

Abstract
Decellularized human adipose tissue has potential clinical utility as a processed biological scaffold for soft tissue cosmesis, grafting and reconstruction. Adipose tissue decellularization has been accomplished using enzymatic-, detergent-, and/or solvent-based methods. To examine the hypothesis that distinct decellularization processes may yield scaffolds with differing compositions, the current study employed mass spectrometry to compare the proteomes of human adipose-derived matrices generated through three independent methods combining enzymatic-, detergent-, and/or solvent-based steps. In addition to protein content, bioscaffolds were evaluated for DNA depletion, ECM composition, and physical structure using optical density, histochemical staining, and scanning electron microscopy (SEM). Mass spectrometry (MS) based proteomic analyses identified 25 proteins (having at least two peptide sequences detected) in the scaffolds generated with an enzymatic approach, 143 with the detergent approach, and 102 with the solvent approach, as compared to 155 detected in unprocessed native human fat. Immunohistochemical detection confirmed the presence of the structural proteins actin, collagen type VI, fibrillin, laminin, and vimentin. Subsequent in vivo analysis of the predominantly enzymatic- and detergent-based decellularized scaffolds following subcutaneous implantation in GFP+ transgenic mice demonstrated that the matrices generated with both approaches supported the ingrowth of host-derived adipocyte progenitors and vasculature in a time dependent manner. Together, these results determine that decellularization methods influence the protein composition of adipose tissue-derived bioscaffolds.
Keywords: Adipose Tissue, Bioscaffold, Decellularization, Extracellular Matrix, Mass Spectrometry Proteomics, Regenerative Medicine
INTRODUCTION
Currently, plastic surgeons employ human adipose tissue as a surgical graft in wound repair, cosmetic and reconstructive surgeries; however, it may have broader applicability as an adjunct therapeutic [1]. More specifically, additional applications may include serving as a vehicle promoting the delivery of drugs, growth factors and/or stem cells, as well as representing an abundant and expendable extracellular matrix (ECM) source for generating bioscaffolds for both autologous and allogeneic transplantation to promote tissue regeneration in patients requiring cosmetic or reconstructive surgery for genetic defects of the breast and chest wall (Poland syndrome) [2], facial deformities (Treacher Collins syndrome) [3], facial lipoatrophy [4] or traumatic scar repair [5, 6]. Researchers have begun to develop decellularized adipose tissue products in efforts to improve long-term graft acceptance, differentiation, survival of transplanted stem cells and tissue regeneration [7-32]. Indeed, recent studies have demonstrated that decellularized adipose tissue derived bioscaffolds can promote the proliferation and differentiation of exogenous stromal/stem cells as evidenced by the expression of adipogenic lineage-specific genes in vitro, including PPARγ, C/EBPα, and LPL [8, 12, 29].
To extend these outcomes in a reproducible and reliable manner, it will be necessary to further define the composition of decellularized human adipose tissue products. One of the most desirable features of a decellularized tissue product would be its retention of bioactive ECM components such as proteoglycans and proteins including collagens, laminins, fibronectin, and elastin [33]. Decellularization processes will inevitably cause disruption in the ECM resulting in the loss of some proteins in addition to lipids, membranes and nucleic acids. Although these decellularization processes vary, most include enzymatic digestion, mechanical, and/or chemical extractions. The existing literature concerning protein analyses of decellularized tissues, including adipose tissue, describes the occurrence of specific ECM proteins such as collagens and elastin, whose presence has been verified by biochemical and immunohistochemical assays [8, 12, 34].
Characterization of the adipose tissue-derived biological scaffold at the protein level is essential to understanding its utility, activity, and functionality. The current study compares adipose-derived bioscaffolds prepared using enzymatic-, detergent-, and solvent-based methods. The bioscaffolds were analyzed using three complementary in vitro approaches; histological and spectrophotometric evaluation of decellularization, qualitative ultrastructure, and ECM protein identification by mass spectrometry. In addition, an in vivo analysis of the functionality of the enzymatic and solvent based bioscaffolds was performed.
METHODS
All methods involving human samples were carried out under a protocol approved by the Pennington Biomedical Institutional Review Board (Baton Rouge, LA; PBRC#23040) or the Western Institutional Review Board (Puyallup WA: WIRB Protocol #20130449). Samples were obtained from male and female donors (n = 6) aged 43+/-9.3, with an average BMI of 23.3+/-2.9 kg/m2. All subjects gave written informed consent prior to collection of their tissues. The hexafluorisopropanol (HFIP)-treated silk scaffolds were obtained from the Tissue Engineering Resource Center, Tufts University (http://ase.tufts.edu/terc/) and implanted as previously described [35].
Decellularization
The adipose tissue samples were divided and processed using three different decellularization methods.
Enzymatic-based Method 1 (M1)
Tissue samples were decellularized using a method developed and published by Flynn [8]. Intact adipose tissue was cut into 20-25 g pieces and subjected to three cycles of freeze-thaw (-80°C to 37°C) in Freezing Buffer Solution (10 mM Tris buffer (pH 8.0), 5 mM ethylenediaminetetraacetic acid (EDTA). The tissue was transferred into Enzymatic Digestion Solution #1 (0.25% trypsin/0.1% EDTA), and incubated with agitation overnight for ~16 hrs followed by a 48 hr polar solvent extraction in absolute isopropanol to remove lipid content. Next the tissue was rinsed three times for 30 minutes in Rinsing Buffer Solution (8 g/L NaCl, 200 mg/L KCl, 1 g/L Na2HPO4, and 200 mg/L KH2PO4 (pH 8.0)), and then incubated for 6 hrs in fresh Enzymatic Digestion Solution #1. Following enzyme digestion, the samples were washed 3 times in Rinsing Buffer Solution. Next, the samples were transferred into Enzymatic Digestion Solution #2 (55 mM Na2HPO4, 17 mM KH2PO4, 4.9 mM MgSO4·7H2O containing 150 U/ml DNase Type II (from bovine pancreas), 0.125 mg/ml RNase Type III A (from bovine pancreas), and 20 Units/ml Lipase Type VI-S (from porcine pancreas)) for 16 hrs of processing (overnight). The next day, samples were rinsed 3 times for 30 minutes in the Rinsing Buffer solution. The samples were subjected to a final polar solvent extraction in absolute isopropanol for 8 hrs, rinsed 3 times for 30 minutes in the Rinsing Buffer solution and rinsed 3 times for 30 minutes in 70% ethanol. Finally, the samples were stored in sterile PBS supplemented with 1% antibiotic/antimycotic solution (ABAM) at 4°C.
Detergent-based Method 2 (M2)
Tissue samples were decellularized using a modified detergent-based and xenoprotein-free method [12]. In this process, the tissue was washed in distilled water (1 g/mL), by shaking, for approximately 10 minutes until the wash liquid was clear. The sample was centrifuged at 1800 X g for 5 minutes at room temperature, and the upper oil layer was discarded. The viscous suspension was treated with a buffered 1 M solution of NaCl (0.0584 g/mL in 10 mM Tris), diluted 1:1 with the tissue, overnight in a 37°C shaker. The following day, the samples were centrifuged at 29.3 X g for 5 minutes at 4°C. The supernatant was decanted and the pelleted samples were rinsed with distilled water for 24 hrs at 4°C with gentle shaking. The medium was replaced with fresh distilled water the next morning, followed by continued gentle shaking at 4°C for 2 hours. The residue was incubated in 1 mM EDTA overnight, at room temperature. The next day the samples were centrifuged and washed with distilled water for 24 hours at 37°C while gently shaking. Finally, the samples were subjected to lysis buffer (1% tergitol type NP-40, 0.1% SDS, 5 mM EDTA, 0.4 M NaCl, 50 mM Tris-HCl pH 8, 1 mM phenylmethylsulfonyl fluoride (PMSF)) (1:1 vol∷vol) overnight at 4° C to remove any remaining intact cells, centrifuged for 10 min, and washed with distilled water. Samples were stored in sterile distilled water at 4°C.
