

Cells coinfected with viruses that possess a multipartite or segmented genome reassort to produce progeny viruses that contain a combination of gene segments from each parent. Reassortment places new pairs of genes together, generating viruses in which mismatched proteins must function together. To test if such forced pairing of proteins that form the virus shell or capsid alters the function of the particle, we investigated properties of reovirus variants in which the σ1 attachment protein and the λ2 protein that anchors σ1 on the particle are mismatched. Our studies demonstrate that a σ1-λ2 mismatch produces particles with lower levels of encapsidated σ1, consequently decreasing virus attachment and infectivity. The mismatch between σ1 and λ2 also altered the capacity of the viral capsid to undergo conformational changes required for cell entry. These studies reveal new functions of reovirus capsid proteins and illuminate both predictable and novel implications of reassortment.
KEYWORDS: reassortment, reovirus, virus entry
ABSTRACT
Following attachment to host receptors via σ1, reovirus particles are endocytosed and disassembled to generate infectious subvirion particles (ISVPs). ISVPs undergo conformational changes to form ISVP*, releasing σ1 and membrane-targeting peptides from the viral μ1 protein. ISVP* formation is required for delivery of the viral core into the cytoplasm for replication. We characterized the properties of T3DF/T3DCS1, an S1 gene monoreassortant between two laboratory isolates of prototype reovirus strain T3D: T3DF and T3DC. T3DF/T3DCS1 is poorly infectious. This deficiency is a consequence of inefficient encapsidation of S1-encoded σ1 on T3DF/T3DCS1 virions. Additionally, compared to T3DF, T3DF/T3DCS1 undergoes ISVP-to-ISVP* conversion more readily, revealing an unexpected role for σ1 in regulating ISVP* formation. The σ1 protein is held within turrets formed by the λ2 protein. To test if the altered properties of T3DF/T3DCS1 are due to a mismatch between σ1 and λ2 proteins from T3DF and T3DC, properties of T3DF/T3DCL2 and T3DF/T3DCS1L2, which express a T3DC-derived λ2, were compared. The presence of T3DC λ2 allowed more efficient σ1 incorporation, producing particles that exhibit T3DF-like infectivity. Compared to T3DF, T3DF/T3DCL2 prematurely converts to ISVP*, uncovering a role for λ2 in regulating ISVP* formation. Importantly, a virus with matching σ1 and λ2 displayed a more regulated conversion to ISVP* than either T3DF/T3DCS1 or T3DF/T3DCL2. In addition to identifying new regulators of ISVP* formation, our results highlight that protein mismatches produced by reassortment can alter virus assembly and thereby influence subsequent functions of the virus capsid.
IMPORTANCE Cells coinfected with viruses that possess a multipartite or segmented genome reassort to produce progeny viruses that contain a combination of gene segments from each parent. Reassortment places new pairs of genes together, generating viruses in which mismatched proteins must function together. To test if such forced pairing of proteins that form the virus shell or capsid alters the function of the particle, we investigated properties of reovirus variants in which the σ1 attachment protein and the λ2 protein that anchors σ1 on the particle are mismatched. Our studies demonstrate that a σ1-λ2 mismatch produces particles with lower levels of encapsidated σ1, consequently decreasing virus attachment and infectivity. The mismatch between σ1 and λ2 also altered the capacity of the viral capsid to undergo conformational changes required for cell entry. These studies reveal new functions of reovirus capsid proteins and illuminate both predictable and novel implications of reassortment.
INTRODUCTION
For segmented RNA viruses, such as influenza virus, rotavirus, and bluetongue virus, genetic reassortment is a crucial evolutionary mechanism (1). Reassortment of gene segments allows viruses to acquire novel genetic markers that are necessary to overcome host defenses (2). Reassortment can also lead to emergence of strains with superior replicative fitness or those with a capacity to infect new hosts (3). One well-studied segmented RNA virus that undergoes reassortment, both in cell culture and in infected animals, is mammalian orthoreovirus (reovirus) (4, 5). The double-stranded RNA genome of reovirus consists of 10 segments. Studies of reassortants formed by coinfection of prototype reovirus strains T1L and T3D have led to the identification of determinants that control efficiency of viral replication in cell culture and those that influence viral pathogenesis in a mouse model (6–12). As with other segmented viruses (13–18), reovirus reassortment is not random and certain gene combinations are favored, whereas others are excluded (5, 19). Although the mechanisms driving preferred selection of parental alleles are still not fully elucidated, one factor that governs the generation and recovery of a reassortant is the maintenance of functional protein-protein interactions in the capsid proteins or replication machinery of the virus (1).
The genome segments of reovirus are encapsidated within two concentric protein shells, the core and the outer capsid (20). During replication of the virus, viral mRNAs are packaged within the core along with viral polymerase and NTPase (λ3 and μ2, respectively) (20). The core is a turreted icosahedron formed from λ1, λ2, and σ2 (21). Pentamers of the λ2 protein form the turret, and this structure can only be formed in the presence of λ1 and σ2 (22, 23). μ1 and σ3, major components of the outer capsid, form heterohexamers (24). The core is connected to the μ1-σ3 layer via interaction of μ1 with both σ2 and λ2 (25, 26). Trimers of σ1 are anchored within the turret by the flap domains of λ2 (21, 25). The minor outer capsid protein σ1 is only found in particles that also contain σ3 and μ1 in their native state (27–29). This effect may be a consequence of changes in the conformation of the λ2 flap due to the presence of the adjacent μ1 protein (26, 30, 31). Fully formed virions initiate infection of new host cells, and this process requires the function of multiple capsid proteins. Attachment of host cells occurs via the σ1 protein (32). Proteolytic degradation of σ3 within the endosome results in formation of ISVPs (33). ISVPs undergo further conformational changes to form ISVP*s, resulting in changes to the λ2 and μ1 structures that facilitate the release of σ1 trimers and μ1-derived peptides (27, 29). The μ1 peptides facilitate the delivery of cores into the cytoplasm (29, 34, 35). Conformational changes in the capsid also activate the viral transcriptional machinery, which allows expression of the viral mRNA and completion of the remainder of the viral replication cycle (27). Analogous to other viral systems, it is anticipated that the protein-protein interactions that govern the proper assembly of stable virus capsids influence properties of the virus capsid.
Two laboratory isolates of prototype reovirus strain T3D, T3DF (F is for Fields) and T3DC (C is for Cashdollar) display differences in their infectious properties, including in the morphology of viral replication factories, cell-killing capacity, and in vivo replication efficiency (36–38). Here, we characterized the properties of capsids of T3DF and T3DF/T3DCS1, a monoreassortant bearing the S1 gene from T3DC in an otherwise T3DF virus. We found that compared to T3DF, particles of T3DF/T3DCS1 display an assembly defect, encapsidating less σ1. Particles of T3DF/T3DCS1 therefore exhibit a diminished capacity to attach and infect cells. Surprisingly, compared to T3DF, capsids of T3DF/T3DCS1 undergo conformational changes characteristic of ISVP-to-ISVP* conversion without an appropriate trigger. The effects of T3DCS1 on the attachment and ISVP* conversion efficiency of T3DF could be overcome by introduction of a matched λ2-encoding T3DC L2 gene. In addition to highlighting changes in σ1 that influence its encapsidation, these studies identify a previously unknown role for σ1 and λ2 in controlling conformational changes required for cell entry. These findings provide new insights into understanding how interaction and matches between proteins that form viral capsids influence properties of the capsid and may influence the generation or replicative capacity of reassortant viruses.
(This article was submitted to an online preprint archive 39.)
RESULTS
The infectivity of T3DF is compromised by introduction of the T3DC σ1 protein.
A single-gene reassortant between prototype reovirus strains T1L and T3D, which contains the μ1-encoding M2 gene segment from T3D in an otherwise T1L genetic background, exhibits enhanced attachment to host cells (40). Reovirus attachment is a function of the σ1 protein (32, 41). The μ1 protein does not make physical contact with σ1, therefore the effect of μ1 on σ1 function is unexpected (26, 40, 42). Curiously, the μ1 proteins of T1L and T3D display ∼98% identity with the two proteins, differing in only 15 out of 708 residues, which are scattered throughout the primary sequence of the protein (43). Thus, it appears that even a minimal difference in the properties of analogous proteins from two different parents can influence the phenotype of reassortant progeny. To determine whether this unforeseen phenotype of reassortment extends to other gene combinations and other virus strains, we characterized the properties of T3DF/T3DCS1, an S1 gene monoreassortant between two laboratory isolates of strain T3D: T3DF and T3DC. The S1 gene reassortant T3DF/T3DCS1 is ideal, since unlike prototype reovirus strains, such as T1L, T2J, and T3D, where the S1 gene sequences are highly divergent, the S1 genes of T3DF and T3DC differ minimally (36). The S1 gene encodes two proteins from overlapping reading frames, σ1 and σ1s (44, 45). The σ1 proteins of T3DF and T3DC differ at amino acid residues 22 and 408, resulting in a valine-to-alanine change at residue 22 and a threonine-to-alanine change at residue 408 (36). Because the 5′ end of S1 that produces a polymorphism in σ1 at position 22 also encodes σ1s in an alternate reading frame, it results in a glutamine-to-histidine change at residue 3 in σ1s (36).
During initial characterization of T3DF and T3DF/T3DCS1, we observed that compared to the parental strain T3DF, T3DF/T3DCS1 shows plaques with a predominantly diminished size in L929 cells (Fig. 1A and B). Plaque morphology is a measure of the capacity of the virus to efficiently complete all stages of the viral replication cycle and infect neighboring cells. It is also affected by the host response to virus infection. To compare the capacity of T3DF and T3DF/T3DCS1 to establish infection in host cells, L929 cells were infected with 3,000 virions/cell, and relative infectivity was determined at 18 h postinfection using quantitative indirect immunofluorescence (46). We observed that compared to T3DF, T3DF/T3DCS1 displayed significantly lower infectivity (Fig. 1C). Evaluation of the reovirus antigen-positive cells using a fluorescent focus assay indicated that a significantly greater number of cells is infected with T3DF than T3DF/T3DCS1 (Fig. 1D). In some tissues, ISVPs are formed extracellularly (47–49). Under these conditions, infection of host cells is initiated by ISVPs. As observed with the infection initiated by virions, chymotrypsin-generated ISVPs of T3DF infected L929 cells to a greater extent than similarly generated ISVPs of T3DF/T3DCS1 (Fig. 1E). Thus, our data indicate that the presence of the T3DCS1 gene segment in an otherwise T3DF background produces a virus that has a diminished capacity to establish infection in host cells.
FIG 1.

T3DF/T3DCS1 exhibits small-plaque morphology and reduced infectivity in L929 cells. (A) Particles of T3DF or T3DF/T3DCS1 diluted in PBS were subjected to plaque assay. (B) Sizes of 20 randomly selected plaques and means are shown. Error bars indicate standard deviations (SD). *, P < 0.05 as determined by Student's t test compared to T3DF. L929 cells were adsorbed with 3,000 particles/cell of either T3DF or T3DF/T3DCS1 (C), 300 particles/cell of T3DF or T3DF/T3DCS1 (D), or 300 ISVPs/cell of either T3DF or T3DF/T3DCS1 (E) at room temperature for 1 h. (C and E) After incubation at 37°C for 18 h, the cells were subjected to indirect immunofluorescence assay using a LI-COR Odyssey scanner. (C and E) Relative infectivity was determined by calculating intensity ratios at 800 nm (green fluorescence), representing viral antigen, and 700 nm (red fluorescence), representing the cell monolayer. The infectivity index for each independent infection and the sample means are shown. Error bars indicate SD. *, P < 0.05 as determined by Student's t test compared to T3DF. (D) Number of antigen-positive cells or fluorescent focus units (FFU) in the entire well were manually quantified. FFU/well for each well and sample mean are shown. Error bars indicate SD. *, P < 0.05 as determined by Student's t test compared to T3DF.
