

SUMMARY
Adaptation of viruses to their hosts can result in specialization and a restricted host range. Species specific polymorphisms in the influenza virus polymerase restrict its host range during transmission from birds to mammals. ANP32A was recently identified as a cellular co-factor affecting polymerase adaption and activity. Avian influenza polymerases require ANP32A containing an insertion resulting from an exon duplication uniquely encoded in birds. Here we find that natural splice variants surrounding this exon create avian ANP32A proteins with distinct effects on polymerase activity. We demonstrate species-independent direct interactions between all ANP32A variants and the PB2 polymerase subunit. This interaction is enhanced in the presence of viral genomic RNA. In contrast, only avian ANP32A restored ribonucleoprotein complex assembly for a restricted polymerase by enhancing RNA synthesis. Our data suggest that ANP32A splicing variation among birds differentially affects viral replication, polymerase adaption, and the potential of avian hosts to be reservoirs.
In Brief
Influenza virus circulating in diverse bird species requires the host co-factor ANP32A. Alternative splicing of an aviansignature exon in ANP32A differentially stimulates the influenza polymerase. Baker et al. demonstrate the precise step at which ANP32A rescues an otherwise restricted polymerase and suggest that splice preferences can affect influenza virus evolution.
Graphical Abstract
INTRODUCTION
Influenza A viruses circulate in diverse host species. Wild aquatic waterfowl are the natural viral reservoir, and zoonoses can occur either directly from birds or through an intermediate host such as swine. Ecological overlap between the major hosts—birds, swine, and humans—creates repeated opportunities for crossspecies virus transmission, yet only a minor fraction of these are successful. Influenza virus must overcome multiple biological barriers for successful cross-species transmission. The viral polymerase is a major determinant of host range (Almond, 1977; Subbarao et al., 1993). Avian-origin polymerases function efficiently in avian cells, but their activity is heavily restricted in human cells (Labadie et al., 2007; Mehle and Doudna, 2008). Restricted polymerases rapidly evolve adaptive mutations enabling efficient function as viruses jump from avian to mammalian hosts.
The influenza polymerase is a heterotrimeric enzyme composed of the subunits PB2,PB1, and PA. The polymerase assembles with viral RNA encapsidated by oligomeric nucleoprotein (NP) to form the viral ribonucleoprotein (vRNP) complex. The polymerase transcribes viral mRNAs via cap snatching and replicates the minus-sense genomic vRNA through a plus-sense cRNA intermediate. Avian-origin polymerases are restricted in mammalian hosts with defects in both replication and transcription (Mehle and Doudna, 2008). The PB2 subunit has long been recognized as a main determinant of species-specific polymerase activity and host range (Almond, 1977; Subbarao et al., 1993). The prototypical adaptive mutation in the PB2 subunit occurs at amino acid 627 located within the eponymous 627 domain (Tarendeau et al., 2008), where an avian-signature glutamic acid is changed to a mammalian-signature lysine (Subbarao et al., 1993). Adaptive mutations increase replication, pathogenicity, and transmission of avian-origin viruses in mammalian hosts. Structural analyses have revealed that portions of PB2, including the 627 domain, remain solvent exposed in the holoenzyme and undergo large-scale conformational reorganization depending on whether the polymerase is replicating or transcribing (Hengrung et al., 2015; Pflug et al., 2014; Reich et al., 2014; Thierry et al., 2016). These data raise the possibility that adaptive mutations in PB2 may be important for intra- or inter-molecular protein:protein interactions and conformational rearrangements.
Viral polymerase activity during infection is regulated by both essential host co-factors as well as restriction factors that antagonize function (Kirui et al., 2016a). Acidic nuclear phosphoprotein 32 family member A (ANP32A, pp32) associates with the influenza A virus polymerase and stimulates vRNA synthesis from a cRNA template in vitro (Bradel-Tretheway et al., 2011; Sugiyama et al., 2015). More recently, ANP32A has been shown to affect the host range of influenza virus as a species-specific co-factor of the viral polymerase (Long et al., 2016). The restriction of avian-origin polymerases in mammalian cells is overcome by expressing avian ANP32A in these cells. Compared with mammalian ANP32A, which does not enhance avian polymerase activity, ANP32A encoded by most Aves species has a partial duplication of exon 4, resulting in an insertion between the N- and C-terminal domains. This insertion is necessary and sufficient to enable ANP32A to rescue restricted avian polymerases in mammalian cells (Long et al., 2016). Although the genetic evidence strongly implicates ANP32A as a host-range factor, it remains unclear how ANP32A stimulates polymerase activity and how avian ANP32A selectively rescues restricted avian polymerases.
Here we dissect the processes by which ANP32A engages the viral polymerase and affects its function. We identify naturally occurring splice variants of ANP32A in avian species that reduce the size of the repeat insertion from 33 to 29 amino acids, removing a SUMO interaction motif (SIM)-like sequence located upstream of the repeat. Both full-length chicken ANP32A (chANP32A33) and the splice isoform lacking the SIM (chANP32A29) rescue activity of a restricted polymerase, with chANP32A33 exhibiting a more potent phenotype. We show that ANP32A interacts with the viral polymerase, binding directly to the 627 domain, and this interaction was enhanced in the presence of viral genomic RNA. However, binding between ANP32A and the polymerase was not species specific and was unaffected by the identity of PB2 residue 627. By contrast, our data reveal that chANP32A29 functions in a species-specific fashion to stimulate the intrinsic enzymatic activity of a restricted avian polymerase. Together, these data elucidate a critical step at which chANP32A rescues polymerase activity and show that ANP32A splice variants present in birds affect its potency, with potential impacts on influenza adaptation and replication in these different hosts.
RESULTS
Differential Splicing Creates Three Predominant Isoforms of Avian ANP32A with Differing Impacts on Avian Influenza Polymerase Activity
Given that species-specific differences in ANP32A affect its function during infection, we analyzed ANP32A expression in diverse avian species. Most avian ANP32A encode a partial duplication and insertion of exon 4 that repeats a portion of the leucine-rich repeat capping motif in the expressed protein (Figure 1A). This duplication is absent in mammals and the avian Palaeognathae clade containing ostriches and tinamous. The chANP32A that was originally shown to enhance polymerase activity contained a 33 amino acid insertion (chANP32A33). Analysis of RNA sequencing (RNA-seq) datasets revealed alternative splicing surrounding the duplicated exons, including splicing to a downstream splice acceptor site to create chANP32A29 (Figures 1B and1C). chANP32A29 lacks four hydrophobic residues from the N terminus of the repeat that compose the SIM present in the 33 amino acid insert (Domingues and Hale, 2017). The ratio of transcripts encoding 33 or 29 amino acid inserts varied across species (Figures 1B and 1C; Table S1): pigeons (Columba livia) express almost exclusively ANP32A33, chickens (Gallus gallus) express ~3-fold more ANP32A33 transcripts than ANP32A29, and starlings (Sturnus vulgaris) express almost exclusively ANP32A29 transcripts. Intriguingly, examples were found in transcripts from many avian species in which the duplicated exon was skipped altogether to create a human-style ANP32A. This was most pronounced in the swan goose (Anser cygnoides), in which >70% of all transcripts skipped the duplicated exon. ANP32A splicing patterns were unaffected by influenza virus infection in chickens and did not differ between inbred lines that are susceptible or partially resistant to influenza A virus (Table S1). Furthermore, there was no obvious difference in ANP32A splicing in chicken cells when interferon (IFN) signaling was activated or inhibited or when they were infected with influenza virus (Table S1).
