

Abstract
Numerous microRNAs and their target mRNAs are co-expressed across diverse cell types. However, it is unknown whether they are regulated in a cellular context-independent or -dependent manner. Here, we explored transcriptome-wide targeting and gene regulation by miR-155, whose activation-induced expression plays important roles in innate and adaptive immunity. Through mapping of miR-155 targets using differential iCLIP, mRNA quantification with RNA-Seq, and 3′UTR usage analysis using polyadenylation (polyA)-Seq in activated miR-155-sufficient and -deficient macrophages, dendritic cells, T and B lymphocytes, we identified numerous targets differentially bound by miR-155. While alternative cleavage and polyadenylation (ApA) contributed to differential miR-155 binding to some transcripts, in a majority of cases identical 3′UTR isoforms were differentially regulated across cell types, suggesting ApA-independent and cellular context-dependent miR-155-mediated gene regulation. Our study provides comprehensive maps of miR-155 regulatory networks and offers a valuable resource for dissecting context-dependent and -independent miRNA-mediated gene regulation in key immune cell types.
Introduction
MicroRNA (miRNA) mediated post-transcriptional regulation of gene expression plays an important role in the immune system1, 2. miRNAs, 20–24 nucleotide in length, direct RNA-induced silencing complex (RISC) to the 3′ untranslated region (3′UTR) of their targets to facilitate degradation and inhibit translation of target mRNAs3, 4. Argonaute (Ago) proteins serve as key components of the RISC complex essential for miRNA targeting and post-transcriptional repression5. The complementarity of mRNA binding sites in the 3′UTR to the position 2–7 (6-mer) seed at the 5′ end of miRNAs can be sufficient for repression, with efficiency increased by additional matches and by relative position within the 3′UTR3. In addition to the canonical binding sites with a perfect 6–8-mer seed match, widespread non-canonical Ago binding sites have been reported. The latter are subject to overall weaker regulation in comparison to mRNA targets harboring canonical sites6, 7. Genome-wide analyses of miRNA targeting using UV cross-linking-enabled immunoprecipitation of Ago-RNA complexes (CLIP) followed by high-throughput sequencing enabled unequivocal identification of miRNA target sites, both in 3′UTRs and in coding regions, although the latter confer minimal regulation6, 8, 9, 10. These biochemical studies revealed that a single miRNA regulates numerous transcripts, which often belong to particular gene regulatory pathways8, 11.
It must be noted that cell type-specific regulation of gene expression, frequently mediated by commonly expressed sequence-specific transcription factors, is the foundational principle in developmental biology. Like transcriptional regulators, miRNAs with a role in cellular function and their mRNA targets can be found in multiple cell types. In the immune system, a prime example of such a miRNA is miR-155, whose expression is observed in functionally distinct T cell subsets, B cells, NK cells, macrophages, and dendritic cells, where it is induced in an activation or a differentiation stage-specific manner12, 13. miR-155 is also highly expressed in myeloid and lymphoid malignancies, where it plays an oncogenic role14, 15. Our recent study showed that miR-155 mediated regulation of an inducible target gene, Socs1, has widely differing cell type- and biological context-dependent functional significance in distinct types of lymphocytes16, suggesting a potential context-dependent regulation of gene expression by miR-155.
However, recent analyses of immortalized human cell lines of different tissue origin showed that the majority of computationally predicted target mRNAs of miR-155 and miR-124 are repressed in a cellular context-independent manner; differential regulation of a minor subset of miRNA targets observed in these cells was attributed to alternative 3′UTR isoform usage17. While these experiments relied on overexpression of ectopic miRNAs, gene array and 3′UTR-seq analyses of mRNA expression in six different organs from miR-22-deficient and -sufficient mice were consistent with these results17. It can be argued, however, that differential regulation of mRNA targets by an endogenous miRNA is more likely to be encountered in differentiated cell types of common developmental origin in response to a challenge or a developmental cue. Indeed, both endogenous cellular miRNAs and miRNAs encoded by Kaposi’s sarcoma-associated herpes virus were found to regulate the expression of a sizable fraction of targets in a context-dependent manner18. However, the contribution of alternative 3′UTR isoform usage to miRNA-mediated regulation of gene expression was not considered in this study18. Thus, it remains unknown whether endogenously expressed miRNA are capable of regulation of commonly expressed target genes in a cell context-dependent manner.
We sought to address this question through computational and comparative genome-wide analyses of miR-155 binding, 3′UTR usage, and miR-155-dependent repression in four key immune cell types- activated macrophages, dendritic cells, B cells, and CD4+ T cells- isolated from miR-155-sufficient and miR-155-deficient mice. Our analyses revealed cellular context-dependent miR-155 targeting and regulation of gene expression. While ApA contributed to differential miR-155 binding for some transcripts, identical 3′UTR isoforms were also differentially regulated across cell types. These results suggest ApA-independent and cellular context-dependent miR-155-mediated post-transcriptional regulation of gene expression reminiscent of transcriptional regulation by sequence-specific transcription factors.