T3DF/T3DCS1 attaches to host cells less efficiently.
The S1 gene segment-encoded σ1 protein mediates attachment of type 3 reovirus strains to cell surface receptors (50–52). Since the S1 genes of T3DF and T3DC differ in their primary sequence, we asked if viruses containing each of these proteins differ in their capacity to attach to host cells. We compared the attachment of T3DF and T3DF/T3DCS1 to adherent L929 cells using a fluorescence-based quantitative binding assay (40). We observed that compared to T3DF, T3DF/T3DCS1 bound to cells less efficiently. These data suggest that the presence of T3DC σ1 in the T3DF virus diminishes the capacity of the virus to bind host cells (Fig. 2A).
FIG 2.

T3DF/T3DCS1 attaches poorly to L929 cells. (A) Confluent monolayers of L929 cells grown in 96-well plates were adsorbed with 5 × 104 particles/cell of either T3DF or T3DF/T3DCS1 at 4°C for 1 h. Cell attachment was determined by indirect immunofluorescence of cell-associated particles using a LI-COR Odyssey scanner. Binding index was determined by calculating intensity ratios at 800 nm (green fluorescence), representing viral antigen, and 700 nm (red fluorescence), representing the cell monolayer. The binding index for each independent infection and the sample means are shown. Error bars indicate SD. *, P < 0.05 as determined by Student's t test compared to T3DF. (B) To examine the effect of 9BG5 MAb on virus infectivity, 1 × 1011 particles/ml of either T3DF or T3DF/T3DCS1 were incubated at 4°C with 0 or 500 ng/ml of 9BG5 MAb hybridoma supernatant overnight. This mixture was used to initiate infection of L929 cells in 96-well plates. (C) To examine the role of glycans on infection, L929 cells were pretreated with 0 or 10 mU/ml neuraminidase. Cells were adsorbed with 3,000 particles/cell of either T3DF or T3DF/T3DCS1 at room temperature for 1 h. (B and C) After incubation at 37°C for 18 h, the cells were subjected to indirect immunofluorescence assay using a LI-COR Odyssey scanner. Relative infectivity was determined by calculating intensity ratios at 800 nm (green fluorescence), representing viral antigen, and 700 nm (red fluorescence), representing the cell monolayer. For each virus strain, the infectivity index for untreated samples was set to 100%. Percent infectivity for each independent infection and the sample mean are shown. Error bars indicate SD. *, P < 0.05 as determined by Student's t test compared to T3DF. **, P < 0.05 compared to similarly treated T3DF-infected cells.
The T3D σ1 protein engages JAM-A via its globular head domain, whereas it interacts with sialic acid via regions within the body domain (53, 54). To confirm that both T3DF and T3DF/T3DCS1 rely on the usual σ1-receptor interactions for type 3 reovirus to attach and infect cells, we assessed the capacity of reagents that diminish interaction of reovirus with each type of receptor to influence the infectivity of T3DF and T3DF/T3DCS1. Incubation of virions with T3D σ1 head-specific neutralizing monoclonal antibody (MAb), 9BG5, which prevents engagement with JAM-A (55, 56), diminished infection by both virus strains (Fig. 2B). While we note a small but statistically significant difference in sensitivity of T3DF and T3DF/T3DCS1 to 9BG5, we did not further explore this difference for the current study. Pretreatment of cells with Arthrobacter ureafaciens neuraminidase to remove cell surface sialic acid also diminished infection by both viruses (Fig. 2C). Based on the evidence that both T3DF and T3DF/T3DCS1 require JAM-A and sialic acid to efficiently establish infection in host cells, we think the difference in the attachment and infectivity of T3DF and T3DF/T3DCS1 are not related to the use of alternate receptors.
A lower level of σ1 is present on T3DF/T3DCS1 particles.
A reovirus particle can bear a maximum of 12 trimers of the σ1 protein (57). Thus, one possible reason for the lower attachment efficiency of T3DF and T3DF/T3DCS1 is the lower levels of particle-associated σ1 protein. To test this idea, we compared the level of σ1 relative to another capsid protein, μ1, using quantitative immunoblotting of particles from three independent, freshly CsCl-purified virus preparations. We found that compared to T3DF, the σ1/μ1 ratio was significantly lower for T3DF/T3DCS1 (Fig. 3A).
FIG 3.
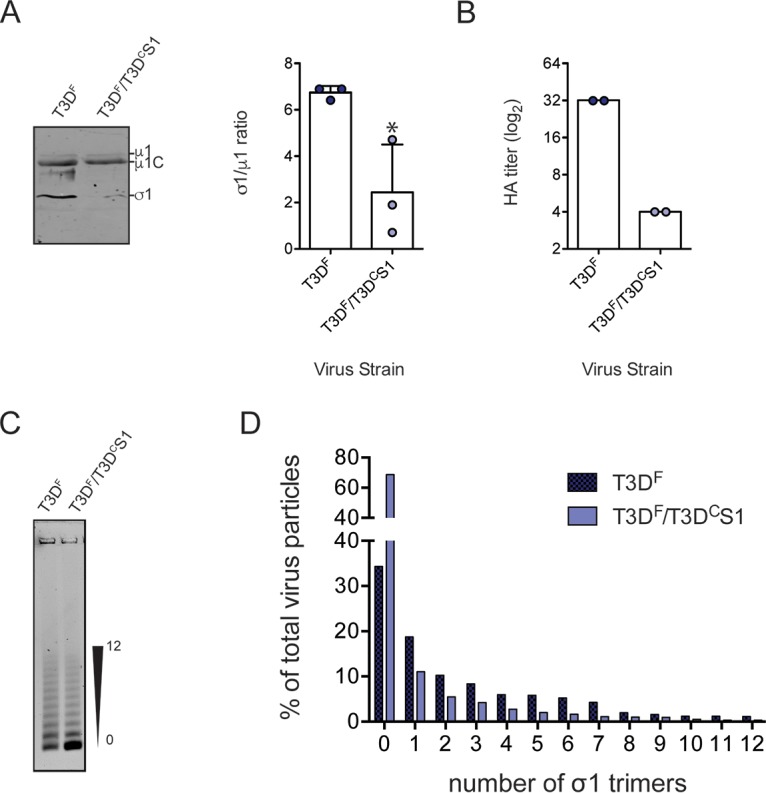
Lower level of σ1 is present on T3DF/T3DCS1 particles. (A) Purified T3DF or T3DF/T3DCS1 (2 × 1010 particles) virions from 3 independent viral preparations were subjected to immunoblotting using antibodies directed against reovirus μ1 protein and T3D σ1 head. Membranes were scanned on a LI-COR Odyssey scanner to determine σ1 and μ1 band intensities. The σ1/μ1 ratios of each independent virus preparation and means are shown. Error bars indicate SD. *, P < 0.05 as determined by Student's t test compared to T3DF. (B) Purified T3DF or T3DF/T3DCS1 virions (1 × 1011 particles) were serially diluted in PBS and incubated with bovine erythrocytes at 4°C overnight. HA titer was expressed as 2 × 1011 particles divided by the number of particles per HA unit for each independent sample, and the mean values are shown. One HA unit is equal to the number of particles of virus sufficient to produce HA. (C and D) Virions (1 × 1011 particles) were resolved on an agarose gel, stained with a colloidal blue staining kit, and scanned using a LI-COR Odyssey scanner. (C) The position of particles with the lowest and highest numbers of σ1 trimers is shown. (D) The band intensity of each virion species was quantified. The abundance of each species as a percentage of all 13 virion species present in the sample is shown.
Type 3 reovirus can agglutinate bovine red blood cells via interaction of σ1 with glycophorins on the surface of cells (58). Hemagglutination (HA) capacity can therefore vary with virus concentration. When virus particle concentration is held constant, hemagglutination capacity is indicative of σ1 levels on the particle. We observed that compared to that for T3DF, a higher concentration of T3DF/T3DCS1 was required to agglutinate red blood cells (Fig. 3B). These data confirm that compared to T3DF, particles of T3DF/T3DCS1 are decorated with lower levels of σ1.
Purified reovirus preparations consist of a conglomeration of virus particles with various amounts of σ1 trimers (59, 60). The experiments described above (Fig. 3A and B) cannot reveal if all T3DF/T3DCS1 particles encapsidate a lower level of σ1 than T3DF or if a subpopulation of virus particles lack σ1. To distinguish between these possibilities, we took advantage of a method to resolve purified virions on agarose gels based on the level of incorporated σ1 trimers (59). As expected, the preparation of T3DF contained particles bearing a varied number of σ1 molecules. Compared to those of T3DF, virions of T3DF/T3DCS1 consisted of higher numbers of virus particles that encapsidate no σ1 trimers (Fig. 3C and D). The absence of σ1 from a large proportion of T3DF/T3DCS1 particles likely explains the poor attachment capacity and infectivity of virions of this strain (Fig. 1C).
σ1 is inefficiently encapsidated on T3DF/T3DCS1 virions.
The data presented thus far describe a defect in attachment of T3DF/T3DCS1 that is produced by changes in properties of the σ1 protein. In addition to the amino acid changes in the σ1 reading frame, polymorphisms in the S1 sequence also result in a change in the σ1s reading frame (36). The nonstructural protein σ1s from T3DF is linked to pathogenesis in vivo but can alter viral protein synthesis, host cell cycle blockade, and cell death induction in cell culture (61, 62). To determine whether differences in the sequence of σ1s in T3DF and T3DF/T3DCS1 influences viral replication and whether T3DF/T3DCS1 was compromised at stages of replication other than attachment, we identified conditions under which attachment of T3DF and T3DF/T3DCS1 were equivalent. We observed that when 2-fold more particles of T3DF/T3DCS1 were incubated with cells, the attachment of T3DF and T3DF/T3DCS1 was equivalent (Fig. 4A). The 2-fold difference in attachment efficiency is consistent with the ∼2-fold difference in particle-to-PFU ratios of T3DF and T3DF/T3DCS1 preparations used for our experiments. When infection was initiated with 2-fold more T3DF/T3DCS1, the infectivity of T3DF and T3DF/T3DCS1 was equivalent (Fig. 4B). Using 2-fold more T3DF/T3DCS1, equivalent infectivity of T3DF and T3DF/T3DCS1 was observed over a wide range of multiplicities of infection (MOIs). These data suggest that when equivalent amounts of virus attach to host cells, other events in the replication cycle that lead to the expression of viral proteins, and therefore detection by our quantitative infectivity assay, are equivalent. These data also rule out the possibility that the antisera used for detection of infected cells display different reactivities to T3DF and T3DF/T3DCS1.
FIG 4.
