

DNA hydroxymethylation is a distinctive epigenetic regulator of human inhibitory neurons, potentially affecting brain diseases.
Abstract
Brain function depends on interaction of diverse cell types whose gene expression and identity are defined, in part, by epigenetic mechanisms. Neuronal DNA contains two major epigenetic modifications, methylcytosine (mC) and hydroxymethylcytosine (hmC), yet their cell type–specific landscapes and relationship with gene expression are poorly understood. We report high-resolution (h)mC analyses, together with transcriptome and histone modification profiling, in three major cell types in human prefrontal cortex: glutamatergic excitatory neurons, medial ganglionic eminence–derived γ-aminobutyric acid (GABA)ergic inhibitory neurons, and oligodendrocytes. We detected a unique association between hmC and gene expression in inhibitory neurons that differed significantly from the pattern in excitatory neurons and oligodendrocytes. We also found that risk loci associated with neuropsychiatric diseases were enriched near regions of reduced hmC in excitatory neurons and reduced mC in inhibitory neurons. Our findings indicate differential roles for mC and hmC in regulation of gene expression in different brain cell types, with implications for the etiology of human brain diseases.
INTRODUCTION
The human brain contains dozens of subtypes of neuronal and glial cells (1). The identity of brain cell types is established and maintained through spatiotemporal regulation of gene expression that, in turn, is defined by cell type–specific epigenomic marks, including DNA methylation and histone modifications. Cytosine methylation is the major form of DNA methylation in mammalian cells (2) and is extensively remodeled during development and differs across tissues and cells (3). In the adult human brain, ~80% of CG and 1.5% of non-CG (CH, where H = A, T, or C) sites are methylated and can be converted to hydroxymethylcytosine (hmC) and further demethylated (4–6). In postmitotic neuronal genomes, non-CG methylcytosine (mCH) and hmC accumulate to a significantly higher level than in other differentiated tissues (7, 8), which is a distinct feature of the brain’s epigenome (9–12). However, the genome-wide distribution and function of these epigenetic marks in specific neuronal and glial cells in the human brain remain poorly understood.
Single-nucleus DNA methylation sequencing was recently used to classify neuronal populations in the human and mouse brain, including multiple subtypes of excitatory and inhibitory neurons with distinct cell type–specific epigenetic signatures (13). Despite the power of single-cell sequencing for unbiased identification of cell types, there is currently no efficient method to examine hmC or specific chromatin modifications in single neurons with sufficient throughput to investigate cell type–specific modifications (14, 15). In mouse, fluorescent labeling and affinity purification of nuclei from defined neuronal types enable cell type–specific epigenome and chromatin accessibility profiling (16, 17). Analysis of nuclear preparations of mouse cerebellum enriched for granule cells indicated a role for hmC in functional demethylation in this cell type (18). However, the cell type–specific distribution of hmC has not been investigated with single-base resolution in the human brain.
Here, we used a flow cytometry–based protocol (19, 20) to isolate nuclei from the major neuronal and glial populations in the human adult autopsied prefrontal cortex (PFC). We then used these preparations for single-base resolution (hydroxy)methylome, transcriptome, and histone modification analyses. The joint analysis of these multiomics data uncovered a complex relationship between cell type–specific epigenomic marks and gene expression and identified a unique role for hmC in epigenetic regulation of the PFC inhibitory neurons.
RESULTS
Major cell types of the human PFC contain unique distributions of hmC and mCH
Cell structure is not preserved in frozen autopsy brain specimens; the nuclei, however, remain intact. We developed a multicolor fluorescence-activated nuclei sorting (FANS) protocol to isolate the medial ganglionic eminence (MGE)–derived inhibitory GABAergic [GABA (γ-aminobutyric acid)–mediated] interneurons (MGE-GABA), excitatory glutamatergic neurons (Glu), and oligodendrocytes (OLIG) from the autopsied human adult PFC (Fig. 1A and table S1) (10, 19, 20). The MGE-derived GABA neurons comprise ~60 to 70% of all neocortical GABAergic neurons and contain the parvalbumin (PV)– and somatostatin (SST)–expressing interneurons (21). OLIG cells are the major glial cell type in the central nervous system that provides support and myelin-based insulation to axons (22).
Fig. 1. Unique distributions of hmC and mCH in Glu, MGE-GABA, and OLIG cells from the human PFC.
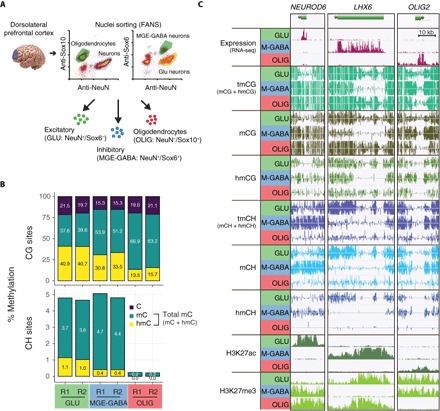
(A) Method for cell type–specific nuclei isolation for transcriptomic and epigenomic analyses. (B) Genome-wide analysis of mC (OxBS) and hmC (BS-OxBS) shows substantial but significantly different levels of hmCG in both neuronal subtypes (Glu and MGE-GABA) and OLIG. Neurons are enriched in mCH, with a small proportion of hmCH. R1 and R2, biological replicates. (C) Examples of marker genes expressed in Glu (NEUROD6), MGE-GABA (LHX6), or OLIG (OLIG2) cells, together with their DNA methylation and histone modification profiles. Glu (GLU), glutamatergic neurons; MGE-GABA (M-GABA), MGE-derived GABAergic interneurons.
Transcriptional profiles obtained using RNA sequencing (RNA-seq) of the sorted nuclei were highly consistent among replicates within the same cell type (r ≥ 0.94) (fig. S1A) and showed higher correlation between the two neuronal data sets (r = 0.91) compared with Glu versus OLIG (r = 0.66) or MGE-GABA versus OLIG (r = 0.68) profiles (fig. S1C). Principal components analysis separated samples from the three studied cell types, linking 22.8% of the RNA expression variability to separation of neurons from OLIGs and 8.3% to neuronal subtype identity (fig. S1B). Known cell type–specific markers were highly enriched in purified nuclei of the respective populations (for example, SLC17A7, SATB2, and TBR1 in Glu neurons; SLC32A1, SST, and LHX6 in MGE-GABA neurons; PLP1, MBP, and MAG in OLIG cells), whereas markers of other lineages were depleted, thus validating the identities of the corresponding populations (fig. S1C). In total, 9068 genes were differentially expressed (DE) [fold change (FC) ≥ 2, false discovery rate (FDR) < 0.05] in at least one pairwise comparison between the cell types (table S2). Notably, DNA methylation–related enzymes were found to be DE among the cell types (fig. S1D). We found significantly higher expression of the methyltransferase DNMT3B in MGE-GABA neurons. All three TET genes (TET1, TET2, and TET3), which are involved in hydroxymethylation, were enriched in OLIGs compared with neuronal subtypes.
We then obtained the genome-wide profiles of total modified cytosine [tmC = mC + hmC, bisulfite sequencing (BS-seq)] and of methylcytosine [mC, oxidative bisulfite sequencing (OxBS-seq)] (23), in parallel with histone marks [using chromatin immunoprecipitation sequencing (ChIP-seq)] of active enhancer and promoter regions (H3K27ac) and Polycomb-repressed regions (H3K27me3) (table S3). The level of hmC was calculated as the difference between the BS and OxBS signals at sites covered by at least five reads in both data sets. In the CG context, the genome-wide level of tmCG was ~5% higher in MGE-GABA compared with Glu or OLIG cells (Fig. 1B), in agreement with our previous findings using HM450K methylation array, reduced-representation bisulfite sequencing assays, and single-nucleus BS-seq data from the human frontal cortex (13, 19). In contrast with tmCG, there were large differences in genome-wide levels of hmCG and mCG between the cell types, with the highest levels of hmCG in Glu (~41%), followed by MGE-GABA (~32%) and OLIG (~14%) cells (Fig. 1B). These data show that, although hmCG and mCG together constitute a relatively constant fraction of all CG sites, hmCG may play a distinct role in each cell type.
Unique among differentiated cell types, adult neurons contain abundant DNA methylation at non-CG genomic positions (mCH), whereas significantly lower mCH levels are detected in glial cells (9–12). However, previous studies did not specifically examine mCH in OLIG cells. We found mCH to be significantly more abundant in neurons compared with OLIGs (Fig. 1B). In addition, total CH methylation (tmCH) level was consistent with measurements in single neuronal nuclei (fig. S1E) (13). Our OxBS data also allowed us to investigate whether a proportion of total mCH modification existed in the form of hmCH, a question that has remained controversial (9, 18). We found a small but reliably measurable fraction of hmCH in neuronal cells that differed between Glu (~1.0% of all cytosines) and MGE-GABA (~0.4%) subtypes. This level of hmCH corresponds to 22% of tmCH methylation in Glu cells and 8.0% in MGE-GABA cells. By contrast, hmCH was not detected in OLIGs (<0.02%).