Results
iCLIP analysis of miR-155 targets
To comprehensively characterize the miR-155 regulatory network, we used individual-nucleotide resolution CLIP (iCLIP19) to precisely map the miR-155 target sites, RNA-Seq to measure the repression efficiency, and PolyA-Seq20 to map and quantify 3′UTR isoforms in B cells, dendritic cells, macrophages and CD4+ T cells extracted from both wild-type and miR-155-deficient mice (Fig. 1a). As previously reported21, 22, 23, 24, miR-155 expression was significantly increased upon immune activation in all four cell types, with peak induction expression observed at 24 h and extending to 48 h (Supplementary Fig. 1a). We used Argonaute 2 antibody to immunoprecipitate RISC-bound RNA from cells activated for 48 h (Supplementary Fig. 1b) and generated iCLIP libraries from the isolated RNA that captured both microRNAs and their mRNA target sequences. Cellular abundances of mature microRNAs were estimated from reads aligned to the corresponding loci in primary microRNA sequences, which confirmed that miR-155 was the only major microRNA with significant change in expression between wild-type and miR-155-deficient cells (Supplementary Fig. 1c and Supplementary Table 1). By applying our CLIPanalyze CLIP processing pipeline to the genomic alignments after removal of potential PCR duplicates, we first identified peak regions in the combined read coverage track (wild-type and miR-155-deficient cell replicates) from all cell types and counted the number of reads within peaks from each iCLIP library. Peaks within RefSeq transcripts constitute ~10–40% of all uniquely mapped iCLIP reads (Supplementary Table 2), and the read counts are generally reproducible between biological replicates of the same cell type and genotype (Pearson correlation coefficient ~0.7–0.9) (Supplementary Fig. 1d). We then modeled the read counts within peaks using negative binomial generalized linear models25 with Trimmed Mean of M-values (TMM) normalization26. We determined the miR-155 dependent sites as peaks within RefSeq transcripts; containing sequence complementary to the miR-155 6-mer seed (nucleotide 2–7); and significantly higher read counts in wild-type samples than miR-155-deficient samples (Benjamini-Hochberg adjusted P < 0.025). In total, 1,200 such sites were found in 999 genes across four cell types, including 796 (66.3%) in 3′UTRs, 386 (32.2%) in CDS (coding sequence), and 18 (1.5%) in 5′UTRs (Supplementary Fig. 1e). In particular, ~20–75% of miR-155 target sites were found to be cell-type specific in pairwise comparisons (Supplementary Table 3), suggesting a prominent cellular context-dependent regulation by miR-155.
Figure 1.

miR-155 mediated Argonaute binding occurs at distinct sites in four immune cell types. (a) Examples of universally bound and differentially bound miR-155 sites across 4 cell types. Normalized read coverage of iCLIP, RNA-Seq and PolyA-Seq libraries are shown with dark colors for wild-type (WT) and light colors for miR-155 knockout (KO) tracks. miR-155 seed-containing iCLIP peaks are highlighted with grey rectangles with asterisks designating significant (FDR < 2.5%) difference between WT and KO coverage. (b) Summary of miR-155 dependent sites in co-expressed genes, including 3′UTR, CDS, and 5′UTR sites, identified by differential iCLIP. Each row represents 250 bp around a miR-155 6mer seed match with colors that demonstrate the log2 ratios of normalized WT to miR-155 KO iCLIP coverage. Heatmap of RNA expression (WT RNA-Seq log10 FPKM, normalized by row) of the same genes containing the miR-155 sites is shown side-by-side. Sites are categorized according to their binding specificity across 4 cell types, while the order within each category are determined by hierarchical clustering of RNA-Seq FPKM values for corresponding genes. (c) Venn diagram of miR-155 dependent iCLIP sites in co-expressed genes. (d) Seed-type composition of miR-155 dependent sites in co-expressed genes. (e) PhastCons scores (for multiple genome alignments between mouse and other 39 placental mammals) of miR-155 dependent sites in co-expressed genes. Analyses of data from independent iCLIP (n = 4), RNA-Seq (n = 3), and PolyA-Seq (n = 4) experiments are shown.
Target and miR-155 levels do not fully account for selective regulation
One obvious explanation for the observed context specificity is that some of the cell-type specific miR-155 target genes may not be expressed or are expressed at very low abundance in the other cell types. Indeed, when a gene contains a miR-155 target specific to one cell type, its mRNA expression in that cell type also tended to be higher than in those where the target did not show differential iCLIP signal (Supplementary Fig. 2a). When we restricted the comparison between cell types to co-expressed genes (RNA-Seq FPKM > 1 in all cell types and < 16-fold difference between any two cell types), 931 target sites in 778 co-expressed genes remained and most of the context specificity was preserved (Fig. 1b,c). Therefore, the cell-type specific expression of mRNAs alone does not account for the observed cell context-specific expression.
The difference in miR-155 abundance after immune stimulation across cell types (Supplementary Fig. 1a) can also partially explain the cell context specificity of miR-155 targeting – the largest number of cell-type specific target sites were found in dendritic cells, where miR-155 expression was also the highest (Fig. 1b). The number of miR-155 dependent sites identified in each cell type is consistent with relative miR-155 expression (Fig. 1c), suggesting that some context-specific sites may have weaker affinity for miR-155 and, therefore, can only be regulated in the presence of higher miR-155 amounts or other cellular factors. Indeed, when we categorized the miR-155 targets by the number of cell types in which they are present, the proportion of sites with only 6-mer complementarity was significantly lower for target sites present in more cell types than those present in fewer cell types (Fisher’s exact test P < 2.57e-10), and the proportion of sites with 8-mer complementarity significantly higher (Fisher’s exact test P < 1.79e-9; Fig. 1d). Similar to previous observations18, the miR-155 seed match sequences of sites present in three or four cell types also showed significantly higher evolutionary conservation than the more cell-type specific sites present in one or two cell types (Fig. 1e, P < 1.92e-6, one-sided KS test). Nevertheless, many context-specific targets are still present in cell types with lower miR-155 expression, suggesting that other cellular factors or potentially alternative cleavage and polyadenylation (ApA) may play a role in cell-type specific targeting.
miR-155 mediated regulation is context-specific
Next, we analyzed the extent of regulation induced by miR-155 dependent targets identified by differential iCLIP. Although miRNAs regulate gene expression through degradation of mRNA targets and inhibition of translation, it has been shown that dominant effect of mammalian miRNAs is at the level of mRNA degradation27. Therefore, we used mRNA expression changes between wild-type and miR-155-deficient cells to estimate the extent of miR-155 regulation per gene (Supplementary Table 4). Consistent with previous studies28, the significance of miR-155 dependent iCLIP sites in 3′UTRs correlated with the extent of regulation of corresponding genes, which was not the case for CDS sites (Supplementary Fig. 2b). Therefore, for further analyses of the effect of miR-155 on gene regulation we only considered miR-155 targets in 3′UTRs.