Figure 1.
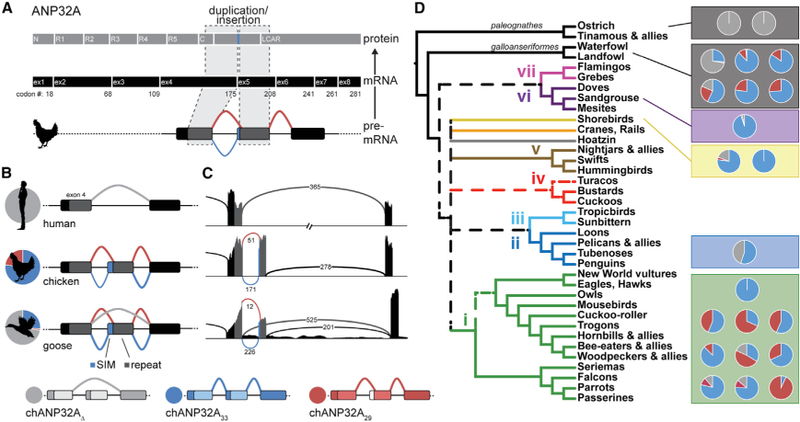
Natural Variation in ANP32A Splicing Patterns in Aves Duplication and insertion of ANP32A exon 4 results in three major splice isoforms in birds. Schematic of chANP32A protein and exonic (ex) mRNA organization indicating duplicated domains. C, C-cap; N, N-cap; R1–R5, leucine-rich regions 1–5. Exon numbering is based on chANP32A33. All human ANP32A transcripts lack the exon duplication (light gray). Schematics of chicken and goose transcripts show splicing upstream to capture codingsequence for the SIM (blue), splicing downstream to omit the SIM (red), or in some cases skipping the repeated exon to create a mammalian-like transcript (light gray). The relative abundance of each splice isoform in RNA-seq data is indicated by the pie charts. SIM, SUMO-interacting motif. Sashimi plots of ANP32A corresponding to examples in (B) and colored similarly. The abundance of each splice variant is indicated on the lines corresponding to the intron-spanning reads. ANP32A splice patterns in diverse bird species overlaid on the Aves consensus phylogeny (dashed lines represent branches that were in two of three consensus trees from Reddy et al., 2017). Pie charts represent relative transcript abundance from RNA-seq datasets (listed in Table S1).
To assess the impact of ANP32A splice variants, we performed polymerase activity assays in human or avian cells. Avian-style polymerases (PB2 E627) were heavily restricted in human cells, whereas co-expression of chANP32A33 restored activity in a dose-dependent fashion (Figure 2A), consistent with earlier reports (Long et al., 2016). The enhancing activity of chANP32A33 was extremely potent, increasing polymerase activity even when its expression was below the limit of detection in our western blots. Expressing chANP32A29 in restrictive human cells also rescued the activity of an avian-style polymerase in a dose-dependent manner (Figure 2B). chANP32A33 and chANP3229 did not significantly affect the activity of a human influenza virus polymerase in human cells (Figures 2A and2B). chANP32A29 also did not affect either human or avian virus polymerases in avian cells (Figure 2B). Expressing human ANP32A (huANP32A) in avian cells exhibited only minimal effects on both polymerases. Blotting confirmed that the increased polymerase activity was independent of changes in viral protein levels.
Figure 2.
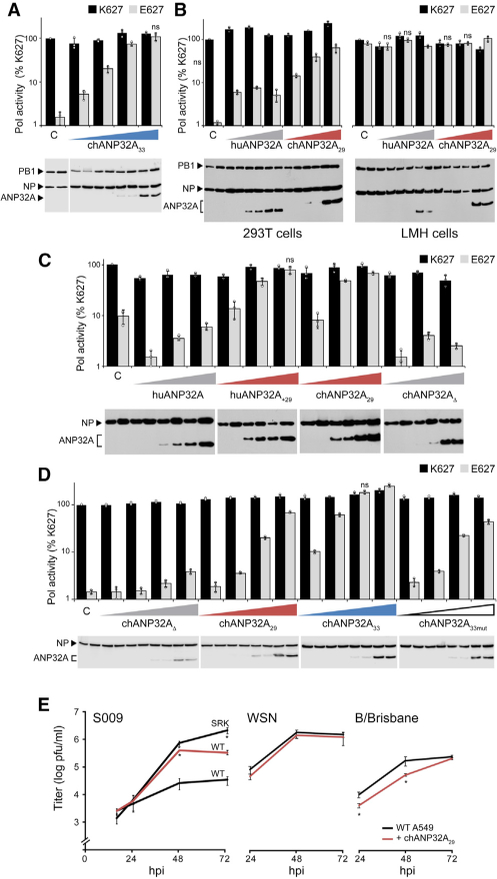
Avian ANP32A29 Is Sufficient to Restore SpeciesRestricted Avian Polymerase Activity and Replication Activity of human-style (PB2 K627) and avian-style (PB2 E627) polymerases was measured in the presence of increasing concentrations of the indicated ANP32A proteins. Protein expression was assessed via western blot. ANP32A33 selectively rescues restricted avian polymerase activity in human 293T cells. ANP32A29 is sufficient to restore polymerase activity in human cells (left) but does not significantly affect human viral polymerase in human cells oreither polymerase in chicken LMH cells (right). Insertion of the avian 29 amino repeat into huANP32A (huANP32A+29) enhances activity, whereas deletion of the repeat in chANP32A (chANP32AD) disables its function. chANP32A33 is the most potent enhancer of avian polymerase activity in human cells compared with chANP32A29 or chANP32A33mut. For all assays, polymerase activity was normalized to an internal control and compared with PB2 K627 polymerase in the absence of ANP32A. Data are shown as mean (columns) of n = 3 technical replicates (dots) ± SD derived from representative results of at least three independent biological replicates. C, empty vector control. In (A)–(D), pairwise comparisons between PB2 K627 and E627 at each condition were significant (p < 0.05, Student’s t test) except where indicated as not significant (ns). Replication kinetics of influenza virus encoding the avian S009 RNP (WT) ora human human-adapted mutant (SRK), WSN, or B/Brisbane. WT A549 cells or those stably expressing chANP32A were infected (MOI = 0.1), and viral titers were determined at the indicated time points. Data are shown as average of n = 3 ± SD. *p < 0.05, one-way ANOVA with post hoc Tukey honestly significant difference (HSD) test compared with WT A549 cells. See also Figures S1 and S2.
We created ANP32A domain swap hybrids to determine if this shortened insert is sufficient to enhance polymerase activity (Figure 2C). Inserting the 29 amino acid repeat into huANP32A (huANP32A+29) conferred enhancing activity, whereas removing the insert from chANP32A (chANP32AD) eliminated its enhancing activity. The 4 amino acids that are absent in the insert of ANP32A29 have been identified as a SIM-like sequence important for the pro-viral activity of chANP32A33 (Domingues and Hale, 2017). We thus tested the relative activity of chANP32A variants (Figures 2D and S1A). Both splice variants of chANP32A increased activity of an avian polymerase in human cells, although the dose-dependent assay revealed that chANP32A33 is more than 10-fold more effective than a similar amount of chANP32A29. This difference in activity was dependent upon the SIM sequence, as simply restoring the insert length to 33 amino acids by adding four glycine residues to create ANP32A33mut did not increase activity above that of chANP32A29 (Figure 2D). Most avian species that we analyzed expressed multiple splice isoforms of ANP32A. We mimicked this mixed expression pattern by testing whether the polymerase enhancing activity of different ANP32A isoforms were compatible with each other (Figure S1B). Polymerase activity was assessed in human cells, expressing endogenous huANP32A, along with differing combinations of ectopic chANP32A29 and chANP32A33. Rescue of avian polymerase activity was more pronounced as the proportion of chANP32A33 increased, consistent with our prior results demonstrating that chANP32A33 is 10-fold more potent than chANP32A29. Moreover, there was no detectable interference or competition between the different ANP32A isoforms.
