

Abstract
Protein prenylation involves the attachment of a farnesyl or geranylgeranyl group onto a cysteine residue located near the C-terminus of a protein, recognized via a specific prenylation motif and results in the formation of a thioether bond. To identify putative prenylated proteins and investigate changes in their levels of expression, metabolic labeling and subsequent bioorthogonal labeling has become one of the methods of choice. In that strategy, synthetic analogues of biosynthetic precursors for post-translational modification bearing bioorthogonal functionality are added to the growth media from which they enter cells and become incorporated into proteins. Subsequently, the cells are lysed and proteins bearing the analogues are then covalently modified using selective chemical reagents that react via bioorthogonal processes allowing a variety probes for visualization or enrichment to be attached for subsequent analysis. Here, we describe protocols for synthesizing several different isoprenoid analogues and describe how they are metabolically incorporated into mammalian cells and the incorporation into prenylated proteins visualized via in-gel fluorescence analysis.
Keywords: Alkyne-containing analogue, farnesyltransferase, farnesyl diphosphate, geranylgeranyltransferase, geranylgeranyl diphosphate, isoprenoid, metabolic labeling, protein prenylation
INTRODUCTION
Protein lipid modifications, including prenylation, myristoylation and palmitoylation, are important for directing and stably anchoring proteins to cellular membranes as well as for protein trafficking and the facilitation of protein-protein interactions involved in cell signaling. In particular, protein prenylation involves the attachment of an isoprenoid onto a cysteine residue located near the C-terminus of a protein, recognized via a specific prenylation motif and results in the formation of a thioether bond (F. L. Zhang & Casey, 1996). Following prenylation, the protein undergoes further processing namely proteolysis of the C-terminal peptide and carboxymethylation to afford the mature protein (Fig. 1A). Many protein substrates for prenylation have a tetrapeptide CaaX box motif that is either modified with a farnesyl or geranylgeranyl group from farnesyl- (FPP) and geranylgeranyl diphosphate (GGPP), catalyzed by farnesyltransferase (FTase) and geranylgeranyltransferase type I (GGTase-I), respectively (Casey, Solski, Der, & Buss, 1989; Casey, Thissen, & Moomaw, 1991). Dual geranylgeranylation on two proximal cysteine residues near the C-terminus also occurs for some proteins with –CCXX, -CXC, or –CC motifs (Kinsella & Maltese, 1992). This process is catalyzed by geranylgeranyltransferase type II (GGTase-II) also known as Rab GGTase, in a process facilitated by rab escort protein (REP). Protein prenylation has been a target of interest for developing anti-tumor drugs, particularly in inhibiting the prenylation of Ras which is one of the most commonly mutated proteins observed in cancer (Lerner et al., 1997). Changes in protein prenylation have also been implicated in a number of diseases including neurodegenerative disorders and progeria, and has served as an attractive drug target against microbial and viral infections (Palsuledesai & Distefano, 2015).
Figure 1.
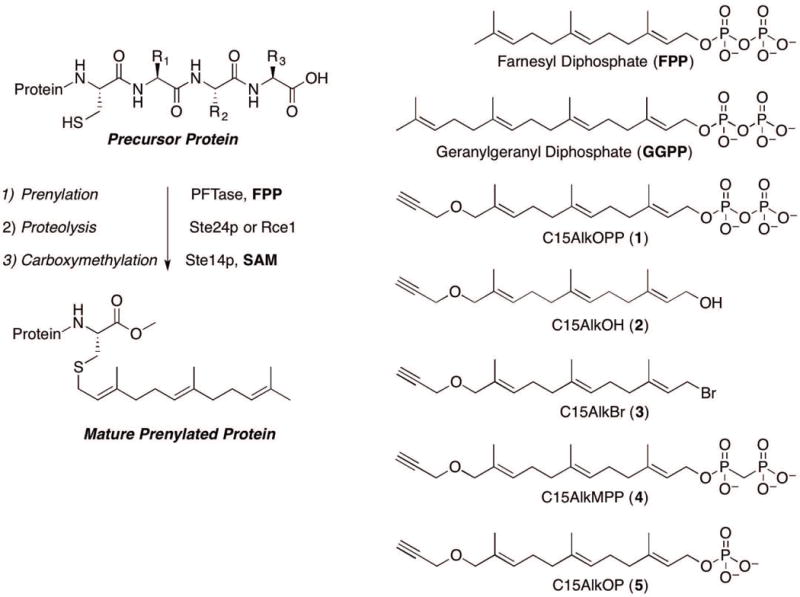
Schematic representation of the maturation of a farnesylated protein from a substrate with a CaaX box (A) and parent and analogue forms of isoprenoids described in this protocol (B). The maturation of geranylgeranylated proteins follows a similar pathway although not all such proteins are processed beyond initial prenylation.
Several biochemical strategies have been developed to study prenylation. A number of these approaches rely on the use of chemically modified isoprenoid analogues that introduce bioorthogonal groups suitable for both in vitro and in vivo investigations (Wang & Distefano, 2016). Reports on functional groups used to modify isoprenoids include azides (Rose et al., 2005) and alkynes (Hosokawa et al., 2007) for copper-catalyzed cycloaddition (click) reactions, aldehydes for oxime ligation (Rashidian, Dozier, Lenevich, & Distefano, 2010) and photoactivatable benzophenones (Henry et al., 2009) and diazirines (Vervacke et al., 2014) for labeling of enzyme active sites. Some of these isoprenoid analogues have proved to be useful for biotechnology applications including enzymatic labeling for bioconjugation of proteins, DNA and nanostructures (Duckworth et al., 2007; Rashidian et al., 2013; Y. Zhang et al., 2015). Aldehyde-functionalized isoprenoids not only allow site-specific labeling of proteins but also facilitate protein purification via capture and release strategies (Mahmoodi, Rashidian, Dozier, & Distefano, 2013; Rashidian, Song, Pricer, & Distefano, 2012).
To identify putative prenylated proteins and investigate changes in their levels of expression, metabolic labeling and subsequent bioorthogonal labeling has become one of the methods of choice (Saxon & Bertozzi, 2000). In that strategy, synthetic analogues of biosynthetic precursors for post-translational modification bearing bioorthogonal functionality are added to the growth media from which they enter cells and become incorporated into proteins. Subsequently, the cells may be lysed and proteins bearing the analogues are then covalently modified using selective chemical reagents that react via bioorthogonal processes allowing a variety of probes for visualization or enrichment to be attached for subsequent analysis (Cravatt & Sorensen, 2000). Early work on the metabolic labeling of prenylated proteins employed azide-containing analogues (Kho et al., 2004). However, due to the lower level of background labeling typically obtained with alkynes, more recent studies have used alkyne-containing isoprenoids (Speers & Cravatt, 2004). The synthesis of C15AlkOPP (1) and its incorporation in vitro by protein farnesyltransferase was reported in 2007 (Hosokawa et al., 2007) followed by reports of its use in metabolic labeling in 2010 (DeGraw et al., 2010). Since that time, it has been employed for a variety of experiments including differential electrophoresis to study prenylation inhibitors (Palsuledesai, Ochocki, Markowski, & Distefano, 2014), labeling of proteins sensitive to human pathogens (Charron, Li, MacDonald, & Hang, 2013) and the delineation of the prenylome of Plasmodium falciparum, the causative agent of malaria (Gisselberg, Zhang, Elias, & Yeh, 2017; Suazo, Schaber, Palsuledesai, Odom John, & Distefano, 2016). Recently, it has been demonstrated that a-factor, a farnesylated pheromone from yeast retains full activity when the farnesyl group is substituted with several chemically modified isoprenoids used for metabolic labeling. Those results suggest that such probes can be used without concern that they will cause undesired interference in normal cell physiology (Diaz-Rodriguez et al., 2017).
In this paper, we describe a general protocol for synthesizing several phosphate analogues (Fig. 1B) of C15AlkOH (2) and evaluate their efficiency for labeling prenylated proteins in cultured mammalian cells through in-gel fluorescence analysis. Basic Protocol 1 describes the synthesis of C15AlkBr (3), the main precursor for the subsequent attachment of different phosphate moieties and C15AlkOPP (1). Basic Protocols 2, and 3 describe the syntheses of C15AlkOPCH2P (4) and C15AlkOP (5), respectively. Basic Protocol 4 describes the metabolic labeling using these isoprenoid analogues in COS-7 cells followed by in-lysate fluorophore conjugation via click chemistry and subsequent in-gel fluorescence analysis.
BASIC PROTOCOL 1: SYNTHESIS OF FARNESYL ALKYNE DIPHOSPHATE (1)
The alkyne-modified farnesol analogue C15AlkOH (2) has been commonly used for probing prenylated proteins through metabolic labeling since it is commercially available. A detailed synthesis of this probe can be found in Support Protocol 1. In synthesizing the phosphorylated analogues, C15AlkOH (2) is first converted to the brominated form, C15AlkBr (3, (2E,6E,10E)-12-bromo-2,6,10-trimethyl-1-(prop-2-yn-1-yloxy)dodeca-2,6,10-triene) via the Appel reaction. This compound, which has a limited shelf life, is then immediately transformed into the phosphate analogue using a specific phosphorylating reagent (Figure 2). In this protocol, 3 is produced from 2 by using Tetrabromomethane (CBr4) and triphenylphosphine. The diphosphate analogue C15AlkOPP is subsequently synthesized using the diphosphorylating reagent tris(tetra-n-butylammonium) hydrogen pyrophosphate (Davisson et al., 1986). The product is initially purified using ion-exchange chromatography followed by semi-preparative HPLC purification.
Figure 2.