In all four immune cell types, we first examined the distribution of mRNA expression changes of potential target genes defined by miR-155 seed matches in the 3′UTRs. Consistent with well-known miRNA targeting characteristics, the extent of miR-155 repression increased with higher 3′UTR seed complementarity, from 6-mer to 7-mer-A1/m8 to 8-mer29. Still, genes with miR-155 dependent iCLIP sites in the 3′UTRs displayed stronger repression than predicted targets with 3′UTR 8-mer seed matches (Fig. 2). We also compared the iCLIP-defined target genes to same number of genes containing sites with top context++ scores from TargetScan 7.030. While the extent of regulation in the top ~10% of the distribution was similar for both sets of genes, the iCLIP-defined target genes overall showed significantly stronger regulation compared to TargetScan predictions (Fig. 2). Thus, miR-155 mediated gene regulation across different cellular contexts is more accurately captured by differential iCLIP assays than cell-type agnostic sequence-based predictions.
Figure 2.
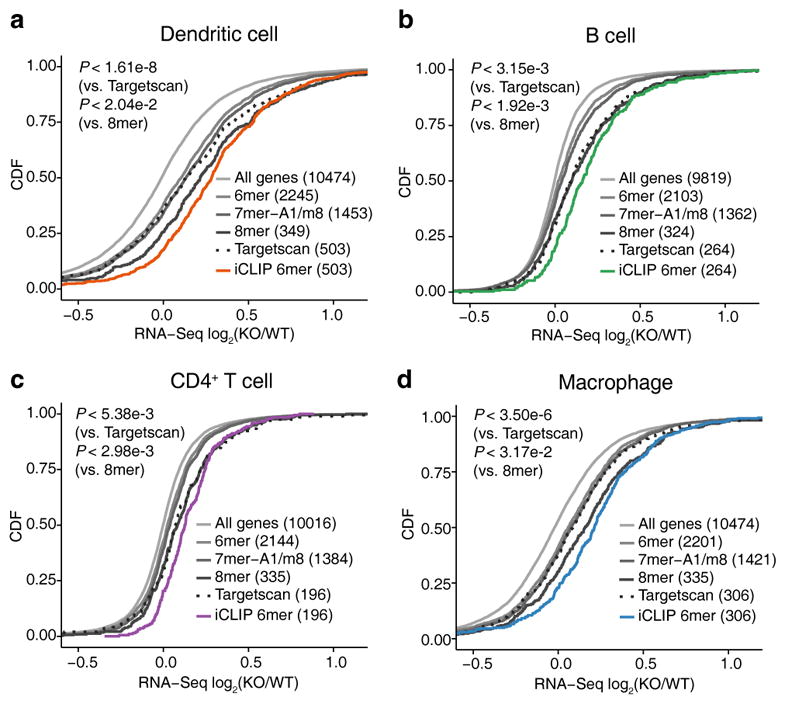
miR-155 represses distinct sets of genes in four immune cell types. In dendritic cells (a), B cells (b), CD4+ T cells (c) and macrophages (d), the distribution of RNA-Seq expression changes between miR-155 KO and WT cells is shown with the cumulative distribution functions (CDFs) for different gene sets. Gene sets include all expressed genes, genes with 3′UTR miR-155 6mer / 7mer-A1 / 7mer-m8 / 8mer seed matches and genes containing 3′UTR miR-155 dependent iCLIP sites with 6mer seed matches (FDR < 2.5%). Predicted miR-155 target genes with top context++ scores from Targetscan 7.0 (same number as the miR-155 target genes identified by differential iCLIP) are also shown. The data represent independent iCLIP (n =4) and RNA-Seq (n = 3) experiments.
In line with previous reports6, 31, 32, 33, 34, we found that 25–45% of identified Ago-bound miR-155 sites were non-canonical (Supplementary Fig. 3a). The majority of non-canonical sites were bound in only one cell type (Supplementary Fig. 3b), consistent with the previous observation that cell-type specific targets tend to have weaker affinity for miR-155 (Fig. 1d). Similarly, when we compared the average iCLIP read coverage around the canonical and non-canonical miR-155 sites, we found that the difference between wild-type and miR-155-deficient libraries was significantly smaller for non-canonical sites (Supplementary Fig. 3c). We found multiple genes significantly repressed by miR-155 with only non-canonical target sites in the 3′UTR (Supplementary Fig. 3d), although the regulation of non-canonical targets was significantly weaker than canonical targets (Supplementary Fig. 3e).
To further dissect the cell-context specificity of miR-155 regulation, we performed pairwise comparisons across the four immune cell types to assess the extent of regulation of common and cell-type specific miR-155 targets (Fig. 3). In each immune cell type, miR-155 target genes identified by differential iCLIP displayed stronger repression than those specific to other cell types, with a few exceptions involving B cells and CD4+ T cells, where fewer cell-type specific targets and weaker regulation were observed. Overall, cell-type specific target genes displayed less pronounced regulation compared to common target genes, consistent with the weaker seed complementarity and lower sequence conservation associated with cell-type specific target sites (Fig. 1d,e).
Figure 3.

Context-specific miR-155 targeting leads to differences in gene regulation between cell types. For all six pairwise comparisons across four immune cells, de-repression of genes containing common (solid lines) and cell-type specific (dotted lines) 3′UTR miR-155 dependent iCLIP sites is shown in the form of CDFs. Genes with 3′UTR miR-155 seed matches are also shown as reference. Only co-expressed genes (WT RNA-Seq FPKM > 1 and difference < 16 fold) are included in each pairwise comparison. In each plot, two P-values from one-sided KS tests are shown. First KS test corresponds to the comparison between all miR-155 target genes identified in this cell type and genes only targeted in the other cell, while the second one corresponds to the comparison between the common target genes and target genes specific to this cell type. Results of four independent iCLIP and three independent RNA-Seq experiments are shown.
To validate cell type-dependent miR-155 gene repression, we performed luciferase reporter assays for several cell context-dependent gene targets, including Actr10 and Terf1, identified by iCLIP as B cell-specific targets, and Hif1a and Jarid2, which were bound by miR-155 in both B cells and dendritic cells, but preferentially repressed in B cells. Enhanced repression of Actr10, Hif1a, Jarid2, and Terf1 was observed in B cells compared to dendritic cells (Fig. 4). Although Terf1 was identified by differential iCLIP as a B cell-specific target, significant Terf1 repression was also detected in dendritic cells, which might be the result of overexpression of exogenous Terf1 in the luciferase assay. Consistently, dendritic cell-specific miR-155 targets Tbca, Uqcrfs1, and Zfp277 exhibited higher repression by miR-155 in dendritic cells than B cells (Fig. 4), even though miR-155 regulation of Uqcrfs1 and Zfp277 reporters was also evident in B cells, possibly again due to limitations of the reporter assay. Overall, these results are consistent with our genomic analyses supporting cell type-specific regulation by miR-155.
