

Abstract
Gemcitabine combined with birinapant, an inhibitor of apoptosis protein antagonist, acts synergistically to reduce pancreatic cancer cell proliferation. A large‐scale proteomics dataset provided rich time‐series data on proteome‐level changes that reflect the underlying biological system and mechanisms of action of these drugs. A multiscale network model was developed to link the signaling pathways of cell cycle regulation, DNA damage response, DNA repair, apoptosis, nuclear factor‐kappa β (NF‐κβ), and mitogen‐activated protein kinase (MAPK)‐p38 to cell cycle progression, proliferation, and death. After validating the network model under different conditions, the Sobol Sensitivity Analysis was applied to identify promising targets to enhance gemcitabine efficacy. The effects of p53 silencing and combining curcumin with gemcitabine were also tested with the developed model. Merging proteomics analysis with systems modeling facilitates the characterization of quantitative relations among relevant signaling pathways in drug action and resistance, and such multiscale network models could be applied for prediction of combination efficacy and target selection.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑Cell signaling‐based network models can provide a quantitative basis for exploring treatment targets in pancreatic cancer. However, existing QSP or network models often lack rich supporting datasets, diminishing the capability for identifying the model parameters and weakening confidence in the quantitative relationships developed.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑Gemcitabine‐based therapy is the standard of care in pancreatic cancer treatment. However, the mechanisms of action of gemcitabine have not been characterized quantitatively or comprehensively and the selection of agents for combination therapy has been empirical. The systems network model is a promising tool to fully utilize up‐to‐date knowledge and emerging information about gemcitabine and provide quantitative guidance for selection of potential combinations.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑This study provides an approach to utilize large‐scale proteomics data to support systems network model development and balance the biology‐driven and data‐driven features, with fewer requirements for complicated mathematical algorithms. The developed network model quantitatively captured key drug‐relevant underlying biological systems, focusing on cell proliferation, apoptosis, and survival signaling pathways in pancreatic cancer cells, and characterized the mechanisms of action and interactions of gemcitabine and birinapant.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑Promising drug targets are suggested that may enhance gemcitabine‐based treatments based on the quantitative network model. Approaches used in this study can be extended to assess other gemcitabine‐based treatments and identify more combinations that are efficacious.
Pancreatic cancer is one of the most fatal cancers in the United States. With incidence and death rates increasing, it is projected to become the second leading cause of cancer death by 2030.1 It is characterized by late‐stage diagnosis, early metastasis, and drug resistance. Underlying clinical challenges include deregulated signaling pathways, large interindividual variability, intratumor heterogeneity, and complex influences of the tumor microenvironment.2 Comprehensive, high‐throughput sequencing of the pancreatic cancer genome reported >1,000 mutations, categorized into 14 core signaling pathways, including DNA damage control and apoptosis.3 Therefore, combination therapies targeting multiple pathways represent a reasonable paradigm for pancreatic cancer treatment. However, mechanisms of action of the standard‐of‐care drug gemcitabine have not been characterized comprehensively and, as a result, the selection of agents for combination therapy has been empirical.
Cell‐based network modeling, as a Quantitative and Systems Pharmacology (QSP) approach, has yielded important advances in recent years. Such network models merge quantitative information on protein‐protein and drug‐target interactions to describe the biological systems and mechanisms of action of existing drugs.4 A well‐developed model can assist in identifying promising drug targets,5 and model‐based analysis and simulation can also predict which drugs will work together to achieve the desired pharmacological end points.6 In addition, factors underlying variability in efficacy can be explored, making it possible to identify patients who will respond to and benefit from a given therapy.7 Therefore, the QSP approach with a cell signaling‐based network model may resolve the current difficulties in selecting efficacious treatments for pancreatic cancer. However, existing network models have limitations that we have sought to address. Model parameter values often are taken from published literature, rather than derived from experimental data under consistent conditions, and model validation usually relies on the responses of limited end points, for example, the expression status of a handful of protein nodes.
Gemcitabine (2′,2′‐difluorodeoxycytidine) is a nucleoside analog, and birinapant is a bivalent antagonist of inhibitor of apoptosis protein (IAP) that promotes apoptosis. In previous in vitro studies, we demonstrated that gemcitabine and birinapant displayed synergistic interactions,8 and we used proteomic analysis to characterize numerous signaling and functional pathways that might underlie the improved efficacy.9 This report describes a QSP approach to assess quantitatively the biological status of signaling networks, as well as drug actions that perturb those systems. This study utilizes comprehensive, quantitative proteomic data to support the protein interaction relationships and constrain parameter values in the development of a network model. The final model provides an opportunity to improve gemcitabine‐based combination therapy in pancreatic cancer.
Methods
Cell culture and cell‐based experiments
The human pancreatic cancer cell line PANC‐1 was used. Based on previous analysis of concentration‐dependent effects on cell proliferation,8 cells were exposed for 0–72 hours to gemcitabine (20 nM) and birinapant (100 nM), alone or in combination, harvested, and analyzed at intervals. Cell culture and counting, cell cycle analysis using propidium iodide staining, and apoptosis analysis with Annexin V/7‐AAD (aminoactinomycin D) were described previously.8 Liquid chromatography‐mass spectrometry (LC‐MS) analysis of total extracted proteins used a nano‐flow liquid chromatography (LC; Eksigent, Dublin, CA), an Orbitrap Fusion mass spectrometry (MS) analyzer (ThermoFisher, Waltham, MA), and the Ionstar workflow to provide accurate, sensitive, and high‐throughput identification and quantification of proteins, as described previously.9, 10, 11, 12, 13 Analysis of the same protein samples by Western blotting provided supporting information and validation.
Development of the multi‐scale network model
Figure 1 shows a schematic of the QSP model. Details of model components and equations are provided in Appendix S1, aspects of model fitting are in Appendices [Link], [Link], [Link], model code is in Appendix S5, and model parameters are summarized in Table 1. Sequential model fitting approaches were used. In the first stage, the dynamic changes in protein abundance were linked quantitatively, and the relevant parameters were estimated simultaneously. In the second stage, the parameters describing the proteins were fixed as a driving force for the changes in cell cycle distribution, apoptosis, and cell numbers.Most protein data were obtained from the proteomic and follow‐up Western blotting analysis. Treatment‐mediated protein abundance changes were normalized by a control group for each time point, and data normalization is discussed in Appendix S3. Data capturing the dynamic changes in p38 phosphorylation at high gemcitabine concentration,14 gemcitabine‐induced secretion of Fas ligand,15 and the temporal profile of TRAF‐bound cIAP2 degradation16 (Figure S1 in Appendix S2) were obtained from the literature. Data for changes in cell numbers and cell distribution in different cell cycle phases and in apoptosis were obtained from our previous study.8
Figure 1.