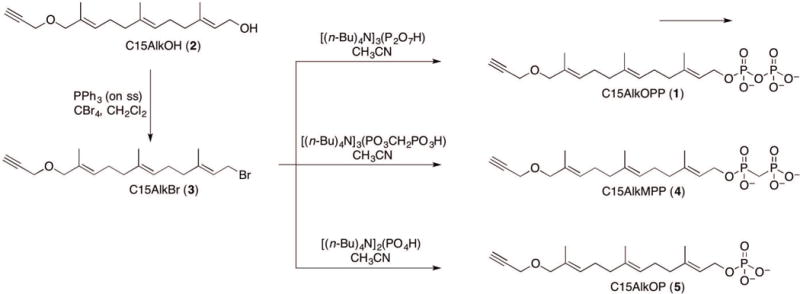
Schematic representation of the synthesis of the isoprenoid phosphate analogues starting from C15AlkOH (2) and the common precursor C15AlkBr (3).
Materials
Alkyne-farnesol (Echelon)
Anhydrous dichloromethane, CH2Cl2
Triphenylphosphine, polymer-bound (Sigma-Aldrich)
Tetrabromomethane, CBr4
Tris(tetra-n-butylammonium) hydrogen pyrophosphate (Sigma-Aldrich)
Anhydrous acetonitrile, CH3CN
Molecular sieves 4A, 8-12 mesh (Acros Organics)
AG 50W-X8(100–200 mesh hydrogen form) ion exchange resin (Sigma-Aldrich)
Ammonium hydroxide (NH4OH)
Ammonium bicarbonate (NH4HCO3)
Isopropanol
Heavy water (D2O)
Ammonium deuteroxide ND4OD (Aldrich)
250-mL and 25-mL round-bottom flasks
Magnetic stir plate and stir bar.
TLC plates
20-mL scintillation vial
30 × 1.5-cm glass column for ion exchange
Filter paper and funnel
Büchi Rotavapor model R-114 or equivalent rotary evaporator
0.45 μm syringe filter
C18 analytical chromatography column with a 5-cm guard column (Luna, 10 μm, 250 × 4 mm, Phenomenex)
C18 semi-preparative chromatography column (Zorbax 300SB, 5 μm, 250 × 9.4 mm)
HPLC instrument (Beckman model 127⁄166) equipped with a UV detector
Electro Spray Ionization Mass Spectrometer (ESI-MS; Bruker BioTOF II)
500 MHz 1H NMR instrument (Oxford. VI-500 MHz)
Synthesis of farnesyl alkyne bromide, C15AlkBr (3)
Dissolve 100 mg (0.36 mmol) of C15AlkOH (2) in 6.75 mL anhydrous CH2Cl2.
Add 240 mg (0.72 mmol) of triphenylphosphine (polymer-bound beads 100-200 mesh, extent of labeling: ~3 mmol/g triphenylphosphine loading).
Stir the resulting suspension for 30 min at room temperature to allow beads to swell.
Add 179 mg of CBr4 (0.54 mmol)
Stir the resulting suspension for 24 h at room temperature or until TLC analysis shows full conversion to product (C15AlkOH Rf ~ 0.30, C15AlkBr Rf ~ 0.60, 5:1 (v/v) hexanes/EtOAc).
Filter off the polymer-bound beads using a standard filtration setup and wash with additional CH2Cl2.
-
Remove solvent with a rotatory evaporator to provide 125 mg (99%) of compound C15AlkBr.
Column chromatography was not performed for two reasons: 1) the allylic bromide is unstable on silica 2) TLC analysis only revealed one spot.
Characterization of C15AlkBr (3)
1H-NMR of farnesyl alkyne bromide 3. 1H-NMR (500 MHz, CDCl3): δ 1.60 (s, 3H), 1.65 (s, 3H), 1.72 (s, 3H), 2.02–2.21 (m, 8H), 2.41 (t, 1H, J = 2.4 Hz), 3.93 (s, 2H), 4.03 (d, 2H, J = 8.4 Hz), 4.08 (d, 2H, J = 2.4 Hz), 5.09 (t, 1H, J = 6.8 Hz), 5.42 (t, 1H, J = 6.8 Hz), 5.53 (t, 1H, J = 6.4 Hz).
Synthesis of diphosphate analogue C15AlkOPP (1)
8. Dissolve 1.4 g (1.56 mmol) of tris(tetra-n-butylammonium) hydrogen pyrophosphate into 15 mL of anhydrous CH3CN in a scintillation vial and add molecular sieves and leave overnight to remove water.
9. Remove molecular sieves and transfer the solution to a flame-dried 50-mL round bottom flask equipped with a magnetic stir bar.
10. Dissolve 420 mg (1.24 mmol) of freshly prepared C15AlkBr (3) in 15 mL of anhydrous CH3CN in a separate flame dried flask.
11. Transfer the resulting mixture containing the pyrophosphate reagent using a 20-mL syringe with an oven-dried metal needle.
12. Stir the resulting solution at room temperature under nitrogen for 24 h.
13. Remove solvent with a rotary evaporator to provide a crude brown residue.
14. Load 30 g of ion exchange resin on a 30 × 1.5-cm column. Wash the resin with three column volumes of 3:1 (v/v) H2O/conc. NH4OH to convert it to the ammonium form, and the equilibrate with three bed volumes of the elution solvent 49:1 (v/v) 25 mM aqueous NH4HCO3/isopropanol.
-
15. Dissolve the crude material in 15 mL of the elution solvent and load it on to the column. Elute with 30 ml of elution solvent and collect fractions.
Ion exchange chromatography is preformed to remove tetra-n-butylammonium salts which impede the following RP-HPLC purification. If the flow rate through the column slows significantly, the percent of isopropanol in the elution solvent can be increased.
16. Pool fractions, freeze in liquid nitrogen, and lyophilize to obtain a crude brown olid.
Purification of diphosphate analogue C15AlkOPP (1)
17. Dissolve the crude material into 15 mL of 25 mM aqueous NH4HCO3 and filter the resulting solution through a 0.45 μm syringe filter. Purify by RP-HPLC using a semi-preparative column under the following conditions:
detection at 220 nm
flow rate: 5.0 mL/min
5-mL injection loop
solvent A, 25 mM aqueous NH4HCO3
solvent B, CH3CN
elution time program:
gradient 0% solvent B for 5 min
0% to 20% solvent B in 15 min
20% to 35% solvent B in 45 min
35% to 100% solvent B in 10 min
-
100% solvent B for 10 min
Compound 5 elutes from 20% to 35% solvent B. Collect fractions every minute in a new test tube.
18. Analyze all collected fractions by ESI-MS. In negative ionization mode, search for a calculated mass of C18H29O8P2, [M-H]− calc. 436.1.
19. Pool fractions contacting the desired mass, freeze in liquid nitrogen, and lyophilize to furnish pure C15AlkOPP (1) as a white powder. Yield: 80 mg (15%) after quantification (see Support Protocol 2).
Characterization of C15AlkOPP (1)
1H-NMR (500 MHz, ND4OD/D2O pH = 8): δ 1.40 (s, 3H), 1.46 (s, 3H), 1.50 (s, 3H), 1.83–1.98 (m, 8H), 2.61 (t, 1H, J = 2.0), 3.73 (s, 2H), 3.84 (d, 2H, J = 2.0), 4.23 (t, 2H, J = 10.0), 4.96 (d, 1H), 5.22 (d, 2H); 31P-NMR (121 MHz, ND4OD/D2O pH = 8): δ −9.87 (d, J = 21), −6.08 (d, J = 21).
BASIC PROTOCOL 2: SYNTHESIS OF FARNESYL ALKYNE METHYLENEBISPHOSPHONATE (4)
Equipped with the C15AlkBr (2) precursor, the hydrolytically resistant analogue C15AlkMPP (4) can be synthesized. Here we describe the synthesis of the novel compound C15AlkMPP, guided by related procedures (Davisson et al., 1986; Stremler & Poulter, 1987). The methyenebisphosphonate reagent is first synthesized by combining methane diphosphonic acid and tetra-n-butyl-ammonium hydroxide and subsequently used to phosphorylate C15AlkBr (3).
Materials
Methane diphosphonic acid (Aldrich)
Deionized water
Tetra-n-butyl-ammonium hydroxide, 40% wt.% aqueous solution (Aldrich)
Liquid nitrogen
Acetonitrile, CH3CN
Methanol, MeOH
P2O5 on silica (Aldrich)
Nitrogen gas
250-mL and 25-mL round-bottom flasks
Magnetic stir plate and stir bar.
pH meter
Glass wool
Synthesis of tris(tetra-n-butylammonium) hydrogen methyenebisphosphonate
Dissolve 1.0 g (5.68 mmol) of methane diphosphonic acid in 20 ml of deionized water.
Stir the solution and monitor the pH with a pH meter.
Add tetra-n-butyl-ammonium hydroxide (40% wt.% aqueous solution) until the pH reaches 10.0 (~12 mL).
Freeze the resulting solution in a liquid nitrogen bath.
Remove water by lyophilization to obtain an oily hydroscopic solid.
Add 50 ml of CH3CN and remove solvent through rotary evaporation. Repeat this process three times to azeotropically remove residual water and provide 4.98 g (97%) of the desired product as a white solid.
Store at −20 °C
Characterization of [(n-Bu)4N]3[PO3CH2PO3H]
1H-NMR (500 MHz, D2O) δ 0.74 (t, 12H, J = 7.4 Hz), 1.18 (sextet, 8H, J = 7.4 Hz), 1.44-1.50 (m, 8H), 1.76 (t, 2H, J = 19.0 Hz), 3.02 (t, 8H, J = 8.5 Hz); 31P-NMR (162 MHz, D2O) δ 16.59 (s, 2P).
