

Zika virus (ZIKV) infection, which causes congenital malformations, including microcephaly and other neurological disorders, has attracted global attention. We observed that several NSAIDs significantly inhibited ZIKV infection. Based on our observations, we propose a novel mechanism of action of antiviral compounds which involves the blockade of virus entry via degradation of the entry cofactor. Furthermore, NSAIDs can be practically used for preventing ZIKV infection in pregnant women, as certain NSAIDs, including ibuprofen and acetaminophen, are considered clinically safe.
KEYWORDS: AXL, NSAID, ZIKV, ubiquitination
ABSTRACT
Zika virus (ZIKV) is genetically and biologically related to other Flaviviridae family members and has disseminated to many countries. It is associated with severe consequences, including the abnormal development of the neural system in fetuses and neurological diseases in adults. Therefore, the development of anti-ZIKV drugs is of paramount importance. Screening of generic drugs revealed that several nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin, ibuprofen, naproxen, acetaminophen, and lornoxicam, potently inhibited the entry of Zika virus Env/HIV-1-pseudotyped viruses. They also significantly inhibited the replication of wild-type ZIKV both in cell lines and in primary human fetal endothelial cells. Interestingly, the NSAIDs exerted this inhibitory effect by potently reducing the expression of AXL, the entry cofactor of ZIKV. Further studies showed that the NSAIDs downregulated the prostaglandin E2/prostaglandin E receptor 2 (EP2)/cAMP/protein kinase A (PKA) signaling pathway and reduced PKA-dependent CDC37 phosphorylation and the interaction between CDC37 and HSP90, which subsequently facilitated CHIP/ubiquitination/proteasome-mediated AXL degradation. Taken together, our results highlight a new mechanism of action of antiviral agents which may assist in designing a convenient strategy for treating ZIKV-infected patients.
IMPORTANCE Zika virus (ZIKV) infection, which causes congenital malformations, including microcephaly and other neurological disorders, has attracted global attention. We observed that several NSAIDs significantly inhibited ZIKV infection. Based on our observations, we propose a novel mechanism of action of antiviral compounds which involves the blockade of virus entry via degradation of the entry cofactor. Furthermore, NSAIDs can be practically used for preventing ZIKV infection in pregnant women, as certain NSAIDs, including ibuprofen and acetaminophen, are considered clinically safe.
INTRODUCTION
Zika virus (ZIKV) has a single positive-stranded RNA genome and belongs to the family Flaviviridae. It is genetically and biologically related to other Flaviviridae members, such as dengue, West Nile, yellow fever, and Japanese encephalitis viruses, and infects humans mainly via Aedes mosquitoes (1). Accumulating evidence indicates that ZIKV infection causes neurological disorders in both fetuses and adults (2–6). Various clinical disorders, including microcephaly, intrauterine growth restriction, fetal demise, eye malformations, and Guillain-Barré syndrome, have been observed to be associated with ZIKV infection by many studies (7–9). ZIKV can be transmitted via multiple routes, such as sexual or vertical routes, as well as through blood transfusion, which poses a considerable challenge in controlling epidemics (10, 11). Until now, ZIKV-specific vaccines or antiviral inhibitors have not been available in the clinic, and because of this, ZIKV infection is a matter of public health concern.
The identification of ZIKV entry-related factors represents a major challenge in the understanding of ZIKV tropism and pathogenesis. ZIKV has been detected in the placenta, amniotic fluid, and blood of newborns (12, 13). However, an upsurge of cases and studies indicating a causal relationship between ZIKV infection and fetal microcephaly make ZIKV infection a matter of immediate concern. Accumulating data suggest that the placenta and its barrier cells are infected by ZIKV, which results in the development of brain lesions in mice, pigtail macaques, and humans (3, 5, 7, 14–16). Human umbilical vein endothelial cells (HUVECs), which are one of the major placental barrier cell types, can be infected by ZIKV, suggesting that fetal endothelial cells might not be an effective barrier to ZIKV (14, 17).
In this study, we developed a high-throughput model for screening anti-ZIKV entrance inhibitors. We screened generic drugs and observed that several nonsteroidal anti-inflammatory drugs (NSAIDs) specifically induced the degradation of the ZIKV entry cofactor AXL and potently inhibited ZIKV infection. Our study reveals a new mechanism of action of antiviral agents and provides insights into the relationship between unique NSAID inhibitors and ZIKV infection, which may result in a novel treatment modality to prevent the development of fetal microcephaly and other brain lesions.
RESULTS
Identification of NSAIDs as inhibitors for the entry of ZIKV by high-throughput screening.
To identify specific ZIKV entrance inhibitors, we generated Zika virus Env/HIV-1-pseudotyped viruses which were able to infect several ZIKV-sensitive cells, such as A549 and Vero cells (Fig. 1A and B). From a library of 1,600 generic drugs, we identified 10 compounds, 50 μM concentrations of which were specifically able to inhibit the entry of Zika virus Env/HIV-1-pseudotyped viruses but not that of vesicular stomatitis virus (VSV) Env/HIV-1-pseudotyped viruses (see Table S3 in the supplemental material). Among them, we found several members of the NSAIDs, including aspirin, ibuprofen, naproxen, acetaminophen, and lornoxicam (Table S4). They inhibited the entry of Zika virus Env/HIV-1-pseudotyped viruses both in A549 cells and in Vero cells but not the entrance of dengue virus (DENV) Env/HIV-1- or VSV G/HIV-1-pseudotyped viruses (Fig. 1C). They also potently inhibited the replication of wild-type ZIKV (ZIKV strain SYSU/2016) in A549 cells at different time points (Fig. 1D). We isolated the ZIKV SYSU/2016 strain from the urine of a man who had traveled from Venezuela to China in March 2016 and amplified it in C6/36 cells or the brains of suckling mice after intracerebroventricular injection. Phylogenetic analysis of the whole genome indicated that this ZIKV strain is closely related to Brazilian and other South American isolates and belongs to the Asian lineage rather than to the African lineage (Fig. 1E). We also used two other ZIKV strains, ZIKV MR766 (Rhesus/1947/Uganda) and ZIKV PRVABC59, to confirm the inhibitory effects of NSAIDs (Fig. 1F and G). However, NSAIDs had no effect on wild-type dengue virus serotype 2 (DENV2) (Fig. 1H). The 50% inhibitory concentrations (IC50s) of the NSAIDs aspirin, ibuprofen, naproxen, acetaminophen, and lornoxicam for inhibition of wild-type ZIKV replication were 3.11 μM, 0.77 μM, 0.48 μM, 1.63 μM, and 0.21 μM, respectively (Fig. 1I). These concentrations are considerably lower than those detected in human blood plasma (which range from 100 to 400 μM) after administration of regular doses of NSAIDs (18). Notably, the 50% cytotoxic concentrations of the NSAIDs were more than 200 μM (Fig. 1J).
FIG 1.

Identification of NSAIDs as inhibitors for the entry of ZIKV by high-throughput sequencing. (A) Schematic representation of the high-throughput sequencing method. (B) Luciferase activity of different pseudotyped viruses following the assay performed as described in panel A with A549 and Vero cells. (C) A549 cells were infected with HIV-1 Luc/Zika virus Env-, HIV-1 Luc/DENV Env-, or HIV-1 Luc/VSV G-pseudotyped viruses and NSAIDs were added. After 72 h, the cells were harvested and luciferase activity was measured. (D) A549 cells were infected with the wild-type ZIKV SYSU/2016 strain (MOI = 1), and then the NSAIDs were added at 50 μM. The cell supernatants were harvested at 0 h, 24 h, 48 h, 72 h, and 96 h for plaque assay. (E) Analysis of the phylogenetic relationships among ZIKV SYSU/2016 (GenBank accession no. KY379148) and other ZIKV strains by whole-genome sequence analysis. The maximum likelihood phylogenetic tree was inferred with sequences of the whole genomes of Zika viruses with 1,000 bootstrap replications using MEGA5 software. These nucleotide sequences included those of 5 ZIKV strains from Guangdong, China, and 26 reference ZIKV sequences from GenBank. The red dots indicate strain ZIKV SYSU/2016/Guangdong, isolated in our cooperative labs, and two other strains, PRVABC59 and MR766, used in this project. The GenBank accession numbers are provided to the left of the strain names. Bootstrap support values are indicated at the nodes. The scale indicating the number of substitutions per site is shown at the bottom. (F) A549 cells were infected with the wild-type MR766 strain (MOI = 1), and then the NSAIDs were added at 50 μM. After 48 h, the cell supernatants were harvested for plaque assay. (G) A549 cells were infected with the wild-type PRVABC59 strain (MOI = 1), and then the NSAIDs were added at 50 μM. After 48 h, the cell supernatants were harvested for plaque assay. (H) A549 cells were infected with a wild-type DENV2 strain (MOI = 1), and then the NSAIDs were added at 50 μM. After 48 h, the cell supernatants were harvested for plaque assay. Data are representative of those from at least three independent experiments. The means ± SEM from three independent experiments are shown. (I) A549 cells were infected with the wild-type ZIKV SYSU/2016 strain, and then the NSAIDs were added at various concentrations. The replication of ZIKV was measured by qRT-PCR at 72 h postinfection. The IC50 was then calculated using GraphPad Prism software. (J) A549 cells were seeded in a 96-well plate and treated with NSAIDs at 200 μM. After 48 h, cell toxicity was detected by measuring CCK8 activity. Asp, aspirin; Ibu, ibuprofen; Nap, naproxen; Ace, acetaminophen; Lor, lornoxicam. *, P < 0.05; **, P < 0.01, ***, P < 0.001; ns, not significant.