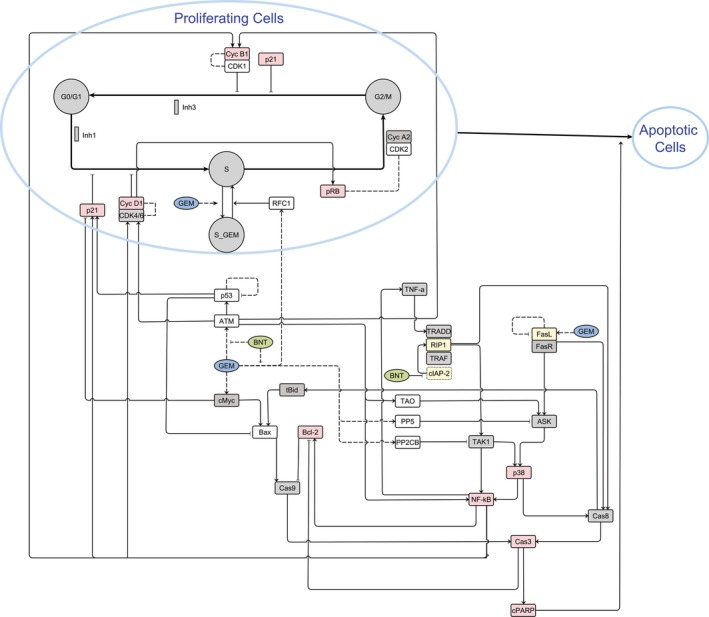
Multi‐scale network model integrating systems information and drug treatment‐mediated effects on cell cycle regulation, DNA damage response, DNA repair, apoptosis, NF‐κB, and MAPK‐p38, signaling pathways, as well as cell cycle distributions (G0/G1‐, S‐, and G2/M‐phases) and apoptosis. The data sources were: proteomic quantification (open boxes), western blot analysis (red boxes), and published literature (yellow boxes). Quantitative information not available is denoted by gray boxes.
Table 1.
Parameter values and coefficients of variation (%CV) estimated in the developed network model (Figure 1) using a two‐stage approach. In the first stage, the dynamics of all protein nodes were estimated simultaneously; in the second stage, the parameters describing protein dynamics were fixed as a driving force for modeling the changes in cell cycle distribution, apoptosis, and cell number
| Parameter (units) | Annotation | Estimate | CV% |
|---|---|---|---|
| First stage | |||
| k atm (h−1) | Turnover rate of protein ATM | 0.126 | 18.5 |
| k g,atm (nM−1) | Induction of protein ATM by gemcitabine | 0.404 | 18.4 |
| k b,atm (nM−1) | Reduction of gemcitabine‐induced ATM by birinapant | 0.00430 | 18.3 |
| k p53 (h−1) | Turnover rate of protein p53 | 0.0301 | 20.6 |
| k p53,fb (h−1) | Rate of negative self‐feedback on protein p53 | 0.792 | 244 |
| H p53,fb | Coefficient for modification of negative self‐feedback on protein p53 | 5.00 | Fixed |
| k fasl (h−1) | Turnover rate of Fas ligand | 0.178 | 8.75 |
| k g,fasl (nM−1) | Induction of Fas ligand by gemcitabine | 6.62 | 13.2 |
| k fasl,fb | Coefficient for modification of negative self‐feedback on Fas ligand | 0.308 | 17.5 |
| k tao (h−1) | Turnover rate of protein for TAO | 0.0137 | 30.2 |
| k pp2cb (h−1) | Turnover rate of protein for PP2CB | 0.101 | 20.6 |
| k g,pp2cb (nM−1) | Induction of protein for PP2CB by gemcitabine | 0.666 | 29.9 |
| k b,pp2cb (nM−1) | Reduction of gemcitabine‐induced PP2CB by birinapant | 0.0103 | 5.49 |
| k pp5 (h−1) | Turnover rate of protein for PP5 | 0.163 | 24.7 |
| k g,pp5 (nM−1) | Induction of protein for PP5 by gemcitabine | 0.439 | 19.4 |
| k b,pp5 (nM−1) | Reduction of gemcitabine‐induced PP5 by birinapant | 0.00791 | 8.74 |
| k tnf (h−1) | Rate of TNF‐α induction by NF‐κB | 0.200 | Fixed |
| k b,iap | Coefficient for cIAP2 degradation induced by birinapant | 0.061 | Fixeda |
| k iap (h−1) | Turnover rate of protein cIAP2 | 0.173 | Fixeda |
| k tnfs (h−1) | Activation rate of TAK1 or Caspase‐8 by TNF‐α | 0.0857 | 17.3 |
| k b,rip | Coefficient for induced RIP1 recruitment by cIAPs degradation | 12.3 | 92.4 |
| k inh,iap | Coefficient for inhibition of TAK1 by cIAPs degradation | 0.600 | Fixed |
| k sti,iap | Coefficient for activation of Caspase‐8 by cIAPs degradation | 0.0925 | 72.8 |
| k tak,p38 | Coefficient for activation of p38 by TNF‐α mediated via TAK1 | 0.0213 | 126 |
| k ask,p38 | Coefficient for activation of p38 via FasL mediated via ASK1 | 0.0323 | 20.5 |
| k tak,p65 | Coefficient for activation of p65 by TNF‐α mediated via TAK1 | 0.129 | 94.7 |
| k atm,p65 | Coefficient for activation of p65 by ATM | 0.280 | 31.4 |
| k p38,p65 | Coefficient for activation of p65 by p38 | 0.497 | 48.4 |
| H fasl | Coefficient for modification of activation of Caspase‐8 by FasL | 0.0567 | 63.4 |
| k g,myc (nM−1) | Induction of Myc activity by gemcitabine | 0.0500 | Fixed |
| k cas8,bax | Induction of Bax by extrinsic apoptotic pathway mediated by tBid | 1.05 | 31.9 |
| H p53,bax | Coefficient for modification of negative regulation of mp53 on Bax | 2.00 | Fixed |
| k apo,bcl | Coefficient for Bcl‐2 degradation induced by Caspase‐3 mediated apoptosis | 0.747 | 18.9 |
| k apo,ind (h−1) | Turnover rate of cleaved PARP | 0.0785 | 16.1 |
| k cycD (h−1) | Turnover rate of cyclin D1 | 0.0160 | 56.9 |
| k p65,cycD | Coefficient for induction of cyclin D1 by NF‐κB | 9.33 | 130 |
| k cycB (h−1) | Turnover rate of cyclin B1 | 0.0795 | 24.6 |
| k p65,cycB | Coefficient for induction of cyclin B1 by NF‐κB | 0.413 | 59.9 |
| k p65,p21 | Coefficient for induction of p21 by NF‐κB | 0.914 | 20.8 |
| k rb (h−1) | Turnover rate of protein Rb | 0.104 | 70.2 |
| k rfc (h−1) | Turnover rate of protein for RFC1 | 0.0619 | 29.8 |
| k g,rfc (nM−1) | Induction of protein for RFC1 by gemcitabine | 0.593 | 33.7 |
| k b,rfc (nM−1) | Reduction of gemcitabine‐induced RFC1 by birinapant | 0.00815 | 9.81 |
| k rb,CDK2 | Coefficient for the positive correlation between activated CDK2 and protein Rb | 1.31 | 27.6 |
| k cycB,CDK1 | Coefficient for the positive correlation between activated CDK1 and cyclin B1 | 0.979 | 19.9 |
| H g | Coefficient for modification of the relationship of gemcitabine concentration vs. protein changes | 0.287 | 16.7 |
| Second Stage | |||
| k inh,G1 | Inhibitory effect of overexpressed cyclin D1 and p21 on G1 phase transition | 0.212 | 27.3 |
| k inh,S | Arrest in S phase regulated by checkpoint proteins represented by protein Rb phosphorylation | 0.0581 | 140 |
| k inh,G2 | Inhibitory effect of overexpressed cyclin B1 and p21 on G2/M phase transition | 0.214 | 16.6 |
| k sti,g,S | Instantaneous S‐phase arrest induced by gemcitabine directly | 0.112 | 6.40 |
| k rfc,S | Effect of protein for RFC1 in reversing the direct gemcitabine‐induced S‐phase arrest | 1.71 | 15.6 |
| T repair,d (h) | Delay in the initiation of DNA repair process | 31.4 | 3.81 |
| R 0 | Initial number of total cells in the culture system | 2.25 × 105 | 1.64 |
| k 1 (h−1) | Rate constant for transition from G0/G1 to S phase | 0.604 | 39.3 |
| k 2 (h−1) | Rate constant for transition from S to G2/M phase | 0.111 | 3.34 |
| k 3 (h−1) | Rate constant for transition from G2/M to G0/G1 phase (mitosis) | 0.339 | 35.6 |
| k apo (h−1) | Rate constant for progression to apoptosis | 0.00530 | 5.16 |
