

Abstract
Herein, excipients are investigated to ameliorate the deleterious effects of lyophilization on peptide-polymer nano-polyplex (NP) morphology, cellular uptake, and bioactivity. The NPs are a previously-described platform technology for intracellular peptide delivery and are formulated from a cationic therapeutic peptide and the anionic, pH-responsive, endosomolytic polymer poly(propylacrylic acid) (PPAA). These NPs are effective when formulated and immediately used for delivery into cells and tissue, but they are not amenable to reconstitution following storage as a lyophilized powder due to aggregation. To develop a lyophilized NP format that facilitates longer-term storage and ease of use, MAPKAP kinase 2 inhibitory peptide-based NPs (MK2i-NPs) were prepared in the presence of a range of concentrations of the excipients sucrose, trehalose, and lactosucrose prior to lyophilization and storage. All excipients improved particle morphology post-lyophilization and significantly improved MK2i-NP uptake in human coronary artery smooth muscle cells relative to lyophilized NPs without excipient. In particular, MK2i-NPs lyophilized with 300 mM lactosucrose as an excipient demonstrated a 5.23 fold increase in cellular uptake (p < 0.001), a 2.52 fold increase in endosomal disruption (p < 0.05), and a 2.39 fold increase in ex vivo bioactivity (p < 0.01) compared to MK2i-NPs lyophilized without excipients. In sum, these data suggest that addition of excipients, particularly lactosucrose, maintains and even improves the uptake and therapeutic efficacy of peptide-polymer NPs post-lyophilization relative to freshly-made formulations. Thus, the use of excipients as lyoprotectants is a promising approach for the long-term storage of biotherapeutic NPs and poises the NP platform for clinical translation.
Keywords: Drug delivery, nanoparticle, polyplex, excipient, lyoprotection, peptide
Graphical abstract
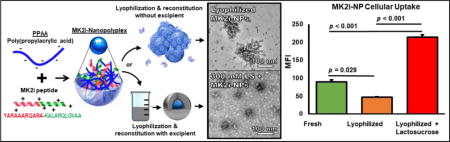
Introduction
The use of peptides in basic science and as therapeutics has continuously increased as a result of the elucidation of protein structures and the role of specific peptide sequences/motifs in protein function and disease etiology [1–4]. Based on this information, peptides can be derived that recapitulate the biological activity of or bind to and block a specific active site of a protein. This rational design opportunity enables development of peptides that modulate kinase activity or disrupt protein-protein interactions with high specificity, both of which are often not possible with conventional small molecule drugs [5]. Despite holding advantages over small molecule drugs in terms of solubility, potency, and specificity, peptides face a number of in vivo pharmacokinetic and metabolic barriers such as short circulatory half-life and susceptibility to proteases, limiting widespread clinical translation of peptide therapeutics. In addition, peptides with intracellular targets are hindered by poor cellular uptake and entrapment and degradation within endolysosomal compartments, leading to poor intracellular bioavailability [3, 6].
Current approaches to overcome peptide pharmacokinetic limitations include fusion with cell penetrating peptide (CPP) sequences [e.g., the trans-activating transcriptional factor (TAT) CPP derived from the human immunodeficiency virus type 1), modification of peptide chemistry and/or structure (e.g., retro-inverso isomerization and peptide stapling), and the use of drug delivery systems (e.g., nanoparticles and polycationic agents) [7–10]. We previously developed a peptide delivery platform consisting of the anionic, pH-responsive polymer poly(propylacrylic acid) (PPAA) that forms electrostatically complexed nano-polyplexes (NPs) with cationic CPP modified peptides. This NP platform was specifically designed to enhance cellular uptake and facilitate pH-triggered escape from acidic endo-lysosomal compartments.
The NPs have been successfully applied to deliver an anti-inflammatory MAPKAP Kinase 2 inhibitory (MK2i) peptide to prevent vascular graft intimal hyperplasia (IH). Binding of MK2i to MK2 prevents phosphorylation and activation of its downstream effectors including heat shock protein 27 (HSP 27), LIM kinase (LIMK), heterogenous nuclear ribonucleoprotein A0 (hnRNP A0), and cAMP (adenosine 3′,5′ monophosphate) response element-binding protein (CREB). Phosphorylated HSP27 associates with and stabilizes filamentous (F) actin by preventing its depolymerization [11]. Phosphorylated LIMK also results in decreased depolymerization of F-actin by phosphorylating and deactivating the actin disassembling protein cofilin [12]. Activated hnRNP A0 increases inflammatory cytokine production by stabilizing inflammatory cytokine mRNA, and CREB binds to cAMP-responsive elements to promote expression of inflammatory cytokines and genes that contribute to smooth muscle cell migration and proliferation [13, 14]. Thus, MK2i-mediated downregulation of these effectors promotes cytoskeletal actin depolymerization (thereby promoting vasorelaxation) and inhibits vascular smooth muscle cell migration, proliferation, and inflammatory cytokine production - key factors in pathological vasoconstriction and the onset and progression of IH [15, 16]. Tissue treatment with freshly-made MK2i-NPs increased MK2i peptide bioactivity by approximately an order of magnitude in human vascular tissue ex vivo [17–19].
The NP platform not only offers distinct benefits in improving the therapeutic efficacy of intracellular-acting peptides such as MK2i, but also has significant potential for clinical translation due to its simplicity and scalability. It is anticipated that large scale production of MK2i-NPs will be preferred over fresh, onsite preparation at clinical facilities due to increased quality control, ease of implementation, and reduced batch-to-batch variability [20]. However, large-scale production must be coupled with measures to ensure long-term stability during distribution and storage and facile reconstitution into a bioactive product. The critical factors to consider for long-term storage are physical instability (i.e., particle aggregation or fusion) and chemical instability [i.e., hydrolysis or chemical modification of the polymer and/or active pharmaceutical ingredient (API) during storage] [21]. Lyophilization is the de facto method for preparing peptide and protein therapeutics within the pharmaceutical industry; however, the process of lyophilization can have deleterious effects on nanoparticle formulations. Namely, lyophilization subjects the nanoparticles to the stresses of freezing and drying, which alter the environment of the particles through exposure to ice-liquid interfaces. These stresses include pH shifts and mechanical damage due to buffer crystallization and the loss of stabilizing hydration shells due to the removal of water. These stresses often lead to particle aggregation or fusion and result in detrimental effects on the bioactivity of the API [22, 23].
To mitigate the deleterious effects of lyophilization, excipients are commonly utilized to shield the particles from the stresses associated with the freezing and drying processes, especially for peptide and protein based biologic drugs, because they often lack long-term stability. Of the stability challenges faced by biologics, preventing aggregation is critical to maintaining the bioactivity of lyophilized peptide or protein based formulations [24]. Excipients such as salts, polyols, and sugars can act as cryoprotectants and/or lyoprotectants when added to protein solutions prior to freeze-drying. Historically, sugars have been widely employed as cryoprotectants in nanoparticle systems, albeit mainly for nucleic acid based formulations [25–27]. Because their vitrification points (i.e., the glass transition temperature due to concentration by freezing, or Tg’) are reached during lyophilization, sugars such as sucrose and trehalose are ideal candidates for lyoprotection of nanoparticle formulations. It is hypothesized that, during lyophilization, these sugars form a glass-like matrix that suspends the nanoparticles and reduces their interaction with ice crystals and each other, thereby preventing mechanical damage and aggregation [21]. Herein, we explore the use of the sugars sucrose, trehalose, and lactosucrose as lyoprotectants for the previously developed and clinically-promising MK2i-NPs.