Figure 4.

Verification of cell type-dependent miR-155 mediated repression. Reporters carrying 3′UTR of genes displaying context-specific targeting were expressed in B cells and dendritic cells. Results are shown for Hif1a and Jarid2 (preferentially repressed in B cells), Actr10 and Terf1 (B cell-specific targets), and Tbca, Uqcrfs1, and Zfp277 (dendritic cell-specific targets). Fold repression was determined from ratio of normalized luciferase activities of mutant and wild-type 3′UTR reporters. Error bar displays standard error from at least three biologically independent samples; P-value was measured by two-sided t-test. *, P < 0.05; **, P < 0.01.
Many functionally important miR-155 cellular targets, including Aicda in B cells, Spi1 in B cells, dendritic cells and macrophages, Inpp5d in both B cells and macrophages, and Tab2 in dendritic cells, have been previously reported (Supplementary Table 5). In our study, most previously identified miR-155 target genes were revealed as repressed miR-155-bound targets by differential iCLIP and RNA-Seq. Very few exceptions most likely were due to the use of particular cell lines instead of primary cells in the related previous studies (Supplementary Table 5). We further confirmed miR-155 mediated regulation by demonstrating reduced protein expression of Aicda, Inpp5d, Spi1, and Tab2 in respective cell types (Supplementary Fig. 4). Furthermore, B cell-specific repression of Jarid2 and Hif1a, and dendritic cell-specific repression of Uqcrfs1 were also observed at the protein level (Supplementary Fig. 4). These results provide additional support for our genomic data analyses of miR-155-mediated regulation of gene expression.
Endogenous RNA competition is unlikely to influence miR-155 targeting
The “competitive endogenous RNA (ceRNA)” hypothesis35 proposes that transcripts with common miRNA target sites compete with each other for regulation, which may explain the biological function of some long non-coding RNAs. There has been growing experimental evidence that certain long non-coding RNAs36 and circular RNAs37, 38 can contain large numbers of miRNA target sites (i.e. >70 miR-7 sites in mammalian circular RNA Cdr1as38) and may function as miRNA “sponges”, particularly in neurons. Thus, it was possible that differentially expressed miR-155 sponges could contribute to cell context-dependent miR-155 mediated regulation of weaker targets. However, when we examined miR-155 target sites in mRNAs along with ones within intronic regions and annotated non-coding RNAs (Supplementary Table 6), we found the vast majority of coding and non-coding RNAs only contained one or two miR-155 target sites in any of the four cell types (Supplementary Fig. 5a), with a maximum of six sites found in a single gene, Picalm. As circular RNAs are generally formed by back-splicing of consecutive exons39, we did not identify candidates that appear likely to act as circular RNAs “sponges” for miR-155 in these four immune cell types.
We also attempted to estimate the fraction of miR-155-Ago complex bound by a given transcript in each cell. Assuming that iCLIP counts are a reasonable proxy for miR-155-Ago binding, we estimate that the most-bound transcript in a given cell binds ~3–10% of the transcript bound complex (Supplementary Fig. 5b). This estimate suggests that these rare already highly expressed transcripts would need to be substantially up-regulated to significantly affect overall miR-155 binding within the cell. Interestingly, the most-bound targets are different for each cell type, even between these closely related immune cells. Among the predominant target miR-155 genes in dendritic cells was Cd274, encoding the inhibitory receptor ligand PD-L1, and in macrophages Msr1, encoding macrophage scavenger receptor 1 (Supplementary Fig. 5b). Thus, our analysis reveals no evidence of a competing RNA acting as a miR-155 regulator.
Limited ApA contribution to cell-type specific miR-155 targeting
Another potential explanation for the observed cell type-dependent regulation of gene expression by miR-155 is alternative polyadenylation. Previous studies40, 41 have shown that genes with multiple 3′UTRs increase the usage of shorter isoforms in activated immune cells, specifically T lymphocytes, simultaneously with the increase in miR-155 expression, which has been proposed as a potential mechanism to evade miRNA-mediated regulation. We performed PolyA-Seq in naïve CD4+ T cells as well as their activated counterparts after in vitro stimulation with CD3 and CD28 antibodies for 24 h and 48 h (Fig. 5a and Supplementary Table 7). Although differential analysis42 indeed revealed widespread changes in 3′UTR isoform usage with a significant shift towards shorter isoforms in activated cells both at 24 h (paired Wilcoxon test P < 2.87e-8, Supplementary Fig. 6a) and 48 h (paired Wilcoxon test P < 6.15e-3, Fig. 5b), markedly increased usage of longer isoforms in activated cells was also observed for a sizable group of transcripts (~40% of those with differential usage). A focused analysis of the two-isoform 3′UTRs targeted by miR-155 did not suggest selective shortening of the mRNAs that contained a miR-155 binding site in the long isoform (Fig. 5c). Changes in 3′UTR length thus did not appear to significantly relieve miR-155 mediated targeting upon T cell activation.
Figure 5.

PolyA-Seq captures change in 3′UTR isoform usage during CD4+ T cell activation. (a) Two examples of 3′UTRs with significant (FDR < 5%) changes in isoform usage during CD4 T cells activation. Tracks represent normalized PolyA-Seq read coverage at 0 h, 24 h and 48 h after activation. (b) The changes in 3′UTR isoform usage for 3′UTRs with two major isoforms at 48 h after CD4+ T cell activation. Highlighted genes displayed significant (FDR < 5%) changes in 3′UTR usage. (c) Same as (b), but highlighting the two-isoform 3′UTRs containing target sites of miR-155. 3′UTRs containing proximal (solid shapes) and distal (hollow shapes) miR-155 target sites are highlighted. The results of three independent PolyA-Seq experiments are shown.