NSAIDs directly downregulate AXL expression.
We first preincubated the compounds with Zika virus Env/HIV-1-pseudotyped viruses to identify the possible targets of these NSAIDs but observed no inhibition of infection (Fig. 2A). In contrast, pretreatment of the target cells with the compounds led to the significant inhibition of virus infection, indicating that NSAIDs exerted their inhibitory effect in the target cells (Fig. 2B). It is well-known that NSAIDs mainly inhibit the enzymatic activity of cyclooxygenase 1 (COX-1) and COX-2 and result in a decrease in prostaglandin E2 (PGE2) production. Based on a report that PGE2 can induce tyrosine phosphorylation of AXL (19), a potent Zika virus entry cofactor (20–22), we examined the possible effects of NSAIDs on AXL phosphorylation. Unexpectedly, a significant decrease in the AXL protein level, initially detected as the input control for AXL tyrosine phosphorylation, was observed with all NSAIDs tested (Fig. 2C). This effect was dose dependent (Fig. 2D) and increased with the incubation time (Fig. 2E). In addition, the NSAIDs also exerted a similar inhibitory effect in two other human glioma cell lines, SNB19 and LN229 (Fig. 2F). This was not due to inhibition at the transcriptional level, as the AXL mRNA level was not significantly affected (Fig. 2G). As AXL functions as a viral entry factor on the cell surface, we developed a biotin-labeled enrichment technique for cell surface proteins and observed that AXL protein expression on the cell membrane decreased (Fig. 2H). We further quantified AXL cell surface expression on A549 cells by flow cytometry (Fig. 2I) and confocal microscopy (Fig. 2J) to confirm that AXL was targeted for degradation after drug treatment. A pulse-chase experiment indicated that the NSAIDs promoted the proteolytic shedding of AXL and facilitated its decay (Fig. 2K and L). Besides, we found that both the viral envelope (E) protein and AXL were simultaneously suppressed by NSAIDs in ZIKV-infected A549 cells (Fig. 2M). As a control, ZIKV could not infect an AXL-knockout (KO) A549 cell line generated using the CRISPR technology (Fig. 2N). Notably, NSAIDs did not affect the expression level of transduced MERTK, TIM-1, or TYRO3 in A549 cells (Fig. 2O). Although it has been reported that the overexpressed MERTK, TIM-1, and TYRO3 could function as the receptors of Zika virus, the knockdown of TIM-1 with small interfering RNA (siRNA) did not affect the replication of ZIKV but did affect the replication of dengue viruses in A549 cells (Fig. 2P) (23). To investigate the underlying cellular mechanism of ZIKV infection, we performed time-of-addition experiments in A549 cells. Ibuprofen effectively inhibited ZIKV infection only when it was added 1 h prior to inoculation (Fig. 2Q). Furthermore, the reduction in the ZIKV RNA level after ibuprofen treatment was apparent either in the entry phase (0 to 4 h after infection) or at the replication phase (4 to 24 h) of the infection cycle (Fig. 2R). Taken together, these results indicate that NSAIDs inhibit ZIKV infection at the entry stage.
FIG 2.

NSAIDs directly downregulate the expression of AXL. (A) HIV-1 p24-normalized (50 ng) HIV-1 Luc/Zika virus Env-pseudotyped viruses were incubated with 50 μM NSAIDs in a 96-well plate at 37°C for 12 h. Then, the compound-virus mixtures were transferred into a Microcon 30-kDa centrifugal filter device (Millipore) and centrifuged (7,000 × g) at 4°C for 15 min. Afterwards, fresh medium was added onto the filter device twice to wash the compound-virus mixtures. Then, 0.5 ml DMEM was used to suspend the compound-virus mixtures, the filter device was centrifuged in reverse (500 × g) at 4°C for 5 min, and the solution was collected and used to infect HEK293T cells after HIV-1 p24 normalization. After 72 h, the intracellular luciferase activity was measured. (B) A549 cells were incubated with the various NSAIDs for 12 h. After washing, the cells were then infected with HIV-1 Luc/Zika virus Env-pseudotyped viruses. Intracellular luciferase activity was measured after 72 h. The assay performed during treatment was the same as the antiviral entrance assay. The results are representative of those from at least three independent experiments. (C) A549 cells were incubated with NSAIDs for 48 h, and then the expression of AXL was detected by Western blotting. (D) A549 cells were incubated with NSAIDs at various concentrations. After 48 h, the expression of AXL was detected by Western blotting. (E) A549 cells were incubated with NSAIDs at 50 μM. The expression of AXL was detected by Western blotting at several time points. (F) Human SNB19 glioma cells and LN229 glioblastoma cells were incubated with the NSAIDs at 50 μM. After 48 h, the expression of AXL was detected by Western blotting with an anti-AXL antibody. (G) A549 cells were incubated with the NSAIDs at 50 μM. After 48 h, total RNA was extracted with the TRIzol reagent and the mRNA expression of AXL was detected by qRT-PCR. (H) A549 cells were treated with NSAIDs for 48 h, and then the surface proteins were biotinylated. Comparable amounts of lysates were immunoprecipitated with streptavidin-coated agarose and detected by Western blotting with anti-AXL antibody. (I) A549 cells were treated with NSAIDs for 48 h, and then the surface AXL proteins were detected by flow cytometry. (J) A549 cells were treated with NSAIDs for 48 h, and then the surface AXL proteins were detected by confocal microscopy. Bars, 10 μm. (K) Pulse-chase experiments. Cells were pulsed with [35S]methionine for 15 min and then chased in the presence of DMSO or 50 μM ibuprofen for the indicated times. The levels of amino acids incorporated into the immunoprecipitated (IP) AXL are shown. The relative amount of AXL was calculated at several time points and normalized. (L) Pulse-chase experiments. Cells were pulsed with [35S]methionine for 15 min and then chased in the presence of DMSO, 50 μM ibuprofen, 50 μM ibuprofen plus 10 μM MG132, or 10 μM MG132 for the indicated times. The levels of amino acids incorporated into the immunoprecipitated AXL are shown. The relative amount of AXL was calculated and normalized. (M) A549 cells were infected with the wild-type ZIKV SYSU/2016 strain, and then the NSAIDs were added at 50 μM. After 48 h, the cells were harvested for Western blotting with primary antibodies against the AXL and ZIKV E proteins. (N) AXL−/− A549 cells were generated by CRISPR/Cas9 gene editing. The A549 cells or AXL−/− A549 cells were infected with the wild-type ZIKV SYSU/2016 strain (MOI = 1). After 48 h, the cells were harvested for Western blotting with primary antibodies against the AXL and ZIKV E proteins. (O) A549 cells were transfected with TIM-1–FLAG-, TYRO3-FLAG-, and MERTK-FLAG-expressing plasmids and then incubated with the NSAIDs at 50 μM. After 48 h, the expression of TIM-1, TYRO3, and MERTK was detected by Western blotting with an anti-FLAG antibody. (P) A549 cells were transfected with siRNA against AXL or TIM-1, and then these cells were infected with wild-type ZIKV or DENV2. After 48 h, cells were collected for Western blotting with primary antibodies against the AXL, TIM-1, ZIKV E, and DENV NS1 proteins. (Q) (Top) Schematic illustration of a time-of-addition experiment for treatment with ibuprofen (WB, Western blotting); (middle) representative images of Western blots of A549 cells after treatment with 50 μM ibuprofen for 1 h before or 4 h after infection with MR766 (MOI = 1); (bottom) quantification of ZIKV E protein band intensities relative to those for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (n = 3 cultures). Data were normalized to those obtained for DMSO-treated cells. Values represent the mean ± SD. (R) A549 cells were treated with 50 μM ibuprofen or DMSO for 1 h prior to infection. Cells were incubated with the viral inoculum at an MOI of 1 for 2 h on ice and then at 37°C (denoted the 0-h time point). Cells were washed with PBS, and total RNA was collected and purified at each time point for qRT-PCR analysis of ZIKV RNA. The relative ZIKV RNA levels after normalization to the GAPDH RNA levels in the same sample are plotted. P values were determined by one-way ANOVA for comparison with the DMSO treatment (n = 3 cultures). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
The CHIP E3 ligase-mediated ubiquitin pathway is involved in AXL degradation.