Materials and Methods
Materials
The MK2i peptide (sequence: YARAAARQARAKALARQLGVAA), 2-propylacrylic acid monomer, and poly(propylacrylic acid) (PPAA) were synthesized and characterized as previously described [18]. Sucrose (99.5% purity) and trehalose dihydrate were purchased from Sigma Aldrich. Lactosyl fructoside (lactosucrose, 95% purity) was purchased from Wako Chemicals. All other materials were purchased from Sigma Aldrich unless otherwise noted.
Cell Culture
Primary human coronary artery vascular smooth muscle cells (HCAVSMCs) were obtained from Lonza. Cells were cultured in complete growth medium [vascular cell basal medium (ATCC) supplemented with 5% FBS, human basic fibroblast growth factor (bFGF, 5ng/mL), human insulin (5 μg/mL), ascorbic acid (50 μg/mL), L-glutamine (10mM), human epidermal growth factor (EGF, 5 ng/mL), 1% penicillin-streptomycin, and 50 μg/mL plasmocin (Invivogen)] in a sterile incubator maintained at 37 °C with a humidified, 5% CO2 atmosphere.
Cell cultures were maintained on a 75 cm2 polystyrene tissue culture flasks (BD Falcon), and culture media was replaced every other day. Cells were seeded at densities specific to each experiment, and, prior to harvest and passage, cells were grown to 80-90% confluence. Only cells from early passages (i.e., 3-8) were used in experiments.
Preparation and Characterization of Nano-polyplex Formulations
PPAA was added to phosphate buffered saline (PBS, without calcium or magnesium) and 1M NaOH was slowly added until the polymer was completely solubilized and a stable pH of 8 was obtained. Purified MK2i peptide was dissolved in pH 8 phosphate buffered saline. 1M stock solutions of excipients were formed by dissolving 0.1 moles of excipient in 100 mL of deionized water. MK2i peptide was then mixed with the PPAA polymer at a charge ratio [NH3+ peptide]:[COO−PPAA] = 1:3 (i.e., a peptide to polymer mass ratio of 1:1.2) to form MK2i-NPs. MK2i-NPs were syringe filtered through a 0.45μm PTFE filter and diluted into solutions of sucrose, trehalose, or lactosucrose to final excipient concentrations of either 150 mM, 300 mM, or 600 mM. MK2i-NP/excipient solutions were then frozen at −80 °C and lyophilized. Lyophilized MK2i-NPs without excipients and freshly prepared MK2i-NPs were utilized as controls. Upon resuspension in deionized water, MK2i-NPs lyophilized with or without excipient were equilibrated for 45 minutes prior to use.
MK2i-NP hydrodynamic diameter and ζ-potential were characterized on a Malvern Zetasizer Nano-ZS with a reusable dip cell kit (Malvern Instruments Ltd., Worcestershire, U.K.). A 1:3 charge ratio was chosen as optimal based on previous work and was utilized in all in vitro and ex vivo experiments [17]. To verify sizes indicated by DLS analysis, MK2i-NP-excipient formulations were visualized using transmission electron microscopy (TEM). Samples for TEM were prepared by inverting carbon film-backed copper grids (Ted Pella) on 20 uL of aqueous polyplex suspensions (.1 mg/mL) and allowing sample to adsorb for 3 minutes before they were blotted dry. Samples were then inverted on 20 uL droplets of .5% uranyl acetate and stained for 10 seconds. Samples were then desiccated in vacuo for 24 hours prior to imaging on a FEI Tecnai Osiris TEM.
Flow Cytometry Assay for Cellular Internalization of MK2i Formulations
An amine-reactive Alexa-488 succinimidyl ester (Life Technologies) was dissolved in DMSO and mixed at a 1 to 3 molar ratio with the MK2i peptide in 100 mM sodium bicarbonate buffer (pH = 8.3) and allowed to react for 4 hours while protected from light. Unreacted fluorophore, N-hydroxysuccinimide salts, and organic solvent were removed using a PD-10 Miditrap G-10 desalting column, and the purified, fluorescently-labeled MK2i peptide was lyophilized. PPAA polymer was mixed with fluorescently labeled MK2i peptide at a charge ratio = 1:3 and syringe filtered through a 0.45 μm PTFE filter to form fluorescent MK2i-NPs. Fluorescent MK2i-NPs were then diluted into excipient solutions, frozen, and lyophilized prior to reconstitution and use. HCAVSMCs were grown to 80-90% confluence, harvested, and seeded at 60,000 cells/well in a 12 well plate and allowed to adhere overnight. HCAVSMCs were treated with MK2i peptide, freshly prepared MK2i-NPs, MK2i-NPs lyophilized without excipients, MK2i-NPs lyophilized with excipients, or PBS -/- as a vehicle control at a concentration of 10 μM Alexa-488 labeled peptide in Opti-MEM medium supplemented with 1% penicillin-streptomycin and 1% FBS for 30 minutes. Following treatment, cells were washed 2× in PBS -/-, harvested with 0.05% trypsin-EDTA, centrifuged, and resuspended in 300 μL of 0.05% Trypan blue in PBS (-/-) for analysis on a FACSCalibur flow cytometer (Becton Dickinson) with BD CellQuest™ Pro software (V 5.2). Data were exported and analyzed with FlowJo software (V 10.1). All samples were run in triplicate in two independent runs.
Fluorescence Confocal Microscopy Imaging of Cellular Internalization of MK2i Formulations
Fluorescent MK2i-NPs formulations were fabricated using the same methods as for flow cytometry. HCAVSMCs were seeded at a density of 5,000 cells/well in a Lab-Tek II 8-well chambered coverglass. Prior to treatment, the media was switched from DMEM with 10% FBS and 1% penicillin-streptomycin to Opti-Mem with 1% FBS and 1% penicillin-streptomycin. Cells were imaged prior to treatment and then treated with MK2i, fresh MK2i-NPs, MK2i-NPs lyophilized without excipients, or MK2i-NPs lyophilized with excipients. All samples were treated with an equivalent dose of 10 μM peptide. Treatments were removed after 30 minutes and cells were then stained with Hoechst solution (1:5000 dilution in FluoroBrite Imaging Media with 1% FBS) for 10 minutes before imaging on a Nikon Eclipse Ti confocal microscope with NIS Elements imaging software.
Hemolysis Assay for Endosome Escape Potential
To evaluate the pH-dependent membrane disruptive capability of the MK2i-NP formulations as a surrogate measure for endosomal disruption and escape potential, an ex vivo red blood cell hemolysis assay was performed as previously described [28]. Following approval by the Vanderbilt University Medical Center Institutional Review Board, whole human blood was drawn from an anonymous donor. Erythrocytes were isolated from plasma by washing three times with 150 mM NaCl. Isolated erythrocytes were then resuspended in phosphate buffers corresponding to physiologic/extracellular, early endosomal, late endosomal, and lysosomal environments (pH 7.4, pH 6.8, and pH 6.2 respectively). MK2i-NPs lyophilized in excipient solutions, MK2i-NPs lyophilized without excipients, and fresh MK2i-NPs (10 μL at MK2i concentrations of 20, 100, and 800 μg/mL) were added to 190 μL of erythrocyte suspensions at each pH to achieve final concentration of 1, 5, and 40 μg/mL of polymer and incubated at 37 °C for 1 hour in 96 well conical-bottom plates. PBS and 1% Triton X-100 were utilized as negative and positive controls, respectively. The plates were then centrifuged to sediment intact red blood cells, and the supernatant was transferred to a new 96-well plate. Hemoglobin release into the supernatant due to pH-dependent, treatment-induced red blood cell membrane disruption was then spectrophotometrically quantified by measuring the absorbance of the supernatant at 541 nm. Percent hemolysis was calculated relative to positive and negative controls.