To investigate whether alternative polyadenylation contributed to cell-type specific targeting we performed PolyA-Seq in all four immune cell types. The PolyA-Seq FPM was well correlated with RNA-Seq FPKM for single-UTR genes (Supplementary Fig. 6b), suggesting that PolyA-Seq is capable of quantifying 3′UTR isoform expression. Differential analysis42 in all four cell types showed that 2,703 out of 3,460 co-expressed multi-UTR genes displayed some extent of alternative polyadenylation (Fig. 6a and Supplementary Table 8). miR-155 targets were significantly enriched in differentially used multi-UTR genes compared to the other genes (Fisher’s exact test P < 2.2e-16, Fig. 6b). Since PolyA-Seq libraries were generated for both wild-type and miR-155-deficient cells, the data also allowed us to assess miR-155 regulation at the level of 3′UTR isoforms. In agreement with previous observations29, regulation of a 3′UTR isoform by a given miR-155 target site negatively correlated with its distance from the 3′UTR end (Supplementary Fig. 6c,d), suggesting the potential of ApA as a mechanism for context-specific miR-155 regulation. Indeed, in multi-isoform 3′UTRs, we observed that the extent of gene-level miR-155 regulation generally increases with higher usage of ApA isoforms containing miR-155 target sites in individual cell types (Supplementary Fig. 7a), as reported by previous studies17. We also observed examples of co-occurrence of ApA and context-specific miR-155 binding through pairwise comparison between cell types (Fig. 6c). However, in most cases, the change in isoform usage between cell types was less than 10%, while overall expression changes of miR-155 and target mRNAs had a much larger dynamic range. Therefore, the majority of the observed context-specific targeting cannot be attributed to alternative polyadenylation (Fig. 6d and Supplementary Fig. 7b).
Figure 6.

The role of alternative polyadenylation in cellular context dependent regulation of gene expression by miR-155. (a) A heatmap showing the usage changes in multi-isoform 3′UTRs across all four cell-types. The usage index (UI) represents the percentage of the shorter isoform usage for two-isoform 3′UTRs, while for 3′UTRs with more isoforms it represents the usage of the short isoform with the most significant usage change. “Elongation” corresponds to the genes with significantly higher usage of longer isoforms in one cell type compared to the rest, whereas “shortening” corresponds to the genes with significantly higher usage of shorter isoforms in one cell type compared to the rest. (b) Number of 3′UTRs containing miR-155 targets and displaying cell type specific ApA differences. (c) iCLIP, RNA-Seq and PolyA-Seq read coverage tracks in dendritic cell and CD4+ T cell for Rbm33. (d) Venn diagram showing the number of shared and cell-specific miR-155 target genes in dendritic cell and B cell, before and after removing genes with cell type specific ApA differences. The data are representative of four independent PolyA-Seq experiments.
Functional target sites of other miRNAs
Our Ago iCLIP data also allowed characterization of target sites for other miRNAs expressed in the four immune cells. The latter relied on computational seed sequence analysis within iCLIP peaks in the absence of a genetic control, i.e. iCLIP and RNA-Seq analysis of corresponding miRNA-deficient cells. When we ranked iCLIP peaks containing miR-155 6-mer seed matches by the normalized read counts in wild-type libraries, ~75–95% of the top 10% of peaks overlapped with miR-155 dependent sites defined by differential iCLIP (Supplementary Fig. 8). We therefore reasoned that stringent read count cutoffs could yield reliable sets of targets for miRNAs other than miR-155. Using the wild-type libraries, we defined the top target sites for miR-142a-3p and miR-27a-3p, which both play key regulatory roles in immune cells43, 44, 45 and were highly expressed in the four immune cells (Supplementary Table 1). When we used publically available gene expression data with perturbed miR-142a43, 44 and miR-27a45 expression in mouse immune cells, we found that similar to miR-155, the target genes defined by 3′UTR iCLIP sites with top read counts in wild-type libraries showed significantly stronger repression than cell-type agnostic sequence-based predictions (Fig. 7a–c, one-sided KS test), which suggests that they indeed defined an accurate set of top miRNA targets in the respective cellular context. Thus, our Ago iCLIP datasets are useful in characterizing targets of other miRNAs.
Figure 7.

Top iCLIP target sites of other miRNAs induce significant gene repression. mRNA expression changes in B cells (a) and dendritic cells (b) with miR-142a KO and in CD4+ T cells with miR-27a overexpression (c) are shown as CDFs for different gene sets. Gene sets consist of all expressed genes, genes with 3′UTR seed matches (6mer, 7mer-A1, 7mer-m8, and 8mer), and genes containing 3′UTR iCLIP sites with 6mer seed matches and most reads in wild-type libraries. Predicted miRNA target genes with top context++ scores from TargetScan 7.0 (same number as the target genes defined by wild-type iCLIP) are also shown. Analyses were shown from independent iCLIP samples (n = 4), miR-142 array data in B cell (GSE61919, n = 3) and dendritic cell (GSE42325, n = 2), and miR-27a RNA-Seq experiments (GSE75909, n = 3).
Discussion
Differential cell type-specific regulation of gene expression enables nuanced responses of cells with distinct function to diverse intrinsic and extrinsic cues. It is well appreciated that DNA-binding transcription factors, whose expression is shared between multiple cell types, can control transcriptional responses in a cell type-specific manner46. Our studies revealed cell-context dependent miRNA-mediated regulation of gene expression in four developmentally related immune cell types. Through differential iCLIP analyses, we identified hundreds of miR-155 cellular target mRNAs, the majority of which were expressed in all cell types yet bound by miR-155 in a cell-type specific manner. It is noteworthy that genes with 3′UTR sites identified by differential iCLIP displayed stronger regulation at the mRNA level in each cell type than genes identified through cell-type agnostic computational predictions. These results suggest a notable role for cellular context in determining functional miRNA targets.
