass concentration value for the human genomic sample that is significantly lower than the mass concentration values derived from the double beam spectrophotometer absorbance values. (The p-value was less than 0.001 with an unpaired t-test using the data in Table 4.) The high sensitivity Qubit assay with the lambda standard or a calf thymus standard showed values that were closer. An AccuGreen assay was used with the supplied calf thymus standard (single point calibration); the compact fluorimeter gave mass concentration results that were comparable to the absorbance values (Table 4). The compact fluorimeter uses a single point calibration, but when we used a dilution series with the supplied standard, the assays gave linear results (results not shown).
We also used two AccuBlue HiRange dye binding assays (A and B, Table 4) with a fluorescent plate reader. The mass concentration values for the human gDNA samples differed by approximately 9 %. (Comparison of the two values using an unpaired t test resulted in a p-value of less than 0.05). Serial dilutions of the supplied calf thymus standards provided by the kit manufacturers gave linear results for both assays (results not shown). An AccuClear assay was also used with the fluorescent plate reader. The AccuClear assay gave linear results with a dilution series of the manufacturer supplied calf thymus DNA, but the values were lower compared to the other assays (Table 4). The difference between the fluorescent dye binding assays using calf thymus calibration was approximately 14%.
The results from the assays could be due to differences in the way that the fluorescent dyes interact with human genomic DNA and calf thymus DNA. The GC content of the human genome is approximately 41%, however the distribution of GC content is not uniform (21). The intensity of PicoGreen fluorescence has been shown to be the same when bound to poly(dA)·poly(dT) and poly(dG)·poly(dC) homopolymers (22). The GC content and complexity of the standard used for an assay should match the target. In the case of human DNA measurements, a standard consisting of well-characterized human DNA would be best. We cannot rule out that differences in the quantitation of the dye-binding assays may be due the way the dyes interact with the calf thymus and human DNA, but a likely explanation is that the values of the standard samples supplied with the kits to calibrate the assay are the source of the differences. We did not attempt to characterize the standard materials included in the kits as standards and used them at the manufacture’s supplied values.
We also tested the human genomic DNA sample for the presence of RNA. The genomic DNA sample and a sample of the tenfold concentrated stock of the same genomic DNA did not contain detectable amounts of RNA (results not shown).
Droplet digital PCR measurements
Human genomic DNA samples were analyzed by six different digital PCR assays that target different chromosomal locations (Table 1). These assays were developed for the certification of NIST cancer biomarker standards (23,24). The calculation of copy number measurements from droplet digital PCR requires an accurate measurement of the droplet size. A droplet size (0.7681 nl, uncertainty 2.3%, k=2) was previously published (9), but a more recent measurement using the same measurement method but a different lot of master mix had droplet sizes of 0.7349 nl (uncertainty 2.3 % k=2) (14), showing the effect of different reaction conditions on the resulting droplet size. The effect of droplet size on the resulting calculated DNA mass concentrations using the manufacture’s default droplet size (0.85 nl), the 0.7681 nl value, and the most recent NIST value (0.7349 nl) are in Table 5. The decreased droplet sizes resulted in an increase in the calculated copy number of approximately 16% (Table 5).
Table 5.
Mass concentration of the human genomic DNA sample calculated from droplet digital PCR data with different assumptions of the droplet size.
| Size (Base pairs) | Genome mass (pg) | Droplet size (nl) | Copies/μl (SD) | Mass concentration (ng/μl) |
|---|---|---|---|---|
| 3.01 × 109 | 3.3 | 0.7349* (uncertainty 2.3%, k = 2) | 16,323 (759, N = 81) | 53.9 (2.5) |
| 3.01 × 109 | 3.3 | 0.7681* (uncertainty 2.3%, k = 2) | 15,610 (715, N = 81) | 51.5 (2.4) |
| 3.01 × 109 | 3.3 | 0.85 | 14,107 (646, N = 81) | 46.6 (2.1) |
The values in parentheses are 1 standard deviation of the data from the MET, DCK, and EIF5B gene assays (data shown in Figure 4). The average haploid genome mass was calculated using an average molecular weight of the base pairs of 660, as detailed in the text.
