

We discovered a new HSV-1 protein, UL24.5, which corresponds to the C-terminal portion of UL24. In contrast to the replication defects observed with HSV-1 strains that do not express full-length UL24, the absence of UL24.5 did not affect viral replication in cell culture. Moreover, in mice, the absence of UL24.5 did not affect viral titers in epithelia or trigeminal ganglia during acute infection; however, it was associated with a prolonged persistence of signs of inflammation. Strikingly, the absence of UL24.5 also led to an increase in the incidence of severe neurological impairment compared to results for wild-type control viruses. This increase in pathogenicity is in stark contrast to the reduction in clinical signs associated with the absence of full-length UL24. Bioinformatic analyses suggest that UL24.5 is conserved among all human alphaherpesviruses and in some nonhuman alphaherpesviruses. Thus, we have identified UL24.5 as a new HSV-1 determinant of pathogenesis.
KEYWORDS: HSV-1, UL24.5, viral pathogenesis, viral neuropathogenesis, herpes simplex virus
ABSTRACT
Herpes simplex virus 1 (HSV-1) infects the host via epithelia and establishes latency in sensory neurons. The UL24 gene is conserved throughout the Herpesviridae family, and the UL24 protein is important for efficient viral replication and pathogenesis. Multiple transcripts are expressed from the UL24 gene. The presence of a transcription initiation site inside the open reading frame of UL24 and an ATG start codon in the same open reading frame led us to suspect that another protein was expressed from the UL24 locus. To test our hypothesis, we constructed a recombinant virus that expresses a hemagglutinin tag at the C terminus of UL24. Western blot analysis revealed the expression of an 18-kDa protein that is not a degradation product of the full-length UL24, which we refer to as UL24.5. Ectopically expressed UL24.5 did not induce the dispersal of nucleolar proteins, as seen for UL24. In order to characterize the role of UL24.5, we constructed a mutant virus encoding a substitution of the predicted initiation methionine to a valine. This substitution eliminated the expression of the 18-kDa polypeptide. Unlike the UL24-null mutant (UL24X), which exhibits reduced viral yields, the UL24.5-null mutant exhibited the same replication phenotype in cell culture as the parental strain. However, in a murine ocular infection model, we observed an increase in the incidence of neurological disorders with the UL24.5 mutant. Alignment of amino acid sequences for various herpesviruses revealed that the initiation site of UL24.5 is conserved among HSV-1 strains and is present in many herpesviruses.
IMPORTANCE We discovered a new HSV-1 protein, UL24.5, which corresponds to the C-terminal portion of UL24. In contrast to the replication defects observed with HSV-1 strains that do not express full-length UL24, the absence of UL24.5 did not affect viral replication in cell culture. Moreover, in mice, the absence of UL24.5 did not affect viral titers in epithelia or trigeminal ganglia during acute infection; however, it was associated with a prolonged persistence of signs of inflammation. Strikingly, the absence of UL24.5 also led to an increase in the incidence of severe neurological impairment compared to results for wild-type control viruses. This increase in pathogenicity is in stark contrast to the reduction in clinical signs associated with the absence of full-length UL24. Bioinformatic analyses suggest that UL24.5 is conserved among all human alphaherpesviruses and in some nonhuman alphaherpesviruses. Thus, we have identified UL24.5 as a new HSV-1 determinant of pathogenesis.
INTRODUCTION
The genome of herpes simplex virus 1 (HSV-1) encodes at least 84 different proteins (1), including one known as UL24. The UL24 gene is conserved throughout all three subfamilies of Herpesviridae (2), suggesting an important role for UL24 in infection. Notably, UL24 of HSV-1 is required for efficient virus replication in vitro (3) and in vivo (4). Moreover, in a mouse model of ocular infection, a UL24-deficient HSV-1 strain (UL24X) exhibits a reduced capacity for the infection to spread from the eye to neurons of the trigeminal ganglia (TG) (5), few, if any, clinical signs (6), and reduced incidence of reactivating TG in ex vivo assays (4). Thus, UL24 is a determinant of HSV-1 pathogenesis.
UL24 is a highly basic protein of 269 amino acids (aa) which is expressed with leaky-late kinetics (7). In infected cells, UL24 protein is detected in the nucleus, where it transiently localizes to nucleoli, and in the cytoplasm (8). UL24 is involved in dispersal of nucleolin and B23 during infection (8, 9), and the N terminus of UL24 is sufficient to induce the redistribution of these proteins throughout the nucleoplasm when transiently expressed in transfection experiments (10). Some UL24 mutant viruses have a syncytial plaque phenotype, suggesting a role for UL24 in events at the cell membrane. We previously found that the absence of UL24 leads to an altered subcellular distribution of viral glycoproteins involved in fusion (11). More recently we discovered that HSV-1 UL24 negatively affects viral gene expression during infection (12). Consistent with the link between UL24 and pathogenesis, Xu et al. recently found that UL24 inhibits signaling via the stimulator of interferon genes (STING) pathway at the level of NF-κB (13).
The UL24 transcription unit is complex. There are multiple transcripts expressed from the UL24 gene during infection, and the reason for this degree of complexity is unclear (7). Among the six UL24 transcripts expressed in infected cells, four transcripts (5.6, 5.4, 1.4, and 1.2 kb) are produced from the first two transcription initiation sites for UL24 and contain the entire open reading frame (ORF). The third set of transcripts (5.2 and 0.9 kb) originates from a transcription start site within the UL24 ORF. Furthermore, several ATG codons are present downstream of this third transcription start site. These features prompted us to test the hypothesis that there is at least one other polypeptide expressed from the UL24 locus, which would be encoded by the 3′ portion of UL24, and thus explain the reason for the third transcription start site in UL24.
RESULTS
Identification of UL24.5, a novel HSV-1-encoded protein.
The UL24 gene is contained within the UL region of the HSV-1 genome (Fig. 1A). Previous studies identified three transcription start sites, with two located upstream of the UL24 ORF and the third start site located within the UL24 ORF (Fig. 1B) (7). Thus, the first and second transcription start sites result in mRNAs that can be translated into full-length UL24 protein; however, no proteins have been shown to be expressed from the transcripts originating from the third start site. Analysis of the UL24 ORF revealed the presence of four in-frame start codons in addition to the initiating MET codon, as well as three out-of-frame ATG sequences. Interestingly, the third in-frame ATG is located 25 nucleotides (nt) downstream from the third transcription start site (Fig. 1B). These data raise the possibility that mRNA originating from the last transcription start site leads to the production of a new HSV-1 protein (Fig. 1C).
FIG 1.

Diagram of the UL24 locus of HSV-1 and vBAC_UL24HA. (a) Schematic representation of the HSV-1 genome showing the unique long (UL) and the unique short (US) regions, each flanked by terminal and internal repeats (TRL, IRL, IRS, and TRS). (b) UL24 ORF with the in-frame ATG (uppercase) and out-of-frame atg (lowercase) methionine codons indicated. Arrows above the UL23 and UL24 ORFs represent the transcription start sites previously identified for UL24. The position of the previously described PD-(D/E)XK endonuclease motif is indicated below the thick arrow. The position of codon 43 in which the UL24X mutation is located is indicated above the thick arrow. (c) Predicted UL24.5 ORF, starting at the third in-frame ATG within UL24 and ending coterminal with full-length UL24. (d) UL24 ORF of vBAC_UL24HA, where the HA epitope tag has been added at the C terminus of UL24.