Synthesis of methyenebisphosphonate analogue C15AlkMPP (4)
8. Add 1.3 g (1.4 mmol) of tris(tetra-n-butylammonium) hydrogen methyenebisphosphonate and a magnetic stir bar to 3 mL of distilled water in a 25 mL round bottom flask.
9. Freeze in liquid nitrogen bath while rotating the flask to distribute the frozen solid evenly onto the walls of the flask.
-
10. Fill the bottom of a lyophilization jar with a 3 cm layer of P2O5 on silica. Then add a 2 cm layer of glasswool. Place the flask containing the frozen tris(tetra-n-butylammonium) hydrogen methyenebisphosphonate and lyophilize to provide the anhydrous dried salt.
P2O5 on silica is a drying agent that increases the drying efficiency of this lyophilization procedure. Drying of the tris(tetra-n-butylammonium) hydrogen methyenebisphosphonate salt prior to use is crucial in the success of this highly water sensitive reaction.
11. Immediately place the dried tris(tetra-n-butylammonium) hydrogen methyenebisphosphonate salt under a nitrogen atmosphere.
12. Dissolve 125 mg (0.36 mmol) of C15AlkBr (3) in 0.8 mL of anhydrous CH3CN in a separate flame-dried flask.
13. Add the resulting solution to the flask containing the dried tris(tetra-n-butylammonium) hydrogen methyenebisphosphonate salt with a syringe with an oven-dried needle.
14. Stir the resulting solution at room temperature under nitrogen for 24 h.
15. Remove the solvent with a rotary evaporator to provide a crude brown residue.
16. Perform ion-exchange chromatography on the product as described in Basic Protocol 1 step 15-16.
17. Dissolve the crude material into 15 mL of 25 mM aqueous NH4HCO3 and filter the resultant solution through a 0.45 μm syringe filter. Purify by RP-HPLC using a semi-preparative column under the conditions described in Basic Protocol 1 step 17.
18. Analyze all collected fractions by ESI-MS. In negative ionization mode, search for a calculated mass of C19H31O7P2, [M-H]− calc. 433.1.
19. Pool fractions contacting the desired mass, freeze in liquid nitrogen, and lyophilize to furnish pure C15AlkMPP (4). Yield: 10.9 mg (7%) after quantification (see Support Protocol 2).
Characterization of C15AlkMPP (4)
1H-NMR (400 MHz, D2O, ND4OD) δ 1.54 (s, 2×3H), 1.62 (s, 3H), 1.86-2.12 (m, 8H), 3.91 (s, 2H), 4.02 (d, 2H, J = 1.9), 4.31 (t, 2H, J = 6.5 Hz), 4.82 (s, 2H), 5.12 (t, 1H, J = 6.2 Hz), 5.35 (t, 1H, J = 6.2 Hz), 5.41 (t, 1H, J = 6.6 Hz). 31P-NMR (162 MHz, D2O, ND4OD) δ 22.10 (d, 1P, J = 10.0 Hz), 11.64 (d, 1P, J = 9.9 Hz).
BASIC PROTOCOL 3: SYNTHESIS OF FARNESYL ALKYNE MONOPHOSPHATE (5)
The synthesis of the diphosphate analogue C15AlkOPP (1) described above is commonly contaminated with the corresponding monophosphate form, C15AlkOP (5), due to the susceptibility of C15AlkOPP to hydrolysis. C15AlkOP (5) is produced in significant amounts and is challenging to separate from C15AlkOPP as both products coelute in several fractions during HPLC purification. Since a substantial amount of the C15AlkOPP (1) is contaminated with C15AlkOP (5), we decided to evaluate whether this analogue could potentially function in metabolic labeling experiments. If C15AlkOP can be used, the purification of C15AlkOPP will be greatly simplified and yields will be higher, as no separation of the two probes will be required. However, isolation of the pure fractions of monophosphate from diphosphate-monophosphate mixture does not yield sufficient amounts for metabolic experiments. This protocol describes the synthesis of C15AlkOP from C15AlkBr with no contamination with 1. The monophosphate reagent is first generated from phosphoric acid and tetra-n-butyl-ammonium hydroxide and subsequently reacted with C15AlkBr (Feld & Weiss, 2006).
Materials
Deionized water
85% phosphoric acid
Tetra-n-butyl-ammonium hydroxide, 40% wt.% aqueous solution (Aldrich)
Anhydrous acetonitrile, CH3CN
Nitrogen gas
50-mL Erlenmeyer flask
10-mL round-bottom flasks
Magnetic stir plate and stir bar.
Glass pipette
50-mL falcon tube
20-mL scintillation vial
0.45 μm syringe filter
Synthesis of compound bis(tetra-n-butylammonium) hydrogen phosphate
Add 1 mL of 85% phosphoric acid to 5 mL deionized water in a 50-mL Erlenmeyer flask.
Stir the solution with a magnetic stirrer.
Slowly add 20 mL of tetra-n-butylammonium hydroxide dropwise using a glass pipette while monitoring the pH. The final pH will be around 9.8
Transfer the solution to a 50-mL falcon tube. Flash freeze the solution in a liquid nitrogen bath.
Lyophilize the solution to obtain a sticky and hygroscopic solid.
Add 30 mL of CH3CN and remove solvent in vacuo using a rotary evaporator. Repeat this process three times to obtain a white solid product (7.0 g, 95%).
Store at −20 °C.
Characterization of [(n-Bu)4N]2[PO4H]
1H-NMR (500 MHz, D2O) δ 0.86 (t, 12H, J = 7.3 Hz), 1.27 (sextet, 8H, J = 7.4 Hz), 1.52-1.60 (m, 8H), 3.11 (t, 8H, J = 8.6 Hz); 31P-NMR (162 MHz, D2O) δ 0.71 (s, 1P).
Synthesis of farnesyl alkyne monophosphate (C15AlkOP, 5)
8. Dissolve 0.5 g (1.0 mmol) of dried bis(tetra-n-butylammonium) hydrogen phosphate and a magnetic stir bar in 1 mL of CH3CN in a 20-mL scintillation vial.
9. Dissolve 70 mg (0.20 mmol) of 3 in 1 mL of anhydrous CH3CN in a 10-mL round-bottomed flask.
10. Add the monophosphate solution to the solution containing C15AlkBr.
11. Stir to react under nitrogen at room temperature overnight.
12. Remove the solvent in vacuo by rotary evaporation.
13. Perform ion-exchange chromatography on the product as described in Basic Protocol 1 step 15-16.
14. Dissolve the crude material into 15 mL of 25 mM aqueous NH4HCO3 and filter the resulting solution through a 0.45 μm syringe filter. Purify by RP-HPLC using a semi-preparative column under the following conditions described in Basic Protocol 1 step 17.
15. Analyze all collected fractions by ESI-MS. In negative ionization mode, search for a calculated mass of C18H28O5P [M-H]− calc. 356.2.
16. Pool fractions contacting the desired mass, freeze in liquid nitrogen, and lyophilize to furnish pure compound 5. Yield: 8.34 mg (11%) after quantification (see Support Protocol 2).
Characterization of C15AlkOP (5)
1H-NMR (500 MHz, CD3OD): δ 1.63 (s, 3H), 1.65 (s, 3H), 1.70 (s, 3H), 1.99–2.22 (m, 8H), 4.07 (s, 2H), 3.94 (s, 2H), 4.42 (t, 1H, J = 5.6 Hz), 5.17 (t, 1H, J = 6.6 Hz), 5.44 (t, 1H, J = 7.1 Hz), 5.48 (t, 1H, J = 7.2 Hz); 31P-NMR (121 MHz, CD3OD, NH4HCO3, pH = 9): δ 5.719 (s).
BASIC PROTOCOL 4: METABOLIC LABELING OF ISOPRENOID ANALOGUES TO MAMMALIAN CELL CULTURES AND SUBSEQUENT CLICK REACTIONS IN-GEL FLUORESCENCE ANALYSIS
The success of using alkyne-modified isoprenoid probes in metabolic labeling experiments is initially evaluated via click reaction with an azide-modified fluorophore followed by in-gel fluorescence analysis. Labeled proteins appear as bands in the fluorescence scan due to the conjugated fluorophore. Additionally, treatment with inhibitors of the isoprenoid biosynthesis such as lovastatin (Palsuledesai et al., 2016) and fosmidomycin (Suazo et al., 2016) are employed to reduce the pool of endogenous FPP and GGPP, thereby enhancing the incorporation of the isoprenoid probe to the prenylated proteins. This protocol describes metabolic labeling using the isoprenoid analogues C15AlkOH (2), C15AlkOPP (1), C15AlkMPP (4) and C15AlkOP (5) in COS-7 cells in the presence or absence of lovastatin. In each case, the lysates obtained are subjected to click reaction with TAMRA-N3 and proteins resolved using an SDS-PAGE gel and subjected to in-gel fluorescence analysis (Fig. 3). Dose-dependent (Fig. 4) and time course experiments (Fig. 4) delineate the optimal concentration and incubation for the probes.
Figure 3.