Live Cell Galectin-8 Recruitment Measure of Endosome Disruption
Embryonic rat aortic smooth muscle cells (A7R5 cells) stably expressing Galectin-8-Yellow Fluorescent Protein (Gal8-YFP) were generated as previously described as a tool for visualizing endosome disruption levels and kinetics [29]. Gal8-YFP A7R5 cells were seeded at a density of 5,000 cells/well in a Lab-Tek II 8-well chambered coverglass. Prior to treatment, media was switched from DMEM with 10% FBS and 1% penicillin-streptomycin to Opti-Mem with 1% FBS and 1% penicillin-streptomycin. Cells were monitored for 5 min to measure baseline Gal8-YFP fluorescence. Cells were then treated with MK2i peptide, freshly prepared MK2i-NPs, MK2i-NPs lyophilized without excipients, or MK2i-NPs lyophilized with excipients, all at an equivalent dose of 10 μM peptide, while control groups were treated with poly(acrylic acid) or poly(propylacrylic acid) alone at a dose of 83 μg/mL. Cells were immediately imaged using confocal microscopy following the addition of treatments. Images were taken every minute for 30 min to monitor galectin-8 YFP recruitment over time. Image processing was performed using ImageJ software, utilizing a spot counting technique to quantify the average number of disrupted endosomes per cell for each treatment group. Briefly, pixel intensity thresholds were determined by taking the mean pixel intensity of each image and adding two standard deviations of the pixel intensity distribution to set a positive threshold for disrupted endosomes. The average number of disrupted endosomes per cell was calculated from n = 10 cells from each treatment group 30 minutes after treatment.
Cytotoxicity Assay
HCAVSMCs were seeded onto 96-well plates at a density of 10,000 cells/well to yield approximately 70% confluence and allowed to adhere overnight. Cells were then treated with 100 μM peptide dose of freshly prepared MK2i-NPs, MK2i-NPs lyophilized without excipient, MK2i-NPs lyophilized with excipients, MK2i peptide alone, or PBS -/- (vehicle control) for 2 hours in Opti-MEM medium supplemented with 1% penicillin-streptomycin and 1% FBS. Note that this is a 10-fold higher dose than used for flow cytometry studies and 4-fold higher dose than used for tissue bioactivity studies. Treatments were subsequently removed, and the cells were cultured in complete growth medium for 24 hours. Cells were then washed 2× with PBS +/+, and cell viability was determined by a CytoTox-ONE Homogenous Membrane Integrity assay (Promega) according to the manufacturer’s instructions. Briefly, 100 μL of Ambion KDalert Lysis Buffer was added to each well, and then 100 μL of freshly prepared CytoTox-ONE reagent was added to each well. After 10 minutes of incubation at room temperature in the absence of light, 50 μL of stop solution was added and the fluorescence of each well (λex = 560 nm, λem = 590 nm) was determined with a TECAN Infinite M1000 Pro plate reader. Cell viability was calculated relative to untreated control cells.
Human Saphenous Vein Preparation
Under a protocol approved by the Vanderbilt University Medical Center Institutional Review Board (IRB), de-identified, discarded segments of human saphenous vein (HSV) were collected from consenting patients undergoing coronary or peripheral vascular bypass surgeries. Following surgical resection, HSV segments were stored in buffered salt solutions until the conclusion of the procedure, at which time they were placed in cold transplant harvest buffer (100 mM potassium lactobionate, 25 mM KH2PO4, 5 mM MgSO4, 30 mM raffinose, 5 mM adenosine, 3 mM glutathione, 1 mM allopurinol, 50 g/L hydroxyethyl starch, pH 7.4). HSV segments were used within 24 hours of harvest. HSV segments were transferred to a 60 mm Petri dish utilizing sterile technique. Both ends of the segments (0.5 mm) were removed with a blade, and excess adventitial and adipose tissue was removed with minimal tissue manipulation. Vein segments were then cut into rings with an approximate width of 1.0 mm.
HSV viability was confirmed prior to performing physiology experiments. The weight and length of each ring segment was recorded, and the rings were then suspended in a muscle bath containing bicarbonate buffer (120mM NaCl, 4.7 mM KCl, 1.0 mM MgSO4, 1.0 mM NaH2PO4, 10 mM glucose, 1.5 mM CaCl2, and 25 mM Na2HCO3, pH 7.4) equilibrated with 95% O2 and 5% CO2 at 37 °C. The rings were stretched, and length was adjusted until maximal-tension was achieved. Reactivity was normalized by calculating the passive length-tension relationship for each individual ring. Rings were maintained in a bath at 1 g resting tension, which was previously determined to maximize vessel response to contractile agonists and equilibrated for 2 hours in buffer. Force measurements were obtained using a Radnoti Glass Technology (Monrovia, CA) force transducer (159901A) interfaced with a Powerlab data acquisition system and LabChart software (AD Instruments, Colorado Springs, CO).
Rings were isometrically contracted with 110 mM KCl (with equimolar replacement of NaCl in bicarbonate buffer), and the generated force was measured to confirm vein viability. 110 mM KCl causes complete membrane depolarization, which results in contraction in functionally viable smooth muscle.
HSV Smooth Muscle Physiology Therapeutic Vasorelaxation Studies
Vasorelaxation was used as a readout for MK2i bioactivity. Viable HSV rings were washed and equilibrated in bicarbonate solution for 30 min and then contracted with phenylephrine (PE, 1 μM). Rings were then relaxed with sodium nitroprusside (SNP, 0.1-10 μM) administered in a cumulative log dose, and the decrease in contractile force was recorded over time. Rings were then washed and equilibrated in buffer until baseline tension was achieved. Rings were then incubated with MK2i peptide, freshly prepared MK2i-NPs, MK2i-NPs lyophilized without excipients, MK2i-NPs lyophilized with excipient solution, excipients alone, or buffer alone for 1 hour. Following incubation, rings were again contracted and subsequently relaxed with the same doses of PE and SNP, and the generated forces were recorded. Force was normalized to ring weight and length and percent inhibition of contraction was calculated by dividing post-treatment force by pre-treatment force, with 0% relaxation defined by the pretreatment force generated with 1 μM PE.
Statistics
Statistical analyses were performed with a one-way ANOVA followed by Tukey’s posthoc test to compare experimental groups. Analyses were performed with Graphpad Prism Software Version 7.02 (La Jolla, California). Statistical significance was accepted within a 95% confidence limit. Results are presented are arithmetic mean ± SEM graphically, and p values are included within the figures or figure captions.
Results
Physiochemical Characterization of Lyophilized MK2i-NPs
To determine the effects of lyophilization on NP size, morphology, and surface charge, MK2i-NPs were lyophilized in solutions of lactosucrose, sucrose, and trehalose at excipient concentrations of 150 mM, 300 mM, and 600 mM prior to reconstitution and analysis. Lyophilization was shown to increase MK2i-NP diameter and polydispersity from Dh = 119 nm and PDI = 0.19 to Dh = 180 nm and PDI= 0.23, respectively (p = 0.032; Fig. 1A). With the exception of an apparent concentration dependent increase in size for MK2i-NPs lyophilized with lactosucrose, there were no clear excipient concentration dependent trends on modulation of NP size. Notably, MK2i-NPs lyophilized in 150 mM lactosucrose demonstrated the best preservation of MK2i-NP size relative to fresh MK2i-NPs, while MK2i-NPs lyophilized in sucrose solutions tended to have lower dispersity (Table S1). Interestingly, sucrose alone was shown to contain nanoparticulates around 200 nm in size (Fig. 1), possibly due to high molecular weight impurities that are commonly retained after commercial sugar refinement processes [30]. TEM imaging further demonstrated that 150 and 300 mM lactosucrose maintained well-defined NP morphology compared to MK2i-NPs lyophilized without excipient (Fig. 1B). In contrast, lyophilization without excipients led to marked aggregation/deformation of the particles (Fig. 1B). Although uranyl acetate is commonly utilized as a negative stain, the negative charge of the PPAA polymer and resulting NPs results in positive uranyl acetate staining [31]. Overall, TEM images of the NPs lyophilized at all excipient concentrations were consistent with the trends measured with DLS (Fig. S1). Following 10-week storage, MK2i-NPs lyophilized in 300 mM lactosucrose were able to retain particle size, while MK2i-NPs lyophilized with no excipient or stored for the during in PBS at 4 °C displayed aggregation (Fig. S2).