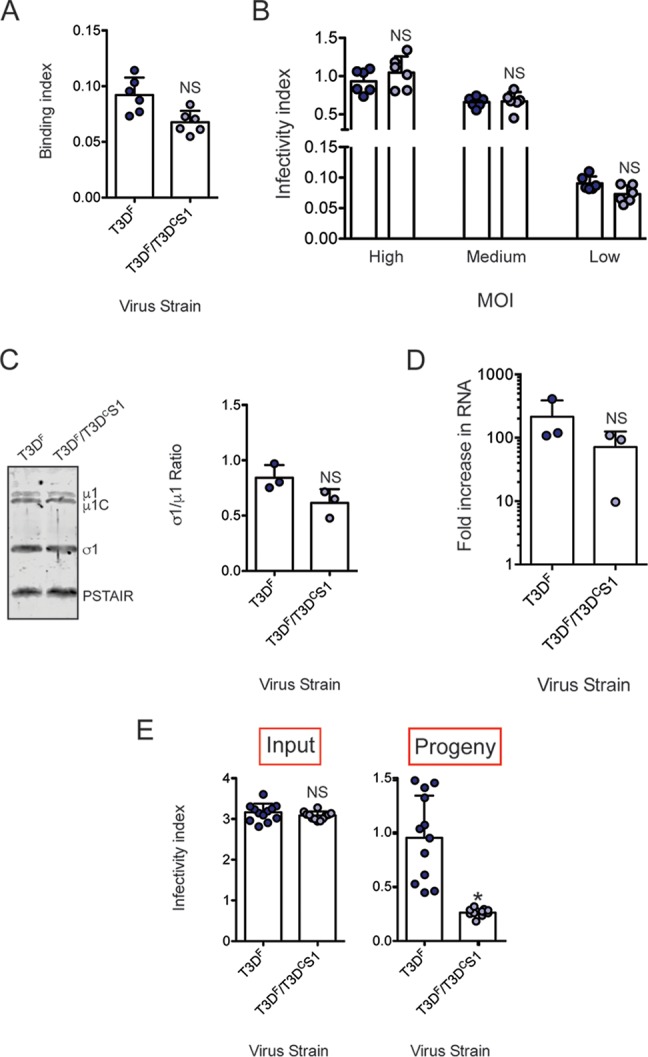
Inefficient assembly contributes to lower level of σ1 on T3DF/T3DCS1 particles. (A) L929 cells grown in 96-well plates were chilled at 4°C for 15 min and then adsorbed with 5 × 104 particles/cell of T3DF or 1 × 105 particles/cell of T3DF/T3DCS1 at 4°C for 1 h. Cell attachment was determined by indirect immunofluorescence of cell-associated particles using a LI-COR Odyssey scanner. Binding index was determined by calculating intensity ratios at 800 nm (green fluorescence), representing viral antigen, and 700 nm (red fluorescence), representing the cell monolayer. The binding index for each independent infection and the sample means are shown. Error bars indicate SD. NS, P > 0.05 as determined by Student's t test compared to T3DF. (B) L929 cells were adsorbed with 3,000, 300, and 30 particles/cell of either T3DF or 6,000, 600, and 60 particles/cell of T3DF/T3DCS1 at room temperature for 1 h. These conditions are designated high, medium, and low MOI, respectively. After incubation at 37°C for 18 h, the cells were subjected to indirect immunofluorescence assay using a LI-COR Odyssey scanner. Relative infectivity was determined by calculating intensity ratios at 800 nm (green fluorescence), representing viral antigen, and 700 nm (red fluorescence), representing the cell monolayer. The infectivity index for each independent infection and the sample mean are shown. Error bars indicate SD. NS, P > 0.05 as determined by Student's t test compared to T3DF at equivalent attachment units. (C) Whole-cell lysates of cells infected with 3,000 particles/cell T3DF or 6,000 particles/cell of T3DF/T3DCS1 were subjected to immunoblotting using antibodies directed against reovirus μ1 protein and T3D σ1 head. Membranes were scanned on a LI-COR Odyssey scanner to determine σ1 and μ1 band intensities. σ1/μ1 ratios for each independent infection and the sample mean are shown. Error bars indicate SD. NS, P > 0.05 as determined by Student's t test compared to T3DF. (D) RNA extracted from cells infected with 3,000 particles/cell T3DF or 6,000 particles/cell of T3DF/T3DCS1 at 0 and 24 h postinfection was subjected to RT-PCR to measure the level of minus strand for the viral M2 gene relative to cellular GAPDH mRNA. The fold increase in the minus strand for each independent infection and sample means are shown. Error bars indicate SD. NS, P > 0.05 as determined by Student's t test compared to T3DF. (E) L929 cells were adsorbed with 30 particles/cell of T3DF or 60 particles/cell of T3DF/T3DCS1 at room temperature for 1 h. After incubation at 37°C for 24 h, medium supernatant was used to infect fresh L929 cells. Relative infectivity of input virus (in the original plate) and that of progeny virus (in a fresh plate) was determined by calculating intensity ratios at 800 nm (green fluorescence), representing viral antigen, and 700 nm (red fluorescence), representing the cell monolayer. The infectivity index for each independent infection and the sample mean are shown. Error bars indicate SD. NS, P > 0.05 as determined by Student's t test compared to T3DF. *, P < 0.05 as determined by Student's t test compared to T3DF.
Our data indicate that differences in attachment of T3DF and T3DF/T3DCS1 are due to differences in the levels of particle-associated σ1. Differences in amounts of σ1 on the particle can be due to changes in steady-state levels of σ1 in infected cells or to defective encapsidation or retention of σ1 on virions. To distinguish between these possibilities, we infected cells with equivalent attachment units of T3DF and T3DF/T3DCS1 (3,000 and 6,000 particles/cell, respectively) and determined the levels of intracellular σ1 protein by immunoblot analysis. As a control, the level of μ1 in infected cells was also evaluated. The σ1/μ1 ratio in lysates of infected cells was similar, indicating equivalent expression of viral proteins in infected cells (Fig. 4C). Synthesis of minus-strand RNA, which requires assembly of progeny core particles, also occurs with equal efficiency following infection with T3DF and T3DF/T3DCS1 (Fig. 4D). Thus, these data indicate that inefficient encapsidation of σ1 on T3DF/T3DCS1 particles is not related to insufficient accumulation of T3DC σ1 in infected cells but due to a later-stage defect in morphogenesis of infectious progeny particles.
In our experiments shown in Fig. 3, we used CsCl-purified viruses to demonstrate that a lower level of σ1 is present on the virus particle. To rule out the possibility that the lower level of particle-associated σ1 on purified particles is a consequence of σ1 ejection due to the method of viral purification, we sought to measure the infectivity of unpurified, released viral progeny. Toward this goal, we used an assay recently described to identify host factors required for assembly and release of infectious progeny viruses (63). L929 cells were infected with equivalent attachment units of T3DF or T3DF/T3DCS1 for 24 h. Consistent with our data shown in Fig. 4B, the infectivity index of T3DF and T3DF/T3DCS1 was equivalent (Fig. 4E, input infectivity). The infectivity of the progeny virus produced from this infection was assessed by evaluating the capacity of the released virus present in the medium supernatant to establish infection. We found that compared to medium from T3DF-infected cells, the medium from cells infected with T3DF/T3DCS1 showed significantly lower infectivity (Fig. 4E, progeny infectivity). These data indicate that assembly and/or release of progeny viruses containing sufficient levels of σ1 is decreased in T3DF/T3DCS1 even when virions are not subjected to sonication and gradient purification procedures (64). Previous work indicates that intracellular yields of T3DF and T3DF/T3DCS1 at 24 h following infection are equivalent (36). We have also observed that an equivalent number of virus particles (∼1013 particles from 4 × 108 cells) can be purified from cells infected with T3DF and T3DF/T3DCS1 (data not shown). Along with the other data presented above about the intracellular levels of other capsid proteins and the assembly of progeny cores, these findings suggest that compared to those of T3DF, particles of T3DF/T3DCS1 contain a lower level of σ1 due to defects in σ1 encapsidation.
T3DF/T3DCS1 displays increased efficiency of ISVP-to-ISVP* conversion.
During cell entry, ISVPs undergo conformational transitions to generate ISVP*s (27). These transitions are likely facilitated by host lipids (65, 66). ISVP-to-ISVP* conversion involves major conformational changes in the particle, including (i) reorganization of the μ1 and λ2 proteins, (ii) release of the μ1-derived pore-forming peptides, μ1N and ϕ, and (iii) ejection of the σ1 attachment protein (29, 34). If σ1 release is a prerequisite for ISVP-to-ISVP* conversion, a particle with a lower level of encapsidated σ1, or one in which σ1 is held with lower affinity, can be expected to undergo this structural change more readily. To determine if either the amount or the nature of particle-associated σ1 influences the efficiency of ISVP-to-ISVP* conversion, we incubated in vitro-generated ISVPs of T3DF and T3DF/T3DCS1 at increasing temperatures and assessed the trypsin sensitivity of δ fragment as a measure of ISVP* conversion (67). We found that compared to T3DF, which converts to ISVP*s at 42°C (Fig. 5A), T3DF/T3DCS1 undergoes ISVP-to-ISVP* transition at a significantly lower temperature, ∼28°C (Fig. 5B).
FIG 5.

T3DF/T3DCS1 shows increased efficiency of ISVP-to-ISVP* conversion. ISVPs (2 × 1012 particles/ml) of T3DF or T3DF/T3DCS1 were divided into aliquots of equivalent volumes and incubated at the indicated temperatures for 20 min (RT, room temperature). (A and B) The reaction mixtures were chilled on ice and digested with 0.10 mg/ml trypsin for 30 min on ice. Following addition of loading dye, the samples were subjected to SDS-PAGE analysis. The gels shown are representative of at least 3 independent experiments. (C and D) The reaction mixtures were chilled on ice and used to initiate infection of L929 cells at 300 particles/cell. After incubation at 37°C for 18 h, the cells were subjected to indirect immunofluorescence assay using a LI-COR Odyssey scanner. Relative infectivity was determined by calculating intensity ratios at 800 nm (green fluorescence), representing viral antigen, and 700 nm (red fluorescence), representing the cell monolayer. The infectivity index of ISVPs maintained at 0°C was set to 100%. Percent infectivity for each independent infection and the sample means are shown. Samples with a calculated infectivity below 0% are not shown. Error bars indicate SD. *, P < 0.05 as determined by 1-way ANOVA with Bonferroni's multiple-comparison test compared to samples maintained at 0°C.
As a consequence of loss of molecules required for cell entry, ISVP* formation results in diminished infectivity of the particle (68). The temperature at which infectivity is lost therefore serves as a measure of ISVP-to-ISVP* conversion efficiency (66). As an alternative way to assess ISVP* formation, in vitro-generated ISVPs of T3DF and T3DF/T3DCS1 were heated at increasing temperatures, and the infectivity of the samples on L929 cells was monitored using the quantitative indirect immunofluorescence assay. We observed that while T3DF lost infectivity at 42°C (Fig. 5C), T3DF/T3DCS1 lost infectivity at a significantly lower temperature (∼28°C) (Fig. 5D). These data indicate that changes to the sequence of the σ1 protein impact the capacity of the particle to undergo ISVP-to-ISVP* transition. These data identify a previously unknown link between the properties of particle-associated σ1 and the propensity for undergoing entry-associated conformational changes.
