

Abstract
Many of the newly discovered therapeutic peptides and molecules are limited by their inability to cross the cell membrane. In the present study we employed a cell penetrating peptide (CPP), VPTLK, derived from Ku70 protein, to facilitate the entry of a mini-chaperone across the cell membrane. Our previous studies suggest that the mini-chaperone peptide representing the chaperone site in αA-crystallin, which can inhibit protein aggregation associated with proteopathies, has therapeutic potential. We have prepared a synthetic mini-chaperone by fusing the VPTLK sequence to N-terminus of mini-chaperone (FVIFLDVKHFSPEDLTVKGRD) to get VPTLKFVIFLDVKHFSPEDLTVKGRD peptide, which we call "CPPGRD." The amino acids, GRD, were added to increase the solubility of the peptide. The chaperone-like function of CPPGRD was measured using unfolding conditions for alcohol dehydrogenase and α-lactalbumin. The anti-apoptotic action of the peptide chaperone was evaluated using H2O2-induced Cos-7 and ARPE-19 cell apoptosis assays. The results show that the CPPGRD has both chaperone function and anti-apoptotic activity. Additionally, the CPPGRD was found to prevent β-amyloid fibril formation and suppress β-amyloid toxicity. The present study demonstrates that the CPPGRD protects unfolding proteins from aggregation and prevents cellular apoptosis. Therefore, the CPPGRD is a mini-chaperone with potential to become a therapeutic agent for protein aggregation diseases.
Keywords: Crystallin, chaperone, aggregation, apoptosis
Graphical abstract
This peptide chaperone encapsulates the chaperone site of small heat shock protein- alpha-A-crystallin. In this study the effects of the peptide on unfolding proteins and cells subjected to oxidative stress is investigated. In presence of the chaperone peptide suppression of unfolding protein aggregation and prevention of H2O2-induced cellular apoptosis is observed.

1. Introduction
Mini-chaperone is a peptide derived from αA-crystallin, corresponding to the 70 to 88 region, and functions like a molecular chaperone similar to full-length αA-crystallin [1]. The mini-chaperone peptide has been widely investigated by many researchers as a means of preventing protein aggregation associated with various stress conditions, including ultraviolet light-induced aggregation of γ-crystallin [2], heat-induced aggregation of alcohol dehydrogenase [2], and dithiothreitol (DTT)–mediated unfolding and aggregation of insulin [3] and α-lactalbumin [4]. Further, the mini-chaperone peptide has been shown to possess the ability to suppress β-amyloid–mediated toxicity in pheochromocytoma (PC12) cell lines [5]. The peptide has also been shown to rescue the mutant αAG98R protein’s stability and chaperone function [6]. Further, the mini-chaperone has been demonstrated to prevent oxidative stress-induced cell apoptosis in retinal pigment epithelium (RPE) cells [7] and suppress selenite-induced cataract formation in an experimental rat animal model [8]. Recently the mini-chaperone peptide was demonstrated to protect human γD-crystallin from thermal- and chemical-induced unfolding and aggregation [9]. These studies altogether suggest that the mini-chaperone peptide can be used as a therapeutic agent for diseases associated with protein aggregation. However, the mini-chaperone amino acid sequence DFVIFLDVKHFSPEDLTVK is a β-sheet-forming hydrophobic peptide and, on incubation at 37°C, forms amyloid fibril-like aggregates when subjected to shaking [10]. We undertook a study to fine-tune the mini-chaperone peptide to improve its chaperone activity, as described in a recent review article [11]. Our initial effort led to the development of mini-αA-GRD peptide (DFVIFLDVKHFSPEDLTVKGRD), with about 30% enhanced chaperone activity and without compromising its solubility in aqueous buffer. The GRD sequence was added to assist the solubility of the chaperone-substrate protein complex.
Recent publications in the field of peptide research promote many peptides as important therapeutic molecules for a wide range of diseases [12]. However, unlike traditional drugs, the cellular uptake of larger peptides is a challenging process, making the delivery system an important consideration in the development of peptide drugs to target cells. At the same time, a delivery system must be efficient, safe and nontoxic. Different delivery methods are currently being utilized, including nanoparticles, microinjection, electroporation, liposomal construction and viral-based vectors, but are associated with such drawbacks as poor specificity, low efficiency and high toxicity to cells [13]. An alternative method is to use protein transduction domains, also referred as cell-penetrating peptides (CPP), to deliver the bioactive peptides/proteins. CPPs are a group of sequences formed of amino acids that have positive charge and are amphipathic in nature (having hydrophilic and hydrophobic properties). The concept of CPP peptides first emerged nearly 30 years ago from studies of the cellular uptake of Tat (trans-activator of transcription) protein from human immunodeficiency virus 1 (HIV-1) [14]. A number of other peptides and proteins have been shown to possess translocation activity, including transportan [15], synthetic arginine-enriched sequences [16] and amphipathic peptide [17].
Over the last 25 years, more than 100 CPPs have been identified and used to transport a variety of biologically active molecules across the cell membranes. Gomez et al designed several CPP peptides derived from Bax-binding domain of ku70, including VPTLK and KLPVM [18]. We used the CPP tag, VPTLK, to fuse with mini-chaperone peptide at the N-terminal, to get VPTLKDFVIFLDVKHFSPEDLTVKGRD (referred to as CPPGRD), and rapid transfer of the chaperone peptide across the cell membranes. This study describes the properties of cell penetrating chaperone peptide.
2. Results
2.1.Characterization of the Cell-Penetrating Chaperone Peptide
The CPPGRD chaperone is a 26 amino acid length peptide with the theoretical mass of 3001.52 Da and pI 6.73. The synthetic peptide appears as a white lyophilized powder and is soluble in water. The secondary structure during far-UV CD analysis showed negative ellipticity at 214 nm, indicating that the peptide has a predominant β-sheet conformation at 25°C (Figure 1A). To determine the hydrophobicity of the chaperone peptide, we investigated the interaction of the hydrophobic probe bis-ANS, which increases in fluorescence intensity upon binding to the hydrophobic peptide or proteins. The bis-ANS profile in the presence of the peptide indicated that the chaperone peptide has an appreciable bis-ANS binding capacity, with the emission maximum for the dye at 498 nm (Figure 1B).
Figure 1. A. Far-UV CD spectra of CPPGRD peptide.