Figure 1. Physiochemical characterization of lyophilized MK2i-NP formulations with and without excipients.
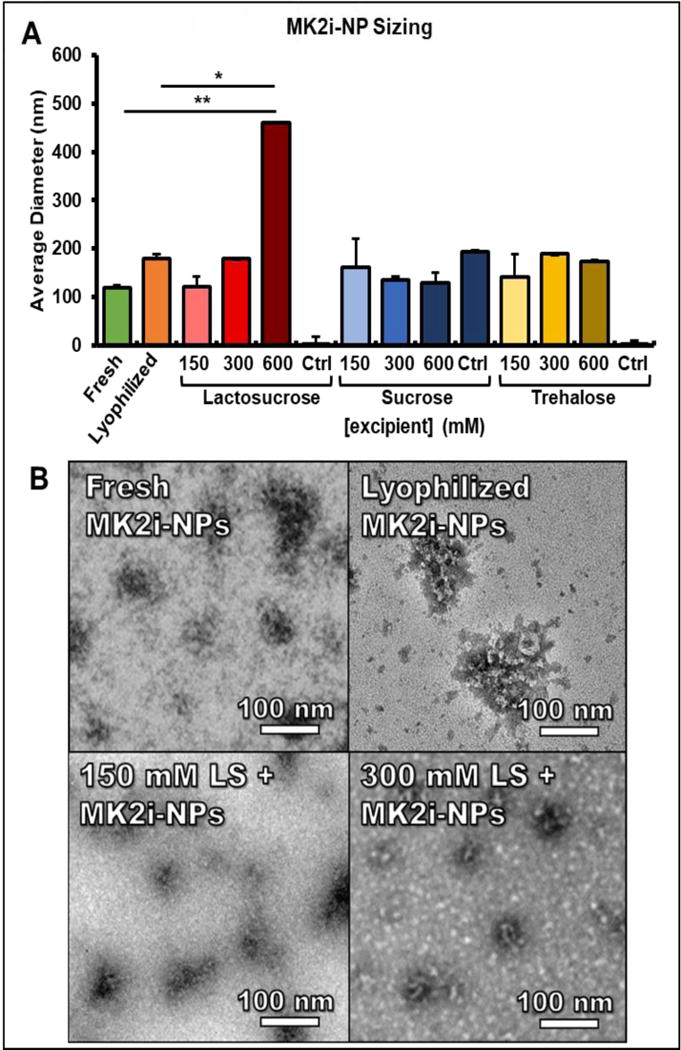
A) The concentration dependent-effects of various excipients on the size of lyophilized MK2i-NPs. For A, n = 3 independent MK2i-NP formulations and measurements. (* p < .005, ** p < .001) B) TEM images for freshly prepared MK2i-NPs, lyophilized MK2i-NPs without excipient, and MK2i-NPs lyophilized in 150 mM and 300 mM lactosucrose. Ctrl = excipient without polyplexes
MK2i-NP Cellular Uptake
MK2i-NPs lyophilized without excipient showed a 48% decrease in peptide internalization in HCAVSMCs compared to freshly prepared MK2i-NPs (p = 0.029; Fig. 2A). However, when lyophilized in excipient solutions, MK2i-NP internalization was restored to and in some cases increased over the level of uptake achieved with freshly prepared MK2i-NPs. Specifically, MK2i-NPs lyophilized in solutions containing either lactosucrose or sucrose showed statistically significant increases in peptide internalization when compared to freshly prepared MK2i-NPs (p = 0.0393 for 150 mM lactosucrose, p < 0.0001 for 300 mM and 600 mM lactosucrose, and p = 0.0083 for 150 mM sucrose). Although there is an apparent dose response in peptide uptake with respect to lactosucrose that appears to saturate at 300 mM, sucrose and trehalose showed no clear trends in excipient concentration-dependent effects on uptake. MK2i-NPs lyophilized in 300 mM lactosucrose solutions consistently demonstrated a greater than 2-fold increase in peptide internalization over freshly prepared NPs. Notably, freshly prepared MK2i-NPs in 300 mM lactosucrose displayed no increase in cellular uptake, indicating that the increase of MK2i-NP formulations lyophilized in 300 mM lactosucrose was a result of the lyophilization process of these components together and not a nonspecific effect of the excipient lactosucrose alone. Representative flow histograms demonstrate the relative shifts in fluorescent peptide cell uptake between each group, and the relative delivery differences were also visualized using confocal microscopy (Fig. 2B, C). Considering the optimal uptake demonstrated for MK2i-NPs lyophilized in 300 mM lactosucrose, MK2i-NPs lyophilized in 300 mM lactosucrose were utilized in all subsequent experiments. Importantly, it was confirmed that control solutions of excipients alone did not contribute to cellular fluorescence.
Figure 2. Excipients Improve the Cellular Uptake of MK2i-NPs in HCAVSMCs.
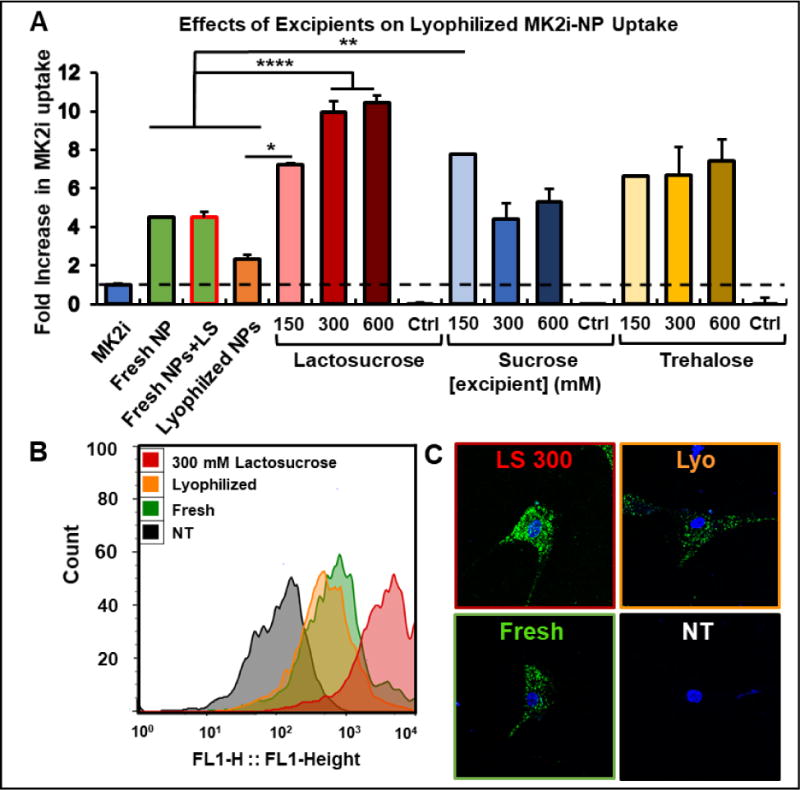
A) Concentration dependent effects of excipients on the uptake of fluorescent, lyophilized MK2i-NPs in HCAVSMCs (n = 3; * p < 0.05, ** p < 0.01, *** p < 0.005, **** p < 0.001) B) Representative histograms of the cellular uptake of freshly prepared and lyophilized MK2i-NP formulations with and without 300 mM lactosucrose in HCAVSMCs. C) Representative confocal microscopy images of the cellular uptake of freshly prepared and lyophilized MK2i-NP formulations with and without 300 mM lactosucrose in HCAVSMCs. NT = no treatment, Ctrl = excipient without polyplexes.