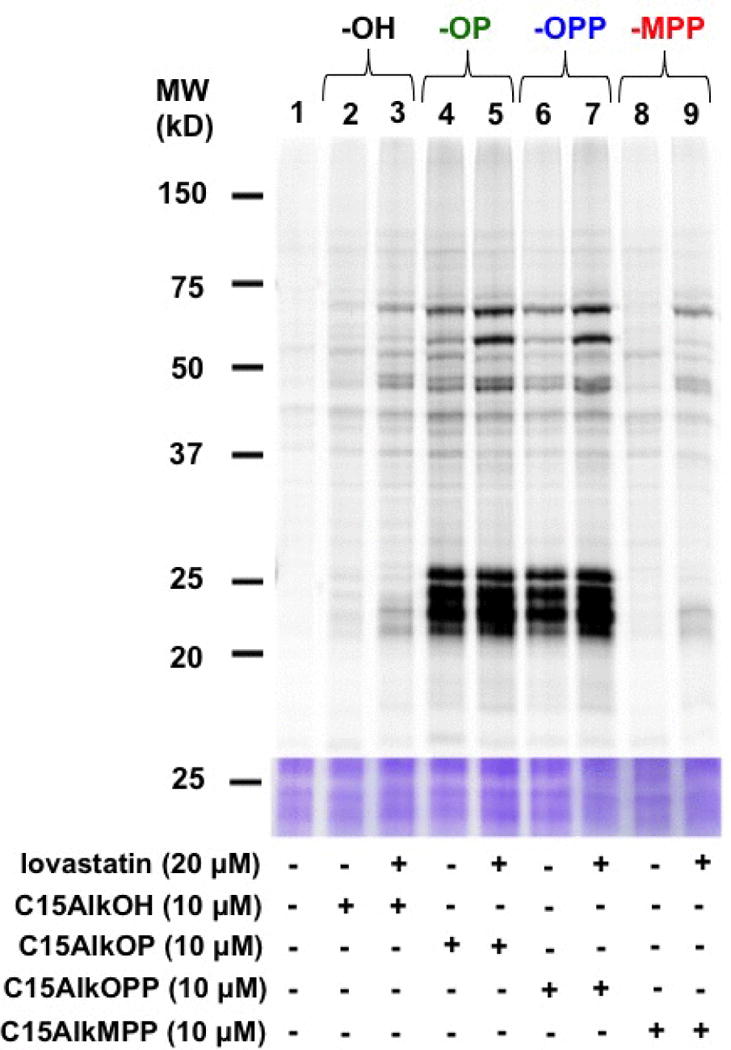
In-gel fluorescence analysis of prenylated proteins from COS-7 cells metabolically labeled with C15AlkOH (2) and the isoprenoid phosphate analogues C15AlkOP (5), C15AlkOPP (1), and C15AlkMPP (4).
Figure 4.
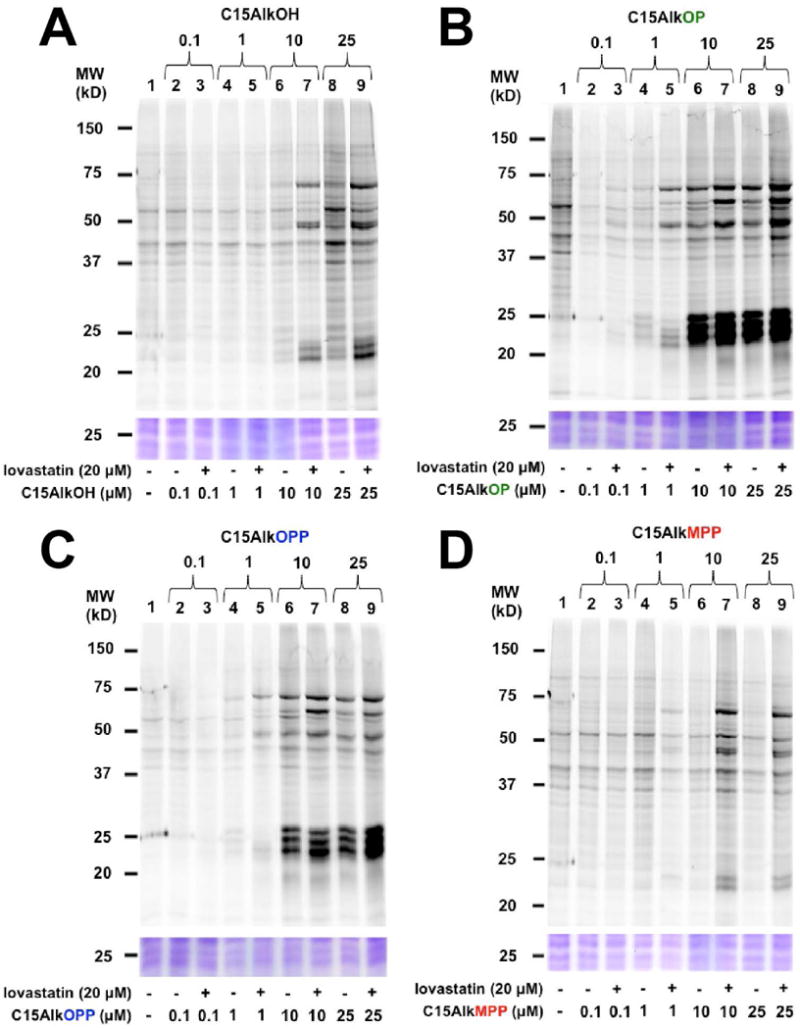
In-gel fluorescence analysis on the dose-dependent metabolic labeling of COS-7 cells with probes C15AlkOH (2, A), C15AlkOP (5, B), C15AlkOPP (2, C), and C15AlkMPP (4, D) at concentrations of 0.1, 1, 10, and 25 μM. Probes are incubated with cells for 24 h after the cells are first pre-incubated in the presence or absence of 20 μM of lovastatin for 6 h.
Materials
DMEM media with 10% fetal bovine serum (FBS, Gibco) and 1% PenStrep (Gibco)
1X PBS (sterile)
Lovastatin, 25 mM in DMSO (Fisher Scientific)
Isoprenoid probes (see BASIC PROTOCOLS 1-3)
BCA protein assay kit (Thermo Scientific)
Lysis buffer: Per 300 μL of 1X PBS with 1% SDS, add 5 μL of protease inhibitor cocktail (Sigma-Aldrich), 0.64 μL of 1 mM phenylmethylsulfonyl fluoride dissolved in DMSO (PMSF, Sigma-Aldrich), and 0.26 μL of 250 units/μL of benzonase nuclease (Sigma-Aldrich)
1X PBS with 1% SDS
TAMRA-PEG3-N3 (BroadPharm)
Tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Sigma-Aldrich)
Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA, Sigma-Aldrich)
CuSO4
ProteoExtract® protein precipitation kit (Calbiochem, Cat. No.: 539180)
1X Laemmli buffer
12% SDS-PAGE gel
Coomassie staining and destaining solutions
100-mm tissue culture dish (Fisher Scientific)
Incubator equipped with CO2 gas source
Glass pipettes
Vacuum system
Cell scraper
1-mL pipette
1.5-mL microcentrifuge tubes
Stick sonicator
Heating block
Fluorescence scanner (Typhoon FLA 9500, GE Healthcare)
Simple document scanner
Metabolic labeling in cultured cells
Grow mammalian cells (800,000 cell count) on a 100-mm dish in 10 mL media overnight at 37 °C in an incubator under 5% CO2.
Remove media with vacuum using a glass pipette. Wash cells by adding 5 mL of sterile 1X PBS and swirling gently. Remove the buffer and wash the cells once more.
Add 5 mL of the media. For lovastatin pre-treatment, add 4 μL of 25 mM lovastatin (20 μM final concentration). Incubate for 6 h at 37 °C.
Without replacing the media, add appropriate volumes of isoprenoid probes to generate 0.1, 1, 10, and 25 μM final concentrations. Incubate at 37 °C overnight.
For time course experiments, pre-treat cells with 2 μL of lovastatin (10 μM final concentration) for 6 h and add 2 μL of 25 mM stock solution of isoprenoid probes (10 μM final concentration). Incubate at 37 °C for different times: 6, 12, 24, and 36 hours.
After the probe treatment is complete, place the plates on ice for cell collection. Three to four plates can be collected at a time.
Remove the media by vacuum using a glass pipette. Wash the cells gently with 5 mL of cold 1X PBS. Repeat washing one more time.
Add 1 mL of 1X PBS to each plate. While the plate lies flat on ice, scrape the cells using a cell scraper in an up and down motion while moving horizontally across the plate. Rotate the plate 90° and scrape cells in the same manner.
Hold the plate vertically and scrape the cell suspension to the edge of the plate. Let stand for 1-2 min. During this time, start scraping the other plates.
Using a 1 mL pipette, collect the cell suspensions into pre-chilled 1.5-mL microcentrifuge tubes.
Centrifuge the tubes at 150 × g for 5 minutes. The cells should appear as pellets at the bottom of the tubes.
Carefully remove the supernatentby suction without disturbing the cell pellet. Store the cell pellets at −80 °C or proceed to lysis.
To lyse the cells, suspend the cell pellets in 300 μL of lysis buffer.
While on ice, lyse the cells by sonication with a stick sonicator 6 to 8 times for 2 seconds at 10-second intervals.
Determine the protein concentration in the lysates using a BCA assay kit with BSA as a standard. Follow the manufacturer’s protocol for this assay.
In-gel fluorescence analysis
16. After the determining the concentration of the protein lysates, obtain an aliquot containing 100 μg of protein and dilute to 92.5 μL with 1X PBS + 1% SDS in a 1.5-mL microcentrifuge tube.
17. Add 2.5 μL of 1 mM TAMRA PEG3-N3 (25 μM final), 2 μL of 50 mM TCEP (1 mM final), and 1 μL of 10 mM TBTA (0.1 mM final). Vortex the mixtures briefly.