Our comparative analysis of miR-155-bound sites in macrophages, dendritic cells, CD4+ T and B cells showed that miR-155 sites that were shared across all four cell types were likely to exhibit more extensive seed complementarity, while sites that were restricted to fewer cell types tended to have weaker seed matches. In line with our previous observation6, we found a sizeable fraction of non-canonical miR-155 sites, i.e. sites lacking a 6-mer seed match, and these were more likely to be cell-type restricted. Further analysis showed that several mRNAs were significantly repressed in the presence of non-canonical sites in the 3′UTR, which is in contrast to the notion that non-canonical miRNA binding sites are ineffective30. Considering the variation in the target sites’ affinity for miR-155, it is reasonable to suggest that differences in inducible miR-155 expression between cell types likely contribute to context specificity of miR-155 regulation. Indeed, we found that immune cells with the highest miR-155 expression also had the largest number of binding sites. Therefore, a number of the observed cell-type specific miR-155-mediated targeting and regulation of gene expression occurred at weaker binding sites, detectable only with higher miRNA expression. Nevertheless, some of the top-ranked miR-155 targets based on iCLIP read coverage were also cell-type dependent. Moreover, immune cell types with relatively low miR-155 abundance and overall fewer miR-155 binding sites also displayed cell-type specific regulation, pointing to additional more complex context-specific mechanisms of miRNA-mediated regulation.
A majority of mammalian mRNAs generate isoforms with different 3′UTR lengths as the result of ApA20. A previous study raised the possibility that ApA could play an important role in the cell-type specific miRNA binding to their targets, and shortening of 3′UTRs through ApA induced upon CD4+ T cell activation was suggested to result in a loss of miRNA binding sites and target derepression40. However, our analysis of polyA site usage in activated CD4+ T cells showed that although the 3′UTRs of mRNAs did exhibit a tendency to shortening in proliferating cells, many genes exhibited increased usage of longer isoforms containing miR-155 binding sites, thereby acquiring miR-155-mediated regulation. Overall, we observed modest cell type-specific differences in 3′UTR usage, which could not explain the majority of cell-type specific miRNA binding. Thus, it appears that many miRNA targets are regulated in a cell-context dependent but ApA-independent manner.
Given the modest impact of ApA on cell-type dependent miR-155-mediated gene regulation, it seems likely that additional factors contribute to cell-context dependent repression by miR-155. Potential factors and mechanisms may include, but are not limited to, differentially expressed RNA-binding proteins (RBPs) and the effects of mRNA modifications on the binding and function of miRNA-bound RISC complexes. In this regard, several RNA binding proteins have been shown to substantially influence the outcome of miRNA-mediated regulation of gene expression. For instance, HuR (AU-rich element binding protein) relieved miR-122 repression of Slc7a1 mRNA47; and Pumilio proteins enhanced E2f3 repression by miR-125b and miR-50348. Moreover, adenosine methylation on mRNAs (N6-methyladenosine, m6A) appears to favor last exons, significantly overlapping with Argonaute binding sites49, suggesting that m6A modifications might influence miRNA function. These findings imply that local context could confer the cell context-dependent miRNA binding and function. Thus, comprehensive unbiased studies are needed for further mechanistic understanding of cell-type specific miRNA binding.
In conclusion, our study demonstrates cellular context-dependent regulation of gene expression by miR-155 in macrophages, dendritic cells, T cells and B cells and provides a complete map of miR-155 regulatory networks in these key immune cell types. Furthermore, genome-wide analyses of RISC binding in these cells enables comprehensive identification of targets of other miRNAs and, therefore, provides a valuable resource for dissecting miRNA-mediated gene regulation in immune systems.
Online Methods
Animals
Mice (C57BL/6J (B6) and miR-155 KO of B6 background) were purchased from The Jackson Laboratory, and usage of mice followed the guidelines of Animal Care Committee at Memorial Sloan Kettering Cancer Center.
Cell purification and culture
Primary dendritic cells, B cells, CD4+ T cells, and macrophages from C57BL/6J wild-type and miR-155 KO mice were cultured in RPMI medium with 10% FBS. Prior to harvesting primary dendritic cells, mice were subcutaneously injected with 1 × 106 B16 melanoma cells expressing Flt3 ligand for about two weeks. After purification of splenic CD11c+ dendritic cells by CD11c microbeads (Miltenyi Biotec), dendritic cells were activated in a medium containing 100 ng/ml LPS (SIGMA) and 20 ng/ml GM-CSF (Tonbo). Splenic primary B cells were purified by negative selection using Dynabeads Mouse CD43 (Invitrogen), and activated in a medium containing 25 μg/ml LPS and 6.5 ng/ml mIL-4 (PeproTech). CD4+ T cells from lymph node and spleen were purified with Dynabeads FlowComp Kit (Invitrogen). CD4+CD25−CD44− T cells were then activated with Dynabeads Mouse T-Activator CD3/CD28 (Invitrogen). Intraperitoneal macrophages, induced by thioglycollate injection, were harvested and activated with 100 ng/ml LPS.
iCLIP
Individual-nucleotide resolution UV-crosslinking and immuoprecipitation (iCLIP) was performed as described with minor modifications50. In brief, primary cells, activated for 48 h, were irradiated with UV with 400 mJ, followed by 200 mJ, and then an additional 200 mJ. Cell lysate was incubated at 37 °C for 5 min in the presence of 10 U RNase I (Ambion), and immunoprecipitated at 4 °C for 2 h using Argonaute 2 antibody as described6. The amount of RNase I in the reaction was determined as described50. Immuoprecipitated RNAs were first dephosphorylated at 3′ ends by PNK and then ligated to 3′ adapters (L32) using T4 RNA ligase 2, truncated KQ (NEB). The Argonaute-RNA complexes were resolved on SDS-PAGE gels, and complexes with a molecular weight corresponding to ~110–160 kDa were purified. Isolated RNAs (~40 to 200 nucleotides in length) were reverse-transcribed using barcoded primers. Purified cDNA libraries were circularized and re-linearized, and PCR amplified iCLIP libraries were submitted for high-throughput sequencing. Sequences of all primers are listed in Supplementary Table 9. For complete iCLIP protocol see Supplementary Methods.