In our previous studies on HSV-1 UL24, we detected the protein using an antiserum directed against the N-terminal third of the protein or due to a hemagglutinin (HA) epitope tag fused to the N terminus of UL24 (7, 8). Therefore, in order to test the hypothesis that a protein is expressed from transcripts originating at the third transcription start site, we constructed a recombinant virus where the HA tag is encoded at the C terminus of UL24 (Fig. 1D). We used the bacterial artificial chromosome (BAC) system to generate recombinant viruses based on the Red recombination system as described by Tischer et al. (14, 15). Because UL24 and the potential new protein we call UL24.5 share the same ORF, the resulting virus, vBAC_UL24HA, would ensure the presence of an HA tag on all proteins in frame and coterminal with full-length UL24.
Two independent isolates of vBAC_UL24HA were constructed. Vero cells were infected in duplicate, and cell lysates were prepared 10 h postinfection (hpi). In addition to detecting the 29-kDa UL24-HA protein, Western blot analysis using a primary antibody directed against the HA epitope revealed expression of an 18-kDa protein that we named UL24.5 (Fig. 2). We also detected a fainter band of approximately 16 kDa. These data support the hypothesis that smaller polypeptides coterminal with full-length UL24 are present in HSV-1-infected cells.
FIG 2.

Expression of an 18-kDa protein, UL24.5, in HSV-1-infected cells. Lysates from Vero cells infected with one or the other of two independent isolates of vBAC_UL24HA were harvested 10 h postinfection (hpi). Lysates were subjected to SDS-PAGE and analyzed by immunoblotting using an anti-HA MAb. The positions of polypeptides recognized by the antibody are indicated to the right, including a 16-kDa polypeptide identified by an asterisk. α-Tubulin served as a loading control.
UL24.5 is not a degradation product of full-length UL24.
To test if the UL24.5 protein was a degradation product of full-length UL24 and not a separate translation product, we analyzed the expression of UL24.5 from a UL24-null strain. The UL24-null strain, UL24X (4), contains stop codons in all three reading frames at codon 43 of HSV-1 UL24. We inserted a C-terminal HA epitope tag in-frame at the end of the UL24 ORF, generating the strain vUL24X-HA. We then compared proteins from cells infected for 12 h with vHA-UL24 (where the HA tag is located at the N terminus of UL24) or with vUL24X-HA (where the HA tag is located at the C terminus of UL24). Western blot analysis revealed that even in the absence of expression of full-length UL24 protein, we detected the 18-kDa UL24.5 protein (Fig. 3). At this exposure, we also noted the presence of a band just above UL24.5, the nature of which is presently unclear. The expression of the UL24.5 protein in the absence of full-length UL24 protein demonstrates that UL24.5 is not a degradation product of full-length UL24.
FIG 3.
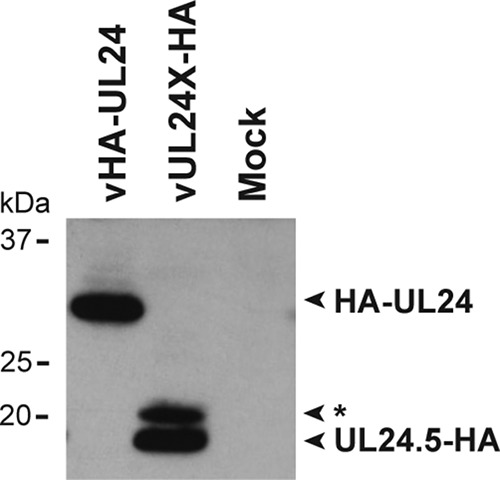
UL24.5 is not a degradation product of full-length UL24. Lysates from mock-infected cells or cells infected with either vHA-UL24 (which expresses an HA epitope tag at the N terminus of UL24) or vUL24X-HA (which expresses an HA tag at the C terminus of UL24) were prepared from cells harvested 12 hpi. Lysates were subjected to SDS-PAGE and analyzed by immunoblotting with an anti-HA MAb. The positions of polypeptides that are recognized by the anti-HA antibody are indicated to the right. The asterisk indicates a minor band migrating slightly above UL24.5.
UL24.5 is a late protein.
Based on the estimated size of UL24.5, as determined by its migration in SDS-PAGE and on the amino acid sequence of UL24, we predicted that the initiation codon for UL24.5 corresponds to the MET ATG codon 122. In order to verify if M122 was indeed the initiation codon for the UL24.5 protein, we introduced a substitution mutation leading to the ATG 122 codon being replace by a GTG codon for valine (Fig. 4A) in the context of a UL24 gene encoding a C-terminal HA epitope tag. The corresponding rescue virus was also produced. Recombinant viruses were generated using the BAC system (14, 15). DNA sequencing confirmed that the desired mutations or restored wild-type sequences in UL24 were present in the recombinant viruses. We also verified that there were no major genomic rearrangements in the new recombinant viruses. The EcoRV restriction digest pattern of the different viral genomes did not reveal any major genomic alterations (Fig. 4B).
FIG 4.

UL24.5 is expressed with late kinetics. (A) Diagram of the UL24 gene of the vBAC_UL24.5negHA mutant and rescue virus. The HA tag is located immediately before the stop codon. The third in-frame ATG of UL24 was mutated to GTG to inhibit expression of the UL24.5 protein. (B) EcoRV digestion patterns of the indicated viral genomes. The digestion products were resolved on a 0.7% agarose gel and stained with ethidium bromide. (C) Mock-, vBAC_KOS-, vBAC_UL24.5negHA-, or vBAC_UL24.5negHA-Rescue-infected cells were infected in parallel either in the presence or in the absence of 400 μg/ml of the viral DNA replication inhibitor PAA. Cell lysates harvested at 5, 10, and 15 hpi were subjected to SDS-PAGE and immunoblotted with anti-HA MAb. The blot was then stripped and reprobed with anti-gC PAbs or anti-γ-tubulin PAbs. Positions of the various proteins are indicated to the right by arrows. An asterisk marks the position of a 15-kDa polypeptide whose identity remains to be determined. The true late viral protein gC served as a late gene control for the inhibition of viral DNA synthesis. γ-Tubulin served as a loading control.
Analysis of UL24-related proteins expressed from the UL24.5 negative strain was carried out by Western blotting (Fig. 4C). Vero cells were either mock infected or infected with vBAC_UL24.5negHA or vBAC_UL24.5negHARescue. In order to determine the kinetics of expression of UL24.5, cells were infected in parallel in the presence of 400 μg/ml of phosphonoacetic acid (PAA), an inhibitor of viral DNA replication. At the indicated times p.i., total cell lysates were harvested, subjected to SDS-PAGE, and analyzed by immunoblotting with an anti-HA MAb. As predicted, for vBAC_UL24.5negHA, we detected expression of the full-length UL24 protein (10 and 15 hpi) but not of the 18-kDa UL24.5 protein. Expression of UL24.5 was restored with the vBAC_UL24.5negHARescue virus. Similar to UL24, UL24.5 was detected at 10 and 15 hpi. We also detected a faster-migrating band a few kilodaltons smaller than UL24.5. We suspect that this UL24-related polypeptide is due to translation from the second ATG downstream of the third UL24 transcription start site (Fig. 1), which would be favored in the context of mutating M122. The intensity of this band varied somewhat between experiments. The blocking of viral DNA replication by PAA treatment resulted in a decrease in steady-state levels of full-length UL24, which is consistent with the previously characterized leaky-late kinetics of this protein (7). Treatment with PAA abolished detection of UL24.5. As a control, the membrane was incubated with antibody directed against gC, a product of a true late gene whose expression is strictly dependent on viral DNA replication. We concluded that UL24.5 protein expression is largely dependent on viral replication, which is consistent with late kinetics.