| f 1 | Ratio of rate constants for progression to apoptosis and cleared from the system | 0.211 | 152 |
| k other (h−1) | Rate constant for progression to non‐apoptotic cell death | 0.000436 | 3.82 |
| f(G1) 0 | Initial fraction of cells in the cell cycle of G0/G1 phase | 47.3 | 1.31 |
| f(S) 0 | Initial fraction of cells in the cell cycle of S phase | 11.6 | 4.83 |
| f(apo)0 | Initial fraction of cells undergoing apoptosis | 5.00 | Fixedb |
| f(other)0 | Initial fraction of cells undergoing non‐apoptotic cell death | 1.50 | Fixedb |
| IR50 | Number of cells that cause half maximal growth restriction | 6.47 × 104 | 48.6 |
| I max3 | Ratio of growth restriction on transition rate k 3 vs. transition rate k 1 | 0.830 | 8.47 |
| K other,g | Nonlinear coefficient for induction of non‐apoptotic cell death by gemcitabine | 0.00001 | Fixedb |
| H other,g | Coefficient modifying the induced non‐apoptotic cell death by gemcitabine | 0.1 | Fixedb |
| K other,b | Nonlinear coefficient for induction of non‐apoptotic cell death by birinapant | 0.0045 | Fixedb |
| H other,b | Coefficient modifying the induced non‐apoptotic cell death by birinapant | 0.8 | Fixedb |
Data analysis
Model fitting and parameter estimation was performed using ADAPT 5 with the maximum likelihood algorithm.17 Naïve‐pooled data from all replicate studies were analyzed using the variance model:
| (1) |
where V i is the variance of the response at the ith time point (t i), and Y(t i) is the predicted response at time t i. The variance parameters intercept and slope were estimated together with system parameters. Model performance was evaluated by goodness‐of‐fit criteria, including visual inspection of the fitted curves, sum of squared residuals, Akaike information criteria, Schwarz criterion, and coefficients of variation (CV%) of the estimated parameters.
Global sensitivity analysis
Global Sobol Sensitivity Analysis was implemented via the IQM Tools (IntiQuan, Basel, Switzerland) based on MATLAB (successor of SBPOP/SBTOOLBOX2) and was used to determine the sensitivity of model outputs to changes in the parameters. The parameter lower and upper bounds were set to values fivefold lower and higher than their estimated values. The mean values were based on bootstrapping with 5,000 resamples.
Results
Mechanisms of drug action and interactions that support the model structure
Our proteomic analysis identified and quantified 1,481 proteins that were changed significantly by gemcitabine and birinapant.9 Together with published literature, they provide data for key signaling pathways relevant to drug mechanisms of action and interactions. Mechanistic information for pathways regulating cell cycle progression, DNA damage response (DDR), DNA repair, mitogen‐activated protein kinase (MAPK)‐p38, nuclear factor‐kappa β (NF‐κβ), and intrinsic and extrinsic apoptosis was summarized to support the development of the system network model (Figure 1).
Cell cycle regulation and DNA damage response
The primary mechanism assumed for gemcitabine‐induced S‐phase arrest is by direct gemcitabine action (Figure S2 in Appendix S2).18 Replication stress by gemcitabine induces DDR. The DDR further activates upstream checkpoint proteins, alters the expression of downstream cell cycle regulators, and perturbs cell cycle progression through all phases.19, 20 DNA repair, also activated as a part of the DDR, reverses the direct gemcitabine‐induced S‐phase arrest and restores cell cycle progression.21 Significant gemcitabine‐induced changes in proteins related to DDR regulation and DNA repair, which may contribute to cell survival and drug resistance, were diminished in the presence of birinapant (Figure S3 in Appendix S2).9 Birinapant also activated cyclins and CDKs, and induced protein p21,9 and these effects most likely are mediated by NF‐κβ.22, 23
MAPK‐p38 and NF‐κB
The MAPK‐p38 pathway is activated by gemcitabine‐induced DDR9 via the TAO protein.24 This pathway may also be activated by tumor necrosis factor α (TNF‐α) via TAK1,25 or by FasL through ASK1,26 and negatively regulated by phosphatases regulating DDRs, such as PP2CB and PP5.25, 27 MAPK‐p38 activation by gemcitabine was reported to contribute to caspase‐8 mediated apoptosis.14 Gemcitabine‐induced NF‐κβ activation (Figure S4 in Appendix S2) is part of the DDR and is considered a cell survival pathway contributing to gemcitabine resistance.28 This activation could be mediated by the TAK129, 30 and MAPK‐p3831 pathways, and is countered by protein PP2CB.25 Birinapant interrupts the MAPK‐p38 and NF‐κβ pathways through degradation of TRAF2‐bound cIAP1 and cIAP2.16 In the presence of TNF‐α, birinapant blocks TNF‐α‐mediated activation of TAK1 and reduces activity of NF‐κβ.16 However, we observed that birinapant increased phosphorylation of p65 in the absence of TNF‐α (Figure S4 in Appendix S2). It is possible that rapid degradation of cIAP1 leads to rapid recruitment of RIP1 to TNF‐R1 with further TAK1 activation and p65 phosphorylation.9, 32 The increased NF‐κβ activity would be expected to induce secretion of TNF‐α in a positive‐feedback manner.33
Apoptosis
Both intrinsic and extrinsic apoptosis may be induced by DDR, mediated via p53, c‐Myc, and TAO. Protein p53 is an important switch in the balance between cell survival and death following DNA damage.34, 35 Normally, it can promote apoptosis through upregulation of Bcl‐2 family members, such as Bax. However, p53 is mutated (mp53) in the PANC‐1 cell line, and mp53 may negatively influence the intrinsic apoptosis pathway.36 Anti‐apoptotic Bcl‐2 could be induced by NF‐κβ,23 but is cleaved by activated caspases during apoptosis.37 In addition to the intrinsic apoptosis pathway, gemcitabine was reported to induce Fas ligand expression15, 34; Fas ligation can induce apoptosis directly,34 or further activate the MAPK‐p38 pathway and induce apoptosis.26 Birinapant‐induced degradation of cIAPs leads to formation of a RIPK1:caspase‐8 protein complex and increased caspase‐8 activation through activation of TNFR1 and TRAIL receptors, and switches the TNF‐α/TAK1/NF‐κβ signaling into caspase‐8‐mediated apoptosis.16 The intrinsic apoptosis pathway is also activated by birinapant, mediated by tBid.38 Therefore, strong apoptotic signaling was induced in the presence of birinapant, as confirmed by cleaved PARP (Figure S5 in Appendix S2) and caspase‐3.9