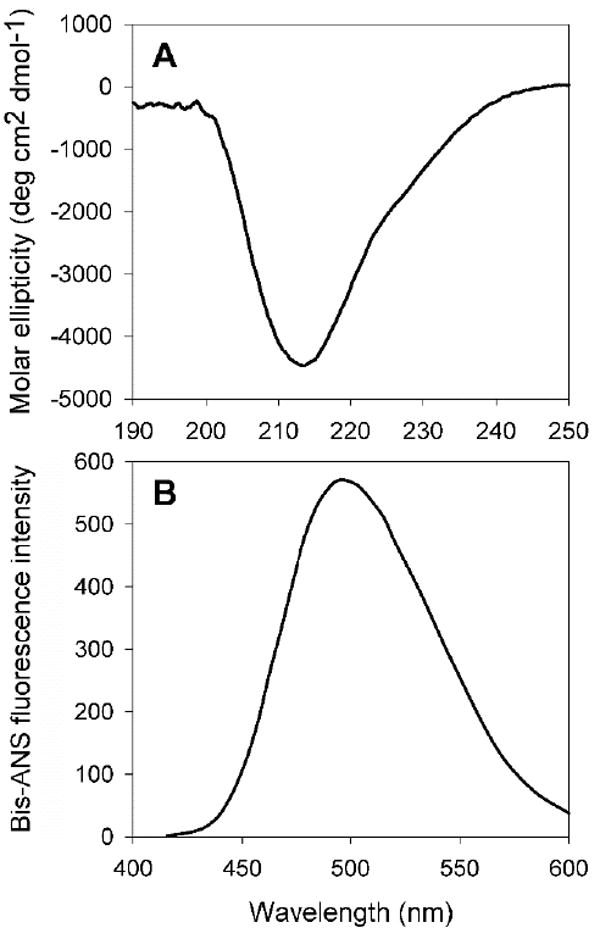
Peptide (33 μM) was used in 350μl of 50 mM phosphate buffer (pH 7.2) at room temperature. The given spectrum is an average of 5 scans. The far-UV CD spectrum shows negative ellipticity at 214 nm, suggesting β sheet conformation.
B) Bis-ANS binding assay of CPPGRD. Bis-ANS stock, 10 μL (10 mM), was added to 33 μM of CPPGRD peptide in 50 mM phosphate buffer (pH 7.2) and incubated at 37°C for 30 min. The sample was excited at 385 nm and the emission was recorded from 400 to 600 nm. The graph shown is representative of 3 independent assays.
2.2.Chaperone Activity
The increased hydrophobicity of some α-crystallin mutant proteins correlates with increased chaperone-like activities [19]. Since the bis-ANS fluorescence increased in the presence of CPPGRD (Figure 1 B) or mini-αA [1], we tested the chaperone-like function of the peptide against EDTA-induced unfolding alcohol dehydrogenase (ADH). Overall, we observed a dose-dependent suppression of ADH aggregation by the peptide chaperone (Figure 2A). The CPPGRD was about 10-20 percent more effective as a chaperone peptide as compared to the parent mini-αA-crystallin. Similarly, the CPPGRD also showed better chaperone activity than the mini-αA-crystallin when unfolding α-lactalbumin was used as substrate protein (Figure 2B). In another experiment we investigated whether the CPPGRD has the ability to prevent ADH aggregation already under way in the absence of chaperone peptide. In this experiment, ADH was allowed to aggregate for up to 20 min or 30 min, at which time CPPGRD was added to the reaction mixture. Reaction kinetic recordings demonstrated that the addition of the CPPGRD during the course of the reaction resulted in a significant reduction of additional aggregation by ADH (Figure 3A). However, the chaperone peptide failed to re-solubilize the preformed aggregates. Similar to the results of the ADH aggregation assay, the DTT-induced α-lactalbumin aggregation assay revealed that the addition of CPPGRD, after 40 min and 50 min of the start of the aggregation assay, led to suppression of further aggregation of α-lactalbumin (Figure 3B).
Figure 2. Chaperone assay in the presence of different concentrations of CPPGRD and mini-αA.
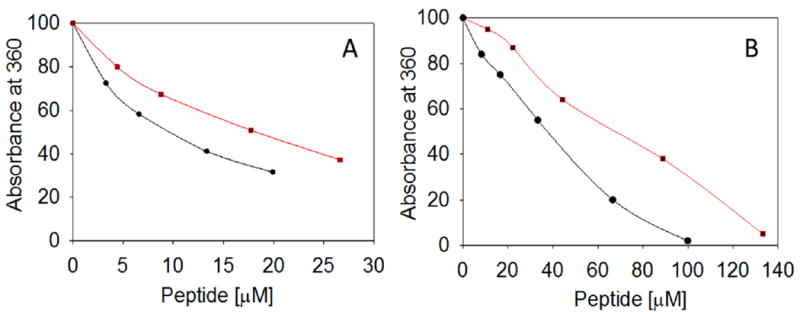
A) The ADH aggregation assay was performed using ADH (6.7 μM) in 50 mM phosphate buffer [150 mM NaCl and 100 mM EDTA (pH 7.2)] in the absence and presence of 0 to 28 μM of CPPGRD (●) or mini-αA (■) peptide at 37°C. The extent of aggregation was monitored at 360 nm in a Shimadzu spectrophotometer. B) α-Lactalbumin (85 μM) aggregation assay was initiated by addition of DTT (10 mM) to of the protein in presence and absence of 0 to 135 μM of CPPGRD or mini-αA at 37°C and the light scattering was recorded as described under methods. The profile shown is representative of 3 repeats.
Figure 3. Effect of the addition CPPGRD during ADH and α-lactalbumin aggregation.
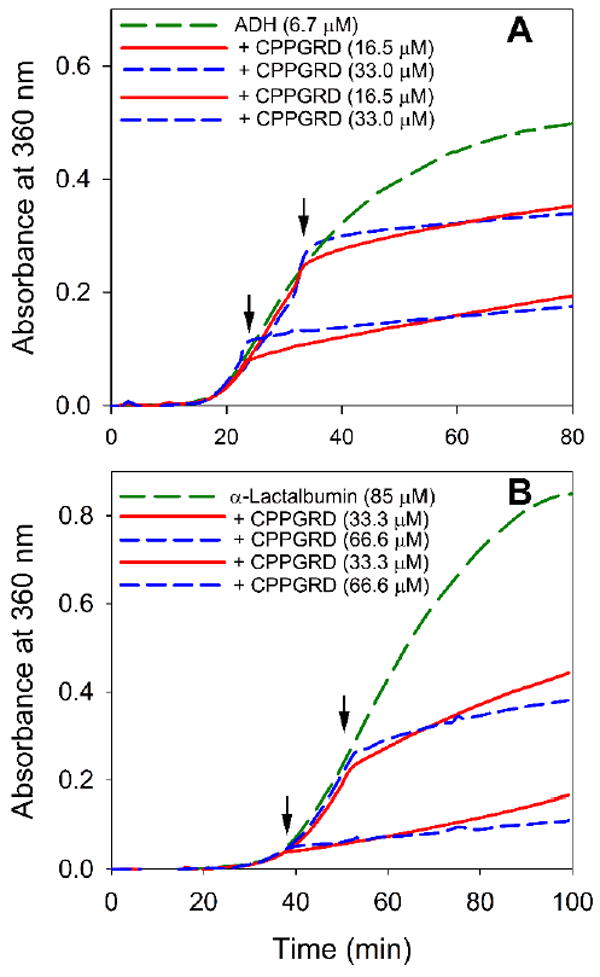
A) To investigate the effect of the CPPGRD on the aggregation of ADH (6.7 μM) during the chaperone assay, the CPPGRD (16.5 μM or 33 μM) was added either 20 min or 30min after the initiation of aggregation (shown by arrow) and the assay was continued up to 90 min.