UL24.5 is not required for HSV-1 replication in epithelial cells in cell culture.
The ability of the UL24.5-null virus to replicate in cell culture was tested in a multistep replication assay. Vero cells were infected with vBAC-KOS, the UL24-deficient virus (vUL24X), vBAC_UL24.5negHA, or vBAC_UL24.5negHA-Rescue at a multiplicity of infection (MOI) of 0.01 (Fig. 5A). We found that viral yields were not affected by the absence of UL24.5, in contrast to the reduction in viral titers observed for vUL24X (4). In cell culture, HSV-1 strains that do not express UL24 exhibit a syncytial plaque phenotype that is more pronounced at high temperatures (3). Vero cells were infected with the indicated viruses and incubated at 37°C or at 39°C for 48 h. In contrast to vUL24X, we found that vUL24.5negHA produced nonsyncytial plaques at both 37°C and 39°C, similar to vBAC-KOS and the rescue virus (Fig. 5B). Thus, UL24.5 does not affect viral yield or plaque morphology in cell culture. Moreover, these results suggest that the M122V substitution used to block expression of UL24.5 does not block the function of full-length UL24.
FIG 5.

UL24.5 is not important for replication in cell culture. (A) The impact of the absence of UL24.5 was assessed in a multistep replication assay. Vero cells were infected in duplicate with the indicated viruses at an MOI of 0.01. At the indicated times postinfection, cells and supernatant were harvested to determine the total production of infectious particles. Error bars represent the standard errors of the means (SEM). (B) Comparison of plaque morphology for the indicated viruses on Vero cells at 2 days postinfection (dpi) at 37°C and 39°C.
UL24.5 is mainly cytoplasmic and does not alter nucleolin localization.
UL24 is sufficient to induce the redistribution of the major nucleolar proteins nucleolin and B23 (9, 10). To test if UL24.5 shares this function with UL24, we constructed mammalian expression plasmids using two different strategies to express UL24.5 with a C-terminal HA epitope tag (Fig. 6A). In the first, the initiating ATG of full-length UL24, as well as the second ATG in the same frame, were mutated to GTG. In the second strategy, we deleted the 5′ sequence of UL24 up to and including the A residue of the second UL24 in-frame ATG, thereby ensuring that for this construct the first ATG encountered by the scanning ribosome is the predicted UL24.5 ATG. Lysates from cells transfected with the vectors pCGPfl-UL24-HA-1,2GTG and pCGPfl-UL24-Δ5′-HA were harvested, analyzed by SDS-PAGE, and subjected to immunoblotting with anti-HA MAb. In both cases we were able to detect the expression of the 18-kDa UL24.5 (Fig. 6B). Expression from pCGPfl-UL24-HA-1,2GTG was weaker than that from the construct containing a 5′ deletion of UL24. This difference may be due the cytomegalovirus (CMV) promoter in the plasmid being stronger than the endogenous UL24 promoter for the third transcription start site, if indeed the endogenous promoter is used for pCGPfl-UL24-HA-1,2GTG. Moreover, the shorter 5′-untranslated region of the deletion construct should reduce the possibility of extensive secondary structure hindering scanning by the ribosome. Two faster-migrating bands were also detected, one corresponding to approximately 15 kDa and another corresponding to 10 kDa. The 15-kDa protein may be due to translation from one of the two other in-frame ATG codons within the UL24.5 coding region. At present, the exact nature of these smaller proteins is unclear. We compared the localization of ectopically expressed UL24.5 to that of UL24 by confocal microscopy. As we have seen previously (10), UL24 was distributed throughout the nucleoplasm and in the cytoplasmic compartment. In contrast, UL24.5 was mainly cytoplasmic, although we did detect a small amount of UL24.5 in nucleoli, which was similar to the minor staining pattern we have seen previously for full-length UL24 (10). Moreover, we found that strong foci of nucleolar staining for nucleolin remained in cells expressing UL24.5, indicating that the latter did not lead to the dispersal of nucleolin. We quantified the effect of UL24 and UL24.5 on the staining pattern for nucleolin. In order to validate that the position of HA tag does not impact UL24 function, we also created a plasmid, pCGPfl-UL24-HA, in which the HA tag is fused to the C terminus of UL24. We found that at 24 hours posttransfection (hpt), expression of either HA-UL24 or UL24-HA led to a similar altered distribution pattern of nucleolin in the majority of transfected cells (Fig. 6C). In contrast, in cells transiently expressing UL24.5 (expressed from the plasmid pCGPfl-UL24-Δ5′-HA), nucleolin remained in distinct foci. We observed no significant difference in the nucleolin distribution compared to that seen in mock-transfected cells. UL24 also induces the redistribution of the nucleolar protein B23 (9). In similar experiments, we found that UL24.5 did not induce the redistribution of B23 either (data not shown). Thus, although a small portion of UL24.5 accumulates in nucleoli, it appears that unlike UL24, UL24.5 does not alter the localization of nucleolar proteins.
FIG 6.

UL24.5 is mainly cytoplasmic and does not alter nucleolin localization. (A) Diagram of the UL24.5 gene from the pCGPfl-UL24-HA-1,2GTG (top) and pCGPfl-UL24-Δ5′-HA (bottom) vectors. UL24.5 transcription is under the control of a CMV promoter (PCMV). (B) Lysates from cells transfected with the indicated UL24.5 expression vector were prepared at 24 h posttransfection, subjected to SDS-PAGE, and analyzed by immunoblotting with anti-HA MAbs. Positions of the various proteins are indicated to the right by arrows. The positions of polypeptides smaller than UL24.5 are indicated by asterisks. (C) Confocal images of Vero cells transiently expressing the full-length UL24 protein with either an N-terminal (HA-UL24) or C-terminal (UL24-HA) HA epitope tag or UL24.5 with a C-terminal HA tag (UL24.5-HA). Twenty-four hours posttransfection, cells were coimmunostained for HA (green) and nucleolin (red). DNA was stained with Hoechst 33342 (blue). Arrowheads show the foci of nucleolin that persist in cells expressing UL24.5. Scale bars correspond to 10 μm. To the right of the confocal images is a graph quantifying the distribution of nucleolin in transfected cells expressing HA-UL24, UL24-HA, or UL24.5-HA or in mock-transfected cells. The graph shows the percentage of cells expressing UL24 or UL24.5 that exhibited altered nucleolin staining. In the case of mock-transfected cells, the value was determined for cells from within a random field of view. Each result represents the average from two experiments where more than 100 cells were analyzed for each condition. Error bars represent the standard errors of the means. ***, P < 0.001; **, 0.001 < P < 0.01; ns, nonsignificant difference in Student's t test comparisons with mock-transfected cells.