MK2i-NP pH-dependent Membrane Disruptive Activity
An ex vivo human red blood cell hemolysis assay was utilized to measure the pH-dependent membrane disruptive activity of lyophilized MK2i-NP formulations as an indicator for endosomal escape capability (Fig. 3A). The pH-responsive PPAA polymer alone displayed no hemolytic activity at physiologic pH but displayed increasing membrane disruptive activity as the pH decreased from early to late endosomal values (6.8 and 6.2, respectively). Freshly prepared MK2i-NPs displayed switch-like membrane disruptive behavior between early endosomal (pH 6.8) to late endosomal (pH 6.2) environments, demonstrating that complexation of PPAA with peptide slightly shifts the onset of pH dependent membrane disruptive activity to not occur until slightly lower pH. Lyophilization with or without excipient did not alter the pH-dependent membrane disruption of the MK2i-NPs except for a decrease at pH 5.6 for 600 mM lactosucrose formulations (Fig. S3). Within MK2i-NP excipient groups, there were no apparent trends between excipient concentration and membrane disruptive activity. Importantly, none of the excipients tested significantly reduced the pH dependent membrane disruptive activity of the NP formulations, indicating that the use of excipients maintains the endosomal escape capability of MK2i-NPs following lyophilization. None of the excipients alone displayed any hemolytic activity, verifying that membrane disruptive activity is due to the pH-responsive polymer PPAA.
Figure 3. Excipients Rescue Endosomal Escape of MK2i-NPs In Vitro.
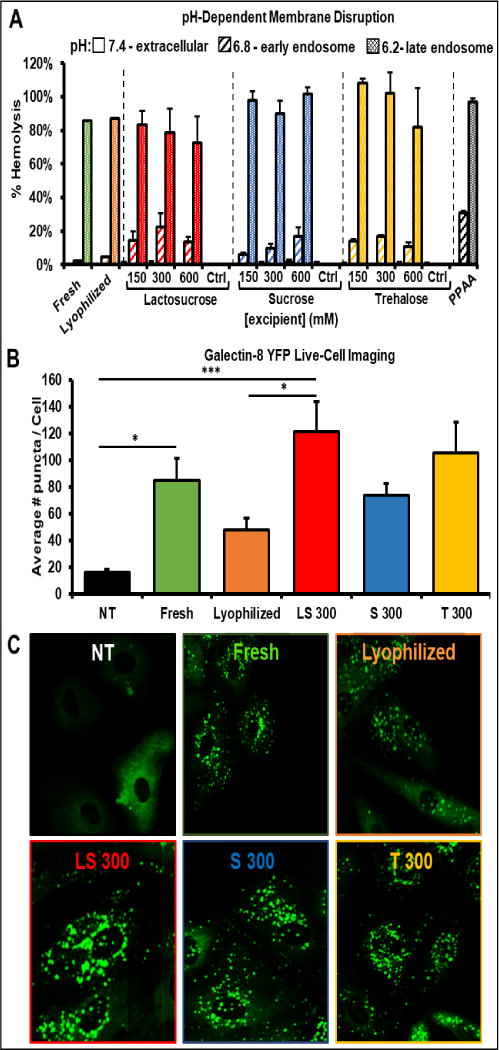
A) Concentration dependent effects of excipients on the pH-dependent membrane disruptive activity of lyophilized MK2i-NP formulations. B) Galectin 8 YFP recruitment in live A7R5 cells as an in vitro assessment of endosomal disruption mediated by lyophilized MK2i-NP formulations (n = 10 cells for each sample). (* p < .05, ** p <.01, *** p < .005) C) Representative confocal fluorescence microscopy images of galectin 8 YFP recruitment in A7R5 rat cells treated with MK2i-NP formulations. NT = no treatment, Ctrl = excipient without polyplexes.
MK2i-NP Endosomal Disruption in Live Cells
To determine the extent of endosomal escape in vitro, a novel live-cell fluorescent imaging technique in a cell line engineered to express fluorescent Galectin-8 was used as previously described [29]. Galectin-8 is a β-galactoside-binding lectin of the galectin family that is normally diffusely spread throughout the cytosol of the cell and functions as part of the innate immune system. Galectin 8 binds to glycans located on the inner leaflet of the endosomal membrane that are only accessible if the endosomal compartment has been disrupted (e.g., by an infectious agent). The recruitment of galectin 8 fused with yellow fluorescent protein (Galectin-8 YFP) enables visualization of disrupted endosomes as fluorescent puncta. Cells treated with freshly-prepared MK2i-NPs displayed a significant increase in galectin-8 recruitment compared to untreated cells (Fig. 3B). Treatment with MK2i-NPs lyophilized without excipient resulted in an apparent decrease in Gal-8 recruitment compared to freshly prepared particles, but this difference was not statistically significant (p = 0.53). MK2-NPs lyophilized in 300 mM lactosucrose and 300 mM trehalose showed significantly more galectin-8 puncta compared to untreated cells (p = 0.0001, p = 0.0016 respectively; Fig. 3B). MK2i-NPs lyophilized with trehalose and lactosucrose demonstrated statistically insignificant increases in galectin-8 recruitment over freshly prepared MK2i-NPs, whereas sucrose demonstrated a statistically insignificant decrease in galectin-8 recruitment. These galectin-8 recruitment data correlate with cellular MK2i-NP uptake as measured by flow cytometry (Fig 2A). Importantly, cells treated with the peptide alone or with a non-endosomolytic control polymer poly(acrylic acid) (PAA, which has a pKa = 4.3 that is at a pH below the range encountered during endo-lysosomal trafficking) demonstrated no galectin-8 recruitment (Fig. S4). In contrast, cells treated with the endosomolytic polymer PPAA alone demonstrated robust galectin-8 recruitment, verifying that the endosomal escape activity demonstrated by MK2i-NP treated cells is due to incorporation of PPAA into the NP formulation. Although the representative images shown in Figure 3C were taken 30 minutes post-treatment administration, robust endosomal disruption was evident as early as five minutes in cells treated with MK2i-NP-excipient formulations (not shown).
MK2i-NP Cytocompatibility
The cytotoxicity of the lyophilized MK2i-NP-excipient formulations were compared to freshly prepared MK2i-NPs, MK2i-NPs lyophilized without an excipient, and free MK2i peptide in HCAVSMCs in vitro. HCAVSMCs were treated with MK2i-NP formulations or MK2i peptide alone at a dose of 100 μM peptide. All treatments were well tolerated by smooth muscle cells at all doses: no significant cytotoxicity was demonstrated for any treatment group and cell viability was maintained above 90% across all groups 24 hours post-treatment (Fig. 4).
Figure 4. Excipients are Biocompatible with HCAVSMCs.

HCAVSMCs were treated for two hours with MK2i-NP formulations or MK2i peptide alone at a dose of 100 uM and subsequently cultured in fresh medium for an additional 24 hours. Cell viability was then assessed through a lactate dehydrogenase (LDH) based cytotoxicity assay and normalized to untreated control cells (n = 3). NT = no treatment, Ctrl = excipient without polyplexes.