Multi‐scale network model development
Based on these drug‐related responses and signaling mechanisms, a network model incorporating the key pathways were developed (Figure 1). Several assumptions were made to simplify the mechanisms as implemented in the model. First, gemcitabine was assumed to induce rapid S‐phase cell cycle arrest directly. It was also assumed that cyclin D1 and p21 regulate G1‐phase progression that the accumulation of phosphorylated Rb protein represents S‐phase arrest, and that cyclin B1 and p21 regulate G2‐phase progression. The RFC1 protein was selected as a marker for DNA repair because its profile followed the general pattern of other DNA repair proteins (Figure S3 in Appendix S2).9, 39 Binding of TNF‐α to TNFR1, mediated by TAK1, or DNA damage protein TAO and binding of FasL, mediated by ASK1, were assumed to induce the phosphorylation of p38. Proteins PP5 and PP2CB were assumed to regulate ASK1‐mediated p38 activation negatively. Checkpoint protein ATM, the binding of TNF‐α to TNFR1 in the presence of cIAPs, and p38 phosphorylation were assumed to further activate NF‐κβ. The mitochondria‐mediated intrinsic apoptosis pathway was assumed to be induced by Bax and antagonized by Bcl‐2. In the model, Bax is induced by c‐Myc and tBid, with tBid transferring signaling from the caspase‐8‐mediated pathway to the intrinsic pathway. Bcl‐2 was assumed to be activated by NF‐κβ and degraded by the activated caspases. We also assumed that gemcitabine can upregulate mp53, but instead of activating the intrinsic apoptosis pathway, mp53 blocks the upregulation of pro‐apoptotic Bax.40 Extrinsic apoptosis is mediated by caspase‐8 and activated by the binding of TNF‐α to TNFR1 in the absence of cIAPs, the binding of Fas ligand, and p38 phosphorylation. Additional details explaining each node and interaction are provided in Appendix S1. The quantitative relationships among protein‐level drug responses were described by 75 equations, listed in Appendix S1, and the references supporting each equation are provided. The first stage of model development quantitatively and simultaneously linked 21 protein nodes and estimated 46 parameters. In the second stage, protein changes were fixed to drive the changes in cell population distribution and proliferation, and 23 parameters were estimated or fixed based on prior knowledge. A rich dataset, consisting of time‐series quantification of 19 proteins by proteomics, Western blot analysis, and literature sources, as well as changes in cell distribution and cell numbers from our previous studies,8 supported the identifiability of the parameters. The observed data were characterized well (Figure 2), and the parameters were estimated with reasonable CV%, as defined and summarized in Table 1.
Figure 2.
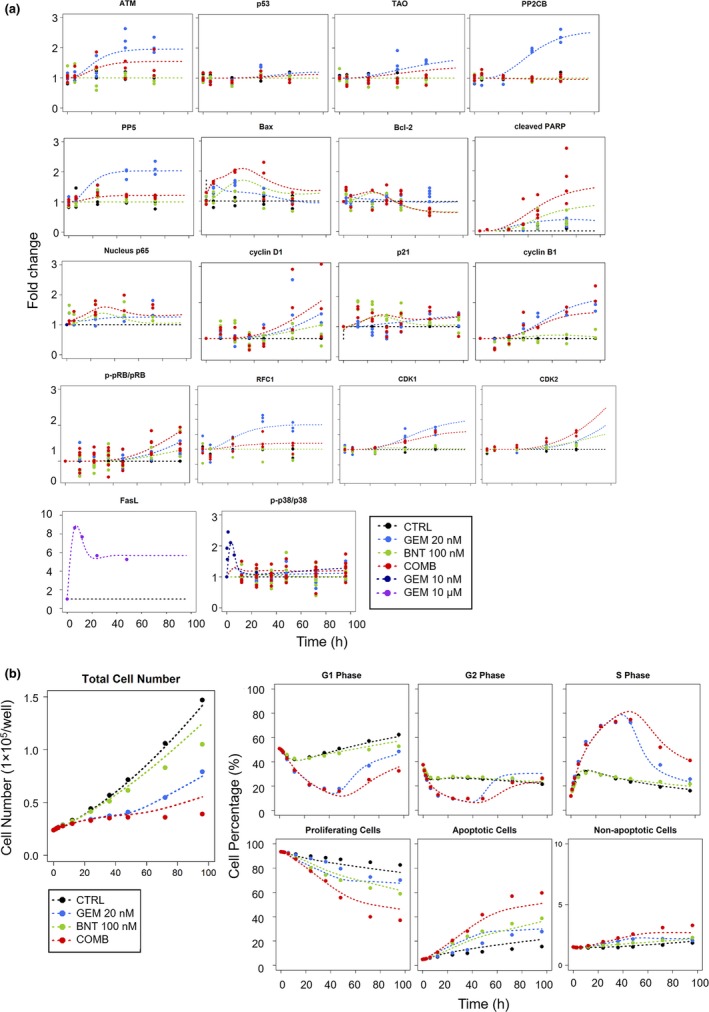
Observations in PANC‐1 cells and fittings based on the network model of Figure 1. Treatment groups included control (CTRL, black), 10 nM, 20 nM, 100 μM gemcitabine (GEM, blue), 100 nM birinapant (BNT, green), and 20 nM/100 nM gemcitabine/birinapant combination (COMB, red). (a) Temporal fold‐change in the abundance of selected protein nodes, and (b) total cell numbers (proliferation) and cell cycle distributions in G0/G1‐, S‐, G2/M‐phases, and in apoptosis. Experimental observations are represented by symbols, and fittings based on the multi‐scale network model (Figure 1) are indicated by curves. Model parameters are listed in Table 1.
Model validation
Several simulations were made under different conditions and compared with experimental observations: (i) the efficacy of gemcitabine in wild‐type p53 (wtp53) vs. mp53, (ii) apoptosis signaling with and without TNF‐α blockade, (iii) transient gemcitabine‐induced S‐phase arrest, and (iv) cell proliferation with simultaneous vs. sequential drug exposure. Consistency of the model predictions with experimental observations increases confidence in the current model and provides model validation. The validated assumptions in mechanisms also provide mechanistic interpretations and insights into previously unexplained drug responses. Appendices S3 and S4 provide further insights into approaches used in modeling this complex system.
Role of p53 mutation
Mutant p53 proteins occur in 50–75% of pancreatic cancers and are involved in regulation of cell survival and drug resistance. The mp53 proteins are incapable of recognizing wtp53 DNA binding sites and can exert oncogenic effects.41 A Kaplan‐Meier plot of 184 patients with pancreatic cancer in PAAD study with mp53 vs. wtp53was generated from The Cancer Genome Atlas database,42 and patients with wtp53 showed higher survival (Figure 3a). Given that gemcitabine is the standard‐of‐care for patients with pancreatic cancer, the impact of p53 mutations on the antiproliferative effects of gemcitabine was explored using the systems model. It was assumed that wtp53 activates Bax,34 whereas mp53 blocks Bax activation.40 Simulations showed that cell proliferation after gemcitabine treatment was similar in the mp53 and wtp53 groups at early times, but at later times, the wtp53 group showed a drastic decrease in proliferation, whereas proliferation remained the same with mp53 (Figure 3a). This demonstrates that the model can capture the diminished efficacy of gemcitabine when p53 mutations are present.
Figure 3.