Absence of the UL24.5 protein does not affect viral titers in the cornea or TG during acute infection.
We tested the importance of UL24.5 in vivo in a murine model of HSV-1 ocular infection that we have used previously (6, 16, 17). Mice were infected with 1 × 106 PFU per eye with the wild-type vBAC_KOS virus, vBAC_UL24.5negHA, vBAC_UL24.5negHA-Rescue, UL24X, and vUL24X-Rescue. Viral titers in tear films were measured for days 1, 2, and 3 postinfection (Fig. 7A). We found that viral titers were similar for all viruses tested except for UL24X, which exhibited an approximately 10-fold reduction, consistent with previous reports (4). Viral titers in TG were evaluated at 3 days postinfection (dpi) (Fig. 7B). Here again we found that viral titers were similar for all of the viruses tested except for UL24X, which exhibited the previously described drastic reduction (up to 4 log10) of viral titers in TG (4). Likewise, we found that vBAC_UL24.5negHA was able to reactivate from latency ex vivo, and there was no statistical difference between the numbers of TG that were positive for viral reactivation for the UL24.5 mutant and the wild-type or rescue viruses (Table 1). We concluded that UL24.5 does not play an important role early during acute infection in the cornea and TG, and it is not required for reactivation ex vivo.
FIG 7.
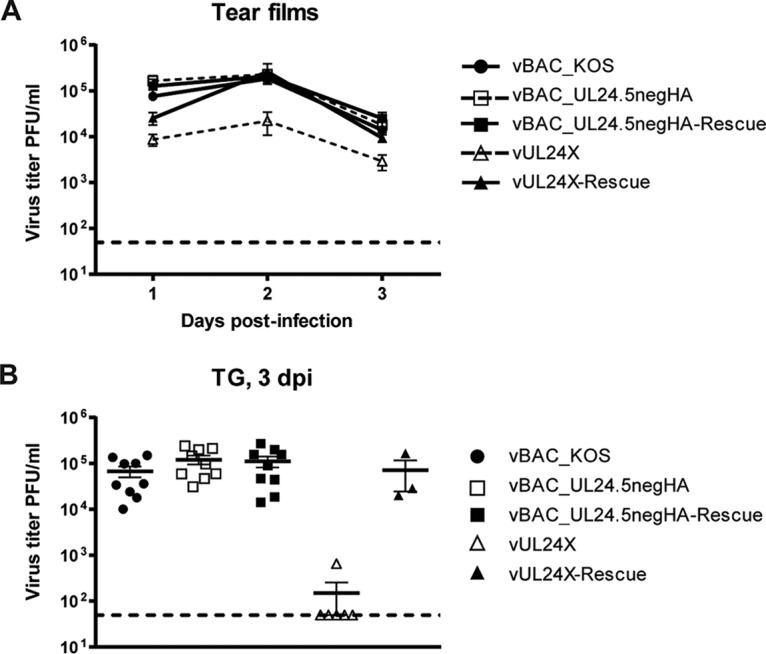
Absence of UL24.5 protein does not affect viral titers in tear films or in trigeminal ganglia (TG) during acute ocular infection of mice. Quantification of viral titers during acute infection in vBAC_KOS-, vBAC_UL24.5negHA-, vBAC_UL24.5negHA-Rescue-, vUL24X-, and vUL24X-Rescue-infected mice. (A) Titers of infectious virus present in tear films of mice at 1, 2, and 3 dpi. Data shown are pooled from four independent experiments. (B) Titers of infectious virus present in TG at 3 dpi. Data shown are pooled from three independent experiments. Each symbol represents an individual mouse. The midpoints of the bars in the graph represent the means. All error bars represent the standard errors of the means (SEM).
TABLE 1.
UL24.5 is not required for HSV-1 reactivation ex vivo
| Virus | No. of reactivated TG/total TG (%) |
|---|---|
| vBAC_KOS | 10/10 (100) |
| vBAC_UL24.5negHA | 10/11 (90.9) |
| vBAC_UL24.5negHA-Rescue | 14/14 (100) |
| vUL24X | 2/4 (50) |
| vUL24X-Rescue | 4/4 (100) |
| Mock | 0/8 (0) |
Increased clinical signs in mice infected with UL24.5 mutants.
During our in vivo analyses, we observed a striking difference in the ability of vUL24.5negHA to cause disease. Because of the unexpected results, we included an independent isolate of the UL24.5neg virus in these experiments. The two strains are referred to as vBAC_UL24.5negHA(1) and vBAC_UL24.5negHA(2), and each has its respective rescue strain. To assess the impact of the absence of UL24.5 on disease, we infected mice with the wild-type virus vBAC_KOS, two independent isolates of the vBAC_UL24.5negHA, the corresponding rescue viruses, UL24X, or vUL24X-Rescue. We scored for the severity of disease by daily visual inspection up to 25 dpi. Assessment of disease was done in a blinded manner. As expected, we observed little periocular disease in mice infected with UL24X, while we observed periocular disease characterized by inflammation, hair loss, and lesions beginning at 4 dpi for the wild-type virus and rescue strains (Fig. 8), consistent with our previous results (6). We observed similar signs in mice infected with the vUL24.5negHA strains. From approximately 13 dpi, we observed a decrease in periocular disease for the wild-type virus and for the respective rescue viruses. Surprisingly, pronounced signs of periocular disease persisted several days longer for mice infected with either of the two independent UL24.5-null strains and were not completely resolved by 25 dpi. We also assessed signs of neurological impairment in a blinded manner (Fig. 9). We observed similar trends for both isolates of the UL24.5-deficient virus. For vBAC_UL24.5negHA(1), 8 out of 9 mice exhibited severe neurological impairment, a difference which was significant compared to the value for the parental strain KOS or for the rescue virus. For the second isolate, 2 out of 6 mice also exhibited signs of severe neurological impairment. The day the maximal score was attained for the vUL24.5neg-infected mice varied between 14 and 25 dpi. The two mice with a score of 4 were euthanized because they had reached the limit points (data not shown). In these experiments, for all other viruses tested we did not observe any signs of neurological problems. As expected, UL24X did not cause neurological symptoms in infected mice (6). Thus, our results demonstrate that the absence of UL24.5 during infection in vivo leads to a prolonged period of periocular disease and an increase in the incidence of severe neurological impairment.
FIG 8.

Absence of UL24.5 protein prolongs the period of periocular disease. Following infection of mice with the indicated viruses, the mean disease score, as determined on a scale of 0 to 4, was assessed each day from 4 to 25 dpi. The graph shows the average scores for data pooled from three independent experiments (n = 9 for vBAC_KOS, vBAC_UL24.5negHA [1], and vBAC_UL24.5negHA-Rescue [1] and n = 6 for vBAC_UL24.5negHA [2], vBAC_UL24.5negHA-Rescue [2], vUL24X, and vUL24X-Rescue), where 1 and 2 represent independent isolates of the indicated strain. The value indicated for each day is the mean value obtained for each animal analyzed on that day postinfection. Statistical significance was evaluated using the nonparametric Mann-Whitney test. For the purposes of statistical analyses, values of 0 were replaced by values of 0.1. Clinical scores were assessed in a blinded manner.