iCLIP sequencing libraries were first de-multiplexed and then preprocessed using the cutadapt51 software to remove the adaptor and low-quality bases. The remaining reads were aligned to the mouse genome (mm9) using the BWA aligner52. Multiple reads aligned to identical coordinates with the same random 7-mer in the barcode were considered PCR duplicates and were merged into a single read. We then ran our peak-calling algorithm CLIPanalyze (https://bitbucket.org/leslielab/clipanalyze, manuscript in preparation) on the combined read coverage from all samples. The algorithm identified peaks by convolving the read coverage signal with the second derivative of a Gaussian filter. The locations where the convolved signal crosses zero correspond to the rising and falling edges in the original signal and these are used as boundaries for the peaks. Each peak was annotated with the corresponding gene name and its location within the gene (i.e. intron, CDS, 5′UTR, 3′UTR). Peaks within intergenic regions further than 5 kb downstream and 1 kb upstream from annotated genes were excluded from subsequent analysis. The peaks were quantified by counting the number of uniquely aligned reads mapped within peak boundaries in each library. To focus on high confidence peaks, peaks without supporting reads in at least 4 out of 8 samples in at least one cell type were removed. For each individual cell type, we did a second round of filtering and only kept the peaks with total read counts within the top 10 percentiles for the differential analysis. We then used negative binomial generalized linear models25 with TMM normalization26 to fit the read counts in each peak, and the significance of the difference in read counts between wild-type and miR-155 KO samples was evaluated using the likelihood ratio test.
Gene expression
RNA from cells activated for 48 h was used for RNA-Seq library preparation. PolyA-Seq libraries were generated as described20. We preprocessed the paired-end reads using cutadapt to remove the adaptors and low-quality bases. The processed reads were then aligned to the mouse genome (mm9) using the STAR aligner53. To account for the variation in 3′UTR usage, we only counted the reads aligned to CDS for coding genes. The read counts per gene were further normalized as fragments per kilobase per million (FPKM) to represent the mRNA abundance.
Differential gene expression analysis was performed for microarray datasets of miR-142a-sufficient and -deficient B cells (GSE61919)43 and bone marrow derived miR-142a-sufficient and -deficient dendritic cells (GSE42325)44 using limma54. We only used probes whose average intensities were above median. When there are still multiple probes mapped to the same gene, we used the median log2 fold change to represent the gene-level regulation. To estimate gene regulation mediated by miR-27a, differential gene expression analysis was performed on a RNA-Seq dataset of wild-type and miR-27a-overexpressed CD4+ T cells (GSE75909)45 in the same way as we analyzed our RNA-Seq data.
PolyA-Seq
Briefly, polyadenylated RNA from cells activated for 48 h (0, 24, and 48 h for CD4+ T cells) was selected by using Sera-Mag oligo-(dT)14 (GE Healthcare), and reverse-transcribed to cDNA using RT primer (RT_1). The complementary (2nd) strand of cDNA was synthesized using the Klenow fragment (exo-, NEB) and extension primer (Ext_primer_1). Barcoded libraries, amplified by PCR from the double-stranded cDNA pool, were submitted for high-throughput sequencing. Nucleic acid sequences of primers are listed in Supplementary Table 9.
The preprocessing, alignment, peak calling and quantification steps for the PolyA-Seq libraries were performed in the same way as the iCLIP libraries. Internally primed peaks were removed as previously described55. Read counts were fitted using the DEXSeq42 model in order to identify cell-specific differences in 3′UTR isoform usage.
Luciferase reporter assay
Primary cells, activated for 24 h, were transfected with psiCheck2 (Promega) carrying 3′UTR of interested gene (without significant ApA usage changes) by using Nucleofector Kits (Lonza) for 8 h. The activity of Renilla luciferase and Firefly luciferase was measured using Dual-Luciferase Reporter Assay System (Promega). Renilla signal was first normalized to Firefly (internal control), and fold repression was calculated from the ratio of normalized Renilla signals of mutant versus wild-type 3′UTR reporter. In mutant reporters, nucleotides 3–6 of miR-155 seed match were mutated to CGTA. Significance was assessed using a two-sided t-test. Reporter constructs contained major 3′UTR isoforms. The nucleic acid sequences of cloning primers and synthesized 3′UTR (Genewiz) are listed in Supplementary Table 10.
RT-qPCR
miR-155 abundance was quantified using Mir-X kit (Clontech). First-strand cDNA synthesis of RNAs was performed as described in the kit manual, and miR-155 transcripts were measured by qPCR and normalized to U6 RNA abundance.
Immunoblotting
The following antibodies were used for immunoblotting: SHIP1 (Inpp5d) (BioLegend, #656601), PU.1 (Spi1) (Cell Signaling, #2258S), Tab2 (LifeSpan, #LS-C498046), Jarid2 (Cell Signaling, #13594T), Uqcrfs1 (LifeSpan, #LS-C185466), HIF-1α (Hif1a) (SIGMA, #ABE279), and β-Actin (Actb) (Sigma, #A3853) antibodies. AID (Aicda) specific antibody is a gift from J. Chaudhuri (MSKCC).
Reporting Summary
More information on experimental design is available in the Life Sciences Reporting Summary.
Data and Code Availability
Accession code of this study is GSE116561. Individual dataset accession is as follows. RNA-Seq dataset (GSE116348); Differential iCLIP dataset (GSE116466); PolyA-Seq dataset (GSE116468). Codes of CLIPanalyze can be downloaded from https://bitbucket.org/leslielab/clipanalyze, and are available upon request.
Supplementary Material
Acknowledgments
We thank A. Chaudhry and other lab members for experimental assistance and discussions. This work was supported by National Institutes of Health grants Al034206 (A.Y.R.), HG007893 (A.Y.R. and C.S.L.), and CA164190 (C.S.L.), P30 CA008748, as well as the Hilton-Ludwig Cancer Prevention Initiative funded by the Conrad N. Hilton Foundation and Ludwig Cancer Research. J.-P. H. was supported by an Irvington Fellowship of the Cancer Research Institute. A.Y.R. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
Author contributions
J.-P.H., G.B.L. and A.Y.R. designed the study. J.-P.H. carried out all experiments. Y.L. and C.S.L. designed analytical tools and performed data analyses. J.-P.H., Y.L., A.Y.R. wrote the manuscript; all the authors edited and approved the final manuscript.
Competing interests
The authors declare no competing interests.