B) α-Lactalbumin (85 μM) assay was performed as described under methods; the CPPGRD (33.3 μM or 66.6 μM) was added either 40 min or 50 min after the initiation of aggregation (shown by arrow) and the assay was continued for an additional 60 min. The graphs show that the CPPGRD suppresses the further aggregation of ADH and lactalbumin as soon as it is added. The graph shown is representative of 3 repeats.
2.3.Thioflavin T Binding Assay and TEM Study
Thioflavin T (ThT) selectively binds to amyloid-like fibrils and the bound ThT shows increased fluorescence. This phenomenon can be used to distinguish between amyloid and non-amyloid forms of protein aggregates. ThT binding assay was used to test the activity of the CPPGRD on the amyloid aggregation processes. β-Amyloid peptide (1-42) was incubated for 24 hr in the presence and absence of three different concentrations (6.6, 8.3 and 16.6 μM) of CPPGRD. We found that there was a decrease in ThT interaction with β-amyloid treated CPPGRD prior to 24 hrs incubation, as evidenced by lower ThT fluorescence (Figure 4). Use of the CPPGRD and β- amyloid peptide in a 1: 2.6 ratio resulted in 60 ± 5 percent suppression of ThT binding. Further, the addition of an increased amount (to obtain 1:1.7 and 1: 1.3 molar ratios) of CPPGRD to the reaction mixture containing β-amyloid resulted in 80 ± 2 percent suppression of ThT fluorescence (Figure 4). However, addition of CPPGRD to β-amyloid after 24 hrs incubation to form fibrils did not suppress ThT binding. When β-amyloid peptide alone was incubated for 24 hr at room temperature and observed under TEM, its distinct fibrillar structure was seen (Figure 5A). However, when CPPGRD was co-incubated with β-amyloid at 1:1 molar ratio, nearly complete suppression of fibril formation was observed (compare Figures 5A and 5B). The CPPGRD peptide by itself did not form amyloid fibrils under the experimental conditions used in this study (Figure 5D).
Figure 4. ThT binding assay to assess the activity of CPPGRD on β-amyloid aggregation.
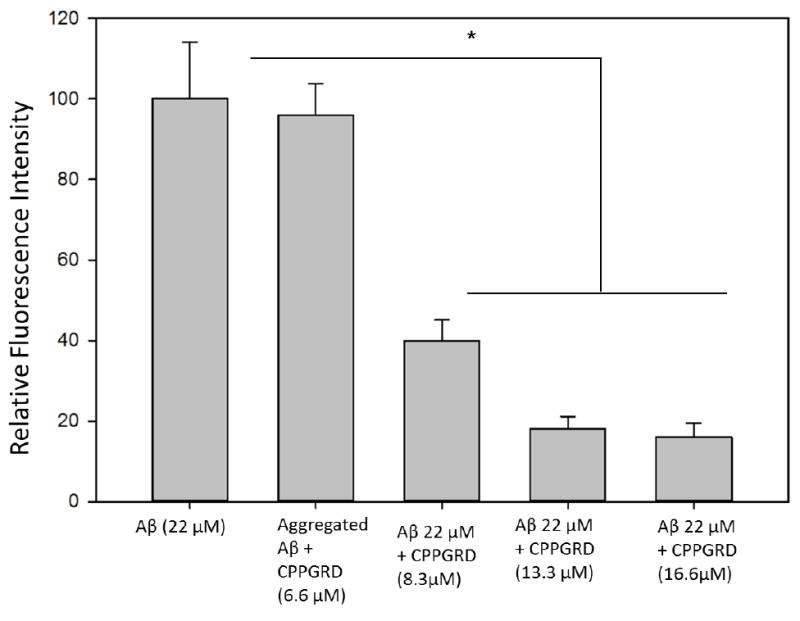
β-Amyloid peptide (22 μM) was incubated with or without 6.6 to 16.6 μM CPPGRD peptide in 50 mM phosphate buffer (pH7.2) at 37°C for 24 hrs prior to the addition of ThT (5 μM final concentration). The mixture was excited at 450 nm and the fluorescence emission was recorded at 484 nm. In a separate experiment, 6.6 μM CPPGRD peptide was added after fibrils were formed by β-Amyloid peptide (22 μM). ThT was added just before the fluorescence was measured. The values shown in the graph are average of triplicates. The asterisk (*) indicates a significant difference (p ≤ 0.05) in the ThT fluorescence due to reduction in fibril formation by β-amyloid in the presence of CPPGRD as compared to β-amyloid incubated without CPPGRD or CPPGRD added after the fibrils are formed.
Figure 5. TEM micrographs of β-amyloid (Aβ) peptide in the presence and absence of CPPGRD.
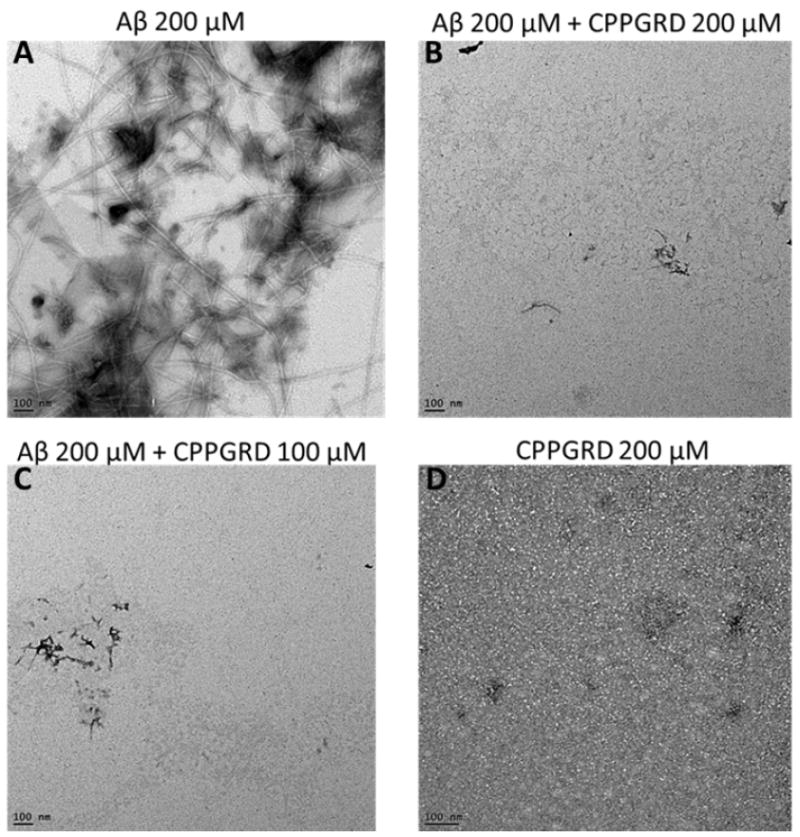
The efficacy of CPPGRD in inhibiting fibril formation was evaluated as described under methods. A) Aβ peptide (200 μM); B) Aβ (200 μM) + CPPGRD (200 μM); C) Aβ (200 μM) + CPPGRD (100 μM); D) CPPGRD (200 μM). Bar in the TEM micrographs = 100 nm. The TEM micrographs show that the CPPGRD prevents fibril formation at both molar ratio. The figure shown is representative of 3 repeats.