To determine if the observed changes in polymerase activity increase viral replication, we performed multi-cycle replication assays in human cells expressing endogenous huANP32A or stably expressing chANP32A29. The avian influenza virus isolate S009 (PB2 E627) is restricted in human cells (Mehle and Doudna, 2009). Virus growth kinetics showed that expression of chANP32A29 increased replication of the restricted S009 wildtype (WT) approximately 10-fold (Figure 2E). Cells were also infected with an S009 mutant encoding a fully humanized PB2 containing S590, R591, and K627 (S009 SRK) (Mehle and Doudna, 2009). As expected, S009 SRK overcame the restriction phenotype in human cells. Restoring replication by expressing chANP32A29 in the target cells or by introducing adaptive mutations into PB2 produced similar viral titers at early time points, although the adapted virus reproducibly achieved higher final titers. It is possible that the more potent chANP32A33 could further enhance the final titers of the non-adapted WT S009 to levels comparable with S009 SRK virus or that the precise amount of chANP32A is critical and this aspect is not fully captured in stable expression cell lines. Infections were repeated with mammalian influenza A (A/WSN) and B (B/Brisbane) viruses (Figure 2E), and chANP32A29 had no significant effect on WSN replication and only minor effects on replication at early times for B/Brisbane. To address if ANP32A alters the subcellular localization of PB2 in a species-specific manner, cells expressing huANP32A or chANP32A29 were infected with A/WSN virus containing PB2 K627 or E627 (Figure S2). PB2 import was unchanged in all conditions, and infection did not cause obvious relocalization of ANP32A. These data suggest that chANP32A33 containing the SIM sequence is the more potent enhancer of avian polymerase activity while also revealing that the shorter splice variant chANP32A29 supports polymerase activity and replication of an avian-style virus.
ANP32A Interacts Directly with the 627 Domain of PB2
We performed a series of experiments to dissect potential interactions between ANP32A and the viral RNP. The remainder of our experiments focused on chANP32A29, which lacks the SIM motif, allowing us to segregate SIM-related activities from the effects of the common repeat sequence. We tested interactions between PB2 and ANP32A in human cells using human and avian versions of both proteins (Figure 3). We used WSN PB2 variants that differed only at the adaptive residue 627 and chANP32A29 or a variant in which the repeat has been deleted (chANP32AD), controlling for other amino acid differences between human and avian versions of these proteins. Co-immunoprecipitations detected the known interaction between NP and PB2 but failed to detect stable binary interactions between ANP32A and PB2 (Figure 3A). By contrast, ANP32A specifically co-precipitated with PB2 when the subunits PB1 and PA were also present (Figure 3B), suggesting that ANP32A preferentially interacts with the trimeric polymerase and in agreement with prior work (Bradel-Tretheway et al., 2011; Sugiyama et al., 2015). We confirmed these results in the context of infected cells in which PB2 specifically co-precipitated ANP32A (Figure 3C). Notably, although there was minor experiment-to-experiment variability, there was no striking evidence for a species specificity in these interactions. In addition, the results show that the SIM was not required for interactions between chANP32A and the polymerase. Thus, although only chANP32A rescues restricted avian polymerases in human cells (Figure 2), the selectivity of this enhancement does not appear to occur at the level of protein:protein interactions.
Figure 3.
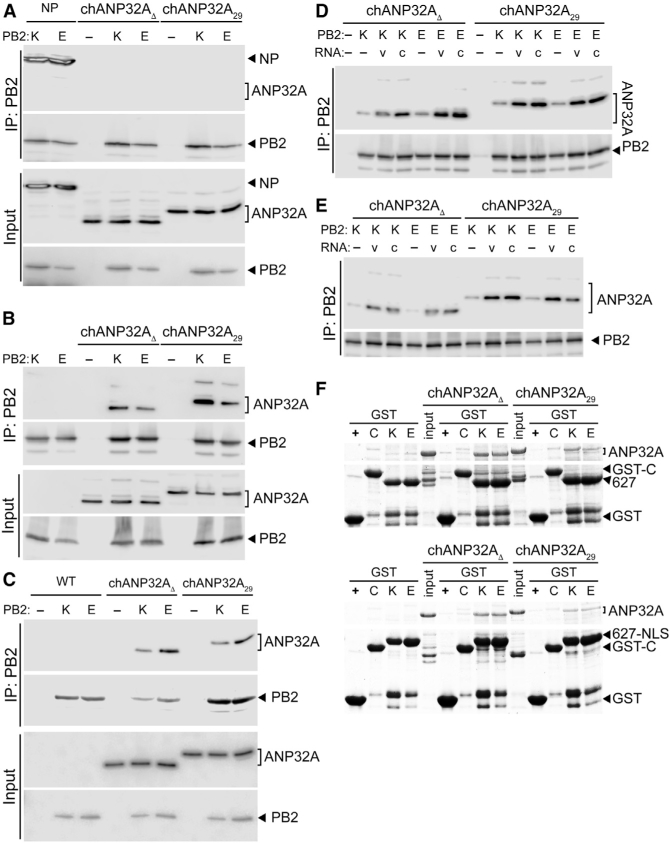
ANP32A Binds Directly to the PB2 627 Domain in a Species-Independent Fashion (A) ANP32A does not stably interact with the PB2 subunit when it is expressed alone. PB2 K627 or E627 was immunoprecipitated (IP) from cells co-expressing ANP32A or the positive control NP. (B) Both chANP32A29 and the humanized chANP32AD interact with the polymerase trimer. Immunoprecipitations were performed from cells expressing ANP32A and the trimeric polymerase containing PB2 K627 or E627. (C) ANP32A interacts with the viral polymerase during infection. WT A549 cells and those stably expressing ANP32A were infected, and PB2 was immunoprecipitated. (D and E) Genomic RNA increases PB2-ANP32A interactions. Immunoprecipitations were performed from cells expressing ANP32A, a vRNA (v) or cRNA (c) genomic segment, and the trimeric polymerase containing PB2 K627 or E627. Binding was measured with catalytically active polymerase (D) or an inactive PB1a mutant polymerase (E). (F) ANP32A binds directly to the PB2 627 domain. In vitro binding was measured between chANP32A variants and GST-tagged PB2 627 domain (top) or PB2 627-NLS (bottom) and visualized by Coomassie staining. GST alone (+) or an irrelevant GST fusion (C) were included as specificity controls.
The viral polymerase, and PB2 in particular, adopts multiple conformations depending on the function of the enzyme and co-factors present. We co-expressed vRNA or cRNA genomic templates with the polymerase and ANP32A to shift the polymerase toward an RNA-bound conformation (Figure 3D). NP was intentionally excluded to prevent genome replication, which would result in a mixture of vRNA and cRNA templates. As above, PB2 K627 and E627 co-precipitated both chANP32A29 and chANP32A∆. Co-expressing either vRNA or cRNA significantly increased co-precipitation of chANP32A29 and chANP32A∆. This increase was similar for plus- or minussense templates and human or avian PB2. To ensure that nascent products are not being produced, we repeated the interaction assays using a catalytically defective PB1 (PB1a), yielding similar results (Figure 3E). Although the identity and structures for all of the proteins and RNAs present in ANP32A-containing complexes is not yet known, these findings suggest that ANP32A interacts more efficiently with RNA-bound conformations of the polymerase and these interactions do not require catalysis.
Most adaptive mutations in avian polymerases arise within the conformationally dynamic PB2 627 and NLS domains. To evaluate if ANP32A interacts directly with the 627 domain of PB2, in vitro binding was tested using recombinant proteins. chANP32A29 and chANP32AD were captured by the PB2 627 domain, but not by an irrelevant control GST fusion protein or GST alone (Figure 3F, top). Similar results were obtained using PB2 containing the 627 and NLS domains (Figure 3F, bottom). Once more, these interactions were not dependent on the presence of the repeat insert in ANP32A or the PB2 variant used. We note that these binding assays were performed at steady state, and minor differences in binding kinetics may not be detectable via this approach. ANP32A interacted with domains of PB2 (Figure 3F) but not the full-length protein (Figure 3A). PB2 exhibits a high degree of structural plasticity (Thierry et al., 2016); the full-length protein in cells may assume a conformation that is not favorable for ANP32A binding, whereas this conformation is absent when measuring interaction between isolated domains in vitro. Moreover, these findings do not exclude additional binding interfaces between the polymerase trimer and ANP32A. Together, our assays show that ANP32A directly binds the 627 domain of PB2 and that this interaction is enhanced when the polymerase adopts template-bound conformations.
The Molecular Defects of PB2 627E Polymerase in Human Cells Are Nullified by chANP32A
Avian polymerases cannot efficiently assemble RNPs in mammalian cells (Mehle and Doudna, 2008). We thus evaluated if RNP formation was affected by ANP32A (Figure 4A). Polymerase, NP, and vRNA were expressed in human cells, and coprecipitation of PB2 by NP was used as a proxy for RNP formation. Avian-style polymerase exhibited the characteristic defect in RNP formation (Figure 4A). Co-expressing chANP32A29 overcame assembly defects for avian-style polymerases as evidenced by an increase in PB2 co-immunoprecipitation. Coexpressing chANP32AD did not restore RNP formation, demonstrating specificity of the enhancement. RNP assembly for human polymerases was unaffected by chANP32A. These results demonstrate that the rescue phenotype observed in viral polymerase activity assays (Figure 2) are in part due to the ability of chANP32A29 to enhance PB2 627E RNP formation.
Figure 4.
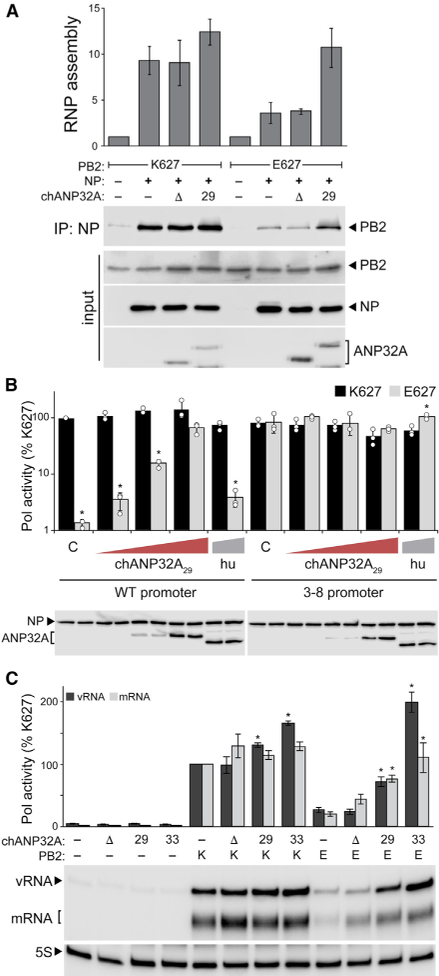
Molecular Defects of a Restricted Avian Polymerase Are Rescued by chANP32A29 (A) chANP32A29 enhances RNP formation for an avian-style polymerase. RNPs were reconstituted in human cells by co-expressing the indicated polymerase, NP, a genomic RNA, and chANP32A variants. NP was immunoprecipitated, and co-precipitating PB2 was measured as a proxy for RNP formation. RNP assembly was quantified from two independent assays, normalized to controls, and represented as the mean ± SD. chANP32A29 functions redundantly with enhancing promoter mutations. Polymerase activity assays were conducted with the indicated ANP32A proteins and either WT or 3–8 mutant vRNA reporters. Proteins were detected by western blot. C, empty vector control. Data are shown as mean (column) of technical triplicates (dots) ± SD. *p < 0.05 (Student’s t test) between PB2 K627 and E627 at each condition. chANP32A increases the enzymatic activity of a restricted polymerase independent of RNP formation. NP-independent polymerase activity on a micro-gene vRNA template was measured by primer extension. chANP32A variants were co-expressed where indicated. mRNA and vRNA products were quantified by phosphorimaging from three independent experiments, normalized to PB2 K627 in the absence of ANP32A, and presented as mean ± SD. *p < 0.05, one-way ANOVA with post hoc Tukey HSD test. Tests were performed separately for vRNA and mRNA levels, and comparisons were made to PB2 K627 or E627 polymerase activity in the absence of ANP32A.
Defects in RNP formation by restricted polymerases are proposed to result from decreased production or stabilization of replication products by the polymerase itself rather than changes to downstream events in the assembly process (Cauld well et al., 2013; Paterson et al., 2014). Mutations in the 3′ vRNA promoter at positions 3 and 8 that increase RNA synthesis by the polymerase also overcame restriction for avian polymerases (Figure 4B; Cauldwell et al., 2013; Paterson et al., 2014). Co-expressing chANP32A29 with a 3–8 mutant vRNA reporter did not further increase polymerase activity. The absence of additive effects suggests that the promoter mutant and chANP32A29 alter polymerase activity at a similar step, possibly through the same pathway (Figure 4B). We next eliminated NP and asked if ANP32A affects the polymerase itself to change synthesis or accumulation of RNA products. Although the viral polymerase requires NP for synthesis of full-length genomic segments, NP is not necessary to replicate and transcribe micro-vRNA-like templates (76 nt or less) (Turrell et al., 2013). Human viral polymerase produced mRNA and vRNA products from a micro-vRNA template, but an avian-style polymerase remained restricted with significant reductions in all RNAs (Figure 4C). Co-expressing chANP32AD slightly increased synthesis of vRNA and mRNA, yet avian-style polymerase remained restricted. However, co-expressing chANP32A29 restored avian-style polymerase product accumulation. chANP32A33 further increased the levels of vRNA and mRNA to those produced by the human polymerase. Thus, the SIMcontaining isoform enhances polymerase activity through a mechanism common to both chANP32A isoforms, and similar to polymerase activity assays with NP (Figure 2), is a more potent enhancer. Together these data suggest that chANP32A29 as well as the more common isoform chANP32A33 restore RNA production resulting in RNP formation and subsequent viral gene expression.