To examine the relationship between different DNA modifications and gene expression, we correlated cell type–specific RNA abundance with each epigenomic mark. Key cell type–specific transcription factors NEUROD6 (Glu), LHX6 (MGE-GABA), and OLIG2 (OLIG) had corresponding patterns of expression and epigenomic regulation (Fig. 1C). Total methylation (tmCG and tmCH) was specifically depleted in the cell type that expressed the gene, whereas extensive H3K27ac peaks indicated the presence of super-enhancers throughout the respective gene bodies (24). High levels of the Polycomb-dependent histone modification, H3K27me3, as well as mCG and mCH correlated with repression in the inactive cell types at these loci. Whereas these patterns exemplify well-known cell type–specific markers with the strongest differential expression, we hypothesized that thousands of DE genes detected with RNA-seq (fig. S1C and table S2) may use distinct epigenetic pathways to regulate expression.
Gene body DNA methylation associates with gene expression and differential expression across brain cell types
We first examined DNA methylation in gene bodies, which has been linked to altered neuronal gene expression following disruption of the DNA methylation reader MeCP2 (25). We examined the abundance of each form of methylation within gene bodies as a function of expression (Fig. 2, A and B, and fig. S2, A and C) and differential expression (Fig. 2, C to E, and fig. S2, B and D). Although Glu neurons had the highest proportion of hmCG (see Fig. 1B), the range of hmCG levels between genes with low [transcripts per million (TPM) = 1] and high (TPM = 100) expression was smaller in Glu than in MGE-GABA neurons (Fig. 2A). In both neuronal subtypes, mCG and hmCG had negative and positive correlations with gene expression (P < 10−10 for all associations), respectively, leading to negligible differences in total methylation (tmCG) over the same range of expression levels. In contrast with CG methylation, CH methylation in both neuron subtypes was more than twice as abundant within the gene bodies of low-expressed compared with actively transcribed genes (Fig. 2B). These data illustrate the substantial differences in dynamic range for different epigenomic marks between the brain cell types.
Fig. 2. Cell type–specific gene body DNA methylation patterns associate with expression and differential expression.
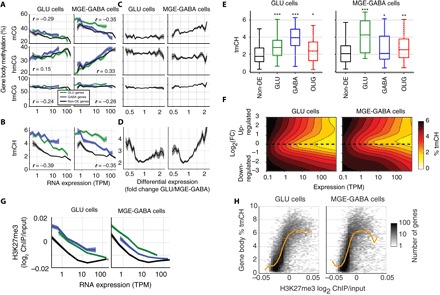
(A) Gene body hmCG has a stronger positive relation with expression in MGE-GABA than Glu cells. r, Spearman correlation (P < 10−10). (B) Gene body mCH is negatively related to expression and enriched in cell type–specific up- and down-regulated genes. (C) CG methylation is not related to differential expression after controlling for expression, except for mCG in MGE-GABA neurons. Only genes with expression in the range (10 < TPM < 100) were included. (D to F) mCH is nonmonotonically related to differential expression: Both up- and down-regulated genes have increased mCH compared with non-DE genes at the same level of expression. In (D), only genes with expression in the range (10 < TPM < 100) were included. In (E), only genes with expression in the range (50 < TPM < 100) were included. Asterisks indicate significant differences in tmCH for each set of DE genes compared with non-DE genes (*P < 0.05; **P < 0.01; ***P < 0.001, t test, Bonferroni-corrected). (G) Polycomb repression (H3K27me3) is negatively correlated with expression and enriched in cell type–specific up- and down-regulated genes. (H) Polycomb repression and mCH are positively correlated for most genes (yellow line, median).
We next explored whether these associations are equally manifested in genes with cell type–specific compared with cell type–independent (non-DE) expression (Fig. 2, B to E, and fig. S2, B and C). Total non-CG DNA methylation (tmCH) was up to 140% higher in the bodies of down-regulated compared with non-DE genes after controlling for expression (Fig. 2D). Remarkably, tmCH was also strongly enriched (up to 80%) within the bodies of genes differentially up-regulated compared with the other cell type (P < 0.001, Glu cells; P < 0.05, MGE-GABA cells). These findings reflect a complex, nonmonotonic relationship between gene expression, differential expression, and tmCH (Fig. 2F) in Glu and MGE-GABA neurons. In contrast with tmCH, CG methylation (mCG, hmCG, and tmCG) was not related to DE in Glu cells (Spearman correlation r = −0.003, −0.022, and −0.003 and P = 0.81, 0.057, and 0.79, respectively) (Fig. 2C). MGE-GABA cells did show a modest increase in mCG (r = 0.12, P < 0.001), but not in hmCG (r = −0.011, P = 0.32), for Glu-specific genes.
DNA methylation acts in concert with histone modifications (26), including the Polycomb repression–associated mark H3K27me3. We observed that Polycomb marks in gene bodies had a similar relationship to expression as tmCH, with the strongest H3K27me3 signal in low-expressed genes and in genes that are differentially up- or down-regulated in Glu or MGE-GABA cells (Fig. 2G). These two epigenetic marks were positively correlated across genes in both neuron types (Fig. 2H; P < 10−10). These observations indicate that mCH and H3K27me3 support the specialized identity of neurons and are in line with recent findings in mouse brain that suggested that the role of mCH is to fine-tune the cell type–specific transcription of neuronal genes (27).
OLIG cells showed a relationship between gene body DNA methylation and gene expression that is similar to that of MGE-GABA neurons (fig. S2, C and D). In particular, we found that gene body hmCG in OLIG cells was strongly modulated by expression, whereas there was no strong effect of differential expression. These findings indicate that gene expression in MGE-GABA and OLIG cells is associated with the conversion of gene body mCG to hmCG. However, OLIG cells lack substantial mCH, and we found little or no relationship between mCH and gene expression in these cells. Notably, using different DNA methylation marks in linear regression to model gene expression in each of the three cell types, we demonstrated that individual marks, hmCG and mCG, provide a basis for better gene expression predictions compared to tmCG (fig. S2E). Collectively, our results indicate that multiple forms of gene body DNA methylation are differentially associated with gene expression in different brain cell types.
Brain cell types differ in association between promoter epigenetic modifications and gene expression
DNA methylation at promoters, particularly in those overlapping with CG islands (CGIs), is known to repress gene expression (28). In both neuronal subtypes, we found the strongest negative correlation (FDR < 0.001) between DNA methylation and expression in the region downstream of the transcription start site (TSS) within the first 1000 base pairs (bp) of the gene body (fig. S3A). This correlation was notably stronger and more localized to the TSS for non-CGI–containing promoters compared with CGI-containing promoters. Notably, Glu DE genes were depleted for CGIs (fig. S3B). Promoters of the Glu-enriched genes were also more strongly methylated in MGE-GABA compared with Glu cells; in contrast, we observed little difference in methylation between Glu and MGE-GABA cells at MGE-GABA–enriched genes (fig. S3, C and D). Together with the gene body methylation patterns, these results demonstrate a greater dynamic range of DNA methylation in MGE-GABA compared with Glu cells.
In all three cell types, there was a strong positive correlation between gene expression and the active histone modification H3K27ac at both non-CGI and CGI promoters (fig. S3E). However, comparing genes with the same level of expression, we found many more H3K27ac peaks at promoters that overlapped a CGI. Thus, H3K27ac might play a more important role in the regulation of the expression of CGI-containing genes.
Brain cell type–specific enhancers have distinct patterns of hmCG
Distal gene regulatory elements such as enhancers account for a substantial source of epigenetic diversity between tissues and cell types and typically have intermediate-to-low levels of DNA methylation compared to the flanking regions (29). Earlier studies showed that hmC accumulates at enhancer regions in brain tissue, suggesting that active DNA demethylation may have a regulatory role (9, 11). We performed H3K27ac ChIP-seq analysis (30) and found 65,645 putative active enhancers (peaks located >2 kb distal from the nearest TSS) and 27,899 active promoters (TSS-proximal peaks) across the three cell types (Fig. 3A). Whereas many active promoters (34.0%) were marked by H3K27ac in all three cell types (common promoters), the vast majority (76.8%) of enhancers were specific to a single cell type, confirming the greater diversity of enhancers versus promoters across tissues and cell types (31).