2.4. CPPGRD Shows Increased Cell Penetration Compared to Mini-αA peptide
We used Cos-7 cells to test the cell-penetrating efficiency of the mini-αA peptide and CPPPRD. Cos-7 cells were treated with either fluorescein isothiocyanate (FITC)–labeled CPPGRD or FITC-labeled mini-αA peptide for 20 min. Fluorescence intensity of the peptide chaperones taken up by the cells was recorded using the confocal microscope with FITC filter. The recorded micrographs were analyzed using image J software (http://imagej.nih.gov). Figure 6A shows the efficacy of FITC-labeled peptide uptake in Cos-7 cells. The increased uptake of CPPGRD-FITC compared to mini-αA-FITC by Cos-7 cells is shown in Figure 6B and summarized in Figure 6A. The results show that after 20 minutes of treatment the CPPGRD penetrated Cos-7 cells 3 times more (1.47E+5 fluorescence units) than did the mini-αA peptide (5.0E+4 fluorescence units). In another experiment, we measured the biological activity of the CPPGRD by investigating its cytoprotective effect against hydrogen peroxide–induced cell apoptosis of ARPE-19 cells (Figures 6C and D). We found that 20 minutes of treatment was sufficient to demonstrate a better protective effect of CPPGRD peptide compared to similar amounts of mini-αA. While there was only 6 ± 0.6 percent of ARPE-19 cell death in presence of CPPGRD there was 9.2 ± 1.2 percent cell death in presence of mini-α (Figure 6C). The cells that were not exposed to H2O2 or peptide chaperone showed 6.1 ± 0.6 percent death, comparable to those treated with was CPPGRD peptide. The ARPE-19 cells that were treated with H2O2 showed 19 ± 0.6 percent cell death suggesting that CPPGRD protects the ARPE-19 cells from H2O2 effect.
Figure 6. A comparison between CPPGRD and miniα-A chaperone.
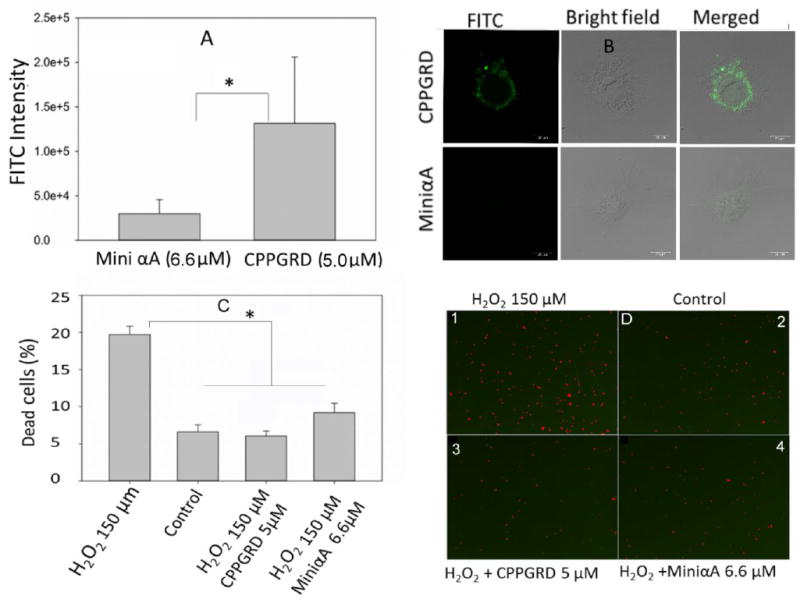
A) The efficacy of cellular uptake of peptide chaperones B) Cytoprotective effect against H2O2 induced cell apoptosis.
A) The Cos-7 cells were incubated in presence and absence of either CPPGRD or mini-αA peptide at 37°C for 20 min. Then the cells were fixed with 4% PFA (Paraformaldehyde) for 1 hr at room temperature. The fixed cells were observed under confocal microscope and images were recorded using digital camera. The fluorescence intensity of FITC-labeled peptide was calculated using image J software. The given intensities are an average of 20 micrographs. * =p<0.05.
B) Representative micrographs showing labeled peptides (green). C) ARPE-19 cells were cultured 80 percent confluency then the cells were treated with CPPGRD (5 μM) or mini-αA (6.6 μM) for 20 min. Peptide treated cells were challenged with H2O2 (150 μM) in serum free medium for overnight. The next day, cells were treated with live and dead cell reagents and analyzed using SpectraMax i3 (Molecular Devices Inc). * = p< 0.05. The bar diagram shows an average of 3 replicates. D) Micrographs 3 and 4 show significant reduction in dead cells (red) in wells treated with chaperone peptides and H2O2 compared to cells in micrograph, treated with H2O2. Cells in control did not receive H2O2. The 6B figures were captured at 20X magnification and the bar is 20 μm. The images in D were captured at 4X magnification.
2.5. Anti-Apoptotic Activity of CPPGRD
From our earlier studies we know that the mini-αA chaperone protects against apoptosis in cells subjected to oxidative stress [7]. Therefore, we tested whether CPPGRD offers protection against oxidative stress in mammalian cell lines that were exposed to the peptide chaperone. We used Cos-7 cells for the study and allowed the cells to grow 80% confluence in Dulbecco’s Modified Eagle’s Medium (DMEM). The cells were treated with 10 μM of the CPPGRD peptide for 4 hr in serum-free medium, after which the cells were washed twice with phosphate buffer and treated with 150 μM H2O2 in serum-free medium. A positive and negative control was set up simultaneously. Apoptotic cell death was assessed by the TUNEL assay. As Figure 7 shows, Cos-7 cells treated with CPPGRD were protected from apoptotic cell death as compared to the cells treated with H2O2 without the chaperone peptide. While 5.5 ± 1.25 percent of the Cos-7 cells were found to be TUNEL positive in presence of H2O2, only 1.13 ± 0.3 percent of the cells treated with CPPGRD + H2O2 were either apoptotic or dead, a value comparable to control cells (1.25 ± 0.37 percent) that were not exposed to either chaperone peptide or H2O2. To confirm the TUNEL results, in a separate experiment, Cos-7 cells were first treated with peptide and challenged with 150 μM H2O2. The early and late apoptotic markers were detected using FITC-labeled annexin V and nuclear stain propidium iodide (PI). Analysis of the stained cells by flow cytometry demonstrated that CPPGRD protects against H2O2-induced cell apoptosis (Figure 8), consistent with the results obtained by TUNEL assay. When ARPE-19 cells were exposed to H2O2 (150 μM) for 24 hr in the presence and absence of 10 μM CPPGRD, the cells treated with the chaperone peptide prior to H2O2 treatment, as expected, showed significantly higher (96 ± 1 percent) survival as compared to the cells that were not treated with the chaperone peptide (82 ± 2 percent) prior to H2O2 exposure (Figure 8). The cells cultured without H2O2 exposure or CPPGRD treatment showed 98 ± 2 percent survival under experimental conditions used. Similarly, ARPE-19 cells treated with the CPPGRD and β-amyloid (100 μM each) exhibited increased survival (81 ± 2 percent) as compared to ARPE-19 cells treated with 100 μM β-amyloid (54 ± 2 percent) (Figure 9). The cells that were treated with 100 μM CPPGRD were 92 ± 1 percent viable under the experimental conditions.