Effects of Lyophilization on MK2i-NP Bioactivity in Human Vascular Tissue
MK2 plays a role in modulating actin dynamics in vascular smooth muscle by phosphorylating and activating downstream effectors such as heat shock protein 27 (HSP27) and LIM kinase (LIMK). Phosphorylation of HSP27 results in the capping of filamentous actin (F-actin), thereby inhibiting actin depolymerization and vasorelaxation. Phosphorylation of LIMK results in the phosphorylation and deactivation of the actin depolymerizing protein cofilin, preventing F-actin degradation and inhibiting vasorelaxation. The MK2 inhibitory peptide (MK2i) binds MK2, preventing MK2 from activating these downstream effectors and, thus, promotes vasorelaxation [17, 18]. To measure the effect of lyophilization with and without excipients on MK2i-NP bioactivity, an ex vivo HSV smooth muscle physiology tissue model was utilized (Fig. 5A). MK2i-NPs lyophilized in 300 mM lactosucrose were chosen as a lead candidate formulation based on physiochemical characterization and in vitro uptake and endosomal escape data. Vein segments were incubated for 1 hour with either fresh MK2i-NPs, lyophilized MK2i-NPs with and without 300 mM lactosucrose, MK2i peptide alone, lactosucrose alone, or left untreated prior to contraction and subsequent relaxation with phenylephrine (PE) and sodium nitroprusside (SNP), respectively. Sample force tracing with and without MK2i-NP treatment is shown (Fig. 5B). Vasorelaxation of untreated vessels was subtracted as a baseline from treated vessel groups. Fresh MK2i-NPs and MK2i-NPs lyophilized without excipient enhanced vasorelaxation by only 2.52-fold (p > 0.05) and 1.63-fold (p > 0.05) compared to free MK2i peptide, respectively (Fig. 5C). Strikingly, MK2i-NPs lyophilized in 300 mM lactosucrose enhanced vasorelaxation by 2.39-fold (p < .01) and 3.89-fold (p < .005) compared to MK2i-NPs lyophilized without excipient and free MK2i peptide, respectively.
Figure 5. Lactosucrose Improves the Bioactivity of MK2i-NPs in Human Vascular Tissue.
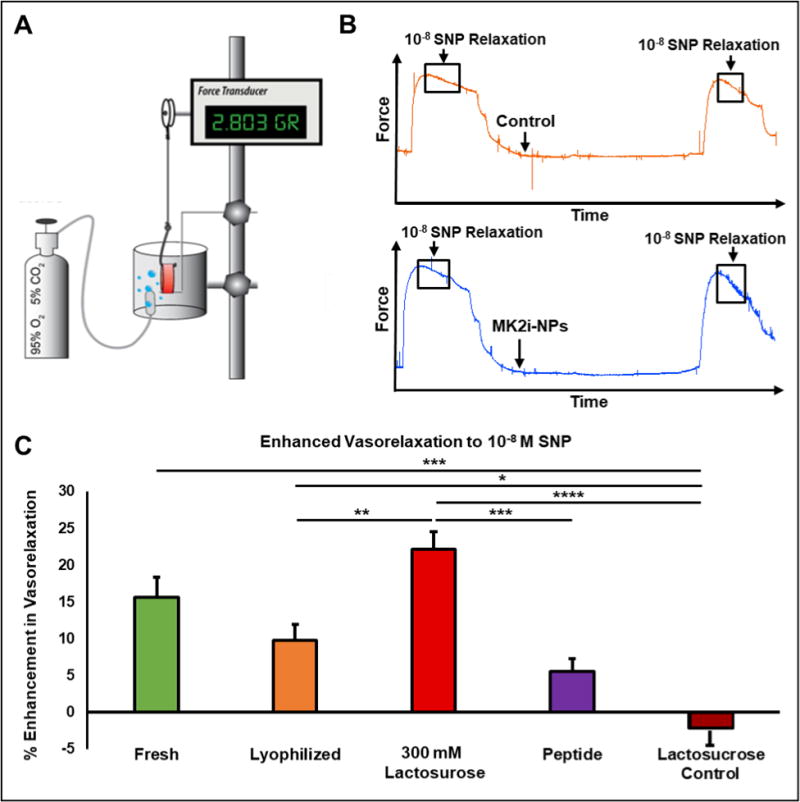
A) Experimental setup for testing MK2i-NP + excipient bioactivity in an ex vivo human saphenous vein (HSV) model. B) Representative force traces of HSV relaxation before and after treatment with and without MK2i-NPs. C) Enhancement in relaxation of contracted human saphenous vein rings in response to treatment with 10−8 M sodium nitroprusside following incubation with MK2i-NP formulations at a peptide dose of 25 μM. All data were normalized to untreated samples (n = 6; * p < 0.05, ** p< 0.01, *** p < 0.005, **** p < 0.001; Lactosucrose control is treatment with 300 mM lactosucrose alone).
Discussion
Lyophilization of MK2i-NPs without excipient significantly increased NP size, while freezing and lyophilization of MK2i-NPs in the presence of excipients was shown to generally maintain particle size relative to fresh MK2i-NPs. In particular, lyophilization in 150 and 300 mM lactosucrose optimally preserved particle size and morphology, which was severely disrupted in MK2i-NPs lyophilized without excipients, as qualitatively revealed by TEM imaging (Fig. 1B). MK2i-NP destabilization during lyophilization in the absence excipients is likely driven by removal of the adsorbed hydration shell, leading to inter-particle associations, aggregation, and structural destabilization. In an excipient solution, as the hydration shell is sublimated, excipient molecules substitute for water as hydrogen bonding partners, helping to stabilize the electrostatic complexes during the drying process [32].
Morphology stabilization is typically beneficial for promoting NP interactions with cell membranes, thereby improving cell internalization [33–35]. To this end, flow cytometric analysis of fluorescently labeled MK2i peptide uptake showed that all excipients tested restored losses in cell uptake of MK2i-NPs that occurred due to lyophilization, and in some cases generated lyophilized formulations with cell uptake levels that exceeded freshly prepared MK2i-NPs (Fig. 2). MK2i-NPs lyophilized in 300 and 600 mM lactosucrose demonstrated the highest uptake out of all excipient formulations tested. Importantly, uptake of fresh MK2i-NPs prepared in 300 mM lactosucrose was equivalent (99.6%) to fresh MK2i-NPs prepared in PBS, indicating that the increased uptake of lyophilized MK2i-NPs with excipients is dependent on lyophilization in the presence of excipients and not due to a cellular effect of the excipient. Previous studies utilizing sucrose (277 mM or 138.5 mM) and lactosucrose (277 mM or 138.5 mM) as lyoprotectants for plasmid-LPEI polyplexes similarly demonstrated the ability to restore lyophilized polyplex uptake and luciferase knockdown relative to freshly prepared polyplexes [22, 36]. There is also precedent for polyplexes lyophilized with excipients having improved activity over the freshly prepared parent formulation. For example, Zhang et al. demonstrated a trehalose dose-dependent increase in transfection efficiency for lyophilized poly(amine-co-ester-co-orthoester) (oPACE) polyplexes for gene delivery, with polyplexes lyophilized in 158 mM trehalose nearly doubling the transfection efficiency relative to fresh oPACE-pDNA polyplexes [37]. Thus, our results suggest that results from polyplexes used for gene delivery are relevant and can be extrapolated to the PPAA-based polyplexes designed for delivery of cationic peptides. The 600 mM lactosucrose formulation had the highest cellular uptake levels in two-dimensional cultures in vitro, but DLS analysis revealed presence of large aggregates in this formulation (Fig. 1A). Because there is an inverse relationship between particle size and tissue penetration [38, 39], we selected the 300 mM lactosucrose as our lead formulation that balances minimal particle size with maximal uptake for subsequent ex vivo smooth muscle physiology-based bioactivity assays in human vascular tissue.