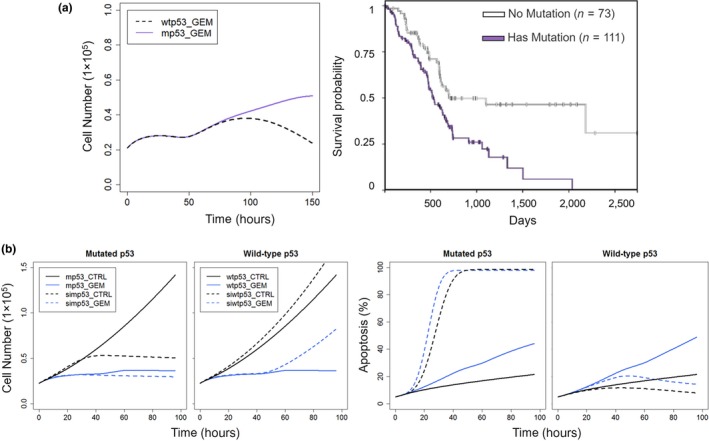
Model‐based predictions of impact of p53 mutations and silencing on gemcitabine‐mediated inhibition of cell proliferation of PANC‐1 cells. (a) Left: Simulations of cell proliferation during exposure to 20 nM gemcitabine in cells with mutant p53 (mp53; purple) and wild‐type p53 (wtp53; black). Right: Kaplan Meier plot showing survival of 184 pancreatic cancer patients in PAAD study with mp53 (purple) and wtp53 (black), generated from the TCGA database using UCSC Xena (https://xenabrowser.net/heatmap/). Abscissa represents days of survival from diagnosis. (b) Silencing p53 shows opposing effects upon cell proliferation and induction of apoptosis by gemcitabine in cells with mp53 vs. wtp53. Profiles of cell numbers (left) and percentage of apoptotic cells (right) in cells with mp53 (solid line) and silenced mp53 (simp53; dashed line) or wtp53 (solid line) and silenced wtp53 (siwtp53; dashed line) in control cells (black) and the presence of 20 nM gemcitabine (blue).
Functional role of TNF‐α
The TNF‐α/NF‐κβ pathway is downstream of the birinapant target (IAP) and NF‐κβ is a key player in the signaling pathways related to gemcitabine mechanisms based on our analysis of upstream regulators.9 Therefore, the TNF‐α/NF‐κβ pathway was investigated in the network model. One challenge was that endogenous TNF‐α concentrations could not be quantified reliably by enzyme‐linked immunosorbent assay, making it difficult to identify the role of this pathway in the observed drug responses. Therefore, PANC‐1 cells were incubated with the anti‐TNF‐α antibody infliximab to investigate the impact of lowering TNF‐α concentrations during drug exposure. In the control‐alone and gemcitabine‐alone groups, addition of infliximab did not change significantly the percentage of apoptotic cells after 96 hours, whereas in the birinapant and gemcitabine/birinapant combination groups, TNF‐α blockage reduced apoptotic cells by 25–30% (Figure 4a), supporting the role of TNF‐α in driving birinapant‐mediated apoptosis. In parallel, the role of TNF‐α was investigated in the network model. By assuming that infliximab reduced TNF‐α by 90%, the model‐predicted percentage of apoptotic cells was similar to the observed results for the four treatment groups (Figure 4b), suggesting that the model captured well the role of TNF‐α.
Figure 4.
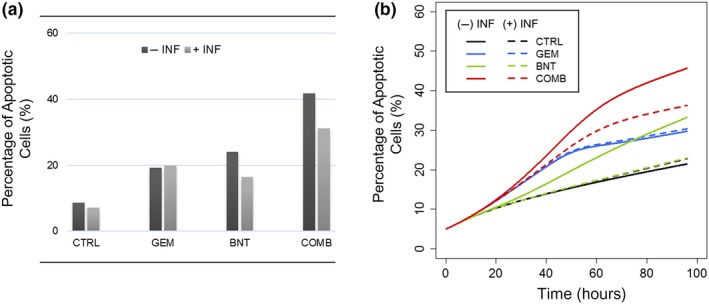
The functional role of TNF‐α, revealed by the effect of anti‐TNF‐α antibody infliximab upon drug‐mediated apoptosis in PANC‐1 cells. Treatment groups included control (CTRL), 20 nM gemcitabine (GEM), 100 nM birinapant (BNT) and 20 nM/100 nM gemcitabine/birinapant combined (COMB) treatment for 96 hours, with (light gray bar) or without (dark gray bar) 1 μg/ml infliximab (INF). Percentage of apoptotic cells (a) measured by annexin V/7‐AAD assay in PANC‐1 cells after treatments of 96 hours, or (b) simulated based on the network model.
Transient gemcitabine‐induced cell cycle arrest and the effect of sequential drug exposure
Previously, gemcitabine was observed to induce rapid but transient S‐phase cell cycle arrest.8 Moreover, with ≥24 hours of gemcitabine exposure, the magnitude and duration of S‐phase arrest became insensitive to duration of gemcitabine exposure (Figure 5a). The network model suggested that delayed DNA repair induced by gemcitabine could explain these findings. In the model, a DNA polymerase accessory protein for RFC1 was used as a pharmacodynamic marker of DNA repair, and induction of RFC1 protein was estimated to occur 30 hours after initiation of gemcitabine exposure. Simulations based on this model showed a decline in S‐phase arrest after 30 hours, which was insensitive to further exposure times, and the model‐predicted S phase temporal distribution profiles (Figure 5b) were similar to observed data (Figure 5a), confirming gemcitabine‐mediated delayed DNA repair as a mechanistic interpretation of this previously unexplained phenomena. Delayed DNA repair after gemcitabine exposure may also explain mechanistically the schedule‐dependent antiproliferative effects of the gemcitabine/birinapant combination observed previously: initiating birinapant after ≥24 hours of gemcitabine exposure produced greater efficacy than simultaneous exposure.8 The assumption was tested with the current network model. Cell proliferation profiles were simulated for a sequential schedule, in which birinapant exposure was initiated after 30 hours of gemcitabine exposure, the time at which activated DNA repair was estimated. At low birinapant concentrations (100 nM), sequential and simultaneous exposure showed similar efficacy (Figure S6 in Appendix S2). However, a higher birinapant concentration (200 nM) was predicted to mediate greater efficacy with sequential exposure (Figure 5c,d). Thus, the network model explained mechanistically the greater efficacy of birinapant sequenced after gemcitabine and supported our previous assumptions, although it slightly underpredicted the efficacy of lower birinapant concentrations combined with gemcitabine.
Figure 5.
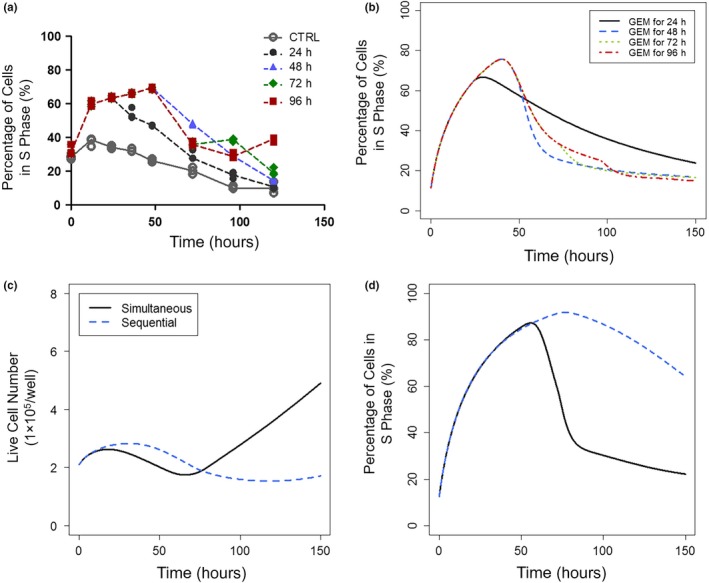
Transient gemcitabine‐induced cell cycle arrest and sequential drug exposure effects. Gemcitabine induced transient cell cycle arrest that was alleviated by delayed initiation of DNA repair. Panels show percentage of cells in S‐phase after incubation with 20 nM gemcitabine for different durations (24, 48, 72, and 96 hours) (a) measured by cell cycle analysis, or (b) predicted by the network model, and cell cycle arrest in S‐phase became insensitive to continuing gemcitabine exposure with ≥24 hours of treatment. Model‐based simulation of cell proliferation and cell cycle arrest with exposure to 20 nM gemcitabine and 200 nM birinapant initiated either simultaneously (solid/black) or sequentially (birinapant initiated 30 hours after gemcitabine; dashed/blue). (c) Sustained suppression of cell proliferation by sequenced exposure, and (d) percentage of cells in S phase with simultaneous vs. sequenced drug exposure. Model prediction recapitulated experimental data showing greater efficacy of gemcitabine/birinapant treatment with sequenced drug exposure.8
Model applications
Several applications of the validated network model were explored, focusing on the selection of potential targets to enhance antitumor efficacy and the impact of genetic variations on therapeutic outcomes.