Figure 7. TUNEL assay of Cos-7 cells treated with H2O2 and the CPPGRD.
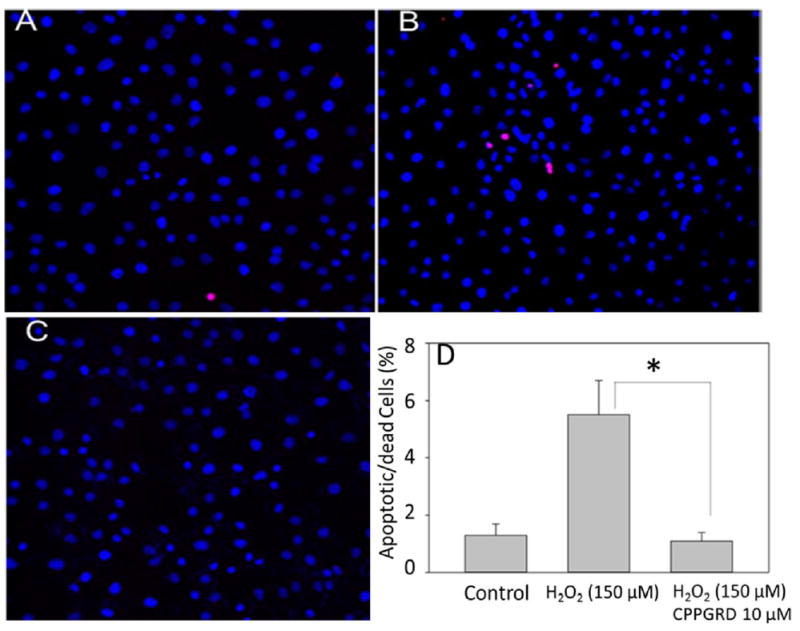
TUNEL assay of H2O2-treated Cos-7 cells was performed according to the manufacturer’s protocol and the images were recorded under a fluorescence microscope. A) Control, B) H2O2 (150 μM), C) CPPGRD (10 μM) + H2O2 (150 μM). Blue represents DAPI staining and Red represents TUNEL- positive cells. The bar graphs on the right depict the summary of flow cytometry data from 3 replicates. The asterisk (* = p<0.05) in the bar graph indicates significant change p ≤ 0.001. CPPGRD by itself had no effect on Cos-7 cells (data not shown).
Figure 8. Flow cytometry analysis of ARPE-19 cells treated with H2O2 and CPPGRD.
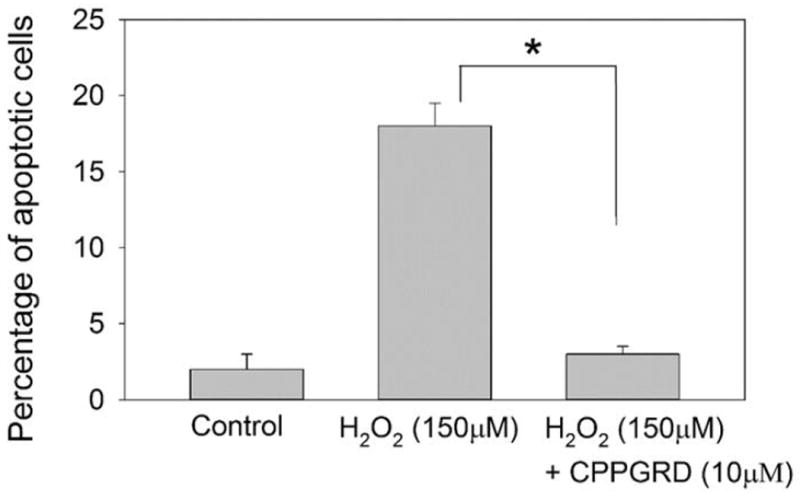
To determine the effect of the CPPGRD on H2O2-induced ARPE-19 cell apoptosis, a FITCAnnexin-V apoptosis detection kit was used. ARPE-19 cells were exposed to H2O2 (150 μM) for 24 hr in the presence and absence of CPPGRD (10 μM). The treated cells were harvested and resuspended in binding buffer containing FITC conjugated annexin-V protein and propidium iodides (PI) stain. The uptake of annexin binding and PI staining was determined by FACSCalibur flow cytometry. The asterisk (* = p < 0.05) indicates a significant difference in the protection against H2O2-induced cell apoptosis in the presence of CPPGRD. n=3.
Figure 9. The effect of CPPGRD on beta amyloid peptide induced cell apoptotic in ARPE - 19 cells.
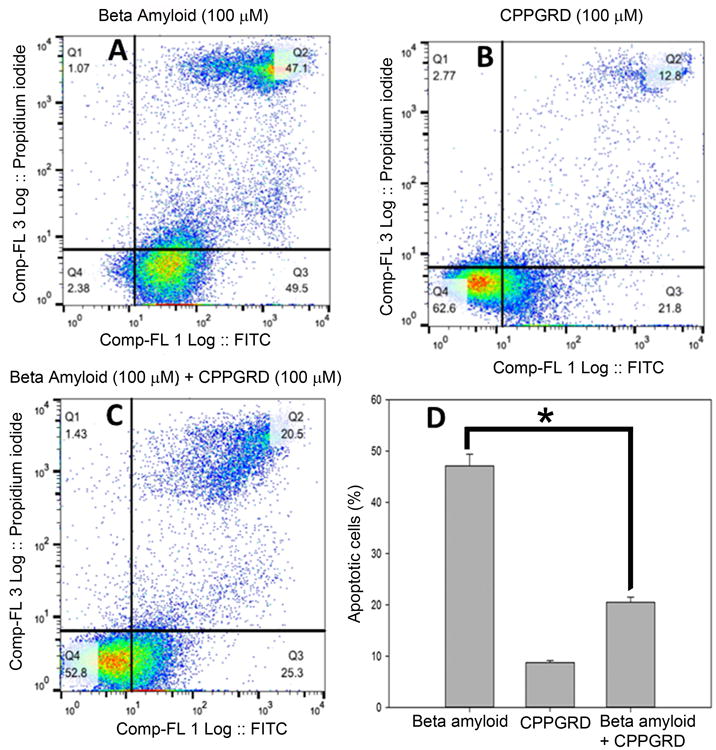
A-C representative Annexin V FITC vs Propidium iodide - contour plots. D) The percentage of apoptotic cells in A-C. One hundred μM of CPPGRD and β-amyloid were used in the study. CPPGRD peptide + β-amyloid treated cells showed significantly (* = p< 0.05) higher degree of survival compared to the cells that were treated with β-amyloid. The bar graphs represent results from three replicates.