Figure 4 shows the copy number results of the six digital PCR assays calculated using the NIST measured droplet sizes. An ANOVA test and a Newman-Keuls multiple comparison test were used to perform pairwise comparisons (p = 0.05, significance level) between the assay data to determine specific pairwise differences. Based on this analysis, the MET, DCK, and the EIF5B assays had statistically comparable results, while the PMM1 and RPS27A assay results were statistically higher and the EGFR results were statistically lower compared with the other assays. The results from the MET, DCK, and EIF5B assays were used to calculate the resulting mass concentration values for the human genomic DNA sample (Tables 4 and 5).
Figure 4. Droplet digital PCR results from six genes for the human genomic DNA sample.
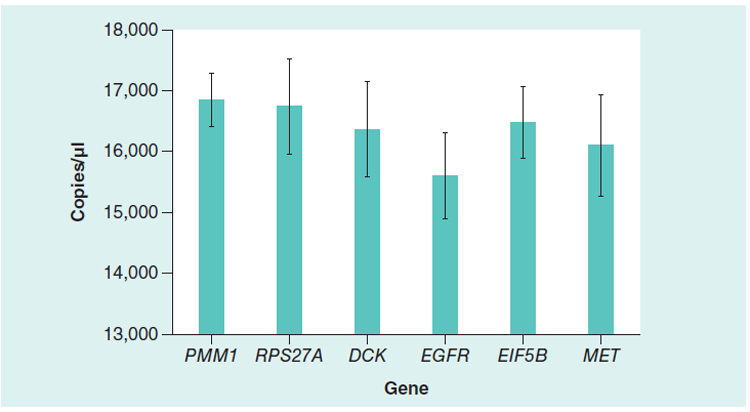
The error bars are 1 standard deviation (N = 27). A one-way ANOVA was performed on the data and a Newman-Keuls multiple comparison test was used to perform pairwise comparisons. The data from the MET, DCK, and EIF5B gene assays were determined to be statistically comparable and used for the calculations shown in Table 4 and 5.
To calculate a mass concentration from the droplet digital PCR data also requires an accurate measurement of the molecular weight of the human genome. The latest estimate of the average size of the human haploid genome based on the sequences of the individual chromosomes is 3.01 × 109 base pairs (25,26). Conversion of the genome size to pg of DNA was done using the average molecular weight of 660 for a DNA base pair. This value was calculated using the average molecular weights of the deoxy-nucleotides (27) by adding the molecular weight of two sodium atoms, subtracting the molecular weight of two hydrogens, and subtracting the molecular weights of one water molecule due to the formation of the DNA chain (27). This value is higher than the molecular weight frequently used, but it is reasonable that the DNA precipitated from solution with the sodium salt used in the solutions. Using these values for human genome size and average base pair molecular weight, the mass of the average human haploid genome was calculated to be approximately 3.3 pg.
The mass of the human genomic DNA sample will also contain the contribution from mitochondrial DNA since the DNA was extracted from whole cells grown in culture. The copy number ratio of the mitochondrial DNA to genomic DNA in the sample was 321.9 (standard deviation 4.5) based on the ratio of the two droplet PCR assays for a mitochondrial marker and a nuclear DNA marker. The size of the human mitochondrial genome is 16,569 base pairs (28). Compared to one copy of the genomic DNA (3.01 billion base pairs), adding 322 copies of the mitochondrial DNA to the extracted DNA would only add 0.16 % of the mass of the nuclear genome, so the contribution of mitochondrial DNA to the mass concentration of the human genomic DNA sample was not significant.
An important consideration for the measurement methods is the physical form of the DNA. Single-stranded DNA has a different UV absorbance extinction coefficient than double-stranded DNA. Also fluorescent DNA dye binding assays are specific for single- or double-stranded nucleic acids, and when double-stranded DNA is converted to the single-stranded form, the copy numbers will double when analyzed by digital PCR. NIST SRM 2372, a human DNA quantitation standard designed for forensic applications, was recertified because the absorbance had increased over years of storage conditions, but the mass of the DNA had not changed, indicating a small amount of single-stranded DNA in the samples resulting in the small increase in absorbance (29). For recertification, analytical samples were prepared by converting the p redominately double-stranded DNA to entirely single-stranded form by denaturation in NaOH, and using the conversion factor that 1 absorbance unit at 260 nm and 1 cm pathlength equals 37 ng/μl, resulting in agreement with the quantitative PCR assays (29). The absorbance values for the human genomic DNA sample used in this study gave consistent mass concentration values for the native samples and the sample denatured by NaOH (Table 4).
Absorbance measurements of human genomic DNA and synthetic oligonucleotides can be done quickly and directly with spectrophotometers that are readily calibrated. The main caveat with absorbance measurements is that the nucleic acid samples must be purified to remove interfering materials. SRM 2082 has significant advantages over potassium dichromate-based absorbance standards for measuring absorbance pathlengths. Potassium dichromate is highly toxic and classified as a human carcinogen (30). The components of SRM 2082 are non-toxic, relatively stable (at least 3 months at 4°C and years at −20°C), and their spectra are close to the most commonly used analytes, proteins, and nucleic acids.
The PicoGreen assay was shown to be susceptible to interference by transfer RNA, proteins, and some organic contaminants (5), and this assay was found to be sensitive to the molecular weight (degradation) of the DNA target (5,31). A major caveat is that the fluorescent dye binding assays require nucleic acid standards that match the samples being measured. Calf thymus DNA (31) and salmon testis DNA (5) are commonly used as standards for human genomic DNA. The double-stranded 48,502 base pair DNA from bacteriophage lambda (32), has been proposed as a standard for traceability to the International System of Units (10). The copy number measurements (determined by digital PCR) of different lots of lambda DNA were compared to UV absorbance and PicoGreen measurements (10). They showed that the copy number measurements could be used for an accurate estimation of the mass concentration of the lambda DNA samples (10). However, lambda DNA is not a good model for the complexity of the human genomic DNA samples.
Digital PCR has the advantages of increased sensitivity and specificity compared to the absorbance and fluorescent dye binding methods. The major caveats of using digital PCR are that the assays used will only measure intact and accessible targets, the presence of single-stranded versus double-stranded DNA should be known, and the samples must be free of PCR inhibitors. Measuring “absolute” concentration using digital PCR requires an accurate determination of the droplet size measured using the same conditions. Recent measurements have shown the importance of accurate measurements of the droplet size to calculate copy numbers and the effects of the reaction conditions (including different lots and types master mix) (9,14) and the mode of droplet formation (33) on the resulting droplet sizes. The use of digital PCR for concentration measurements requires more expensive instru ments, additional labor, and the reagents and assays used must be adequately validated to ensure specificity and efficiency. However with care, digital PCR technology has the potential to be used for traceable measurements to the International System of Units for DNA mass measurements (8). Our measurements in this study with the digital PCR assays had CVs that were approximately 5%. However, conversion to mass concentrations also requires accurate measurements of the droplet size values and the molecular weight of the human genome.
This study shows that absorbance measurements with the new generation of MV spectrophotometers, plate readers, and short pathlengths yield reliable results for nucleic acid samples that are free of contaminants. Fluorescent dye binding assays have increased sensitivity and tolerance of some contaminants. (This should be confirmed.) We realize that the DNA samples used for many applications may be degraded in molecular weight, such as circulating cell-free DNA. We are currently developing DNA materials that can be used to test the DNA quantitation of degraded samples, which will be the subject of further studies.
The MV spectrophotometers and plate reader have absorbance measurements that are sufficiently reproducible and accurate for many applications. The MV instruments have the significant advantages of using small amounts of valuable samples, the measurements are rapid, and they require minimal sample preparation. NIST SRM 2082 is a useful standard to benchmark absorbance measurements to ensure reproducible results.
Acknowledgments
We wish to thank Margaret Kline at NIST for helpful advice on droplet digital PCR measurements and measurements on mitochondrial DNA.
Footnotes
Endnote
Certain commercial equipment, instruments, or materials are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
References
- 1.Hall JM, LeDuc CA, Watson AR, and Roter AH. 1996. An approach to high-throughput genotyping. Genome Res 6:781–790. [DOI] [PubMed] [Google Scholar]
- 2.Lee SB, Crouse CA, and Kline MC. 2013. Chapter 2. Optimizing Storage and Handling of DNA Extracts, p. 19–38. In Shewale JG, and Liu RH (Eds.), Forensic DNA Analysis, Current Practices and Emerging Technologies. CRC Press, Boca Raton, FL, USA. [Google Scholar]
- 3.Sambrook J, Fritsch EF, and Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, New York, NY, USA [Google Scholar]
- 4.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, and Struhl K. 2004. Current Protocols in Molecular Biology John Wiley & Sons, Hoboken, New Jersey, NJ, USA [Google Scholar]
- 5.Li X, Wu Y, Zhang L, Cao Y, Li Y, Li J, Zhu L, and Wu G. 2014. Comparison of three common DNA concentration measurement methods. Anal. Biochem 451:18–24. [DOI] [PubMed] [Google Scholar]
- 6.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, et al. 2011. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem 83:8604–8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kline MC, Romsos EL, and Duewer DL. 2016. Evaluating Digital PCR for the Quantification of Human Genomic DNA: Accessible Amplifiable Targets. Anal. Chem 88:2132–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kline MC and Duewer DL. 2017. Evaluating Droplet Digital Polymerase Chain Reaction for the Quantification of Human Genomic DNA: Lifting the Traceability Fog Anal. Chem 89:4648–4654. [DOI] [PubMed] [Google Scholar]
- 9.Dagata JA, Farkas N, and Kramer JA. 2016. Method for Measuring the Volume of Nominally 100 μm Diameter Spherical Water-in-Oil Emulsion Droplets, NIST Special Publication 260–184(http://nvlpubs.nist.gov/nistpubs/SpecialPublications/NIST.SP.260–184.pdf) National Institute of Standards and Technology, Washington, DC, 2016. [Google Scholar]
- 10.Bhat S, Curach N, Mostyn T, Singh Bains G, Griffiths KR, and Emslie KR. 2010. Comparison of methods for accurate quantification of DNA mass concentration with traceability to the international system of units. Anal. Chem 82:7185–7192. [DOI] [PubMed] [Google Scholar]
- 11.Huggett JF, Foy CA, Benes V, Emslie K, Garson JA, Haynes R, Hellemans J, Kubista M, et al. 2013. Guidelines for minimum information for publication of quantitative digital PCR experiments. Clinical Chem 59:1–12. [DOI] [PubMed] [Google Scholar]
- 12.Zook JM, Catoe D, McDaniel J, Vang L, Spies N, Sidow A, Weng Z, Liu Y, et al. 2016. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data 3:160025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lang B, Urbas A, DeRose P, Liu H-K, Travis JC, Choquette SJ, and Cole KD. 2017. Development of NIST SRM 2082, a Pathlength Standard for Measurements in the UV. NIST J Research 122:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farkas N 2017. NIST Measurement Report.
- 15.Walker JA, Garber RK, Hedges DJ, Kilroy GE, Xing J, and Batzer MA. 2004. Resolution of mixed human DNA samples using mitochondrial DNA sequence variants. Anal. Biochem 325:171–173. [DOI] [PubMed] [Google Scholar]
- 16.Timken MD, Swango KL, Orrego C, and Buoncristiani MR. 2005. A duplex real-time qPCR assay for the quantification of human nuclear and mitochondrial DNA in forensic samples: implications for quantifying DNA in degraded samples. J. Forensic Sci 50:1044–1060. [PubMed] [Google Scholar]
- 17.Glasel JA 1995. Validity of nucleic acid purities monitored by 260nm/280nm absorbance ratios. BioTech. 18:62–63. [PubMed] [Google Scholar]
- 18.Johnson I and Spence MTZ. 2010. Chapter 8 — Nucleic Acid Detection and Analysis p. 326–348. In Johnson I, and Spence MTZ (Eds.), Molecular Probes™ Handbook, A Guide to Fluorescent Probes and Labeling Technologies, 11th Edition. [Google Scholar]
- 19.Dragan AI, Casas-Finet JR, Bishop ES, Strouse RJ, Schenerman MA, and Geddes CD. 2010. Characterization of PicoGreen Interaction with dsDNA and the Origin of Its Fluorescence Enhancement upon Binding. Biophy. J 99:3010–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romppanen E-L, Savolainen K, and Mononen I. 2000. Optimal Use of the Fluorescent PicoGreen Dye for Quantitative Analysis of Amplified Polymerase Chain Reaction Products on Microplate. Anal. Biochem 279:111–114. [DOI] [PubMed] [Google Scholar]
- 21.Romiguier J, Ranwez V, Douzery EJP, and Galtier N. 2010. Contrasting GC-content dynamics across 33 mammalian genomes: Relationship with life-history traits and chromosome sizes. Genome Res 20:1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singer VL, Jones LJ, Yue ST, and Haugland RP. 1997. Characterization of PicoGreen Reagent and Development of a Fluorescence-Based Solution Assay for Double-Stranded DNA Quantitation. Anal. Biochem 249:228–238. [DOI] [PubMed] [Google Scholar]
- 23.Lih C-J, Harrington RD, Harper K, Sims DJ, McGregor P, Camalier C, Si H, Das B, et al. 2016. Certified DNA Reference Materials to Compare HER2 Gene Amplification Measurements Using Next Generation Sequencing Methods. Journal of Molecular Diagnosis. J. Mol. Diag 18:753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He H-J, Almeida JL, Lund S, Steffen CR, Choquette S, and Cole KD. 2016. Development of NIST Standard Reference Material 2373: Genomic DNA Standards for HER2 Measurements. Biomolecular Detection and Quantification 8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, Murphy TD, Pruitt KD, et al. 2017. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Research 27:849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consortium GR 2017. Human Genome Assembly GRCh38.p11, https://www.ncbi.nlm.nih.gov/grc/human/data.
- 27.Doležel J, B. J, Voglmayr H, and Greil-huber J. 2003. Letter to the Editor. Cytometry Part A 51A:127–128. [Google Scholar]
- 28.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, et al. 1981. Sequence and organization of the human mitochondrial genome. Nature 290:457–465. [DOI] [PubMed] [Google Scholar]
- 29.Vallone PM, Butts ELR, Duewer D, and Kline MC. 2013. Recertification of the NIST Standard Reference Material® 2372, human DNA quantitation standard. For. Sci. Inter. Gen. Suppl, Series 4:e256–e257. [Google Scholar]
- 30.National Center for Biotechnology Information, PubChem Open Chemistry Database, p. CID=24502, https://pubchem.ncbi.nlm.nih.gov/compound/24502 (accessed 17 January 2018).
- 31.Georgiou CD and Papapostolou I. 2006. Assay for the quantification of intact/fragmented genomic DNA. Anal. Biochem 358:247–256. [DOI] [PubMed] [Google Scholar]
- 32.Becker A and Murialdo H. 1990. Bacteriophage lambda DNA: the beginning of the end. J. Bacteriol 172:2819–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Košir AB, Divieto C, Pavšič J, Pavarelli S, Dobnik D, Dreo T, Bellotti R, Sassi MP, and Žel J. 2017. Droplet volume variability as a critical factor for accuracy of absolute quantification using droplet digital PCR. Anal. Bioanal. Chem doi: 10.1007/s00216–017–0625-y:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]