Introduction of a matched L2 gene from T3DC restores σ1 encapsidation and infectivity of T3DF/T3DCS1.
The σ1 protein is anchored to the particle within turrets formed by the λ2 protein (26, 69). Thus, the encapsidation or retention of σ1 on the particle can be affected by the nature of the σ1-λ2 interaction. The outer surface of the λ2 protein also makes contact with the μ1 protein that forms a lattice on the surface of the ISVPs (26). Because the λ2 protein is an intermediary between the σ1 and μ1 proteins (26), a change in the σ1-λ2 interaction could transduce a signal to μ1 and alter its properties. Since both the attachment and ISVP* phenotype of T3DF/T3DCS1 could be explained by σ1-λ2 interaction, we decided to determine whether a mismatch between T3DC σ1 and T3DF λ2 accounts for the phenotypes observed. The λ2 proteins of T3DF and T3DC differ at residue 504, resulting in a glycine-to-glutamate change, and at residue 509, resulting in a glycine-to-arginine change (70). Thus, to test our idea about the contribution of σ1-λ2 mismatch, we generated two additional viruses, T3DF/T3DCL2 (a monoreassortant that contains an L2 gene segment from strain T3DC in an otherwise T3DF background) and T3DF/T3DCS1L2 (a double reassortant that contains both the S1 and L2 gene segments from strain T3DC in an otherwise T3DF background). Among these, σ1 and λ2 remain mismatched in T3DF/T3DCL2, whereas they are matched in T3DF/T3DCS1L2. Compared to those of T3DF, plaques formed by T3DF/T3DCL2 and T3DF/T3DCS1L2 were predominantly smaller. Notably, the plaque size of both viruses and, most notably, T3DF/T3DCS1L2 were marginally larger than those formed by T3DF/T3DCS1 (compare Fig. 6A and B to 1A and B).
FIG 6.
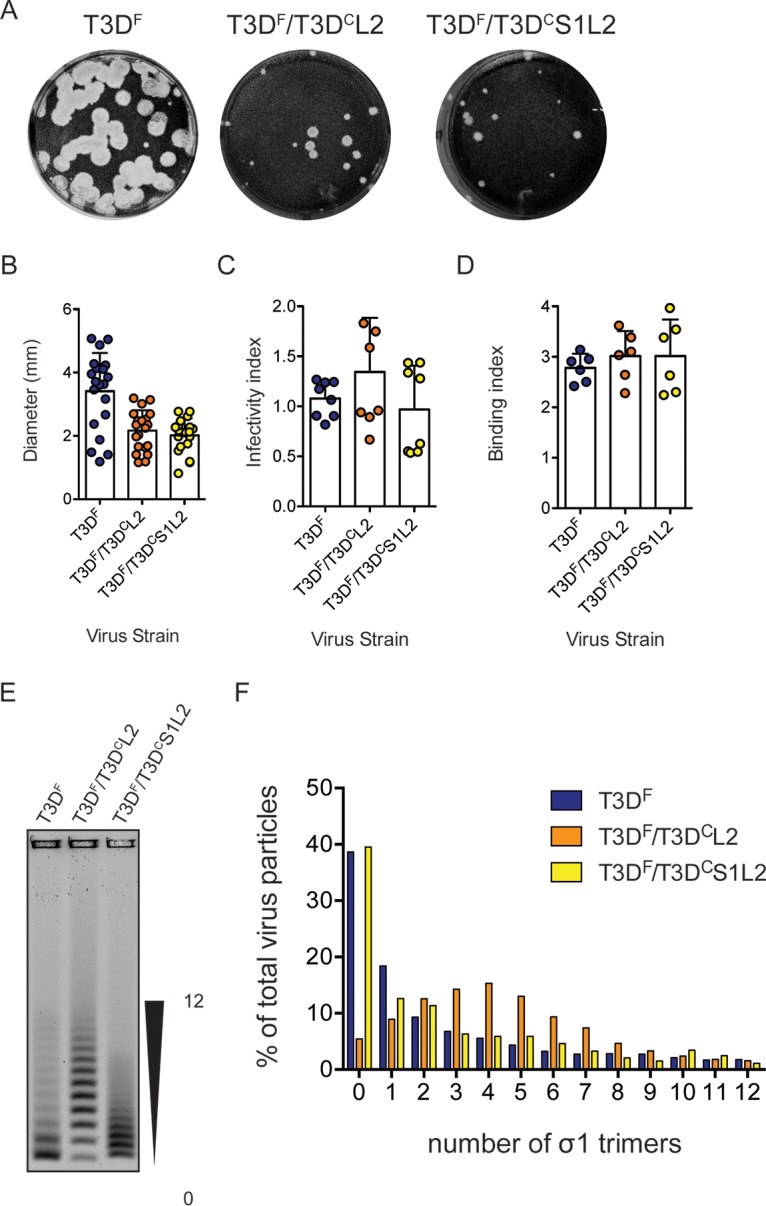
Matched L2 gene from T3DC restores σ1 encapsidation defect in T3DF/T3DCS1. (A) Particles of either T3DF, T3DF/T3DCL2, or T3DF/T3DCS1L2 diluted in PBS were subjected to plaque assay on L929 cells. This plaque assay was performed in parallel with one shown in Fig. 1. (B) Size of 20 randomly selected plaques and means are shown. Error bars indicate SD. *, P < 0.05 as determined by Student's t test compared to T3DF. The same data for T3DF as that shown in Fig. 1 were used. (C) L929 cells were adsorbed with 3,000 particles/cell of either T3DF, T3DF/T3DCL2, or T3DF/T3DCS1L2. After incubation at 37°C for 18 h, the cells were subjected to indirect immunofluorescence assay using a LI-COR Odyssey scanner. Relative infectivity was determined by calculating intensity ratios at 800 nm (green fluorescence), representing viral antigen, and 700 nm (red fluorescence), representing the cell monolayer. The infectivity index for each independent infection and the sample means are shown. Error bars indicate SD. NS, P > 0.05 as determined by 1-way ANOVA with Bonferroni's multiple-comparison test compared to T3DF. (D) Confluent monolayers of L929 cells grown in 96-well plates were adsorbed with 5 × 104 particles/cell of either T3DF, T3DF/T3DCL2, or T3DF/T3DCS1L2 at 4°C for 1 h. Cell attachment was determined by indirect immunofluorescence of cell-associated particles using a LI-COR Odyssey scanner. The binding index was determined by calculating intensity ratios at 800 nm (green fluorescence), representing viral antigen, and 700 nm (red fluorescence), representing the cell monolayer. Binding index for each independent infection and the sample mean are shown. Error bars indicate SD. NS, P > 0.05 as determined by 1-way ANOVA with Bonferroni's multiple-comparison test compared to T3DF. (E and F) Virions (1 × 1011 particles) were resolved on an agarose gel, stained with a colloidal blue staining kit, and scanned using a LI-COR Odyssey scanner. (E) The position of virions with the lowest and highest numbers of σ1 trimers is shown. (F) The band intensity of each virion species was quantified. The abundance of each species as a percentage of all 13 virion species present in the sample is shown.
To further evaluate the phenotype of T3DF/T3DCL2 and T3DF/T3DCS1L2, we infected L929 cells with 3,000 particles/cell of each virus and assessed their infectivity using indirect immunofluorescence. We found that T3DF/T3DCL2 and T3DF/T3DCS1L2 established infection as efficiently as T3DF (Fig. 6C). These data indicate that changing the L2 sequence through reassortment of a T3DC L2 gene alone does not influence the capacity of virus to establish infection. Additionally, based on the comparison between the infectivity of T3DF/T3DCS1 (Fig. 1) and T3DF/T3DCS1L2, our data indicate that introduction of a matched T3DC L2 gene segment into T3DF/T3DCS1 restores the infectivity of the virus to wild-type levels.
Lower infectivity of T3DF/T3DCS1 is a consequence of reduced capacity of this virus strain to attach host cells. To examine the cell attachment efficiency of T3DF/T3DCL2 and T3DF/T3DCS1L2, we used a plate-based quantitative attachment assay. We found that T3DF/T3DCL2 and T3DF/T3DCS1L2 displayed attachment comparable to that of T3DF (Fig. 6D). These data indicate that the attachment phenotype produced by the introduction of T3DC σ1 into T3DF is overcome by simultaneous introduction of a matched T3DC-derived λ2 protein.
Based on the attachment properties of T3DF/T3DCL2 and T3DF/T3DCS1L2, it was expected that virions of this strain would exhibit T3DF-like σ1 encapsidation. To directly test this possibility, purified virus particles were resolved on agarose gels. We observed that compared to that of T3DF, the pattern of distribution of viruses based on σ1 trimer encapsidation was different (Fig. 6E). The T3DF/T3DCL2 preparation contained a higher proportion of virions with more σ1 (Fig. 6F). Because of the efficient encapsidation of σ1 on the particle, T3DF/T3DCL2 is able to efficiently attach and infect host cells. The preparation of T3DF/T3DCS1L2 contained virions with various levels of σ1 trimers. Although the T3DF/T3DCS1L2 preparation does not contain a large proportion of virions with higher numbers of σ1 trimers observed for T3DF, it contains sufficient numbers of particles with the needed level of σ1 trimers to allow T3DF-like infection (Fig. 6E and F). Importantly, despite the shared T3DC S1 allele, the presence of a large proportion of virions with no σ1 trimers noted for T3DF/T3DCS1 is not observed in the preparation of T3DF/T3DCS1L2 (compare Fig. 6 to 3). These data indicate that the natures of both σ1 and λ2 influence the encapsidation of σ1 trimers on the particle. Furthermore, these data indicate that the encapsidation defect produced by reassortment of the T3DC S1 into T3DF can be overcome by the presence of a T3DC-derived matched λ2 protein.
Properties of the λ2 protein influence ISVP-to-ISVP* conversion.
In addition to the effect of T3DC S1 on attachment and infection, we also observed that compared to T3DF, T3DF/T3DCS1 forms ISVP*s much more readily under less stringent conditions (Fig. 5). Based on the evidence that inclusion of T3DC λ2 restored the attachment phenotype of T3DF/T3DCS1, we determined the capacity of T3DF/T3DCL2 and T3DF/T3DCS1L2 to undergo ISVP-to-ISVP* conversion. Chymotrypsin-generated ISVPs of each virus were incubated at increasing temperature, and the conditions under which the δ fragment of μ1 attains a trypsin-sensitive conformation was determined. We found that compared to T3DF ISVPs, which display a trypsin-sensitive conformer of δ at 42°C (Fig. 7A), the T3DF/T3DCL2 δ fragment attains a trypsin-sensitive conformation at a lower temperature of 32°C (Fig. 7B). These data indicate that properties of the λ2 protein influence the propensity with which ISVP*s are generated. The temperature at which T3DF/T3DCS1L2 converted to ISVP* was higher (34°C) than that of T3DF/T3DCL2 (Fig. 7C). Importantly, although T3DF/T3DCS1L2 converted to ISVP* in a more facile manner than the parental T3DF strain, its propensity to form ISVP*s was lower than that of mismatched viruses T3DF/T3DCS1 and T3DF/T3DCL2, which convert to ISVP* at ∼28°C and 32°C, respectively (compare Fig. 7B to 5B). These data indicate that placement of the T3DC L2 in T3DF/T3DCS1 raises the temperature at which ISVPs undergo ISVP-to-ISVP* conversion. To assess ISVP* formation by monitoring loss of infectivity of ISVPs, in vitro-generated ISVPs of T3DF, T3DF/T3DCL2, and T3DF/T3DCS1L2 were heated at increasing temperatures, and the infectivity of the samples on L929 cells was monitored using the quantitative indirect immunofluorescence assay. We observed that while T3DF lost infectivity at 42°C, T3DF/T3DCL2 and T3DF/T3DCS1L2 lost infectivity at 30°C and 32°C, respectively (Fig. 7D, E, and F). Importantly, the temperature at which T3DF/T3DCS1L2 loses infectivity is higher than that for T3DF/T3DCS1 and T3DF/T3DCL2 (28°C and 30°C, respectively) (Fig. 5). Thus, our results highlight a previously unknown effect of λ2 on the efficiency of ISVP-to-ISVP* conversion. Further, our data also indicate that a match between the σ1 and λ2 proteins controls conformational changes in virus capsid that are required for bypassing cellular membranes during cell entry.