3. Discussion
Proteins have both a structural and a functional role in biological systems. Disruption of the normal structure of proteins due to mutations can lead to a number of disease conditions. While some mutations lead to loss of function, others can result in toxic gain of function [20]. Abnormal interactions between functional proteins can also lead to diseases [21], as in the case of cataracts, Alzheimer’s dementia, Parkinson’s disease, amyloidosis and Huntington’s disease. Many of the abnormal properties of proteins can be controlled by molecular chaperones that are either protein-based chaperones or chemical chaperones [22]. The chaperones rescue the functions of mutant proteins or prevent the abnormal interactions of mutant proteins.
Similar to chemical chaperones (e.g. trimethylamine-N-oxide) that suppress abnormal protein interactions, protein chaperones can suppress disease-causing interactions in mutant proteins as well as proteins that show pathological interactions. We have shown that mini-αA-chaperone, a peptide representing the chaperone-binding site in αA-crystallin, prevents destabilized protein aggregation [1]. We also have shown that mini-chaperone interacts with β-amyloid and reduces its toxicity [5]. However, delivering the mini-chaperone peptide to the sites that most require would not be easy because of cell membrane barrier and the potential of endogenous peptidases cleaving the peptide before it reaches the target site. Therefore, we added a cell-penetrating tag to the mini-chaperone peptide and created the CPPGRD peptide so that it will enter the cells rapidly. The major goal of the present study was to assess the CPPGRD for its chaperone function, its cell-penetrating ability and its efficacy in preventing apoptotic cell death in cells subjected to oxidative stress.
In our study the CPPGRD was characterized by different methods. The CPPGRD is a hydrophobic peptide with significant bis-ANS binding property like the native mini-αA peptide chaperone [1]. This suggests that the CPPGRD peptide has a high degree of exposed, clustered hydrophobicity surface, an important property for its chaperone-like function. Circular dichroism studies showed that the peptide chaperone exhibits a predominantly β-sheet structure, similar to that observed with the parent mini-chaperone peptide [1]. This characteristic indicates that the addition of the CPP tag and incorporation of GRD residues to the C-terminal region have little effect on the overall chaperone peptide structure. In previous studies, we observed that the addition of the EEKPTSAPSS sequence to the C-terminal of mini-chaperone results in the loss of β-sheet formation [23]. The chaperone activity of the CPPGRD against unfolding ADH was about 10-20 percent higher than that of the original mini-chaperone, demonstrating that the cell penetrating sequence did not diminish the chaperone activity of the peptide. As with mini-αA, the CPPGRD suppressed aggregation of α-lactalbumin unfolded by reducing agent (Figures 2A and 2B). When we added the chaperone peptide to an unfolding protein aggregation system, such as that of ADH or α-lactalbumin, we found that immediately after the addition of the peptide chaperone, further aggregation of unfolding protein in the assay was suppressed, to the extent of 90 to 95 percent (Figure 3). This suggests that if the chaperone peptide can be delivered to the target cell that is showing protein aggregation, further aggregation of proteins could potentially be prevented. This opens up the opportunity to test the ability of the CPPGRD in vivo and to determine whether it can suppress the ongoing aggregation of mutant proteins.
The capacity of the CPPGRD to prevent protein aggregation was not limited to reactions involving unfolding proteins. When the CPPGRD was added to fibril-forming β-amyloid peptide (1-42), we found that there was noticeable suppression of fibril formation, as evidenced by TEM studies (Figure 5). This result was corroborated by ThT binding studies, which demonstrated decreased ThT binding to β-amyloid peptide that was co-incubated with the chaperone peptide (Figure 4). Earlier we reported that the parent mini-chaperone suppresses β-amyloid fibril formation [5]. In the present study, we now see that the CPPGRD exhibits preventive properties against β-amyloid fibril formation in spite of the addition of the cell-penetrating sequence to the N-terminus and the GRD sequence to the C-terminus of the parent mini-chaperone. Specific peptides interacting with and preventing the fibril formation by β-amyloid has been reported earlier [24]. Therefore we hypothesize that a direct interaction between β-amyloid and CPPGRD may be responsible for the suppression of fibril assembly by β-amyloid peptide. Further studies are required to pinpoint the amino acid residues involved in these interactions. Alternately, transient interaction between CPPGRD and β-amyloid may also lead to suppression of β-amyloid fibril formation in presence of CPPGRD peptide as seen with αB-crystallin and α-lactalbumin [25]. Studies have shown that β-amyloid is toxic to cells [26,27], and our previous studies using PC-12 cells showed that mini-chaperone protects the PC-12 cells from β-amyloid toxicity [5]. The results of flow cytometry (Figure 9) show that the CPPGRD protected the ARPE-19 cells from β-amyloid induced toxicity and that the addition of the cell-penetrating sequence did not abolish the toxicity-neutralizing property of the chaperone peptide.
Treatment of Cos-7 or ARPE19 cells with CPPGRD showed no cytotoxic effects. The oxidative stress assay involving H2O2 treatment of Cos-7 cells revealed that CPPGRD exerts cytoprotective properties against hydrogen peroxide–induced oxidative damage (Figure 7), similar to the observation we made earlier with mini-chaperone and ARPE-19 cells [23]. This finding suggests that the addition of the cell-penetrating sequence does not diminish the cell protection property of the mini-chaperone. Therefore, the present study shows that the cell penetrating mini-chaperone has both chaperone activity and cytoprotective activity.
In cells, membranes play an important role in protecting cells and transporting molecules in and out of the cell, depending on the molecular size, polarity, charge and concentration inside and outside of the cells. On the other hand, cellular receptors located on the membranes selectively allow certain molecules to enter and exit the cell through the membrane. Further studies are required to determine the mechanism involved in the transport of CPPGRD across Cos-7 cell membrane.