Urea-based Solvent Method 3 (M3)
Tissue decellularization matched steps for the preparation of the commercially available ECM product, Matrigel™ as described by Kleinman [36]. One hundred grams of frozen adipose tissue were thawed in 200 mL of 3.4 M NaCl buffer at room temperature. Individual samples from 2-3 donors were then homogenized for dispersement, followed by centrifugation at 8,000 x g for 15 minutes at 4°C. The supernatant was discarded, the homogenization step was repeated, and 100 mL of 2 M urea buffer (buffered with Tris) was added to the homogenate before repeating the homogenization step. The samples were stirred overnight at 4°C, centrifuged at 23,000 x g at 4°C for 20 minutes, and the thick supernatant was saved on wet ice. The pellets were homogenized in 2 M urea buffer, centrifuged at 23,000 x g at 4°C for 20 minutes, and the supernatant collected. The successive supernatants from a single donor were combined and the pellets discarded. Next, the samples were dialyzed against two liters of tris buffered saline (TBS) (50 mM Tris-Cl, 150 mM NaCl pH 7.5 supplemented with 1% chloroform to minimize bacteria or spore contamination) for ≥ 4 hours. Samples were then dialyzed twice more for ≥ 2 hours each time, against 2 liters of TBS, then dialyzed once more against 2 liters of PBS. In a sterile hood, the exterior of the dialysis bag was rinsed with 70% ethanol before cutting one end. The dialyzed contents were transferred into sterile conical tubes, on ice, and stored at 4°C until further use.
Bioscaffold Analyses
DNA extraction and quantitation
Following decellularization, all samples (n = 3 to 4 donors) were assayed for DNA content. In a 1.5 ml micro-centrifuge tube, a 300 mg sample was combined with 700 μL of Lysis Buffer (50 mL of 1 M Tris at pH 8, 20 mL of 5 M NaCl, 5 mL of 0.5 M EDTA, 100 mL of 1% SDS, and distilled deionized (DD) water to a final volume of 500 mL) or Proteinase Digestion Buffer (100 mM Tris pH 8.0, 200 mM NaCl, 5 mM EDTA, 0.2% SDS) containing 10 μl proteinase K (10 mg/ml Roche) and incubated overnight at 55°C. The next day, the samples were extracted with 700 μL phenol:chloroform:isoamyl alcohol (25:24:1), vortexed and centrifuged at 16,873 X g for 10 minutes at room temperature. 500 μL of the supernatant was transferred to a fresh 1.5 mL micro-centrifuge tube, combined with 500 μL absolute ethanol, and stored at −20°C for at least 2 hours. The samples were then centrifuged at maximum speed for 15 minutes at 4°C and the supernatant was discarded. Next, the resulting DNA pellet was washed with 900 μL of 70% ethanol, air dried, and re-suspended in 100 μL elution buffer (EB). Samples were stored at −80°C until use. DNA concentration was measured via optical density, on a Nanodrop 1000. The final concentration was calculated as:
Histochemistry and Immunohistochemistry
To confirm decellularization and assess microscopic tissue structure and composition, histochemistry and immunohistochemistry were performed using modifications of previously published methods [37, 38]. Bioscaffold samples were fixed in 4% paraformaldehyde (PFA) and processed on a Thermo Scientific Excelsior ES Tissue processor. Serial sections of 5 microns were taken. Paraffin sections were stained with Hematoxylin and Eosin (H&E) (Leica Microsystems, Buffalo Grove, IL) and a Masson’s Trichrome Staining Kit (Poly Scientific, Bay Shore, NY) according to the kit specifications. The H&E staining was performed using a Leica St 5020 Autostainer. Slides were scanned using a Hamamatsu Nanozoomer Digital Pathology (NDP) system (Hamamatsu City, Japan.)
Immunohistochemical staining for collagen VI (rabbit polyclonal IgG (1:100 dilution) Novus Biologicals NB120-6588), alpha smooth muscle actin (mouse monoclonal IgG2 (1200 dilution) Millipore MAB1522), and vitronectin (mouse monoclonal IgG (1:100 dilution) Millipore MAB1926) was performed as follows. Slides were warmed on a heating platform at 57°C for 30 minutes immediately prior to deparaffinization. For deparaffinization, slides were rinsed in the following solutions: Xylene - 2 times for 5 minutes, 100% EtOH - 2 times for 2 minutes, 95% EtOH - 2 times for 2 minutes, 70% EtOH - 1 time for 2 minutes, 50% EtOH - 1 time for 2 minutes, and finally DD water - 1 time for 2 minutes. For antigen retrieval, tissue sections were washed twice for 5 minutes in 20 mM Tris-HCl (pH 8.0) and incubated in a 0.4 mg/mL proteinase K solution (in Tris-HCl, pH 8.0) for 15 minutes at 37°C in a humidified chamber. The slides were then rinsed liberally with water, followed by two 5-minute washes in Tris-buffered saline with tween (TBS-T) (50 mM Tris, 150 mM NaCl, 0.05% Tween in DD water, pH 7.6). For antibody staining, the sections were covered with TBS + 10% normal serum (secondary antibody host) + 1% BSA and incubated for 2 hours at room temperature in a humidified chamber. Next the primary antibody was applied and the sections were incubated overnight at 4°C in a humidified chamber. The next day the slides were rinsed 2 times for 5 minutes in TBS-T. The secondary antibody (either goat anti-rabbit IgG or goat anti-mouse IgG at 1:200 dilution in TBS plus 1% BSA) was applied and the sections were incubated for 1 hour at room temperature in a humidified chamber protected from light. The slides were then rinsed 2 times for 5 minutes in TBS followed by a liberal water rinse. Coverslips were added with the fluorescence-maintaining mounting medium, Prolong+ DAPI (Life Technologies). Sections were imaged using a Nikon Eclipse TE300 with an Olympus DP70 color camera in the Microbiology and Immunology microscopy core at the Tulane University School of Medicine.
Triglyceride Measurement
To assess residual lipid content following decellularization, measurements were made using a Triglyceride Colorimetric Assay kit (#10010303; Cayman Chemical Inc., Ann Arbor, MI). 350-400 mg of cryomilled tissue samples were minced in 2 mL of the diluted Standard Diluent containing Halt™ protease inhibitor cocktail (Thermo Scientific™). The samples were then centrifuged at 10,000 x g for 10 minutes at 4°C. The supernatant was transferred to a second tube and stored on ice. Samples were diluted 1:10, 1:25, 1:50, and 1:100 using the diluted Standard Diluent. Assays were prepared using 10 μL of standard or sample in each well of a 96 well plate. The reaction was initiated by the addition of 150 μL of diluted Enzyme Buffer solution to each well. The microtiter plate was shaken to mix the contents of the wells, covered, and incubated for 15 minutes at room temperature. Absorbance was measured at 540 nm on a BioRad Benchmark Plus™ plate reader.
Scanning Electron Microscopy
To assess tissue ultrastructure, electron microscopy was performed using a modification of published methods [8, 11]. In preparation for imaging under SEM, a ~15 mg piece of each bioscaffold was fixed in 0.2 M cacodylate at room temperature overnight, followed by dehydration in an increasing acetone concentration solution series (30% to 100%). The sample was then dried by replacing acetone with CO2 gas and sputter-coated with platinum for 4 minutes. The samples were then imaged in the Socolofsky Microscopy Center, Louisiana State University on a JSM-6610LV scanning electron microscope.
Mass Spectrometry and Peptide Identification
Mass spectrometry and peptide analyses were performed using a modification of published methods [39].
Trypsinization
Protein samples were digested with sequencing grade modified trypsin (Promega Corp) according to the manufacturer’s instructions. Briefly, 45 μL of 200 mM triethyl ammonium bicarbonate (TEAB) was added to aliquots of 100 μg of protein sample and the final volume was adjusted to 100 μL with ultrapure water. To solubilize complex protein mixtures, 5 uL of 2% SDS was added before adjusting to final volume. Five microliters of 200 mM Tris (2-carboxyethyl) phosphine (TCEP) was added and the resulting mixture was incubated for 1 h at 55° C, then 5 μL of 375 mM iodoacetamide was added and the mixture was incubated for 30 minutes without light. After incubation, 1 mL of pre-chilled acetone was added and the precipitation was allowed to proceed overnight at −20° C. The acetone-precipitated protein pellets were suspended with 100 μL of 200 mM TEAB and 2.5 μg of trypsin was added to digest the sample overnight at 37 °C.
Fractionation of Labeled Peptide Mixture Using a Strong Cation Exchange Column
The peptide mixture was fractionated with a strong cation exchange column (SCX) (Thermo Scientific) on a Shimadzu 2010 HPLC equipped with a UV detector (Shimadzu, Columbus, MD). Mobile phase consisted of buffer A (5 mM KH2PO4, 25% acetonitrile, pH 2.8) and buffer B (buffer A plus 350 mM KCl). The column was equilibrated with Buffer A for 30 minutes before sample injection. The mobile phase gradient was set at a flow rate of 1.0 mL/minute as follows: (a) 0 to 10 minutes: 0% buffer B; (b) 10 to 40 min: 0% to 25% Buffer B, (c) 40 to 45 min: 25% to 100% Buffer B; (d) 45 to 50 minutes: 100% buffer B; (e) 50 to 60 minutes: 100% to 0% buffer B; (f) 60 minutes to 90 minutes: 0% buffer B. A total of 60 fractions were initially collected, lyophilized and combined into 15 final fractions based on SCX chromatographic peaks.
Desalination of Fractionated Samples
A C18 solid-phase extraction (SPE) column (Hyper-Sep SPE Columns, Thermo-Fisher Scientific) was used to desalt all collected fractions. The combined 15 fractions were each adjusted to 1 ml final volume containing 0.25% (v/v in water) trifluoroacetic acid (TFA, Sigma). The C18 SPE columns were conditioned before use by filling them with 1 mL acetonitrile and allowing the solvent to pass through the column slowly (~3 minutes). The columns were then rinsed three times with 1 mL 0.25% (v/v in water) TFA solution. The fractions were loaded on to the top of the SPE cartridge and allowed to elute slowly. Columns were washed four times with 1 mL 0.25% TFA aliquots before the peptides were eluted with 3 × 400 μL of 80% acetonitrile/0.1% formic acid (aqueous).
LC-MS/MS Analysis on LTQ-Orbitrap
Peptides were analyzed on an LTQ-Orbitrap XL instrument (Thermo-Fisher Scientific) coupled to an Ultimate 3000 Dionex nanoflow LC system (Dionex, Sunnyvale, CA). High mass resolution was used for peptide identification and high energy collision dissociation (HCD) was employed for reporter ion quantification. The RP-LC system consisted of a peptide Cap-Trap cartridge (0.5 × 2 mm) (Michrom BioResources, Auburn, CA) and a prepacked BioBasic C18 PicoFrit analytical column (75 μm i.d. × 15 cm length, New Objective, Woburn, MA) fitted with a FortisTip emitter tip. Samples were loaded onto the trap cartridge and washed with mobile phase A (98% H2O, 2% acetonitrile and 0.1% formic acid) for concentration and desalting. Subsequently, peptides were eluted over 180 minutes from the analytical column via the trap cartridge using a linear gradient of 6–100% mobile phase B (20% H2O, 80% acetonitrile and 0.1% formic acid) at a flow-rate of 0.3 μl/minutes using the following gradient: 6% B for 5 minutes; 6–60% B for 125 minutes; 60–100% B for 5 minutes; hold at 100% B for 5 minutes; 100–6% B in 2 minutes; hold at 6% B for 38 minutes.
The LTQ-Orbitrap tandem mass spectrometer was operated in a data-dependent mode. Briefly, each full MS scan (60,000 resolving power) was followed by six MS/MS scans where the three most abundant molecular ions were dynamically selected and fragmented by collision-induced dissociation (CID) using a normalized collision energy of 35%, and the same three molecular ions were also scanned three times by HCD-MS2 with collision energy of 45%. MS scans were acquired in profile mode and MS/MS scans in centroid mode. LTQ-Orbitrap settings were as follows: spray voltage 2.0 kV, 1 microscan for MS1 scans at 60,000 resolution (fwhm at m/z 400), microscans for MS2 at 7500 resolution (fwhm at m/z 400); full MS mass range, m/z 400–1400; MS/MS mass range, m/z 100–2000. The “FT master scan preview mode,” “Charge state screening,” “Monoisotopic precursor selection,” and “Charge state rejection” were enabled so that only the 2+, 3+, and 4+ ions were selected and fragmented by CID and HCD.
Database Search
The protein search algorithm used was Mascot v2.3.01 (Matrix Science, Boston, Ma). Mascot format files were generated by the Proteome Discoverer 1.2 software (Thermo-Fisher Scientific) using the following criteria: database, IPI_Human.fasta.v3.77 (containing 89,422 entries and concatenated with the reversed versions of all sequences.); enzyme, trypsin; maximum missed cleavages, 2; Static modifications, carbamidomethylation (+57 Da), N-terminal TMT6plex (+229 Da), lysyl TMT6plex (+229 Da). Dynamic modifications, N-terminal Clnpyro- Glu (+17Da); methionine oxidation (+16 Da); STY phosphorylation (+80 Da); Precursor mass tolerance was set at 20 ppm; fragment match tolerance was set at 0.8 Da. Peptides reported by the search engine were accepted only if they met the false discovery rate of p < 0.05 (target decoy database), a Mascot ion score ≥30 for peptide identifications was required.
Gene Ontology Analysis
Identification of retained proteins and associated molecular mechanisms was performed through the free online data base DAVID Bioinformatics [40, 41] . Protein identifiers where converted to gene name prior to analysis and GOTERM BP-FAT was used for each decellularization type.
In Vivo Implantation
Murine studies examining the implantation of the decellularized adipose tissue (DAT) scaffolds were conducted in accordance with IACUC Protocol #4302 which had been reviewed and approved by the Tulane University Institutional Animal Care and Use Committee (IACUC) prior to the initiation of this work.
The DAT was cut into sections with the dimensions of 10 × 10 × 5 mm or 500 μL total volume, rinsed repeatedly in 70% ethanol, and rehydrated twice in sterile PBS. Prior to implantation, GFP+C57Bl/6 mice were anesthetized with 2% isoflurane in 2 L/minute of O2. The dorsal hair was shaved and one 1 cm incision was placed in the midline dorsal skin of the mice. The M1 and M2 decellularized adipose ECM as well as silk scaffolds were carefully placed in subcutaneous pockets on the back of each animal using tweezers as described in [42]. Each mouse received 4 implants (two per side bilaterally adjacent to the spinal cord), with duplicate scaffolds prepared for each of three donors and a total of 12 mice in the study. All animals were housed in separate cages using standard husbandry conditions of 12 h light/12 h dark, room temperature of 20° to 21°C and unlimited access to food and water. The animals were sacrificed after three, six and nine weeks post-implantation and implanted scaffolds were collected within their surrounding tissues. The explanted scaffold samples were fixed in 4% paraformaldehyde and processed on a tissue processor (Thermo Scientific Excelsior ES Tissue processor). Serial sections of 5 μm were stained with Hematoxylin and Eosin (H&E) (Leica Microsystems, Buffalo Grove, IL and Leica St 5020 Autostainer). For the immunohistochemistry study, the paraffin embedded and fixed sections were stained using GFP (Anti-GFP rabbit polyclonal antibody unconjugated 2 mg/mL 100 uL, Invitrogen Molecular Probes, Eugene, Oregon, USA), perilipin (Rb pAb to Perilipin A 110 uL, Abcam, Cambridge, MA, USA) and CD31 (DSHB Hybridoma Product 2H8, Ames, Iowa, USA) antibodies according to the protocol described in the Histochemistry and Immunohistochemistry section above. Sections were imaged using Scan Scope Console – Leica, Version 10.2.0.2314 and Image Scan Scope – Leica, Biosystems, 2006-2013, Version 12.1.0.5029 and Aperio eSlide Manager online desktop Scan Scope Console software.
Statistics
Outcomes are reported as the mean ± standard deviation unless otherwise indicated. Comparisons are based on the student t-test with significance defined as p < 0.05 performed with Excel (Microsoft, Redmond WA).
RESULTS
Decellularization and Physical Characterization
The three decellularized scaffold preparations, enzymatic-based Method 1 (M1), detergent-based Method 2 (M2), and solvent-based Method 3- Matrigel™ (M3), underwent characterization studies to determine DNA content removal, resulting physical structure, and retained protein composition. Following the decellularization steps, the bioscaffolds displayed the following overall macroscopic appearances: whitish color with retention of tissue volume (M1); yellowish color with contracted tissue volume (M2) and; whitish color with loss of tissue architecture and appearance of a suspension (M3). Staining with H&E and Masson’s trichrome was performed to qualitatively assess cell nuclei and tissue structure in the untreated adipose tissue relative to the decellularized adipose tissues. Histochemical staining documented the presence of nuclei in the untreated tissues, and a lack of nuclei following the M1, M2 and M3 decellularization procedures (Figure 1). To further evaluate the presence of retained nuclear content, optical density readings were taken for quantitative measurements of genomic DNA remaining in the decellularized bioscaffolds relative to untreated tissue (Table 1). Relative to the genomic DNA isolated from the untreated control (281 ± 152 ng/μL), the M1 (47 ± 27 ng/μL) and M2 (37 ± 9 ng/μL) decellularization methods significantly reduced the genomic DNA content while the M3 method (237 ± 221 ng/μL) did not. To evaluate residual lipid content following decellularization, lipid vacuole shape and triglyceride content were next evaluated. Lipid vacuoles appeared as imperfect spheres in M1 and M2 bioscaffolds, similar to that of native tissue; however, these structures were less apparent within M3 scaffolds (H&E staining; Figure 1); this may reflect the presence of urea in the M3 protocol or, alternatively, may be an artifact of sectioning. Triglyceride measurements indicated that all three decellularization methods retained less than fifty-percent of the triglyceride content relative to the untreated native tissue (Table 1).
Figure 1.
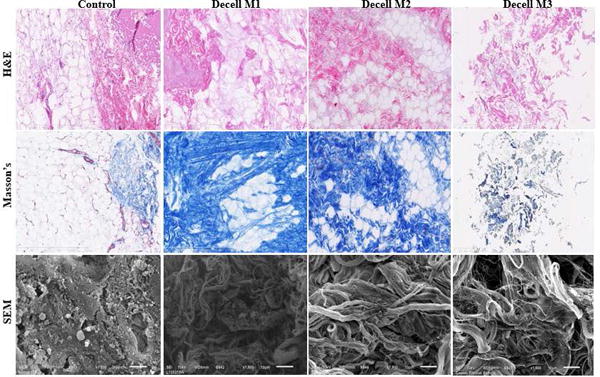
Microscopic Analysis of Untreated and Decellularized Adipose Tissue Samples. Untreated control tissue and the products of the M1, M2, and M3 decellularization procedures were evaluated by in H&E staining (top), Masson’s Trichrome stain (middle), and Scanning Electron Microscopy (SEM) (shown at 1500 X magnification). Individual images are representative of at least n = 3.
Table 1.
Genomic DNA and Triglyceride Levels in Decellularized Human Adipose-Derived Scaffolds
| Untreated | Decell M1 | Decell M2 | Decell M3 | |
|---|---|---|---|---|
| Genomic DNA (ng/μl) | 281 ± 152 | 47 ± 21* | 37 ± 9* | 237 ± 221 |
| Triglyceride (mg/dl) | 56.8 ± 0.2 | 16.9 ± 0.1 | 23.7 ± 0.1 | 16.8 ± 0.1 |
Values are reported as the mean ± standard deviation of n = 3 samples from individual donors. Significance was calculated based on a one tailed student t-test relative to the untreated control where (*) represents p value < 0.05.
To further evaluate the effect of decellularization, scaffolds prepared with all Methods were stained with Masson’s Trichrome (Figure 1). The level of Masson’s Trichrome staining was enriched following M1 and M2 decellularization methods but not M3 relative to the untreated control. To qualitatively assess the collagen fiber structure in the processed samples, scanning electron microscopy (SEM) was performed (Figure 1). SEM under M2 and M3 revealed a complex fibrous network with varied patterns and densities on the surface of the bioscaffolds relative to the untreated control tissue. In contrast, SEM under M1 appeared to display smaller collagen bundles; however, this may reflect regional differences within the fixed specimen. To further determine the retained cell and matrix composition, immunofluorescent staining for collagen VI, actin, and vitronectin was performed using M1 and M2 scaffolds (Figure 2). Consistent with mass spectrometry results, collagen VI gave the strongest positive signal in control and decellularized tissues. In contrast, actin signal was reduced in both M1 and M2 while vitronectin, although present in M1, was nearly absent in M2 relative to the untreated tissue control. The M3 scaffolds were not examined due to failure of the bioscaffold suspension to properly mount onto the slides.
Figure 2.
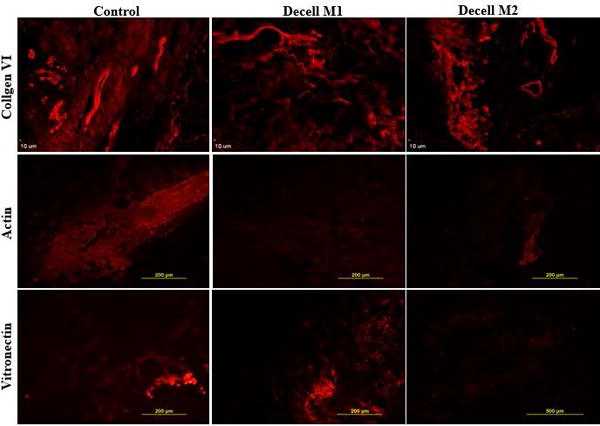
Immunofluorescent staining for collagen VI, actin, and vitronectin. Paraffin fixed slides prepared with tissue from untreated controls or scaffolds decellularized using methods M1 and M2 were stained with fluorochrome labeled antibodies to collagen VI, actin, and vitronectin. Images are representative of n =3.
Mass Spectrometry and Immunohistochemical Proteomic Characterization
Mass spectrometry proteomic analyses provided an unbiased global assessment of the protein content of the bioscaffolds. The untreated tissue sample had a reading of 281 peptides; of these, 155 were identified by the detection of at least two (2+) peptides and 29 were associated with the detection of seven (7+) peptides (Table 2 and Supplementary Table 1). The M1 sample had a reading of 67 total peptides identified, with 25 having 2+ peptide sequences detected and only one having 7+ peptides detected (Supplementary Table 2). The M2 scaffold had a reading of 296 total peptides, 143 of which had 2+ peptides detected, and 30 of these had 7+ peptides detected (Supplementary Table 3). The M3 scaffold sample had 243 total peptides, with 102 having 2+ peptides detected and 15 having 7+ peptides detected (Supplementary Table 4). We evaluated the top twenty peptides (by number of peptides detected) from each sample for commonalities (Supplemental Table 5), as well as protein localization comparisons (Figure 3). Comparisons of the top twenty peptides identified in the untreated tissue and decellularized scaffolds by the different methods revealed that these were localized to the blood, nucleus, cytoskeleton, extracellular space, other various intracellular locales, and those whose locations were unidentified. Inquiries were performed to identify key factors in the ECM composition and to determine if they were associated with a pathological biomarker or pathway. No markers or pathways were identified that associated with particular diseases or pathologies. DAVID bioinformatics was used to identify gene ontology analysis based on the genes assumed or known to be associated with the proteins identified by LC MS/MS, to determine functionality of the components of the final scaffold products. Analysis of total protein content determined that constituents remaining in all three scaffold preparation method products showed association with cell adhesion, biological adhesion, wound response, protein complex assembly, and macromolecular complex assembly- among others (Table 3). Full gene ontology lists of the individual scaffold preparation methods can be found in the supplemental material (Supplemental Table 6). Identification of protein ontology uniquely identified only with tissue decellularization and not present in native tissue, demonstrated enrichment for proteins associated with biological and cell adhesion as well as lipid metabolism and DNA packaging. Proteins included within this are: Isoform 1 of Myosin-9, Basement membrane-specific heparan sulfate proteoglycan core protein, Histone H3.3, Isoform 2 of Hormone-sensitive lipase, collagen alpha-3(VI) chain isoform 4 precursor, and Possible J 56 gene segment (Fragment). Finally, comparisons across all decellularization methods to identify proteins uniquely enriched between the three methods demonstrated that M1 and M2 uniquely shared 66 proteins not found in either M3 or native tissue. M3 shared one protein uniquely with M2 (Ig kappa chain V-III region B6) and 2 proteins uniquely with M1 (Alpha-1-acid glycoprotein 2 and Annexin A5).
Table 2.
LC-MS/MS Peptides Identified with each Decellularization Method
| Untreated | Decell M1 | Decell M2 | Decell M3 | |
|---|---|---|---|---|
| # Identified Peptides | 281 | 66 | 296 | 242 |
| # Identified with 2 Peptides | 155 | 25 | 143 | 102 |
| # Identified with 7 Peptides | 29 | 1 | 30 | 15 |
Figure 3.
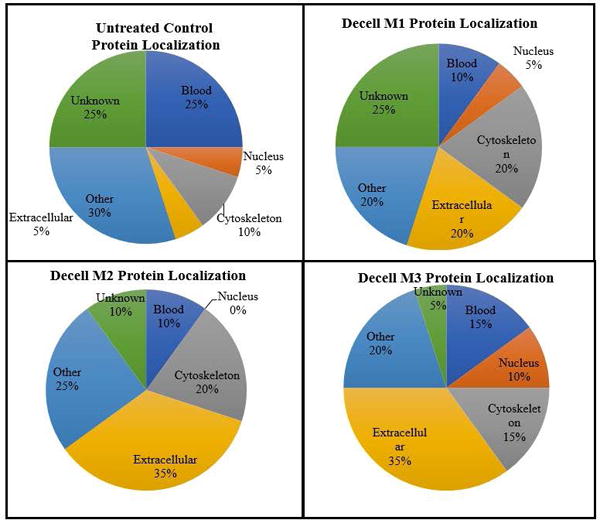
Subcellular Localization of the Top Twenty Peptides (based on number of hits) in Each Decellularized Scaffold Relative to Untreated Tissue Control.
Table 3.
Gene Ontology Analysis of Decellularization Methods 1, 2, & 3 Scaffolds
| Decell Method | M1 | M2 | M3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Category | Freq | % | P Value | Freq | % | P Value | Freq | % | P Value |
| Cell Adhesion | 8 | 21.6 | 0.0018 | 32 | 16.2 | 3 × 10−9 | 35 | 20.6 | 1.1 × 10−133 |
| Biological Adhesion | 8 | 21.6 | 0.0018 | 32 | 16.2 | 4 × 10−9 | 35 | 20.6 | 1.1 × 10−13 |
| Response to Wounding | 7 | 18.9 | 0.0022 | 19 | 9.6 | 2 × 10−4 | 23 | 12.9 | 3.1 × 10−8 |
| Protein Complex Assembly | 6 | 16.2 | 0.0091 | 18 | 9.1 | 4.7 × 10−4 | 37 | 10 | 0.0003 |
| Macromolecular Complex Assembly | 9 | 24.3 | 0.00024 | 25 | 12.6 | 9.8 × 10−6 | 24 | 14.1 | 8.7 × 10−7 |
Five functionality traits found in all three scaffold preparations. Category-= Gene Ontology (GO) term function description; Freq (Frequency) = number of genes represented which are related to the GO term function; p values = probability of presence of number of genes (frequency) out of total number of genes of the particular function, related to the whole genome.
Bioscaffold implantation in vivo allowed repopulation with mature adipocytes
Subcutaneous implantation was performed using the decellularized M1 and M2 adipose scaffolds to determine their support of adipose tissue formation in vivo since they retained intact ECM proteins and architecture in vitro; however, comparable studies were not pursued using the M3 scaffolds due to the suspension nature of the M3 bioscaffold and its loss of the intact tissue architecture during the decellularization process. The M1 and M2 scaffolds were implanted subcutaneously into transgenic C57Bl/6 GFP+ mice (Figure 4 and Supplemental Figure 1). The scaffolds were harvested at serial intervals (3, 6 and 9 weeks). At necropsy, gross visualization of the decellularized adipose scaffolds indicated adipose tissue formation equivalent to or more robust than that obtained with a silk scaffold control (Supplemental Figure 1). Of note, the M1 and M2 decellularized adipose scaffolds retained an architecture similar to native adipose tissue, while the control silk scaffolds constructs were more fibrotic in appearance. This was confirmed histologically based on H&E staining (Data not shown). The presence of functional adipose cells increased in a time dependent manner after implantation based on staining for perilipin, a lipid vacuole membrane associated protein (Figure 4). The newly formed adipocytes stained positive for GFP, consistent with the ingrowth and repopulation of the scaffold by host origin cells (Figure 4). Additionally, the scaffolds contained cells staining positive for CD31, a biomarker primarily but not exclusively associated with endothelial cells (Figure 5). An intact, native fat pad from transgenic GFP+ C57Bl/6 mice served as a positive control (Figures 4 and 5).
Figure 4.
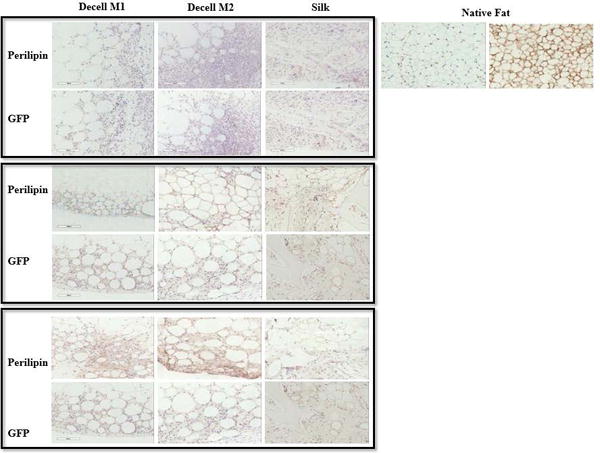
Immunohistochemical Detection of Adipogenic Biomarker (Perilipin) and Host Cells (GFP) in M1 and M2 Tissue Scaffold In Vivo Implants. Scaffolds were implanted into C57BL/6 mice transgenic for ubiquitous expression of the green fluorescent protein (GFP) for periods of 3, 6, or 9 weeks (top, middle, or bottom panels). A native adipose tissue served as positive controls for both antibodies.
Figure 5.
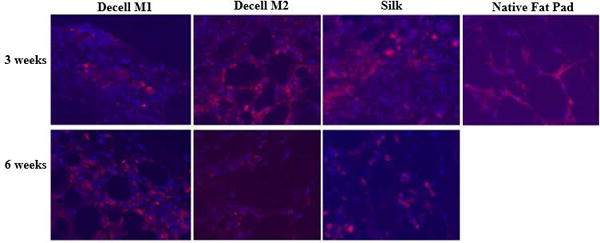
Immunofluorescent Detection of CD 31. demonstrated the vascularization of the implanted scaffolds denoting their integration. Scaffolds were implanted into C57BL/6 mice transgenic for ubiquitous expression of the green fluorescent protein (GFP) for periods of 3 or 6 weeks. A silk scaffold implant and native adipose tissue served as positive controls. Sections were stained with DAPI for detection of nuclei (blue) or anti-CD31 fluorochrome labeled antibodies (red).
DISCUSSION
Recent studies have reported the preparation of decellularized adipose tissue extracellular matrices; a comprehensive review of this literature has recently been published by Banyard et al. [25]. While the majority of studies reported on a single decellularization approach, the work by Brown et al., stands out as an exception. This publication compared three independent methods distinguished based on their predominant reliance on enzymatic, chemical or physical methods of decellularizing porcine adipose tissue [30]. They concluded that the individual methods resulted in decellularized matrices that were distinct based on biochemical and structural features. The current study has confirmed and extended these approaches by evaluating human adipose tissues processed with three decellularization methods relying on enzymatic, detergent, or solvent agents, respectively. The resulting products were examined and compared based on in vitro and in vivo outcomes. Consistent with recent mass spectrometry analyses of detergent-decellularized lung tissues, the current findings suggest that the proteomic composition of adipose tissue-derived bioscaffolds is affected by the chosen decellularization method and this may have implications with respect to their utility and favorability for specific clinical translational applications [34].
Despite these differences in the complexity of the decellularized tissue proteome, the physical structure of the decellularized adipose product using two methods (M1 and M2) was highly similar based on SEM as well as histochemical analyses, including fibrous networks indicative of collagen bundles. Gene ontology analysis of the mass spectroscopic proteomic profile was employed to explore the possible functionality of the extracellular proteins remaining post-decellularization. All three scaffold preparations displayed common functional features related to cell adhesion, biological adhesion, and response to wounding. All of the aforementioned functions may prove to be desirable traits for a reconstructive or regenerative bioscaffold product designed for a particular or unique application.
Intact genomic DNA retained in decellularized tissues may prove to induce an immune response and foreign body reaction by the host to scaffold implants. As such, Gilbert et al. have suggested that 50 ng/mg dry weight is a part of the minimum criteria for effective decellularization in terms of the amount of genomic DNA remaining in decellularized biological scaffolds [43]. This value was approached with both M1 and M2 based on spectrophotometric readings; however, Method 3 genomic DNA content exceeded this threshold value. The absence or presence of visible nuclear structures based on H&E staining complemented the spectrophotometric detection of genomic DNA.
Adipose tissue is distinguished from other tissue types due to its relatively high triglyceride content. The decellularization methods were all expected to deplete triglycerides, albeit through different mechanisms. Indeed, triglyceride content was found to be highest in scaffolds prepared using the detergent based M2. The relative triglyceride content was reduced using either enzymatic (M1) or solvent (M3) based approaches. The triglyceride retention in the M2 product suggests that the method is less stringent with respect to lipid removal. It remains be determined whether the triglyceride content of the decellularized adipose product will impact its properties and utility for cosmetic and reconstructive surgery. As triglycerides can act to induce adipogenesis at the expense of osteogenesis, the final triglyceride content of the product may have potential benefits with respect to soft tissue augmentation and reconstruction while being detrimental to bone repair [44].
An important feature of an ideal scaffold product is its retention of the ECM protein content and the relative depletion of cytoplasmic and nuclear protein content [43, 45]. The cellularized scaffolds displayed an enrichment of ECM proteins based on analyses of the mass spectrometry proteomics. This is consistent with the findings based on histochemical and SEM analyses, which suggested a well-maintained ECM following decellularization independent of the method. Since a critical goal of decellularization is to yield a defined ECM product, it was necessary to confirm that the major ECM structural proteins- collagen, laminin, fibronectin and elastin were preserved following decellularization. Mass spectrometry-based proteomic data from all three decellularization procedures revealed the ubiquitous presence of type VI collagen, and a varying presence of laminin and fibronectin. While elastin was not detected by mass spectrometry, this does not necessarily rule out its presence since certain peptides may be masked by others and thus may not be uncovered under this type of analysis. Indeed, the fact that high abundance proteins may interfere with the detection of other protein features detected by protein labeling presents a possible limitation to the current study. This can lead to a lack of labeling or a false minimal labeling with subsequent low detection levels for low abundance proteins. Thus, the LC-MS/MS findings herein represent only a subset of the total proteome contained within these biological scaffolds. Future studies may need to selectively remove known high abundance proteins to improve sensitivity. Such an approach, using selective antibody depletion of high abundance proteins identified in the top twenty hits, has been employed successfully in analyses of human serum [46].
Based on mass spectrometry, both the M2 and M3 scaffold preparations retained the largest variety of extracellular matrix proteins relative to those detected within intact adipose tissue. Additionally, M2 scaffold showed minimal retention of nuclear proteins. In contrast, M1 displayed a depletion not only of cytoplasmic and nuclear proteins, but resulted in a more restricted subset of ECM proteins relative to those found in intact adipose tissue. In this respect, M1 yielded the most “homogenous” decellularization product based on the diversity of its ECM protein profile as compared to M2 and M3; however, the depletion of cytoplasmic and nuclear associated proteins components could alter the biological features of the M1 product. Consistent with this observation, Calle et al., using a quantitative [C13] labeling mass spectrometry approach, found that the milder detergent based decellularization methods differentially extracted and enriched lung matrix proteins [34]. Their comparison found that the milder SDS/Triton X-100 detergent increased the yield of basement membrane proteins, proteoglycans, and laminin relative to the harsher CHAPS based decellularization [34]. Thus, the selection of a particular decellularization protocol can significantly influence the composition of final extracellular matrix product. Furthermore, the use of mass spectrometry modified to allow for quantitative measurements has merit for future studies of the decellularized adipose matrix.
Consistent with previously published studies using adipose-derived ECM prepared using a variety of decellularization protocols [10, 11, 14, 15, 17, 20, 28, 32], implantation of scaffolds prepared using M1 and M2 demonstrated the ability to promote formation of an adipose depot in vivo. The implants displayed a time dependent infiltration of GFP+ adipocytes and vascular cells from the adjacent tissues that was accompanied by the presence of characteristic mature adipocytes. While only a few scattered cell nuclei were detected within the M1 and M2 scaffolds at earlier time points (3 weeks), the number of nuclei present based on H&E staining increased considerably at later time points (9 weeks). These preliminary in vivo studies validate the ability of M1 and M2 scaffolds to promote native cell migration and de novo formation of soft tissue, confirming previous reports in the literature [12, 17].
The current study has several limitations that merit consideration with respect to future experimentation. First, the MS-based proteomic methods employed were qualitative rather than quantitative. The use of [13C] isotope labeled standards, as employed by Calle et al. in analyses of the decellularized lung, could address this issue [34]. Additionally, label free mass spectrometry methods have been developed which calculate the total protein injected into the assay system and use this as a basis for quantifying individual peptide concentrations [47]. Second, the current study examined only subcutaneous adipose tissue from a limited number of lean (BMI < 25) donors. Future studies will need to compare the mass spectrometrically identified proteome of subcutaneous to visceral and other adipose depots from lean, overweight, and obese individuals. Baker et al. have pioneered such studies by documenting the effect of decellularized extracellular matrix from visceral adipose tissue on the metabolism of cultured pre-adipocytes from diabetic and non-diabetic subjects [48]. It will be informative to determine whether correlations exist between the metabolic outcomes and the decellularized adipose ECM proteomes as a function of patient disease. Finally, the current study did not address the biophysical and mechanical properties of the decellularized adipose ECM as had been earlier addressed by Omidi et al. [18]. Studies correlating the elasticity, stiffness and compressive properties of the bioscaffolds to the proteome as a function of donor and depot origin would extend the scope of the current data.
In summary, the decellularization methods explored in this study were chosen based on fundamental differences in processing- enzymatic, detergent, and solvent. Each method resulted in products with promising features. M1 generated a product with a more narrowly focused subset of ECM proteins, yielding a product containing fewer identified cytoplasmic and nuclear proteins relative to the other decellularization Methods. M2 successfully generated a bioscaffold product isolated without the use of xenogeneic enzymes. This has the potential to reduce the likelihood of a subsequent adverse immunologic response or foreign body response to the implant by the patient or host. The M3 process was explored since it mimics the process used to manufacture commercially available Matrigel™. Since Matrigel™ has desirable thermolabile and biomaterial features, such as maintaining a liquid form at low temperatures and a solid form at body temperature, a human adipose tissue-derived bioscaffold would be desirable from a clinical translation perspective for treatments requiring small volume injections; however, degradation of the matrix may interfere with its utility in larger volume applications. Each of the individual methods has features which may provide economies of scale in bioprocessing human adipose tissue commercially. Likewise, each method has elements that may prove disadvantageous during the scale-up process. Nevertheless, while further work will be necessary to define an optimal decellularization method for large scale bioscaffold production, the current study demonstrates that comparable ECM products can be obtained with varying combinations of enzymatic, detergent, and solvent based decellularization approaches. Eventually, it may prove useful to develop a robust and reliable quantitative mass spectrometry lot release assay for the characterization and validation of large scale produced decellularized adipose tissue matrices.
CONCLUSIONS
The objective of the current study was to compare the proteome of human adipose tissue decellularized by multiple methods and to explore its potential application as a scaffold promoting adipogenesis in vivo. Based on histological results, human adipose tissue can be successfully decellularized using any of three methods involving a combination of detergents, enzymes, and/or solvents. Based on mass spectrometric analyses, the resulting scaffolds retain a common subset of extracellular matrix proteins; however, the retention of cytoplasmic and nuclear proteins varies depending on the extraction methodology. In confirmation of prior publications, two of the matrices displayed the ability to promote in vivo adipose depot formation through the recruitment of host progenitor cells and vasculature. In conclusion, multiple decellularization methods can be applied to human adipose tissue to create a product with potential tissue engineering utility; however, the resulting proteome of the decellularized matrix varies dependent on the decellularization method.
Supplementary Material
Supplemental Figure 1. Gross Morphoglogy of Representative M1 and M2 Implants. (A) Photographic image of M1, M2, and silk scaffolds in situ after 6 and 9 weeks of implantation. (B) Higher magnification image of the M1 and M2 scaffolds before implantation and at 6 or 9 weeks after implantation in the murine host.
Supplemental Table 1. Proteins identified by mass spectrometry in native (unprocessed) adipose tissue.
Supplemental Table 2. Proteins identified by mass spectrometry in adipose tissue decellularized by Method 1.
Supplemental Table 3. Proteins identified by mass spectrometry in adipose tissue decellularized by Method 2.
Supplemental Table 4. Proteins identified by mass spectrometry in adipose tissue decellularized by Method 3.
Supplemental Table 5. Top twenty most abundant peptides identified in each sample. Column 1 (far left) lists peptide names and molecular weights. Peptides common among samples are noted in column 2, 3, and 4. Subcellular locations of peptides are listed in column 5.
Supplemental Table 6. Gene Ontology Analyses of Mass Spectrometrically Identified Proteins in Decellularized Adipose Tissue Prepared by Methods M1, M2, and M3. The total proteome as determined by LC/MS-MS is evaluated by Gene Ontology in the M1, M2, and M3 decellularized matrices.
Acknowledgments
DISCLOSURES:
The authors thank Dr. James Wade and his staff and patients (Baton Rouge LA) for their support of and participation in this study. Additionally, the authors thank Dr. David Kaplan and Dr. Rosalyn Abbott at the Tufts University Tissue Engineering Resource Center for providing the HFIP silk scaffolds. This work utilized the facilities of the Cell Biology and Bioimaging Core that are supported in part by COBRE (NIH 8 P20-GM103528) and NORC (NIH 2P30-DK072476) center grants from the National Institutes of Health. CT-P, TF, BAB, and JMG were supported in part by funding from the National Institues of Health (R21DK094254 and R21DK094254S1) and the Musculoskeletal Transplant Foundation (Established Investigator Award). JL received financial support from the China Scholarship Council (CSC), No. 201406240118. FZ received financial support from CAPES, Brazil (Process BEX 1524/15-1). MB and GW were supported by funding from NIMHD (2G12MD007595). JMG, XW, and TF are co-owners of Obatala Sciences, Inc. where TF serves as President and CEO while JMG and XW are co-owners, as well as Chief Scientific Officer and Vice President of Research & Development of LaCell LLC, respectively; FS and TF were LaCell LLC employees at the time of this study. CT-P performed this study as partial fullfillment of her PhD thesis requirements at Tulane University School of Medicine.
References
- 1.Kaufman MR, Bradley JP, Dickinson B, Heller JB, Wasson K, O’Hara C, Huang C, Gabbay J, Ghadjar K, Miller TA. Autologous fat transfer national consensus survey: trends in techniques for harvest, preparation, and application, and perception of short- and long-term results. Plastic and reconstructive surgery. 2007;119(1):323–31. doi: 10.1097/01.prs.0000244903.51440.8c. [DOI] [PubMed] [Google Scholar]
- 2.Coudurier J, Ho Quoc C, Ismail M, Dlimi C, Tourasse C, Delay E. Long-term outcome of lipomodeling in Poland’s syndrome: about our first case with an eleven-years’ follow-up. Ann Chir Plast Esthet. 2015;60(1):65–9. doi: 10.1016/j.anplas.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 3.Marra KG, Rubin JP. The potential of adipose-derived stem cells in craniofacial repair and regeneration. Birth Defects Res C Embryo Today. 2012;96(1):95–7. doi: 10.1002/bdrc.21001. [DOI] [PubMed] [Google Scholar]
- 4.Yoshimura K, Sato K, Aoi N, Kurita M, Inoue K, Suga H, Eto H, Kato H, Hirohi T, Harii K. Cell-assisted lipotransfer for facial lipoatrophy: efficacy of clinical use of adipose-derived stem cells. Dermatologic surgery: official publication for American Society for Dermatologic Surgery [et al] 2008;34(9):1178–85. doi: 10.1111/j.1524-4725.2008.34256.x. [DOI] [PubMed] [Google Scholar]
- 5.Pallua N, Baroncini A, Alharbi Z, Stromps JP. Improvement of facial scar appearance and microcirculation by autologous lipofilling. Journal of plastic, reconstructive & aesthetic surgery: JPRAS. 2014;67(8):1033–7. doi: 10.1016/j.bjps.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 6.Gentile P, De Angelis B, Pasin M, Cervelli G, Curcio CB, Floris M, Di Pasquali C, Bocchini I, Balzani A, Nicoli F, Insalaco C, Tati E, Lucarini L, Palla L, Pascali M, De Logu P, Di Segni C, Bottini DJ, Cervelli V. Adipose-derived stromal vascular fraction cells and platelet-rich plasma: basic and clinical evaluation for cell-based therapies in patients with scars on the face. The Journal of craniofacial surgery. 2014;25(1):267–72. doi: 10.1097/01.scs.0000436746.21031.ba. [DOI] [PubMed] [Google Scholar]
- 7.O’Brien FJ. Biomaterials and scaffolds for tissue engineering. Materials Today. 2011 Mar;14:88–95. [Google Scholar]
- 8.Flynn LE. The use of decellularized adipose tissue to provide an inductive microenvironment for the adipogenic differentiation of human adipose-derived stem cells. Biomaterials. 2010;31(17):4715–24. doi: 10.1016/j.biomaterials.2010.02.046. [DOI] [PubMed] [Google Scholar]
- 9.Turner AE, Flynn LE. Design and characterization of tissue-specific extracellular matrix-derived microcarriers. Tissue engineering Part C, Methods. 2012;18(3):186–97. doi: 10.1089/ten.TEC.2011.0246. [DOI] [PubMed] [Google Scholar]
- 10.Turner AE, Yu C, Bianco J, Watkins JF, Flynn LE. The performance of decellularized adipose tissue microcarriers as an inductive substrate for human adipose-derived stem cells. Biomaterials. 2012;33(18):4490–9. doi: 10.1016/j.biomaterials.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 11.Yu C, Bianco J, Brown C, Fuetterer L, Watkins JF, Samani A, Flynn LE. Porous decellularized adipose tissue foams for soft tissue regeneration. Biomaterials. 2013;34(13):3290–302. doi: 10.1016/j.biomaterials.2013.01.056. [DOI] [PubMed] [Google Scholar]
- 12.Choi JS, Kim BS, Kim JY, Kim JD, Choi YC, Yang HJ, Park K, Lee HY, Cho YW. Decellularized extracellular matrix derived from human adipose tissue as a potential scaffold for allograft tissue engineering. Journal of biomedical materials research Part A. 2011;97(3):292–9. doi: 10.1002/jbm.a.33056. [DOI] [PubMed] [Google Scholar]
- 13.Kim EJ, Choi JS, Kim JS, Choi YC, Cho YW. Injectable and Thermosensitive Soluble Extracellular Matrix and Methylcellulose Hydrogels for Stem Cell Delivery in Skin Wounds. Biomacromolecules. 2016;17(1):4–11. doi: 10.1021/acs.biomac.5b01566. [DOI] [PubMed] [Google Scholar]
- 14.Brown CF, Yan J, Han TT, Marecak DM, Amsden BG, Flynn LE. Effect of decellularized adipose tissue particle size and cell density on adipose-derived stem cell proliferation and adipogenic differentiation in composite methacrylated chondroitin sulphate hydrogels. Biomed Mater. 2015;10(4):045010. doi: 10.1088/1748-6041/10/4/045010. [DOI] [PubMed] [Google Scholar]
- 15.Cheung HK, Han TT, Marecak DM, Watkins JF, Amsden BG, Flynn LE. Composite hydrogel scaffolds incorporating decellularized adipose tissue for soft tissue engineering with adipose-derived stem cells. Biomaterials. 2014;35(6):1914–23. doi: 10.1016/j.biomaterials.2013.11.067. [DOI] [PubMed] [Google Scholar]
- 16.Haddad SM, Omidi E, Flynn LE, Samani A. Comparative biomechanical study of using decellularized human adipose tissues for post-mastectomy and post-lumpectomy breast reconstruction. J Mech Behav Biomed Mater. 2016;57:235–45. doi: 10.1016/j.jmbbm.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Han TT, Toutounji S, Amsden BG, Flynn LE. Adipose-derived stromal cells mediate in vivo adipogenesis, angiogenesis and inflammation in decellularized adipose tissue bioscaffolds. Biomaterials. 2015;72:125–37. doi: 10.1016/j.biomaterials.2015.08.053. [DOI] [PubMed] [Google Scholar]
- 18.Omidi E, Fuetterer L, Reza Mousavi S, Armstrong RC, Flynn LE, Samani A. Characterization and assessment of hyperelastic and elastic properties of decellularized human adipose tissues. J Biomech. 2014;47(15):3657–63. doi: 10.1016/j.jbiomech.2014.09.035. [DOI] [PubMed] [Google Scholar]
- 19.Shridhar A, Gillies E, Amsden BG, Flynn LE. Composite Bioscaffolds Incorporating Decellularized ECM as a Cell-Instructive Component Within Hydrogels as In Vitro Models and Cell Delivery Systems. Methods in molecular biology. 2017 doi: 10.1007/7651_2017_36. [DOI] [PubMed] [Google Scholar]
- 20.Tan QW, Zhang Y, Luo JC, Zhang D, Xiong BJ, Yang JQ, Xie HQ, Lv Q. Hydrogel derived from decellularized porcine adipose tissue as a promising biomaterial for soft tissue augmentation. Journal of biomedical materials research Part A. 2017;105(6):1756–1764. doi: 10.1002/jbm.a.36025. [DOI] [PubMed] [Google Scholar]
- 21.Yu C, Kornmuller A, Brown C, Hoare T, Flynn LE. Decellularized adipose tissue microcarriers as a dynamic culture platform for human adipose-derived stem/stromal cell expansion. Biomaterials. 2017;120:66–80. doi: 10.1016/j.biomaterials.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 22.Roehm KD, Hornberger J, Madihally SV. In vitro characterization of acelluar porcine adipose tissue matrix for use as a tissue regenerative scaffold. Journal of biomedical materials research Part A. 2016;104(12):3127–3136. doi: 10.1002/jbm.a.35844. [DOI] [PubMed] [Google Scholar]
- 23.Zhang S, Lu Q, Cao T, Toh WS. Adipose Tissue and Extracellular Matrix Development by Injectable Decellularized Adipose Matrix Loaded with Basic Fibroblast Growth Factor. Plastic and reconstructive surgery. 2016;137(4):1171–80. doi: 10.1097/PRS.0000000000002019. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Q, Johnson JA, Dunne LW, Chen Y, Iyyanki T, Wu Y, Chang EI, Branch-Brooks CD, Robb GL, Butler CE. Decellularized skin/adipose tissue flap matrix for engineering vascularized composite soft tissue flaps. Acta biomaterialia. 2016;35:166–84. doi: 10.1016/j.actbio.2016.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banyard DA, Borad V, Amezcua E, Wirth GA, Evans GR, Widgerow AD. Preparation, Characterization, and Clinical Implications of Human Decellularized Adipose Tissue Extracellular Matrix (hDAM): A Comprehensive Review. Aesthetic surgery journal/the American Society for Aesthetic Plastic surgery. 2016;36(3):349–57. doi: 10.1093/asj/sjv170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunne LW, Huang Z, Meng W, Fan X, Zhang N, Zhang Q, An Z. Human decellularized adipose tissue scaffold as a model for breast cancer cell growth and drug treatments. Biomaterials. 2014;35(18):4940–9. doi: 10.1016/j.biomaterials.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Adam Young D, Bajaj V, Christman KL. Award winner for outstanding research in the PhD category, 2014 Society for Biomaterials annual meeting and exposition, Denver, Colorado, April 16-19, 2014: Decellularized adipose matrix hydrogels stimulate in vivo neovascularization and adipose formation. Journal of biomedical materials research Part A. 2014;102(6):1641–51. doi: 10.1002/jbm.a.35109. [DOI] [PubMed] [Google Scholar]
- 28.Wang L, Johnson JA, Zhang Q, Beahm EK. Combining decellularized human adipose tissue extracellular matrix and adipose-derived stem cells for adipose tissue engineering. Acta biomaterialia. 2013;9(11):8921–31. doi: 10.1016/j.actbio.2013.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi YC, Choi JS, Kim BS, Kim JD, Yoon HI, Cho YW. Decellularized extracellular matrix derived from porcine adipose tissue as a xenogeneic biomaterial for tissue engineering. Tissue engineering Part C, Methods. 2012;18(11):866–76. doi: 10.1089/ten.tec.2012.0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown BN, Freund JM, Han L, Rubin JP, Reing JE, Jeffries EM, Wolf MT, Tottey S, Barnes CA, Ratner BD, Badylak SF. Comparison of three methods for the derivation of a biologic scaffold composed of adipose tissue extracellular matrix. Tissue engineering Part C, Methods. 2011;17(4):411–21. doi: 10.1089/ten.tec.2010.0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu I, Nahas Z, Kimmerling KA, Rosson GD, Elisseeff JH. An injectable adipose matrix for soft-tissue reconstruction. Plastic and reconstructive surgery. 2012;129(6):1247–57. doi: 10.1097/PRS.0b013e31824ec3dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young DA, Ibrahim DO, Hu D, Christman KL. Injectable hydrogel scaffold from decellularized human lipoaspirate. Acta biomaterialia. 2011;7(3):1040–9. doi: 10.1016/j.actbio.2010.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. Journal of cell science. 2010;123(Pt 24):4195–200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calle EA, Hill RC, Leiby KL, Le AV, Gard AL, Madri JA, Hansen KC, Niklason LE. Targeted proteomics effectively quantifies differences between native lung and detergent-decellularized lung extracellular matrices. Acta biomaterialia. 2016;46:91–100. doi: 10.1016/j.actbio.2016.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frazier TP, Bowles A, Lee S, Abbott R, Tucker HA, Kaplan D, Wang M, Strong A, Brown Q, He J, Bunnell BA, Gimble JM. Serially Transplanted Nonpericytic CD146(-) Adipose Stromal/Stem Cells in Silk Bioscaffolds Regenerate Adipose Tissue In Vivo. Stem cells. 2016;34(4):1097–111. doi: 10.1002/stem.2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kleinman HK. Preparation of basement membrane components from EHS tumors. 1998 doi: 10.1002/0471143030.cb1002s00. [DOI] [PubMed] [Google Scholar]
- 37.Scarritt ME, Bonvillain RW, Burkett BJ, Wang G, Glotser EY, Zhang Q, Sammarco MC, Betancourt AM, Sullivan DE, Bunnell BA. Hypertensive rat lungs retain hallmarks of vascular disease upon decellularization but support the growth of mesenchymal stem cells. Tissue engineering Part A. 2014;20(9–10):1426–43. doi: 10.1089/ten.tea.2013.0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pashos NC, Scarritt ME, Eagle ZR, Gimble JM, Chaffin AE, Bunnell BA. Characterization of an Acellular Scaffold for a Tissue Engineering Approach to the Nipple-Areolar Complex Reconstruction. Cells, tissues, organs. 2017;203(3):183–193. doi: 10.1159/000455070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou C, Zhong Q, Rhodes LV, Townley I, Bratton MR, Zhang Q, Martin EC, Elliott S, Collins-Burow BM, Burow ME, Wang G. Proteomic analysis of acquired tamoxifen resistance in MCF-7 cells reveals expression signatures associated with enhanced migration. Breast cancer research: BCR. 2012;14(2):R45. doi: 10.1186/bcr3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(5):3. [PubMed] [Google Scholar]
- 41.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 42.B A, Frazier TP, Lee S, Abbott R, Tucker HA, Kaplan D, Wang M, Strong A, Brown Q, Heb J, Bunnell BA, Gimble JM. Serially transplanted non-pericytic CD146- Adipose Stromal/Stem Cells in silk bioscaffolds regenerate adipose tissue in vivo Stem cells. 2016 doi: 10.1002/stem.2325. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilbert TW, Freund JM, Badylak SF. Quantification of DNA in biologic scaffold materials. The Journal of surgical research. 2009;152(1):135–9. doi: 10.1016/j.jss.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diascro DD, Jr, Vogel RL, Johnson TE, Witherup KM, Pitzenberger SM, Rutledge SJ, Prescott DJ, Rodan GA, Schmidt A. High fatty acid content in rabbit serum is responsible for the differentiation of osteoblasts into adipocyte-like cells. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 1998;13(1):96–106. doi: 10.1359/jbmr.1998.13.1.96. [DOI] [PubMed] [Google Scholar]
- 45.Lin CH, Kao YC, Lin YH, Ma H, Tsay RY. A fiber-progressive-engagement model to evaluate the composition, microstructure, and nonlinear pseudoelastic behavior of porcine arteries and decellularized derivatives. Acta biomaterialia. 2016;46:101–111. doi: 10.1016/j.actbio.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 46.Adkins JN, Varnum SM, Auberry KJ, Moore RJ, Angell NH, Smith RD, Springer DL, Pounds JG. Toward a human blood serum proteome: analysis by multidimensional separation coupled with mass spectrometry. Molecular & cellular proteomics: MCP. 2002;1(12):947–55. doi: 10.1074/mcp.m200066-mcp200. [DOI] [PubMed] [Google Scholar]
- 47.Schubert OT, Rost HL, Collins BC, Rosenberger G, Aebersold R. Quantitative proteomics: challenges and opportunities in basic and applied research. Nature protocols. 2017;12(7):1289–1294. doi: 10.1038/nprot.2017.040. [DOI] [PubMed] [Google Scholar]
- 48.Baker NA, Muir LA, Washabaugh AR, Neeley CK, Chen SY, Flesher CG, Vorwald J, Finks JF, Ghaferi AA, Mulholland MW, Varban OA, Lumeng CN, O’Rourke RW. Diabetes-Specific Regulation of Adipocyte Metabolism by the Adipose Tissue Extracellular Matrix. The Journal of clinical endocrinology and metabolism. 2017;102(3):1032–1043. doi: 10.1210/jc.2016-2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Gross Morphoglogy of Representative M1 and M2 Implants. (A) Photographic image of M1, M2, and silk scaffolds in situ after 6 and 9 weeks of implantation. (B) Higher magnification image of the M1 and M2 scaffolds before implantation and at 6 or 9 weeks after implantation in the murine host.
Supplemental Table 1. Proteins identified by mass spectrometry in native (unprocessed) adipose tissue.
Supplemental Table 2. Proteins identified by mass spectrometry in adipose tissue decellularized by Method 1.
Supplemental Table 3. Proteins identified by mass spectrometry in adipose tissue decellularized by Method 2.
Supplemental Table 4. Proteins identified by mass spectrometry in adipose tissue decellularized by Method 3.
Supplemental Table 5. Top twenty most abundant peptides identified in each sample. Column 1 (far left) lists peptide names and molecular weights. Peptides common among samples are noted in column 2, 3, and 4. Subcellular locations of peptides are listed in column 5.
Supplemental Table 6. Gene Ontology Analyses of Mass Spectrometrically Identified Proteins in Decellularized Adipose Tissue Prepared by Methods M1, M2, and M3. The total proteome as determined by LC/MS-MS is evaluated by Gene Ontology in the M1, M2, and M3 decellularized matrices.