18. Add 2 μL of 50 mM CuSO4 (1 mM final).
19. Incubate in the dark with gentle shaking at room temperature for 1 h.
20. Precipitate out proteins using a ProteoExtract® protein precipitation kit by following the manufacturer’s protocol.
21. Redissolve the protein pellets in 40 μL of 1X Laemmli buffer by pipetting it in and out.
22. Heat the samples using a heating block at 95 °C for 5 min. Centrifuge briefly to drive down the condensate to the bottom of the tubes.
23. Load 20 μL of the sample (50 μg proteins) into each lane of a 12% SDS-PAGE gel (3″ × 4″ gel). Run at 120 V until the tracking dye runs off.
24. Place the gel in water and immediately scan for TAMRA fluorescence (542/568 nm excitation/emission) using a fluorescence scanner.
25. Stain the gel with Coomassie stain for 20 min and destain with destaining solution for 3 h.
26. Scan the stained gel on a document scanner.
27. For formatting of gel images, adjust the brightness and contrast in ImageJ.
SUPPORT PROTOCOL 1: SYNTHESIS OF FARNESYL ALKYNE ALCOHOL, C15AlkOH (2)
This protocol describes the synthesis of the alkyne-modified farnesol (C15AlkOH, 2) as shown in Figure 6 (Hosokawa et al., 2007).
Figure 6.
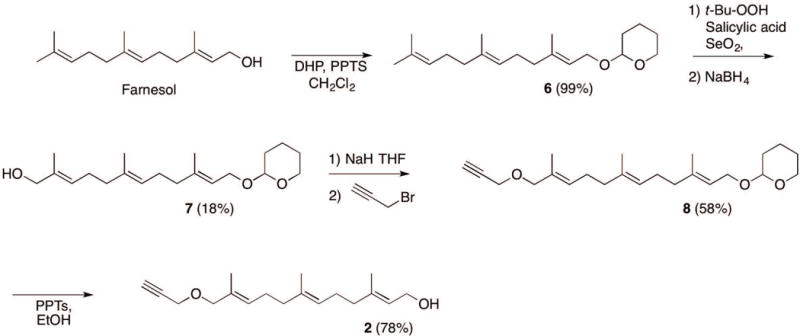
Synthesis of alkyne-modified farnesol (C15AlkOH, 2) from farnesol.
Materials
Farnesol (Aldrich)
Dichloromethane (CH2Cl2)
3,4-Dihydropyran (DHP; Aldrich)
Pyridinium p-toluenesulfonate (PPTS)
Sodium bicarbonate (NaHCO3)
Brine
Sodium sulfate (Na2SO4)
Silica gel
70% tert-Butyl hydroperoxide aqueous solution (Aldrich)
Salicylic acid (Aldrich)
Selenium dioxide (SeO2; Aldrich)
Toluene
Methanol (MeOH)
Sodium borohydride (NaBH4)
Diethyl ether (Et2O)
Hexanes
Ethyl acetate (EtOAc)
Tetrahydrofuran (THF)
Sodium hydride 60 % dispersion in mineral oil (Aldrich)
Propargyl bromide 80 wt.% solution in toluene (Aldrich)
Ethanol (EtOH)
Triphenylphosphine (polymer-bound 100-200 mesh, extent of labeling: ~3 mmol/g triphenylphosphine loading; Aldrich)
Carbontetrabromide (CBr4; Aldrich)
500-ml, 250-ml, 50-ml, and 25-ml round-bottom flasks
Magnetic stir plate with heating capabilities and stir bar.
Büchi Rotavapor model R-114 or equivalent rotary evaporator
Separatory funnel
Ice bath
TLC plates (silica gel 60 F-254; Merck)
Buchner funnel
Chromatography columns (30 × 1.5-cm) and (25.4 × 5-cm)
Sealed tube with a threaded type A plug (Ace Glass)
Oil bath
500 MHz 1H NMR instrument (Oxford. VI-500 MHz)
Synthesis of compound 6
Dissolve 3.00 g (13.5 mmol) of farnesol in 30 mL dichloromethane in a round bottom flask containing a magnetic stir bar.
Add 1.8. mL (20.3 mmol) 3,4-dihydropyran.
Add 338 mg (1.4 mmol) pyridinium p-toluenesulfonate.
Stir the resulting solution for 24 h at room temperature using a magnetic stirrer.
Quench the reaction by adding 15 ml of saturated aqueous NaHCO3.
Extract the organic layer and separate layers in a separatory funnel. Extract the aqueous layer two additional times, each time with 20 mL of CH2Cl2.
Combine organic layers, wash with 50 mL of brine and dry over Na2SO4. Filter off drying agent with a Buchner funnel.
Remove solvent under reduced pressure in a rotary evaporator to yield 4.12 g (99%) of compound 6 as a colorless oil.
Characterization of (6)
1H-NMR (500 MHz, CDCl3): δ 1.60 (s, 6 H), 1.68 (s, 6 H), 1.50-1.89 (m, 6H), 1.94-2.16 (m, 8H), 3.47-3.93 (m, 2H), 4.06 (dd, 1H, J = 12.0 Hz and 7.5 Hz), 4.24 (dd, 1H, J = 12.0 Hz and 6.5 Hz), 4.63 (t, 1H, J = 4.0 Hz), 5.10-5.13 (m, 2H), and 5.36 (t, 1H, J = 6.5 Hz),
Synthesis of compound 7
9. Dissolve 4.12 g (13.5 mmol) of compound 6 in 50 mL CH2Cl2 in a round bottom flask.
10. Add 5.6 mL (30 mmol) 70% tert-butyl hydroperoxide aqueous solution.
Add 186 mg (1.4 mmol) salicylic acid.
12. Add 150 mg (1.4 mmol) SeO2.
13. Stir the resulting solution for 24 h at room temperature with a magnetic stirrer.
14. Remove the CH2Cl2 under reduced pressure by rotary evaporation.
-
15. Add 50 mL toluene to the remaining solution and evaporate solvent by rotary evaporation. Repeat this process four times to ensure removal of excess tert-butyl hydroperoxide.
Toluene and tert-butyl hydroperoxide form a low boiling point azeotrope, which expedites the evaporation of tert-butyl hydroperoxide under reduced pressure.
16. Dissolve the resulting residue in 30 mL CH3OH and cool in an ice bath.
17. Add 400 mg (10 mmol) NaBH4 incrementally over the course of ten min.
18. Stir the resulting solution for 2 h at 0 °C with a magnetic stirrer.
19. Quench the reaction by slowly adding 30 mL cold H2O.
20. Add 100 mL of Et2O and transfer to a separatory funnel. Separate the layers and extract the aqueous layer two additional times, each time with 100 mL of Et2O.
21. Combine organic layers, wash with 200 mL of brine, and dry over Na2SO4. Filter off drying agent.
22. Remove solvent under reduced pressure with a rotatory evaporator to afford a pale yellow oil.
23. Load 100 g silica gel in a 25.4 × 5-cm column. Equilibrate the column with hexanes.
24. Dissolve the crude reaction sample in a minimal amount of hexanes (a few drops of EtOAc may be added to improve solubility) and load it onto the column. Elute with the following gradient:
100 mL hexanes
200 mL of 9:1 (v/v) hexanes/EtOAc
200 mL of 8:2 (v/v) hexanes/EtOAc
200 mL of 7:3 (v/v) hexanes/EtOAc
6:5 (v/v) hexanes/EtOAc until TLC analyses verifies the product has fully eluted (Rf ~ 0.25, 5:1 (v/v) hexanes/EtOAc).
Pool fractions that contain pure product.
25. Concentrate pooled fractions with a rotatory evaporator to yield 760 mg (18%) of compound 7 as a clear oil.
Characterization of (7)
1H-NMR (500 MHz, CDCl3): δ 1.60 (s, 3H), 1.66 (s, 3H) 1.68 (s, 3H), 1.50-1.75 (m, 6H), 2.00-2.14 (m, 8H), 3.49-3.53 (m, 1H), 3.87-3.92 (m, 1H), 3.99 (s, 2 H), 4.03 (dd, 1H, J = 12.0 Hz and 7.5 Hz), 4.22 (dd, 1H, J = 12.0 Hz and 6.5 Hz), 4.63 (t, 1H, J = 4.0 Hz), 5.12 (1H, t, J = 6.5 Hz), and 5.41-5.34 (m, 2H).
Synthesis of compound 8
26. Dissolve 500 mg (1.4 mmol) of compound 7 in 4 mL anhydrous THF in a sealed tube with a threaded type A plug and cool in an ice bath.
27. Add 217 mg sodium hydride 60 % dispersion in mineral oil (5.4 mmol) slowly over the course of five min.
28. Stir the resulting solution at 0 °C for 90 min.
29. Add 2 mL propargyl bromide 80 wt.% solution in toluene (13.95 mmol) pre-chilled to 0 °C over the span of one min.
30. Seal tube securely, allow to stir at 0 °C for 30 min, then place reaction tube into an oil bath. Slowly increase the temperature to 85 °C and stir for two days.
Heating the reaction above 90 °C induces product degradation and results in a lower yield.
31. Quench the reaction with 10 mL of H2O, extract three times with 20 mL of EtOAc.
32. Combine organic layers, wash with 30 mL of brine, and dry over Na2SO4. Filter off drying agent.
33. Remove solvent by rotary evaporation to obtain a brown oil.
34. Load 100 g silica gel in a 30 × 1.5-cm column. Equilibrate with 5:1 (v/v) hexanes/EtOAc.
35. Dissolve crude sample in a minimal amount of 5:1 (v/v) hexanes/EtOAc and load it onto the column. Elute the column with 5:1 (v/v) hexanes/EtOAc until TLC analysis shows the product has eluted off the column (Rf ~ 0.75, 5:1 (v/v) hexanes/EtOAc). Pool fractions that contain pure product.