Endosomal escape has been identified as a critical barrier for the successful cytosolic delivery of biologic therapeutics with intracellular pharmacological activity [40]. The MK2i-NP formulation was specifically designed to address this barrier by incorporating the pH-responsive, membrane disruptive polymer PPAA. Our previous work highlighted that MK2i-NP formulations facilitate escape from endolysosomal trafficking and also significantly increase peptide uptake and longevity of action [18]. The pH-dependent membrane disruptive activity of the PPAA polymer, freshly prepared MK2i-NPs, and all lyophilized MK2i-NPs was consistent with our previous findings. All MK2i-NP formulations produced no membrane disruptive activity at physiologic pH (< 5% hemolysis) but increasing membrane disruption as the pH progressed from early (pH 6.8) to late endosomal (pH 6.2) environments (> 60% hemolysis; Fig 3A). Preservation of pH-dependent, endosome-disruptive behavior to facilitate escape from intracellular endolysosomal trafficking was detected in live cells in vitro using a galectin-8 YFP imaging technique previously utilized by our lab [29]. Upon exposure to intracellular glycans, such as those found on the luminal surfaces of endosomes, diffuse cytosolic galectin-8 will cluster around the exposed glycans and form a punctate fluorescence pattern within the cell that allows for a quantifiable, real-time measure of endosomal disruption. Intracellular galectin-8 YFP spot counting following treatment demonstrated a 43% decrease in galectin-8 recruitment for MK2i-NPs lyophilized without excipient relative to fresh MK2i-NPs. Conversely, relative to MK2i-NPs lyophilized without excipient, MK2i-NPs lyophilized with lactosucrose created a 152% higher (p < 0.05) galectin-8 recruitment (Fig. 3B,C). Consistent with a lack of hemolysis at pH 7.4, no cytotoxicity was seen during galectin-8 live cell imaging or in vitro cell viability screen relative to vehicle control or fresh MK2i-NPs (Fig. 4). As expected, the galectin-8 YFP recruitment results match the trends seen in cellular uptake (Fig. 2A), as membrane disruptive activity is expected to be proportional to the quantity of internalized polymer. In addition, excipient-mediated preservation of NP morphology as demonstrated by TEM imaging and DLS analysis likely contributes to endosomal membrane interactions and disruption by maintaining higher surface area to volume ratios intrinsic to smaller, non-aggregated NPs.
It is anticipated that maximal bioactivity will be achieved based on optimizing both cell internalization and endosomal escape into the cytoplasm were active MK2 is found [17, 18]. Here, MK2i-NP bioactivity was assessed ex vivo in HSV based on physiologic relaxation. Smooth muscle physiology is a relevant and established readout [11] because MK2i peptide blocks activation of substrates downstream of MK2 that inhibit vasorelaxation by stabilizing actin stress fibers and inhibiting their depolymerization. At a dose of 25 μM MK2i peptide, the fresh MK2i-NPs increased HSV vasorelaxation 2.79-fold compared to free MK2i peptide, whereas MK2i-NPs lyophilized without excipient only resulted in a 1.75-fold increase. This finding confirms that the deleterious effects of lyophilization on NP size, morphology, and cellular uptake functionally reduce therapeutic bioactivity. Strikingly, incorporation of 300 mM lactosucrose as a lyoprotectant was found to not only restore but also enhance lyophilized MK2i-NP bioactivity compared to freshly prepared MK2i-NPs (3.96-fold enhancement over free MK2i). The exact mechanism by which these excipients enhance NP uptake and bioactivity is not definitively known, but may be due to formation of a stable, concentrated coating of lactosucrose on the NP surface that continues to facilitate particle stability following reconstitution.
Initially, the MK2i-NPs were optimized for ex vivo treatment of harvested vascular grafts immediately prior to transplantation. However, iterative designs of our nanopolyplex based system would be required to extrapolate this NP approach to in vivo applications that require systemic administration (e.g., intravenous administration) as these NPs would likely be unstable in blood due to competing interactions with blood substituents. For example, surface functionalization (e.g., PEGylation) would be a logical step toward increasing MK2i-NP colloidal stability and stealth.
Conclusions
The ability to lyophilize biologic therapeutics is key for long-term storage, distribution, and quality control. MK2i-NPs are a promising therapeutic for reducing vascular graft intimal hyperplasia and preventing pathological vasospasm, but lyophilization of MK2i-NPs significantly increased particle size and decreased nanoparticle uptake and bioactivity in human vascular smooth muscle cells and vascular tissue, respectively. The addition of excipients as lyoprotectants prior to lyophilization, particularly lactosucrose, preserves and/or improves particle morphology while also increasing cellular uptake, maintaining endosomal escape functionality, and enhancing the bioactivity of lyophilized MK2i-NPs over both NP formulations lyophilized without excipients and freshly prepared NPs.
Supplementary Material
Article Highlights.
Nano-Polyplexes (NPs) aggregate and lose bioactivity post-lyophilization
Excipients preserve NP size and morphology post-lyophilization
NPs lyophilized with lactosucrose have higher cellular uptake than even fresh NPs
NPs lyophilized with lactosucrose retain effective endosomal disruption ability
NPs lyophilized with lactosucrose have potent bioactivity in intact human vein
Acknowledgments
Funding
This work was supported by funding from the Vanderbilt University School of Engineering Summer Research Program (VUSE), National Institutes of Health (R01HL122347) and the National Science Foundation (Phase I STTR Award Number 1622828).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
C.L.D, C.M.B., and B.C.E. conceived and directed the project. B.C.E. synthesized the PPAA polymer and provided input on the experimental study design. B.B. generated preliminary data demonstrating the deleterious effects of lyophilization on the nano-polyplex formulation. A.J.M. synthesized, formulated, and characterized NPs and performed in vitro assays, flow cytometry, smooth muscle physiology experiments, image analysis, and statistical analysis. E.A.D. performed transmission electron microscopy. K.V.K. performed fluorescence confocal microscopy. J.C.F. aided in experimental study design and smooth muscle physiology studies. A.J.M. wrote the manuscript with the aid of B.C.E. and received feedback from and final approval from all authors.
Data and materials availability: All data are in the manuscript and all materials are available.