FIG 7.

λ2 protein influences ISVP-to-ISVP* conversion. ISVPs (2 × 1012 particles/ml) of T3DF, T3DF/T3DCL2, or T3DF/T3DCS1L2 were divided into aliquots of equivalent volumes and incubated at the indicated temperatures for 20 min (RT, room temperature). (A, B, and C) The reaction mixtures were chilled on ice and digested with 0.10 mg/ml trypsin for 30 min on ice. Following addition of loading dye, the samples were subjected to SDS-PAGE analysis. The gels shown are representative of at least 3 independent experiments. (D, E, and F) The reaction mixtures were chilled on ice and used to initiate infection of L929 cells at 300 particles/cell. After incubation at 37°C for 18 h, the cells were subjected to indirect immunofluorescence assay using a LI-COR Odyssey scanner. Relative infectivity was determined by calculating intensity ratios at 800 nm (green fluorescence), representing viral antigen, and 700 nm (red fluorescence), representing the cell monolayer. Infectivity index of ISVPs maintained at 0°C was set to 100%. Percent infectivity for each independent infection and the sample mean are shown. Samples with a calculated infectivity below 0% are not shown. Error bars indicate SD. *, P < 0.05 as determined by 1-way ANOVA with Bonferroni's multiple-comparison test compared to samples maintained at 0°C.
DISCUSSION
To test the idea that new protein-protein interactions forced by placement of alleles from two parent viruses in a reassortant virus alter the infectious properties of virus capsids, we compared the T3DF parent with T3DF/T3DCS1, a monoreassortant containing the S1 gene from laboratory isolate T3DC. Compared to T3DF, T3DF/T3DCS1 encapsidates the S1-encoded σ1 protein less efficiently, resulting in a population of particles that have no σ1. Given the function of σ1 in attachment, this defect results in a lower capacity of T3DF/T3DCS1 to attach and infect host cells. Cointroduction of the T3DC-derived L2 gene, which encodes the λ2 protein that anchors fibers of the σ1 protein into the virion, allowed better encapsidation of σ1 and restored the attachment and infectivity defect observed for T3DF/T3DCS1. These data indicate that reassortment could influence how adjacent capsid proteins fit with each other and consequently influence the function of the capsid.
The σ1 protein folds into a homotrimeric structure with a distinct head, body, and tail domain (71, 72). The N-terminal end of the protein is predicted to form an α-helical coiled coil (71, 72). This region transitions into a body domain that is formed from β-spiral repeats (73). The C-terminal portion of the protein forms a globular head structure with an eight-stranded β-barrel (73). σ1 is anchored to the particles via turrets formed by the λ2 protein (26, 69). While structural evidence highlighting contacts between σ1 and λ2 is lacking, mutational analyses indicate that a σ1 region comprised of amino acids 3 to 34, which includes a hydrophobic stretch, is necessary for anchoring into λ2 (74, 75). The T3DF-T3DC polymorphism at residue 22 in σ1 produces a valine-to-alanine change, which could alter the hydrophobicity of the σ1 N terminus and influence its encapsidation on the particle. The second T3DF-T3DC polymorphism at residue 408 lies within the head domain of σ1. This region is not thought to engage any portion of the virus and thus is not expected to directly influence encapsidation of σ1 on the virus particle. However, because the structure of σ1 on the virion is unknown, the contribution of the head domain of σ1 on encapsidation of σ1 cannot be ruled out. Our results indicate that the attachment defect produced by the presence of the T3DC σ1 protein is overcome by introduction of the T3DC λ2 protein. Although σ1 encapsidation on T3DF/T3DCL2 and T3DF/T3DCS1L2 is not identical to that observed for T3DF (Fig. 6), this difference does not appear to influence the capacity of the virus to attach and infect cells. This result is consistent with previous observations indicating that a threshold number of σ1 on particles is sufficient for the virus to establish infection and that particles with less σ1 than the parental strain retain their infectivity (59, 60).
The λ2 protein forms a pentameric turret at the 5-fold vertices of the virus (21). The hollow cavity formed by λ2 is lined by enzymatic domains that are required for capping viral mRNA (21). Additionally, in virions and ISVPs, the λ2 pentamer holds the σ1 fiber via the flaps formed by the last 250 amino acid residues of λ2 (21, 26, 42). Thus, the T3DC σ1 encapsidation defect may be a consequence of a mismatch between T3DC σ1 and T3DF λ2. The amino acid sequence of T3DF and T3DC λ2 proteins differs at two residues, 504 and 509, that constitute the methylase-1 domain (21). No changes are present in the flap region of λ2 that is proposed to interact with σ1. Thus, why an allelic match between σ1 and λ2 is required for optimal attachment is not immediately obvious. The arginine at position 509 is in a location where it able to interact with the underside of the flap domain comprised of residues 1263 to 1267 (Fig. 8). Such an interaction may alter the conformation of the λ2 flaps and alter its capacity to assemble or retain σ1 efficiently. Our observations with T3DF/T3DCL2 indicate that even if changes at residues 504 and 509 do influence the local structure of λ2, its effects are only manifested in the presence of T3DC σ1. Whether this is due to the greater hydrophobicity of the T3DF σ1 protein or some other effects is not known. It should be noted that even though the parental virus T3DC contains a matched σ1-λ2 pair, similar to T3DF/T3DCS1L2, it encapsidates more σ1 than T3DF (36). Thus, σ1 encapsidation may also be influenced by the properties of other capsid proteins. Indeed, we have previously observed that a μ1 variant contains different levels of particle-associated σ1 (76). Another study has also suggested that compatibility between μ1 and σ1 confers optimal σ1 encapsidation and function on the particle (70). The difference in σ1 encapsidation between T3DF and T3DC may be governed by the function of μ1 or yet another protein.
FIG 8.

Position of λ2 polymorphisms on λ2 pentamer. (A) Top view of the λ2 pentamer rendered using UCSF Chimera from PDB entry 2CSE is shown with each λ2 monomer in a different color (94). The positions of residue 504 (orange) and 509 (red) and the σ1 trimer are shown using an asterisk. The yellow ribbon indicates the position of the underside of the λ2 flap domain. (B and C) A side of the same molecule (B) and a magnification of the inset from panel B (C) are shown.
Capsids of T3DF and T3DF/T3DCS1 differ in at least one unexpected way. Unlike T3DF, T3DF/T3DCS1 undergoes ISVP-to-ISVP* conformational changes that are required for entry into cells more readily. As described above, the only known interaction between σ1 and the particle is via the λ2 pentamer (26, 69). As a consequence of ISVP-to-ISVP* transition, λ2 adopts an open conformation similar to the one found in core particles, consequently allowing the release of the σ1 protein and the derepression of viral transcriptase activity (27). The δ fragment derived from the μ1 protein also undergoes a structural transition during ISVP* formation, generating a protease-sensitive conformer (27, 77). The outer side of the λ2 turret contacts δ in ISVP particles (26). Thus, changes to structure of λ2 likely influence its interaction with δ. It is possible that changes to the structure of each of the proteins is hierarchical, with one structural change being a prerequisite for another. Alternatively, the different components of the capsid may function like an interlocking gear set, where changes to one protein (for example, λ2) cannot occur without a concurrent change to another part of the capsid (for example, δ). We hypothesize that the presence of σ1 serves as a lock such that a lower level of this protein in the λ2 pentamer, or one that is loosely held (such as in T3DF/T3DCS1), would favor conformational changes in the remaining capsid. Our work using T3DF/T3DCL2 and T3DF/T3DCS1L2 indicates that a virus that bears matching T3DC σ1 and λ2 proteins displays a more regulated conversion to ISVP* than either mismatched virus. Because the levels of σ1 on T3DF/T3DCL2 and T3DF/T3DCS1L2 are higher than those on T3DF/T3DCS1, the level of σ1 does not appear to correlate with the capacity of the virus to form ISVP*. Based on our characterization of T3DF/T3DCL2, our studies also revealed for the first time that the nature of λ2 influences the efficiency of ISVP-to-ISVP* conversion. As stated above, the amino acid residues that are different between T3DF and T3DC λ2 are proximal to the inner cavity of the λ2 pentamer proteins and are not in a position to influence interaction with δ on the outer side of λ2. Thus, one possibility is that there is a significant structural difference in the T3DF and T3DC λ2 pentamers. However, we favor a different possibility to explain our results. This idea is an extension of our hypothesis about the locking structures in the virus. Reovirus ISVPs are metastable structures that are poised to undergo structural changes (27, 68). The metastability of ISVPs is maintained by interactions between capsid proteins (43, 78, 79). When such interactions are altered by placing together alleles that did not coevolve, the particles are rendered more metastable and, thus, more likely to readily convert to ISVP*. We think this is an underappreciated effect of reassortment.
Our studies indicate that T3DF/T3DCS1, T3DF/T3DCL2, and T3DF/T3DCS1L2 display a smaller plaque size than the T3DF parent (Fig. 1 and 6). Because plaque size is dependent on efficient completion of all stages of virus replication, the exact basis for which defect results in smaller plaque size cannot be easily identified. Nonetheless, each of the variants used in this study share a phenotype, i.e., the capacity to undergo ISVP-to-ISVP* transition more easily (Fig. 5 and 7). Interestingly, we have also previously noted an inverse correlation between plaque size and enhanced capacity for ISVP* formation (78, 79). Thus, it is possible that the multicycle replication defect that results in formation of a small plaque is related to the capacity of these viruses to produce ISVP*s more efficiently.