FIG 9.
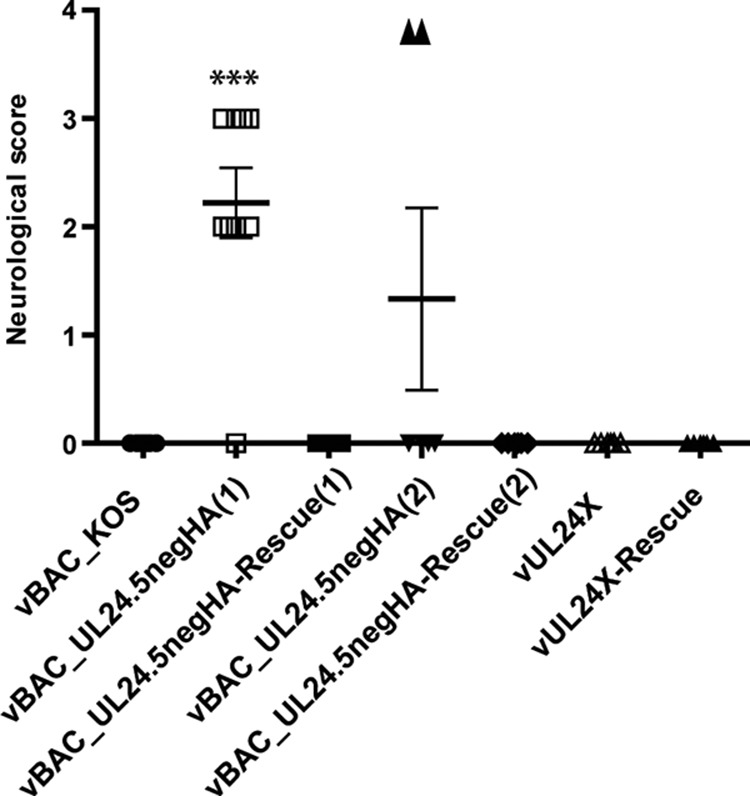
Absence of UL24.5 results in an increased incidence of severe neurological signs. Following infection of mice with the indicated viruses, the mean neurological score, as determined on a scale of 0 to 4, was assessed each day from 4 to 25 dpi. The graph shows the maximum score reached by each mouse over that time period. Data shown were pooled from two or three independent experiments (n = 9 for vBAC_KOS, vBAC_UL24.5negHA [1], and vBAC_UL24.5negHA-Rescue [1] and n = 6 for vBAC_UL24.5negHA [2], vBAC_UL24.5negHA-Rescue [2], vUL24X, and vUL24X-Rescue). Clinical scores were assessed in a blinded manner. The midpoint of the bars in the graph represent the means. Error bars represent the standard errors of the means (SEM). Statistical analysis was carried out using a nonparametric Mann-Whitney test (***, P < 0.001). For the second isolate of the UL24.5-deficient virus, the increase in the mean compared to that of controls was not significant. Scores of 0 were replaced by 0.1 for the purpose of the statistical analyses.
Conservation of methionine 122 among alphaherpesviruses.
To determine if it is likely that UL24.5 orthologs are present in other herpesviruses, we undertook a bioinformatic analysis to assess the degree of conservation of the M122 codon. We aligned the UL24 amino acid sequences of eight HSV-1 strains whose full genome sequences are in NCBI (Fig. 10A and Table 2). The results revealed that the M122 codon is present in all of the HSV-1 UL24 protein sequences studied. These results support the notion that UL24.5 is a genuine HSV-1 protein and not specific to the strain KOS. We noted that M180, an in-frame methionine codon downstream of M122, is conserved among the HSV-1 UL24 sequences, while M191, another in-frame methionine codon, was found in 5 of 8 of the strains. When we analyzed UL24 homologs of all human alphaherpesviruses, we found that M122 was also conserved for HSV-2 and for varicella-zoster virus (VZV) and in several nonhuman alpha herpesviruses (Fig. 10B and Table 2). In contrast, in the beta- and gammaherpesviruses M122 is not conserved. In most instances, a leucine is present in the place of M122. Because leucine codons can serve as initiation codons, their presence is compatible with the expression of UL24.5. For a small number of viruses, no equivalent was found for M122 (e.g., Epstein-Barr virus). In conclusion, our results suggest that UL24.5 is a bona fide HSV-1 protein and likely is also expressed by other alphaherpesviruses. Expression in beta- and gammaherpesviruses would necessarily imply translation initiation from an alternative initiation codon.
FIG 10.

Methionine 122 of HSV-1, the initiation codon of UL24.5, is conserved among several alphaherpesvirus UL24 orthologs. (A) Comparison of UL24 protein sequences across multiple HSV-1 strains as indicated. (B) Analysis of UL24 protein sequences among the human alpha-, beta-, and gammaherpesviruses and among several nonhuman alpha-, beta-, and gammaherpesviruses. The in-frame methionines of HSV-1 UL24 and the equivalent residues in the other herpesviruses are indicated in boldface, including leucine residues, which may function as alternative initiation codons.
TABLE 2.
Accession numbers for sequences used in analyses presented in Fig. 10
DISCUSSION
In this study, we investigated the possibility that the third transcription initiation site for the UL24 gene, which is located within the UL24 ORF, leads to the expression of a new HSV-1 protein. By generating a virus and expression constructs designed to fuse an HA epitope to the C terminus of UL24 we identified a protein, UL24.5, which is coded in frame with the carboxy end of UL24 and whose N terminus corresponds to M122 of full-length UL24. There are sets of both early and late UL24 transcripts. We found that like full-length UL24 (7), UL24.5 is expressed with late kinetics, thus the function of the early UL24 transcripts remains a mystery. Our findings are consistent with early reports describing HSV-1 gene expression where Northern blot analysis of polysome-associated RNAs from infected cells identified 5.6- and 5.2-kb transcripts corresponding to the UL24 locus (18), with the 5.2-kb species likely originating from the third transcription start site of UL24 (19). Consistent with the late kinetics of the 5.2-kb UL24 transcript, there is a TATA box located 26 nucleotides upstream of the start site of initiation. The leader sequence for the 5.4- and 1.2-kb UL24 transcripts is predicted to be only 25 nucleotides. Although this length is relatively short, there are examples in infected cells where leader sequences have been shown to have little impact on translation. Namely, Alonso-Caplen et al. (20) created recombinant adenoviruses engineered to express the influenza NP gene under the control of a late adeno promoter in which they inserted various lengths of the NP tripartite leader sequence (from 0 to 200 nt). In this context, the leader sequence was not required for efficient translation, although it did affect transcript accumulation. Thus, it may be that in the context of viral infection certain translation regulatory sequences are less important than they are in uninfected cells.
There have been other reports of sets of coterminal polypeptides expressed from HSV-1 genes. For example, HSV-1 Us1 encodes the immediate-early protein ICP22 as well as the N-terminally truncated form, US1.5 (21). Interestingly, ICP22 and US1.5 share certain functions in cell culture, but only ICP22 appears to affect viral infection in vivo (22). Thus, in the case of both UL24/UL24.5 and ICP22/US1.5, the additional coding capacity of the viral genome has an impact on pathogenicity.