DISCUSSION
Avian influenza virus polymerases are heavily restricted in mammalian cells. This is due in part to the species-specific dependence on the host factor ANP32A. An exon duplication and insertion present in most Aves species confers replication competence to influenza virus encoding an avian-style polymerase. Here we have shown that this duplicated exon is alternatively spliced in bird species creating ANP32A proteins with differential ability to support avian influenza polymerases. chANP32A33, the most prevalent variant in common reservoir hosts such as ducks and chickens, was the most potent isoform. The chANP32A29 variant, which lacks the SIM sequence, was sufficient to rescue avian polymerase function in mammalian cells. In contrast, transcripts that skipped the duplicated exon altogether produced chANP32AD that mimicked mammalian ANP32A and failed to rescue polymerase activity. ANP32A bound directly to the PB2 627 domain and binding in cells was enhanced by the presence of genomic RNA, yet binding lacked species specificity. Species specificity became apparent only when investigating the intrinsic activity of the viral polymerase. chANP32A selectively enhanced production of RNA products by restricted avian-style polymerases. Together these data show that natural splice variants in ANP32A affect its ability to selectively enhance the production of RNA products by restricted polymerases.
The precise step(s) at which avian polymerases are restricted in mammals, and at which ANP32A functions to alleviate restriction, is not well understood. Complementation assays during infection suggest that restricted polymerases exhibit defects in replication, but are largely competent for transcription (Ma¨ nz et al., 2012). Restricted polymerases express, localize, assemble into holoenzymes, and bind to genomic RNA similarly to functional polymerases (Mehle and Doudna, 2008; Nilsson et al., 2017). The use of promoter mutants to bypass restriction has also shown that restricted polymerases do not have defects in elongation (Crescenzo-Chaigne et al., 2002; Paterson et al., 2014). When restricted polymerases are purified from human cells, they retain full activity in in vitro assays, indicating that components in the cellular environment alter their activity (Paterson et al., 2014). Thus, restriction appears to occur at the earliest steps in the catalytic process, after template binding and before elongation. This is also the stage at which our data show enhanced ANP32A:polymerase interactions and species-specific enhancement of RNA synthesis (Figures 2, 3, and 4). 3–8 promoter mutants can also function at this step to drive increased production of viral RNAs. Our demonstration that chANP32A showed no additive effects with the 3–8 promoter mutant provides further support that ANP32A affects these early steps in RNA synthesis. This supports a model whereby ANP32A stimulates intrinsic activity of restricted polymerases by favoring a conformation poised for replication or by recruiting necessary trans-acting or trans-activating polymerases.
Our data raise the possibility that host-specific expression and splicing patterns of ANP32A apply distinct selective pressures on influenza virus. Ostrich ANP32A does not encode a duplicated exon 4 (Long et al., 2016). Instead, they express a human-like ANP32A, which likely explains why experimental infection of ostriches with an avian influenza virus resulted in acquisition of mammalian adaptive mutations in PB2 (Shinya et al., 2009; Yamada et al., 2010). Similar dynamics may have been in play during a highly pathogenic avian influenza outbreak at Qinghai Lake in 2005. Avian viruses isolated from this outbreak encoded the prototypical human adaptation PB2 E627K (Chen et al., 2005, 2006; Liu et al., 2005). Bar-headed geese (Anser indicus) were the index species for this outbreak. RNA-seq analysis from the closely related swan goose (Anser cygnoides domesticus) revealed stark differences in ANP32A splicing patterns, with >70% of transcripts skipping the repeated exon to express a human-like ANP32A (Figure 1; Table S1). Our mechanistic data show that this pattern of ANP32A expression would pressure the emergence of human-signature adaptations in PB2. This might be especially relevant in passerine birds and their close relatives, which tend to express higher proportions of the less potent chANP32A29 than the typical influenza hosts, migratory aquatic birds. Thus, inherent host differences in splicing of ANP32A transcripts have the potential to shape the evolution of PB2 in natural avian hosts and poise viruses for cross-species transmission by ‘‘pre-adapting’’ them for replication in mammals. In summary, we show that chANP32A isoforms differentially stimulate the enzymatic activity of restricted polymerases and posit that host-specific ANP32A splicing patterns affect replication within and transmission from avian reservoirs.
STAR⋆METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Mehle (amehle@wisc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Mammalian cells were grown in DMEM supplemented with 10% FBS: 293T, \; A549, _; MDBK, _; MDCK, \. Chicken cells were grown in DMEM/F-12 with 5% FBS: LMH, _. All cells were maintained at 37C, 5% CO2. Cell stocks were routinely tested for mycoplasma (MycoAlert, Lonza). Human cell lines were authenticated by STR analysis (UAGC).
Viruses
Viruses and plasmid clones were derived from A/WSN/33 (H1N1; WSN) and A/green-winged teal/Ohio/175/1986 (H2N1; S009) (Mehle and Doudna, 2009, 2008). Recombinant virus was rescued by transfecting co-cultures of 293T and MDCK cells with pTM DRNP (encoding WSN vRNA segments HA, NA, M, and NS), the bi-directional pBD plasmids (encoding vRNA and mRNA) PB1, PA, NP, and the indicated PB2 mutants (Mehle and Doudna, 2008; Neumann et al., 2005). S009 WT and SRK viruses contain NP and polymerase genes from S009 and the remaining segments from WSN (Mehle and Doudna, 2009). WSN PB2-FLAG-143 virus (hereafter referred to as WSN PB2-FLAG) was generated as previously described (Kirui et al., 2016b; Dos Santos Afonso et al., 2005). WSN PB2-K627E-FLAG virus was generated similarly. Viral stocks were amplified on MDBK cells and titrated by plaque assay on MDCK cells. Influenza virus infections were performed by inoculating cells with stocks diluted in virus growth media (VGM): DMEM supplemented with penicillin/streptomycin, 25 mM HEPES, 0.3% BSA, and 0.25 – 0.5 mg/ml TPCK-trypsin. Multi-cycle growth curves were performed by inoculating A549 cells at a multiplicity of infection (MOI) of 0.1 at 33C, collecting supernatants at the indicated times postinfection, and titering samples by plaque assay on MDCK cells.
VSV-G pseudotyped lentivirus was prepared by transfecting 293T cells with pLX304, psPAX2 and pMD2.G. Resultant viruses were used to transduce A549 cells. Cells were selected with blasticidin to obtain lines stably expressing ANP32A.
METHOD DETAILS
RNA sequencing analysis
RNA-seq datasets detailed in Table S1 were aligned to ANP32A genomic DNA assemblies from their respective species. Gene indices were built using HISAT2 (function -build; -q -f) (Kim et al., 2015). Indices for Aptenodytes forsteri and Sturnus vulgaris were insufficient for initial alignment. These regions were re-built using a custom Python script (available upon request) to search RNA-seq files for reads mapping proximal to gene regions with poor alignment and using these to complete the gene assembly. Genomic DNA sequence was unavailable for Leucophaeus atricilla, so reads were aligned to Calidris Pugnax and as above, iteratively
corrected to achieve alignment. RNA-seq datasets were first quality assessed using FastQC (Andrews, 2010), trimmed with BBDuk (parameters ktrim = r, k = 21, mink = 10, hdist = 1) (Bushnell, 2015), aligned using HISAT2 (option -U -S), and sorted and indexed with Samtools (Li et al., 2009). Splicing events were visualized in IGV (sashimi plot function) to enumerate the intron-spanning reads between avian exons 4, 5, and 6 (Katz et al., 2015).