Fig. 3. Increased hmCG at MGE-GABA–specific distal gene regulatory elements.
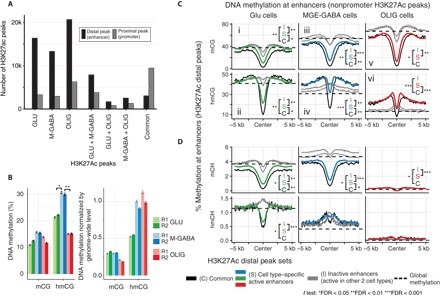
(A) H3K27ac ChIP-seq data identify ~15,000 to 25,000 cell type–specific distal peaks in Glu, MGE-GABA, and OLIG cells. (B) DNA methylation at the center of cell type–specific enhancers. MGE-GABA cells are enriched for hmCG at cell type–specific peaks. R1 and R2, biological replicates. Error bars denote SEM. Significant differences of methylation levels are indicated with asterisks (pairwise t test). (C and D) Mean profile of DNA methylation marks at cell type–specific distal H3K27ac peaks. Genome-wide methylation levels (as in Fig. 1B) are shown in horizontal dashed lines. Differences of methylation levels in the center of the regions were tested between common peaks (C), cell type–specific peaks (S), and inactive peaks (I), and significances are indicated with asterisks (pairwise t test).
Focusing on H3K27ac-marked active enhancers, we again found profound differences in the DNA methylation patterns in different brain cell types. Although hmCG and mCG were depleted relative to each cell’s genome-wide background level in the center of enhancers that were active in all three cell types (common enhancers), hmCG was depleted at cell type–specific enhancers only in Glu cells [Fig. 3, B and C (ii), and fig. S4, A and B]. By contrast, MGE-GABA and OLIG cells retained hmCG at their cell type–specific enhancers at levels nearly equivalent to the genome-wide background for each cell type [Fig. 3, B and C (iv and vi), and fig. S4C]. Notably, non-CG hydroxymethylation (hmCH), but not mCH, was also significantly depleted in Glu-specific but elevated in MGE-GABA–specific enhancers compared to genome-wide levels (Fig. 3D).
Compared with cell type–specific active enhancers, inactive enhancers (regions corresponding to active enhancers in one of the other two cell types) remain highly methylated, with mCG and hmCG levels near the genomic background level. In MGE-GABA and OLIG (but not in Glu) cells, the largest hmC differences between active and inactive enhancers were detected in the ~5-kb-wide flanking regions, which showed elevated hmC levels in active enhancers compared to the genome-wide background. These findings reveal distinct cell type–specific patterns of hmC, with notably high hmC in both CG and CH contexts in the regions of active MGE-GABA–specific enhancers [Fig. 3C (iv) and D].
Because cell type specificity of enhancers could be related to their distance from the TSS, we profiled DNA methylation marks of H3K27ac peaks situated at 2 to 10 kb, 10 to 50 kb, and >50 kb from the TSS but observed no significant differences in relation to cell specificity (fig. S5).
We next compared DNA methylation between enhancers that are active in both the adult and early embryo brain and enhancers that are inactive in early embryo but are activated later on. We accomplished this by overlapping distal H3K27ac peaks in adult Glu and MGE-GABA neurons obtained in our study with regions marked as open chromatin in the fetal brain [post-conception week (PCW) 15 to 17] on the basis of the recently published Assay for Transposase-Accessible Chromatin sequencing (ATAC-seq) data (32). We detected a significant overlap that was larger for common enhancers (78.1% at PCW17) as compared with cell type–specific enhancers (37.2 to 42.1% at PCW17) (fig. S6A). We next examined DNA methylation marks in the overlapping (“early” enhancers) and nonoverlapping (“late” enhancers) H3K27ac peaks. In common enhancers, tmCG was significantly lower in early versus late enhancers (P < 0.01), mainly because of the difference in hmCG (fig. S6B). Similarly, tmCG was lower in active MGE-GABA–specific early versus late enhancers (P < 0.05). In contrast, tmCG did not differ appreciably between active Glu-specific early and late enhancers (fig. S6C).
Collectively, our data show that the enhancers that are active early in brain development (before PCW17) have very low DNA methylation (mC and tmC) in adulthood, with the early common and Glu-specific (but not MGE-GABA–specific) enhancers also marked by low hmC. The enhancers that are activated later on (after PCW17) also lose mCG; however, the late common and MGE-GABA–specific (but not Glu-specific) enhancers retain a substantial amount of hmCG in the adult brain. Thus, hmCG in adult active enhancers might be a signature of the late developmental activation of these regions. The high level of hmCG at MGE-GABA–specific enhancers (Fig. 3, B and C) could thus reflect their relatively late activation compared with Glu enhancers.
OxBS-Seq data identifies thousands of new differentially methylated regions
Using our BS-seq data, we identified 202,823 differentially methylated regions (tmCG DMRs, adjusted for the global level of tmCG in each cell type; see Materials and Methods) when comparing pairs of cell types (Fig. 4A and table S4). There was a substantial overlap between tmCG DMRs and distal H3K27ac peaks, consistent with an active regulatory role for many DMRs. Notably, tmCG DMRs had a nonuniform distribution across cell types: There were approximately four times more regions with low methylation in Glu (Glu DMRs, 54,178) and OLIG (OLIG DMRs, 64,435) compared to MGE-GABA (MGE-GABA DMRs, 14,310). A preponderance of Glu relative to MGE-GABA DMRs has also been observed in mouse cortical neurons (17) and in human single-neuron methylomes (13). In both studies, only tmCG was assessed by BS-seq. We reasoned that this imbalance could reflect the different roles of mCG and hmCG in each cell type, and called mCG DMRs using OxBS-seq measurements, while adjusting for the different global level of mCG in each cell type. We identified thousands of mCG DMRs, with a greater number in MGE-GABA (40,644) compared to Glu (9998) neurons (Fig. 4A). Notably, many mCG DMRs were not tmCG DMRs, highlighting the added value of OxBS data and suggesting that higher hmCG partially compensates for low mCG in these regions. More such regions overlapped enhancers in MGE-GABA than in Glu cells, consistent with a greater role for hmCG in enhancers that are active in inhibitory neurons. Together, the total number of regions with differential methylation (mCG or tmCG DMRs) was similar in Glu (59,995) and MGE-GABA (48,828) cells.
Fig. 4. Enrichment of cell type–specific DMRs in disease-associated variants.
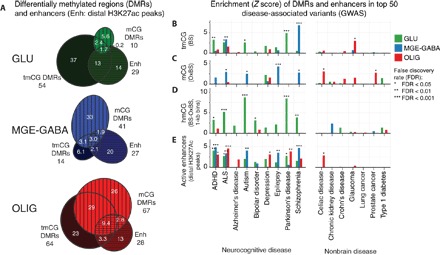
(A) The number of DMRs (labeled in thousands; methylation difference ≥ 0.3) detected using tmCG (BS-seq) or mCG (OxBS-seq) data (green, Glu < MGE-GABA; blue, MGE-GABA < Glu; red, OLIG < Glu and MGE-GABA). Many Glu DMRs may be driven by lower hmC levels, whereas MGE-GABA DMRs are mainly driven by lower mCG. (B to D) Enrichment of DMRs near disease-associated genetic variants from GWAS. For each disease, the top 50 most significant regions were retained. Schizophrenia loci were strongly enriched in MGE-GABA DMRs. By contrast, other neuropsychiatric diseases such as attention-deficit hyperactivity disorder (ADHD), autism, and Parkinson’s disease were associated with Glu DMRs (tmCG) that may be driven by lower hmCG. (E) Cell type–specific enhancers for all three brain cell types were enriched in many brain disease–associated variants.
We directly compared these DMRs with the published list of DMRs from single-neuron methylC-seq in the human frontal cortex (13) (fig. S7). Our results were consistent, with most of the tmCG DMRs from our study also appearing in the corresponding cell types in single-cell data. As expected, the single-neuron data revealed additional DMRs that may represent differences between subtypes of Glu (for example, L2/L3 versus L4) or MGE-GABA (for example, PV+ versus SST+) cells. In addition, we also identified thousands of new regulatory elements. In particular, our OxBS-seq analysis identified 34,903 novel DMRs with lower mCG in MGE-GABA neurons compared with Glu neurons. These mCG DMRs did not overlap tmCG DMRs, indicating that hmCG might compensate for the reduced mCG in MGE-GABA cells at these sites. For this reason, these DMRs might have remained undetected by single-nucleus BS-seq, which cannot distinguish mC and hmC (13).