In addition to the UL24.5 protein, we detected two other smaller proteins coded in frame with full-length UL24, a protein of approximately 15 to 16 kDa and one of 10 kDa (Fig. 4). These proteins likely arise from the use of M180 and M191 as initiating methionines (Fig. 10). Mutagenesis experiments targeting these potential initiation codons would be required to confirm that they function as such. Very recently, several different isoforms of the UL24 homolog in HHV-8 (ORF20) were identified (23). The shortest of the isoforms, ORF20B (257 aa), corresponds to full-length UL24 in HSV-1, and no equivalent of UL24.5 has been described. Their finding is consistent with our bioinformatics analysis. We found that the initiation MET codon for UL24.5 was conserved in all HSV-1 strains analyzed, as well as in HSV-2 and VZV reference strains and a subset of nonhuman alphaherpesviruses. In contrast, there was no equivalent MET codon for any beta- or gammaherpesviruses we analyzed. Thus, we propose that UL24.5 is important specifically for members of the Alphaherpesvirinae subfamily.
The primary sequence of UL24.5 begins within the 4th homology domain of UL24, thus it has an intact 5th homology domain and contains the entirety of the less well-conserved C-terminal domain (Fig. 1). The N-terminal portion of UL24 (aa 1 to 190) is sufficient to induce the dispersal of nucleolin; furthermore, deletion of any one of the homology domains abrogates this activity (10). Thus, it was not surprising to find that ectopically expressed UL24.5 did not induce the redistribution of either nucleolin or B23. Nevertheless, we found that UL24.5 was still able to accumulate in the nucleolus to some degree, even though the protein was mainly cytoplasmic. This result may be related to the predicted nuclear localization signal in the C terminus that has been described but not yet validated. A sequence capable of targeting a heterologous protein to nucleoli has been located in the first 60 aa of HSV-1 UL24 (10). The expression of UL24.5 may explain the presence of nuclear/nucleolar targeting sequences in both the N- and C-terminal domains of the UL24 ORF. Instead of simple redundancy in the full-length UL24 protein, the nuclear targeting signal near the C terminus may be there as a requirement for the proper localization of UL24.5.
We found that the absence of UL24.5 during infection in cell culture had no effect on viral yield or on plaque morphology. Moreover, in a mouse model of ocular infection, no reduction in viral titers was found in the eye or in TG during acute infection. This result is in stark contrast to the impact of the absence of full-length UL24 on HSV-1 replication in vivo, which results in a decrease in viral titers of 1 log10 in epithelial cells of the cornea and of up to 4 log10 reduction of viral titers in TG (4, 6). These results are further evidence that UL24 and UL24.5 have distinct roles in the host cell.
Where we did observe an impact of the loss of UL24.5 during infection was with regard to pathogenesis. A blind assessment of periocular disease carried out between 4 and 25 days postinfection revealed that signs of disease persisted longer in mice infected with either of the two vUL24.5negHA independent isolates than with the parental strain or the rescue viruses. It will be important to determine if the prolonged period of periocular disease reflects a prolonged period of viral replication in the eye or TG or a delay in the adaptive immune response necessary to ultimately clear the infection. The M122V substitution mutation that blocked expression of UL24.5 also necessarily introduced the M122V substitution in full-length UL24. Although we cannot formally rule out that this substitution is the cause of the in vivo phenotypes we observed, it is unlikely that the substitution has a negative impact on the function of full-length UL24, because no replication defect was detected in cell culture or during acute infection of mice 1 to 3 dpi. Interestingly, a previous report found that the absence of ORF20, the homolog of HSV-1 UL24 in MHV-68, resulted in prolonged persistence of viremia in the lungs following intranasal infection (24), which is similar to our observations with vUL24.5negHA. However, there is no M122 equivalent in MHV-68, suggesting that if there is a similar mechanism at play, then in some herpesviruses full-length UL24 itself is responsible for the functions dedicated to UL24.5 in other viruses. In addition to prolonged periocular disease, we observed an increase in the incidence of severe neurological disorders in mice infected with UL24.5-deficient viruses. Presently, it is unclear if these symptoms are due to increased viral titers in the brain at late times in infection or due to some form of dysregulation of the immune response to infection leading to neurological damage. Regardless, our results point to UL24.5 involvement in limiting neurological disease during acute infection.
To conclude, we have discovered a new HSV-1 protein, UL24.5. UL24.5 appears to have different functions during infection than UL24. Although a virus deficient in UL24.5 did not exhibit any replication defects in cell culture, in vivo, such a mutant exhibited a more severe pathology, namely, prolonged signs of periocular disease and an increased incidence of severe neurological disorders. Further studies are required to elucidate the role of UL24.5 in vivo and to determine if it affects the host immune response.
MATERIALS AND METHODS
Cells.
Vero cells (an African green monkey kidney epithelial cell line) were grown in Dulbecco's modified Eagle medium (DMEM) with 4.5 g/liter glucose, supplemented with 5% newborn calf serum (NCS), 50 U/ml penicillin, and 50 μg/ml streptomycin. Cells were maintained in an incubator at 37°C with 5% CO2.
Plasmid construction.
All oligonucleotide sequences are listed in Table 3. The UL24-HA gene was amplified by PCR using primers 419 and 420. The resulting PCR product flanked by BglII and BamHI restriction sites was then subcloned into the pBlueScript II SK+ vector. The sequence of interest was excised with BglII and BamHI and then inserted in place of the corresponding 3′ end of the UL24 gene contained in the mammalian expression vector pCGPfl-UL24 (10). The resulting vector is named pCGPfl-UL24-HA. The pCGPfl-UL24-Δ5′-HA vector was constructed by amplification of the last 504 nucleotides of the UL24 gene, carried out using primers 543 and 420. The resulting PCR product, flanked by two BamHI restriction sites, was then subcloned into the pBlueScript II SK+ vector. The resulting DNA sequence of interest, i.e., a fragment of UL24 lacking the first 306 nucleotides, was then ligated into the pCGPfl vector using BamHI restriction sites. To generate the vector pCGPfl-UL24-HA-1,2GTG, site-directed mutagenesis using overlap extension PCR was carried out on pCGPfl-HA-UL24 with primers 420, 440, 487, and 489 to change the second in-frame ATG of UL24 to GTG. The resulting plasmid, pCGPfl-UL24-HA-2GTG, was then used as a template for the second site-directed mutagenesis step to change the first ATG of UL24 to GTG using the primers 542 and 420. The resulting PCR product was then digested with BamH1 and ligated into the pCGPfl vector using BamHI restriction sites. The pCGPfl-HA-UL24 vector was described previously (10). All intermediate and final constructs were confirmed by sequencing, which was carried out by the McGill University and Genome Innovation Center sequencing service.
TABLE 3.
Primers used in this study
| Primer | Sequence, 5′ to 3′a |
|---|---|
| 419 BglII5′UL24top | CCC AGA TCT GCG GCA CGC TG |
| 420 BamHI_UL24c-HAbot | CCC GGA TCC TCA AGC GTA ATC TGG AAC ATC GTA TGG GTA TTC GGA GGC GGC TCG |
| 440 Pfl23II_HSV1-UL24_F | TAT CGT ACG ATG GCC GCG AGA ACG CGC |
| 487 HSVUL24_2eATGenGTG_rev | ATG GCT TCG TAC CCC TGC CAC CAA CAC |
| 489 HSVUL24_2eATG_fwd | GCA GGG GTA CGA AGC CAT ACG CGC TTC |
| 542 HSVUL24_mut1erATGenGTG_fwd(2) | ATA GGA TCC GTG GCC GCG AGA ACG CG |
| 543 HSVUL24_without306nuc5′ | ATA GGA TCC TGC AAA TAT ATT TCT TCC GGG GAC |
| 602 BAC_KOS_UL24.5negHA_Rescue_F | CAA ATA TATTTC TTC CGG GGA CAC CGC CAG CAA ACG CGA GCA ACG GGC CAC GGG GAT GAA GCA GCT GCG CCA CTC CCT GAA GGA TGA CGA CGA TAA GTA |
| 603 BAC_KOS_UL24.5negHA_Rescue_R | CAC CCG GAG GCG CGA GGG ACT GCA GGA GCT TCA GGG AGT GGC GCA GCT GCT TCA TCC CCG TGG CCC GTT GCT CGC GTT TGC AAC CAA TTA ACC AAT TCT GA |
| 592 BAC_KOS_UL24.5negHA_F | CAA ATA TAT TTC TTC CGG GGA CAC CGC CAG CAA ACG CGA GCA ACG GGC CAC GGG GGT GAA GCA GCT GCG CCA CTC CCT GAA GGA TGA CGA CGA TAA GTA |
| 593 BAC_KOS_UL24.5negHA_R | CAC CCG GAG GCG CGA GGG ACT GCA GGA GCT TCA GGG AGT GGC GCA GCT GCT TCA CCC CCG TGG CCC GTT GCT CGC GTT TGC AAC CAA TTA ACC AAT TCT GA |
Restriction enzyme sites used for cloning are underlined.
Viruses.
The HSV-1 strains KOS and UL24X (4) were originally provided by D. M. Coen (Harvard Medical School, Boston, MA). UL24X is in the KOS background. vBAC_KOS HSV-1 was described previously (12). vUL24X-Rescue was generated by homologous recombination, as described previously (25). The viral strains vBAC_UL24.5negHA and vBAC_UL24.5negHA-Rescue were created using the Red recombination system as we have used previously (12). The primers used to create the viruses are listed in Table 3. The complete UL24 genes for both of the vUL24.5neg independent isolates were sequenced to confirm the presence of the mutation and that no undesired mutations were present. For the rescue viruses, correction of the mutation was confirmed by sequencing. In contrast to vBAC_KOS HSV-1, which expresses untagged forms of UL24 and UL24.5, vBAC_UL24.5negHA and vBAC_UL24.5negHA-Rescue express UL24 or UL24.5 proteins with a hemagglutinin (HA) epitope tag fused at the C terminus.
Transfections.
A total of 2.5 × 104 Vero cells/well were plated in 24-well plates. Twenty-four hours later, cells were cotransfected with 1 μg of total plasmid DNA per well, as indicated, using Lipofectamine transfection reagent (Life Technologies).
Infection and Western blotting.
To assess expression of the HA-tagged UL24 proteins, 1 × 106 Vero cells were seeded per well in 6-well plates. The following day, cells were infected with the indicated virus at an MOI of 5 and lysed at 5, 10, or 15 h postinfection (hpi) in 150 μl of radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM Tris [pH 8.0], 1% Triton X-100, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate, and 500 mM NaCl). Cellular debris and lysates were collected, insoluble cellular debris was pelleted using a microcentrifuge, and the supernatant was retained. Protein samples were resolved by polyacrylamide gel electrophoresis on a denaturing sodium dodecyl sulfate 12.5% gel. Proteins were transferred to a polyvinylidene difluoride membrane (PVDF) and analyzed by Western blotting. The primary antibodies were used under the following conditions: a monoclonal mouse antibody (MAb) directed against HA (Covance) (1:250) for 1 h, an anti-γ-tubulin rabbit polyclonal antibody (PAb) (Sigma) (1:2,000) for 1 h, and an HSV-1 gC MAb (Fitzgerald) (1:150) overnight. Incubation with the indicated peroxidase-conjugated secondary antibodies was carried out at room temperature for 1 h. An anti-mouse secondary antibody was used to detect the expression of HA (1:1,000) and gC HSV-1 protein (1:5,000) (Jackson Immuno Research), and an anti-rabbit secondary antibody was used for the detection of the expression of γ-tubulin (Jackson Immuno Research) (1:5,000). Detection was done by enhanced chemiluminescence using ECL Plus reagent (GE-Amersham).
Murine model of ocular infection.
Experiments to test HSV-1 mutants in vivo were conducted using an ocular model of HSV-1 infection essentially as described previously (16, 17). Six- to 8-week-old CD-1 males were deeply anesthetized by intraperitoneal injection with a mixture of ketamine (75 mg/kg of body weight; Bioniche) and xylazine (10 mg/kg; Bajer) in saline solution. Corneas of anesthetized mice were infected as described previously with 1 × 106 PFU per eye (6). For strains vBAC_UL24.5negHA, vBAC_UL24.5negHA-Rescue, and vBAC_KOS, viral titers in the eye and in TG were determined for at least three mice each. For strains vUL24X and vUL24X-Rescue, viral titers in the eye and in TG were determined for at least two mice each. For ex vivo reactivation assays, TG were removed immediately postmortem, dissociated enzymatically, and overlaid on a monolayer of Vero cells. Reactivation was detected by observing the monolayer daily for 10 days, with the presence of the cytopathic effect of the Vero cells used to establish that viral reactivation had occurred. Backtiters of inocula were verified following infection of the mice. All animal experiments were performed at the INRS-Center for Experimental Biology in accordance with institutional good animal care practices. Data points below the level of detection in the experiments were treated as the limit values for calculation of means.
Disease scoring. (i) Inflammation.
Periocular disease was assessed visually on a daily basis and in a blind manner over the indicated period of time and scored on a scale of 0 to 4: 0, no change; 1, mild inflammation (swelling); 2, moderate inflammation and mild periocular hair loss; 3, severe inflammation, moderate periocular hair loss, and mild skin lesions; 4, severe inflammation, severe periocular hair loss, and severe skin lesions.
(ii) Neurological impairment.
Neurological signs were assessed visually on a daily basis and in a blind manner over the indicated period of time. Neurological signs were scored on a scale of 0 to 4: 0, no neurological signs; 1, slow, creeping movement; 2, slow movement, mild hemiataxia on the left or right side; 3, difficulty moving, severe hemiataxia on the left or right side leading to circular movement; 4, hemiplegia on the left or right side, pronounced difficulty moving, difficulty breathing.
Immunofluorescence microscopy.