36. Evaporate solvent with a rotary evaporator to afford 319 mg (58%) compound 8 as a clear oil.
Characterization of (8)
1H-NMR (500 MHz, CDCl3): δ 1.61 (s, 3H), 1.66 (s, 3H) 1.69 (s, 3H), 1.46-1.95 (m, 6H), 1.95-2.27 (m, 8H), 2.42 (t, 1H, J = 2.3 Hz), 3.48-3.57 (m, 1H), 3.86-3.94 (m, 1H), 3.98-4.16 (m, 1H), 3.95 (s, 2 H), 4.09 (s, 2H), 4.20-4.30 (m, 1H), 4.65 (t, 1H, J = 4.0 Hz), 5.13 (1H, t, J = 6.7 Hz), 5.38 (1H, t, J = 6.5 Hz) and 5.44 (1H, t, J = 7.4 Hz).
Synthesis of compound 2
37. Dissolve 331 mg (0.88 mmol) of compound 8 in 5 mL EtOH.
38. Add 22 mg (0.09 mmol) of pyridinium p-toluenesulfonate.
39. Stir the resulting solution at 55 °C for 24 h.
40. Remove solvent by rotary evaporation to obtain a clear oil.
41. Load 100 g silica gel into a 30 × 1.5-cm column. Equilibrate with 5:1 (v/v) hexanes/EtOAc.
42. Dissolve crude sample in a minimal amount 5:1 (v/v) hexanes/EtOAc and load it onto the column. Elute the column with 5:1 (v/v) hexanes/EtOAc until TLC analysis shows the product is off the column (Rf ~ 0.30, 5:1 (v/v) hexanes/EtOAc). Pool fractions that contain pure product.
43. Evaporate solvent with a rotary evaporator to afford 190 mg (78%) of compound 2 as a clear oil.
Characterization of C15AlkOH (2)
1H-NMR (500 MHz, CDCl3): δ 1.63 (s, 3H), 1.67 (s, 3H), 1.70 (s, 3H), 2.01–2.23 (m, 8H), 2.43 (t, 1H, J = 2.4 Hz), 3.95 (s, 2H), 4.09 (d, 2H, J = 2.4 Hz), 4.16 (d, 2H, J = 6.9 Hz), 5.13 (t, 1H), 5.38 (t, 1H), 5.41 (t, 1H).
SUPPORT PROTOCOL 2: Determination of the concentration of isoprenoid analogues
The preparation of stock solutions of C15AlkOH is usually performed by dissolving known quantities, measured by weighing, in DMSO. Unlike C15AlkOH, the isoprenoid phosphate analogues can be conveniently quantified through 31P NMR via the detection of the phosphorus in the phosphate moiety (Lenevich & Distefano, 2011). Here we describe the accurate determination of the concentration of the isoprenoid phosphate analogues as solutions in deuterium oxide. For the purpose of preparing a quantified stock solution of C15AlkOH to generate accurate comparison of its labeling efficiency with the phosphate analogues, we also describe a method to quantify its concentration in deuterated chloroform using 1H NMR.
Isoprenoid probes (see BASIC PROTOCOLS 1-3)
Deuterium oxide (D2O)
Ammonium deuteroxide (ND4OD)
Sodium hydrogen phosphate (Na2HPO4)
Deuterated chloroform (CDCl3)
500 MHz 1H and 31P NMR instrument (Oxford. VI-500 MHz)
Determination of the concentration of isoprenoid analogues
Dissolve the pure isoprenoid analogue powder in 650 μL D2O and add one drop of ND4OD.
This solution will be used to determine the concentration of C15AlkOPP, C15AlkOP and C15AlkMPP, and serve as the stock solution for future experiments with these probes.
Prepare a 10 mM solution of Na2HPO4 in D2O. This solution will be used as an internal standard for determining the concentration of C15AlkOPP, C15AlkOP and C15AlkMPP.
Combine 100 μL of the isoprenoid analogue stock solution with 300 μL of the Na2HPO4 internal standard solution in an NMR tube.
-
Acquire a phosphorus NMR with 124 scans and a relaxation time of 45 sec between pulses. Use the integration ratio between the isoprenoid phosphate analogue and the internal standard [δ 2.60 (s, 1P)] to calculate the concentration of farnesyl alkyne methyenebisphosphonate.
When preforming this calculation, each peak corresponds to one phosphorus atom. Average the integration values of the two doublets attributed to phosphorus atoms in 1 and 4 when determining the ratio.
Determination of the concentration of C15AlkOH
5. Dissolve sufficient amounts of C15AlkOH in CDCl3.
6. Prepare 0.1 M solution of 4-methylbenzonitrile in CDCl3 as the standard.
7. Add 10 μL aliquot of each of the C15AlkOH and 4-methylbenzonitrile solutions to 500 μL of CDCl3. Transfer the resulting solution to an NMR tube.
8. Acquire a 1H NMR with 16 scans and a relaxation time of 45 sec between pulses. Use the integration ratio between the vinylic protons of C15AlkOH (δ 5.43-5.46 ppm, m, 2H) and the methyl group of the standard (δ 2.45 ppm, s, 3H) to calculate the concentration of C15AlkOH.
9. Evaporate the CDCl3 to dryness and redissolve the residue with an exact amount of DMSO to prepare a stock solution (e.g. 25 mM) suitable for metabolic labeling experiments.
COMMENTARY
Background Information
Early metabolic labeling work in the prenylation field used an azide-functionalized isoprenoid for metabolic labeling. Subsequent protein labeling in crude lysate was performed via Staudinger ligation reaction with a biotinylated phosphine reagent (Kho et al., 2004). Due to the slow nature of the Staudinger ligation, more recent efforts have focused on using the copper catalyzed alkyne azide cycloaddition; in that case the alkyne group can be present either on the probe for metabolic labeling or on the reagent used for biotinylation. Interestingly, it has generally been found that protein labeling with an azide-containing probe followed by derivatization with excess alkyne reagent leads to greater nonspecific background labeling than when the converse is performed (Speers & Cravatt, 2004). Those observations led us to develop alkyne-containing probes including C15AlkOPP (1). Not surprisingly, we observed lower background labeling using C15AlkOPP compared with the corresponding azide (DeGraw et al., 2010).
A second issue in the design of probes for metabolic labeling concerns their ability to freely enter cells and become incorporated into target proteins. For lipid modifications such as palmitoylation, probes are introduced as free fatty acids that must be activated within cells to their CoA-esters to be substrates for N-myristoyltransferase since the CoA-esters themselves are not cell permeable (Hang et al., 2007). In contrast, isoprenoid analogues exhibit different behavior. While the mechanism is unclear, farnesyl diphosphate and related analogues are apparently able to directly enter cells even though they are negatively charged at physiological pH. Despite this fact, most metabolic labeling experiments have been performed using alcohol forms of alkyne analogues based on well established reports that farnesol is readily incorporated into prenylated proteins (Crick, Andres, & Waechter, 1995); in that case, it is believed that such isoprenoid alcohols are converted by microsomal and peroxisomal farnesol kinase and farnesyl diphosphate kinase (Bentinger, Grünler, Peterson, Swiezewska, & Dallner, 1998) to the metabolically active diphosphate form. The main reason why the alcohols have been used more often is that the synthesis of the diphosphates is low yielding and inconvenient. The diphosphate products are water-soluble and require reversed-phase HPLC for their purification. In addition, the diphosphates often undergo some hydrolysis to the corresponding monophosphates, generating mixtures that are difficult to separate. As a final point, it should be noted that the alcohol forms of isoprenoid analogues appear to exhibit some toxicity that limits the concentrations that they can be employed at and which in turn limits the efficiency of metabolic incorporation (Gisselberg et al., 2017).
Critical Parameters
Reagent preparation and storage
Allylic bromides are susceptible to hydrolysis. Farnesyl alkyne bromide 2 should be used immediately after being synthesized or stored at −20 °C for no more than a month.
Prepare stock solutions of isoprenoids in DMSO (for C15AlkOH) or 25 mM NH4HCO3 (for phosphorylated analogues). For NMR analysis, add in a few drops of ND4OD to phosphorylated analogues dissolved in D2O.
All click reagents including TCEP, TBTA and TAMRA-N3 are dissolved in DMSO. TCEP oxidizes over time especially after several freeze-and-thaw cycles. It is desirable to use freshly prepared TCEP solutions for click reactions.
Performing experiment
In the synthesis of compound 7, three major products are formed: the alcohol at the terminal position (desired product, Rf ~0.25), a regioisomer (Rf ~0.35), and an aldehyde form of the desired product due to overoxidation (Rf ~0.50). When reducing the aldehyde to the alcohol form using NaBH4, constantly monitor the reaction until the spot for the aldehyde product disappears. Add more NaBH4 if necessarily to drive the reaction forward. Combine only fractions that contain the desired product but are free from the regioisomer since it will react in successive steps and generate an isoprenoid analogue that may interfere during metabolic labeling. Change the solvent gradients if needed to better separate the isomers.
The bis(tetra-n-butylammonium) hydrogen phosphate is extremely hygroscopic. Prior to use, weigh a certain amount of the reagent then dissolve in 5 mL chloroform in a pre-weighed 20-mL scintillation vial, evaporate, then repeat three times. Dissolve the residue in 5 mL acetonitrile and evaporate to dryness. Repeat the process three times. Weigh the dry product plus vial immediately and dissolve in DMF. Obtain an aliquot of the desired amount to be used in the experiment.
When precipitating out the proteins using the ProteoExtract kit, do not leave the reaction in the precipitating reagent overnight even at −20 °C, as this results in high background labeling with the TAMRA-N3. It is generally preferred to have the proteins incubated for 1 hour before pelleting the proteins.