Plasmids
Gallus gallus ANP32A encoding a 29 amino acid insert (XM_004943928, chANP32A29) was codon-optimized, synthesized (IDT, Inc.) and cloned into pENTR/TEV/D-TOPO without a stop codon (Invitrogen). This construct was used as a template for inverse PCR to create chANP32A encoding a 33 amino acid insert (XM_413932, chANP32A33). pDONR221-ANP32A encoding human ANP32A (huANP32A) was acquired from the DNASU Plasmid Repository (HsCD00042415). PCR was used to insert the 29 amino acid Gallus repeat into the human gene to create pDONR221-huANP32A+29, to remove the repeat from the Gallus construct to create pENTRchANP32AD, and to introduce a stop codon for generation of untagged ANP32A vectors. Coding sequences for the WSN PB2 627 domain (amino acids 538–676) or the 627 domain plus the NLS (amino acids 538–759) were cloned into pENTR/TEV/D-TOPO. V5-tagged expression constructs for cell culture were created by Gateway recombination (Invitrogen) into pcDNA6.2 and pLX304 (Addgene 25890) (Yang et al., 2011). Bacterial expression constructs were created by recombination into pHGGWA and pHMGWA (Busso et al., 2005). Influenza virus reporter gene and micro gene constructs were previously described (Kirui et al., 2016b).
Polymerase activity assays
RNP complexes were reconstituted by co-transfecting cells with p3X-1T to express PB2, PB1-TAP, and PA, pcDNA6.2 NP-V5 to express NP, and human or chicken pol I-driven vNA-Luc reporters (Mehle and Doudna, 2008). The G3A, C8U (3–8) mutant in the 3′ vRNA UTR was previously described (Neumann and Hobom, 1995) and incorporated into a vNA-Luc reporter by PCR mutagenesis. The relative amount of ANP32A plasmid DNA in the dose escalations series was: Figure 2A and 2D) 10-fold increases starting at 0.004% of total DNA, 2B) 5-fold increases starting at 0.08% of total DNA, 2C) 10-fold increases starting at 0.04% of total DNA, and S1A) 10-fold increases starting at 0.4% total DNA. The sum of ANP32A plasmid DNA in Figure S1B was 0.1% of the total DNA. pRL-SV40 (Promega) constitutively expressing Renilla luciferase was included as a control to normalize gene expression. Cells were lysed one day post-transfection, luciferase activities were measured, and firefly luciferase was normalized to the internal Renilla control. Protein levels in lysate were evaluated by western blotting using anti-V5-HRP (Sigma, 1:10,000) to simultaneously detect PB1-TAP, NP-V5 and ANP32A-V5. At least 3 biological replicate experiments were performed each with R 3 technical replicates per experiment. Results represent mean ± SD of the replicates within one representative experiment.
Immunofluorescence
ANP32A-expressing A549 cells were grown on coverslips and infected with WSN PB2-FLAG at an MOI of 1 for 5 h. Monolayers were fixed and permeabilized (4% paraformaldehyde, 0.1% Triton X-100 in PBS for 10 min each), and blocked at 4C overnight in 3% BSA/PBS. The following primary and secondary antibodies were sequentially incubated for 1 h each at room temperature at 1 mg/ ml in blocking buffer: mouse anti-FLAG (Sigma, F1804) and rabbit anti-V5 (Bethyl, A190–120A) followed by goat anti-mouse Alexa Fluor 594 (Invitrogen, A-11032) and goat anti-rabbit Alexa Fluor 488 (Invitrogen, A-11008). Mounted coverslips were imaged at 40 X using a Zeiss Axio Imager.M2 and captured with an AxioCam MRm. Images were processed identically and pseudocolored in Adobe Photoshop CC.
Co-immunoprecipitations
Plasmids expressing PB1, PA, PB2-FLAG (WT or K627E) and ANP32A-V5 were co-transfected into 293T cells. The catalytically inactive PB1a (D445A/D446A) mutant was created by PCR as previously described (Vreede et al., 2004). To test the effect of viral RNA on polymerase:ANP32A interactions, plasmids expressing vNA-Luc or cNA-Luc reporters were included when indicated. Cells were lysed two days post-transfection in co-immunoprecipitation (co-IP) buffer (50 mM Tris, pH7.5; 150 mM NaCl; 0.5% NP40) and clarified by centrifugation. Lysates were pre-cleared with protein A agarose (Santa Cruz) for 1 h followed by affinity capture with antiFLAG agarose resin (M2, Sigma) overnight. Immunoprecipitates were recovered, washed four times with co-IP buffer and eluted by boiling in Laemmli sample buffer. Immunoprecipitates and input samples were separated by SDS-PAGE, semi-dry transferred to PVDF membranes in Bjerrum Schafer-Nielsen buffer, and probed for ANP32A with anti-V5 HRP (Sigma) or PB2 with rabbit polyclonal anti-PB2 (Mehle and Doudna, 2008). Chemiluminescent images were captured on an Odyssey Fc Imager and quantified using Image Studio v5.2.5 (LI-COR). At least 3 biological replicate experiments were performed.
RNP assembly in the presence or absence of ANP32A was measured as previously described (Mondal et al., 2015). To test protein interactions during infection, wild-type A549 cells or those stably expressing ANP32A were inoculated with WSN PB2-FLAG or PB2-K627E-FLAG (MOI 0.2). Cells were lysed 18–24 hpi and immunoprecipitations were performed as above. NP was detected by western blot with anti-RNP antibody (BEI NR-3133). 2 biological replicate experiments were performed.
Purification and in vitro protein interactions
Rosetta 2(DE3)pLysS E. coli were transformed with pHMGWA-ANP32A or pHGGWA-PB2 constructs, grown to OD 0.8 and induced with 0.5 mM IPTG at 16C for 18–20 h. HisMBP-ANP32A proteins were purified as previously described and dialyzed into 50 mM HEPES pH 7.9, 150 mM KCl, 1 mM DTT and 10% glycerol (Sugiyama et al., 2015). HisGST-PB2 proteins were purified as before and dialyzed into 10 mM Tris-HCl pH 7.0, 200 mM NaCl, 10% glycerol (Tarendeau et al., 2008). In vitro protein interactions were performed by mixing 20 mg each of bait and prey proteins in 100 ml total interaction buffer (10 mM HEPES pH 7.5, 100 mM KCl, 10 mM NaCl, 5 mM MgCl2) followed by rotating for 1 h at room temperature (Habrukowich et al., 2010). Bait proteins were then captured with 10 ml MagneGST glutathione particles (Promega). Pull-down complexes were washed four times with interaction buffer, eluted by boiling in Laemmli sample buffer, separated by SDS-PAGE and detected by Coomassie staining. Input lanes contain 5 mg protein.
Primer extension
NP-independent transcription and replication of a micro-gene vRNA template (77 nt derived from NP gene segment) was performed in transfected 293T cells (Kirui et al., 2016b; Turrell et al., 2013). Total RNA was isolated from cells two days post-transfection using TRIzol (Invitrogen) and the levels of mRNA and vRNA were quantified by primer extension (Fodor and Smith, 2004; Mehle and Doudna, 2008). Primers that recognize NP77 in the plus sense (5′-TGATTTCGATGTCACTCTGTGAGT-3′) or minus sense (5′-GCAGGGTAGATAATCACTGACAGAG-3′) or 5 s ribosomal RNA (5′-ACCCTGCTTAGCTTCCGAGA-3′) were radio-labeled and used in the reaction. These primers are expected to generate products indicative of mRNA (approximately 56–60 nt), vRNA (70 nt) and 5 s RNA (62 nt). Products were resolved by denaturing PAGE (12% acrylamide, 7 M urea, 0.5x TBE), dried and detected by phosphorimaging.