References
- 1.Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009;136:26–36. doi: 10.1016/j.cell.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 2.O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 3.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 2013;14:475–488. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- 5.Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet. 2013;14:447–459. doi: 10.1038/nrg3462. [DOI] [PubMed] [Google Scholar]
- 6.Loeb GB, et al. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol Cell. 2012;48:760–770. doi: 10.1016/j.molcel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chi SW, Hannon GJ, Darnell RB. An alternative mode of microRNA target recognition. Nat Struct Mol Biol. 2012;19:321–327. doi: 10.1038/nsmb.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hafner M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zisoulis DG, et al. Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat Struct Mol Biol. 2010;17:173–179. doi: 10.1038/nsmb.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farazi TA, et al. Identification of distinct miRNA target regulation between breast cancer molecular subtypes using AGO2-PAR-CLIP and patient datasets. Genome Biol. 2014;15:R9. doi: 10.1186/gb-2014-15-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vigorito E, Kohlhaas S, Lu D, Leyland R. miR-155: an ancient regulator of the immune system. Immunol Rev. 2013;253:146–157. doi: 10.1111/imr.12057. [DOI] [PubMed] [Google Scholar]
- 13.Mashima R. Physiological roles of miR-155. Immunology. 2015;145:323–333. doi: 10.1111/imm.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eis PS, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seddiki N, Brezar V, Ruffin N, Levy Y, Swaminathan S. Role of miR-155 in the regulation of lymphocyte immune function and disease. Immunology. 2014;142:32–38. doi: 10.1111/imm.12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu LF, et al. A Single miRNA-mRNA Interaction Affects the Immune Response in a Context- and Cell-Type-Specific Manner. Immunity. 2015;43:52–64. doi: 10.1016/j.immuni.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nam JW, et al. Global analyses of the effect of different cellular contexts on microRNA targeting. Mol Cell. 2014;53:1031–1043. doi: 10.1016/j.molcel.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erhard F, et al. Widespread context dependency of microRNA-mediated regulation. Genome Res. 2014;24:906–919. doi: 10.1101/gr.166702.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Konig J, et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Derti A, et al. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012;22:1173–1183. doi: 10.1101/gr.132563.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haasch D, et al. T cell activation induces a noncoding RNA transcript sensitive to inhibition by immunosuppressant drugs and encoded by the proto-oncogene, BIC. Cell Immunol. 2002;217:78–86. doi: 10.1016/s0008-8749(02)00506-3. [DOI] [PubMed] [Google Scholar]
- 22.van den Berg A, et al. High expression of B-cell receptor inducible gene BIC in all subtypes of Hodgkin lymphoma. Genes Chromosomes Cancer. 2003;37:20–28. doi: 10.1002/gcc.10186. [DOI] [PubMed] [Google Scholar]
- 23.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ceppi M, et al. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci U S A. 2009;106:2735–2740. doi: 10.1073/pnas.0811073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartel DP. Metazoan MicroRNAs. Cell. 2018;173:20–51. doi: 10.1016/j.cell.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hausser J, Syed AP, Bilen B, Zavolan M. Analysis of CDS-located miRNA target sites suggests that they can effectively inhibit translation. Genome Res. 2013;23:604–615. doi: 10.1101/gr.139758.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grimson A, et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4 doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grosswendt S, et al. Unambiguous identification of miRNA:target site interactions by different types of ligation reactions. Mol Cell. 2014;54:1042–1054. doi: 10.1016/j.molcel.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broughton JP, Lovci MT, Huang JL, Yeo GW, Pasquinelli AE. Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol Cell. 2016;64:320–333. doi: 10.1016/j.molcel.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore MJ, et al. miRNA-target chimeras reveal miRNA 3′-end pairing as a major determinant of Argonaute target specificity. Nat Commun. 2015;6:8864. doi: 10.1038/ncomms9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, et al. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev Cell. 2013;25:69–80. doi: 10.1016/j.devcel.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 37.Memczak S, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 38.Piwecka M, et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science. 2017;357 doi: 10.1126/science.aam8526. [DOI] [PubMed] [Google Scholar]
- 39.Barrett SP, Salzman J. Circular RNAs: analysis, expression and potential functions. Development. 2016;143:1838–1847. doi: 10.1242/dev.128074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gruber AR, et al. Global 3′ UTR shortening has a limited effect on protein abundance in proliferating T cells. Nat Commun. 2014;5:5465. doi: 10.1038/ncomms6465. [DOI] [PubMed] [Google Scholar]
- 42.Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kramer NJ, et al. Altered lymphopoiesis and immunodeficiency in miR-142 null mice. Blood. 2015;125:3720–3730. doi: 10.1182/blood-2014-10-603951. [DOI] [PubMed] [Google Scholar]
- 44.Mildner A, et al. Mononuclear phagocyte miRNome analysis identifies miR-142 as critical regulator of murine dendritic cell homeostasis. Blood. 2013;121:1016–1027. doi: 10.1182/blood-2012-07-445999. [DOI] [PubMed] [Google Scholar]
- 45.Cho S, et al. miR-23 approximately 27 approximately 24 clusters control effector T cell differentiation and function. J Exp Med. 2016;213:235–249. doi: 10.1084/jem.20150990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 48.Miles WO, Tschop K, Herr A, Ji JY, Dyson NJ. Pumilio facilitates miRNA regulation of the E2F3 oncogene. Genes Dev. 2012;26:356–368. doi: 10.1101/gad.182568.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ke S, et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015;29:2037–2053. doi: 10.1101/gad.269415.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huppertz I, et al. iCLIP: protein-RNA interactions at nucleotide resolution. Methods. 2014;65:274–287. doi: 10.1016/j.ymeth.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:10. [Google Scholar]
- 52.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lianoglou S, Garg V, Yang JL, Leslie CS, Mayr C. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 2013;27:2380–2396. doi: 10.1101/gad.229328.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession code of this study is GSE116561. Individual dataset accession is as follows. RNA-Seq dataset (GSE116348); Differential iCLIP dataset (GSE116466); PolyA-Seq dataset (GSE116468). Codes of CLIPanalyze can be downloaded from https://bitbucket.org/leslielab/clipanalyze, and are available upon request.