Following intranasal inoculation, compared to T3DF, T3DC more efficiently replicates in the lungs and disseminates to secondary sites (36). Based on the evidence that T3DF/T3DCS1 replicates more efficiently at secondary sites of infection than T3DF, this difference was attributed to the S1 gene segment, and a role for both σ1 and σ1s was described (36). T3DC incorporates significantly more σ1 than T3DF (36). However, based on our evidence that T3DF/T3DCS1 incorporates less σ1 than T3DF, we think it is unlikely that the level of σ1 controls the difference in the in vivo properties of T3DF and T3DC or T3DF/T3DCS1. Compared to T3DF, T3DF/T3DCS1 is less sensitive to inactivation by proteases (36). Because how this compares to T3DC is not known, the contribution of protease sensitivity to superior replication of T3DF/T3DCS1 in vivo is undefined. Why the lower level of σ1 incorporation does not compromise replication of T3DF/T3DCS1 in vivo is puzzling. We speculate that differences in the level of host cell receptors in different tissues accessed by the virus following intranasal inoculation or the use of alternate receptors, such as Ngr1, which may engage parts of the virus other than σ1, allow the virus to replicate efficiently in vivo (80). Our previous work indicates that mutants that are impaired in the capacity to convert to ISVP*s are compromised for replication, at least in the central nervous system (81). Whether replication in other tissues is influenced by ISVP-to-ISVP* conversion efficiency remains to be determined. Determinants of viral replication in different mouse tissues are different, and it is possible that the impact of this stage of virus replication is only evident in some tissues (82–85).
MATERIALS AND METHODS
Cells.
Spinner-adapted murine L929 cells were maintained in Joklik's minimal essential medium (MEM) (Lonza) supplemented to contain 5% fetal bovine serum (FBS) (Sigma-Aldrich), 2 mM l-glutamine (Invitrogen), 100 U/ml penicillin (Invitrogen), 100 μg/ml streptomycin (Invitrogen), and 25 ng/ml amphotericin B (Sigma-Aldrich). Spinner-adapted L929 cells were used for cultivating, purifying, and determining titers of viruses. L929 cells, obtained from the ATCC, were maintained in Eagle's MEM (Lonza) supplemented to contain FBS (Sigma-Aldrich) and 2 mM l-glutamine (Invitrogen). Experiments to measure attachment and infectivity were done using L929 cells from the ATCC.
Generation of recombinant viruses.
Changes to the sequence of pT7-T3D S1 and pT7-T3D L2 were engineered using a QuikChange lightning site-directed mutagenesis kit (Agilent). Plasmid sequences were confirmed by sequencing. Recombinant strains T3DF/T3DCS1, which contains the T3DF S1 gene with mutations (V22A and T408A), T3DF/T3DCL2, which contains a mutated T3DF L2 gene (G504E and G509R), and T3DF/T3DCS1L2, which contains the T3DF S1 gene with mutations (V22A and T408A) and with mutated T3DF L2 gene (G504E and G509R) in an otherwise T3DF background, were generated by using a plasmid-based reverse-genetics strategy (86, 87). To confirm sequences of mutant viruses, viral RNA was extracted from infected cells or virus particles and subjected to reverse transcription-PCR (RT-PCR) using S1-specific or L2-specific primers. PCR products were resolved on Tris-acetate-EDTA (TAE) agarose gels, purified, and confirmed by sequence analysis. Primer sequences will be shared upon request.
Purification of viruses.
Purified reovirus virions were generated using second- or third-passage L-cell lysate stocks of reovirus. Viral particles were Vertrel-XF (Dupont) extracted from infected cell lysates, layered onto 1.2- to 1.4-g/cm3 CsCl gradients, and centrifuged at 187,183 × g for 4 h. Bands corresponding to virions (1.36 g/cm3) were collected and dialyzed in virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.4]) (64). The concentration of reovirus virions in purified preparations was determined from an equivalence of one unit of optical density at 260 nm being 2.1 × 1012 virions/ml (88). Virus titer was determined by plaque assay on spinner-adapted L929 cells. At least three preparations each of T3DF, T3DF/T3DCS1, T3DF/T3DCL2, and T3DF/T3DCS1L2 were used for this study.
Assessment of plaque morphology.
Plaque assays were conducted in spinner-adapted L929 cells plated in 6-well plates (Greiner Bio-One). Cells were adsorbed with dilutions of virus in phosphate-buffered saline (PBS). Cells were overlaid with a molten mixture comprised of 1× medium 199 and 1% Bacto agar supplemented with 10 μg/ml chymotrypsin. Seven days following infection, the monolayers were fixed by addition of 4% formaldehyde solution in PBS and incubated overnight. The agar overlay was peeled off, and the monolayers were stained with 1% crystal violet stain in 5% ethanol for 5 h at room temperature. The monolayers were washed with water. Plaque sizes were quantified using ImageJ software (89).
Generation of ISVPs in vitro.
ISVPs were generated in vitro by incubation of 2 × 1012 virions with 200 μg/ml of Nα-p-tosyl-l-lysine chloromethyl ketone (TLCK)-treated chymotrypsin at 32°C in virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.4]) for 20 or 60 min. Proteolysis was terminated by addition of 2 mM phenylmethylsulfonyl fluoride (PMSF) and incubation of reactions on ice. Generation of ISVPs was confirmed by SDS-PAGE and Coomassie brilliant blue staining.
Assessment of infectivity by indirect immunofluorescence.
Monolayers of L929 cells (4 × 104) in clear-bottom tissue culture-treated 96-well plates (Corning) were washed with PBS and infected with virions or ISVPs of the indicated reovirus strain at 4°C for 1 h. In experiments to examine the role of glycans on infection, the cells were pretreated with serum-free Eagle's MEM containing 10 mU/ml neuraminidase (Roche) at 37°C for 1 h prior to virus attachment. In experiments to examine the effect of 9BG5 MAb on virus infectivity, 1 × 1011 virus particles/ml were incubated at 4°C with 500 ng/ml of hybridoma supernatant containing 9BG5 MAb overnight (55, 90). This mixture was used to initiate infection as described above. Monolayers were fixed with methanol at −20°C for a minimum of 30 min and washed with PBS containing 0.5% Tween 20 (DPBS-T). Cells were then incubated with polyclonal rabbit antireovirus serum at a 1:1,000 dilution in PBS with 1% bovine serum albumin (DPBS-BSA) at 37°C for 60 min (91). Monolayers were washed twice with DPBS-T and incubated with DPBS-BSA for 37°C for 60 min, followed by two washes with DPBS-T. Cells were stained with a 1:1,000 dilution of LI-COR CW 800 anti-rabbit immunoglobulin G, 1:1,000 dilution of Sapphire 700 (LI-COR), and DRAQ5 (Cell Signaling Technology) at a concentration of 1:10,000 for 37°C for 60 min. Monolayers were washed thrice with DPBS-T, and fluorescence intensity was measured using the Odyssey Imaging System and Image Studio Lite software (LI-COR). For each well, the ratios of fluorescence at 800 nm (green fluorescence; for infected cells) and 700 nm (red fluorescence; for total cells) were quantified. Relative infectivity, in arbitrary units, was quantified using the following formula: relative infectivity = (green fluorescence/red fluorescence)infected − (green fluorescence/red fluorescence)uninfected. To quantify the number of infected cells, in each well, images of the wells were imported into the ImageJ software, and the number of antigen-positive cells was quantified manually and reported as fluorescent focus units (FFU) per well.
Assessment of reovirus attachment.
L929 cells (4 × 104 cell per well) grown in 96-well plates were chilled at 4°C for 15 min and then adsorbed with particles of the indicated virus strains at 4°C for 1 h. Cells were washed with chilled PBS and blocked with PBS-BSA at 4°C for 15 min. Cells were then incubated with polyclonal rabbit antireovirus serum at a 1:2,500 dilution in PBS-BSA at 4°C for 30 min. The cells were washed twice with PBS-BSA, followed by incubation with a 1:1,000 dilution of Alexa Fluor 750-labeled goat anti-rabbit antibody at 4°C for 30 min. After two washes with PBS-BSA, cells were stained with a 1:1,000 dilution of the DNA stain DRAQ5 (Cell Signaling Technology) at 4°C for 5 min. Cells were washed and then fixed with 4% formaldehyde at room temperature for 20 min. Fluorescence intensity was measured using the Odyssey Imaging System and Image Studio Lite software (LI-COR). For each well, the ratio of fluorescence at 800 nm (for attached reovirus) and 700 nm (for total cells) was quantified. Binding index in arbitrary units was quantified using the following formula: binding index = (green fluorescence/red fluorescence)infected − (green fluorescence/red fluorescence)uninfected.
Analysis of protein levels by immunoblotting.
The samples were purified virions or whole-cell lysates of infected cells prepared using radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM NaCl, 1 mM EDTA at pH 8, 50 mM Tris at pH 7.5, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitor cocktail (Roche) and 500 μM PMSF, and they were resolved on 10% SDS-PAGE gels and transferred to nitrocellulose membranes. For immunoblotting using polyclonal rabbit antireovirus serum, the membranes were blocked with 5% milk in Tris-buffered saline (TBS) at room temperature for 1 h. For immunoblotting using 4A3 mouse anti-μ1 MAb and rabbit anti-T3D σ1 head antibody, membranes were blocked with T20 Starting Block (ThermoFisher Scientific) (55, 92). Following blocking, rabbit anti-reovirus serum (1:1,000), 4A3 mouse anti-μ1 MAb (1:500), and rabbit anti T3D σ1 head antibody (1:750) were incubated with the membrane in appropriate blocking buffer at room temperature for 1 h. The membranes were washed with TBS supplemented with 0.1% Tween 20 (TBS-T) twice for 15 min and then incubated with Alexa Fluor-conjugated anti-rabbit IgG or anti-mouse IgG in blocking buffer. Following three washes, membranes were scanned using an Odyssey infrared imager (LI-COR), and intensities of μ1C and σ1 bands were quantified using Image Studio Lite software (LI-COR).
HA assay.
Purified T3DF or T3DF/T3DCS1 virions (1 × 1011 particles) were serially diluted in 50 μl of PBS in 96-well V-bottom microtiter plates (Corning-Costar). Bovine erythrocytes (Colorado Serum Company) were washed twice with chilled PBS and were resuspended at a concentration of 1% (vol/vol) in PBS. Washed erythrocytes (50 μl) were added to wells containing virus and incubated at 4°C overnight. HA titer was expressed as 2 × 1011 particles divided by the number of particles per HA unit. One HA unit is equal to the number of particles of virus sufficient to produce HA (51).
Agarose gel separation of reovirus particles by σ1 content.
A total of 1 × 1011 virus particles were resuspended in dialysis buffer, mixed with 2× gel loading dye (NEB), and resolved on 1% ultrapure agarose gel (Invitrogen) in 1× TAE, pH 7.2, at a constant voltage of 25 V for 18 h. The gel was stained with a Novex colloidal blue staining kit (Invitrogen) for 6 h and destained overnight in water. The gel was scanned using an Odyssey infrared imager (LI-COR). The intensity of each band, representing particles with a specific number of σ1 trimers, was quantified using Image Studio Lite software. The data were represented as the percentage of that species within the virus preparation.
Measurement of minus-strand synthesis.
RNA from infected cells was isolated using TRIzol (Thermo Fisher) extraction. RNA (0.5 to 2 μg) was reverse transcribed with a forward primer specific for T3D M2 and a reverse primer specific for murine glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using a high-capacity cDNA reverse transcription kit (Applied Biosystems). The cDNA was subjected to PCR using SYBR Select master mix (Applied Biosystems) using T3D M2 and murine GAPDH-specific primers. The levels of the M2 transcript relative that of GAPDH was measured for each time interval. The increase in minus-strand synthesis compared to that at 0 h following infection was determined by the ΔΔCT method (where CT is threshold cycle) (93). The following primer sequences were used: T3D M2 forward primer, GCTGAAGACAGCTCATTTACCG; T3D M2 reverse primer, GGCATCCCAAACATCAGGCT; GAPDH forward primer, ACCCAGAAGACTGTGGATGG; GAPDH reverse primer, GGATGCAGGGATGATGTTCT.