Hydroxymethylation marks CGIs near promoters of expressed cell type–specific genes
To further connect cell type–specific DNA methylation to gene expression, we focused on DMRs located near CGIs, in the shore (0 to 2 kb) or shelf (2 to 4 kb) regions, which demonstrate enrichment of tmCG DMRs (Fig. 5A) and are likely to regulate the nearest gene promoter. The vast majority (88%) of these regions had lower tmCG in the cell type with higher expression of the nearest gene (Fig. 5B). However, we found a small group of genes with tmCG DMRs showing the opposite pattern, that is, higher tmCG in the more active cell type. These genes include essential cell type–specific transcription factors as well as key markers of functional identity of MGE-GABA (DLX2, VAX1, and GAD2; Fig. 5F) and Glu (NEUROD2, EMX1, PAX6, BDNF, and SLC17A7/6; Fig. 5G) neurons. Notably, using our previously published data (9), we found that DNA methylation is low at these sites in the fetal cortex (20 weeks after conception, Fig. 5C), consistent with the role of these DMRs in developmental regulation. These DMRs had higher hmCG in the active cell type (Fig. 5D), whereas H3K27me3 was enriched in the body of nearby genes in the less active cell type (Fig. 5E). These adult neuronal DMRs likely correspond to a recently described subset of DNA methylation valleys (DMVs), which, in contrast with most DMVs, demonstrate differences in DNA methylation across tissues or cell types and a positive correlation of DNA methylation with expression of associated genes (17, 33, 34). Our findings using BS-seq and OxBS-seq reveal that this DNA methylation is largely composed of hmCG and suggest that hmCG is a stable epigenetic modification with a distinct function at CGI-associated regions in a number of neuronal genes. Notably, the genes associated with hmCG-enriched DMRs include not only early transcriptional regulators that are mostly active in development (for example, PAX6 and DLX2) but also genes that are important for the function of the mature neuronal subtypes and that are highly expressed in adult brain in a neuron subtype–specific manner (for example, GAD2 and SLC17A7/6).
Fig. 5. CGI-associated regions of elevated tmCG near promoters of key neuron subtype–specific genes are enriched in hmCG and depleted of H3K27me3.
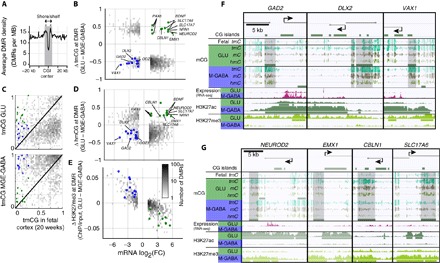
(A) Glu versus MGE-GABA tmCG DMR density around CpG islands. CpG shores and shelves (<4 kb away from CpG islands) are indicated in the shaded area. The enrichment of DMRs was tested using DMR density in the flanking region (>10 kb away) as a background control. Asterisks indicate significant enrichments (P < 0.05). (B) Cell type–specific tmCG DMRs within CGIs, shores, and shelves (CGI ± 4 kb) (gray dots). Most tmCG DMRs are negatively related to expression of the nearest gene. A subset of genes has tmCG DMRs with positive association with gene expression (green, Glu-enriched genes; blue, MGE-GABA–enriched genes). (C) tmCG DMRs that demonstrate positive association with gene expression have low tmCG levels in the fetal cortex. These tmCG DMRs demonstrate neuron subtype–specific enrichment in hmCG (D) and H3K27me3 depletion in the gene body of the nearest gene (E). (F and G) Examples of DMRs positively associated with gene expression of nearest MGE-GABA–specific (F) (GAD2, DLX2, and VAX1) or Glu-specific (G) (NEUROD2, EMX1, CBLN1, and SLC17A6) genes. MB, million base pairs or megabase.
Cell type–specific DMRs are enriched in disease-associated variants
Many candidate disease-related genetic variants identified in genome-wide association studies (GWAS) fall outside protein-coding regions and could reflect vulnerable noncoding sites of gene regulation (35). Using a recently described approach (36), we examined the overlap of cell type–specific DMRs and enhancers with the top GWAS loci that were associated with a range of diseases [genome-wide significant single-nucleotide polymorphisms (SNPs), P < 10−6]. We found significant enrichment of cell type–specific tmCG and mCG DMRs as well as enhancers in the top GWAS loci for a panel of brain-related diseases (Fig. 4B), but few significant associations with similarly powered nonbrain control GWAS. In particular, MGE-GABA tmCG DMRs were highly enriched in sites associated with schizophrenia (FDR < 0.001), consistent with a distinctive role for GABAergic circuitry in this disease (37). We also detected strong enrichment of MGE-GABA mCG (but not tmCG) DMRs at epilepsy risk loci (Fig. 4C). By contrast, Glu tmCG DMRs were enriched at risk loci for Parkinson’s disease. Because our analysis directly probed tmCG or mCG, we used the difference between BS-seq and OxBS-seq signals in 1-kb bins to examine the enrichment of cell type–specific hmCG DMRs (see Materials and Methods). Regions with lower hmCG in Glu versus MGE-GABA cells (Glu hmCG DMRs) were strongly enriched in risk loci for autism, amyotrophic lateral sclerosis (ALS), and Parkinson’s disease (Fig. 4D), suggesting that these risk loci are marked by hmCG in MGE-GABA cells. We also detected strong enrichment of active enhancers in all three cell types for many brain diseases, providing independent validation for the DNA methylation–based disease associations (Fig. 4E).
DISCUSSION
A major challenge in understanding epigenomic regulation of complex tissues, such as the human brain, is the ambiguity of measurements on mixed cell types and of assays that conflate multiple epigenomic marks (that is, total DNA methylation). Our study offers a fine-grained perspective on the epigenome of the human brain, specifically focusing on the PFC, the brain region that is critical for cognition, memory, and executive function and is broadly implicated in neuropsychiatric illness. We simultaneously analyzed three major PFC cell types (excitatory glutamatergic neurons, MGE-derived GABAergic interneurons, and OLIG cells) and two major DNA modifications (mC and hmC), in parallel with high-resolution transcriptome and histone modification data. Our analyses show that, although all brain cells share general features of epigenomic regulation such as the negative correlation of mCG and mCH with gene expression (9), the quantitative relationship between mC, hmC, and RNA expression is significantly different among Glu neurons, MGE-GABA neurons, and OLIG cells. In particular, we discovered a unique role for hmC in epigenetic regulation of the human MGE-GABA neurons in the PFC. The altered function of these PFC inhibitory interneurons have been previously associated with several psychiatric and neurological disorders, including schizophrenia, major depression disorder, autism spectrum disorders, and epilepsy (38, 39).
Our study builds on previous reports that used whole-genome, base-resolution epigenomic assays to examine mouse brain cell types via cell type purification (17, 18) or single-cell sequencing (13). Compared with previous purification-based studies, our work offers a comprehensive multiomics approach and is focused on the human brain. Notably, whereas the emerging field of single-cell transcriptome and epigenome sequencing enables unbiased identification of brain cell types (1, 13), there are currently no efficient methods for measuring hmC or chromatin modifications in single cells with sufficient throughput to reliably assess human neuronal diversity (14, 15).
We found that although the genome-wide level of the total mCG was only slightly different between the cell types (~5% higher in MGE-GABA versus Glu or OLIG cells), the hmCG content varied markedly across different brain cells. The highest hmCG levels were found in Glu neurons (~40%) and the lowest hmCG levels were found in OLIG cells (~14%). These differences were not readily explained by the differences in the expression levels of DNA methylation–modifying enzymes (fig. S1D), suggesting that other regulators of DNA methylation (for example, cell type–specific DNA binding proteins) might play a pivotal role in the establishment and maintenance of hmCG patterns (40). We also detected a significant proportion of hmCH in neuronal cells (1% in Glu and 0.4% in MGE-GABA). This finding, which strongly suggests a similar demethylation route for mCH and mCG, has remained controversial (41). In particular, whereas an early work by Lister et al. (9) did not detect hmCH in the mouse brain, a recent study by Mellén et al. (18) reported low but reliably measured hmCH (0.57%) in the mouse cerebellar granule cells. Notably, in contrast to Lister et al., who used the Tet-assisted BS-seq method, both Mellén et al. and our group used OxBS-seq, a complementary technique with potentially different sensitivity and specificity for detecting hmC. Moreover, our study examined human cortical neurons, which have much higher levels of mCH compared with adult mouse brain tissue (9). Cortical Glu and MGE-GABA neurons also have significantly more mCH (3.6 to 4.7%, Fig. 1A) than cerebellar granule cells (0.94%) (18). In addition, we observed distinct hmCH and hmCG profiles within distal regulatory elements that differ between MGE-GABA and Glu cells (with unexpected elevation in MGE-GABA–specific enhancers, Fig. 3D), suggesting that the detected hmCH patterns are unlikely to be experimental artifacts.