4. Conclusions
Overall, we have developed a cell-penetrating mini-chaperone peptide (CPPGRD) which has the capacity to prevent the aggregation of unfolding proteins and to protect cells from apoptosis induced by oxidative stress. The encapsulated dual property of the peptide—namely, the chaperone activity and cell penetration capacity—may make it a potential platform for the development of therapeutic molecules to treat many debilitating protein aggregation diseases.
5. Experimental Section
Peptide and Protein Procurement
We designed the CPPGRD (VPTLKFVIFLDVKHFSPEDLTVKGRD) and had it synthesized by GenScript Corp (Piscataway, NJ). High-performance liquid chromatography (HPLC) and mass spectrometry analysis showed that the synthesized peptide was >95 percent pure. Dried peptide, 2 mg/vial, was stored at −80°C. The peptide was dissolved in sterile water at 2mg/ml concentration. ADH and other chemicals were procured from Sigma-Aldrich (St. Louis, MO). Thioflavin T was purchased from Sigma (St. Louis, MO), and β-amyloid peptide [1-42] was obtained from EZBiolab Inc (Carmel, IN). Circular Dichroism (CD) Spectroscopy: The secondary structure of the CPPGRD was determined by far-ultraviolet (UV) spectra analysis using a Jasco J-815 CD spectrophotometer (Easton, MD). The CPPGRD, was dissolved in 350 μl of phosphate buffer (5 mM) to get 95 μM peptide concentration and far-UV CD measurements were carried out with a 2-mm path length cuvette over the wavelength of 190 -250 nm, with a bandwidth of 0.5 nm and a scan speed of 10 nm/min at 25°C.
Bis-ANS Binding Assay
The hydrophobic probe bis-ANS (1,1′-bi-[4-anilino]naphthalene-5,5′-disulfonic acid) (Molecular Probes, Inc., Eugene, OR) was prepared as stock of 10 mM strength in 95 percent ethanol. We diluted 10 μl of bis-ANS stock solution in 1 ml of 50 mM phosphate buffer, pH containing 100 μg of CPPGRD. The reaction mixture (33 μM peptide) was incubated at 37°C for 30 min. The sample was excited at 385 nm and the intensity of bis-ANS was measured by recording the emission spectra between 400 to 600 nm in a Jasco Spectrofluorimeter FP-750 (Jasco Corporation, Tokyo, Japan).
Thioflavin T Binding Assay
Thioflavin T (ThT) was prepared in 50 mM glycine-NaOH (pH 8.4) buffer at a concentration of 1 mM, passed through a 0.45 μm filter and stored at 4°C. β-amyloid peptide (100 μM) was incubated at 37°C in the presence and absence of CPPGRD (6.3, 8.3 and 16.6 μM) for 24 hrs. At the end of the incubation, ThT was added to get a final concentration of 5 μM, and the mixtures were transferred to 1-mL cuvettes with a 1-cm path length. The samples were excited at 450 nm (bandwidth 5 nm) and the emission was recorded at 482 nm using a Jasco FP750 spectrofluorometer (Jasco Corporation, Tokyo, Japan).
TEM of β-Amyloid Fibrils
To examine the efficacy of the CPPGRD in inhibiting β-amyloid fibril formation, 200 μM of β-amyloid was incubated in the presence and absence of CPPGRD in 50 mM phosphate buffer (pH 7.2) at 37°C for 24 hrs. At the end of the incubation, a 5-μl sample was placed on a carbon-coated copper grid and left for 1 min. Excess solution was wicked away with filter paper and the grid was stained for 10 min with 5 μl of freshly prepared 5 percent uranyl acetate solution. The negatively stained grid was air-dried and then observed under transmission electron microscopy (TEM) (JEOL 1400 (120kV). The TEM micrographs were captured on a digital camera at 25K magnification.
Chaperone Assay
To measure the chaperone activity of CPPGRD, the ADH aggregation assay was performed using ADH (6.7 μM) in 1 ml of 50 mM phosphate buffer containing150 mM NaCl and 50 mM EDTA (pH7.2) at 37°C in the presence of different concentrations of CPPGRD. The assay was monitored for up to 90 min and 120 min for light scattering at 360 nm, using a Shimadzu spectrophotometer equipped with a temperature controller and a multi-cell transporter. In another experiment, the CPPGRD was added after 20 or 30 min of the start of the assay to determine whether the addition of the peptide prevents continued aggregation of unfolding ADH and light scattering was measured for up to 90 min. The tubes containing ADH control also received buffer in place of peptide. We also tested the chaperone efficacy of the CPPGRD peptide against α-lactalbumin aggregation. In this experiment the chaperone peptide was added at 40 and 50 min after the start of the assay and light scattering was monitored for up to 100 min.
Cell Culture Experiments
Cos-7 cells were cultured in Gibco cell culture media (DMEM), from Thermo Fisher Scientific, supplemented with 100 μg/mL streptomycin, 100 units/mL penicillin and 5 percent fetal bovine serum, at 37°C in an incubator with 5 percent CO2 atmosphere. Cells (25,000 cells/cm2) were seeded in an 8-well slide chamber and allowed to grow at 37°C. After 80 percent confluency, the cells were treated with CPPGRD (10 μM) in serum-free medium for 4 hr. Then the cells were washed twice with serum-free media and challenged with 150 μM H2O2 overnight. On the next day the cells were processed for TUNEL assay. To confirm the TUNEL data, we performed flow cytometry analysis using ARPE-19 cells. The effect of the CPPGRD (10 μM) on H2O2-induced cell apoptosis (150 μM of H2O2) was studied as described earlier [23]. ARPE-19 cells were also used to test the toxic effect of β-amyloid and the protection of the cells by the peptide chaperone. To accomplish this, the ARPE-19 cells were first treated with 100 μM of peptide chaperone and then β-amyloid (100 μM final concentrations) was added. After 24 hr of incubation the cells were subjected to flow cytometric analysis.
TUNEL Assay
TdT-Mediated digoxigenin-dUTP nick end labeling (TUNEL) assays were performed using ApopTag Red in situ apoptosis detection kit (Chemicon, Temecula, CA), according to the manufacturer’s protocol. In brief, CPPGRD treated cells were fixed in 4 percent paraformaldehyde overnight at 4°C. Fixed cells were washed twice with phosphate-buffered saline for 5 min and post fixed in an ethanol/acetic acid mixture (2:1) for reagent permeability. Anti-digoxigenin-rhodamine solution was used to detect TUNEL labeling, whereas counter stain DAPI (4-,6-diamidino-1-phenylindole) was used to detect the nucleus. Stained cells were observed under a fluorescence microscope (Leica DMR) and the images were recorded using an Optonics digital camera. The percentage of apoptotic cells was calculated by counting TUNEL positive cells divided by the total number of cells visualized in a given area.
