

Abstract
Objective
Ectopic lipid accumulation in the liver and kidneys is a hallmark of metabolic diseases leading to non-alcoholic fatty liver disease (NAFLD) and chronic kidney disease (CKD). Moreover, recent data have highlighted a strong correlation between NAFLD and CKD incidences. In this study, we use two mouse models of hepatic steatosis or CKD, each initiated independently of the other upon the suppression of glucose production specifically in the liver or kidneys, to elucidate the mechanisms underlying the development of CKD in the context of NAFLD-like pathology.
Methods
Mice with a deletion of G6pc, encoding glucose-6 phosphatase catalytic subunit, specifically in the liver (L.G6pc−/− mice) or the kidneys (K.G6pc−/− mice), were fed with either a standard diet or a high fat/high sucrose (HF/HS) diet during 9 months. These mice represent two original models of a rare metabolic disease named Glycogen Storage Disease Type Ia (GSDIa) that is characterized by both NAFLD-like pathology and CKD. Two other groups of L.G6pc−/− and K.G6pc−/− mice were fed a standard diet for 6 months and then treated with fenofibrate for 3 months. Lipid and glucose metabolisms were characterized, and NAFLD-like and CKD damages were evaluated.
Results
Lipid depot exacerbation upon high-calorie diet strongly accelerated hepatic and renal pathologies induced by the G6pc-deficiency. In L.G6pc−/− mice, HF/HS diet increased liver injuries, characterized by higher levels of plasmatic transaminases and increased hepatic tumor incidence. In K.G6pc−/− mice, HF/HS diet increased urinary albumin and lipocalin 2 excretion and aggravated renal fibrosis. In both cases, the worsening of NAFLD-like injuries and CKD was independent of glycogen content. Furthermore, fenofibrate, via the activation of lipid oxidation significantly decreased the hepatic or renal lipid accumulations and prevented liver or kidney damages in L.G6pc−/− and K.G6pc−/− mice, respectively. Finally, we show that L.G6pc−/− mice and K.G6pc−/− mice developed NAFLD-like pathology and CKD independently.
Conclusions
This study highlights the crucial role that lipids play in the independent development of both NAFLD and CKD and demonstrates the importance of lipid-lowering treatments in various metabolic diseases featured by lipid load, from the “rare” GSDIa to the “epidemic” morbid obesity or type 2 diabetes.
Keywords: Metabolic diseases, Lipids, Glycogen, Fenofibrate
Abbreviations: AST, aspartate transaminase; ALT, alanine transaminase; CKD, Chronic Kidney Disease; EMT, epithelial-mesenchymal transition; G6P, glucose-6 phosphate; G6Pase, glucose-6 phosphatase; GSDI, Glycogen Storage Disease type I; HCA, hepatocellular adenomas; HCC, hepatocellular carcinomas; HF/HS, high fat/high sucrose; PPARα, Peroxisome Proliferator Activated Receptor α; STD diet, standard diet; TG, triglyceride
Graphical abstract
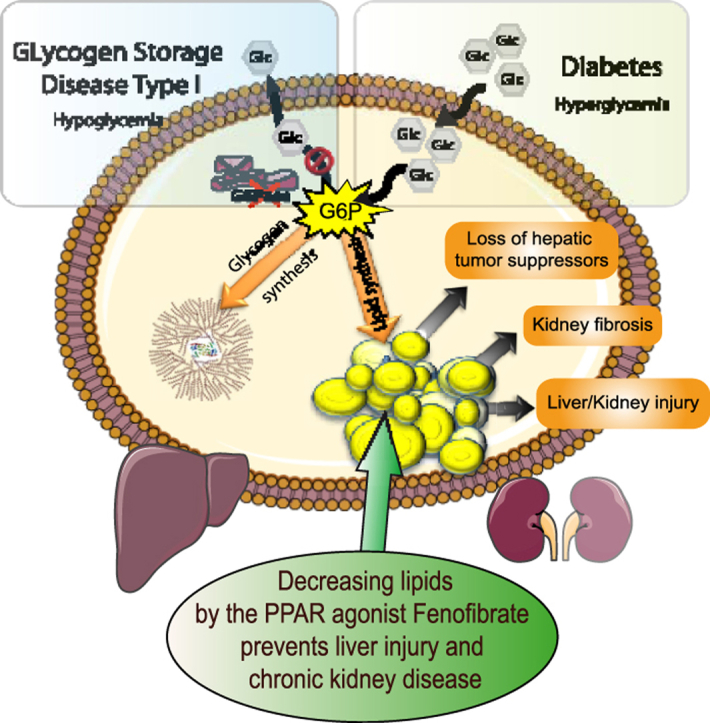
Highlights
-
•
Exacerbating lipid accumulation aggravates liver/kidney injury in GSDI.
-
•
Fenofibrate-mediated PPARα activation induces hepatic and renal lipid turnover.
-
•
Increased lipid turnover prevents glycogen synthesis and accumulation.
-
•
PPARα–mediated metabolic reprograming prevents hepatic and renal GSDI complications.
-
•
NAFLD and CKD develop independently.
1. Introduction
The rise in global epidemics of obesity and type 2 diabetes in westernized countries contributes to a worrying increase in the incidence of non-alcoholic fatty liver disease (NAFLD) and chronic kidney disease (CKD). Both pathologies are associated with high morbidity and mortality, and thus represent serious public health problems. Indeed, NAFLD and CKD have been associated with long-term complications such as hepatic tumorigenesis and renal failure, respectively [1], [2]. A common metabolic feature of these complications is ectopic lipid accumulation, which is suspected to induce organ damage and dysfunction [3]. Indeed, abnormal lipid content in non-adipose tissues is often considered as a major factor involved in cell injury, inflammation, necrosis, and activation of pathological pathways [4], [5], [6], [7], [8], [9].
It is noteworthy that ectopic accumulation of lipids in both the liver and kidneys is an important hallmark of a rare disease named Glycogen Storage Disease type I (GSDI) [10], [11]. GSDI is caused by glucose-6 phosphatase (G6Pase) deficiency, leading to severe hypoglycemia during short fasts [12], [13], [14]. Mutations in G6PC, encoding the catalytic subunit of G6Pase, are responsible for GSD type Ia (GSDIa), while mutations in SLC37A4 encoding the transport subunit of the G6Pase, are responsible for GSD type Ib (GSDIb). G6Pase operates the hydrolysis of glucose-6 phosphate (G6P) in glucose thus allowing liver and kidneys, the main organs responsible for endogenous glucose production, to release glucose in the blood and regulate plasma glucose concentration [15]. The lack of G6Pase induces G6P accumulation in the liver and kidneys, leading to metabolic reprogramming in these organs [16]. The first consequence of G6P increase is abnormal accumulation of glycogen in both organs [11], [14], [17], [18], which has given its name to the disease. However, hepatic and renal lipid metabolism are also markedly altered, characterized by a G6P-dependent increase in de novo lipogenesis and fatty acid chain elongation [10], [11], [17], [19], [20], [21]. Thus, GSDI patients suffer from a combined hypertriglyceridemia and hypercholesterolemia associated with hepatic steatosis, characterized by a low-inflammatory state without fibrosis [22], [23], [24] and abnormal lipid deposition in the kidney cortex [17]. Interestingly, GSDI patients exhibit all NAFLD features, concomitantly with an elevated incidence of hepatocellular adenomas (HCA), which can later transform in hepatocellular carcinomas (HCC) [25], [26]. Moreover, GSDI patients exhibit the first signs of CKD quite early during their youth, which can progress to renal failure with age [10], [13], [14].
For several years, we have used contrasting obesity/diabetes (characterized by over- or unrepressed production of glucose) and GSDI (characterized by a loss of glucose production) as a strategy to reciprocally unravel these two “mirror” pathologies, with special focus on liver and kidneys. This strategy has already allowed us to emphasize the striking similarities between hepatic and renal metabolism in GSDI and diabetes, an increased metabolism downstream of G6P being a common causal feature [20]. For that, we use mouse models of obesity/diabetes and two mouse models with targeted deletions of G6PC either in the liver (L.G6pc−/− mice) or in the kidney (K.G6pc−/− mice). These mice develop GSDIa hallmarks, including deregulated lipid metabolism in the targeted organ exclusively [11], [17]. More precisely, the loss of G6Pase in the liver leads to severe hepatic steatosis, in the absence of fibrosis, and later to the development of HCA/HCC [11]. In kidneys, this leads to tubular damages and then to the development of interstitial fibrosis and glomerulosclerosis, associated with a thickening of the basal glomerular membrane, a loss of podocytes/pedicels and proliferation of mesangial cells, finally responsible for the loss of the renal filtration function [10], [17].
It is noteworthy that, in the obesity/diabetes field, recent data have highlighted a strong correlation between NAFLD and CKD incidences. Moreover, arguments have suggested suggest that NAFLD may play a causal role in CKD development [27], [28], [29]. However, studies firmly unravelling a causal relationship or a mechanism to account for a link between these two pathologies are currently lacking.
In this study, to decipher the role of triglycerides (TG) in the mechanisms underlying both hepatic and renal complications leading to NAFLD or CKD, we first investigated whether and how a diet enriched in lipids could accelerate liver or kidney injuries in L.G6pc−/− and K.G6pc−/− mice. In parallel, using the same mouse models, we examined whether a drug activating intracellular lipid catabolism could prevent or delay NAFLD and CKD, respectively. Finally, a part of this study also allowed us to document whether NAFLD can influence the development of CKD or not.