Assessment of infectivity of input and progeny virus.
In experiments to determine infectivity of virus released from infected cells, L929 cells were adsorbed with virions at room temperature for 1 h. After incubation at 37°C for 24 h, 25 μl of the medium supernatants from infected wells was used to initiate infection of fresh L929 cells plated in 96-well plates. Plate 1 was fixed with cold methanol. The infection in plate 2 was allowed to proceed at 37°C for 24 h. Infectivity was measured using the indirect fluorescence assay described above.
Analysis of ISVP-to-ISVP* conversion.
ISVPs (2 × 1012 particles/ml) of the indicated viral strains were divided into aliquots of equivalent volumes and heated at temperatures ranging from 0°C to 42°C for 20 min. The reaction mixtures were cooled on ice and then digested with 0.10 mg/ml trypsin (Sigma-Aldrich) for 30 min on ice. Following addition of the SDS-PAGE loading dye, the samples were subjected to SDS-PAGE analysis. For analysis by quantitative infectivity assay, the heated samples were used to initiate infection of L929 cells.
Statistical analysis.
Statistical significance between two experimental groups was determined using the Student t test function of GraphPad Prism software. When an experimental group was compared to more than one other experimental group, 1-way analysis of variance (ANOVA) with Bonferroni's multiple-comparison test function of GraphPad Prism software was used.
ACKNOWLEDGMENTS
We thank Karl Boehme, Tuli Mukhopadhyay, John Patton, and Anthony Snyder, along with members of our laboratory, for helpful suggestions and reviews of the manuscript.
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number 1R01AI110637 (to P.D.) and by Indiana University Bloomington.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
REFERENCES
- 1.McDonald SM, Nelson MI, Turner PE, Patton JT. 2016. Reassortment in segmented RNA viruses: mechanisms and outcomes. Nat Rev Microbiol 14:448–460. doi: 10.1038/nrmicro.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vijaykrishna D, Mukerji R, Smith GJ. 2015. RNA virus reassortment: an evolutionary mechanism for host jumps and immune evasion. PLoS Pathog 11:e1004902. doi: 10.1371/journal.ppat.1004902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neumann G, Noda T, Kawaoka Y. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939. doi: 10.1038/nature08157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cross RK, Fields BN. 1976. Use of an aberrant polypeptide as a marker in three-factor crosses: further evidence for independent reassortment as the mechanism of recombination between temperature-sensitive mutants of reovirus type 3. Virology 74:345–362. doi: 10.1016/0042-6822(76)90341-X. [DOI] [PubMed] [Google Scholar]
- 5.Wenske EA, Chanock SJ, Krata L, Fields BN. 1985. Genetic reassortment of mammalian reoviruses in mice. J Virol 56:613–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keroack M, Fields BN. 1986. Viral shedding and transmission between hosts determined by reovirus L2 gene. Science 232:1635–1638. doi: 10.1126/science.3012780. [DOI] [PubMed] [Google Scholar]
- 7.Rubin DH, Fields BN. 1980. Molecular basis of reovirus virulence: role of the M2 gene. J Exp Med 152:853–868. doi: 10.1084/jem.152.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiner HL, Powers ML, Fields BN. 1980. Absolute linkage of virulence and central nervous system tropism of reoviruses to viral hemagglutinin. J Infect Dis 141:609–616. doi: 10.1093/infdis/141.5.609. [DOI] [PubMed] [Google Scholar]
- 9.Wilson GA, Morrison LA, Fields BN. 1994. Association of the reovirus S1 gene with serotype 3-induced biliary atresia in mice. J Virol 68:6458–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tardieu M, Weiner HL. 1982. Viral receptors on isolated murine and human ependymal cells. Science 215:419–421. doi: 10.1126/science.6276976. [DOI] [PubMed] [Google Scholar]
- 11.Mbisa JL, Becker MM, Zou S, Dermody TS, Brown EG. 2000. Reovirus m2 protein determines strain-specific differences in the rate of viral inclusion formation in L929 cells. Virology 272:16–26. doi: 10.1006/viro.2000.0362. [DOI] [PubMed] [Google Scholar]
- 12.Tyler KL, Squier MK, Rodgers SE, Schneider BE, Oberhaus SM, Grdina TA, Cohen JJ, Dermody TS. 1995. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein sigma 1. J Virol 69:6972–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lubeck MD, Palese P, Schulman JL. 1979. Nonrandom association of parental genes in influenza A virus recombinants. Virology 95:269–274. doi: 10.1016/0042-6822(79)90430-6. [DOI] [PubMed] [Google Scholar]
- 14.Urquidi V, Bishop DH. 1992. Non-random reassortment between the tripartite RNA genomes of La Crosse and snowshoe hare viruses. J Gen Virol 73(Part 9):2255–2265. doi: 10.1099/0022-1317-73-9-2255. [DOI] [PubMed] [Google Scholar]
- 15.Pringle CR, Lees JF, Clark W, Elliott RM. 1984. Genome subunit reassortment among Bunyaviruses analysed by dot hybridization using molecularly cloned complementary DNA probes. Virology 135:244–256. doi: 10.1016/0042-6822(84)90134-X. [DOI] [PubMed] [Google Scholar]
- 16.Gombold JL, Ramig RF. 1986. Analysis of reassortment of genome segments in mice mixedly infected with rotaviruses SA11 and RRV. J Virol 57:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graham A, Kudesia G, Allen AM, Desselberger U. 1987. Reassortment of human rotavirus possessing genome rearrangements with bovine rotavirus: evidence for host cell selection. J Gen Virol 68:115–122. doi: 10.1099/0022-1317-68-1-115. [DOI] [PubMed] [Google Scholar]
- 18.Stott JL, Oberst RD, Channell MB, Osburn BI. 1987. Genome segment reassortment between two serotypes of bluetongue virus in a natural host. J Virol 61:2670–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nibert ML, Margraf RL, Coombs KM. 1996. Nonrandom segregation of parental alleles in reovirus reassortants. J Virol 70:7295–7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dermody TS, Parker JC, Sherry B. 2013. Orthoreoviruses. In Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 21.Reinisch KM, Nibert ML, Harrison SC. 2000. Structure of the reovirus core at 3.6 Å resolution. Nature 404:960–967. doi: 10.1038/35010041. [DOI] [PubMed] [Google Scholar]
- 22.Mao ZX, Joklik WK. 1991. Isolation and enzymatic characterization of protein l2, the reovirus guanylytransferase. Virology 185:377–386. doi: 10.1016/0042-6822(91)90785-A. [DOI] [PubMed] [Google Scholar]
- 23.Kim J, Zhang X, Centonze VE, Bowman VD, Noble S, Baker TS, Nibert ML. 2002. The hydrophilic amino-terminal arm of reovirus core shell protein lambda1 is dispensable for particle assembly. J Virol 76:12211–12222. doi: 10.1128/JVI.76.23.12211-12222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liemann S, Chandran K, Baker TS, Nibert ML, Harrison SC. 2002. Structure of the reovirus membrane-penetration protein, m1, in a complex with its protector protein, s3. Cell 108:283–295. doi: 10.1016/S0092-8674(02)00612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Ji Y, Zhang L, Harrison SC, Marinescu DC, Nibert ML, Baker TS. 2005. Features of reovirus outer capsid protein m1 revealed by electron cryomicroscopy and image reconstruction of the virion at 7.0 Å resolution. Structure 13:1545–1557. doi: 10.1016/j.str.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dryden KA, Wang G, Yeager M, Nibert ML, Coombs KM, Furlong DB, Fields BN, Baker TS. 1993. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J Cell Biol 122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chandran K, Farsetta DL, Nibert ML. 2002. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein m1 mediates membrane disruption. J Virol 76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chandran K, Zhang X, Olson NH, Walker SB, Chappell JD, Dermody TS, Baker TS, Nibert ML. 2001. Complete in vitro assembly of the reovirus outer capsid produces highly infectious particles suitable for genetic studies of the receptor-binding protein. J Virol 75:5335–5342. doi: 10.1128/JVI.75.11.5335-5342.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivanovic T, Agosto MA, Zhang L, Chandran K, Harrison SC, Nibert ML. 2008. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J 27:1289–1298. doi: 10.1038/emboj.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandran K, Walker SB, Chen Y, Contreras CM, Schiff LA, Baker TS, Nibert ML. 1999. In vitro recoating of reovirus cores with baculovirus-expressed outer-capsid proteins m1 and s3. J Virol 73:3941–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farsetta DL, Chandran K, Nibert ML. 2000. Transcriptional activities of reovirus RNA polymerase in recoated cores. Initiation and elongation are regulated by separate mechanisms. J Biol Chem 275:39693–39701. [DOI] [PubMed] [Google Scholar]
- 32.Lee PWK, Hayes EC, Joklik WK. 1981. Protein s1 is the reovirus cell attachment protein. Virology 108:156–163. doi: 10.1016/0042-6822(81)90535-3. [DOI] [PubMed] [Google Scholar]
- 33.Ebert DH, Deussing J, Peters C, Dermody TS. 2002. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem 277:24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- 34.Agosto MA, Ivanovic T, Nibert ML. 2006. Mammalian reovirus, a nonfusogenic nonenveloped virus, forms size-selective pores in a model membrane. Proc Natl Acad Sci USA 103:16496–16501. doi: 10.1073/pnas.0605835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L, Agosto MA, Ivanovic T, King DS, Nibert ML, Harrison SC. 2009. Requirements for the formation of membrane pores by the reovirus myristoylated micro1N peptide. J Virol 83:7004–7014. doi: 10.1128/JVI.00377-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nygaard RM, Lahti L, Boehme KW, Ikizler M, Doyle JD, Dermody TS, Schiff LA. 2013. Genetic determinants of reovirus pathogenesis in a murine model of respiratory infection. J Virol 87:9279–9289. doi: 10.1128/JVI.00182-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coffey CM, Sheh A, Kim IS, Chandran K, Nibert ML, Parker JS. 2006. Reovirus outer capsid protein m1 induces apoptosis and associates with lipid droplets, endoplasmic reticulum, and mitochondria. J Virol 80:8422–8438. doi: 10.1128/JVI.02601-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parker JS, Broering TJ, Kim J, Higgins DE, Nibert ML. 2002. Reovirus core protein m2 determines the filamentous morphology of viral inclusion bodies by interacting with and stabilizing microtubules. J Virol 76:4483–4496. doi: 10.1128/JVI.76.9.4483-4496.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thete D, Danthi P. 2018. Protein mismatches caused by reassortment influence functions of the reovirus capsid. bioRxiv doi: 10.1101/322651. [DOI] [PMC free article] [PubMed]
- 40.Thete D, Snyder AJ, Mainou B, Danthi P. 2016. Reovirus mu1 protein affects infection by altering virus-receptor interactions. J Virol 90:10951–10962. doi: 10.1128/JVI.01843-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weiner HL, Ault KA, Fields BN. 1980. Interaction of reovirus with cell surface receptors. I. Murine and human lymphocytes have a receptor for the hemagglutinin of reovirus type 3. J Immunol 124:2143–2148. [PubMed] [Google Scholar]