Sobol Sensitivity Analysis for target selection
To test which molecules or pathways may have the greatest impact on gemcitabine efficacy, the Sobol Sensitivity Analysis was performed with the network model perturbed by gemcitabine. The selected end point was the change in viable cell numbers at 100 hours. The five most important factors observed were: (i) the DNA repair proteins represented by RFC1, (ii) negative self‐feedback or developed resistance to Fas pathway43 activation, (iii) the DDR sensor ATM, (iv) p53, the regulator of DDR, and (v) the G1 cell cycle regulator cyclin D1 (Figure 6a). Thus, targeting pathways related to DNA repair and DDR, as well as the death receptor‐mediated extrinsic pathways were predicted to enhance gemcitabine efficacy. Cell proliferation profiles were simulated under: (i) blockade of RFC1 induced by gemcitabine, and (ii) blockade of developed resistance to Fas, or the addition of external Fas ligand (Figure 6b). Simulations predicted lower cell numbers under all scenarios, and that both proposed approaches could enhance the efficacy of gemcitabine.
Figure 6.
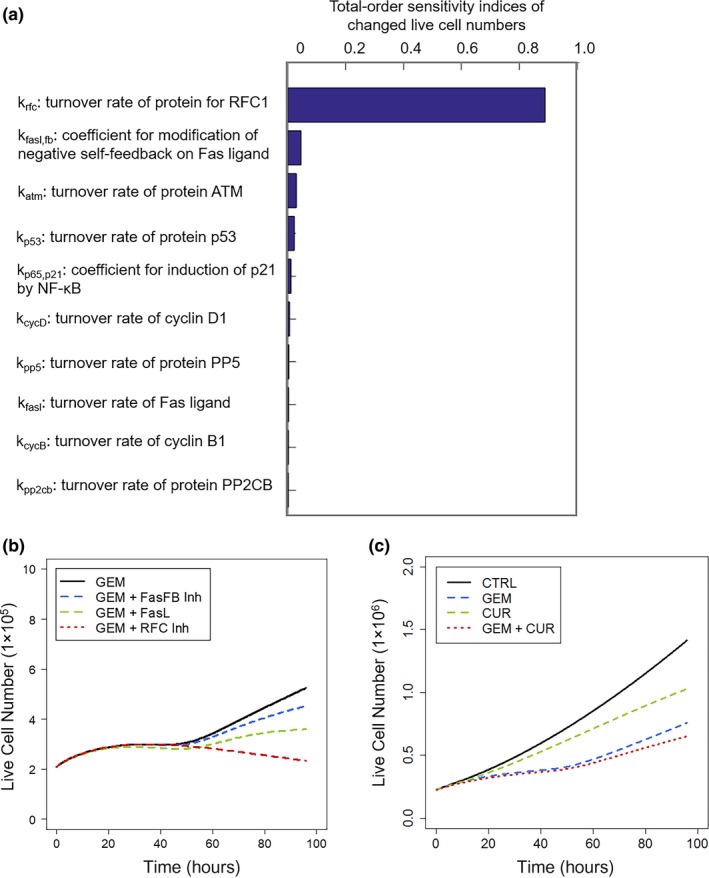
Model‐based analysis and simulations for target selection and prediction of drug combination efficacy. Sobol Sensitivity Analysis assisted in the selection of protein targets that could potentially enhance gemcitabine efficacy. (a) Rank‐order sensitivity indices of model kinetic parameters having the most significant impact on proliferating cell numbers. (b) Simulated profiles of cell proliferation responses to 20 nM gemcitabine alone (GEM; black), development of resistance to Fas pathway signaling (FasFB Inh; blue), Fas ligand exposure (FasL; green), or combined with inhibition of DNA repair (RFC Inh; red). (c) Model‐based simulation of cell proliferation profiles of drug‐free control cells (black) and when treated with gemcitabine (GEM; blue), curcumin (CUR; green), or both (red). Specific model parameters were fixed based upon assumed mechanisms of curcumin action (see text).
Effect of p53 silencing
Because p53 is an important regulator of cell survival and death following DNA damage, gemcitabine effects were investigated under p53 silencing. In the model, the baseline p53 activity was reduced to 10% to mimic p53 silencing, and effects on cell proliferation were simulated for wtp53, wtp53 silencing (siwtp53), mp53, and mp53 silencing (simp53; Figure 3b). Silencing mp53 would relieve its suppression of apoptotic signaling, and the model predicted considerably reduced cell proliferation in the control group (Figure 3b). Despite that, the model predicted little improvement in gemcitabine efficacy with simp53. However, silencing wtp53 reduced the activation of apoptotic signaling, and the efficacy of gemcitabine was reduced considerably (Figure 3b). These simulation‐based hypotheses remain to be tested, but suggest that direct p53 silencing may not have major clinical benefits.
Efficacy of an empirically selected combination
Literature reports suggest that curcumin, a natural compound extracted from turmeric (Curcuma longa), may exert beneficial effects in various cancers via inhibition of constitutively activated NF‐κβ, p53 activation, and downregulation of NF‐κβ‐regulated proteins such as Bcl‐2, IAPs, and also cyclin D1, c‐Myc inhibition.44, 45 One study indicated that curcumin also potentiated the apoptotic effects of gemcitabine and enhanced its antitumor effects.45 However, clinical trials show no benefit of curcumin with gemcitabine‐based therapy,46 and in vitro studies demonstrated antagonistic effects when combining gemcitabine and curcumin (Appendix S6). This combination was investigated mechanistically by simulation with the network model. Curcumin was assumed to reduce the activation of NF‐κβ by 90%, decrease NF‐κβ‐regulated proteins, activate p53, and block the activation of c‐Myc.44, 45 Cell proliferation profiles were simulated for control, gemcitabine, curcumin, and combined gemcitabine/curcumin groups. Although simulations showed that both gemcitabine and curcumin decreased cell proliferation as single agents, the two combined were less efficacious than expected for simple additivity (Figure 6c). This result may reflect the complex roles that the major curcumin targets play in functions related to cell progression and repression. For example, although NF‐κβ induces Bcl‐2 expression and apoptosis resistance, it also affects the expression of cyclins and is partially responsible for cell cycle arrest. Thus, simulations with the model suggest a potential mechanistic rationale that is consistent with clinical and in vitro findings that curcumin does not enhance gemcitabine‐based therapies.