QUANTIFICATION AND STATISTICAL ANALYSIS
A Student’s t test was performed for pairwise comparisons of polymerase activity in the presence of PB2 K627 or PB2 E627. Multiple comparisons were analyzed by one-way ANOVA with post hoc Tukey HSD to identify groups with significant differences.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal V5-HRP (clone V5–10) | Sigma | Cat#: V2260; RRID: AB_261857 |
| Rabbit polyclonal anti-PB2 | Mehle and Doudna, 2008 | N/A |
| Goat polyclonal anti-RNP | BEI Resources | Cat#: NR-3133 |
| Mouse monoclonal anti-FLAG (clone M2) | Sigma | Cat#: F1804; RRID: AB_262044 |
| Rabbit polyclonal anti-V5 | Bethyl | Cat#: A190–120A; RRID: AB_67586 |
| Goat anti-mouse Alexa Fluor 594 | Invitrogen | Cat#: A-11032; RRID: AB_2534091 |
| Goat anti-rabbit Alexa Fluor 488 | Invitrogen | Cat#: A-11008; RRID: AB_143165 |
| Agarose resin: anti-FLAG (clone M2) | Sigma | Cat#: A2220 |
| Bacterial and Virus Strains | ||
| E. coli: strain BL21 Rosetta 2(DE3)pLysS | Novagen | Cat#: 70956 |
| Influenza A virus: A/WSN/33 (H1N1; WSN) | Neumann et al., 2005 | N/A |
| Influenza A virus: A/green-winged teal/Ohio/175/1986 (H2N1; S009) |
Mehle and Doudna, 2009 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Magnetic beads: MagneGST glutathione particles | Promega | Cat#: V8611 |
| MMLV Reverse Transcriptase | Kirui et al., 2016b | N/A |
| T4 Polynucleotide Kinase | NEB | Cat#: M0201L |
| ECL | GE Healthcare | Cat#: GERPN2236 |
| Critical Commercial Assays | ||
| MycoAlert | Lonza | Cat#: LT07–218 |
| Renilla luciferase assay system | Promega | Cat#: E2810 |
| Experimental Models: Cell Lines | ||
| Human: 293T | ATCC | CRL-3216 |
| Human: A549 | ATCC | CCL-185 |
| Cow: MDBK | ATCC | CCL-22 |
| Dog: MDCK | ATCC | CCL-34 |
| Chicken: LMH | ATCC | CRL-2117 |
| Experimental Models: Organisms/Strains | ||
| WSN-PB2-FLAG | Kirui et al., 2016b | N/A |
| WSN-PB2-K627E-FLAG | This paper | N/A |
| S009 SRK | Mehle and Doudna, 2009 | N/A |
| Oligonucleotides | ||
| Primer: NP77F-TGATTTCGATGTCACTCTGTGAGT | Kirui et al., 2016b | N/A |
| Primer: NP77R-GCAGGGTAGATAATCACTGACAGAG | Kirui et al., 2016b | N/A |
| Primer: 5 s- ACCCTGCTTAGCTTCCGAGA | Kirui et al., 2016b | N/A |
| Recombinant DNA | ||
| Plasmid: pENTR chANP32A29 | This paper | N/A |
| Plasmid: pENTR chANP32AD | This paper | N/A |
| Plasmid: pENTR chANP32A33 | This paper | N/A |
| Plasmid: pDONR221 huANP32A | DNASU | HsCD00042415 |
| Plasmid: pDONR221 huANP32A+29 | This paper | N/A |
| Plasmid: pENTR PB2 627 domain | This paper | N/A |
| Plasmid: pENTR PB2 627-NLS | This paper | N/A |
| Plasmid: pcDNA6.2/V5-DEST | Invitrogen | Cat#: 12489027 |
| Plasmid: pENTR/TEV/D-TOPO | Invitrogen | Cat#: K253520 |
| Plasmid: pHGGWA | Busso et al., 2005 | N/A |
| Plasmid: pHMGWA | Busso et al., 2005 | N/A |
| Plasmid: pLX304 | Yang et al., 2011 | Addgene Cat#: 25890 |
| Plasmid: pHH21 vNA-Luc | Mehle and Doudna, 2008 | N/A |
| Plasmid: pHH21 3–8 vNA-Luc | This paper | N/A |
| Plasmid: pHH21 NP-77 | Kirui et al., 2016b | N/A |
| Plasmid: pcDNA PB1a | This paper | N/A |
| Software and Algorithms | ||
| HISAT2 v2.1.0 | Kim et al., 2015 | www.ccb.jhu.edu/people/infphilo |
| BBDuk v37.75 | Bushnell, 2015 | https://sourceforge.net/projects/bbmap/ |
| FastQC v0.11.5 | Andrews, 2010 | https://www.bioinformatics.babraham.ac.uk/ projects/fastqc/ |
| Samtools v1.6 | Li et al., 2009 | http://www.htslib.org/download/ |
| IGV v2.4.4 | Katz et al., 2015 | https://software.broadinstitute.org/software/igv/ |
ANP32A is differentially spliced at the avian-signature exon duplication/insertion
Isoforms of ANP32A differentially stimulate avian-adapted virus polymerase
The polymerase PB2–627 domain engages ANP32A to
stimulate RNA synthesis
ACKNOWLEDGMENTS
Anti-influenza virus RNP antiserum (NR-3133) was obtained through the NIH Biodefense and Emerging Infections Research Resources Repository, National Institute of Allergy and Infectious Diseases (NIAID), NIH. We thank members of the Mehle lab for critical reading of the manuscript. This work was supported by NIH/NIAID grant R01AI125271 and the Greater Milwaukee Foundation Shaw Scientist Award to A.M. S.F.B. was supported by NIAID grant T32AI55397. M.P.L was funded by National Science Foundation (NSF) grant GRFP DGE-1747503. A.M. is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Almond JW (1977). A single gene determines the host range of influenza virus. Nature 270, 617–618. [DOI] [PubMed] [Google Scholar]
- Andrews S (2010). FastQC: A quality control tool for high throughput sequence data http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Bradel-Tretheway BG, Mattiacio JL, Krasnoselsky A, Stevenson C, Purdy D, Dewhurst S, and Katze MG (2011). Comprehensive proteomic analysis of influenza virus polymerase complex reveals a novel association with mitochondrial proteins and RNA polymerase accessory factors. J. Virol 85, 8569–8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B (2015). BBMap (version 37.75) https://sourceforge.net/projects/ bbmap/.