Our multiomics analysis uncovered unique cell type–specific features of the epigenetic landscape of the human PFC and demonstrated the role of discrete epigenetic marks in defining the cell specificity of gene expression. We found the distinct hydroxymethylation signature that marks MGE-derived GABA neurons. Specifically, in gene bodies, the range of hmCG levels between genes with low and high expression was significantly higher in MGE-GABA than in Glu cells, resulting in higher correlation of hmCG with gene expression in inhibitory neurons (see Fig. 2A). Similarly, there was a greater dynamic range of DNA methylation in the promoters of MGE-GABA versus Glu neurons, especially in promoters without CGIs. Also, compared to the genome-wide background, we detected notably higher hmC levels in both CG and CH contexts within regions of active MGE-GABA–specific versus Glu-specific enhancers (see Fig. 3). On the basis of these findings and the previously proposed model of mC and hmC changes during enhancer activation (42), we hypothesize that DNA methylation patterns at brain enhancers develop in stages and reflect trajectories of epigenomic differentiation. In particular, in early development, regions encompassing brain enhancers could initially become hydroxymethylated. At later stages, cell type–specific enhancers could be defined by demethylation, with both hydroxymethylation and demethylation stages being differentially regulated in different neuronal subpopulations and/or enhancer subsets.
Our FANS-separated OLIG population consists of OLIG lineage cells (that is, mature OLIG and OLIG precursors) (20), and there is a diversity of neuronal subpopulations within the purified Glu and MGE-GABA neurons (1, 13). The remaining diversity of cells within these purified populations might obscure epigenomic or transcriptomic signatures of subtypes and is a limitation of our study. In particular, the diversity of interneurons within our MGE-GABA sample could, at least in part, explain the hmC signature of MGE-GABA neurons that we observed. Nevertheless, it appears highly unlikely that these patterns resulted entirely from regulatory diversity within subpopulations of MGE-GABA neurons. Had this been the case, we would expect to see corresponding signatures in the other epigenetic marks, for example, H3K27ac and mC. Instead, we detected more H3K27ac peaks in Glu versus MGE-GABA neurons (Fig. 3A) and did not observe differences in mC levels between the cell types within H3K27ac-marked enhancers (Fig. 3B). In contrast, hmC was significantly higher in MGE-GABA versus Glu neurons in these enhancers. Heterogeneity is a confounding factor for any study of a cell population, including single-cell studies, which rely on an assumption that all cells within a computationally defined “cluster” are homogeneous. Future work using a combination of single-cell and more refined purification strategies will help to dissect the fine distinctions in cellular regulation among the multitude of neuronal and glial brain cell types.
Another notable finding of our study is the discovery of a complex relationship between non-CG methylation and gene expression. Although the repressive function of mCH has been reported previously (9, 17, 19), we found that gene body mCH was robustly associated not only with absolute levels of gene expression but also with differential expression. Notably, the relationship of mCH with differential gene expression was nonmonotonic. In particular, in agreement with findings in mouse brain (27), mCH was significantly higher in the bodies of down-regulated compared with non-DE genes with the same level of expression. However, mCH was also enriched within the bodies of genes that were differentially up-regulated compared with the other neuronal subtype, suggesting that mCH might be involved in maintaining specialized neuronal identities by fine-tuning the expression of specific genes in each neuronal subtype.
Finally, we found that genetic risk variants for several psychiatric and neurological disorders (including autism, schizophrenia, and ALS) are enriched in regions with cell type–specific mC or hmC signatures (DMRs). This finding allows identification of candidate functional risk variants and could shed light on the underlying molecular mechanisms of these diseases. Notably, many of these DMRs do not overlap with promoters or enhancers, and their functional roles remain to be investigated.
It should be emphasized that the cell type–specific patterns reported here could not have been detected in heterogeneous bulk brain specimens containing multiple cell types that have been predominantly used in previous studies of DNA methylation in the human brain (43, 44). Thus, our work provides a rich resource for future investigations of the human brain in both health and mental illness, as well as throughout the life span, to establish the genetic and epigenetic factors shaping the functional diversity of brain circuit components.
MATERIALS AND METHODS
Human brain samples
Postmortem human brain specimens were obtained from the Brain Collection of Y.L.H. (table S1) (45, 46). Brain samples used in the study were from clinically unremarkable adult male Caucasian subjects, not diagnosed with any neurological or psychiatric condition at the time of death and with negative toxicology for common drugs of abuse or for therapeutic agents. Specimens had been collected at autopsy within 24 hours after death by the personnel of the Department of Forensic Medicine (Semmelweis University, Hungary) under approved local ethical guidelines. The cause of death was determined by a forensic pathologist. All subjects used in our study died of non–suicide- and non–drug abuse–related causes, such as accident or cardiac failure. Immediately after autopsy, brains were cut coronally in slabs and kept frozen at −70°C. There were no significant differences in brain pH among specimens. Postmortem intervals for all subjects were <24 hours. Tissue samples were dissected from dorsolateral PFC, Brodmann area 9.
Nuclei isolation
The protocol and reagents for the isolation of brain nuclei before the flow cytometry separation were described previously (10). In short, ~750 mg of tissue was homogenized in lysis buffer, underlaid with high sucrose buffer, and centrifuged for 1 hour at 24,000 rpm. The nuclear pellets were resuspended in the antibody-incubation buffer and incubated with primary antibodies for 1.5 hours. A second centrifugation step was performed, and nuclei were incubated with secondary antibodies for an additional 1 hour. A FANS method was then used to separate nuclei of MGE-GABA neurons, Glu neurons, and OLIG, similar to the protocol described in our previous publication (10), with some modifications. Antibodies against brain cell population markers NeuN, SOX6, and SOX10 were used in the FANS protocol. NeuN (also known as RNA-binding protein RBFOX3) is a well-established marker of neuronal nuclei (47). SOX6 is a transcription factor expressed in MGE-derived GABA neurons during development and into adulthood (48); the use of anti-SOX6 antibodies (1:1500, guinea pig polyclonal) (49) to separate the nuclei of MGE-derived GABA neurons from nuclei of Glu neurons was described by us previously (10). SOX10 is a transcription factor specifically expressed in OLIG. The application of anti-SOX10 antibodies (1:300, goat polyclonal; R&D Systems, AF2864) to isolate the nuclei of OLIG was described by Ernst and colleagues (20). For simultaneous FANS isolation of MGE-GABA, Glu, and OLIG nuclei, we used mouse monoclonal anti-NeuN phycoerythrin (PE)–conjugated antibodies (Millipore, FCMAB317PE, 1:1000), donkey anti-guinea pig AX647-conjugated secondary antibodies (Jackson ImmunoResearch, 1:1500) to detect SOX6-positive nuclei, and donkey anti-goat AX488-conjugated secondary antibodies (Life Technologies, A21447, 1:1500) to detect SOX10-positive nuclei. DNA stain 4′,6-diamidino-2-phenylindole was used to label intact nuclei.
ChIP and ChIP-seq library construction
The native ChIP protocol using chromatin fragmentation with micrococcal nuclease (MNase) was used as described in (50). During nuclei isolation, buffers were supplemented with protease inhibitors (0.1 mM benzamidine and 0.1 mM phenylmethylsulfonyl fluoride). Nuclei (150,000 to 200,000) of each cell type were used per ChIP reaction. Anti-H3K27ac antibody was from Active Motif (catalog #39133; lot #31814008; rabbit polyclonal, 3 μg per sample). The antibodies were prevalidated for specificity using a dot blot assay (AbSurance Histone H3 Antibody Specificity Array Kit, Millipore). ChIP-seq libraries were prepared with the NEBNext Ultra DNA Library Prep Kit (New England BioLabs). The resulting libraries were sequenced on a HiSeq 2500 instrument, using a paired-end 50-cycle protocol, to an average of ~40 million read pairs per sample. For each sample, a matching input control sample obtained from 1 ng of MNase-digested DNA was prepared and sequenced.
RNA isolation and RNA-seq library construction
Nuclei of cell populations were prepared using the FANS-based method as described in our previous publications (10, 51). Nuclei were prepared as described above with the following modifications: During tissue homogenization, proteinase inhibitors were omitted from the buffers; instead, during tissue homogenization and nucleus separation, ribonuclease (RNase) inhibitors (Recombinant RNase Inhibitor, Clontech) were added to all buffers (1:40 during tissue lysis and 1:100 during other steps). Tissue (100 to 200 mg) was used per preparation. Total RNA was prepared from ~40,000 nuclei per sample, using the PicoPure RNA Isolation Kit (Thermo Fisher Scientific). RNA-seq libraries were prepared with the SMARTer Stranded Total RNA-Seq Kit, Pico Input (Clontech) from 10 ng of RNA. Libraries were sequenced on HiSeq 2500, using a paired-end 50-cycle protocol, to an average of ~50 million read pairs per sample.