References
- 1.Wilson TR, Johnston PG, Longley DB. Anti-apoptotic mechanisms of drug resistance in cancer. Curr Cancer Drug Targets. 2009;9(3):307–19. doi: 10.2174/156800909788166547. [DOI] [PubMed] [Google Scholar]
- 2.Alexander JH, et al. Efficacy and safety of edifoligide, an E2F transcription factor decoy, for prevention of vein graft failure following coronary artery bypass graft surgery - PREVENT IV: A randomized controlled trial. Jama-Journal of the American Medical Association. 2005;294(19):2446–2454. doi: 10.1001/jama.294.19.2446. [DOI] [PubMed] [Google Scholar]
- 3.Craik DJ, et al. The Future of Peptide-based Drugs. Chemical Biology & Drug Design. 2013;81(1):136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 4.Ewing MM, et al. Annexin A5 Therapy Attenuates Vascular Inflammation and Remodeling and Improves Endothelial Function in Mice. Arteriosclerosis Thrombosis and Vascular Biology. 2011;31(1):95. doi: 10.1161/ATVBAHA.110.216747. [DOI] [PubMed] [Google Scholar]
- 5.McGregor DP. Discovering and improving novel peptide therapeutics. Current Opinion in Pharmacology. 2008;8(5):616–619. doi: 10.1016/j.coph.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Belting M, Sandgren S, Wittrup A. Nuclear delivery of macromolecules: barriers and carriers. Advanced Drug Delivery Reviews. 2005;57(4):505–527. doi: 10.1016/j.addr.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Richard JP, et al. Cell-penetrating peptides A reevaluation of the mechanism of cellular uptake. Journal of Biological Chemistry. 2003;278(1):585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 8.Lindgren M, et al. Cell-penetrating peptides. Trends in pharmacological sciences. 2000;21(3):99–103. doi: 10.1016/s0165-6147(00)01447-4. [DOI] [PubMed] [Google Scholar]
- 9.Lau YH, et al. Peptide stapling techniques based on different macrocyclisation chemistries. Chemical Society Reviews. 2015;44(1):91–102. doi: 10.1039/c4cs00246f. [DOI] [PubMed] [Google Scholar]
- 10.Fletcher MD, Campbell MM. Partially Modified Retro-Inverso Peptides: Development, Synthesis, and Conformational Behavior. Chemical Reviews. 1998;98(2):763–796. doi: 10.1021/cr970468t. [DOI] [PubMed] [Google Scholar]
- 11.Hedges JC, et al. A role for p38MAPK/HSP27 pathway in smooth muscle cell migration. Journal of Biological Chemistry. 1999;274(34):24211–24219. doi: 10.1074/jbc.274.34.24211. [DOI] [PubMed] [Google Scholar]
- 12.San Martín A, et al. Dual regulation of cofilin activity by LIM kinase and Slingshot-1L phosphatase controls platelet-derived growth factor–induced migration of human aortic smooth muscle cells. Circulation research. 2008;102(4):432–438. doi: 10.1161/CIRCRESAHA.107.158923. [DOI] [PubMed] [Google Scholar]
- 13.Rousseau S, et al. Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP K2 and its interaction with cytokine mRNAs. The EMBO journal. 2002;21(23):6505–6514. doi: 10.1093/emboj/cdf639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molnar P, et al. The cyclic AMP response element-binding protein (CREB) mediates smooth muscle cell proliferation in response to angiotensin II. Journal of cell communication and signaling. 2014;8(1):29–37. doi: 10.1007/s12079-013-0215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brugnano JL, et al. Cell-penetrating peptides can confer biological function: Regulation of inflammatory cytokines in human monocytes by MK2 inhibitor peptides. Journal of Controlled Release. 2011;155(2):128–133. doi: 10.1016/j.jconrel.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Muto A, et al. Inhibition of Mitogen Activated Protein Kinase Activated Protein Kinase II with MMI-0100 reduces intimal hyperplasia ex vivo and in vivo. Vascular Pharmacology. 2012;56(1):47–55. doi: 10.1016/j.vph.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans BC, et al. Endosomolytic Nano-Polyplex Platform Technology for Cytosolic Peptide Delivery To Inhibit Pathological Vasoconstriction. ACS Nano. 2015;9(6):5893–5907. doi: 10.1021/acsnano.5b00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans BC, et al. MK2 inhibitory peptide delivered in nanopolyplexes prevents vascular graft intimal hyperplasia. Science Translational Medicine. 2015;7(291):291ra95–291ra95. doi: 10.1126/scitranslmed.aaa4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans BC, et al. MK2 inhibitory peptide delivered in nanopolyplexes prevents vascular graft intimal hyperplasia. Science translational medicine. 2015;7(291):291ra95. doi: 10.1126/scitranslmed.aaa4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Talsma H, et al. Stabilization of gene delivery systems by freeze-drying. International Journal of Pharmaceutics. 1997;157(2):233–238. doi: 10.1016/s0378-5173(97)00244-5. [DOI] [PubMed] [Google Scholar]
- 21.Abdelwahed W, et al. Freeze-drying of nanoparticles: Formulation, process and storage considerations. Advanced Drug Delivery Reviews. 2006;58(15):1688–1713. doi: 10.1016/j.addr.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 22.Kasper JC, et al. Development of a lyophilized plasmid/LPEI polyplex formulation with long-term stability—A step closer from promising technology to application. Journal of Controlled Release. 2011;151(3):246–255. doi: 10.1016/j.jconrel.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Pikal-Cleland KA, et al. Protein Denaturation during Freezing and Thawing in Phosphate Buffer Systems: Monomeric and Tetrameric β-Galactosidase. Archives of Biochemistry and Biophysics. 2000;384(2):398–406. doi: 10.1006/abbi.2000.2088. [DOI] [PubMed] [Google Scholar]
- 24.Frokjaer S, Otzen DE. Protein drug stability: a formulation challenge. Nat Rev Drug Discov. 2005;4(4):298–306. doi: 10.1038/nrd1695. [DOI] [PubMed] [Google Scholar]
- 25.Cherng JY, et al. Stabilization of polymer-based gene delivery systems. International Journal of Pharmaceutics. 1999;183(1):25–28. doi: 10.1016/s0378-5173(99)00037-x. [DOI] [PubMed] [Google Scholar]
- 26.Anchordoquy TJ, Carpenter JF, Kroll DJ. Maintenance of Transfection Rates and Physical Characterization of Lipid/DNA Complexes after Freeze-Drying and Rehydration. Archives of Biochemistry and Biophysics. 1997;348(1):199–206. doi: 10.1006/abbi.1997.0385. [DOI] [PubMed] [Google Scholar]
- 27.Allison SD, Molina MdC, Anchordoquy TJ. Stabilization of lipid/DNA complexes during the freezing step of the lyophilization process: the particle isolation hypothesis. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2000;1468(1):127–138. doi: 10.1016/s0005-2736(00)00251-0. [DOI] [PubMed] [Google Scholar]
- 28.Evans BC, et al. Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. Journal of visualized experiments JoVE. 2013;73:e50166. doi: 10.3791/50166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kilchrist KV, et al. Mechanism of Enhanced Cellular Uptake and Cytosolic Retention of MK2 Inhibitory Peptide Nano-polyplexes. Cellular and Molecular Bioengineering. 2016;9(3):368–381. doi: 10.1007/s12195-016-0446-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weinbuch D, et al. Nanoparticulate Impurities in Pharmaceutical-Grade Sugars and their Interference with Light Scattering-Based Analysis of Protein Formulations. Pharmaceutical Research. 2015;32(7):2419–2427. doi: 10.1007/s11095-015-1634-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McLendon PM, et al. Interaction of Poly(glycoamidoamine) DNA Delivery Vehicles with Cell-Surface Glycosaminoglycans Leads to Polyplex Internalization in a Manner Not Solely Dependent on Charge. Molecular Pharmaceutics. 2010;7(5):1757–1768. doi: 10.1021/mp100135n. [DOI] [PubMed] [Google Scholar]
- 32.Crowe JH, et al. Stabilization of dry phospholipid bilayers and proteins by sugars. Biochemical Journal. 1987;242(1):1–10. doi: 10.1042/bj2420001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trewyn BG, et al. Biocompatible mesoporous silica nanoparticles with different morphologies for animal cell membrane penetration. Chemical Engineering Journal. 2008;137(1):23–29. [Google Scholar]
- 34.Yan F, et al. The effect of poloxamer 188 on nanoparticle morphology, size, cancer cell uptake, and cytotoxicity. Nanomedicine: Nanotechnology, Biology and Medicine. 2010;6(1):170–178. doi: 10.1016/j.nano.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Zhao W, et al. Polymerization-Induced Self-Assembly (PISA) - Control over the Morphology of 19F-Containing Polymeric Nano-objects for Cell Uptake and Tracking. Biomacromolecules. 2017;18(4):1145–1156. doi: 10.1021/acs.biomac.6b01788. [DOI] [PubMed] [Google Scholar]
- 36.Kasper JC, et al. Formulation development of lyophilized, long-term stable siRNA/oligoaminoamide polyplexes. European Journal of Pharmaceutics and Biopharmaceutics. 2013;85(2):294–305. doi: 10.1016/j.ejpb.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, et al. Multifunctional Poly(amine-co-ester-co-orthoester) for Efficient and Safe Gene Delivery. ACS Biomaterials Science & Engineering. 2016;2(11):2080–2089. doi: 10.1021/acsbiomaterials.6b00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Advanced Drug Delivery Reviews. 2003;55(3):329–347. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- 39.Tong R, et al. Photoswitchable Nanoparticles for Triggered Tissue Penetration and Drug Delivery. Journal of the American Chemical Society. 2012;134(21):8848–8855. doi: 10.1021/ja211888a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varkouhi AK, et al. Endosomal escape pathways for delivery of biologicals. Journal of Controlled Release. 2011;151(3):220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.