- Busso D, Delagoutte-Busso B, and Moras D (2005). Construction of a set Gateway-based destination vectors for high-throughput cloning and expression screening in Escherichia coli. Anal. Biochem 343, 313–321. [DOI] [PubMed] [Google Scholar]
- Cauldwell AV, Moncorge O, and Barclay WS (2013). Unstable polymerase-nucleoprotein interaction is not responsible for avian influenza virus polymerase restriction in human cells. J. Virol 87, 1278–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Smith GJ, Zhang SY, Qin K, Wang J, Li KS, Webster RG, Peiris JS, and Guan Y (2005). Avian flu: H5N1 virus outbreak in migratory waterfowl. Nature 436, 191–192. [DOI] [PubMed] [Google Scholar]
- Chen H, Li Y, Li Z, Shi J, Shinya K, Deng G, Qi Q, Tian G, Fan S, Zhao H, et al. (2006). Properties and dissemination of H5N1 viruses isolated during an influenza outbreak in migratory waterfowl in western China. J. Virol 80, 5976–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crescenzo-Chaigne B, van der Werf S, and Naffakh N (2002). Differential effect of nucleotide substitutions in the 30 arm of the influenza A virus vRNA promoter on transcription/replication by avian and human polymerase complexes is related to the nature of PB2 amino acid 627. Virology 303, 240–252. [DOI] [PubMed] [Google Scholar]
- Domingues P, and Hale BG (2017). Functional insights into ANP32Adependent influenza A virus polymerase host restriction. Cell Rep 20, 2538– 2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos Afonso E, Escriou N, Leclercq I, van der Werf S, and Naffakh N (2005). The generation of recombinant influenza A viruses expressing a PB2 fusion protein requires the conservation of a packaging signal overlapping the coding and noncoding regions at the 50 end of the PB2 segment. Virology 341, 34–46. [DOI] [PubMed] [Google Scholar]
- Fodor E, and Smith M (2004). The PA subunit is required for efficient nuclear accumulation of the PB1 subunit of the influenza A virus RNA polymerase complex. J. Virol 78, 9144–9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habrukowich C, Han DK, Le A, Rezaul K, Pan W, Ghosh M, Li Z, Dodge-Kafka K, Jiang X, Bittman R, and Hla T (2010). Sphingosine interaction with acidic leucine-rich nuclear phosphoprotein-32A (ANP32A) regulates PP2A activity and cyclooxygenase (COX)-2 expression in human endothelial cells. J. Biol. Chem 285, 26825–26831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengrung N, El Omari K, Serna Martin I, Vreede FT, Cusack S, Rambo RP, Vonrhein C, Bricogne G, Stuart DI, Grimes JM, and Fodor E (2015). Crystal structure of the RNA-dependent RNA polymerase from influenza C virus. Nature 527, 114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Silterra J, Schwartz S, Wong B, Thorvaldsdo´ ttir H, Robinson JT, Mesirov JP, Airoldi EM, and Burge CB. (2015). Quantitative visualization of alternative exon expression from RNA-seq data. Bioinformatics 31, 2400–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirui J, Tran V, and Mehle A (2016a). Host factors regulating the influenza virus replication machinery. In Influenza: Current Research, Wang Q and Tao YJ, eds. (Norfolk, UK: Caister Academic Press; ), pp. 77–100. [Google Scholar]
- Kirui J, Mondal A, and Mehle A (2016b). Ubiquitination up-regulates influenza virus polymerase function. J. Virol 90, 10906–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labadie K, Dos Santos Afonso E, Rameix-Welti MA, van der Werf S, and Naffakh N (2007). Host-range determinants on the PB2 protein of influenza A viruses control the interaction between the viral polymerase and nucleoprotein in human cells. Virology 362, 271–282. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xiao H, Lei F, Zhu Q, Qin K, Zhang XW, Zhang XL, Zhao D, Wang G, Feng Y, et al. (2005). Epidemiology: highly pathogenic H5N1 influenza virus infection in migratory birds. Science 309, 1206. [DOI] [PubMed] [Google Scholar]
- Long JS, Giotis ES, Moncorge O, Frise R, Mistry B, James J, Morisson M, Iqbal M, Vignal A, Skinner MA, and Barclay WS (2016). Species difference in ANP32A underlies influenza A virus polymerase host restriction. Nature 529, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma¨ nz B, Brunotte L, Reuther P, and Schwemmle M (2012). Adaptive mutations in NEP compensate for defective H5N1 RNA replication in cultured human cells. Nat. Commun 3, 802. [DOI] [PubMed] [Google Scholar]
- Mehle A, and Doudna JA (2008). An inhibitory activity in human cells restricts the function of an avian-like influenza virus polymerase. Cell Host Microbe 4, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A, and Doudna JA (2009). Adaptive strategies of the influenza virus polymerase for replication in humans. Proc. Natl. Acad. Sci. U S A 106, 21312–21316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal A, Potts GK, Dawson AR, Coon JJ, and Mehle A (2015). Phosphorylation at the homotypic interface regulates nucleoprotein oligomerization and assembly of the influenza virus replication machinery. PLoS Pathog 11, e1004826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann G, and Hobom G (1995). Mutational analysis of influenza virus promoter elements in vivo. J. Gen. Virol 76, 1709–1717. [DOI] [PubMed] [Google Scholar]
- Neumann G, Fujii K, Kino Y, and Kawaoka Y (2005). An improved reverse genetics system for influenza A virus generation and its implications for vaccine production. Proc. Natl. Acad. Sci. U S A 102, 16825–16829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson BE, Te Velthuis AJW, and Fodor E (2017). Role of the PB2 627 domain in influenza A virus polymerase function. J. Virol 91, e02467–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson D, te Velthuis AJW, Vreede FT, and Fodor E (2014). Host restriction of influenza virus polymerase activity by PB2 627E is diminished on short viral templates in a nucleoprotein-independent manner. J. Virol 88, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflug A, Guilligay D, Reich S, and Cusack S (2014). Structure of influenza A polymerase bound to the viral RNA promoter. Nature 516, 355–360. [DOI] [PubMed] [Google Scholar]
- Reddy S, Kimball RT, Pandey A, Hosner PA, Braun MJ, Hackett SJ, Han KL, Harshman J, Huddleston CJ, Kingston S, et al. (2017). Why do phylogenomic data sets yield conflicting trees? Data type influences the avian tree of life more than taxon sampling. Syst. Biol 66, 857–879. [DOI] [PubMed] [Google Scholar]
- Reich S, Guilligay D, Pflug A, Malet H, Berger I, Cre´ pin T, Hart D, Lunardi T, Nanao M, Ruigrok RW, and Cusack S (2014). Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 516, 361–366. [DOI] [PubMed] [Google Scholar]
- Shinya K, Makino A, Ozawa M, Kim JH, Sakai-Tagawa Y, Ito M, Le QM, and Kawaoka Y (2009). Ostrich involvement in the selection of H5N1 influenza virus possessing mammalian-type amino acids in the PB2 protein. J. Virol 83, 13015–13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbarao EK, London W, and Murphy BR (1993). A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J. Virol 67, 1761–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama K, Kawaguchi A, Okuwaki M, and Nagata K (2015). pp32 and APRIL are host cell-derived regulators of influenza virus RNA synthesis from cRNA. eLife 4, e08939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarendeau F, Crepin T, Guilligay D, Ruigrok RWH, Cusack S, and Hart DJ (2008). Host determinant residue lysine 627 lies on the surface of a discrete, folded domain of influenza virus polymerase PB2 subunit. PLoS Pathog 4, e1000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry E, Guilligay D, Kosinski J, Bock T, Gaudon S, Round A, Pflug A, Hengrung N, El Omari K, Baudin F, et al. (2016). Influenza polymerase can adopt an alternative configuration involving a radical repacking of PB2 domains. Mol. Cell 61, 125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrell L, Lyall JW, Tiley LS, Fodor E, and Vreede FT (2013). The role and assembly mechanism of nucleoprotein in influenza A virus ribonucleoprotein complexes. Nat. Commun 4, 1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreede FT, Jung TE, and Brownlee GG (2004). Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. J. Virol 78, 9568–9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Hatta M, Staker BL, Watanabe S, Imai M, Shinya K, SakaiTagawa Y, Ito M, Ozawa M, Watanabe T, et al. (2010). Biological and structural characterization of a host-adapting amino acid in influenza virus. PLoS Pathog 6, e1001034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, et al. (2011). A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 8, 659–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.