DNA methylation assay
Between 350,000 and 700,000 nuclei of Glu neurons, MGE-GABA neurons, and OLIG were isolated by flow cytometry, as described above. High-quality genomic DNA (average fragment length, >20,000 bp) was isolated using the DNeasy Blood and Tissue Kit (Qiagen) and concentrated with a Genomic DNA cleanup kit (Zymo). Genomic DNA (500 ng) was used for BS/OxBS processing and library preparation using the TrueMethyl Whole Genome kit (CEGX). Libraries were sequenced on an Illumina HiSeq 2500 system, using a paired-end 100-cycle protocol. The conversion performance in the oxidation and bisulfite treatment reactions was assessed using tailed CEGX spike-in control oligonucleotides included in the CEGX protocol. All samples passed the conversion quality control criteria suggested by CEGX (see table S3).
Data processing
BS-seq and OxBS-seq reads were trimmed and then mapped to the bisulfite-converted human hg19 reference genome with bowtie2 (52). The calling of unmethylated and methylated base calls was performed by Methylpy (https://github.com/yupenghe/methylpy) (12).
Bisulfite conversion efficiency for each type of cytosines was estimated with CEGX spike-in control sequences (table S3). In each sample, let RC_BS, RmC_BS, and RhmC_BS be the conversion rates (fraction of bases read as thymine in sequencing) for unmethylated Cs, methylated Cs, and hydroxymethylated Cs in the BS-seq data, respectively. Similarly, let RC_OxBS, RmC_OxBS, and RhmC_OxBS be the conversion rates for unmethylated Cs, methylated Cs, and hydroxymethylated Cs in the OxBS-seq data, respectively. Thus, we have
where C′, mC′, and hmC′ are the observed percentages of unmethylated Cs, methylated Cs, and hydroxymethylated Cs in a particular region estimated from the BS-seq and OxBS-seq data, and C, mC, and hmC are the adjusted levels of unmethylated Cs, methylated Cs, and hydroxymethylated Cs, respectively. Nonconversion rate adjustment was done by solving the system of equations above.
DMRs for total methylation (tmC) and true methylation (mC) were defined using Dispersion Shrinkage for Sequencing (DSS) (53) with some modification. Differentially methylated loci (DMLs) were called using a modified “callDML” function, where the difference of mean methylation level between the two samples was compared with the global methylation difference using a Wald test at each CpG site. CpG sites with FDR-corrected P value less than 0.05 were deemed as DMLs. DMRs were then called with the “callDMR” function, and we required each DMR to contain at least three DMLs, to have a minimum length of 50 bp, to have an absolute difference of methylation level greater than 0.3 relative to the global methylation difference in the two cell types in comparison, and to have a detected P value less than 0.05.
Because hmC is indirectly inferred on the basis of the difference between BS and OxBS signals, DSS and other published DMR calling methods are not applicable to hmC in our data sets. To call DMRs for hydroxymethylation (DhMR), the genome was tiled by 1-kb bins. hmCG levels in these bins were estimated and adjusted for nonconversion rate using both BS-seq and OxBS-seq data. DhMRs were then defined as bins that have a difference of hmCG levels greater than the global difference of +0.3 in the two cell types in comparison.
RNA-seq reads were trimmed to remove sequencing adapters and low-quality sequences using Cutadapt (54) in paired-end mode. The first 3 bp that form the 5′ end of read 1 were also removed. Trimmed reads were then mapped to the hg19 genome and the GENCODE annotated transcriptome (V25, coordinates lifted to hg19 with liftOver) with STAR (Spliced Transcripts Alignment to a Reference) (55). Gene expression was estimated using RSEM (RNA-Seq by Expectation Maximization) (56), and DE genes were called using edgeR (57) in exact test mode requiring FDR < 0.05 and FC > 2 (table S2).
ChIP-seq data analysis
FASTQ data files were trimmed to remove adapters and low-quality reads using Scythe and Sickle software tools (https://github.com/vsbuffalo/scythe and https://github.com/najoshi/sickle) and aligned to the hg19 human genome using STAR (55). Non-uniquely mapped and duplicate reads were removed with SAMtools (58) and Picard (http://broadinstitute.github.io/picard/). Alignment files were sorted by read name, and peaks were called using the DFilter software package (59), with command line parameters based on software suggestions for each histone mark. For each sample, a matching input control was included. For H3K27me3 data sets, the following command line parameters were used: “-f=bam -pe -ks=20 -lpval=3 -nonzero”, resulting in 21,000 to 29,000 peaks per sample. For H3K27ac data sets, peaks were called with DFilter using the following parameters: “-f=bam -pe -ks=60 -lpval=4”. For each cell type, H3K27ac peak lists for replicate samples were then overlapped using a custom R script, and peaks that were present in at least half of replicates were preserved for further analysis (peak numbers: 44,519 in MGE-GABA neurons, 46,580 in Glu neurons, and 45,963 in OLIG cells). Peaks detected in Glu, MGE-GABA, and OLIG cells were further overlapped using the bedtools package (60) to obtain Glu-specific (19,697), MGE-GABA–specific (16,297), OLIG-specific (26,975), and common (12,549) peaks, as well as the peaks present in two of three cell types. ChIP-seq enrichment score in a particular region was defined as log2 (RPM pulldown/RPM input) (RPM, reads per million).
Enrichment test of DMRs in risk-associated SNP regions from GWAS data
Analysis of GWAS-associated SNPs in DMRs was adapted from a recent publication (36) with the following modifications. GWAS data were obtained from the National Institutes of Health GRASP (Genome-Wide Repository of Associations Between SNPs and Phenotypes) database (https://grasp.nhlbi.nih.gov/Overview.aspx). Studies with labeled phenotype categories in nine brain-related disorders (ADHD, ALS, Alzheimer’s disease, autism, bipolar disorder, depression, epilepsy, Parkinson’s disease, and schizophrenia) as well as six nonbrain diseases (celiac disease, chronic kidney disease, Crohn’s disease, lung cancer, prostate cancer, and type 1 diabetes) were selected, and only associated SNPs with reported P values of <10−6 (that is, genome-wide significant SNPs) were kept for the following analysis. For each disease category, each SNP was extended to a ±50-kb flanking region, and any regions overlapping each other were merged. The resulting merged regions were ranked in ascending order by the smallest P value of the associated SNPs within the region, and the top 50 regions were defined as the most significant risk-associated SNP regions. Then, the number of overlaps between DMRs and these SNP regions was counted. To test whether the overlaps were enriched or depleted, we compared the observed number of overlaps X with the expected number of overlaps Y if the DMRs were randomly distributed across the genome. Here, Y should follow a binomial distribution with parameters n and p where n is the number of DMRs and p is the coverage of the SNP regions in the genome. With this, the Z scores for each test were calculated, and the empirical two-tailed P values were corrected for multiple comparisons using Benjamini and Hochberg’s FDR method (61).