Vero cells (5 × 104) were seeded on glass coverslips 24 h before transfection with expression plasmids using Lipofectamine transfection reagent (Life Technologies) according to the manufacturer's instructions. Twenty-four hpt, cells were fixed, permeabilized, and immunostained as described previously (8). Anti-HA high affinity (1:100; Roche) and anti-nucleolin (1:200; Abcam) were used as primary antibodies, and anti-rat or anti-rabbit polyclonal antibodies coupled to Alexa-488 or Alexa-568 (Invitrogen) were used as secondary antibodies. Following immunostaining, coverslips were incubated for 1 min with Hoechst 33342 (Molecular Probes) diluted 1:2,000 in phosphate-buffered saline (PBS). The coverslips were then washed three times in PBS and mounted onto glass slides using a Prolong Gold antifade reagent (Invitrogen). The slides were visualized using a Zeiss LSM780 confocal system equipped with a diode laser at 405 nm, an argon multiline laser at 458/488/514 nm, a diode-pumped solid-state laser at 561 nm, and an HeNe laser at 633 nm mounted on a Zeiss Axio Observer Z1 microscope (63× objective; numeric aperture, 1.4) and operated with ZEN 2011 software (Zeiss). Images were prepared using Adobe Illustrator CS5 software.
ACKNOWLEDGMENTS
C.S.-S. and C.E.G.S. were recipients of scholarships from the Fondation Armand-Frappier (FAF). This work was funded by an operating grant from the Canadian Institutes of Health Research to A.P. (MOP 82924).
REFERENCES
- 1.Whitley RJ, Roizman B. 2001. Herpes simplex virus infections. Lancet 357:1513–1518. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- 2.Davison A. 1993. Herpesvirus genes. Rev Med Virol 3:237–244. doi: 10.1002/rmv.1980030407. [DOI] [Google Scholar]
- 3.Jacobson JG, Martin SL, Coen DM. 1989. A conserved open reading frame that overlaps the herpes simplex virus thymidine kinase gene is important for viral growth in cell culture. J Virol 63:1839–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacobson JG, Chen SH, Cook WJ, Kramer MF, Coen DM. 1998. Importance of the herpes simplex virus UL24 gene for productive ganglionic infection in mice. Virology 242:161–169. doi: 10.1006/viro.1997.9012. [DOI] [PubMed] [Google Scholar]
- 5.Rochette PA, Bourget A, Sanabria-Solano C, Lahmidi S, Ouellet Lavallee G, Pearson A. 2015. Mutation of UL24 impedes the dissemination of acute herpes simplex virus 1 infection from the cornea to neurons of the trigeminal ganglia. J Gen Virol 96:2794–2805. doi: 10.1099/vir.0.000189. [DOI] [PubMed] [Google Scholar]
- 6.Leiva-Torres GA, Rochette PA, Pearson A. 2010. Differential importance of highly conserved residues in UL24 for herpes simplex virus 1 replication in vivo and reactivation. J Gen Virol 91:1109–1116. doi: 10.1099/vir.0.017921-0. [DOI] [PubMed] [Google Scholar]
- 7.Pearson A, Coen DM. 2002. Identification, localization, and regulation of expression of the UL24 protein of herpes simplex virus type 1. J Virol 76:10821–10828. doi: 10.1128/JVI.76.21.10821-10828.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lymberopoulos MH, Pearson A. 2007. Involvement of UL24 in herpes-simplex-virus-1-induced dispersal of nucleolin. Virology 363:397–409. doi: 10.1016/j.virol.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 9.Lymberopoulos MH, Bourget A, Ben Abdeljelil N, Pearson A. 2011. Involvement of the UL24 protein in herpes simplex virus 1-induced dispersal of B23 and in nuclear egress. Virology 412:341–348. doi: 10.1016/j.virol.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 10.Bertrand L, Pearson A. 2008. The conserved N-terminal domain of herpes simplex virus 1 UL24 protein is sufficient to induce the spatial redistribution of nucleolin. J Gen Virol 89:1142–1151. doi: 10.1099/vir.0.83573-0. [DOI] [PubMed] [Google Scholar]
- 11.Ben Abdeljelil N, Rochette PA, Pearson A. 2013. The UL24 protein of herpes simplex virus 1 affects the sub-cellular distribution of viral glycoproteins involved in fusion. Virology 444:263–273. doi: 10.1016/j.virol.2013.06.021. [DOI] [PubMed] [Google Scholar]
- 12.Sanabria-Solano C, Gonzalez CE, Richerioux N, Bertrand L, Dridi S, Griffiths A, Langelier Y, Pearson A. 2016. Regulation of viral gene expression by the herpes simplex virus 1UL24 protein (HSV-1UL24 inhibits accumulation of viral transcripts). Virology 495:148–160. doi: 10.1016/j.virol.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Xu H, Su C, Pearson A, Mody CH, Zheng C. 2017. Herpes simplex virus 1 UL24 abrogates the DNA sensing signal pathway by inhibiting NF-κB activation. J Virol 91:e00025-17. doi: 10.1128/JVI.00025-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 15.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 16.Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, Tyler KL, Knipe DM. 1989. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci U S A 86:4736–4740. doi: 10.1073/pnas.86.12.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol 63:2893–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holland LE, Sandri-Goldin RM, Goldin AL, Glorioso JC, Levine M. 1984. Transcriptional and genetic analyses of the herpes simplex virus type 1 genome: coordinates 0.29 to 0.45. J Virol 49:947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cook WJ, Wobbe KK, Boni J, Coen DM. 1996. Regulation of neighboring gene expression by the herpes simplex virus type 1 thymidine kinase gene. Virology 218:193–203. doi: 10.1006/viro.1996.0179. [DOI] [PubMed] [Google Scholar]
- 20.Alonso-Caplen FV, Katze MG, Krug RM. 1988. Efficient transcription, not translation, is dependent on adenovirus tripartite leader sequences at late times of infection. J Virol 62:1606–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carter KL, Roizman B. 1996. The promoter and transcriptional unit of a novel herpes simplex virus 1 alpha gene are contained in, and encode a protein in frame with, the open reading frame of the alpha 22 gene. J Virol 70:172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mostafa HH, Davido DJ. 2013. Herpes simplex virus 1 ICP22 but not US 1.5 is required for efficient acute replication in mice and VICE domain formation. J Virol 87:13510–13519. doi: 10.1128/JVI.02424-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bussey KA, Lau U, Schumann S, Gallo A, Osbelt L, Stempel M, Arnold C, Wissing J, Gad HH, Hartmann R. 2018. The interferon-stimulated gene product oligoadenylate synthetase-like protein enhances replication of Kaposi's sarcoma-associated herpesvirus (KSHV) and interacts with the KSHV ORF20 protein. PLoS Path 14:e1006937. doi: 10.1371/journal.ppat.1006937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nascimento R, Costa H, Dias JD, Parkhouse RM. 2011. MHV-68 open reading frame 20 is a nonessential gene delaying lung viral clearance. Arch Virol 156:375–386. doi: 10.1007/s00705-010-0862-2. [DOI] [PubMed] [Google Scholar]
- 25.Bertrand L, Leiva-Torres GA, Hyjazie H, Pearson A. 2010. Conserved residues in the UL24 protein of herpes simplex virus 1 are important for dispersal of the nucleolar protein nucleolin. J Virol 84:109–118. doi: 10.1128/JVI.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]