Experimental controls
For metabolic experiments, an alternative experimental control can be used where the samples are treated with FPP instead either in the presence or absence of lovastatin. This assures that the only difference in both treated and control samples is the presence of alkyne moiety in the probe. This is especially relevant when further analyses will be performed such as in quantitative proteomic profiling.
TROUBLESHOOTING
Low yield of isoprenoid phosphate analogues
High recovery of the isoprenoid analogues is largely affected by stability of the C15AlkBr precursor. Not only is it imperative to utilize the precursor immediately but also to use anhydrous solvents. Make sure the solvents used are dry as the presence of water results in the degradation of C15AlkBr. If anhydrous solvents are not available, dry solvents with MgSO4 or incubate with molecular sieves overnight prior to use. Despite these issues, it is worth noting that probe 1 has been prepared by numerous undergraduate researchers suggesting that extensive synthetic experience is not required to prepare the compounds described here.
The phosphate reagents used to obtain farnesyl alkyne methyenebisphosphonate 4 and monophosphate 5 are highly water sensitive. Initial attempts to synthesize 4 gave yields of less than 0.5%. Implementation of the lyophilization procedure discussed above improved the yield to 7%. Although this reaction requires following an operationally involved protocol to be successful, the purification of 4 is less challenging in comparison to traditional diphosphate analogs as the methyenebisphosphonate moiety is not susceptible to hydrolysis in the buffered conditions used.
Low protein recovery
The presence of lovastatin inhibits the growth of cells and may pose some level of toxicity. The number of cells collected varies and is usually lower for those treated with statin at low probe concentrations. Before lysis, compare the recovery of cells across different samples. It is necessary to reduce the amount of lysis buffer to be used for those with lower number of cells in order to generate protein lysates with concentrations sufficient to perform the protocol for the in-lysate click reactions described above.
Low protein labeling
If low protein labeling is observed after in-gel fluorescence scan, assess the amount of proteins loaded after coomassie staining. Adjust the amount of proteins to load in the gel if necessary. Make sure a good amount of proteins are recovered after precipitation from the click reaction. Prepare fresh solutions of the click reagents particularly the TAMRA-N3 and TCEP which degrades and oxidizes over time, especially if accidentally left unfrozen.
Anticipated Results
Basic Protocol 1 will provide farnesyl alkyne bromide C15AlkBr (3). This precursor can be transformed into monophosphate (C15AlkOP, 5), diphosphate (C15AlkOPP, 1) and, methyenebisphosphonate (C15AlkMPP, 4) analogs, as described in Basic Protocols 1, 2, and 3. Expected results from Basic Protocol 4 are represented in Figures 3, 4, and, 5. Using 10 μM probe and cell incubation for 24 hours (Fig. 3), protein labeling is apparent with all probes. These proteins may include Ras and Rab proteins near 25 kDa and DnaJ’s and lamins near 50 and 75 kDa, respectively, as profiled from a mammalian macrophage using the C15AlkOH probe (Charron et al., 2013). Although C15AlkOH was able to label these regions in COS-7 cells (lane 2) with dramatic improvement in the presence of lovastatin (lane 3), significantly better labeling is achieved with the isoprenoid analogues C15AlkOP and C15AlkOPP (lanes 4 and 6), which is enhanced by the addition of lovastatin (lanes 5 and 7). In contrast, the C15AlkMPP manifests minimal incorporation (lane 8) and requires lovastatin to induce more effective labeling.
Figure 5.
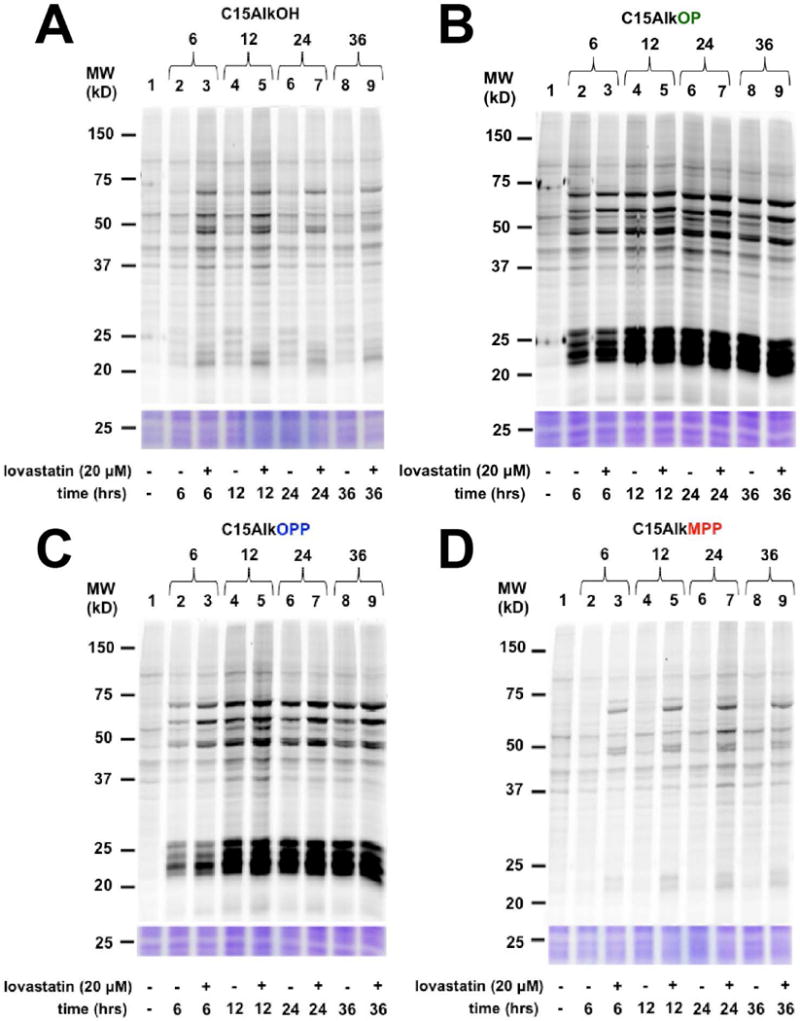
In-gel fluorescence analysis of the time-course of metabolic labeling of COS-7 cells with probes C15AlkOH (2, A), C15AlkOP (5, B), C15AlkOPP (2, C), and C15AlkMPP (4, D) incubated for 6, 12, 24, or 36 h. Probes are added at 10 μM final concentrations for the times indicated in the presence or absence of 10 μM of lovastatin pre-treated for 6 h.
Results from the dose-dependence study (Fig. 4) show that 10 μM probe treatment is optimal for sufficient labeling to be observed across all probes. Better labeling was achieved with C15AlkOP (Fig. 4B) and C15AlkOPP (Fig. 4C), as evidenced by the presence of labeled proteins already observed at 1 μM. Both C15AlkOH (Fig. 4A) and C15AlkMPP (Fig. 4D) do not exhibit obvious protein labeling at this lower concentration. For the time-course experiments, labeling of proteins is already observable after 6 hours across all probes (Fig. 5). The C15AlkOH already achieved its maximum labeling capacity (Fig. 5A) while both C15AlkOP (Fig. 5B) and C15AlkOPP (Fig. 5C) require 12 hours to maximize protein labeling. The C15AlkMPP performs best using a 24-hour incubation (Fig. 5D).
Taken together, these results suggest that the use C15AlkOP and C15AlkOPP function best for metabolic labeling experiments. As both probes label prenylated proteins equally with high efficiency, there is no reason why either C15AlkOP or C15AlkOPP that is contaminated with some C15AlkOP cannot be used for metabolic labeling particularly when the goal of the experiment is simply to identify prenylated proteins (in contrast to experiments where the levels of prenylation are being investigated).
Time Considerations
The transformation of the C15AlkBr from the C15AlkOH and the concurrent synthesis of the phosphate reagents can be accomplished in two days. The phosphate analogues can be synthesized simultaneously in an additional two days plus an additional two days for purification and lyophilization. If the C15AlkOH will be synthesized from farnesol, an additional two weeks should be allotted. Once the probes are available, the metabolic labeling can be accomplished in two days given that an ample amount of cells are prepared prior to the experiment. An additional day or two is needed for the click reaction and subsequent in-gel fluorescence scanning.
Significance Statement.
Protein prenylation is an essential post-translational modification. Methods that allow selective labeling of prenylated proteins are particularly useful since they can be used to identify new target proteins as well as to follow changes in prenylation that occur in various disease states. The method described here, using alkyne-modified isoprenoids, can be used to selectively label prenylated proteins in living cells. The resulting modified proteins can be then be studied using a variety of different methods to identify and quantify their levels.
Acknowledgments
This research was supported by the National Institutes of Health (GM084152 and AG056976) and the National Science Foundation (CHE-1308655).