Supplementary Material
Acknowledgments
We are grateful to W. Doyle, C. Luo, and S. Patkar for helpful discussions. Funding: This work was supported by R01DA043247 (to S.D. and Y.L.H.) and R00NS080911 (to E.A.M.), VA Merit awards BX001829 (to S.D.) and BX002876 (to W.M.B.), and Intramural funds of the U.S. Department of Health and Human Services to the National Library of Medicine (to E.V.K.). ChIP-seq and RNA-seq data were generated as part of the PsychENCODE Consortium, supported by grants U01MH103392, U01MH103365, U01MH103346, U01MH103340, U01MH103339, R21MH109956, R21MH105881, R21MH105853, R21MH103877, R21MH102791, R01MH111721, R01MH110928, R01MH110927, R01MH110926, R01MH110921, R01MH110920, R01MH110905, R01MH109715, R01MH109677, R01MH105898, R01MH105898, R01MH094714, and P50MH106934 awarded to S. Akbarian (Icahn School of Medicine at Mount Sinai), G. Crawford (Duke University), S. Dracheva (Icahn School of Medicine at Mount Sinai), P. Farnham (University of Southern California), M. Gerstein (Yale University), D. Geschwind (University of California, Los Angeles), F. Goes (Johns Hopkins University), T. M. Hyde (Lieber Institute for Brain Development), A. Jaffe (Lieber Institute for Brain Development), J. A. Knowles (University of Southern California), C. Liu (SUNY Upstate Medical University), D. Pinto (Icahn School of Medicine at Mount Sinai), P. Roussos (Icahn School of Medicine at Mount Sinai), S. Sanders (University of California, San Francisco), N. Sestan (Yale University), P. Sklar (Icahn School of Medicine at Mount Sinai), M. State (University of California, San Francisco), P. Sullivan (University of North Carolina), F. Vaccarino (Yale University), D. Weinberger (Lieber Institute for Brain Development), S. Weissman (Yale University), K. White (University of Chicago), J. Willsey (University of California, San Francisco), and P. Zandi (Johns Hopkins University). Author contributions: S.D., E.A.M., P.A., and A.K. conceived and designed the experiments. P.A. and A.K. performed the experiments. J.L., A.K., and E.A.M. analyzed the data. Y.L.H., W.M.B., and M.W. provided study materials. S.D., E.A.M., A.K., J.L., and E.V.K. wrote the manuscript. All authors contributed to discussing the results. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Data are available at Synapse (https://doi.org/10.7303/syn12034263) and through a browser (https://brainome.ucsd.edu/humanBrainSortedCells). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Postmortem tissue samples were used during the analysis. All data obtained from these samples, even if not presented in the paper, will be made available to researchers. Additionally, the authors have only a limited amount of the anti-SOX6 antibodies and cannot fulfill requests for the reagent. All data related to this reagent will be made available as well. For additional information regarding postmortem tissue samples and anti-SOX6 antibodies, please contact Y.L.H. and M.W., respectively. There is enough information provided in the paper to allow for the reproduction of the experimental conditions.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/9/eaau6190/DC1
Fig. S1. RNA-seq validates the identities of FANS-sorted populations.
Fig. S2. Cell type–specific gene body DNA methylation patterns in neurons and OLIG associate with expression and differential expression.
Fig. S3. Association of epigenetic modifications at promoters with gene expression in different brain cell types.
Fig. S4. DNA methylation profiles at cell type–specific distal gene regulatory elements.
Fig. S5. DNA methylation profiles at cell type–specific distal H3K27ac peaks situated at different distances from TSS.
Fig. S6. Comparison of adult enhancers (H3K27ac peaks) with regions of open chromatin (ATAC-seq) in the developing human fetal brain.
Fig. S7. Comparison of cell type–specific DMRs with DMRs from single-nucleus methylC-seq data.
Table S1. Sample information and demographics.
Table S2. Results of RNA-seq analysis (TPMs of all genes in each sample and results of differential expression analyses).
Table S3. Sequencing metrics for BS-seq, OX-BS-seq, and RNA-seq analyses.
Table S4. DMRs for tmCG, mCG, and hmCG.
REFERENCES AND NOTES
- 1.Lake B. B., Ai R., Kaeser G. E., Salathia N. S., Yung Y. C., Liu R., Wildberg A., Gao D., Fung H.-L., Chen S., Vijayaraghavan R., Wong J., Chen A., Sheng X., Kaper F., Shen R., Ronaghi M., Fan J.-B., Wang W., Chun J., Zhang K., Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith Z. D., Meissner A., DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 14, 204–220 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Hemberger M., Dean W., Reik W., Epigenetic dynamics of stem cells and cell lineage commitment: Digging Waddington’s canal. Nat. Rev. Mol. Cell Biol. 10, 526–537 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Tahiliani M., Peng Koh K., Shen Y., Pastor W. A., Bandukwala H., Brudno Y., Agarwal S., Iyer L. M., Liu D. R., Aravind L., Rao A., Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He Y.-F., Li B.-Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., Sun Y., Li X., Dai Q., Song C.-X., Zhang K., He C., Xu G.-L., Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ito S., Shen L., Dai Q., Wu S. C., Collins L. B., Swenberg J. A., He C., Zhang Y., Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kriaucionis S., Heintz N., The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szulwach K. E., Li X., Li Y., Song C.-X., Wu H., Dai Q., Irier H., Upadhyay A. K., Gearing M., Levey A. I., Vasanthakumar A., Godley L. A., Chang Q., Cheng X., He C., Jin P., 5-hmC–mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14, 1607–1616 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lister R., Mukamel E. A., Nery J. R., Urich M., Puddifoot C. A., Johnson N. D., Lucero J., Huang Y., Dwork A. J., Schultz M. D., Yu M., Tonti-Filippini J., Heyn H., Hu S., Wu J. C., Rao A., Esteller M., He C., Haghighi F. G., Sejnowski T. J., Margarita Behrens M., Ecker J. R., Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozlenkov A., Roussos P., Timashpolsky A., Barbu M., Rudchenko S., Bibikova M., Klotzle B., Byne W., Lyddon R., Di Narzo A. F., Hurd Y. L., Koonin E. V., Dracheva S., Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 42, 109–127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wen L., Li X., Yan L., Tan Y., Li R., Zhao Y., Wang Y., Xie J., Zhang Y., Song C., Yu M., Liu X., Zhu P., Li X., Hou Y., Guo H., Wu X., He C., Li R., Tang F., Qiao J., Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 15, R49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schultz M. D., He Y., Whitaker J. W., Hariharan M., Mukamel E. A., Leung D., Rajagopal N., Nery J. R., Urich M. A., Chen H., Lin S., Lin Y., Jung I., Schmitt A. D., Selvaraj S., Ren B., Sejnowski T. J., Wang W., Ecker J. R., Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523, 212–216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo C., Keown C. L., Kurihara L., Zhou J., He Y., Li J., Castanon R., Lucero J., Nery J. R., Sandoval J. P., Bui B., Sejnowski T. J., Harkins T. T., Mukamel E. A., Margarita Behrens M., Ecker J. R., Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rotem A., Ram O., Shoresh N., Sperling R. A., Goren A., Weitz D. A., Bernstein B. E., Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 33, 1165–1172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mooijman D., Dey S. S., Boisset J.-C., Crosetto N., van Oudenaarden A., Single-cell 5hmC sequencing reveals chromosome-wide cell-to-cell variability and enables lineage reconstruction. Nat. Biotechnol. 34, 852–856 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Mellén M., Ayata P., Dewell S., Kriaucionis S., Heintz N., MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151, 1417–1430 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mo A., Mukamel E. A., Davis F. P., Luo C., Henry G. L., Picard S., Urich M. A., Nery J. R., Sejnowski T. J., Lister R., Eddy S. R., Ecker J. R., Nathans J., Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86, 1369–1384 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mellén M., Ayata P., Heintz N., 5-hydroxymethylcytosine accumulation in postmitotic neurons results in functional demethylation of expressed genes. Proc. Natl. Acad. Sci. U.S.A. 114, E7812–E7821 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozlenkov A., Wang M., Roussos P., Rudchenko S., Barbu M., Bibikova M., Klotzle B., Dwork A. J., Zhang B., Hurd Y. L., Koonin E. V., Wegner M., Dracheva S., Substantial DNA methylation differences between two major neuronal subtypes in human brain. Nucleic Acids Res. 44, 2593–2612 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ernst A., Alkass K., Bernard S., Salehpour M., Perl S., Tisdale J., Possnert G., Druid H., Frisén J., Neurogenesis in the striatum of the adult human brain. Cell 156, 1072–1083 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Petilla Interneuron Nomenclature Group, Ascoli G. A., Alonso-Nanclares L., Anderson S. A., Barrionuevo G., Benavides-Piccione R., Burkhalter A., Buzsáki G., Cauli B., Defelipe J., Fairén A., Feldmeyer D., Fishell G., Fregnac Y., Freund T. F., Gardner D., Gardner E. P., Goldberg J. H., Helmstaedter M., Hestrin S., Karube F., Kisvárday Z. F., Lambolez B., Lewis D. A., Marin O., Markram H., Muñoz A., Packer A., Petersen C. C., Rockland K. S., Rossier J., Rudy B., Somogyi P., Staiger J. F., Tamas G., Thomson A. M., Toledo-Rodriguez M., Wang Y., West D. C., Yuste R., Petilla terminology: Nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat. Rev. Neurosci. 