- 42.Zhang X, Tang J, Walker SB, O'Hara D, Nibert ML, Duncan R, Baker TS. 2005. Structure of avian orthoreovirus virion by electron cryomicroscopy and image reconstruction. Virology 343:25–35. doi: 10.1016/j.virol.2005.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarkar P, Danthi P. 2010. Determinants of strain-specific differences in efficiency of reovirus entry. J Virol 84:12723–12732. doi: 10.1128/JVI.01385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarkar G, Pelletier J, Bassel-Duby R, Jayasuriya A, Fields BN, Sonenberg N. 1985. Identification of a new polypeptide coded by reovirus gene S1. J Virol 54:720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ernst H, Shatkin AJ. 1985. Reovirus hemagglutinin mRNA codes for two polypeptides in overlapping reading frames. Proc Natl Acad Sci USA 82:48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iskarpatyoti JA, Willis JZ, Guan J, Morse EA, Ikizler M, Wetzel JD, Dermody TS, Contractor N. 2012. A rapid, automated approach for quantitation of rotavirus and reovirus infectivity. J Virol Methods 184:1–7. doi: 10.1016/j.jviromet.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 47.Borsa J, Morash BD, Sargent MD, Copps TP, Lievaart PA, Szekely JG. 1979. Two modes of entry of reovirus particles into L cells. J Gen Virol 45:161–170. doi: 10.1099/0022-1317-45-1-161. [DOI] [PubMed] [Google Scholar]
- 48.Nygaard RM, Golden JW, Schiff LA. 2012. Impact of host proteases on reovirus infection in the respiratory tract. J Virol 86:1238–1243. doi: 10.1128/JVI.06429-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bass DM, Bodkin D, Dambrauskas R, Trier JS, Fields BN, Wolf JL. 1990. Intraluminal proteolytic activation plays an important role in replication of type 1 reovirus in the intestines of neonatal mice. J Virol 64:1830–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem 276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 51.Dermody TS, Nibert ML, Bassel-Duby R, Fields BN. 1990. A sigma 1 region important for hemagglutination by serotype 3 reovirus strains. J Virol 64:5173–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Campbell JA, Schelling P, Wetzel JD, Johnson EM, Forrest JC, Wilson GA, Aurrand-Lions M, Imhof BA, Stehle T, Dermody TS. 2005. Junctional adhesion molecule a serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J Virol 79:7967–7978. doi: 10.1128/JVI.79.13.7967-7978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kirchner E, Guglielmi KM, Strauss HM, Dermody TS, Stehle T. 2008. Structure of reovirus sigma1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog 4:e1000235. doi: 10.1371/journal.ppat.1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reiter DM, Frierson JM, Halvorson EE, Kobayashi T, Dermody TS, Stehle T. 2011. Crystal structure of reovirus attachment protein sigma1 in complex with sialylated oligosaccharides. PLoS Pathog 7:e1002166. doi: 10.1371/journal.ppat.1002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Virgin HW, Mann IVMA, Fields BN, Tyler KL. 1991. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J Virol 65:6772–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dietrich MH, Ogden KM, Katen SP, Reiss K, Sutherland DM, Carnahan RH, Goff M, Cooper T, Dermody TS, Stehle T. 2017. Structural insights into reovirus sigma1 interactions with two neutralizing antibodies. J Virol 91:e01621-16. doi: 10.1128/JVI.01621-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coombs KM. 1998. Stoichiometry of reovirus structural proteins in virus, ISVP, and core particles. Virology 243:218–228. doi: 10.1006/viro.1998.9061. [DOI] [PubMed] [Google Scholar]
- 58.Lerner AM, Cherry JD, Finland M. 1963. Haemagglutination with reoviruses. Virology 19:58–65. doi: 10.1016/0042-6822(63)90024-2. [DOI] [PubMed] [Google Scholar]
- 59.Larson SM, Antczak JB, Joklik WK. 1994. Reovirus exists in the form of 13 particle species that differ in their content of protein sigma 1. Virology 201:303–311. doi: 10.1006/viro.1994.1295. [DOI] [PubMed] [Google Scholar]
- 60.Mohamed A, Teicher C, Haefliger S, Shmulevitz M. 2015. Reduction of virion-associated sigma1 fibers on oncolytic reovirus variants promotes adaptation toward tumorigenic cells. J Virol 89:4319–4334. doi: 10.1128/JVI.03651-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boehme KW, Hammer K, Tollefson WC, Konopka-Anstadt JL, Kobayashi T, Dermody TS. 2013. Nonstructural protein sigma1s mediates reovirus-induced cell cycle arrest and apoptosis. J Virol 87:12967–12979. doi: 10.1128/JVI.02080-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phillips MB, Stuart JD, Simon EJ, Boehme KW. 10 January 2018. Non-structural protein sigma1s is required for optimal reovirus protein expression. J Virol doi: 10.1128/JVI.02259-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Knowlton JJ, Fernandez de Castro I, Ashbrook AW, Gestaut DR, Zamora PF, Bauer JA, Forrest JC, Frydman J, Risco C, Dermody TS. 2018. The TRiC chaperonin controls reovirus replication through outer-capsid folding. Nat Microbiol 3:481–493. doi: 10.1038/s41564-018-0122-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berard A, Coombs KM. 2009. Mammalian reoviruses: propagation, quantification, and storage. Curr Protoc Microbiol Chapter 15:Unit15C.1. [DOI] [PubMed] [Google Scholar]
- 65.Snyder AJ, Danthi P. 2016. Lipids cooperate with the reovirus membrane penetration peptide to facilitate particle uncoating. J Biol Chem 291:26773–26785. doi: 10.1074/jbc.M116.747477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Snyder AJ, Danthi P. 2015. Lipid membranes facilitate conformational changes required for reovirus cell entry. J Virol 90:2628–2638. doi: 10.1128/JVI.02997-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Snyder AJ, Danthi P. 2018. Infectious subviral particle to membrane penetration active particle (ISVP-to-ISVP*) conversion assay for mammalian orthoreovirus. Bio Protoc 8:e2700. doi: 10.21769/BioProtoc.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Middleton JK, Severson TF, Chandran K, Gillian AL, Yin J, Nibert ML. 2002. Thermostability of reovirus disassembly intermediates (ISVPs) correlates with genetic, biochemical, and thermodynamic properties of major surface protein mu1. J Virol 76:1051–1061. doi: 10.1128/JVI.76.3.1051-1061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol 62:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sandekian V, Lemay G. 2015. Amino acids substitutions in sigma1 and mu1 outer capsid proteins of a Vero cell-adapted mammalian orthoreovirus are required for optimal virus binding and disassembly. Virus Res 196:20–29. doi: 10.1016/j.virusres.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 71.Fraser RDB, Furlong DB, Trus BL, Nibert ML, Fields BN, Steven AC. 1990. Molecular structure of the cell-attachment protein of reovirus: correlation of computer-processed electron micrographs with sequence-based predictions. J Virol 64:2990–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dietrich MH, Ogden KM, Long JM, Ebenhoch R, Thor A, Dermody TS, Stehle T. 2018. Structural and functional features of the reovirus sigma1 tail. J Virol 92:e00336-18. doi: 10.1128/JVI.00336-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chappell JD, Prota AE, Dermody TS, Stehle T. 2002. Crystal structure of reovirus attachment protein sigma1 reveals evolutionary relationship to adenovirus fiber. EMBO J 21:1–11. doi: 10.1093/emboj/21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leone G, Mah DCW, Lee PWK. 1991. The incorporation of reovirus cell attachment protein s1 into virions requires the amino-terminal hydrophobic tail and the adjacent heptad repeat region. Virology 182:346–350. doi: 10.1016/0042-6822(91)90678-5. [DOI] [PubMed] [Google Scholar]
- 75.Mah DCW, Leone G, Jankowski JM, Lee PWK. 1990. The N-terminal quarter of reovirus cell attachment protein s1 possesses intrinsic virion-anchoring function. Virology 179:95–103. doi: 10.1016/0042-6822(90)90278-Y. [DOI] [PubMed] [Google Scholar]
- 76.Snyder AJ, Danthi P. 2018. Cleavage of the C-terminal fragment of reovirus mu1 is required for optimal infectivity. J Virol 92:e01848-17. doi: 10.1128/JVI.01848-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chandran K, Parker JS, Ehrlich M, Kirchhausen T, Nibert ML. 2003. The delta region of outer-capsid protein m1 undergoes conformational change and release from reovirus particles during cell entry. J Virol 77:13361–13375. doi: 10.1128/JVI.77.24.13361-13375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sarkar P, Danthi P. 2013. The mu1 72-96 loop controls conformational transitions during reovirus cell entry. J Virol 87:13532–13542. doi: 10.1128/JVI.01899-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Snyder AJ, Danthi P. 9 August 2017. The reovirus mu1 340-343 loop controls entry related conformational changes. J Virol doi: 10.1128/JVI.00898-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Konopka-Anstadt JL, Mainou BA, Sutherland DM, Sekine Y, Strittmatter SM, Dermody TS. 2014. The Nogo receptor NgR1 mediates infection by mammalian reovirus. Cell Host Microbe 15:681–691. doi: 10.1016/j.chom.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS. 2008. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J Virol 82:161–172. doi: 10.1128/JVI.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bodkin DK, Fields BN. 1989. Growth and survival of reovirus in intestinal tissue: role of the L2 and S1 genes. J Virol 63:1188–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hrdy DB, Rubin DH, Fields BN. 1982. Molecular basis of reovirus neurovirulence: role of the M2 gene in avirulence. Proc Natl Acad Sci USA 79:1298–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sherry B, Fields BN. 1989. The reovirus M1 gene, encoding a viral core protein, is associated with the myocarditic phenotype of a reovirus variant. J Virol 63:4850–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weiner HL, Drayna D, Averill DR Jr, Fields BN. 1977. Molecular basis of reovirus virulence: role of the S1 gene. Proc Natl Acad Sci USA 74:5744–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kobayashi T, Antar AAR, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. 2010. An improved reverse genetics system for mammalian orthoreoviruses. Virology 398:194–200. doi: 10.1016/j.virol.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smith RE, Zweerink HJ, Joklik WK. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791–810. doi: 10.1016/0042-6822(69)90017-8. [DOI] [PubMed] [Google Scholar]
- 89.Rasband WS. 2007. ImageJ. U.S. National Institutes of Health, Bethesda, MD. [Google Scholar]
- 90.Guglielmi KM, Kirchner E, Holm GH, Stehle T, Dermody TS. 2007. Reovirus binding determinants in junctional adhesion molecule-A. J Biol Chem 282:17930–17940. doi: 10.1074/jbc.M702180200. [DOI] [PubMed] [Google Scholar]
- 91.Wetzel JD, Chappell JD, Fogo AB, Dermody TS. 1997. Efficiency of viral entry determines the capacity of murine erythroleukemia cells to support persistent infections by mammalian reoviruses. J Virol 71:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boehme KW, Guglielmi KM, Dermody TS. 2009. Reovirus nonstructural protein sigma1s is required for establishment of viremia and systemic dissemination. Proc Natl Acad Sci U S A 106:19986–19991. doi: 10.1073/pnas.0907412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 94.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]