2. Methods
2.1. Experimental animal models
L.G6pc−/− mice and K.G6pc−/− mice were obtained by deletion of exon 3 of the G6pc specifically in the liver or kidneys (more precisely in the proximal tubules that specifically express G6Pase [30]), respectively, thanks to an inducible CRE-lox strategy, as previously described [11], [17]. Briefly, B6.G6pcex3lox/ex3lox mice were crossed with mice expressing the inducible CREERT2 recombinase under the control of the serum albumin promoter (B6.SACreERT2/w) or under the control of the kidney androgen-regulated protein promoter (B6.KapCreERT2/w) to generate L.G6pc−/− and K.G6pc−/− mice, respectively, after intraperitoneal injections of tamoxifen (five consecutive days in 6–8 weeks old male). Male C57Bl/6J mice (Charles Rivers Laboratories) were also treated with tamoxifen (here referred to as WT mice). Female mice were not used because Kap promoter is under androgenic control.
Mice were housed in the animal facility of Lyon 1 University (ALECS) under temperature controlled (22 °C) conditions and with a 12/12 h light/dark cycle, in enriched environment in groups of 4–6 mice. After tamoxifen treatment, mice were fed either a standard (STD) diet (3.1% fat, 60% carbohydrates, 16.1% proteins, Safe) or a HF/HS diet (36.1% fat, 35% carbohydrates composed by maltodextrine (50%, wt/wt) and sucrose (50%, wt/wt), 19.8% proteins, produced at the “Unité de Préparation des Aliments Expérimentaux”; UE0300 INRA, Jouy-en-Josas, France) for 9 months. Fenofibrate-treated mice were first fed a STD chow diet for 6 months and then a STD diet supplemented with 0.2% fenofibrate (Sigma, wt/wt) (Safe, Augy, France) for additional 3 months. All mice were killed 9 months after tamoxifen treatment by cervical dislocation at 6 h of fasting (with continuous access to water). Tissue was frozen by freeze-clamping in liquid nitrogen and then stored at −80 °C. All the procedures were performed in accordance with the principles and guidelines established by the European Convention for the Protection of Laboratory Animals. The regional animal care committee (C2EA-55, Université Lyon 1, Lyon) approved all the experiments.
2.2. Histological analysis
A piece of fresh liver and a piece of kidney were fixed in formaldehyde and embedded in paraffin. The 4 μm-thick sections were stained with hematoxylin and eosin (H&E) or Masson's Trichrome staining. The slides were examined under a Coolscope microscope (Nikon). For transmission electron microscopy analyses, small pieces of the liver were immediately fixed in 2% glutaraldehyde at 4 °C. The sample was dehydrated in a grade series of ethanol and embedded in an epoxy resin. Tissue was surveyed with a series of 70 nm sections and observed with a Jeol 1400JEM transmission electron microscope equipped with a Orius 100 camera and digital micrograph.
2.3. Urine parameters
K.G6pc−/− mice were housed in individual metabolic cages (Ugo Basile) for urine collection during 24 h. Urea concentration was assessed using a BioAssay Systems colorimetric kit (Hayward, CA, USA). Uric acid concentration was measured with a colorimetric kit (DiaSys, Holzheim, Germany). Albuminuria and lipocalin 2 levels were assessed using a mouse albumin ELISA kit (Neobiotech, Clinisciences, Nanterre, France) and a mouse Lipocalin-2/NGAL ELISA kit (R&D Systems, Lille, France), respectively.
2.4. Plasma parameters
Blood was withdrawn by submandibular bleeding using a lancet after 6 h of fasting. Blood glucose was measured with an Accu-Check Go glucometer (Roche Diagnostic, Meylan, France). Plasma TG, cholesterol, NEFAs, and uric acid concentrations were determined with colorimetric kits (DiaSys, Holzheim, Germany). Β-hydroxybutyrate concentration, aspartate transaminase (AST) and alanine transaminase (ALT) activities were assessed with colorimetric kits (Abcam, Cambridge, UK).
2.5. Glycogen and glucose assay
A piece of 100 mg of frozen tissue was added to 8 volumes of perchloric acid 6% and crushed with the Fast Prep® system (MP Biomedicals). After a centrifugation at 10,000 g for 15 min at 4 °C, the supernatant was neutralized with 3.2 mM K2CO3 and pH was adjusted between 6.5 and 8.5. The solution was then centrifuged at 10,000 g for 5 min at 4 °C and glycogen quantity was then indirectly determined on the supernatant, by using the Keppler and Decker method as previously described [31] Glycogen was partially hydrolyzed with by boiling for 20min in NaOH 0.15 M and then digested into glucose by α-amyloglucosidase for 1 h at 45 °C. Glucose released was measured after the addition of 0.7U of hexokinase and glucose-6-phosphate dehydrogenase in the presence of NADP+. NADPH production was detected at 340 nm.
2.6. Triglyceride assay
Hepatic and renal TG were extracted by the “Folch” procedure. Briefly, a piece of 100 mg of frozen tissue was added to chloroform/methanol 2/1 solution (1.7 mL for 100 mg of tissue) and crushed with the Fast Prep® system. The solution was centrifuged twice at 2,000 g for 10 min at 4 °C. By adding 2 mL of NaCl 0.73% to the supernatant, two phases were created. The inferior organic phase, which contains TG, was kept. After chloroform evaporation, TG were diluted in 100 μL of propanol and measured with a colorimetric kit (DiaSys, Holzheim, Germany).
2.7. Western blots
Western blot analyses were carried out using the whole cell extracts from 50 mg of tissues lysed in lysis buffer (50 mM Tris pH 7.5, 5 mM MgCl2, 100 mM KCl, 1 mM EDTA, 10% glycerol, 1 mM DTT, 1% NP40, protease and phosphatase inhibitors) and homogenized by the FastPrep® system, at 4 °C. Proteins were dosed with Pierce™ BCA Protein Assay Kit. Aliquots of 30 μg proteins, denatured in Laemmli buffer, were separated by 9 or 12%-SDS polyacrylamide gel electrophoresis and transferred to PVDF Immobilon membranes. After 1 h-saturation in TBS/0.2% Tween/5% BSA at room temperature, the membranes were probed (overnight at 4 °C) with rabbit polyclonal antibodies (see Table A.1, Appendices) diluted in TBS/0.2% Tween/5% BSA. After washing with TBST/Tween 0.2%, membranes were incubated (1 h at room temperature) with goat anti-rabbit IgG antibody linked to peroxidase diluted in TBS/Tween 0.2%/BSA 5%. Membranes were rinsed again and exposed to ClarityTM Western ECL Substrate (Bio-Rad). The spots intensity was determined by densitometry with ChemiDoc Software (Bio-Rad) and analysed using the Image Lab™ software (Bio-Rad).
2.8. Gene expression analyses
A piece of 50 mg of frozen tissue was homogenized with the Fast Prep® system and total RNAs were isolated according to the Trizol protocol (Invitrogen Life Technologies). Reverse transcription was done on 1 μg of mRNA using the Qiagen QuantiTect Reverse Transcription kit. Real-time qPCRs were performed using sequence-specific primers with SsoAdvanced™ Universal SYBR® Green Supermix in a CFX Connect™ Real-Time System (Bio-Rad). Primer sequences are indicated in Table A.2 (in Appendices). The expression of mRNA was normalized to the mouse ribosomal protein mL19 transcript (Rpl19) expression using the 2-ΔΔCt method. Western blots were cut to show 3 representative samples of each experimental conditions and/or to remove samples from experimental conditions not used in this paper.
2.9. Statistical analyses
The results are reported as the mean ± s.e.m. (standard error of the mean). Groups were compared using one-way ANOVA followed by Tukey's post hoc test (against all groups) or unpaired two-tailed Student's T test as mentioned in the Legends of figures. Differences were considered to be statistically significant at P-value <0.05.
3. Results
3.1. High fat/high sucrose diet exacerbates ectopic lipid accumulation and aggravates hepatic and renal complications in GSDIa mice
To elucidate the role of TG in hepatic and renal pathologies, we decided to exacerbate lipid accumulation in both the liver and kidneys of L.G6pc−/− and K.G6pc−/− mice with a high fat/high sucrose diet. The progression of the hepatic and renal pathologies with the HF/HS diet was compared with L.G6pc−/− and K.G6pc−/− mice fed a standard diet. As previously observed, L.G6pc−/− mice fed a standard diet developed steatosis as indicated by elevated TG content and lipid droplets in the liver (Figure 1A,Ba), while K.G6pc−/− mice only exhibited a trend to TG accumulation in the kidneys (Figure 1E). As expected, hepatic and renal lipid accumulation was increased when L.G6pc−/− and K.G6pc−/− mice were fed a HF/HS diet, as illustrated by the significant increase in hepatic (Figure 1A–B a and b) and renal TG contents (Figure 1E). Strikingly, HF/HS diet aggravated liver injury and CKD that were characteristic of L.G6pc−/− and K.G6pc−/− mice, respectively (Figure 1). Accordingly, assessment of liver injury markers (ALT, AST) revealed a significant increase in plasmatic transaminase activities in L.G6pc−/− mice fed a HF/HS diet, compared to a STD diet (Figure 1C). In addition, we observed that the development of hepatic tumors in L.G6pc−/− mice was enhanced by HF/HS diet, as mentioned in [32]. Indeed, while none of L.G6pc−/− mice fed a STD diet or WT mice fed a HF/HS diet developed hepatic nodules (data not shown), most (6 out of 7) of L.G6pc−/− mice fed a HF/HS diet for 9 months developed nodules and all of them developed millimetric lesions (Figure 1Bc). The development of nephropathy in K.G6pc−/− mice was also worsened by the HF/HS diet, since urinary excretion of albumin and lipocalin 2, two markers of renal injury, was higher than that observed in K.G6pc−/− fed a STD diet (Figure 1F). In accordance with the loss of renal function, marked fibrosis was observed in kidneys of HF/HS-fed K.G6pc−/− mice, while fibrosis was more discrete in the kidneys of STD-fed K.G6pc−/− mice (Figure 1G). In addition, the renal expression of several genes involved in the development of CKD, i.e. the profibrotic TGF-β1 factor, mesenchymal genes (vimentin, Vim; α-smooth muscle actin 2, Acta2; plasminogen activator inhibitor 1, Pai1; and fibronectin, Fn1), was increased on the HF/HS diet, confirming CKD development aggravation by this diet (Figure 1H). We next analyzed the impact of HF/HS diet on glycogen content, which is known to be elevated in the liver of L.G6pc−/− mice and in the kidneys of K.G6pc−/− mice fed a standard diet. In L.G6pc−/− mice fed a HF/HS diet hepatic glycogen content was similar as that observed in mice fed a standard diet (Figure 1D). Interestingly, renal glycogen content was drastically decreased in K.G6pc−/− mice fed a HF/HS diet, compared to K.G6pc−/− mice fed a standard diet (Figure 1I).