9, 557–568 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baumann N., Pham-Dinh D., Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 81, 871–927 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Booth M. J., Branco M. R., Ficz G., Oxley D., Krueger F., Reik W., Balasubramanian S., Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 336, 934–937 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Whyte W. A., Orlando D. A., Hnisz D., Abraham B. J., Lin C. Y., Kagey M. H., Rahl P. B., Ihn Lee T., Young R. A., Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gabel H. W., Kinde B. Z., Stroud H., Gilbert C. S., Harmin D. A., Kastan N. R., Hemberg M., Ebert D. H., Greenberg M. E., Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu J., Adli M., Zou J. Y., Verstappen G., Coyne M., Zhang X., Durham T., Miri M., Deshpande V., De Jager P. L., Bennett D. A., Houmard J. A., Muoio D. M., Onder T. T., Camahort R., Cowan C. A., Meissner A., Epstein C. B., Shoresh N., Bernstein B. E., Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152, 642–654 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stroud H., Su S. C., Hrvatin S., Greben A. W., Renthal W., Boxer L. D., Nagy M. A., Hochbaum D. R., Kinde B., Gabel H. W., Greenberg M. E., Early-life gene expression in neurons modulates lasting epigenetic states. Cell 171, 1151–1164.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deaton A. M., Bird A., CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stadler M. B., Murr R., Burger L., Ivanek R., Lienert F., Schöler A., van Nimwegen E., Wirbelauer C., Oakeley E. J., Gaidatzis D., Tiwari V. K., Schübeler D., DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Creyghton M. P., Cheng A. W., Grant Welstead G., Kooistra T., Carey B. W., Steine E. J., Hanna J., Lodato M. A., Frampton G. M., Sharp P. A., Boyer L. A., Young R. A., Jaenisch R., Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 107, 21931–21936 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roadmap Epigenomics Consortium, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M. J., Amin V., Whitaker J. W., Schultz M. D., Ward L. D., Sarkar A., Quon G., Sandstrom R. S., Eaton M. L., Wu Y. C., Pfenning A. R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R. A., Shoresh N., Epstein C. B., Gjoneska E., Leung D., Xie W., Hawkins R. D., Lister R., Hong C., Gascard P., Mungall A. J., Moore R., Chuah E., Tam A., Canfield T. K., Hansen R. S., Kaul R., Sabo P. J., Bansal M. S., Carles A., Dixon J. R., Farh K. H., Feizi S., Karlic R., Kim A. R., Kulkarni A., Li D., Lowdon R., Elliott G., Mercer T. R., Neph S. J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R. C., Siebenthall K. T., Sinnott-Armstrong N. A., Stevens M., Thurman R. E., Wu J., Zhang B., Zhou X., Beaudet A. E., Boyer L. A., De Jager P. L., Farnham P. J., Fisher S. J., Haussler D., Jones S. J., Li W., Marra M. A., McManus M. T., Sunyaev S., Thomson J. A., Tlsty T. D., Tsai L. H., Wang W., Waterland R. A., Zhang M. Q., Chadwick L. H., Bernstein B. E., Costello J. F., Ecker J. R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J. A., Wang T., Kellis M., Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de la Torre-Ubieta L., Stein J. L., Won H., Opland C. K., Liang D., Lu D., Geschwind D. H., The dynamic landscape of open chromatin during human cortical neurogenesis. Cell 172, 289–304.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie W., Schultz M. D., Lister R., Hou Z., Rajagopal N., Ray P., Whitaker J. W., Tian S., David Hawkins R., Leung D., Yang H., Wang T., Young Lee A., Swanson S. A., Zhang J., Zhu Y., Kim A., Nery J. R., Urich M. A., Kuan S., Yen C.-a., Klugman S., Yu P., Suknuntha K., Propson N. E., Chen H., Edsall L. E., Wagner U., Li Y., Ye Z., Kulkarni A., Xuan Z., Chung W.-Y., Chi N. C., Antosiewicz-Bourget J. E., Slukvin I., Stewart R., Zhang M. Q., Wang W., Thomson J. A., Ecker J. R., Ren B., Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y., Zheng H., Wang Q., Zhou C., Wei L., Liu X., Zhang W., Zhang Y., Du Z., Wang X., Xie W., Genome-wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Biol. 19, 18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barr C. L., Misener V. L., Decoding the non-coding genome: Elucidating genetic risk outside the coding genome. Genes Brain Behav. 15, 187–204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lake B. B., Chen S., Sos B. C., Fan J., Kaeser G. E., Yung Y. C., Duong T. E., Gao D., Chun J., Kharchenko P. V., Zhang K., Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 36, 70–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis D. A., Inhibitory neurons in human cortical circuits: Substrate for cognitive dysfunction in schizophrenia. Curr. Opin. Neurobiol. 26, 22–26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Magueresse C., Monyer H., GABAergic interneurons shape the functional maturation of the cortex. Neuron 77, 388–405 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Kepecs A., Fishell G., Interneuron cell types are fit to function. Nature 505, 318–326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spruijt C. G., Gnerlich F., Smits A. H., Pfaffeneder T., Jansen P. W., Bauer C., Münzel M., Wagner M., Müller M., Khan F., Eberl H. C., Mensinga A., Brinkman A. B., Lephikov K., Müller U., Walter J., Boelens R., van Ingen H., Leonhardt H., Carell T., Vermeulen M., Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152, 1146–1159 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Wu X., Zhang Y., TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 18, 517–534 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Scott-Browne J. P., Lio C.-W. J., Rao A., TET proteins in natural and induced differentiation. Curr. Opin. Genet. Dev. 46, 202–208 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaffe A. E., Gao Y., Deep-Soboslay A., Tao R., Hyde T. M., Weinberger D. R., Kleinman J. E., Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat. Neurosci. 19, 40–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lunnon K., Hannon E., Smith R. G., Dempster E., Wong C., Burrage J., Troakes C., Al-Sarraj S., Kepa A., Schalkwyk L., Mill J., Variation in 5-hydroxymethylcytosine across human cortex and cerebellum. Genome Biol. 17, 27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drakenberg K., Nikoshkov A., Horváth M. C., Fagergren P., Gharibyan A., Saarelainen K., Rahman S., Nylander I., Bakalkin G., Rajs J., Keller E., Hurd Y. L., μ opioid receptor A118G polymorphism in association with striatal opioid neuropeptide gene expression in heroin abusers. Proc. Natl. Acad. Sci. U.S.A. 103, 7883–7888 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nikoshkov A., Drakenberg K., Wang X., Horvath M. C., Keller E., Hurd Y. L., Opioid neuropeptide genotypes in relation to heroin abuse: Dopamine tone contributes to reversed mesolimbic proenkephalin expression. Proc. Natl. Acad. Sci. U.S.A. 105, 786–791 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang Y., Matevossian A., Huang H.-S., Straubhaar J., Akbarian S., Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 9, 42 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Batista-Brito R., Rossignol E., Hjerling-Leffler J., Denaxa M., Wegner M., Lefebvre V., Pachnis V., Fishell G., The cell-intrinsic requirement of Sox6 for cortical interneuron development. Neuron 63, 466–481 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stolt C. C., Schlierf A., Lommes P., Hillgärtner S., Werner T., Kosian T., Sock E., Kessaris N., Richardson W. D., Lefebvre V., Wegner M., SoxD proteins influence multiple stages of oligodendrocyte development and modulate SoxE protein function. Dev. Cell 11, 697–709 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Brind’Amour J., Liu S., Hudson M., Chen C., Karimi M. M., Lorincz M. C., An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat. Commun. 6, 6033 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Kozlenkov A., Jaffe A. E., Timashpolsky A., Apontes P., Rudchenko S., Barbu M., Byne W., Hurd Y. L., Horvath S., Dracheva S., DNA methylation profiling of human prefrontal cortex neurons in heroin users shows significant difference between genomic contexts of hyper- and hypomethylation and a younger epigenetic age. Genes 8, E152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feng H., Conneely K. N., Wu H., A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 42, e69 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011). [Google Scholar]
- 55.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; Genome Project Data Processing Subgroup , The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar V., Muratani M., Arul Rayan N., Kraus P., Lufkin T., Hui Ng H., Prabhakar S., Uniform, optimal signal processing of mapped deep-sequencing data. Nat. Biotechnol. 31, 615–622 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300 (1995). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/9/eaau6190/DC1
Fig. S1. RNA-seq validates the identities of FANS-sorted populations.
Fig. S2. Cell type–specific gene body DNA methylation patterns in neurons and OLIG associate with expression and differential expression.
Fig. S3. Association of epigenetic modifications at promoters with gene expression in different brain cell types.
Fig. S4. DNA methylation profiles at cell type–specific distal gene regulatory elements.
Fig. S5. DNA methylation profiles at cell type–specific distal H3K27ac peaks situated at different distances from TSS.
Fig. S6. Comparison of adult enhancers (H3K27ac peaks) with regions of open chromatin (ATAC-seq) in the developing human fetal brain.
Fig. S7. Comparison of cell type–specific DMRs with DMRs from single-nucleus methylC-seq data.
Table S1. Sample information and demographics.
Table S2. Results of RNA-seq analysis (TPMs of all genes in each sample and results of differential expression analyses).
Table S3. Sequencing metrics for BS-seq, OX-BS-seq, and RNA-seq analyses.
Table S4. DMRs for tmCG, mCG, and hmCG.