Cellular Uptake of Chaperone Peptides
Cos-7 cells (25,000) were incubated with 0.2 mL DMEM complete medium in 8 well-chamber glass slides. After 24 hrs, the culture medium was replaced with serum-free medium then the cells were treated with FITC-labeled CPPGRD or mini-αA peptide for 10, 20 and 30 min and the cells were washed thoroughly before they were fixed using 4% PFA. On the next day, the cells were washed with PBS buffer thrice and the cells were observed under fluorescence microscope. Cell images were recorded using a digital camera and analyzed for fluorescence intensity using image J software.
Flow Cytometry Analysis
We also analyzed ARPE-19 cell apoptosis by flow cytometry using an FITC annexin V apoptosis detection kit (BioLegend, San Diego, CA), as per the manufacturer’s protocol. In brief, cells were harvested and washed twice with phosphate buffer. The cells were mixed in 200 μl binding buffer, and annexin-V 5 μl (10 μg/mL) and propidium iodine (10 μl) were added. The labeling reaction mixtures were incubated for 15 min at room temperature in darkness. After incubation for 15 min, the cells were analyzed with a flow cytometer, BD FACScalibur, at the University of Missouri Cytology Core Facility.
Statistical Analysis
The graphs and statistical analyses were performed using the Sigma Plot 12.0 software Program (Systat Software Inc., San Jose, CA) and Microsoft Exl spread sheet. The Student-t test and one-sided testing was used to determine statistical significance. The experiments were replicated at least 3 times for each group and the differences were considered significant if P<0.05.
Acknowledgments
The work was supported in part by NIH grant EY 023219 and Beryl J. Ortwerth Professorship to KKS. We thank Sharon Morey for help in the preparation of the manuscript and Dr. Alexander Jurkevich of the Molecular Cytology Core, University of Missouri, for his help with confocal microscopy.
References
- 1.Sharma KK, Kumar RS, Kumar GS, Quinn PT. J Biol Chem. 2000;275:3767. doi: 10.1074/jbc.275.6.3767. [DOI] [PubMed] [Google Scholar]
- 2.Kumar RS, Sharma KK. J Pept Res. 2000;56:157. doi: 10.1034/j.1399-3011.2000.00785.x. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharyya J, Sharma KK. J Pept Res. 2001;57:428. doi: 10.1034/j.1399-3011.2001.00871.x. [DOI] [PubMed] [Google Scholar]
- 4.Sreelakshmi Y, Sharma KK. J Protein Chem. 2001;20:123. doi: 10.1023/a:1011077307262. [DOI] [PubMed] [Google Scholar]
- 5.Santhoshkumar P, Sharma KK. Mol Cell Biochem. 2004;267:147. doi: 10.1023/b:mcbi.0000049373.15558.b8. [DOI] [PubMed] [Google Scholar]
- 6.Raju M, Santhoshkumar P, Sharma KK. PLoS One. 2012;7:e44077. doi: 10.1371/journal.pone.0044077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sreekumar PG, Chothe P, Sharma KK, Baid R, Kompella U, Spee C, Kannan N, Manh C, Ryan SJ, Ganapathy V, Kannan R, Hinton DR. Invest Ophthalmol Vis Sci. 2013;54:2787. doi: 10.1167/iovs.12-11571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nahomi RB, Wang B, Raghavan CT, Voss O, Doseff AI, Santhoshkumar P, Nagaraj RH. J Biol Chem. 2013;288:13022. doi: 10.1074/jbc.M112.440214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banerjee PR, Pande A, Shekhtman A, Pande J. Biochemistry. 2015;54:505. doi: 10.1021/bi5014479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka N, Tanaka R, Tokuhara M, Kunugi S, Lee YF, Hamada D. Biochemistry. 2008;47:2961. doi: 10.1021/bi701823g. [DOI] [PubMed] [Google Scholar]
- 11.Raju M, Santhoshkumar P, Sharma KK. Biochim Biophys Acta. 2016;1860:246. doi: 10.1016/j.bbagen.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dinca A, Chien WM, Chin MT. Int J Mol Sci. 2016;17:263. doi: 10.3390/ijms17020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torchilin VP, Omelyanenko VG, Papisov MI, Bogdanov AA, Trubetskoy VS, Herron JN, Gentry CA. Biochim Biophys Acta. 1994;1195:11. doi: 10.1016/0005-2736(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 14.Frankel AD, Pabo CO. Cell. 1988;55:1189. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 15.Pooga M, Hällbrink M, Zorko M, Langel U. FASEB J. 1998;12:67. doi: 10.1096/fasebj.12.1.67. [DOI] [PubMed] [Google Scholar]
- 16.Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. J Biol Chem. 2001;276:5836. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 17.Oehlke J, Scheller A, Wiesner B, Krause E, Beyermann M, Klauschenz E, Melzig M, Bienert M. Biochim Biophys Acta. 1998;1414:127. doi: 10.1016/s0005-2736(98)00161-8. [DOI] [PubMed] [Google Scholar]
- 18.Gomez JA, Chen J, Ngo J, Hajkova D, Yeh IJ, Gama V, Miyagi M, Matsuyama S. Pharmaceuticals. 2010;3:3594. doi: 10.3390/ph3123594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shroff NP, Bera S, Cherian-Shaw M, Abraham EC. Mol Cell Biochem. 2001;220:127. doi: 10.1023/a:1010834107809. [DOI] [PubMed] [Google Scholar]
- 20.Burgunder JM, Schöls L, Baets J, Andersen P, Gasser T, Szolnoki Z, Fontaine B, Broeckhoven CV, Donato SD, De Jonghe P, Lynch T, Mariotti C, Spinazzola A, Tabrizi SJ, Tallaksen C, Zeviani M, Harbo HF, Finsterer J. Eur J Neurol. 2011;18:207. doi: 10.1111/j.1468-1331.2010.03069.x. [DOI] [PubMed] [Google Scholar]
- 21.Takalo M, Salminen A, Soininen H, Hiltunen M, Haapasalo A. Am J Neurodegener Dis. 2013;2:1. [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao JH, Liu HL, Lin HY, Huang CH, Fang HW, Chen SS, Ho Y, Tsai WB, Chen WY. Perspect Medicin Chem. 2007;1:39. doi: 10.4137/pmc.s212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raju M, Santhoshkumar P, Xie L, Sharma KK. Biochemistry. 2014;53:2615. doi: 10.1021/bi4017268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jjernberg LO, Naslund J, Lindquist F, Johansson J, Karlstrom AR, Thyberg J, Terenius L, Nordstedt C. J Biol Chem. 1996;271:8545. doi: 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- 25.Kulig M, Ecroyd H. Biochem J. 2012;448(3):343. doi: 10.1042/BJ20121187. [DOI] [PubMed] [Google Scholar]
- 26.Manelli AM, Puttfarcken PS. Brain Res Bull. 1995;38:569. doi: 10.1016/0361-9230(95)02034-x. [DOI] [PubMed] [Google Scholar]
- 27.Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML. J Neurosci. 2000;20:1386. doi: 10.1523/JNEUROSCI.20-04-01386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]