LITERATURE CITED
- Bentinger M, Grünler J, Peterson E, Swiezewska E, Dallner G. Phosphorylation of Farnesol in Rat Liver Microsomes: Properties of Farnesol Kinase and Farnesyl Phosphate Kinase. Archives of Biochemistry and Biophysics. 1998;353(2):191–198. doi: 10.1006/abbi.1998.0611. [DOI] [PubMed] [Google Scholar]
- Casey PJ, Solski PA, Der CJ, Buss JE. p21ras is modified by a farnesyl isoprenoid. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(21):8323–8327. doi: 10.1073/pnas.86.21.8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey PJ, Thissen JA, Moomaw JF. Enzymatic modification of proteins with a geranylgeranyl isoprenoid. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(19):8631–8635. doi: 10.1073/pnas.88.19.8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron G, Li MMH, MacDonald MR, Hang HC. Prenylome profiling reveals S-farnesylation is crucial for membrane targeting and antiviral activity of ZAP long-isoform. Proceedings of the National Academy of Sciences. 2013;110(27):11085–11090. doi: 10.1073/pnas.1302564110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Sorensen EJ. Chemical strategies for the global analysis of protein function. Current Opinion in Chemical Biology. 2000;4(6):663–668. doi: 10.1016/S1367-5931(00)00147-2. [DOI] [PubMed] [Google Scholar]
- Crick DC, Andres DA, Waechter CJ. Farnesol Is Utilized for Protein Isoprenylation and the Biosynthesis of Cholesterol in Mammalian Cells. Biochemical and Biophysical Research Communications. 1995;211(2):590–599. doi: 10.1006/bbrc.1995.1854. [DOI] [PubMed] [Google Scholar]
- Davisson VJ, Woodside AB, Neal TR, Stremler KE, Muehlbacher M, Poulter CD. Phosphorylation of isoprenoid alcohols. The Journal of Organic Chemistry. 1986;51(25):4768–4779. doi: 10.1021/jo00375a005. [DOI] [Google Scholar]
- DeGraw AJ, Palsuledesai C, Ochocki JD, Dozier JK, Lenevich S, Rashidian M, Distefano MD. Evaluation of alkyne-modified isoprenoids as chemical reporters of protein prenylation. Chemical Biology and Drug Design. 2010;76(6):460–471. doi: 10.1111/j.1747-0285.2010.01037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Rodriguez V, Hsu ET, Ganusova E, Werst E, Becker JM, Hrycyna CA, Distefano MD. a-Factor analogues containing alkyne- and azide functionalized isoprenoids are efficiently enzymatically processed and retain wild type bioactivity. Bioconjugate Chemistry. 2017 doi: 10.1021/acs.bioconjchem.7b00648. Just Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckworth BP, Chen Y, Wollack JW, Sham Y, Mueller JD, Taton TA, Distefano MD. A Universal Method for the Preparation of Covalent Protein–DNA Conjugates for Use in Creating Protein Nanostructures. Angewandte Chemie International Edition. 2007;46(46):8819–8822. doi: 10.1002/anie.200701942. [DOI] [PubMed] [Google Scholar]
- Feld BK, Weiss GA. Convenient methods for the synthesis of P1-farnesyl-P2-indicator diphosphates. Bioorganic & Medicinal Chemistry Letters. 2006;16(6):1665–1667. doi: 10.1016/j.bmcl.2005.12.016. [DOI] [PubMed] [Google Scholar]
- Gisselberg JE, Zhang L, Elias JE, Yeh E. The Prenylated Proteome of Plasmodium falciparum Reveals Pathogen-specific Prenylation Activity and Drug Mechanism-of-action. Molecular & Cellular Proteomics. 2017;16(4 suppl 1):S54–S64. doi: 10.1074/mcp.M116.064550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang HC, Geutjes EJ, Grotenbreg G, Pollington AM, Bijlmakers MJ, Ploegh HL. Chemical Probes for the Rapid Detection of Fatty-Acylated Proteins in Mammalian Cells. Journal of the American Chemical Society. 2007;129(10):2744–2745. doi: 10.1021/ja0685001. [DOI] [PubMed] [Google Scholar]
- Henry O, Lopez-Gallego F, Agger SA, Schmidt-Dannert C, Sen S, Shintani D, Distefano MD. A versatile photoactivatable probe designed to label the diphosphate binding site of farnesyl diphosphate utilizing enzymes. Bioorganic & Medicinal Chemistry. 2009;17(13):4797–4805. doi: 10.1016/j.bmc.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa A, Wollack JW, Zhang Z, Chen L, Barany G, Distefano MD. Evaluation of an Alkyne-containing Analogue of Farnesyl Diphosphate as a Dual Substrate for Protein-prenyltransferases. International Journal of Peptide Research and Therapeutics. 2007;13(1):345–354. doi: 10.1007/s10989-007-9090-3. [DOI] [Google Scholar]
- Kho Y, Kim SC, Jiang C, Barma D, Kwon SW, Cheng J, Zhao Y. A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(34):12479–12484. doi: 10.1073/pnas.0403413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella BT, Maltese WA. rab GTP-binding proteins with three different carboxyl-terminal cysteine motifs are modified in vivo by 20-carbon isoprenoids. Journal of Biological Chemistry. 1992;267(6):3940–3945. [PubMed] [Google Scholar]
- Lenevich S, Distefano MD. NMR-based quantification of organic diphosphates. Analytical Biochemistry. 2011;408(2):316–320. doi: 10.1016/j.ab.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner EC, Zhang TT, Knowles DB, Qian Y, Hamilton AD, Sebti SM. Inhibition of the prenylation of K-Ras, but not H- or N-Ras, is highly resistant to CAAX peptidomimetics and requires both a farnesyltransferase and a geranylgeranyltransferase I inhibitor in human tumor cell lines. Oncogene. 1997;15:1283. doi: 10.1038/sj.onc.1201296. [DOI] [PubMed] [Google Scholar]
- Mahmoodi MM, Rashidian M, Dozier JK, Distefano MD. Chemoenzymatic Site-Specific Reversible Immobilization and Labeling of Proteins from Crude Cellular Extract Without Prior Purification Using Oxime and Hydrazine Ligation. Current Protocols in Chemical Biology. 2013;5(2):89–109. doi: 10.1002/9780470559277.ch120247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsuledesai CC, Distefano MD. Protein Prenylation: Enzymes, Therapeutics, and Biotechnology Applications. ACS Chemical Biology. 2015;10(1):51–62. doi: 10.1021/cb500791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsuledesai CC, Ochocki JD, Kuhns MM, Wang YC, Warmka JK, Chernick DS, Distefano MD. Metabolic Labeling with an Alkyne-modified Isoprenoid Analog Facilitates Imaging and Quantification of the Prenylome in Cells. ACS Chemical Biology. 2016;11(10):2820–2828. doi: 10.1021/acschembio.6b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsuledesai CC, Ochocki JD, Markowski TW, Distefano MD. A combination of metabolic labeling and 2D-DIGE analysis in response to a farnesyltransferase inhibitor facilitates the discovery of new prenylated proteins. Molecular bioSystems. 2014;10(5):1094–1103. doi: 10.1039/c3mb70593e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashidian M, Dozier JK, Lenevich S, Distefano MD. Selective labeling of polypeptides using protein farnesyltransferase via rapid oxime ligation. Chemical Communications. 2010;46(47):8998–9000. doi: 10.1039/C0CC03305G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashidian M, Kumarapperuma SC, Gabrielse K, Fegan A, Wagner CR, Distefano MD. Simultaneous Dual Protein Labeling Using a Triorthogonal Reagent. Journal of the American Chemical Society. 2013;135(44):16388–16396. doi: 10.1021/ja403813b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashidian M, Song JM, Pricer RE, Distefano MD. Chemoenzymatic Reversible Immobilization and Labeling of Proteins without Prior Purification. Journal of the American Chemical Society. 2012;134(20):8455–8467. doi: 10.1021/ja211308s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MW, Rose ND, Boggs J, Lenevich S, Xu J, Barany G, Distefano MD. Evaluation of geranylazide and farnesylazide diphosphate for incorporation of prenylazides into a CAAX box-containing peptide using protein farnesyltransferase. Journal of Peptide Research. 2005;65(6):529–537. doi: 10.1111/j.1399-3011.2005.00261.x. [DOI] [PubMed] [Google Scholar]
- Saxon E, Bertozzi CR. Cell Surface Engineering by a Modified Staudinger Reaction. Science. 2000;287(5460):2007LP–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- Speers AE, Cravatt BF. Profiling Enzyme Activities In Vivo Using Click Chemistry Methods. Chemistry & Biology. 2004;11(4):535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Stremler KE, Poulter CD. Methane- and difluoromethanediphosphonate analogs of geranyl diphosphate: hydrolysis-inert alternate substrates. Journal of the American Chemical Society. 1987;109(18):5542–5544. doi: 10.1021/ja00252a050. [DOI] [Google Scholar]
- Suazo KF, Schaber C, Palsuledesai CC, Odom John AR, Distefano MD. Global proteomic analysis of prenylated proteins in Plasmodium falciparum using an alkyne-modified isoprenoid analogue. Scientific Reports. 2016;6:38615. doi: 10.1038/srep38615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervacke JS, Funk AL, Wang YC, Strom M, Hrycyna CA, Distefano MD. Diazirine-Containing Photoactivatable Isoprenoid: Synthesis and Application in Studies with Isoprenylcysteine Carboxyl Methyltransferase. The Journal of Organic Chemistry. 2014;79(5):1971–1978. doi: 10.1021/jo402600b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Distefano MD. Synthetic isoprenoid analogues for the study of prenylated proteins: Fluorescent imaging and proteomic applications. Bioorganic Chemistry. 2016;64(Supplement C):59–65. doi: 10.1016/j.bioorg.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang FL, Casey PJ. Protein Prenylation: Molecular Mechanisms and Functional Consequences. Annual Review of Biochemistry. 1996;65(1):241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Blanden MJ, Sudheer C, Gangopadhyay SA, Rashidian M, Hougland JL, Distefano MD. Simultaneous Site-Specific Dual Protein Labeling Using Protein Prenyltransferases. Bioconjugate Chemistry. 2015;26(12):2542–2553. doi: 10.1021/acs.bioconjchem.5b00553. [DOI] [PMC free article] [PubMed] [Google Scholar]