Figure 1.

High fat/high sucrose diet exacerbates hepatic and renal complications in GSDI mice. (A) Hepatic TG content, (B) Histological analyses of livers (HPS staining) and representative image of a liver of L.G6pc−/− mice fed a HF/HS diet, (C) Plasmatic AST and ALT activities, and (D) Glycogen content of livers of WT mice fed a standard (STD) diet (black bars), and L.G6pc−/− fed mice a STD diet (grey bars) or a HF/HS diet (dotted bars) (n = 5–10 mice/group). (E) Renal TG content, (F) Urinary parameters obtained from mouse urine samples collected during 24 h (n = 5–11 mice/group), (G) Histological analyses of Masson's Trichrome staining of the kidneys, (H) Quantitative analyses of Lcn2, Agt, Tgfb1, Fn1, and Pai1 gene expression by RT-qPCR, (I) Glycogen content of kidneys. Data were obtained from WT mice fed a STD diet (black bars), and K.G6pc−/− mice fed a STD (grey bars) or HF/HS diet (dotted bars) (n = 5–10 mice/group). ND = not detected. Data are expressed as the mean ± sem. Significant differences are indicated as ∗ p < 0.05; ∗∗ p < 0.01; ∗∗∗ p < 0.001. Statistical test: One-way ANOVA followed by Tukey's post hoc test.
In conclusion, these results suggest a critical role of ectopic lipid accumulation in the development of hepatic and renal pathologies in GSDIa. In addition, the worsening of hepatic injury and CKD by HF/HS diet was independent of glycogen content, in either L.G6pc−/− mice or K.G6pc−/− mice, respectively.
3.2. Fenofibrate exerts a blood lipid-lowering effect in both L.G6pc−/− and K.G6pc−/− mice
To further assess the role of lipids in the setting of both NAFLD-like and CKD, L.G6pc−/− and K.G6pc−/− mice fed a standard diet were treated with a lipid-lowering drug fenofibrate. Fenofibrate is clinically used to treat dyslipidemia [33]. It acts as an agonist of the Peroxisome Proliferator Activated Receptor α (PPARα), which is known to stimulate lipid oxidation [34], [35], [36]. This treatment was initiated at 6 months after G6pc deletion, the starting point of the first signs of long-term hepatic (i.e. pre-tumoral stage, characterized by severe steatosis, slight inflammation, and absence of HCA/HCC) and renal (i.e. microalbuminuria) pathologies [11], [17], and was continued during 3 months.
The lipid-lowering effect of fenofibrate was confirmed by the assessment of several plasmatic parameters in L.G6pc−/− and K.G6pc−/− mice, compared to WT mice fed a standard diet. While hypertriglyceridemia was observed right after G6pc deletion in L.G6pc−/− mice [11], plasmatic TG levels in L.G6pc−/− mice and K.G6pc−/− mice were similar to those in WT mice after 9 months of G6pc deletion (Table 1), as previously shown [10], [11]. Nevertheless, the levels of plasmatic TG and non-esterified fatty acids (NEFA) were strongly reduced by fenofibrate (Table 1). Finally, as previously shown [37], fenofibrate treatment induced a catabolic phenotype since L.G6pc−/− and K.G6pc−/− mice showed a 20% decrease in body weight during fenofibrate treatment, compared to untreated mice (Table 1). Taken together, these results confirmed the lipid-lowering effect of fenofibrate. Interestingly, cholesterol level in L.G6pc−/− mice was slightly increased under fenofibrate; this could be explained by the fact that fenofibrate is known to increase the synthesis of the HDL cholesterol [38]. Furthermore, as expected, L.G6pc−/− mice exhibited hypoglycemia in the post-prandial state (6 h-food deprivation), since they are unable to mobilize their glycogen stores [11], and fenofibrate treatment did not normalize this parameter (Table 1). K.G6pc−/− mice showed similar blood glucose as WT mice in the absence or presence of fenofibrate. Concomitantly, L.G6pc−/− and K.G6pc−/− mice had increased plasma ketone body concentration, which was further increased with fenofibrate. Finally, plasmatic uric acid concentration, which was slightly increased in K.G6pc−/− mice compared to WT, was further increased with fenofibrate and had a tendency to increase in fenofibrate-treated L.G6pc−/− mice (Table 1).
Table 1.
Body weight and plasmatic parameters of WT, L.G6pc−/− and K.G6pc−/− mice treated or not with fenofibrate.
| WT | L.G6pc−/− | L.G6pc−/−+ fenofibrate | WT | K.G6pc−/− | K.G6pc−/−+ fenofibrate | |
|---|---|---|---|---|---|---|
| TG (g/L) | 0.77 ± 0.05 | 0.75 ± 0.08 | 0.36 ± 0.05 *** ## |
0.86 ± 0.03 | 0.86 ± 0.02 | 0.68 ± 0.01 *** # |
| NEFA (mg/dL) | 26.2 ± 1.8 | 28.2 ± 1.9 | 18.4 ± 1.5 * ## |
27.6 ± 1 | 29.1 ± 0.9 | 21.1 ± 0.9 *** ### |
| Cholesterol (g/L) | 1.7 ± 0.2 | 1.5 ± 0.3 | 2.4 ± 0.2 ## |
1.4 ± 0.1 | 1.2 ± 0.1 | 1.4 ± 0.1 |
| Glucose (mg/dL) | 121 ± 9 | 57 ± 7 *** |
64 ± 4 *** |
149 ± 5 | 138 ± 7 | 133 ± 8 |
| Ketone bodies (nmol/L) | 0.19 ± 0.03 | 0.72 ± 0.15 * |
1.21 ± 0.25 ** |
0.65 ± 0.05 | 1.02 ± 0.06 ** | 1.49 ± 0.05 *** ## |
| Uric acid (mg/L) | 7.8 ± 1.6 | 7.9 ± 0.7 | 10.7 ± 1.6 | 8.3 ± 0.2 | 10.4 ± 0.6 * |
14.1 ± 0.6 *** ### |
| Body weight (g) | 38.7 ± 1.1 | 36.1 ± 0.9 | 29.1 ± 0.4 *** ### |
37.6 ± 1.2 | 33.5 ± 0.6 | 27.6 ± 0.6 *** ### |
WT mice were fed a STD diet and L.G6pc−/− mice were fed a STD diet ± fenofibrate during 9 months. Data were obtained from mice after 6 h of fasting and are expressed as the mean ± S.E.M. (n = 5–14). Significant differences between WT and L.G6pc−/− and between WT and K.G6pc−/− are indicated as * p < 0.05; ** p < 0.01; *** p < 0.001. Significant differences between L.G6pc−/− and fenofibrate-treated L.G6pc−/− and between K.G6pc−/− and fenofibrate-treated K.G6pc−/− are indicated as # p < 0.05; # # p < 0.01; # # # p < 0.001. Statistical test: One-way ANOVA followed by Tukey's post hoc test.
3.3. Fenofibrate normalizes hepatic and renal triglycerides in L.G6pc−/− and K.G6pc−/− mice
Next, lipid metabolism in GSDIa liver and GSDIa kidneys was analyzed to characterize the lipid-lowering effect of fenofibrate in these organs. First, it is noteworthy that the expression of PPARα and Cpt1 was reduced in the liver and kidneys of GSDIa mice, suggesting a decrease in fatty acid oxidation (Figure 2). As expected, lipid catabolism was strongly induced in both L.G6pc−/− livers and K.G6pc−/− kidneys after the treatment with fenofibrate, compared to untreated L.G6pc−/− and K.G6pc−/− mice, respectively (Figure 2). Indeed, we observed a marked PPARα increase in the liver of L.G6pc−/− mice treated with fenofibrate (Figure 2A), associated with a restoration of Ppara gene expression (Figure 2B). Concomitantly, a strong increase in the expression of several genes implicated in lipid catabolism, such as fatty acid binding protein 1 (Fabp1), acyl-CoA oxidase 1 (Acox1) and carnitine palmitoyltransferase 1a (Cpt1a), both involved in mitochondrial fatty acid oxidation, acyl-CoA dehydrogenase long-chain (Acadl) involved in peroxisomal fatty acid oxidation, as well as cytochrome P450 4A10 (Cyp4a10) and cytochrome P450 4A14 (Cyp4a14), both involved in microsomal fatty acid oxidation was observed in the fenofibrate-treated L.G6pc−/− livers (Figure 3B). In addition, the expression of Fgf21, a well-known hepatokine up-regulated by PPARα [39], was highly increased by fenofibrate, further confirming the efficiency of the treatment. Furthermore, in the kidneys of K.G6pc−/− mice treated with fenofibrate, we observed a normalization of PPARα protein levels, the latter being decreased in untreated K.G6pc−/− mice (Figure 2C). Thereby, PPARα increase was accompanied with a rise in the expression of Fabp1, Cyp4a10, and Cyp4a14 (Figure 2D), resulting in a marked activation of renal lipid catabolism.
Figure 2.

Fenofibrate induces strong lipid catabolism in L.G6pc−/−and K.G6pc−/−mice. (A, C) Quantitative analyses of hepatic (A) and renal (C) PPARα by western blot (n = 4–5 mice/group). (B, D) Quantitative analyses of hepatic (B) and renal (D) lipid catabolism by RT-qPCR. The expression of target mRNA of L.G6pc−/− (B) or K.G6pc−/− (D) mice fed a STD diet and treated or not with fenofibrate is expressed relatively to the WT mice fed a STD diet (n = 7–8 mice/group). Data are expressed as the mean ± sem. Significant differences are indicated as ∗ p < 0.05; ∗∗ p < 0.01; ∗∗∗ p < 0.001. Statistical test: One-way ANOVA followed by Tukey's post hoc test.
Figure 3.

Fenofibrate entails an induction of lipid anabolism in L.G6pc−/−and K.G6pc−/−mice. (A–B) Quantitative analyses of hepatic lipid (A) and renal (B) anabolism by RT-qPCR. The expression of target mRNA of L.G6pc−/− (A) or K.G6pc−/− (B) mice fed a STD diet and treated or not with fenofibrate is expressed relatively to the WT mice fed a STD diet (n = 7–8 mice/group). (C–D) Triglycerides (TG) content in the livers (C) and kidneys (D) from WT and L.G6pc−/− (C) or K.G6pc−/− (D) mice treated or not with fenofibrate (n = 5–7 mice/group). Data are expressed as the mean ± sem. Significant differences are indicated as ∗ p < 0.05; ∗∗ p < 0.01; ∗∗∗ p < 0.001. Statistical test: One-way ANOVA followed by Tukey's post hoc test.
Since pharmacological activation of PPARα has also been shown to promote the expression of lipogenic genes [39], we analyzed the expression of the key enzymes of de novo lipogenesis. In the liver of L.G6pc−/− mice and in the kidneys of K.G6pc−/− mice treated with fenofibrate, we observed a high increase in fatty acid synthase (Fasn) and fatty acid elongase 6 (Elovl6), while both enzymes were already up-regulated in untreated KO-mice, compared to WT mice (Figure 3A,B). The expression of stearoyl-CoA desaturase-1 (Scd1) was also increased after fenofibrate treatment (Figure 3A,B). Concomitantly, an increase in the expression of 3-Hydroxy-3-Methylglutaryl-CoA Synthase 2 (Hmgcs2), a key enzyme in ketogenesis, was observed with the fenofibrate treatment in both L.G6pc−/− and K.G6pc−/− mice (Figure 3A,B), in accordance with the increased plasmatic ketone body levels (Table 1). The rise in the expression of genes involved in de novo fatty acid synthesis in L.G6pc−/− and K.G6pc−/− fenofibrate-treated mice might be mediated via the Carbohydrate-responsive element-binding protein (ChREBP) and/or sterol regulatory element-binding protein 1c (SREBP1c), respectively (Figure 3A,B).
Interestingly, in a context of increased oxidation (see above), this increase in lipid anabolism did not induce hepatic or renal lipid accumulation. Indeed, we observed a normalization of TG content in the liver of L.G6pc−/−mice treated with fenofibrate, compared to untreated L.G6pc−/− mice, which exhibited severe hepatic steatosis (Figure 3C). Moreover, TG content was significantly decreased in the kidneys of K.G6pc−/− mice after fenofibrate treatment and was similar to that in WT mice (Figure 3D).
In conclusion, these data suggest a stimulation of lipid turnover by fenofibrate, resulting in a normalization of TG content in both the liver and kidneys of GSDIa mice.
3.4. Fenofibrate decreases glycogen synthesis and prevents hepatic and renal glycogen accumulation
As the modifications of lipid metabolism could have an impact on the whole cell energy homeostasis, we next assessed glycogen metabolism in GSDIa liver and kidneys treated or not treated with fenofibrate. While L.G6pc−/− mice presented excessive glycogen accumulation in the liver, fenofibrate-treated L.G6pc−/− mice exhibited normalized hepatic glycogen content (Figure 4A). In accordance, L.G6pc−/− mice treated with fenofibrate showed a decrease in serine 9 phosphorylation of glycogen synthase kinase 3β (GSK3β), which is known to decrease glycogen synthase activity, thereby decreasing glycogen synthesis (Figure 4B). In addition, glycogen debranching enzyme (AGL), which was increased in untreated L.G6pc−/− mice, was normalized with fenofibrate, suggesting a decrease in glycogen degradation (Figure 4B). These results indicated that the decrease in glycogen content in fenofibrate-treated L.G6pc−/− livers was probably due to a decrease in glycogen synthesis rather than an increase in degradation. Moreover, hepatic glucose content, which was already decreased in L.G6pc−/−mice compared to WT, was further decreased in fenofibrate-treated L.G6pc−/− mice (Figure 4C). Glucose transporter 1 (Slc2a1) expression was decreased in L.G6pc−/− mice treated with fenofibrate, compared to untreated L.G6pc−/− mice, while glucose transporter 2 (Slc2a2) expression remained unchanged (Figure 4D). As previously observed [40], glucokinase (Gck) expression was decreased in L.G6pc−/− mice. Gck expression was further decreased with fenofibrate, which is in line with the observation that Gck is a direct target of PPARα [41] (Figure 4D). These results suggest a decrease in hepatic glucose uptake and phosphorylation, in accordance with the decrease in hepatic glucose levels and in glycogen synthesis.
Figure 4.

Fenofibrate decreases glycogen synthesis and prevents hepatic and renal glycogen accumulation. (A, E) Glycogen content of L.G6pc−/− livers (A) and K.G6pc−/− kidneys (E) treated or not with fenofibrate, compared to WT liver/kidney (n = 5–7 mice/group). (B–F) Quantitative analyses of hepatic (B) and renal (F) GSK3β phosphorylation and AGL by western blot (n = 4–5 mice/group). (C, H) Glucose content of L.G6pc−/− livers (C) and K.G6pc−/− kidneys (G) treated or not with fenofibrate, compared to WT liver/kidney. (n = 5–7 mice/group). (D, H) Quantitative analyses of hepatic (D) and renal (H) glucose utilization by RT-qPCR. The expression of target mRNA in the L.G6pc−/− livers (D) and K.G6pc−/−kidneys treated or not with fenofibrate is expressed relatively to WT liver/kidney (n = 7–8 mice/group). (I) Weight of K.G6pc−/− kidney treated or not with fenofibrate, compared to WT liver/kidney (n = 7–8 mice/group). All mice were fed a STD diet containing or not fenofibrate. Data are expressed as the mean ± sem. Significant differences are indicated as ∗ p < 0.05; ∗∗ p < 0.01; ∗∗∗ p < 0.001. Statistical test: One-way ANOVA followed by Tukey's post hoc test.
Similarly, renal glycogen content in K.G6pc−/− mice was drastically decreased after fenofibrate treatment (Figure 4E). This was associated with a decrease in both glycogen synthesis and degradation, as illustrated by the decrease in phospho-GSK3β at Ser9 and AGL levels (Figure 4F). Renal glucose content was lower in K.G6pc−/− mice than in WT mice and was further decreased after fenofibrate treatment (Figure 4G). This was consistent with concomitant decrease in glucose uptake and phosphorylation, highlighted via the decreased mRNA expression in glucose transporters SLC2A1 and SLC2A2A and in hexokinase enzyme (HK) (Figure 4H). As observed in the liver, these data suggest that the decrease in uptake and phosphorylation of glucose results in a decrease in glycogen synthesis. Interestingly, the decrease in glycogen accumulation in the fenofibrate-treated kidneys was associated with a decrease in nephromegaly, which is a hallmark of GSDIa (Figure 4I). As already shown specifically in rodents [37], [42], fenofibrate induced the increase in liver weight in both L.G6pc−/− and K.G6pc−/− mice (Fig. 1A in Appendices), which could probably be a consequence of the proliferation of peroxisomes in hepatocytes (as observed in Figure 6B, panel c).
Figure 6.

Liver injury is prevented by fenofibrate in L.G6pc−/−mice. (A) Histological analysis of H&E-staining of the livers and (B) Transmission electron microscopy analysis of hepatocytes from WT (a, d), L.G6pc−/−(b, e) and fenofibrate-treated L.G6pc−/−(c, f) mice. Arrows show donut-shaped mitochondria. (C) Quantitative analysis of Plin2 gene expression by RT-qPCR; (D) Plasmatic AST and ALT activities; (E) Quantitative analysis of AMPK phosphorylation (Thr172) by western blot (n = 4–5 mice/group) and (F) Quantitative analyses of Pten gene expression by RT-qPCR from WT and L.G6pc−/− mice treated or not with fenofibrate. The expression of target mRNA in the livers is expressed relative to WT livers (n = 7–8 mice/group). All mice were fed a STD diet containing or not fenofibrate. Data are expressed as the mean ± sem. Significant differences are indicated as ∗ p < 0.05; ∗∗ p < 0.01; ∗∗∗ p < 0.001. Statistical test: One-way ANOVA followed by Tukey's post hoc test.
3.5. Fenofibrate treatment prevents nephropathy development in K.G6pc−/− mice
In order to investigate whether the decrease in renal lipid content affected nephropathy development, renal structure and function were assessed. As mentioned before, K.G6pc−/− mice developed first signs of CKD, i.e. microalbuminuria, associated with an increase in urine excretion and renal expression of lipocalin 2 (Figure 5A,B). In addition, urea and uric acid excretions were slightly increased in K.G6pc−/− mice. Interestingly, renal function was normalized after fenofibrate treatment in K.G6pc−/− mice, with a concomitant decrease in albumin, lipocalin 2, urea, and uric acid excretion (Figure 5A,B). This was in accordance with histological observations of the kidneys. Indeed, H&E staining of K.G6pc−/− kidneys showed a strong tubular clarification due to lipid and glycogen accumulation in the proximal tubules (Figure 5C panel b). Furthermore, strong collagen accumulation was evidenced upon Trichrome Masson's staining, confirming fibrosis development (Figure 5C panel e). On the contrary, histology features of fenofibrate-treated K.G6pc−/− kidneys (Figure 5C panel c) were similar to those in WT mice (Figure 5C panel a). In addition, fibrosis was significantly decreased and even nearly absent in the presence of fenofibrate (Figure 5C, panel f).
Figure 5.

Prevention of nephropathy development by fenofibrate treatment in K.G6pc−/−mice. (A) Urinary parameters of WT mice and K.G6pc−/− mice treated or not with fenofibrate. Data were obtained from mouse urine samples collected during 24 h (n = 5–11 mice/group). (B) Relative lipocalin-2 (Lcn2) gene expression in the kidneys of K.G6pc−/− mice treated or not with fenofibrate, expressed relatively to WT mice. (C) Histological analyses of H&E-staining (panels a–c) and Masson's Trichrome staining (panels d–f) of the kidneys from (a, d) WT, (b, e) K.G6pc−/− and (c, f) fenofibrate-treated K.G6pc−/− mice. (D) Quantitative analyses of renal pro-fibrotic pathways by RT-qPCR (n = 7–8 mice/group) and western blot (n = 4–5 mice/group). The expression of target mRNA of K.G6pc−/− mice treated or not with fenofibrate is expressed relative to the WT mice. All mice were fed a STD diet containing or not fenofibrate. Data are expressed as the mean ± sem. Significant differences are indicated as ∗ p < 0.05; ∗∗ p < 0.01; ∗∗∗ p < 0.001. Statistical test: One-way ANOVA followed by Tukey's post hoc test.
As CKD was prevented in K.G6pc−/− mice treated with fenofibrate, we analyzed molecular mechanisms behind GSDIa nephropathy, which were characterized in previous studies [10], [17]. In K.G6pc−/− mice, the Renin-Angiotensin system (RAS) was induced, which, in turn, increased Tgf-β1 expression, responsible for the activation of the epithelial-mesenchymal transition (EMT) and subsequent fibrosis development. Here, renal Agt expression was significantly decreased in fenofibrate-treated K.G6pc−/− mice, compared to untreated K.G6pc−/− mice (Figure 5D). Consequently, Tgf-β1 expression was normalized with fenofibrate, indicating a decrease in RAS/TGF-β1 signaling. This result was confirmed with the decrease in EMT. Indeed, the expression of epithelial markers, i.e. E-cadherin and β–catenin, which was decreased in untreated K.G6pc−/− kidneys, was restored with fenofibrate (Figure 5D). Furthermore, the expression of the mesenchymal markers, i.e. Vim, Acta2, Pai1 and Fn1 mRNA, which was increased in K.G6pc−/− kidneys, was significantly reduced with the fenofibrate treatment (Figure 5D).
In conclusion, our results strongly report that fenofibrate downregulated the RAS/TGF-β1 pathway of signalization, subsequently inhibiting the EMT process. This inhibition markedly prevented renal fibrosis and thereby maintained the integrity of renal function in fenofibrate-treated K.G6pc−/− mice.
3.6. Liver injury is prevented by fenofibrate in L.G6pc−/− mice
As hepatic steatosis was markedly decreased by fenofibrate in L.G6pc−/− livers, liver structure and function were further characterized. Histological analyses confirmed a marked accumulation of lipid droplets and glycogen in L.G6pc−/− livers (Figure 6A,B, panels b and e), which was strongly reduced with the fenofibrate treatment (Figure 6A,B, panels c and f). Furthermore, TEM revealed a large reduction in the size of the lipid vesicles in the livers of fenofibrate-treated L.G6pc−/− mice (Figure 6B, panel c), compared to untreated L.G6pc−/− mice (Figure 6B, panel b). This observation was in accordance with the increase in Perilipin 2 (Plin2), an important factor in small lipid droplet formation, in the livers of L.G6pc−/− mice treated with fenofibrate (Figure 6C). Interestingly, we observed donut-shaped mitochondria (indicator of cellular stress) in the livers of L.G6pc−/− mice (Figure 6B, panel e) that were observed neither in WT (Figure 6B, panel d), nor in fenofibrate-treated L.G6pc−/− livers (Figure 6B, panel f).
Fenofibrate treatment allowed the normalization of liver injury markers, i.e. AST and ALT activity, compared to untreated L.G6pc−/− mice (Figure 6D). Furthermore, previous studies showed that the metabolic reprogramming occurring in GSDIa livers, as a consequence of the excessive G6P levels, promotes hepatic tumorigenesis [16], [25]. Recent data have shown a decrease in tumor suppressors in L.G6pc−/− livers (unpublished data), in accordance with the pre-neoplastic status of G6pc−/− hepatocytes [16]. Interestingly, tumor suppressors, such as AMP-activated protein kinase (AMPK) and phosphatase and tensin homolog (PTEN), were down-regulated in L.G6pc−/− livers, while fenofibrate restored their expression to the level observed in WT mice (Figure 6E).
Altogether, these data suggest that the normalization in hepatic lipid content in L.G6pc−/− mice results in the restoration of liver function and rescue of tumor suppressor expression.
3.7. Independent development of NAFLD-like damages and CKD
As there is growing recent evidence suggesting an association between NAFLD and CKD [27], [28], [29], we analysed the incidence of NAFLD-like damages in K.G6pc−/− mice and of CKD in L.G6pc−/− mice. This was studied in the context of standard diet, to obviate the possible confounding effects of a HF/HS diet in both organs. On one hand, K.G6pc−/− mice that developed CKD did not seem to exhibit liver injuries. Interestingly, liver weight of K.G6pc−/− mice was slightly reduced compared to WT mice (Figure 7A). Hepatic histology of K.G6pc−/− mice revealed a normal liver structure as well as an absence of liver steatosis in these mice, which was confirmed by the low level of hepatic TG content, despite the development of CKD (Figure 7B–D). In addition, PAS staining and glycogen content assay of K.G6pc−/− liver highlighted a decrease in hepatic glycogen level compared to WT mice (Figure 7C–E). Finally, plasmatic ALT level, a liver injury specific marker was similar in K.G6pc−/− and WT mice. This was consistent with the absence of G6pc targeting in the liver of K.G6pc−/− mice (Figure 7F). On the other hand, the kidneys were not damaged in L.G6pc−/− mice that developed hepatic steatosis and the first signs of liver injury (plasma transaminase increase) (see Figure 1). Indeed, the kidneys weight of L.G6pc−/− is similar as that of WT mice. Moreover, renal lipid metabolism was not altered in L.G6pc−/− mice, suggesting normal renal lipid content in the kidneys of these mice (Figure 7G–H). Finally, it is noteworthy that neither the markers of kidney function nor those of renal fibrosis development were increased in the kidneys of L.G6pc−/− mice, despite the concomitant development of hepatic steatosis (Figure 7I–J). Indeed, the urinary excretion ratio of albumin/creatinine of L.G6pc−/− mice was equal as that in WT mice and was significantly decreased compared to K.G6pc−/− mice while the renal lipocalin 2 expression was unchanged in L.G6pc−/− mice compared to WT mice (Figure 7I). Furthermore, the expression of genes involved in the fibrotic pathway induced by RAS activation in L.G6pc−/− mice was similar as that observed in WT mice (Figure 7J).
Figure 7.

Independent development of NAFLD-like pathology and CKD in L.G6pc−/−and K.G6pc−/−mice, respectively. (A) Liver weight, (B–C) Histological analyses of HPS (B) and PAS (C) stainings of the livers, (D–E) Hepatic TG (D) and glycogen (E) contents, (F) Plasmatic ALT activities of WT and K.G6pc−/− mice fed a STD diet for 9 months; and (G) Kidney weight, (H) Quantitative analyses of Acaca, Scd1, Elovl6, Cpt1 and Cyp1a14 gene expression by RT-qPCR from WT and L.G6pc−/− mice fed a STD diet for 9 months, (I) Albumin excretion of WT, L.G6pc−/− mice and K.G6pc−/− mice fed a STD diet, (I–J) Quantitative analyses of Lcn2(I), Agt, Tgfb1, Fn1 and Pai1(J) gene expression by RT-qPCR from WT and L.G6pc−/− mice fed a STD diet. Data are expressed as the mean ± sem. Significant differences are indicated as ∗ p < 0.05; ∗∗ p < 0.01; ∗∗∗ p < 0.001. Statistical test: Unpaired two-tailed Student's T test for Panels A–H and J, and One-way ANOVA followed by Tukey's post hoc test for Panel I.
These results show that K.G6pc−/− mice that develop CKD did not show any liver injury or steatosis and L.G6pc−/− mice, despite hepatic steatosis and the first signs of hepatic injury, did not show sign of CKD.
4. Discussion
GSDIa is a pathology characterized by abnormal glycogen and lipid accumulation, specifically in the liver and kidneys, possibly leading to hepatic tumor development and renal failure with age. The deficiency in G6Pase and the subsequent G6P accumulation in hepatocytes and renal proximal tubules are at the origin of an important metabolic remodeling [16], [25], [30]. Indeed, the increased utilization of G6P, a common substrate for both lipid and glycogen synthesis, markedly induces these anabolic pathways in GSDIa. Thus, excessive lipid accumulation is mainly due to an increase in de novo lipogenesis [11], [17], [19], [21], [43]. In addition, our results also suggest impaired fatty acid oxidation characterized by a significant decrease in PPARα activity in both the liver and kidneys, in agreement with the decreased mitochondrial oxidative capacity previously observed in GSD1a [44]. It is noteworthy that in diabetes about 60% of TG accumulated in the liver of obese/diabetic patients originates from NEFA, 10% originating from the diet and almost 30% from de novo lipogenesis [45]. Additionally, mitochondrial fatty acid oxidation is defective in this situation. Thus, while ectopic lipid storage is aggravated by lipid spillover from adipose tissue in obesity/insulin resistance, this metabolic mechanism does not seem to be important in GSDIa. However, even though plasmatic NEFA are slightly elevated in GSDIa mice during fasting [46], their contribution to hepatic steatosis has never been addressed. While the origin of lipids accumulated in the liver, and probably in the kidneys, seems to be slightly different, we previously emphasized how the hepatic and renal long-term pathologies of GSDIa strikingly matches what occurs in diabetes [20]. Until now, the specific contribution of lipids to the GSDIa pathology was a subject of speculation, as it is the case for the exact role of ectopic lipids in NAFLD or CKD in diabetes [27]. In this study, we demonstrate that excessive lipid accumulation in the liver and kidneys is a major contributor to the development of long-term complications in both organs, i.e. NAFLD-like complications and CKD. Indeed, the exacerbation of hepatic and renal lipid content by a HF/HS diet significantly accelerated the development of these pathologies. Interestingly, fenofibrate treatment, which led to a normalization of both hepatic and renal lipid stores in L.G6pc−/− and K.G6pc−/− mice, prevented both liver and kidney injuries. Finally, this study points out that CKD can develop in the absence of NAFLD, and more importantly that CKD onset is not a consequence of NAFLD, especially in GSDIa. Since molecular mechanisms behind CKD are similar in GSDIa and diabetes, in particular the activation of RAS [20], we propose that extending these last results obtained in mice with GSDIa to diabetes may be relevant.
Regarding GSDIa pathology, it is noteworthy that submitting L.G6pc−/− mice to a HF/HS diet exacerbated hepatic lipid accumulation, without modification of glycogen content. This led to an acceleration of hepatic injuries with the development of hepatic tumors. In addition, CKD was strongly aggravated by HF/HS diet in K.G6pc−/− mice, whereas renal glycogen stores were decreased. These data suggest an important role for lipids rather that for glycogen in the induction of NAFLD-like and CKD complications in GSDIa. This constitutes a first breakthrough of this study since glycogen was considered as the main responsible for the induction of the physiopathological outcomes of the disease. The central role of lipids was confirmed through decreasing ectopic lipid accumulation using fenofibrate. Indeed, enhanced PPARα activity strongly stimulated the expression of genes involved in lipid catabolism in L.G6pc−/− livers and K.G6pc−/− kidneys, that entailed a decrease in hepatic and renal TG content and prevention of hepatic injuries and CKD. It is important to note that the beneficial effects of fenofibrate can also be attributed to a decrease in glycogen stores (see below).
As for K.G6pc−/− kidneys, this reno-protective effect of lipid lowering was associated with the inhibition of RAS and TGF-β1 pathways, preventing EMT and thereby, renal fibrosis. These results are in accordance with previous data reporting that fenofibrate prevented the induction of RAS/TGF-β1 pathway and fibrosis in diabetic and/or obese mice by decreasing lipids [47], [48], [49], [50], [51], [52]. At the molecular level, it was already proposed that lipid derivatives, such as diacylglycerol and ceramides, are potent activators of protein kinase C, leading to the activation of RAS pathway in kidneys [5], [53].
As for L.G6pc−/− liver, the decrease in steatosis by fenofibrate treatment lowered the risks associated with NAFLD, as previously observed in mouse models of NAFLD [36], [54], [55]. Indeed, L.G6pc−/− treated mice exhibited a decrease in liver injury markers and a preservation of the expression of tumor suppressors, together with a rescue of hepatocyte histology features. Even though we did not examine the effect of fenofibrate on the long-term development of hepatic tumors, a protective effect is likely, given the strong link between hepatic tumorigenesis and NAFLD, as suggested in both GSDIa and obesity/diabetes [8], [32], [56].
Taking into account our results obtained with the HF/HS diet and fenofibrate, lipid lowering is a novel unconventional strategy that should be particularly considered in obesity/diabetes and GSDI to prevent not only hypertriglyceridemia, but also hepatic and renal long-term complications. Currently, fenofibrate treatment was only recommended in GSDI patients who present severe hypertriglyceridemia (generally over 10 g/L), in order to lower plasmatic TGs [13]. Unfortunately, there are no data addressing the effect of fibrates neither on the onset of CKD, nor on hepatic complications, in patients with GSDI. In addition, our results show an increase in uric acid levels under fenofibrate. Therefore, it is important to pay attention to uric acid levels in GSDI patients treated with fenofibrate, even though they are probably also treated with allopurinol. Most patients presenting the first signs of CKD are generally treated with an inhibitor of ACE or an angiotensin receptor analogue [10]. Therefore, the use and the effects of fenofibrate alone in GSDI patients should be further examined, independently of other treatments.
Interestingly, our study also highlights an additional benefit of fenofibrate that is the marked decrease in hepatic and renal glycogen accumulation observed in L.G6pc−/− and K.G6pc−/− mice, respectively. Indeed, fenofibrate treatment markedly induced lipid turnover, since the expression of several genes involved in de novo lipogenesis was enhanced concomitantly with increased lipid catabolism in L.G6pc−/− and K.G6pc−/− mice. This may protect the liver and kidneys against the putative lipotoxic effects of specific intracellular NEFA or their oxidation products, as previously suggested [39]. Moreover, the induction of lipid turnover by the PPARα agonist corrected glycogen metabolism, decreasing glycogen accumulation in both the liver and kidneys of respective GSDIa mouse models. This decrease was not due to an increase in glycogen degradation but rather to an inhibition of glycogen synthesis. Thereby, we suggest that the glycogen metabolism remodeling by fenofibrate derives from the diversion of G6P from glycogen synthesis towards lipid anabolism, thus resulting in a decreased glycogen synthesis. The decrease in glucose uptake (glucose transporters) and phosphorylation (glucokinase/hexokinases) should also greatly contribute to the decrease in G6P availability for glycogen synthesis. This is in keeping with the inhibitory effects of PPARα on glucose uptake and glycolysis and on glycogen synthesis as well [57]. These processes may play a key role in the amelioration of hepatic metabolism or the correction of nephromegaly observed after treatment. The beneficial consequence of fenofibrate on excessive glycogen accumulation is another key breakthrough in the knowledge of the GSDIa pathology.
Finally, L.G6pc−/− and K.G6pc−/− mice represent unique models of NAFLD-like or CKD in that proneness to the diseases can be initiated separately in the respective organs. Importantly, liver injuries and CKD occur independently in these GSDIa mouse models, strongly suggesting that fatty liver does not affect renal function and CKD development can be induced only by an alteration of intra-renal metabolism and without hepatic steatosis, whereas many recent studies suggested a causal relationship between these two pathologies [27], [28], [29], [58]. Thus, an intra-organ altered metabolism resulting in lipid accumulation constitutes an independent worsening factor for both liver injuries and CKD.
In conclusion, the current study shows that the mechanisms involved in the development of hepatic and renal long-term complications of GSDIa are mainly initiated by altered lipid metabolism resulting in lipid accumulation, independently of excessive glycogen contents in these organs. Moreover, it provides evidence that pharmacological activation of PPARα could be a suitable therapeutic strategy to prevent the development of hepatic and renal complications in GSDIa, which could likely be extended to GSDIb. Interestingly, this could be relevant for a variety of other metabolic diseases, including obesity and diabetes. Indeed, some studies in obese and/or diabetic patients already evoked renoprotective effects of fenofibrate or beneficial effects of the drug on liver lipid profile in patients with fatty liver [59], [60], [61], [62], [63], [64]. In addition, PPARα agonists were recently suggested as potentially beneficial in the prevention of NAFLD in human [65]. Moreover, new PPARα and/or δ agonists are currently developed by several pharmas, in keeping with their potential action on NAFLD [66]. Thus this work emphasizes that promoting lipid turnover might represent a potential therapeutic approach to prevent both NAFLD-like and CKD development in the rare disease that is GSDI and in the epidemic diseases that are obesity and type 2 diabetes.
Author contributions
L.M. and M.G. designed, conducted experiments, analyzed the data and wrote the paper. M.S. and V.V. were in charge of mouse breeding and animal experiments. L.I. and A.D conducted experiments. H.G. edited the manuscript. GM wrote the paper. FR supervised the studies and wrote the paper.
Acknowledgements
We would like to thank the members of Animaleries Lyon Est Conventionnelle et SPF (ALECS, Université Lyon 1, SFR Santé Lyon Est) for the animal care, the members of the CIQLE platform (Université Lyon 1, SFR Santé Lyon Est) for the TEM observations and the members of the Anipath platform for the histology experiments (Université Lyon 1). We thank Dr. Catherine Postic for constructive discussions.
This work was supported by research grants from the Agence Nationale de la Recherche (ANR-15-CE14-0026-03 and ANR16-CE14-0022-02), the Association Francophone des Glycogénoses, and the Ligue régionale contre le cancer. LM and MG are recipients of funding of the Fondation pour la Recherche Médicale (FRM grant number ECO20160736048) and the Ligue nationale contre le cancer, respectively.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.molmet.2018.07.006.
Contributor Information
Laure Monteillet, Email: laure.monteillet@laposte.net.
Monika Gjorgjieva, Email: gj_moni@yahoo.com.
Marine Silva, Email: marine.silva@univ-lyon1.fr.
Vincent Verzieux, Email: vincent.verzieux@univ-lyon1.fr.
Linda Imikirene, Email: linda.imikirene@etu.udamail.fr.
Adeline Duchampt, Email: adeline.duchampt@inserm.fr.
Hervé Guillou, Email: herve.guillou@inra.fr.
Gilles Mithieux, Email: gilles.mithieux@univ-lyon1.fr.
Fabienne Rajas, Email: fabienne.rajas@univ-lyon1.fr.
Conflict of interest
The authors declare there is no conflict of interest in relation to this work.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Kazancioğlu R. Risk factors for chronic kidney disease: an update. Kidney International Supplements. 2013;3(4):368–371. doi: 10.1038/kisup.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reeves H.L., Zaki M.Y.W., Day C.P. Hepatocellular carcinoma in obesity, type 2 diabetes, and NAFLD. Digestive Diseases and Sciences. 2016;61(5):1234–1245. doi: 10.1007/s10620-016-4085-6. [DOI] [PubMed] [Google Scholar]
- 3.Schaffer J.E. Lipotoxicity: when tissues overeat. Current Opinion in Lipidology. 2003;14(3):281–287. doi: 10.1097/00041433-200306000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Asrih M., Jornayvaz F.R. Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. Journal of Endocrinology. 2013;218(3):R25–R36. doi: 10.1530/JOE-13-0201. [DOI] [PubMed] [Google Scholar]
- 5.Bobulescu I.A. Renal lipid metabolism and lipotoxicity. Current Opinion in Nephrology and Hypertension. 2010;19(4):393–402. doi: 10.1097/MNH.0b013e32833aa4ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herman-Edelstein M., Scherzer P., Tobar A., Levi M., Gafter U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. Journal of Lipid Research. 2014;55(3):561–572. doi: 10.1194/jlr.P040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park E.J., Lee J.H., Yu G.-Y., He G., Ali S.R., Holzer R.G. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Streba L.A.M., Vere C.C., Rogoveanu I., Streba C.T. Nonalcoholic fatty liver disease, metabolic risk factors, and hepatocellular carcinoma: an open question. World Journal of Gastroenterology – WJG. 2015;21(14):4103–4110. doi: 10.3748/wjg.v21.i14.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Vries A.P.J., Ruggenenti P., Ruan X.Z., Praga M., Cruzado J.M., Bajema I.M. Fatty kidney: emerging role of ectopic lipid in obesity-related renal disease. The Lancet – Diabetes & Endocrinology. 2014;2(5):417–426. doi: 10.1016/S2213-8587(14)70065-8. [DOI] [PubMed] [Google Scholar]
- 10.Gjorgjieva M., Raffin M., Duchampt A., Perry A., Stefanutti A., Brevet M. Progressive development of renal cysts in glycogen storage disease type I. Human Molecular Genetics. 2016;25(17):3784–3797. doi: 10.1093/hmg/ddw224. [DOI] [PubMed] [Google Scholar]
- 11.Mutel E., Abdul-Wahed A., Ramamonjisoa N., Stefanutti A., Houberdon I., Cavassila S. Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. Journal of Hepatology. 2011;54(3):529–537. doi: 10.1016/j.jhep.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 12.Chou J.Y. The molecular basis of type 1 glycogen storage diseases. Current Molecular Medicine. 2001;1(1):25–44. doi: 10.2174/1566524013364112. [DOI] [PubMed] [Google Scholar]
- 13.Froissart R., Piraud M., Boudjemline A.M., Vianey-Saban C., Petit F., Hubert-Buron A. Glucose-6-phosphatase deficiency. Orphanet Journal of Rare Diseases. 2011;6(1):27. doi: 10.1186/1750-1172-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kishnani P.S., Austin S.L., Abdenur J.E., Arn P., Bali D.S., Boney A. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genetics in Medicine – Official Journal of the American College of Medical Genetics. 2014;16(11):e1. doi: 10.1038/gim.2014.128. [DOI] [PubMed] [Google Scholar]
- 15.Soty M., Gautier-Stein A., Rajas F., Mithieux G. Gut-brain glucose signaling in energy homeostasis. Cell Metabolism. 2017;25(6):1231–1242. doi: 10.1016/j.cmet.2017.04.032. [DOI] [PubMed] [Google Scholar]
- 16.Gjorgjieva M., Oosterveer M.H., Mithieux G., Rajas F. Mechanisms by which metabolic reprogramming in GSD1 liver generates a favorable tumorigenic environment. Journal of Inborn Errors of Metabolism and Screening. 2016;4 2326409816679429, (in press), (PMID:29869165) [Google Scholar]
- 17.Clar J., Gri B., Calderaro J., Birling M.-C., Hérault Y., Smit G.P.A. Targeted deletion of kidney glucose-6 phosphatase leads to nephropathy. Kidney International. 2014;86(4):747–756. doi: 10.1038/ki.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rake J.P., Visser G., Labrune P., Leonard J.V., Ullrich K., Smit G.P.A. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I) European Journal of Pediatrics. 2002;161(Suppl. 1):S20–S34. doi: 10.1007/s00431-002-0999-4. [DOI] [PubMed] [Google Scholar]
- 19.Bandsma R.H.J., Prinsen B.H., de Sain-van der Velden M., Rake J.-P., Boer T., Smit G.P.A. Increased de novo lipogenesis and delayed conversion of large VLDL into intermediate density lipoprotein particles contribute to hyperlipidemia in glycogen storage disease type 1a. Pediatric Research. 2008;63(6):702–707. doi: 10.1203/PDR.0b013e31816c9013. [DOI] [PubMed] [Google Scholar]
- 20.Rajas F., Labrune P., Mithieux G. Glycogen storage disease type 1 and diabetes: learning by comparing and contrasting the two disorders. Diabetes & Metabolism. 2013;39(5):377–387. doi: 10.1016/j.diabet.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Reijngoud D.-J. Flux analysis of inborn errors of metabolism. Journal of Inherited Metabolic Disease. 2018;41(3):309–328. doi: 10.1007/s10545-017-0124-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bandsma R.H.J., Smit G.P.A., Kuipers F. Disturbed lipid metabolism in glycogen storage disease type 1. European Journal of Pediatrics. 2014;161(1):S65–S69. doi: 10.1007/s00431-002-1007-8. [DOI] [PubMed] [Google Scholar]
- 23.Derks T.G.J., van Rijn M. Lipids in hepatic glycogen storage diseases: pathophysiology, monitoring of dietary management and future directions. Journal of Inherited Metabolic Disease. 2015;38(3):537–543. doi: 10.1007/s10545-015-9811-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Postic C., Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. Journal of Clinical Investigation. 2008;118(3):829–838. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calderaro J., Labrune P., Morcrette G., Rebouissou S., Franco D., Prévot S. Molecular characterization of hepatocellular adenomas developed in patients with glycogen storage disease type I. Journal of Hepatology. 2013;58(2):350–357. doi: 10.1016/j.jhep.2012.09.030. [DOI] [PubMed] [Google Scholar]
- 26.Labrune P., Trioche P., Duvaltier I., Chevalier P., Odièvre M. Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature. Journal of Pediatric Gastroenterology and Nutrition. 1997;24(3):276–279. doi: 10.1097/00005176-199703000-00008. [DOI] [PubMed] [Google Scholar]
- 27.Marcuccilli M., Chonchol M. NAFLD and chronic kidney disease. International Journal of Molecular Sciences. 2016;17(4):562. doi: 10.3390/ijms17040562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinn D.H., Kang D., Jang H.R., Gu S., Cho S.J., Paik S.W. Development of chronic kidney disease in patients with non-alcoholic fatty liver disease: a cohort study. Journal of Hepatology. 2017;67(6):1274–1280. doi: 10.1016/j.jhep.2017.08.024. [DOI] [PubMed] [Google Scholar]
- 29.Targher G., Byrne C.D. Non-alcoholic fatty liver disease: an emerging driving force in chronic kidney disease. Nature Reviews Nephrology. 2017;13(5):297–310. doi: 10.1038/nrneph.2017.16. [DOI] [PubMed] [Google Scholar]
- 30.Rajas F., Jourdan-Pineau H., Stefanutti A., Mrad E.A., Iynedjian P.B., Mithieux G. Immunocytochemical localization of glucose 6-phosphatase and cytosolic phosphoenolpyruvate carboxykinase in gluconeogenic tissues reveals unsuspected metabolic zonation. Histochemistry and Cell Biology. 2007;127(5):555–565. doi: 10.1007/s00418-006-0263-5. [DOI] [PubMed] [Google Scholar]
- 31.Mithieux G., Guignot L., Bordet J.-C., Wiernsperger N. Intrahepatic mechanisms underlying the effect of metformin in decreasing basal glucose production in rats fed a high-fat diet. Diabetes. 2002;51(1):139–143. doi: 10.2337/diabetes.51.1.139. [DOI] [PubMed] [Google Scholar]
- 32.Rajas F., Clar J., Gautier-Stein A., Mithieux G. Lessons from new mouse models of glycogen storage disease type 1a in relation to the time course and organ specificity of the disease. Journal of Inherited Metabolic Disease. 2015;38(3):521–527. doi: 10.1007/s10545-014-9761-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duval C., Müller M., Kersten S. PPARalpha and dyslipidemia. Biochimica et Biophysica Acta. 2007;1771(8):961–971. doi: 10.1016/j.bbalip.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 34.Kersten S. Integrated physiology and systems biology of PPARα. Molecular Metabolism. 2014;3(4):354–371. doi: 10.1016/j.molmet.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montagner A., Polizzi A., Fouché E., Ducheix S., Lippi Y., Lasserre F. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut. 2016;65(7):1202–1214. doi: 10.1136/gutjnl-2015-310798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pawlak M., Lefebvre P., Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. Journal of Hepatology. 2015;62(3):720–733. doi: 10.1016/j.jhep.2014.10.039. [DOI] [PubMed] [Google Scholar]
- 37.Oosterveer M.H., Grefhorst A., van Dijk T.H., Havinga R., Staels B., Kuipers F. Fenofibrate simultaneously induces hepatic fatty acid oxidation, synthesis, and elongation in mice. Journal of Biological Chemistry. 2009;284(49):34036–34044. doi: 10.1074/jbc.M109.051052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Hoogt C.C., de Haan W., Westerterp M., Hoekstra M., Dallinga-Thie G.M., Romijn J.A. Fenofibrate increases HDL-cholesterol by reducing cholesteryl ester transfer protein expression. Journal of Lipid Research. 2007;48(8):1763–1771. doi: 10.1194/jlr.M700108-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Badman M.K., Pissios P., Kennedy A.R., Koukos G., Flier J.S., Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metabolism. 2007;5(6):426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 40.Hijmans B.S., Boss A., van Dijk T.H., Soty M., Wolters H., Mutel E. Hepatocytes contribute to residual glucose production in a mouse model for glycogen storage disease type Ia. Hepatology (Baltimore, Md.) 2017;66(6):2042–2054. doi: 10.1002/hep.29389. [DOI] [PubMed] [Google Scholar]
- 41.Soltis A.R., Motola S., Vernia S., Ng C.W., Kennedy N.J., Dalin S. Hyper- and hypo- nutrition studies of the hepatic transcriptome and epigenome suggest that PPARα regulates anaerobic glycolysis. Scientific Reports. 2017;7(1):174. doi: 10.1038/s41598-017-00267-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balfour J.A., McTavish D., Heel R.C. Fenofibrate. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in dyslipidaemia. Drugs. 1990;40(2):260–290. doi: 10.2165/00003495-199040020-00007. [DOI] [PubMed] [Google Scholar]
- 43.Abdul-Wahed A., Gautier-Stein A., Casteras S., Soty M., Roussel D., Romestaing C. A link between hepatic glucose production and peripheral energy metabolism via hepatokines. Molecular Metabolism. 2014;3(5):531–543. doi: 10.1016/j.molmet.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farah B.L., Sinha R.A., Wu Y., Singh B.K., Lim A., Hirayama M. Hepatic mitochondrial dysfunction is a feature of glycogen storage disease type Ia (GSDIa) Scientific Reports. 2017;7:44408. doi: 10.1038/srep44408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donnelly K.L., Smith C.I., Schwarzenberg S.J., Jessurun J., Boldt M.D., Parks E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. Journal of Clinical Investigation. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mutel E., Gautier-Stein A., Abdul-Wahed A., Amigó-Correig M., Zitoun C., Stefanutti A. Control of blood glucose in the absence of hepatic glucose production during prolonged fasting in mice. Diabetes. 2011;60(12):3121–3131. doi: 10.2337/db11-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng R., Ding L., He X., Takahashi Y., Ma J.-X. Interaction of PPARα with the canonic wnt pathway in the regulation of renal fibrosis. Diabetes. 2016;65(12):3730–3743. doi: 10.2337/db16-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hong Y.A., Lim J.H., Kim M.Y., Kim T.W., Kim Y., Yang K.S. Fenofibrate improves renal lipotoxicity through activation of AMPK-PGC-1α in db/db mice. PLoS One. 2014;9(5):e96147. doi: 10.1371/journal.pone.0096147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li L., Emmett N., Mann D., Zhao X. Fenofibrate attenuates tubulointerstitial fibrosis and inflammation through suppression of nuclear factor-κB and transforming growth factor-β1/Smad3 in diabetic nephropathy. Experimental Biology and Medicine (Maywood, N.J.) 2010;235(3):383–391. doi: 10.1258/ebm.2009.009218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park C.W., Zhang Y., Zhang X., Wu J., Chen L., Cha D.R. PPARalpha agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney International. 2006;69(9):1511–1517. doi: 10.1038/sj.ki.5000209. [DOI] [PubMed] [Google Scholar]
- 51.Sohn M., Kim K., Uddin M.J., Lee G., Hwang I., Kang H. Delayed treatment with fenofibrate protects against high-fat diet-induced kidney injury in mice: the possible role of AMPK autophagy. American Journal of Physiology – Renal Physiology. 2017;312(2):F323–F334. doi: 10.1152/ajprenal.00596.2015. [DOI] [PubMed] [Google Scholar]
- 52.Tanaka Y., Kume S., Araki S., Isshiki K., Chin-Kanasaki M., Sakaguchi M. Fenofibrate, a PPARα agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney International. 2011;79(8):871–882. doi: 10.1038/ki.2010.530. [DOI] [PubMed] [Google Scholar]
- 53.Koya D., King G.L. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47(6):859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 54.Kostapanos M.S., Kei A., Elisaf M.S. Current role of fenofibrate in the prevention and management of non-alcoholic fatty liver disease. World Journal of Hepatology. 2013;5(9):470–478. doi: 10.4254/wjh.v5.i9.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van der Veen J.N., Lingrell S., Gao X., Takawale A., Kassiri Z., Vance D.E. Fenofibrate, but not ezetimibe, prevents fatty liver disease in mice lacking phosphatidylethanolamine N-methyltransferase. Journal of Lipid Research. 2017;58(4):656–667. doi: 10.1194/jlr.M070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baffy G. Hepatocellular carcinoma in non-alcoholic fatty liver disease: epidemiology, pathogenesis, and prevention. Journal of Clinical and Translational Hepatology. 2013;1(2):131–137. doi: 10.14218/JCTH.2013.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peeters A., Baes M. Role of PPARα in hepatic carbohydrate metabolism. PPAR Research. 2010;2010 doi: 10.1155/2010/572405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Targher G., Francque S.M. A fatty liver leads to decreased kidney function? Journal of Hepatology. 2017;67(6):1137–1139. doi: 10.1016/j.jhep.2017.08.018. [DOI] [PubMed] [Google Scholar]
- 59.Kaysen G.A. Lipid-lowering therapy in CKD: should we use it and in which patients. Blood Purification. 2017;43(1–3):196–199. doi: 10.1159/000452727. [DOI] [PubMed] [Google Scholar]
- 60.Kostapanos M.S., Florentin M., Elisaf M.S. Fenofibrate and the kidney: an overview. European Journal of Clinical Investigation. 2013;43(5):522–531. doi: 10.1111/eci.12068. [DOI] [PubMed] [Google Scholar]
- 61.Musso G., Cassader M., Gambino R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nature Reviews Drug Discovery. 2016;15(4):249–274. doi: 10.1038/nrd.2015.3. [DOI] [PubMed] [Google Scholar]
- 62.Yaghoubi M., Mansell K., Vatanparastc H., Steeves M., Zeng W., Farag M. Effects of pharmacy-based interventions on the control and management of diabetes in adults: a systematic review and meta-analysis. Canadian Journal of Diabetes. 2017;41(6):628–641. doi: 10.1016/j.jcjd.2017.09.014. [DOI] [PubMed] [Google Scholar]
- 63.Lewis D., Wanner C. Diabetes: should we use fibrates in patients with diabetes and mild CKD? Nature Reviews Nephrology. 2012;8(4):201–202. doi: 10.1038/nrneph.2012.37. [DOI] [PubMed] [Google Scholar]
- 64.Ting R.-D., Keech A.C., Drury P.L., Donoghoe M.W., Hedley J., Jenkins A.J. Benefits and safety of long-term fenofibrate therapy in people with type 2 diabetes and renal impairment: the FIELD Study. Diabetes Care. 2012;35(2):218–225. doi: 10.2337/dc11-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kersten S., Stienstra R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie. 2017;136:75–84. doi: 10.1016/j.biochi.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 66.Staels B., Rubenstrunk A., Noel B., Rigou G., Delataille P., Millatt L.J. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology (Baltimore, Md.) 2013;58(6):1941–1952. doi: 10.1002/hep.26461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.