To investigate the mechanism underlying the NSAID-mediated degradation of AXL, we first treated cells with the proteasome inhibitors MG132 and bortezomib (Velcade) or the lysosome inhibitor concanamycin A (ConA). The proteasome inhibitors, but not the lysosome inhibitor, significantly reversed the effects of the NSAIDs (Fig. 3A). To investigate this further, we assessed the ubiquitin conjugation of AXL and observed that the level of ubiquitinated AXL increased with NSAID treatment in the presence of MG132 (Fig. 3B and C). To systematically search for factors that may be involved in NSAID-induced AXL degradation, we performed a cell membrane enzyme-linked immunosorbent assay (ELISA) with the depletion of a number of membrane protein-related E3 ligases or some endocytosis-related factors through the use of a mini-siRNA library. We observed that CHIP participated in AXL degradation (Fig. 3D and E) (24). The knockdown of CHIP with siRNA inhibited the NSAID-induced degradation of AXL, which was rescued by CHIP overexpression (Fig. 3F and G). The interaction of CHIP with AXL was enhanced by NSAID treatment (Fig. 3H). Collectively, our results indicate that the NSAIDs induced AXL degradation through the CHIP/ubiquitination proteasome system (UPS) (25, 26).
FIG 3.

The CHIP E3 ligase-mediated ubiquitin pathway is involved in the degradation of AXL. (A) A549 cells were incubated with NSAIDs, and then 2 μM MG-132, 100 nM bortezomib, or 50 nM ConA was added. At 48 h posttreatment, the cells were collected for Western blotting. (B) A549 cells were transfected with FLAG-tagged AXL and then treated with or without 50 μM NSAIDs for 48 h, and then 2 μM MG-132 was added. After co-IP with AXL antibody, the purified proteins were subjected to Western blotting with primary antibodies to AXL, GAPDH, and ubiquitin (Ub; antibody FK2). (C) Proximity ligation assay to detect the ubiquitin of AXL in A549 cells. A549 cells were treated with DMSO, ibuprofen, or ibuprofen plus MG132 for 48 h. Then, the ubiquitin signals were captured by a Sigma Duolink in situ red starter kit mouse/rabbit (catalog number DUO92101-1KT). Red, ubiquitin (antibody FK2); green, AXL (antibody C89E7); blue, DAPI. Bars, 10 μm. (D) Screening of membrane proteins related to E3 ligase and some endocytosis-related factors with an siRNA library. A549 cells were seeded in a 96-well plate and were transfected with an siRNA library, followed by treatment with 50 μM ibuprofen. After 48 h, cells were detected by a cell membrane ELISA. The assays were repeated 3 times, and the siRNAs enhanced 20% were chosen for use in the next step. (E) A549 cells were transfected with siRNA against VPS25, MARCH-4, and CHIP, followed by treatment with 50 μM ibuprofen. After 48 h, the cells were detected by a cell membrane ELISA. The assays were repeated 3 times, and the siRNAs enhanced 20% were chosen for use in the next step. *, P < 0.05; **, P < 0.01; ns, not significant. (F) A549 cells were first treated with DMSO or ibuprofen and then transfected with siRNA against the negative control (NC) (si-NC) or CHIP (si-CHIP). After 48 h, cell surface AXL expression was detected by flow cytometry. (G) A549 cells were transfected with 50 nM siRNA against NC, 50 nM siRNA against CHIP, or 50 nM siRNA against CHIP plus 500 ng the pcDNA3.1-CHIP or pcDNA3.1 plasmid. Then, the cells were incubated with or without ibuprofen. After 48 h, cells were harvested and the expression of AXL was detected by Western blotting. GFP, green fluorescent protein. (H) A CHIP-HA-expressing plasmid was transfected into HEK293T cells, and the cells were treated with or without ibuprofen. At 48 h posttransfection, cells were collected for co-IP with an anti-HA antibody, followed by Western blotting with an anti-AXL antibody. The plasmid expressing green fluorescent protein-HA served as a negative control.
NSAIDs induce the dephosphorylation of CDC37 via the PGE2/cAMP/PKA pathway.
We further studied the signaling pathway(s) potentially used by NSAIDs for AXL downregulation. PGE2 significantly reversed the effect of NSAIDs, indicating that NSAIDs inhibit AXL expression by decreasing PGE2 synthesis (Fig. 4A). Indeed, NSAIDs significantly inhibited the basal concentrations of PGE2 in A549 cells (Fig. 4B). To further confirm that the effect of NSAIDs was due to COX inhibition, we transfected COX-1- or COX-2-specific siRNAs in A549 cells and observed that both cyclooxygenases were involved in AXL maintenance (Fig. 4C). PGE2 acts by interacting with its receptor, EP, leading to the activation of adenylyl cyclase (AC) and an increase in the cAMP level; subsequently, cAMP activates protein kinase A (PKA) (27). To further investigate if this signaling pathway is involved in AXL degradation, we used EP receptor inhibitors and observed that addition of the EP2 inhibitor PF-04418948 (28) rather than the EP4 inhibitor (29) facilitated AXL degradation (Fig. 4D). Next, we added forskolin, a specific activator of AC, to the cell culture (30). The results showed that forskolin effectively counteracted the inhibitory effect of NSAIDs on AXL. However, a specific PKA kinase inhibitor, H89, significantly facilitated AXL degradation (Fig. 4E); a PKA-specific siRNA also exerted a similar effect (Fig. 4F). Interestingly, H89 itself could also significantly inhibit the replication of wild-type ZIKV in A549 cells (Fig. 4G). Overall, these results indicate that NSAIDs promote AXL degradation by downregulating PKA activity via the PGE2/EP2/cAMP/PKA pathway.
FIG 4.

NSAIDs induce the dephosphorylation of CDC37 by the PGE2/cAMP/PKA pathway. (A) A549 cells were incubated with the NSAIDs for 48 h and then were treated with or without 5 μM PGE2 for 24 h before harvest. The cells were subjected to Western blotting. (B) A549 cells were treated with different concentration of NSAIDs, and after 48 h the cells were harvested and lysed for detecting PGE2 via ELISA. P values were determined by one-way ANOVA for comparison with the DMSO treatment (n = 3 cultures). *, P < 0.05; **, P < 0.01. (C) A549 cells were transfected with 50 nM siRNAs against COX-1 and/or COX-2. After 48 h, cells were subjected to Western blotting with an anti-AXL antibody. (D) An EP2 or an EP4 inhibitor was added to the A549 cells. After 48 h, the cells were subjected to Western blotting with an anti-AXL antibody. (E) A549 cells were incubated with the NSAIDs for 48 h, and then the culture was treated with or without 10 μM forskolin (FSK) or 5 μM H89 for 24 h before harvest. Then, the cells were subjected to Western blotting. (F) A549 cells were transfected with 50 nM siRNA against PKA (si-PKA) and were then treated with or without 50 μM NSAIDs. After 48 h, the cells were harvested and subjected to Western blotting with an anti-AXL antibody. (G) A549 cells were infected with the wild-type ZIKV SYSU/2016 strain, and then H89 was added at 5 μM or 10 μM. After 48 h, the cells were harvested for Western blotting with primary antibodies against AXL or the ZIKV E protein.
NSAIDs inhibit the formation of the HSP90-CDC37 complex.
As HSP70, CHIP, HSP90, and CDC37 are involved in the CHIP-mediated pathway, we then investigated the putative PKA phosphorylation site(s) in these proteins (26). CDC37 is an important cochaperone for HSP90, which stabilizes and activates more than half of the human kinome (31–34). We performed phosphorylation mass spectrometry (phosphor-MS) analysis of the above-mentioned proteins, which were immunoprecipitated from the cells treated with or without ibuprofen. The phosphor-MS data revealed a phosphorylated peptide corresponding to amino acid residue serine 97 (S97) of CDC37 in immunoprecipitates prepared from dimethyl sulfoxide (DMSO)-treated cells but not in those prepared from NSAID-treated cells (Fig. 5A and B). Online software predicted that the PKA phosphorylation site was in the S97 region (Fig. 5C). Furthermore, the results of an antiphosphorylation Western blot analysis for wild-type CDC37 and CDC37 with the S97 mutation indicated that S97 was the target of PKA (Fig. 5D). PKA catalyzed the phosphorylation of glutathione S-transferase (GST)-CDC37 but not that of GST-CDC37 with the S97A mutation in vitro (Fig. 5E), which corroborates this observation. Unexpectedly, we found that treatment with the NSAIDs inhibited the HSP90-CDC37 interaction when we prepared the samples for phosphor-MS (Fig. 5F). Further coimmunoprecipitation (co-IP) analysis also supported this finding (Fig. 5G). We also found that NSAID treatment reduced the interaction between HSP90 and AXL (Fig. 5H). The mutation at S97 decreased the interaction between HSP90 and CDC37 (Fig. 5I). Depletion of CDC37 with siRNA decreased the interaction between AXL and HSP90. However, overexpression of wild-type CDC37 but not that of CDC37 with the S97A mutation rescued the AXL-HSP90 interaction (Fig. 5J). Collectively, our results indicate that NSAID-mediated dephosphorylation of CDC37 at S97 inhibits the CDC37-HSP90 interaction. As CDC37 is involved in HSP90 function and bridges the interaction between HSP90 and AXL, this inhibition leads to an incorrect AXL conformation and subsequently facilitates AXL degradation through the HSP90-CHIP-UPS pathway.
FIG 5.

NSAIDs inhibit the formation of the HSP90-CDC37 complex. (A) The phosphorylation of S97 was demonstrated from the MS/MS spectrum of the m/z 708.303324 ion at a retention time (RT) of 34.29 min. FTMS, Fourier transform mass spectrometry; Tol, tolerance. (B) The extracted ion chromatogram of m/z 708.303324 corresponds to the triply charged CDC37 phosphopeptide (amino acids [aa] 97 to 106, pSWEQKLEEMR), which was identified in immunoprecipitated CDC37-FLAG from DMSO-treated cells but not that from ibuprofen-treated cells. NL, normalized level. (C) Online software (http://kinasephos.mbc.nctu.edu.tw/) was used to predict the PKA phosphorylation site on CDC37 and show a common motif (RXXS) in S97. (D) A CDC37-FLAG- or CDC37-S97A-FLAG-expressing plasmid was transfected into A549 cells, and then the cells were treated with or without 50 μM ibuprofen. After 48 h, the cells were immunoprecipitated from total lysates with anti-FLAG antibody-coated beads. (Top) The resulting samples were subjected to Western blotting with a phosphorylation-specific PKA substrate. (Bottom) The stripped membrane was reprobed with an anti-FLAG antibody. (E) Phosphorylation of CDC37 by PKA in vitro. Bacterially produced GST fusion proteins, including wild-type CDC37 or the CDC37-S97A mutant (each at 100 ng), were incubated with or without 2,500 units of the catalytic subunit of PKA in a kinase buffer at 30°C for 1 h. The extent of phosphorylation was assessed by Western blot analysis using the phosphorylation-specific PKA substrate antibody (p-CDC37, phosphorylated CDC37) (top). The membrane was then stripped and reprobed using an anti-CDC37 antibody (bottom). (F) A549 cells were transfected with a CDC37-FLAG-expressing plasmid and were then treated with or without 50 μM ibuprofen. After 48 h, cells were harvested and cell lysates were subjected to immunoprecipitation with anti-FLAG antibody-coated beads, followed by SDS-PAGE. The gel was then stained with Coomassie brilliant blue. The indicated proteins were identified by mass spectrometry. (G) The CDC37-FLAG-expressing plasmid was transfected into A549 cells, and then the cells were treated with or without 50 μM ibuprofen. Cells were collected at 48 h posttransfection for immunoprecipitation with an anti-FLAG antibody, followed by Western blotting with an anti-HSP90 antibody. The green fluorescent protein-FLAG-expressing plasmid served as a negative control. (H) A549 cells were transfected with an HSP90-FLAG-expressing plasmid and then were treated with or without 50 μM ibuprofen. At 48 h posttransfection, cells were harvested for co-IP with FLAG beads, followed by Western blotting with an anti-AXL antibody. (I) A CDC37-FLAG-expressing or CDC37-S97A-FLAG-expressing plasmid was transfected into A549 cells, and then cells were collected at 48 h posttransfection for immunoprecipitation with FLAG beads. The resulting samples were subjected to Western blotting with anti-HSP90 or anti-FLAG antibodies. (J) A549 cells were transfected with the indicated siRNAs and/or plasmids: 50 nM siRNA against NC (si-NC) plus 4 μg HSP90-FLAG plasmid; 50 nM siRNA against CDC37 (si-CDC37) and 4 μg HSP90-FLAG-expressing plasmid; 50 nM siRNA against CDC37, 4 μg HSP90-FLAG-expressing plasmid, and 2 μg CDC37-expressing plasmid; or 50 nM siRNA against CDC37, 4 μg HSP90-FLAG-expressing plasmid, and 2 μg CDC37-S97A-expressing plasmid. After 48 h, cells were collected for co-IP experiments with FLAG beads. The resulting samples were subjected to Western blotting with anti-CDC37, anti-AXL, antiactin, and anti-FLAG (HSP90) antibodies.
NSAIDs inhibit the replication of Zika virus in HUVECs by inducing AXL degradation.
As ZIKV has been reported to cause fetal microcephaly, we then evaluated the inhibitory effect of NSAIDs on ZIKV replication in fetal endothelial cells, which act as a barrier between the maternal and fetal compartments. We observed a significant decrease in the AXL protein level in HUVECs after they were initially treated with several NSAIDs (Fig. 6A). Then, we quantified AXL cell surface expression by flow cytometry after treating HUVECs with NSAIDs and found that the drugs reduced AXL expression on the cell surface (Fig. 6B). Furthermore, the NSAIDs also indeed significantly inhibited the basal concentrations of PGE2 in A549 cells (Fig. 6C). The NSAIDs significantly reduced viral replication in HUVECs infected with ZIKV MR766 (rhesus/1947/Uganda) at a multiplicity of infection (MOI) of 1 (Fig. 6D). Furthermore, we assessed virus growth and RNA copy numbers and observed that the NSAIDs significantly inhibited ZIKV replication in HUVECs (Fig. 6E and F). To determine whether AXL expression on HUVECs contributed to ZIKV infection, we knocked out AXL using the CRISPR/Cas9 system. In agreement with the reduction in AXL expression to undetectable levels, ZIKV infection was almost abolished, as determined by measurement of E protein expression using Western blot analysis (Fig. 6G to H). As AXL is a member of the TYRO3, AXL, and MERTK (TAM) family, we also assessed the roles of MERTK and TYRO3 in ZIKV infection in HUVECs. TAM family-expressing plasmids were transfected into AXL-KO HUVECs individually, followed by infection with ZIKV MR766 (rhesus/1947/Uganda) and ibuprofen treatment. The results showed that overexpression of TAM family members rendered the AXL-KO HUVECs susceptible to ZIKV infection. However, the NSAIDs inhibited ZIKV replication only when AXL was overexpressed and not when MERTK or TYRO3 was overexpressed (Fig. 6I to J). Collectively, these results indicate that AXL is an important factor for ZIKV infection of fetal endothelial cells and that the NSAIDs inhibit ZIKV replication by specifically inducing AXL degradation.
FIG 6.

NSAIDs inhibit the replication of Zika virus in HUVECs by inducing the degradation of the AXL receptor. (A) HUVECs were incubated with the NSAIDs for 48 h, and then the cells were harvested. The expression of AXL was detected by Western blotting. (B) HUVECs were incubated with the NSAIDs for 48 h, and then the cells were harvested. The expression of surface AXL was detected by flow cytometry. (C) HUVECs were treated with different concentration of the NSAIDs, and after 48 h cells were harvested and lysed to detect PGE2 via ELISA. P values were determined by one-way ANOVA for comparison with the DMSO treatment (n = 3 cultures). *, P < 0.05; **, P < 0.01. (D) HUVECs were infected with the wild-type ZIKV MR766 strain (MOI = 1), and then the NSAIDs were added at 50 μM. After 48 h, the cells were harvested for Western blotting with primary antibodies against the ZIKV E protein. (E) HUVECs were infected with the wild-type ZIKV SYSU/2016 strain, and then the NSAIDs were added. The replication of ZIKV was measured at 72 h postinfection by qRT-PCR. **, P < 0.01. (F) HUVECs were infected with the wild-type ZIKV SYSU/2016 strain (MOI = 1), and then the NSAIDs were added at 50 μM. The cell supernatants were harvested at 0 h, 24 h, 48 h, 72 h, and 96 h for plaque assay. (G) The efficiency of AXL gene editing via the CRISPR/Cas9 method, directed by an AXL-specific sgRNA in HUVECs (AXL-KO cells; red) and wild-type HUVEC (control cells; blue), is shown. (H) HUVECs and AXL-KO HUVECs were infected with the wild-type ZIKV MR766 strain. After 48 h, the cells were harvested for Western blotting with primary antibodies against the AXL and ZIKV E proteins. (I) Plasmids expressing AXL, MERTK, and TYRO3 were individually transfected into AXL-KO HUVECs. After 24 h, these cells were infected with the wild-type ZIKV MR766 strain, and then the NSAIDs were added at 50 μM. After 48 h, cells were harvested for Western blotting with primary antibodies against the ZIKV E protein. (J) HUVECs were transfected with AXL-FLAG-, TYRO3-FLAG-, and MERTK-FLAG-expressing plasmids. After 48 h, the expression of TIM-1, TYRO3, and MERTK was detected by Western blotting with an anti-FLAG antibody.
DISCUSSION
Recent reports showed that the expression of several known flavivirus entry-related factors, including AXL, dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN), TIM-1, and TYRO3, may confer sensitivity to ZIKV in susceptible cells (14, 17, 35). In this study, we observed that NSAIDs inhibited ZIKV replication by downregulating the viral entry cofactor AXL (36), which belongs to the TAM family of receptor tyrosine kinases (RTKs), known to regulate diverse cellular processes (37). Consistent with previous results, our data also indicated that the AXL-KO cells were no longer infected with ZIKV (17, 35, 36). NSAIDs inhibited ZIKV replication mediated by overexpressed AXL but not that mediated by overexpressed TYRO3 or MERTK in HUVECs, indicating that the function of NSAIDS is AXL specific. Conversely, as the three TAM family members share high sequence homology and can partially compensate for each others' function, we speculated that TYRO3, which is expressed at low levels in most ZIKV-targeting cells under physiological conditions, may efficiently substitute for AXL and function as a ZIKV entry cofactor under certain circumstances (35). To verify this hypothesis, we detected the mRNA levels of MERTK, TYRO3, and TIM-1 in the spleens of AXL-KO mice. TYRO3 expression was 3- to 4-fold higher in the KO mouse than in the wild-type mouse, although MERTK and TIM-1 expression remained unaltered (data not shown). This result could explain why AXL-knockout mice could still be efficiently infected by ZIKV (21, 38, 39). In addition, although Wells et al. found that the loss of AXL did not affect infectivity or virus-mediated cell death in neural progenitors or cerebral organoids, they indeed observed that TYRO3 expression increased, with TYRO3 possibly acting as a substitute for AXL and functioning as a viral entry cofactor for ZIKV (39). Nevertheless, we observed that the knockout of AXL in HUVECs prevented ZIKV infection, suggesting that AXL plays a major role in mediating ZIKV infection in these human endothelial cells but not in other tissues (17). Considering that human endothelial cells are major placental barrier cells, the potent inhibitory effect of NSAIDs on ZIKV infection via AXL targeting may efficiently prevent the transmission of ZIKV from the maternal compartment to the fetal compartment.
Our findings indicate that NSAIDs potently inhibit the COX-1/2 enzymatic activity and the production of PGE2. Consequently, signal transduction through the EP2 receptor to cAMP decreases and protein kinase A activity is inhibited. Alternatively, to identify E3 ligases or endocytosis-associated factors that may be involved in AXL degradation, we generated a mini-siRNA library for membrane protein degradation-related E3 ligases or endocytosis-related factors using cell membrane ELISA. The results indicated that the CHIP pathway is the major target utilized by NSAIDs to induce AXL degradation. The function of CHIP-HSP70 is closely related to that of HSP90, which maintains newly synthesized proteins in the correct conformation. HSP90 inhibitors facilitate the degradation of client proteins via the HSP70-CHIP-UPS pathway. It is well-known that HSP70, CHIP, and HSP90 are involved in the CHIP-mediated pathway (26). The client protein, if not properly fold by HSP90, is eventually passed to HSP70-CHIP for degradation. It is not surprising that NSAID treatment eliminated AXL on the plasma membrane (PM), as 17-allylamino-17-demethoxygeldanamycin-mediated HSP90 inhibition also showed the same effect (40). CHIP can also act on the PM (41), and the degradation of certain other receptor tyrosine kinases (RTKs), such as HER2/ERBB2 and c-Met, are mediated by CHIP (26). Our results indicate that all pools of AXL, including these newly synthesized AXL pools and AXL on the PM or in endosomes for recycling, are reduced by the HSP90-CHIP system, as they affect the membrane proteins at multiple steps (34). However, as both MG132 and bortezomib significantly prevented the degradation of AXL, we believe that AXL is targeted for NSAID-mediated degradation before it reaches the plasma membrane, and a reduction in its level eventually affects the amount of AXL on the PM.
HSP90 requires various cochaperones to accomplish its function. Among them, CDC37 is an important cofactor of HSP90 that assists in stabilizing and activating more than half of the human kinome and, therefore, is one of the major cofactors that will be targeted for evaluation in further studies (31–34). To further understand the mechanism of NSAID-mediated AXL degradation, we examined the putative PKA phosphorylation site(s) in HSP70, CHIP, HSP90, CDC37, and AXL that could be sensitive to NSAIDs using phosphorylation mass spectrometry, anti-PKA phosphorylation Western blotting, site-specific mutagenesis, and in vitro phosphorylation and identified that S97 of CDC37 is the PKA target that can be inhibited by NSAIDs. As S97 is located in the long coiled-coil structure in the N-terminal domain (NTD) of CDC37, which was recently identified to be the second binding site for CDC37-HSP90 and a bridge during conformational changes associated with the CDC37-HSP90 interaction (32, 42), we hypothesized that the NSAID-mediated reduction in phosphorylation at this site may change the interface between CDC37 and HSP90 and affect their binding affinity. Incorrect interactions between CDC37 and HSP90 certainly reduce their function during multiple ATP hydrolysis events. As a result, AXL accumulates in an abortive folding complex, and HSP70-CHIP eventually mediates its degradation (Fig. 7). An HSP90 ATP hydrolysis inhibitor also potently decreases the expression of AXL expression, which thereby supports our hypothesis (40).
FIG 7.
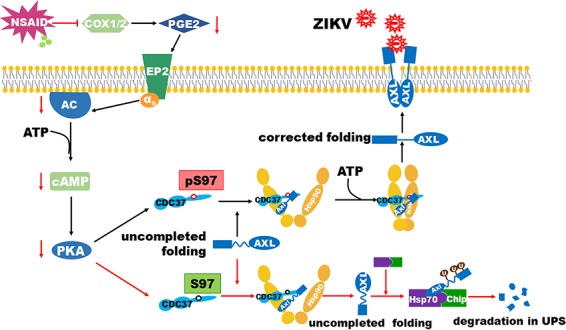
Schematic for NSAID-mediated AXL degradation. NSAIDs potently inhibit the COX-1/2 enzymatic activity and the production of PGE2. The signal through the EP2 receptor to cAMP consequently decreases. As a result, the protein kinase A (PKA) activity is inhibited. The S97 at the N-terminal domain of CDC37 is the specific target site for PKA. Its less phosphorylated status leads to a decreased interaction between CDC37 and HSP90. ZIKV entry cofactor AXL, as the client kinase, is improperly folded and eventually degraded by HSP70-CHIP-UPS.
NSAIDs are widely used in clinics to treat various diseases, including viral infections. As aspirin can trigger aberrant fetal development and cause Reye's syndrome during viral infection, it should not be used for treating ZIKV infection. Nevertheless, certain NSAIDs, such as ibuprofen and acetaminophen, which can be used to treat various diseases during pregnancy, can also be used to protect fetuses from ZIKV infection, especially during the middle stage of pregnancy (43). In conclusion, we have identified a novel target for inhibiting ZIKV replication and suggest a novel treatment modality for controlling ZIKV infection.
MATERIALS AND METHODS
Ethics statement.
All mouse experiments were carried out in strict accordance with good animal practice, as defined by the National Centre for the Replacement, Refinement & Reduction of Animals in Research (NC3Rs), and in the accordance with the Sun Yat-Sen University Laboratory Animal Center guidelines and were approved by the Institutional Animal Care and Use Committee of Sun Yat-Sen University. The Ethics Committee of Animal Experimentation of Sun Yat-Sen University approved all animal work. Animals were bred and housed under conventional conditions. The project license number assigned by the ethics committee is SYSU-2016-053. All efforts were made to minimize animal suffering. The use of human samples from ZIKV-infected patients was also approved by the Guangzhou Women and Children's Medical Center. We received the samples from its collection, and all samples were anonymized. The Zika viruses were isolated from the urine of a ZIKV-infected man who had traveled from Venezuela to China in March 2016. The symptoms and the findings of clinical examinations of this patient were well described in a recent report. This patient was the father (patient 1) of a 4-person ZIKV-infected family. Informed consent to use his urine sample was provided.
Cells and plasmids.
A549, HEK293T, and Vero cells were obtained from ATCC and maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Gibco) at 37°C with 5% CO2. The genotypes of all the cell lines were verified, and the cells were mycoplasma free. HUVECs were maintained in endothelial cell medium (catalog number SC-1001; ScienCell). The DNA sequences of the Zika virus prM/E and dengue virus prM/E proteins were chemically synthesized by Genewiz (Suzhou, China) and inserted into the pcDNA3.1 plasmid. An AXL-expressing plasmid was constructed by amplifying the mRNA of AXL by reverse transcription-PCR (RT-PCR), which was confirmed by sequencing, followed by insertion into plasmids pcDNA3.1 with a hemagglutinin (HA) tag and pcDNA3.1 with a FLAG tag. CHIP-HA-, HSP90-FLAG- and CDC37-FLAG-expressing plasmids were purchased from Vigene Biosciences Company. The siRNA-resistant CHIP- and CDC37-expressing plasmids were generated by mutating the CHIP- and CDC37-specific siRNA target sequences in the CHIP-HA- and CDC37-FLAG-expressing plasmids with multiple silent mutations (the sites of the mutations are underlined): for CHIP, 5′-CTATGAAGGAGGTTATTGA-3′, 5′-AGGCCAAGCACGACAAGTA-3′, and 5′-GGAGATGGAGAGCTATGAT-3′; and for CDC37, 5′-AGACAATCGTCATGCAATT-3′ and 5′-GGCAGTTCTTCACTAAGAT-3′.
Reagents and antibodies.
DMEM, FBS, the Lipofectamine 2000 and 3000 reagents, DAPI (4′,6-diamidino-2-phenylindole), and penicillin-streptomycin were obtained from Gibco. The NSAIDs, MG132, ConA, forskolin, and H89 were purchased from Selleck. PGE2 and balanol were purchased from Sigma. PF-04418948 and MF498 were purchased from MedChem Express. All the smart pools of siRNA were obtained from the RiboBio Company (Guangzhou, China), and the siRNA target sequences are listed in Table S1 in the supplemental material. All the information about the antibodies is also listed in Table S2.
GST-tagged protein purification.
Plasmid pGEX6p-1 expressing the GST-tagged CDC37 or GST-tagged CDC37-S97A gene was transformed into Escherichia coli BL21 competent cells (TaKaRa). After the expression of the proteins was induced by 1 mM isopropyl-thio-β-d-galactoside, the bacterial cells were lysed by sonication. The insoluble fraction was pelleted at 10,000 × g for 10 min, and the supernatant was applied to a glutathione-Sepharose column (GE). After washing, the bound GST fusion proteins were eluted and the concentration was measured by the Bradford method. The samples were subsequently aliquoted and frozen at −80°C.
Cell-based ELISA for detecting cell surface proteins.
The procedures for detecting cell surface proteins described previously were followed, with minor modifications (24). Briefly, A549 cells were seeded into a 96-well plate at 20,000 cells/well and then transfected with various siRNAs per well on the next day. After the cells were treated with the NSAIDs for 48 h, the cells were fixed with 4% paraformaldehyde and blocked with 5% milk, followed by washing with ice-cold phosphate-buffered saline (PBS). Primary antibodies from two different host species were added at 4°C for 1 h. One was specific for the target protein, AXL (rabbit), and the other was specific for IgG protein (mouse), which was used for normalization. Unbound antibody was washed away with PBS. Then, horseradish peroxidase (HRP) and alkaline phosphatase (AP) were used. Two spectrally distinct fluorogenic substrates for HRP or AP were used to simultaneously detect both the target and the normalization proteins in the same well. Fluorescence was measured with excitation at a wavelength of 540 nm and emission at a wavelength 600 nm for the F1 substrate (the HRP-specific substrate). A second reading was taken with excitation at a wavelength of 360 nm and emission at a wavelength 450 nm for the F2 substrate (the AP-specific substrate).The fluorescence of the target protein, AXL, could be adjusted to that for the normalization protein, IgG, to account for well-to-well variations.
Generation of Zika virus Env/HIV-1-pseudotyped viruses and high-throughput screening.
High-throughput screening was performed by following the procedures described in our previous report (44). The pseudotyped viruses were produced by the cotransfection of pHIV-luciferase, pCMV-ΔR8.2, and different envelope (Env)-expressing plasmids into HEK293T cells that were 90% confluent in a 10-cm plate with the Lipofectamine 2000 reagent by following the instructions of the manufacturer (Invitrogen). A 1,600-drug FDA-approved library was purchased from Topscience (Shanghai, China). High-throughput screening was conducted in 96-well plates with 50 μM compounds per well. A549 cells were infected with 200 μl HIV-1 Luc/Zika virus Env-pseudotyped viruses containing 10 μg/ml Polybrene and then incubated with the different compounds. After 72 h, the intracellular luciferase activity was examined with a GloMax 96 microplate luminometer (Promega), and the compounds that inhibited the luciferase activity by more than 50% were selected for secondary screening, which was executed with both HIV-1 Luc/Zika virus Env and HIV-1 Luc/VSV G-pseudotyped viruses by a similar procedure.
Western blotting and coimmunoprecipitation (co-IP).
A549 cells were transfected with the indicated plasmids by use of the Lipofectamine 3000 reagent. After 48 h, cells were lysed at 4°C in radioimmunoprecipitation assay (RIPA) lysis buffer (Millipore), and the lysates were clarified by centrifugation at 10,000 × g for 20 min. Then, the lysates of the proteins were subjected to Western blotting. For immunoprecipitation, comparable amounts of protein-containing lysate were incubated with the appropriate antibody for 3 h or overnight and then with protein A/G-Sepharose for 1 h at 4°C. Antigen-antibody-bead complexes were centrifuged, washed using washing buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Triton X-100), resuspended in the RIPA buffer, and subjected to SDS-PAGE. If these plasmids were HA or FLAG tagged, the immunoprecipitation was simplified with HA or FLAG beads (Sigma).
In vitro PKA phosphorylation assay.
For the in vitro PKA phosphorylation assay, the procedures described previously were followed with minor modifications (45). Briefly, GST-CDC37 and GST-CDC37-S97A fusion proteins were generated as described above. For each of the substrates, 100 ng was incubated with or without 1 μl (2,500 units) of the catalytic subunit of PKA (cAMP-dependent protein kinase [PKA]; New England BioLabs) in kinase buffer (50 mmol/liter Tris-HCl, pH 7.5, 10 mmol/liter MgCl2, 2.5 mmol/liter ATP) in a total volume of 25 μl at 30°C for 1 h. The reactions were terminated by lowering the temperature to 0°C or by adding sample loading buffer. The proteins were then subjected to SDS-PAGE and detected by Western blotting using a phospho-(Ser/Thr) PKA substrate antibody. The membranes were stripped and then reprobed using anti-CDC37 antibody.
Biotinylation of surface proteins.
The procedures for the biotinylation of surface proteins described previously were followed with minor modifications (40). Briefly, A549 cells grown to about 90% confluence were washed twice with ice-cold PBS, and surface proteins were labeled for 30 min using 1 mg/ml EZ-Link sulfo-NHS-SS-biotin (Pierce). The unreacted biotin was removed by washing with 50 mM NH4Cl in PBS. All manipulations were carried out on ice to avoid internalization at these steps. The cells were then lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA). Comparable amounts of proteins were incubated overnight with streptavidin-agarose resin (Pierce), and then the proteins were thoroughly washed, denatured, and analyzed by Western blotting with anti-AXL antibodies.
[35S]methionine-cysteine labeling and pulse-chase analysis.
For [35S]methionine-cysteine labeling and pulse-chase analysis, the procedures described previously were followed with minor modifications (31). Briefly, A549 cells plated in a 60-mm dish at 80% confluence were starved for 1 h in methionine- and cysteine-free DMEM (Sigma) containing 2% dialyzed FBS. The cells were then metabolically labeled with 200 μCi of [35S]methionine-cysteine (Express 35S protein labeling mixture; PerkinElmer Life Sciences) for 15 min in methionine- and cysteine-free medium (pulse); unbound radioactive amino acids were washed and incubated with prewarmed complete medium (chase) in the presence of DMSO or 50 μM ibuprofen. The cells were then disrupted in ice-cold RIPA lysis buffer, and comparable amounts of cell extracts were immunoprecipitated with anti-AXL antibody. After washing 3 times, samples were subjected to SDS-PAGE, followed by transfer onto a nitrocellulose membrane. The labeled proteins were visualized by autoradiography.
In vitro infection.
A549 cells or other cells were seeded in 6-well plates. Then, the cells were infected with ZIKV at an MOI of 1 and treated with NSAIDs. After 48 h, the cells were harvested for Western blotting with the primary antibody to the Zika virus E protein.
LC-MS/MS analysis.
The CDC37-FLAG-, HSP90-FLAG-, HSP70-FLAG-, or CHIP-FLAG-expressing plasmids were transfected into A549 cells. The cell lysates were prepared and incubated with anti-FLAG antibody-conjugated beads. The immunoprecipitated proteins were resolved by SDS-PAGE and stained with Coomassie brilliant blue. Protein bands corresponding to the indicated proteins from ibuprofen-treated and untreated cells were excised, reduced, alkylated, and digested with trypsin (Promega) in 50 mM ammonium bicarbonate for 16 h at 37°C. The resultant peptides were analyzed on an Easy nLC 1000 liquid chromatography (LC) system coupled to an Orbitrap Q Exactive mass spectrometer (Thermo Fisher Scientific). To generate an extracted ion chromatogram, the raw data were processed using Xcalibur software (Thermo Fisher Scientific) and directly analyzed against the Swiss-Prot database, with restriction to Homo sapiens, with the Mascot search engine (version 2.4; Matrix Science).
CRISPR/Cas9-mediated silencing of AXL expression.
The method of CRISPR/Cas9-mediated silencing was performed by following the procedures described previously with minor modifications (46). To generate the AXL gene-edited HUVECs or A549 cells, an AXL-specific single guide RNA (sgRNA; sgRNA sequences, 5′-CACCGCAGAGCCCGTGGACCTACTC-3′ and 5′-AAACGAGTAGGTCCACGGGCTCTGC-3′) was cloned into the lentiCRISPR (version 2) plasmid (catalog number 52961; Addgene). HUVECs or A549 cells were transduced with lentiviruses coexpressing Cas9 and sgRNA and selected with 2 μg/ml puromycin at 48 h postransduction for 2 days. After that, cells were detached and plated for further infection assays and the analysis of AXL expression. To generate a single AXL−/− A549 cell clone, the cells were collected and replated onto 96-well plates at a concentration of approximately 0 to 2 cells/well. One week later, cells from single colonies were selected for further validation by Western blotting and sequencing.
Wild-type ZIKV isolation.
The Zika viruses were isolated from the urine of a ZIKV-infected man who had traveled from Venezuela to China in March 2016. The symptoms and findings of clinical examinations of this patient, who was the father (patient 1) of a 4-person ZIKV-infected family, were well described in a recent report (47). Informed consent to use his urine sample was provided. The urine from this patient was passed through a filter, followed by ultracentrifugation. The concentrated urine was injected intracerebroventricularly into suckling mice. After 14 days, the ZIKV-infected mice were euthanized and the brains were ground and suspended in 1 ml of minimal essential PBS. The wild-type viruses replicated in the brains of suckling mice extensively, which was determined by a TaqMan one-step quantitative RT-PCR (qRT-PCR) on a Bio-Rad CFX96 instrument using standard cycling conditions. The successful isolation was further confirmed by reinoculation in the brains of suckling mice, and amplification was measured by qRT-PCR and a plaque assay. The amount of virus was expressed on a log10 scale as the number of viral RNA equivalents or the number of RNA copies per milliliter after comparison with a standard curve, produced using serial 10-fold dilutions of ZIKV RNA following the procedure described in a previous publication (4). The forward primer was 5′-CCGCTGCCCAACACAAG-3′, the reverse primer was 5′-CCACTAACGTTCTTTTGCAGACAT-3′, and the probe was 5′-FAM-AGCCTACCTTGACAAGCAATCAGACACTCAA-TAMRA-3′ (where FAM is 6-carboxyfluorescein and TAMRA is 6-carboxytetramethylrhodamine) (Life Technologies). Alternatively, viral replication was also measured by infecting A549 cells and Vero cells. The sequence of the ZIKV envelope was determined, and phylogenetic analysis was performed.
Wild-type ZIKV titration and plaque assay.
The titration of wild-type ZIKV was performed by a plaque assay by following the procedures described previously with minor modifications (48). Briefly, Vero cells were seeded in 12-well plates and used for infection when the cells were grown to 100% confluence. The cells were washed with phosphate-buffered saline (PBS) once and infected with a series of dilutions of viruses for 1 h at 37°C with 5% CO2. After the virus inocula were removed, the cells were washed with PBS. The cells were overlaid with agarose-DMEM containing 0.6% bovine serum albumin (BSA) and 1% low-melting-point agarose. The contents of the plates were allowed to settle at 4°C for 5 to 10 min until the agarose medium became solid, followed by culture upside down at 37°C for 96 h. The visible plaques were counted, and the virus titers were determined. The data are shown as the means ± standard errors of the means (SEM) from three independent experiments.
Statistics and graphs.
Statistical analyses were carried out using Prism software (GraphPad). All data are reported as means ± SEM. Differences were calculated by one-way analysis of variance (ANOVA) for comparison with the results obtained with the DMSO treatment and were found to be significant when P was less than 0.05, 0.01, or 0.001, as indicated in the figures. Most graphs were produced using Prism software.
Supplementary Material
ACKNOWLEDGMENTS
We thank Bisi Fu for supplying ZIKV strain MR766 (rhesus/1947/Uganda) and PRVABC59. We also thank Jun Li for supplying the human umbilical vein endothelial cells.
This work was supported by the National Special Research Program of China for Important Infectious Diseases (2018ZX10302103 and 2017ZX10202102), the Important Key Program of the Natural Science Foundation of China (81730060), the International Collaboration Program of the Natural Science Foundation of China and the U.S. NIH (81561128007), the Joint-Innovation Program in Healthcare for Special Scientific Research Projects of Guangzhou (201803040002), and the Introduction of Innovative R&D Team Program of Guangdong Province (2009010058) to H.Z. This work was also supported by the National Natural Science Foundation of China (no. 31500740), the Natural Science Foundation of Guangdong (2015A030310442), the Science and Technology Planning Project of Guangzhou (201704020226), and the Pearl River S&T Nova Program of Guangzhou (201806010118) to T.P. This work was also supported by the National Science and Technology Major Project (2018ZX10101004003001) to J.Z.
We declare that no conflict of interest exists.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01018-18.
REFERENCES
- 1.Petersen LR, Jamieson DJ, Powers AM, Honein MA. 2016. Zika virus. N Engl J Med 374:1552–1563. doi: 10.1056/NEJMra1602113. [DOI] [PubMed] [Google Scholar]
- 2.Broutet N, Krauer F, Riesen M, Khalakdina A, Almiron M, Aldighieri S, Espinal M, Low N, Dye C. 2016. Zika virus as a cause of neurologic disorders. N Engl J Med 374:1506–1509. doi: 10.1056/NEJMp1602708. [DOI] [PubMed] [Google Scholar]
- 3.Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JL, Guimaraes KP, Benazzato C, Almeida N, Pignatari GC, Romero S, Polonio CM, Cunha I, Freitas CL, Brandao WN, Rossato C, Andrade DG, Faria DDP, Garcez AT, Buchpigel CA, Braconi CT, Mendes E, Sall AA, Zanotto PM, Peron JP, Muotri AR, Beltrao-Braga PC. 2016. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534:267–271. doi: 10.1038/nature18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS. 2016. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19:720–730. doi: 10.1016/j.chom.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, Mysorekar IU, Diamond MS. 2016. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 165:1081–1091. doi: 10.1016/j.cell.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yockey LJ, Varela L, Rakib T, Khoury-Hanold W, Fink SL, Stutz B, Szigeti-Buck K, Van den Pol A, Lindenbach BD, Horvath TL, Iwasaki A. 2016. Vaginal exposure to Zika virus during pregnancy leads to fetal brain infection. Cell 166:1247–1256.e1244. doi: 10.1016/j.cell.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mlakar J, Korva M, Tul N, Popovic M, Poljsak-Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodusek V, Vizjak A, Pizem J, Petrovec M, Avsic Zupanc T. 2016. Zika virus associated with microcephaly. N Engl J Med 374:951–958. doi: 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- 8.de Paula Freitas B, de Oliveira Dias JR, Prazeres J, Sacramento GA, Ko AI, Maia M, Belfort R Jr. 2016. Ocular findings in infants with microcephaly associated with presumed Zika virus congenital infection in Salvador, Brazil. JAMA Ophthalmol 134:529–535. doi: 10.1001/jamaophthalmol.2016.0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ventura CV, Maia M, Bravo-Filho V, Gois AL, Belfort R Jr. 2016. Zika virus in Brazil and macular atrophy in a child with microcephaly. Lancet 387:228. doi: 10.1016/S0140-6736(16)00006-4. [DOI] [PubMed] [Google Scholar]
- 10.Deckard DT, Chung WM, Brooks JT, Smith JC, Woldai S, Hennessey M, Kwit N, Mead P. 2016. Male-to-male sexual transmission of Zika virus—Texas, January 2016. MMWR Morb Mortal Wkly Rep 65:372–374. doi: 10.15585/mmwr.mm6514a3. [DOI] [PubMed] [Google Scholar]
- 11.D'Ortenzio E, Matheron S, de Lamballerie X, Hubert B, Piorkowski G, Maquart M, Descamps D, Damond F, Yazdanpanah Y, Leparc-Goffart I. 2016. Evidence of sexual transmission of Zika virus. N Engl J Med 374:2195–2198. doi: 10.1056/NEJMc1604449. [DOI] [PubMed] [Google Scholar]
- 12.Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, Nascimento JM, Brindeiro R, Tanuri A, Rehen SK. 2016. Zika virus impairs growth in human neurospheres and brain organoids. Science 352:816–818. doi: 10.1126/science.aaf6116. [DOI] [PubMed] [Google Scholar]
- 13.Calvet G, Aguiar RS, Melo ASO, Sampaio SA, de Filippis I, Fabri A, Araujo ESM, de Sequeira PC, de Mendonca MCL, de Oliveira L, Tschoeke DA, Schrago CG, Thompson FL, Brasil P, Dos Santos FB, Nogueira RMR, Tanuri A, de Filippis AMB. 2016. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis 16:653–660. doi: 10.1016/S1473-3099(16)00095-5. [DOI] [PubMed] [Google Scholar]
- 14.Tabata T, Petitt M, Puerta-Guardo H, Michlmayr D, Wang C, Fang-Hoover J, Harris E, Pereira L. 2016. Zika virus targets different primary human placental cells, suggesting two routes for vertical transmission. Cell Host Microbe 20:155–166. doi: 10.1016/j.chom.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Driggers RW, Ho CY, Korhonen EM, Kuivanen S, Jaaskelainen AJ, Smura T, Rosenberg A, Hill DA, DeBiasi RL, Vezina G, Timofeev J, Rodriguez FJ, Levanov L, Razak J, Iyengar P, Hennenfent A, Kennedy R, Lanciotti R, du Plessis A, Vapalahti O. 2016. Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N Engl J Med 374:2142–2151. doi: 10.1056/NEJMoa1601824. [DOI] [PubMed] [Google Scholar]
- 16.Adams Waldorf KM, Stencel-Baerenwald JE, Kapur RP, Studholme C, Boldenow E, Vornhagen J, Baldessari A, Dighe MK, Thiel J, Merillat S, Armistead B, Tisoncik-Go J, Green RR, Davis MA, Dewey EC, Fairgrieve MR, Gatenby JC, Richards T, Garden GA, Diamond MS, Juul SE, Grant RF, Kuller L, Shaw DW, Ogle J, Gough GM, Lee W, English C, Hevner RF, Dobyns WB, Gale M Jr, Rajagopal L. 2016. Fetal brain lesions after subcutaneous inoculation of Zika virus in a pregnant nonhuman primate. Nat Med 22:1256–1259. doi: 10.1038/nm.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richard AS, Shim BS, Kwon YC, Zhang R, Otsuka Y, Schmitt K, Berri F, Diamond MS, Choe H. 2017. AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proc Natl Acad Sci U S A 114:2024–2029. doi: 10.1073/pnas.1620558114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marx J, Walls R, Hockberger R. 2013. Rosen's emergency medicine—concepts and clinical practice. Elsevier Health Sciences, Philadelphia, PA. [Google Scholar]
- 19.Ganesh K, Das A, Dickerson R, Khanna S, Parinandi NL, Gordillo GM, Sen CK, Roy S. 2012. Prostaglandin E(2) induces oncostatin M expression in human chronic wound macrophages through Axl receptor tyrosine kinase pathway. J Immunol 189:2563–2573. doi: 10.4049/jimmunol.1102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, John SP, Aker AM, Renzette N, Robbins DR, Guo Z, Green S, Kowalik TF, Brass AL. 2016. Identification of Zika virus and dengue virus dependency factors using functional genomics. Cell Rep 16:232–246. doi: 10.1016/j.celrep.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 21.Miner JJ, Sene A, Richner JM, Smith AM, Santeford A, Ban N, Weger-Lucarelli J, Manzella F, Ruckert C, Govero J, Noguchi KK, Ebel GD, Diamond MS, Apte RS. 2016. Zika virus infection in mice causes panuveitis with shedding of virus in tears. Cell Rep 16:3208–3218. doi: 10.1016/j.celrep.2016.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meertens L, Labeau A, Dejarnac O, Cipriani S, Sinigaglia L, Bonnet-Madin L, Le Charpentier T, Hafirassou ML, Zamborlini A, Cao-Lormeau VM, Coulpier M, Misse D, Jouvenet N, Tabibiazar R, Gressens P, Schwartz O, Amara A. 2017. Axl mediates ZIKA virus entry in human glial cells and modulates innate immune responses. Cell Rep 18:324–333. doi: 10.1016/j.celrep.2016.12.045. [DOI] [PubMed] [Google Scholar]
- 23.Meertens L, Carnec X, Lecoin MP, Ramdasi R, Guivel-Benhassine F, Lew E, Lemke G, Schwartz O, Amara A. 2012. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 12:544–557. doi: 10.1016/j.chom.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, Young JC, Lukacs GL. 2010. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329:805–810. doi: 10.1126/science.1191542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kundrat L, Regan L. 2010. Identification of residues on Hsp70 and Hsp90 ubiquitinated by the cochaperone CHIP. J Mol Biol 395:587–594. doi: 10.1016/j.jmb.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao Z, Li G, Shao Q, Yang G, Zheng L, Zhang T, Zhao Y. 2016. CHIP: a new modulator of human malignant disorders. Oncotarget 7:29864–29874. doi: 10.18632/oncotarget.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugimoto Y, Narumiya S. 2007. Prostaglandin E receptors. J Biol Chem 282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 28.Birrell MA, Nials AT. 2011. At last, a truly selective EP(2) receptor antagonist. Br J Pharmacol 164:1845–1846. doi: 10.1111/j.1476-5381.2011.01494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clark P, Rowland SE, Denis D, Mathieu MC, Stocco R, Poirier H, Burch J, Han Y, Audoly L, Therien AG, Xu D. 2008. MF498 [N-{[4-(5,9-diethoxy-6-oxo-6,8-dihydro-7H-pyrrolo[3,4-g]quinolin-7-yl)-3-methylbenzyl]sulfonyl}-2-(2-methoxyphenyl)acetamide], a selective E prostanoid receptor 4 antagonist, relieves joint inflammation and pain in rodent models of rheumatoid and osteoarthritis. J Pharmacol Exp Ther 325:425–434. doi: 10.1124/jpet.107.134510. [DOI] [PubMed] [Google Scholar]
- 30.Seamon KB, Padgett W, Daly JW. 1981. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci U S A 78:3363–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. 2002. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem 277:39858–39866. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- 32.Verba KA, Wang RY, Arakawa A, Liu Y, Shirouzu M, Yokoyama S, Agard DA. 2016. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352:1542–1547. doi: 10.1126/science.aaf5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karnitz LM, Felts SJ. 2007. Cdc37 regulation of the kinome: when to hold 'em and when to fold 'em. Sci STKE 2007:pe22. doi: 10.1126/stke.3852007pe22. [DOI] [PubMed] [Google Scholar]
- 34.Roe SM, Ali MM, Meyer P, Vaughan CK, Panaretou B, Piper PW, Prodromou C, Pearl LH. 2004. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 116:87–98. doi: 10.1016/S0092-8674(03)01027-4. [DOI] [PubMed] [Google Scholar]
- 35.Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, Cao-Lormeau VM, Choumet V, Briant L, Despres P, Amara A, Yssel H, Misse D. 2015. Biology of Zika virus infection in human skin cells. J Virol 89:8880–8896. doi: 10.1128/JVI.00354-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowakowski TJ, Pollen AA, Di Lullo E, Sandoval-Espinosa C, Bershteyn M, Kriegstein AR. 2016. Expression analysis highlights AXL as a candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell 18:591–596. doi: 10.1016/j.stem.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Linger RM, Keating AK, Earp HS, Graham DK. 2008. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res 100:35–83. doi: 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hastings AK, Yockey LJ, Jagger BW, Hwang J, Uraki R, Gaitsch HF, Parnell LA, Cao B, Mysorekar IU, Rothlin CV, Fikrig E, Diamond MS, Iwasaki A. 2017. TAM receptors are not required for Zika virus infection in mice. Cell Rep 19:558–568. doi: 10.1016/j.celrep.2017.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wells MF, Salick MR, Wiskow O, Ho DJ, Worringer KA, Ihry RJ, Kommineni S, Bilican B, Klim JR, Hill EJ, Kane LT, Ye C, Kaykas A, Eggan K. 2016. Genetic ablation of AXL does not protect human neural progenitor cells and cerebral organoids from Zika virus infection. Cell Stem Cell 19:703–708. doi: 10.1016/j.stem.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 40.Krishnamoorthy GP, Guida T, Alfano L, Avilla E, Santoro M, Carlomagno F, Melillo RM. 2013. Molecular mechanism of 17-allylamino-17-demethoxygeldanamycin (17-AAG)-induced AXL receptor tyrosine kinase degradation. J Biol Chem 288:17481–17494. doi: 10.1074/jbc.M112.439422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Apaja PM, Lukacs GL. 2014. Protein homeostasis at the plasma membrane. Physiology 29:265–277. doi: 10.1152/physiol.00058.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eckl JM, Scherr MJ, Freiburger L, Daake MA, Sattler M, Richter K. 2015. Hsp90 · Cdc37 complexes with protein kinases form cooperatively with multiple distinct interaction sites. J Biol Chem 290:30843–30854. doi: 10.1074/jbc.M115.693150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Savidis G, Perreira JM, Portmann JM, Meraner P, Guo Z, Green S, Brass AL. 2016. The IFITMs inhibit Zika virus replication. Cell Rep 15:2323–2330. doi: 10.1016/j.celrep.2016.05.074. [DOI] [PubMed] [Google Scholar]
- 44.Zhou N, Pan T, Zhang J, Li Q, Zhang X, Bai C, Huang F, Peng T, Zhang J, Liu C, Tao L, Zhang H. 2016. Glycopeptide antibiotics potently inhibit cathepsin L in the late endosome/lysosome and block the entry of Ebola virus, Middle East respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV). J Biol Chem 291:9218–9232. doi: 10.1074/jbc.M116.716100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lei H, Venkatakrishnan A, Yu S, Kazlauskas A. 2007. Protein kinase A-dependent translocation of Hsp90 alpha impairs endothelial nitric-oxide synthase activity in high glucose and diabetes. J Biol Chem 282:9364–9371. doi: 10.1074/jbc.M608985200. [DOI] [PubMed] [Google Scholar]
- 46.Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yin Y, Xu Y, Su L, Zhu X, Chen M, Zhu W, Xia H, Huang X, Gong S. 2016. Epidemiologic investigation of a family cluster of imported ZIKV cases in Guangdong, China: probable human-to-human transmission. Emerg Microb Infect 5:e100. doi: 10.1038/emi.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, Huang F, Tan L, Bai C, Chen B, Liu J, Liang J, Liu C, Zhang S, Lu G, Chen Y, Zhang H. 2016. Host protein Moloney leukemia virus 10 (MOV10) acts as a restriction factor of influenza A virus by inhibiting the nuclear import of the viral nucleoprotein. J Virol 90:3966–3980. doi: 10.1128/JVI.03137-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.