Discussion
A recent study of transcriptomic and genomic data showed little predictive power for selection of efficacious drugs for patients with pancreatic cancer.47 Analysis of chemotherapy responses at the level of the proteome could provide insights that would inform such decisions. The strategy of utilizing proteomic data as the basis for cellular network modeling has been explored previously48; in this study, it provides comprehensive and accurate quantification of the effector molecules in numerous inter‐related pathways. Proteomics also assists in identifying marker proteins within key signaling pathways that reveal the status of the network. Moreover, such a modeling approach allowed quantitative discrimination of the underlying drug‐independent biological systems and drug effects upon these signaling pathways. The key pathways related to gemcitabine and birinapant mechanisms of action and interactions, cell cycle progression, DDR, DNA repair, MAPK, NF‐κβ, and apoptosis, were characterized quantitatively. Based on these systems, new targets or combination therapeutic strategies could be explored. Approaches, such as targeting DDR, DNA repair,49 or Fas/FasL,50 have been reported in preclinical settings; model sensitivity analysis supported the importance of those pathways. Model‐based simulations also indicated the limited additional benefits of direct p53 silencing or use of natural products, like curcumin with current gemcitabine‐based treatments in pancreatic cancer.Systems modeling approaches represent a promising avenue in cancer research.4, 5, 6, 7 However, selection of the protein nodes to be included in a systems model often is not well justified, and parameter values usually are obtained from the literature and adjusted based on limited observations. Several strategies were utilized here to support the quantitative relationships developed, and to limit the biological and quantitative uncertainty. First, proteomic analysis provided mechanistic support that the protein nodes included in the model represent specific and relevant signaling pathways. Moreover, because the modeling approach includes both mechanism‐driven and data‐driven components, only those protein‐protein relations that could characterize well the rich experimental dataset were retained. Therefore, the model development process itself represents an alternative validation of the qualitative relationships in the model, minimizing the biological uncertainty. Second, full temporal profiles were obtained for most proteins comprising the developed model; this supports parameter identifiability and reduces quantitative uncertainty. Third, the model was validated by comparing model‐predicted results with experimental observations, increasing confidence in the developed model.Several limitations of the work require future consideration. (i) Further reduction of quantitative uncertainty: the majority of observed protein responses were obtained for single drug concentrations, because of sample capacity limits of the LC/MS analysis. Therefore, extrapolating to different concentrations or scaling to humans requires additional data. (ii) Comprehensiveness of mechanism: the current model does not include all potentially important mechanisms of gemcitabine action. For example, the PI3K/AKT pathway was not explored because proteomic quantification revealed minimal treatment‐mediated changes in AKT abundance (data not shown). In addition, a single pancreatic cancer cell line was used, limiting the ability to anticipate interindividual variations in the disease. Therefore, refining the model to capture drug responses of other cell lines and in vivo models is a necessary bridge to accurate predictions. Similarly, extension of the analysis to the phosphoproteome would provide an additional layer of insight into drug response mechanisms. Nonetheless, the work provides proof‐of‐concept for using a sensitive, large‐scale proteomic dataset as a means of developing biologically relevant and quantitative relationships. (iii) Limitations of computational approaches: the algorithm used for solving the differential equations is a local method; global methods (e.g., the Global Optimization Toolbox based on MATLAB) would be required for models of comparable complexity but with sparser data.In conclusion, systems network modeling, combined with comprehensive and reproducible proteomics data, has identified reliable quantitative pharmacological relationships, accurately characterized underlying signaling pathways related to cell proliferation and apoptosis, and provides insights into the mechanisms of action and interactions of gemcitabine and birinapant. This paradigm can be applied to identify promising targets for therapy or mechanistically complementary drug combinations, and predict the impact of genetic mutations in pharmacological response networks.
Funding
This work was supported by the National Institutes of Health (NIH) grant GM24211 for WJJ, AI129518 and GM121174 for JQ, and CA198096 for RMS.
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
X.Z., R.M.S., and W.J.J. wrote the manuscript. X.Z. and X.S. designed the research. X.Z. and X.S. performed the research. X.Z. analyzed the data. J.Q. contributed analytical tools.
Supporting information
Acknowledgments
We thank Tetralogic Pharmaceuticals for providing birinapant. Drs Jun Li, Shichen Shen, and Xue Wang provided valuable technical support in proteomics and western blot analysis. Mr Zhicheng Qian contributed data in Figure 4a. We also appreciate the advice and suggestions of Dr David D'Argenio (University of South California) for implementing the model in ADAPT 5.
References
- 1. Rahib, L. , Smith, B.D. , Aizenberg, R. , Rosenzweig, A.B. , Fleshman, J.M. & Matrisian, L.M. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 74, 2913–2921 (2014). [DOI] [PubMed] [Google Scholar]
- 2. Melisi, D. , Calvetti, L. , Frizziero, M. & Tortora, G. Pancreatic cancer: systemic combination therapies for a heterogeneous disease. Curr. Pharm. Des. 20, 6660–6669 (2014). [DOI] [PubMed] [Google Scholar]
- 3. Yachida, S. & Iacobuzio‐Donahue, C. Evolution and dynamics of pancreatic cancer progression. Oncogene 32, 5253–5260 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang, X.‐Y. , Birtwistle, M.R. & Gallo, J.M. A general network pharmacodynamic model‐based design pipeline for customized cancer therapy applied to the VEGFR pathway. CPT Pharmacometrics Syst. Pharmacol. 3, e92 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Geva‐Zatorsky, N. , Dekel, E. , Cohen, A.A. , Danon, T. , Cohen, L. & Alon, U. Protein dynamics in drug combinations: a linear superposition of individual‐drug responses. Cell 140, 643–651 (2010). [DOI] [PubMed] [Google Scholar]
- 6. Kirouac, D.C. et al Computational modeling of ERBB2‐amplified breast cancer identifies combined ErbB2/3 blockade as superior to the combination of MEK and AKT inhibitors. Sci. Signal. 6, ra68 (2013). [DOI] [PubMed] [Google Scholar]
- 7. Gadkar, K. , Budha, N. , Baruch, A. , Davis, J.D. , Fielder, P. & Ramanujan, S. A mechanistic systems pharmacology model for prediction of LDL cholesterol lowering by PCSK9 antagonism in human dyslipidemic populations. CPT Pharmacometrics Syst. Pharmacol. 3, e149 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu, X. , Straubinger, R.M. & Jusko, W.J. Mechanism‐based mathematical modeling of combined gemcitabine and birinapant in pancreatic cancer cells. J. Pharmacokinet Pharmacodyn. 42, 477–496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu, X. , Shen, X. , Qu, J. , Straubinger, R.M. & Jusko, W.J. Proteomic analysis of combined gemcitabine and birinapant in pancreatic cancer cells. Front. Pharmacol. 9, 84 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang, D.W. , Sherman, B.T. & Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 11. Kanehisa, M. , Goto, S. , Sato, Y. , Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large‐scale molecular data sets. Nucleic Acids Res. 40, 109–114 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kramer, A. , Green, J. , Pollard, J. & Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shen, X. et al An IonStar experimental strategy for MS1 ion current‐based quantification using ultrahigh‐field orbitrap: reproducible, in‐depth, and accurate protein measurement in large cohorts. J. Proteome Res. 16, 2445–2456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakashima, M. et al Phosphorylation status of heat shock protein 27 plays a key role in gemcitabine‐induced apoptosis of pancreatic cancer cells. Cancer Lett. 313, 218–225 (2011). [DOI] [PubMed] [Google Scholar]
- 15. Wang, P. et al The serum miR‐21 level serves as a predictor for the chemosensitivity of advanced pancreatic cancer, and miR‐21 expression confers chemoresistance by targeting FasL. Mol. Oncol. 7, 334–345 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Benetatos, C.A. et al Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2‐associated cIAPs, abrogates TNF‐induced NF‐κB activation, and is active in patient‐derived xenograft models. Mol. Cancer Ther. 13, 867–879 (2014). [DOI] [PubMed] [Google Scholar]
- 17. D'Argenio, D.Z. , Schumitzky, A. & Wang, X. ADAPT 5 User's Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software (Biomedical Simulations Resource, Los Angeles, 2009). [Google Scholar]
- 18. Morgan, M.A. , Parsels, L.A. , Parsels, J.D. , Mesiwala, A.K. , Maybaum, J. & Lawrence, T.S. Role of checkpoint kinase 1 in preventing premature mitosis in response to gemcitabine. Cancer Res. 65, 6835–6842 (2005). [DOI] [PubMed] [Google Scholar]
- 19. Dai, Y. & Grant, S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin. Cancer Res. 16, 376–383 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karnitz, L.M. et al Gemcitabine‐induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol. Pharmacol. 68, 1636–1644 (2005). [DOI] [PubMed] [Google Scholar]
- 21. Christmann, M. & Kaina, B. Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res. 41, 8403–8420 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma, S. et al Induction of p21 by p65 in p53 null cells treated with Doxorubicin. Biochim. Biophys. Acta—Mol. Cell Res. 1783, 935–940 (2008). [DOI] [PubMed] [Google Scholar]
- 23. Karin, M. , Cao, Y. , Greten, F.R. & Li, Z.‐W. NF‐κB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer 2, 301–310 (2002). [DOI] [PubMed] [Google Scholar]
- 24. Raman, M. , Earnest, S. , Zhang, K. , Zhao, Y. & Cobb, M.H. TAO kinases mediate activation of p38 in response to DNA damage. EMBO J. 26, 2005–2014 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dai, L. , Aye Thu, C. , Liu, X.‐Y. , Xi, J. & Cheung, P.C.F. TAK1, more than just innate immunity. IUBMB Life 64, 825–834 (2012). [DOI] [PubMed] [Google Scholar]
- 26. Farley, N. et al P38 mitogen‐activated protein kinase mediates the Fas‐induced mitochondrial death pathway in CD8 + T cells. Mol. Cell. Biol. 26, 2118–2129 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morita, K.I. et al Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 20, 6028–6036 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Voutsadakis, I.A. Molecular predictors of gemcitabine response in pancreatic cancer. World J. Gastrointest. Oncol. 3, 153–164 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jin, H.S. , Lee, D.H. , Kim, D.H. , Chung, J.H. , Lee, S.J. & Lee, T. cIAP1, cIAP2, and XIAP act cooperatively via nonredundant pathways to regulate genotoxic stress‐induced nuclear factor‐κB activation. Cancer Res. 69, 1782–1791 (2009). [DOI] [PubMed] [Google Scholar]
- 30. McCool, K.W. & Miyamoto, S. DNA damage‐dependent NF‐κB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 246, 311–326 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kapoor, G.S. et al Abstract 2278: The SMAC‐mimetic birinapant regulates autocrine TNF production by caspase‐8:RIPK1 complex via p38MAPK pathway. Cancer Res. 74, 2278–2278 (2014). [Google Scholar]
- 32. Vince, J.E. et al IAP antagonists target cIAP1 to induce TNFα‐dependent apoptosis. Cell 131, 682–693 (2007). [DOI] [PubMed] [Google Scholar]
- 33. Gyrd‐Hansen, M. & Meier, P. IAPs: from caspase inhibitors to modulators of NF‐κB, inflammation and cancer. Nat. Rev. Cancer 10, 561–574 (2010). [DOI] [PubMed] [Google Scholar]
- 34. Roos, W.P. , Thomas, A.D. & Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 16, 20–33 (2015). [DOI] [PubMed] [Google Scholar]
- 35. Kitano, H. Cancer as a robust system: implications for anticancer therapy. Nat. Rev. Cancer 4, 227–235 (2004). [DOI] [PubMed] [Google Scholar]
- 36. Fiorini, C. , Cordani, M. , Padroni, C. , Blandino, G. , Di Agostino, S. & Donadelli, M. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim. Biophys. Acta 1853, 89–100 (2015). [DOI] [PubMed] [Google Scholar]
- 37. Kirsch, D.G. et al Caspase‐3‐dependent cleavage of Bcl‐2 promotes release of cytochrome c. J. Biol. Chem. 274, 21155–21161 (1999). [DOI] [PubMed] [Google Scholar]
- 38. Czabotar, P.E. , Lessene, G. , Strasser, A. & Adams, J.M. Control of apoptosis by the BCL‐2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63 (2014). [DOI] [PubMed] [Google Scholar]
- 39. Cohen, A.A. et al Dynamic proteomics of individual cancer cells in response to a drug. Science 322, 1511–1516 (2008). [DOI] [PubMed] [Google Scholar]
- 40. Izetti, P. et al PRIMA‐1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Invest. New Drugs 32, 783–794 (2014). 10.1007/s10637-014-0090-9 [DOI] [PubMed] [Google Scholar]
- 41. Muller, P.A.J. & Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 15, 2–8 (2013). [DOI] [PubMed] [Google Scholar]
- 42. Cline, M.S. et al Exploring TCGA pan‐cancer data at the UCSC cancer genomics browser. Sci. Rep. 3, 2652 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kornmann, M. , Ishiwata, T. , Kleeff, J. , Beger, H.G. & Korc, M. Fas and Fas‐ligand expression in human pancreatic cancer. Ann. Surg. 231, 368–379 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aggarwal, B.B. & Sung, B. Pharmacological basis for the role of curcumin in chronic diseases: an age‐old spice with modern targets. Trends Pharmacol. Sci. 30, 85–94 (2009). [DOI] [PubMed] [Google Scholar]
- 45. Kunnumakkara, A.B. , Guha, S. , Krishnan, S. , Diagaradjane, P. , Gelovani, J. & Aggarwal, B.B. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor‐κB‐regulated gene products. Cancer Res. 67, 3853–3861 (2007). [DOI] [PubMed] [Google Scholar]
- 46. Epelbaum, R. , Schaffer, M. , Vizel, B. , Badmaev, V. & Bar‐Sela, G. Curcumin and gemcitabine in patients with advanced pancreatic cancer. Nutr. Cancer 62, 1137–1141 (2010). [DOI] [PubMed] [Google Scholar]
- 47. Witkiewicz, A.K. et al Integrated patient‐derived models delineate individualized therapeutic vulnerabilities of pancreatic cancer. Cell Rep. 16, 2017–2031 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Janes, K.A. & Lauffenburger, D.A. A biological approach to computational models of proteomic networks. Curr. Opin. Chem. Biol. 10, 73–80 (2006). [DOI] [PubMed] [Google Scholar]
- 49. Kelley, M.R. , Logsdon, D. & Fishel, M.L. Targeting DNA repair pathways for cancer treatment: what's new? Future Oncol. 10, 1215–1237 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Villa‐Morales, M. & Fernández‐Piqueras, J. Targeting the Fas/FasL signaling pathway in cancer therapy. Expert Opin. Ther. Targets 16, 85–101 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials