

Human cytomegalovirus (HCMV) establishes a lifelong latent infection in the human host. While the infection is typically asymptomatic in healthy individuals, HCMV infection poses life-threatening disease risk in immunocompromised individuals and is the leading cause of birth defects. Understanding how HCMV controls the lifelong latent infection and reactivation of replication from latency is critical to developing strategies to control HCMV disease. Here, we identify the host factors targeted by a viral protein that is required for reactivation. We define the importance of this virus-host interaction in reactivation from latency, providing new insights into the molecular underpinnings of HCMV latency and reactivation.
KEYWORDS: cytomegalovirus, EGFR, Rab5, UL135, human herpesviruses, latency
ABSTRACT
Human cytomegalovirus, HCMV, is a betaherpesvirus that establishes a lifelong latent infection in its host that is marked by recurrent episodes of reactivation. The molecular mechanisms by which the virus and host regulate entry into and exit from latency remain poorly understood. We have previously reported that UL135 is critical for reactivation, functioning in part by overcoming suppressive effects of the latency determinant UL138. We have demonstrated a role for UL135 in diminishing cell surface levels and targeting epidermal growth factor receptor (EGFR) for turnover. The attenuation of EGFR signaling promotes HCMV reactivation in combination with cellular differentiation. In this study, we sought to define the mechanisms by which UL135 functions in regulating EGFR turnover and viral reactivation. Screens to identify proteins interacting with pUL135 identified two host adaptor proteins, CIN85 and Abi-1, with overlapping activities in regulating EGFR levels in the cell. We mapped the amino acids in pUL135 necessary for interaction with Abi-1 and CIN85 and generated recombinant viruses expressing variants of pUL135 that do not interact with CIN85 or Abi-1. These recombinant viruses replicate in fibroblasts but are defective for reactivation in an experimental model for latency using primary CD34+ hematopoietic progenitor cells (HPCs). These UL135 variants have altered trafficking of EGFR and are defective in targeting EGFR for turnover. These studies demonstrate a requirement for pUL135 interactions with Abi-1 and CIN85 for regulation of EGFR and mechanistically link the regulation of EGFR to reactivation.
IMPORTANCE Human cytomegalovirus (HCMV) establishes a lifelong latent infection in the human host. While the infection is typically asymptomatic in healthy individuals, HCMV infection poses life-threatening disease risk in immunocompromised individuals and is the leading cause of birth defects. Understanding how HCMV controls the lifelong latent infection and reactivation of replication from latency is critical to developing strategies to control HCMV disease. Here, we identify the host factors targeted by a viral protein that is required for reactivation. We define the importance of this virus-host interaction in reactivation from latency, providing new insights into the molecular underpinnings of HCMV latency and reactivation.
INTRODUCTION
Human cytomegalovirus (HCMV) is a betaherpesvirus and an important human pathogen. Like all herpesviruses, HCMV establishes an incurable latent infection and persists for the lifetime of the host. While the latent persistence of HCMV is typically asymptomatic in healthy individuals, reactivation from latency in the immunocompromised poses a life-threatening disease risk. As such, HCMV is a serious disease risk in the context of stem cell and solid organ transplantation (1, 2). Further, infection, reinfection, or reactivation of HCMV during pregnancy can result in cognitive and physical deficits, and HCMV is the leading cause of congenital birth defects (3, 4). Understanding HCMV latency and reactivation at the molecular level is critical to identifying novel strategies to control reactivation from latency and HCMV disease.
HCMV infects the majority of the human population worldwide (3). Within infected individuals, HCMV genomes are detected in undifferentiated hematopoietic populations, including CD34+ hematopoietic progenitor cells (HPCs) (5, 6). Reactivation of virus replication occurs in hematopoietic cells in response to allogeneic stimulation or myeloid differentiation (7, 8). Molecular mechanisms of HCMV latency have been difficult to define as the viral genome is maintained at very low copy numbers, and viral genes are expressed at very low levels in the host (9–12). As HCMV infection and latency are restricted to human cells, experimental models of latency in cultured primary human hematopoietic cells or cell lines have been critical.
The establishment and maintenance of viral latency, as well as reactivation from latency, depend on a number of as yet ill-defined host signaling events associated with cell survival, differentiation, chromatin remodeling, and stress (13, 14). In the herpesvirus field, viral determinants that function to modulate signaling pathways have begun to emerge as mechanisms controlling latency and reactivation. We have previously identified a 3.6-kb polycistronic locus within the ULb′ region of the HCMV genome that is lost upon serial passage of the virus in fibroblasts (15, 16). This locus encodes four genes, UL133, UL135, UL136, and UL138, that play important roles in regulating viral latency and reactivation from latency (16–23). In particular, UL135 is critical for reactivation of the virus from latency (18, 23). Viruses containing a disruption of UL135, either by deletion of the entire coding sequence (CDS) or by insertion of premature stop codons at the 5′ end of the CDS (UL135STOP), cannot be readily reconstituted from transfection of infectious genomes unless expression of UL138 is also disrupted (23). Therefore, UL135 functions, in part, by overcoming the suppressive effects of UL138, a gene important for the establishment or maintenance of latency. pUL135 targets epidermal growth factor receptor (EGFR), reducing surface levels of EGFR by promoting its turnover or impeding its recycling (18). This role is in opposition to the role of UL138 in recycling EGFR back to the cell surface to sustain EGFR signaling (18). UL135 is also important for intracellular membrane organization and viral replication in endothelial cells (24). Perhaps related to these functions, pUL135 interacts with the WAVE complex to prevent the formation of actin stress fibers (25) and further protect HCMV-infected cells from NK and T cells. UL135 has also been implicated in degradation of the actin cytoskeleton modulator ROCK1 (26) in infected cells via an unknown mechanism. The molecular underpinnings linking pUL135 and EGFR have not yet been explored.
We sought to determine the mechanisms by which UL135 mediates EGFR trafficking and turnover and facilitates reactivation from latency. We used two complementary screens to identify proteins that interact with pUL135: immunoprecipitation of pUL135 followed by tandem mass spectrometry (IP/MS) and yeast two-hybrid (Y2H) screen. We identified the Src homology 3 (SH3) domain-containing kinase binding protein 1 (SH3KBP1, also known as SETA, RUK, and CIN85; herein referred to as CIN85) as a pUL135 interactor by IP/MS. In addition, we also identified Abelson-interacting protein-1 (Abi-1, alternatively known as e3B1 [27]) as a pUL135 interactor by Y2H screening. Both CIN85 and Abi-1 contain well-defined SH3 domains, for which the consensus ligands have been mapped; pUL135 contains amino acid sequences similar or identical to the consensus ligands. As Abi-1 and CIN85 have partially overlapping functions in regulating the signaling, trafficking, and turnover of EGFR, we investigated the pUL135 interactions with Abi-1 and CIN85 in parallel. Viruses containing pUL135 mutations that disrupted the interactions between pUL135 and Abi-1 and/or CIN85 replicated in fibroblasts but exhibited differences in their abilities to regulate the trafficking of EGFR and reactivation from latency in CD34+ HPCs. The interactions between pUL135 and Abi-1 and CIN85 contribute to our mechanistic understanding of how UL135 functions to promote viral reactivation, linking the turnover of EGFR to reactivation from latency.
RESULTS
pUL135 interacts with Abi-1 and CIN85 host adaptor proteins.
To determine how pUL135 functions in infection to regulate EGFR turnover and stimulate reactivation, we conducted two proteomic screens to identify interacting partners of pUL135: IP/MS and Y2H. IP/MS can identify both direct and indirect interactions with host or viral proteins in the context of infection. We infected cells with a recombinant HCMV strain TB40/E that expresses endogenous levels of UL135 with a C-terminal 3×FLAG epitope tag (TB40/E UL1353×FLAG). Fusion of epitope tags to the 3′ end of the UL135 open reading frame (ORF) does not affect virus replication relative to that of the wild type (WT) (17). Proteins interacting with pUL1353×FLAG were coprecipitated using a monoclonal FLAG antibody and identified by IP/MS, as previously described (18, 28). As a control for nonspecific interactions, the same IP/MS was performed on fibroblasts infected with WT HCMV, which does not contain a 3×FLAG tag. Proteins identified in both the 3×FLAG and control data sets were considered nonspecific and excluded from further analysis. The results of this screen are shown in Table 1. A list of all interacting proteins identified by IP/MS, without exclusion of interactions also identified in the control pulldown, is provided as Table S1 in the supplemental material.
TABLE 1.
Host-pUL135 interacting proteins identified by IP/MS
| UniProt accession no.a | Symbol | Entrez Gene Name | Locationb | Protein type | Peptide countc | No. of scans | % coverage |
|---|---|---|---|---|---|---|---|
| Q64ET4 | UL135 | Cytomegalovirus UL135 | 14 | 145 | 54.57 | ||
| Q9Y5K6 | CD2AP | CD2-associated protein | Cytoplasm | Other | 5 | 13 | 7.98 |
| P21589 | NT5E | 5′-Nucleotidase ecto | Plasma membrane | Phosphatase | 4 | 8 | 9.58 |
| Q96B97 | SH3KBP1 (CIN85) | SH3 domain-containing kinase binding protein 1, (CIN85) | Cytoplasm | Other | 4 | 11 | 7.37 |
| Q14667 | KIAA0100 | KIAA0100 | Extracellular space | Other | 2 | 4 | 1.30 |
| Q9H882 | MYH14 | Myosin heavy chain 14 | Extracellular space | Enzyme | 2 | 5 | 1.20 |
| O95967 | EFEMP2 | EGF-containing fibulin-like extracellular matrix protein 2 | Extracellular space | Other | 2 | 5 | 5.19 |
| Q07065 | CKAP4 | Cytoskeleton-associated protein 4 | Cytoplasm | Other | 2 | 4 | 5.65 |
| O00391 | QSOX1 | Quiescin sulfhydryl oxidase 1 | Cytoplasm | Enzyme | 2 | 3 | 4.55 |
Candidate pUL135 interacting proteins were identified by IP/MS.
Localization and type defined by Ingenuity Pathway Analysis (Qiagen).
Candidate proteins meet the criteria of at least 2 peptides and a peptide probability of >0.95.
The IP/MS approach is limited to studies in fibroblasts because of the requirement for starting material and sufficient levels of the viral protein of interest; therefore, we cannot translate this approach to identify interactions in HPCs, where undetectable levels of pUL135 exist during latency (17). To circumvent these limitations and to complement the IP/MS, we also conducted a Y2H screen for direct pUL135 interactors. We cloned the entire cytosolic domain of pUL135 (residues 44 to 328) as a Y2H bait construct to screen a normalized universal human cDNA library. We identified 50 unique cDNA clones, representing 13 genes (Fig. 1A). Of the bait constructs identified, the majority (76%) contained fragments of Abelson-interacting protein-1 and -2 (Abi-1 and Abi-2, respectively). Abi-1 regulates signaling pathways, including EGFR and EGF-induced Ras, extracellular signal-regulated kinases (ERK), and phosphatidylinositol 3-kinase (PI3K) pathways (29–33). The interaction between pUL135 and Abi-1 was previously detected in a similar Y2H screen (25). By aligning the cDNAs of Abi-1 prey constructs identified in our Y2H screen, we identified a minimal region of Abi-1 (residues 448 to 508) present in all pUL135 Y2H prey clones of Abi-1 (Fig. 1B). This region encodes the SH3 domain of Abi-1 (residues 450 to 508) (29), implying that pUL135 interacts with the SH3 domain of Abi-1.
FIG 1.

Host pUL135-interacting proteins identified by yeast two-hybrid screen. (A) Host proteins identified as pUL135-interacting proteins, including their subcellular localization as determined by Ingenuity software (Qiagen, Inc.), and the number of clones obtained from the Y2H screen. (B) A schematic showing the alignment of Abi-1 prey cDNA constructs (top, black lines) identified by the Y2H screen with the Abi-1 cDNA sequence. Similar cDNA clones differing by ±5 nucleotides at the 5′ end of the prey cDNA are shown as a single black bar. The Abi-1 CDS is shown as a hollow arrow; vertical lines within the arrow indicate exon splice junctions. The SH3 domain of Abi-1 is labeled.
No overlap between the sets of proteins identified by Y2H and IP/MS screens was observed. The lack of overlap in data sets likely reflects the different nature of each of these approaches. In addition to differences inherent to yeast versus mammalian cells, the Y2H screen uses only the soluble portion of pUL135 whereas pUL135 is membrane bound in the IP/MS (17). Nevertheless, several of the proteins identified stood out for their overlapping roles in regulating EGFR. Like Abi-1, CIN85 and CD2AP regulate trafficking and signaling of receptor tyrosine kinases (RTKs) (34–39) by recruiting endocytic components (e.g., Alix [40], Dab2 [41], clathrin adaptor AP-2 [42], endophilins [43, 44], and Cbl ubiquitin ligase [37, 44–49]). CD2AP is highly homologous to CIN85 in regard to structure and function (39, 45–47, 50–52). Because CD2AP and CIN85 bind to the same polyproline consensus sites and have similar interactomes and similar functions, we cannot genetically distinguish the contribution of CIN85 and CD2AP interactions solely by mutating their common SH3 ligands in pUL135. While both CIN85 and CD2AP were identified with similar coverage by IP/MS, CIN85's interactions with EGFR are better characterized. Therefore, our analyses focus on CIN85 and Abi-1 although the effects we attribute to CIN85 may be partially mediated by interaction with CD2AP.
We confirmed the interactions between pUL135 and CIN85 or Abi-1 by immunoprecipitating myc-tagged viral proteins from fibroblasts infected with either TB40/E UL135myc or UL133myc virus, followed by immunoblotting for the host proteins (Fig. 2). Consistent with the Y2H and IP/MS screens, both CIN85 and Abi-1 coprecipitated with pUL135myc. In contrast, no interaction with endogenous Abi-1 or CIN85 was detected in cells infected with a control virus, TB40/E UL133myc, indicating that the interactions are specific to pUL135. We have previously shown that the multiple bands representing pUL135 are due, in part, to initiation of translation from distinct start codons (23). Also, while the quantity of Abi-1 appears to be increased by infection in Fig. 2, this observation was not consistent over multiple experiments.
FIG 2.
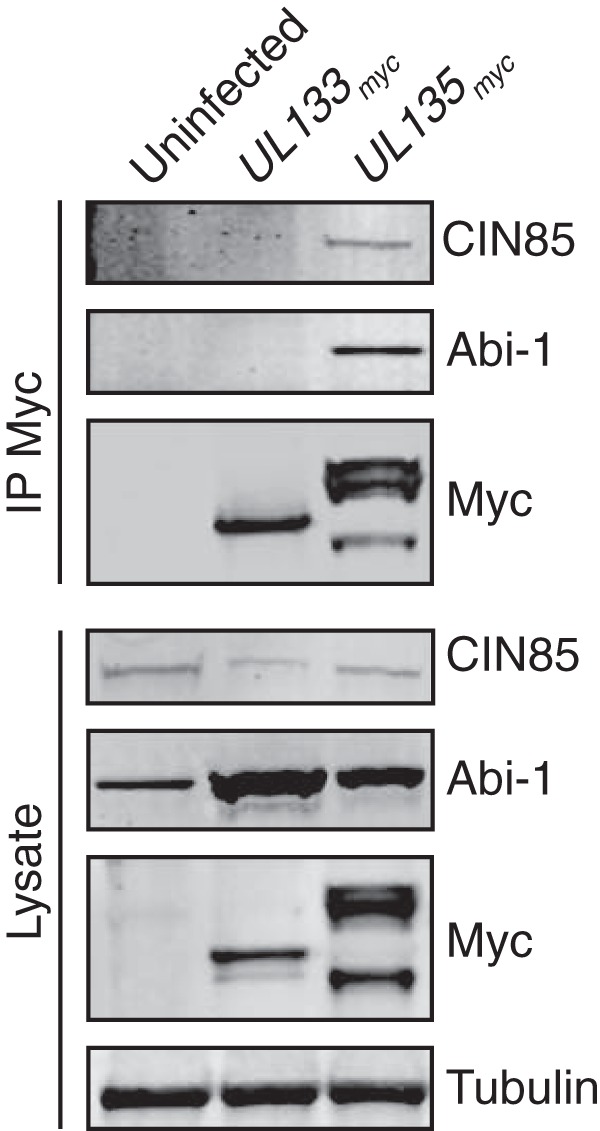
Host proteins Abi-1 and CIN85 associate with pUL135 in infection. MRC-5 fibroblasts were infected with recombinant HCMV TB40/E encoding either myc epitope-tagged UL133 (as a negative control; UL133myc) or UL135 (UL135myc) at an MOI of 2. Myc-tagged proteins were immunoprecipitated from lysates harvested at 48 hpi with Ms anti-Myc antibody and then separated by SDS-PAGE. Abi-1, CIN85, and myc-tagged proteins were detected by Western blotting using monoclonal antibodies (see Table 3). Blots of 40 μg of total cellular lysate are shown with tubulin as a loading control.
pUL135 interacts with host adaptor proteins via discrete polyproline sites.
Many of the protein-protein interactions of Abi-1 and CIN85 are mediated by SH3-polyproline interaction motifs (29, 32, 45). pUL135 is 18% proline and is rich with potential polyproline SH3 ligands. The Y2H screen identified the SH3 domain of Abi-1 as the region that interacts with pUL135 (Fig. 1B). As CIN85 contains three SH3 domains, we hypothesized that the pUL135-CIN85 interaction might also be mediated by SH3-polyproline interactions. We identified consensus SH3 polyproline ligands within pUL135 for CIN85/CD2AP and Abi-1 using MinimotifMiner (Fig. 3A and B) (53). To determine if pUL135 interacts with CIN85 and Abi-1 through SH3-polyproline motif interactions, we used site-directed mutagenesis to substitute alanine residues for key proline residues of potential SH3 ligands in a vector expressing UL135 with a C-terminal myc epitope tag (pCIG-UL135myc) (Fig. 3B).
FIG 3.

Host proteins Abi-1 and CIN85 interact with pUL135 via SH3-type interactions. (A) A schematic showing the SH3 ligands within pUL135. TMD indicates the transmembrane domain on pUL135. Colored blocks correspond to the colors in panel B. M1, M21, and M97 indicate methionine codons where translation of UL135 initiates. (B) SH3 ligand classes, the consensus ligands, and their location within pUL135 are indicated. The WT amino acid sequence and the mutations introduced for each are shown. (C) HEK 293-T cells were transfected with constructs expressing an HA epitope-tagged version of Abi-1 and a UL135 variant or empty vector control. HAAbi-1 was immunoprecipitated from 400 μg of lysate using Ms anti-HA monoclonal antibody, and proteins were separated by SDS-PAGE. Immunoprecipitated proteins were detected by Western blotting using Rb anti-myc and Rb anti-HA monoclonal antibodies. A Western blot of 40 μg of total lysate is shown with tubulin as a loading control. (D) HEK 293-T cells were transfected with constructs expressing an HA epitope-tagged version of CIN85 and a UL135 variant or empty vector control. pUL135myc was immunoprecipitated from 200 μg of lysate using Ms anti-myc antibody, and proteins were separated by SDS-PAGE. Immunoprecipitated proteins were detected by Western blotting using Rb anti-HA or Rb anti-Myc monoclonal antibodies. A Western blot of 40 μg of total lysate is shown with tubulin as a loading control.
The consensus SH3 ligands of Abi-1 and Abi-2 are not of the classical PXXP form but are of the atypical form of PPPXXXP (54). pUL135 contains this atypical ligand sequence, as PPPWSKP (anchor residues are underlined). We mutated key residues to PPASWKA, to create pCIG-UL135mycΔAtypSH3. Additionally, we created expression vectors containing alanine substitutions within an SH3 class I ligand to create pCIG-UL135mycΔSH3cI and an SH3 class II ligand to create pCIG-UL135mycΔSH3cII. Finally, we constructed expression vector pCIG-UL135mycΔSH3x3, which contains mutations to all three of these putative SH3 ligands. The locations of putative Abi-1 interaction sites are shown in Fig. 3A, and the mutagenesis of these motifs is detailed in Fig. 3B.
To determine if the predicted SH3 ligands within pUL135 were the basis of interaction with Abi-1, we transiently transfected 293 cells with plasmids encoding Abi-1 with an N-terminal hemagglutinin (HA) tag (HAAbi-1) and either wild-type UL135myc or UL135myc mutants. We immunoprecipitated HAAbi-1 and immunoblotted the precipitates for pUL135myc. Targeted mutation of the SH3cI ligand disrupted the interaction between HAAbi-1 and pUL135 (Fig. 3C, lanes 4 and 6). Notably, this is the reciprocal immunoprecipitation of the pulldown used to identify host interactions (Fig. 2). Surprisingly, the interaction between Abi-1 and pUL135 did not require the canonical Abi-1 SH3 ligand (atypical SH3, PPPXXXP) as alanine substitutions of key interaction residues of this motif did not abolish the Abi-1–pUL135 interaction (Fig. 3C, lane 3). Finally, the pUL135–Abi-1 interaction did not require the SH3cII site (Fig. 3C, lane 5). A pUL135 mutant containing alanine substitutions in all three putative SH3 ligands (ΔSH3cI, ΔSH3cII, and ΔAtypSH3) also abrogated the interaction with Abi-1. These studies identify the SH3cI ligand of pUL135 (KRPPTPP; essential anchor residues are underlined) as necessary for the interaction with Abi-1.
The consensus ligands of the three SH3 domains within CIN85 have been previously mapped as XPX(P/A)XPRX, XPX(A/P)XPRX, and XPX(P/A)X(P/V)RXX (essential anchor residues are underlined) for the first, second, and third SH3 domains of CIN85, respectively; the consensus ligand of all three SH3 domains can be generalized as PXXXPR (55, 56). pUL135 contains five such motifs, predicted to bind to the SH3 domains of CIN85 (Fig. 3A and B). Because the binding interactions of CIN85's three SH3 domains are often both simultaneous and cooperative (45, 56), we mutated all five putative CIN85 SH3 ligands in UL135myc from PXXXPR to PXXXAA to create expression vector pCIG-UL135mycΔCIN85 (Fig. 3B).
To determine if mutations of the putative CIN85 ligands of pUL135 was sufficient to disrupt the pUL135-CIN85 interaction, we cotransfected 293 cells with vectors pCIG-CIN85HA (CIN85 with a C-terminal HA tag) and either pCIG-UL135myc or pCIG-UL135mycΔCIN85. We immunoprecipitated the myc-tagged proteins from transfected cells and subjected them to Western blotting for CIN85HA. CIN85 coprecipitated with pUL135myc but did not coprecipitate with pUL135mycΔCIN85, indicating that the protein interaction is occurring via one or more of the SH3 domains and SH3 ligands (Fig. 3D).
UL135 interactions with CIN85 and Abi-1 augment viral replication in fibroblasts.
Having identified the residues required for the interactions between pUL135myc, CIN85, and Abi-1, we introduced these mutations into UL135myc in the bacterial artificial chromosome (BAC) clone of TB40/E (17, 57) to create recombinant viruses where pUL135 host interactions had been abrogated. A simian virus 40 (SV40)-green fluorescent protein (GFP) cassette engineered into the TB40/E genome between US34 and TRS1 (17) provides GFP expression as a marker of infection. Mutations of the UL135 SH3cI ligand and the CIN85 interaction ligands were incorporated into the viral genome either individually (creating ΔSH3cI and ΔCIN85 strains, respectively) or in combination (ΔSH3cI/CIN85). These recombinant viruses represent the most robust approach to defining the significance of pUL135-host protein interactions in the context of infection as discrete mutations disrupt specific interactions while preserving the expression of the host protein, thereby avoiding the risk of pleiotropic effects of protein knockdown. Disruption of the entire UL135 ORF results in a virus that is severely defective in reconstituting infectious virus from HCMV BAC transfection in fibroblasts but, once reconstituted, replicates with little to no defect from infection with a virus stock (23). In comparison, transfection of BACs harboring discrete mutations of the SH3cI or CIN85 interaction motif resulted in no defect in reconstitution.
To confirm that the mutated forms of pUL135 did not interact with Abi-1 and/or CIN85 in the context of infection, we immunoprecipitated myc-tagged proteins from cells infected with TB40/E UL133myc (as a nonspecific control), UL135myc, ΔSH3cI, ΔCIN85, and ΔSH3cI/CIN85 viruses. We then performed Western blotting for cellular proteins Abi-1 and CIN85 (Fig. 4A). Mutagenesis of the SH3cI and CIN85 SH3 ligands of pUL135 was sufficient to disrupt the interactions between Abi-1 and CIN85, respectively. Although our immunoprecipitations were not intended to be quantitative, the interaction between Abi-1 and pUL135ΔCIN85 appeared 2- to 3-fold weaker than that with UL135myc over multiple experiments. Therefore, we cannot rule out the possibility that interaction with CIN85 is required for maximal interaction with Abi-1, at least in the context of infection. The double mutant protein, ΔSH3cI/CIN85, interacted with neither Abi-1 nor CIN85 in infected cells.
FIG 4.
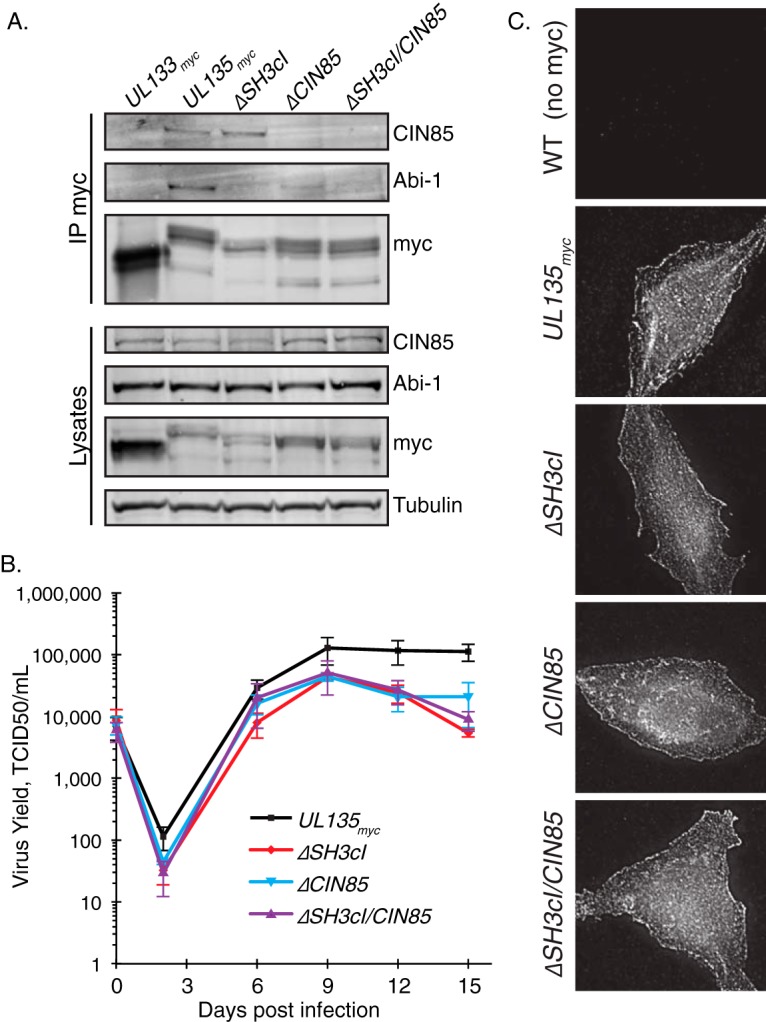
Disruption of UL135-host interactions in recombinant viruses results in a mild replication defect. (A) Recombinant viruses containing disruptions in the SH3cI or CIN85 ligands were generated by BAC recombineering. Fibroblasts were infected at an MOI of 2 with TB40/E CMV encoding UL133myc (a control protein), UL135myc, ΔSH3cI, ΔCIN85, or ΔSH3cI/CIN85. pUL135myc was immunoprecipitated using Ms anti-myc monoclonal antibody from 1 mg of lysates. Interaction with CIN85 or Abi-1 was analyzed by coprecipitation of these proteins and Western blotting. A Western blot of 40 μg of total lysate is shown with tubulin as a loading control. (B) The replication kinetics of recombinant viruses encoding UL135myc, ΔSH3cI, ΔCIN85, and ΔSH3cI/CIN85 were determined by infecting MRC-5 fibroblasts at an MOI of 0.02; virus yields in cell lysates were measured over a time course by 50% tissue culture infective dose (TCID50). Data points represent the average from at least four experiments, and error bars represent the standard errors of the means. (C) Intracellular distribution of pUL135 is not altered by mutagenesis of the SH3 ligands within UL135. MRC-5 fibroblasts were infected at an MOI of 1 with the indicated viruses, fixed at 48 hpi, and immunostained for the myc epitope tag on pUL135myc. Cells infected with a WT virus that lacks the myc tag are shown as a staining control.
We determined the contribution of the interactions between pUL135, Abi-1, and CIN85 to viral fitness by a multistep viral replication assay (Fig. 4B). Compared with the UL135myc parental virus, recombinant ΔSH3cI, ΔCIN85, and ΔSH3cI/CIN85 viruses replicated with kinetics similar to those of WT virus in fibroblasts but with an ∼4.5-fold reduction in final titers. The differences in levels of replication between 0 and 12 days were not statistically significant, leading us to conclude that interactions of pUL135 with CIN85 and/or Abi-1 augment viral replication but are not necessary for viral replication in fibroblasts.
As a control to ensure that mutation of the pUL135 SH3 ligands did not cause mislocalization, we analyzed the localization of myc-tagged, virally expressed pUL135myc, pUL135ΔSH3cI, pUL135ΔCIN85, and pUL135ΔSH3cI/CIN85 in infected fibroblasts. Fibroblasts were infected at a multiplicity of infection (MOI) of 1, fixed at 48 h postinfection (hpi), and stained with antibodies to visualize the virally expressed myc-tagged UL135 proteins (Fig. 4C). As a control we used WT TB40/E, which contains no myc tag. Both WT and alanine-substituted mutants of pUL135 show similar cytoplasmic and cell membrane distributions, suggesting that the mutations did not grossly mislocalize pUL135. Further, all pUL135 mutants were expressed to similar levels, in both immunofluorescence and Western blotting experiments, suggesting that the alanine-substituted variants of pUL135 were stable relative to the parental pUL135myc.
pUL135 interactions with cellular proteins modulate total and surface EGFR levels in fibroblasts.
HCMV infection has previously been shown to decrease surface and total EGFR levels (18, 58, 59). We previously demonstrated a role for UL135 in decreasing the steady-state level of cell surface and total EGFR (18). As both Abi-1 (29, 30) and CIN85 (34–39, 43, 47) have roles in modulating EGFR endocytosis and trafficking, we hypothesized that the interactions between pUL135 and adaptor proteins might allow the virus to control EGFR trafficking and turnover in infected cells.
Steady-state EGFR levels were assayed in fibroblasts infected with UL135myc, ΔSH3cI, ΔCIN85, ΔSH3cI/CIN85, or UL135STOP virus by immunoblotting. Consistent with prior work (18), total EGFR levels were significantly lower in CMV-infected cells (Fig. 5A and C), and the total EGFR levels of cells infected with UL135STOP virus were 2-fold greater than those in cells infected with the UL135myc parental virus (Fig. 5B and D). Infection with ΔSH3cI or ΔCIN85 virus resulted in an intermediate phenotype relative to the phenotypes of infection with UL135myc and UL135STOP viruses, but these differences were not statistically significant. However, infection with the ΔSH3cI/CIN85 mutant virus where both interactions were disrupted increased total levels of EGFR, similarly to infection with UL135STOP virus, suggesting that interactions with both Abi-1 and CIN85 are required for pUL135-mediated turnover of EGFR.
FIG 5.

pUL135 interactions with CIN85 and Abi-1 alter the total and surface levels of EGFR protein in fibroblasts. (A to D) Fibroblasts were infected with UL135myc, ΔSH3cI, ΔCIN85, ΔSH3cI/CIN85, or UL135STOP virus at an MOI of 2. Twenty-four hours after infection, the inoculum was removed, and the medium was replenished. Forty-eight hours after infection, cells were lysed, and 40 μg of protein was analyzed by Western blotting to quantitate total EGFR, normalized to the level of α-tubulin. Representative Western blots from a single experiment show the total levels of EGFR and α-tubulin (A and B). Graphs show the total amount of EGFR, normalized to the level of α-tubulin, for at least five independent experiments for each sample, each utilizing an independent virus preparation. Error bars indicate standard errors of the means. Values for total EGFR in uninfected fibroblasts relative to that in infected fibroblasts (C) and for total EGFR in UL135 mutant viruses relative to that in the UL135myc parental virus (D) are shown. Statistical significance was calculated by a one-way analysis of variance and Tukey's test. **, P < 0.01; *, P < 0.05. (E) Surface levels of EGFR on MRC-5 fibroblasts infected at an MOI of 1 with UL135myc, ΔSH3cI, ΔCIN85, ΔSH3cI/CIN85, or UL135STOP virus were analyzed at 48 hpi by flow cytometry using a fluorescently conjugated Ms anti-EGFR antibody. Student's t test was used to compare the surface levels of each cell type to that with UL135myc virus. **, P < 0.01. Error bars indicate standard errors of the means.
To determine if the pUL135 host interactions impact surface levels of EGFR, we measured the steady-state surface levels of EGFR on cells infected with WT HCMV or our UL135 mutant viruses using flow cytometry. In line with our previous observations, infection with UL135STOP virus had nearly 2-fold more surface EGFR than WT-infected cells (Fig. 5E). Single mutation viruses, ΔCIN85 or ΔSH3cI, had EGFR surface levels indistinguishable from those of WT infection, indicating that neither single mutation was sufficient to phenocopy the total loss of pUL135. However, ΔSH3cI/CIN85 infection restored EGFR surface levels similar to the level with UL135STOP infection and ∼1.7-fold greater than that with WT-infected cells. Together these results indicate, similar to total concentration, that pUL135's interaction with both CIN85 and Abi-1 accounts for decreased EGFR surface levels caused by pUL135. These results suggest a cooperative role of pUL135 interactions with Abi-1 and CIN85 in modulating EGFR levels in infection.
pUL135 interactions with Abi-1 and CIN85 affect intracellular EGFR trafficking.
We next sought to determine how pUL135 interactions with Abi-1 and CIN85 impacted the intracellular trafficking of EGFR. Following endocytosis, EGFR will traffic with Rab5+ early endosomes, from which it can be recycled to the cell surface or traffic toward degradation as Rab5+ endosomes mature into Rab7+ endosomes destined for fusion with the lysosome. We monitored the association and dissociation of fluorescently conjugated EGF (using Alexa Fluor 647 [EGF647]) with Rab5+ following endocytosis. Fibroblasts were labeled at 48 hpi with EGF647 on ice and then returned to 37°C. Over time postinternalization, cells were fixed and stained with a Rab5 antibody, and the association of the EGF ligand with Rab5+ endosomes was imaged by deconvolution microscopy and analyzed as previously described (18, 60). Representative images demonstrating the association of EGF647 with Rab5+ at 15 and 90 min postendocytosis are shown in Fig. 6A. The frequency of colocalization between EGF and Rab5 is calculated as the area of coincident EGF+ and Rab5+ signal divided by the total area of EGF+ signal and was quantified over multiple experiments; results are shown in Fig. 6B. In uninfected cells the majority of EGF647 transiently associated with the Rab5+ early endosome at 15 min postendocytosis, and only a small fraction remained after 90 min (retaining only 19% of the colocalization observed at 15 min) (Fig. 6B and C). In cells infected with UL135myc virus, EGF647 exhibited moderately increased retention with Rab5+ endosomes at 90 min (31% retention). However, in UL135STOP virus-infected cells, a significant portion of the EGF647 remains within the Rab5+ compartment after 90 min (69% retention). This suggests that pUL135 promotes EGFR egress from the early endosome. Cells infected with ΔSH3cI virus showed increased retention of EGF647 that was statistically insignificant from UL135myc virus infection (48% retention). In contrast, cells infected with ΔCIN85 virus showed a significant and sustained defect in clearing EGF647 from the early endosome (64% retention), suggesting that the interaction between pUL135 and CIN85 plays role in regulating EGFR trafficking from the early endosome to the lysosome. Infection with ΔSH3cI/CIN85 virus resulted in retention of EGF647 in Rab5+ endosomes (73% retention) similar to that of infection with UL135STOP virus and the ΔCIN85 mutant virus. These results imply that pUL135 interactions with CIN85 facilitate efflux of EGF/EGFR from the Rab5+ early endosome in infected cells. Because the loss of interaction with Abi-1 does not compound the defect in egress associated with loss of the CIN85 interaction, we cannot ascribe a role for Abi-1 for EGF/EGFR trafficking in the context of HCMV infection.
FIG 6.

Interactions between pUL135 and CIN85 facilitate efflux of EGF/EGFR from Rab5+ endosomes. MRC-5 fibroblasts were infected at an MOI of 1 with UL135myc, ΔSH3cI, ΔCIN85, ΔSH3cI/CIN85, or UL135STOP virus. Cells were transferred to serum-free medium at 24 hpi. At 48 hpi, cells were labeled on ice with EGF647, washed, and transferred to 37°C with 10% serum medium and then fixed at the indicated time points. Rab5 was stained with a rabbit monoclonal antibody, and cells were imaged as z-stacks by deconvolution microscopy. Images were analyzed using the Squassh plug-in for ImageJ, and the colocalization between EGF signal and the Rab5 early endosomal marker was calculated. (A) Representative optical z-slices from an uninfected cell, showing the colocalization of EGF647 within Rab5+ endosomes at 15 min (top) and 90 min (bottom) after stimulation. (B) The efflux of EGF647 from Rab5+ endosomes was calculated by dividing the mean colocalization value for a time point by the 15-min colocalization value. Error bars indicate standard errors of the means over three independent experiments. Significance at the 90-min time point was determined by two-way analysis of variance and Tukey's test. *, P < 0.05. (C) Retention of EGF647 in the early endosome is calculated as the colocalization at 90 min divided by the colocalization at 15 min.
UL135 host interactions modulate surface-level EGFR and latency in CD34+ HPCs.
We have previously demonstrated a requirement for pUL135 in reactivation from latency in primary CD34+ HPCs (18, 23). Given the rarity of these cells (≤1% of the mononuclear faction of cells in bone marrow), primary CD34+ HPCs are not amenable to many of the molecular approaches we have employed in fibroblasts to define the function of UL135 and its interactions. However, we have found that the roles of UL135, UL136, and UL138 in promoting or suppressing replication for latency in CD34+ HPCs are reflected as replication advantages or defects in fibroblasts (17, 19, 23). Therefore, it follows that the function of these proteins is at least partially conserved across cell types, but the impact is greater in CD34+ HPCs where viral gene expression and replication are restricted. Therefore, we use fibroblasts to define basic molecular functions and then confirm that key phenotypes are recapitulated in CD34+ HPCs. Therefore, we next sought to determine the role of pUL135-host interactions in the regulation of EGFR in the context of latency in CD34+ HPCs.
Having previously determined that pUL135 impacts cell surface EGFR levels at 1 day postinfection (dpi) in CD34+ HPCs, but not at 4 or 8 dpi (18), we investigated the effects of UL135 mutant viruses on EGFR surface levels at 1 dpi. We infected CD34+ HPCs at an MOI of 1 for 24 h with UL135myc, UL135STOP, ΔSH3cI, ΔCIN85, or ΔSH3cI/CIN85 virus. Surface EGFR levels were measured by EGF647 binding, as assayed by flow cytometry. In contrast to the reduction of surface EGFR levels observed in fibroblasts upon HCMV infection and consistent with previous results (18), infection of CD34+ HPCs resulted in increased surface EGFR relative to levels on uninfected cells (Fig. 7A). EGFR surface levels were further increased by infection with UL135STOP virus (18). Infection of CD34+ cells with ΔSH3CI or ΔCIN85 virus resulted in surface EGFR levels indistinguishable from the level with the UL135myc parental virus. In contrast, infection with ΔSH3cI/CIN85 virus increased CD34+ surface EGFR to levels statistically indistinguishable from levels with UL135STOP virus; ΔSH3cI/CIN85-infected cells have significantly more surface EGFR than infection with the UL135myc parental virus. From this, we conclude that pUL135 interactions with Abi-1 and CIN85 are both necessary for the UL135-mediated modulation of EGFR levels at the surface of CD34+ HPCs.
FIG 7.

pUL135 interaction with CIN85 and Abi-1 regulates EGFR surface levels and reactivation in CD34+ HPCs. Primary CD34+ cells were infected with UL135myc, ΔSH3cI, ΔCIN85, ΔSH3cI/CIN85, or UL135STOP virus at an MOI of 2. (A) Twenty-four hours after infection, cell surface proteins were stained with EGF647 and BV421 anti-CD34 antibody prior to analysis by flow cytometry. EGF staining intensity was evaluated on CD34+ GFP+ (infected) cells. Data represent three experiments performed using independent CD34+ donors; error bars represent standard errors of the means. Significance levels were calculated by one-way analysis of variance with Tukey's test. *, P < 0.05, for each comparison shown by a bar. The differences in surface EGFR levels between uninfected cells and all infections were statistically significant, and comparison bars are not shown. (B) At 24 hpi, infected (GFP+) CD34+ cells were isolated by FACS and seeded into long-term bone marrow culture in transwells over irradiated stromal cell support for 10 days to prevent differentiation. After 10 days in culture, cells or an equivalent cell lysate (prereactivation control) was seeded by limiting dilution into a coculture with fibroblasts on 96-well dishes in cytokine-rich medium to promote myeloid differentiation and reactivation. Fourteen days after plating, GFP+ wells were scored, and the frequency of infectious centers was calculated using ELDA software. Error bars represent the range of two independent experiments using independent CD34+ cell donors.
UL135 is required for reactivation from latency; UL135STOP virus maintains viral genomes during long-term bone marrow culture in the absence of viral replication but fails to reactivate (23). We have shown that inhibition of EGFR or downstream PI3K stimulates reactivation and rescues the reactivation defect of UL135STOP virus infection (18). To determine if the interactions of pUL135 with Abi-1 and/or CIN85 contribute to reactivation, we assessed ability of ΔSH3cI, ΔCIN85, and ΔSH3cI/CIN85 viruses to establish and reactivate from latency relative to the level with the UL135myc virus. Primary CD34+ cells were infected for 24 h, and then CD34+/GFP+ cells were isolated by fluorescence-activated cell sorting (FACS). Pure CD34+/GFP+ cells were cocultured for 10 days in long-term bone marrow cultures using a bone marrow stromal cell support that maintains hematopoietic cell progenitor phenotype and function (61). This period in long-term bone marrow culture allows for the establishment of HCMV latency. At 10 dpi, half of the cells were cocultured with fibroblasts by limiting dilution in a cytokine-rich medium to promote myeloid cell differentiation and reactivation. The other half of the culture was lysed and added to parallel cultures of fibroblasts in limiting dilution to quantify any virus present at the time of reactivation (62). The ΔSH3cI, ΔCIN85, and ΔSH3cI/CIN85 mutant viruses all exhibited a defect in reactivation similar to the that of the UL135STOP virus (Fig. 7B). Thus, both interactions between pUL135 with CIN85 and Abi-1 are necessary for HCMV reactivation from latency.
DISCUSSION
Complex and poorly understood virus-host interactions regulate the establishment and maintenance of infection and reactivation from latency. We have previously demonstrated a requirement for the HCMV ULb′-encoded UL135 in reactivation from latency (23). In the absence of UL135, latent viral genomes are maintained but cannot be reactivated to produce progeny virus. UL135 opposes the action of UL138, another ULb′ gene whose expression is coordinated with UL135. Both pUL135 and pUL138 interact with EGFR but with opposing effects: pUL138 promotes increased surface levels of EGFR, whereas pUL135 decreases total and cell surface EGFR levels (18). Attenuation of EGFR signaling or of the downstream PI3K enhances reactivation of latent viral infections (18). Thus, the balance between pUL135 and pUL138 in regulating EGFR and its downstream signaling forms a functional switch controlling latency and reactivation. The molecular underpinnings by which pUL135 and pUL138 impact EGFR levels and signaling and, in turn, viral replication and latency are largely unknown. We identified the adaptor proteins CIN85 and Abi-1 as host proteins interacting with pUL135 using two complementary screens to identify interacting proteins. CIN85 and Abi-1 each have roles in modulating the trafficking and turnover of EGFR. We demonstrate a cooperative role for these pUL135 interactions in modulating EGFR trafficking and turnover in the context of HCMV infection and in reactivation from latency. These findings mechanistically link pUL135-mediated turnover of EGFR with its role in reactivation from latency.
CIN85 is a ubiquitously expressed, multifunctional adaptor protein with roles in endocytosis, trafficking of endocytic cargo, and modulation of signaling (reviewed in references 38 and 47). CIN85 contains three N-terminal SH3 domains, a proline-rich region, and a C-terminal coiled-coil domain. Active RTKs such as EGFR phosphorylate Cbl, exposing Cbl's CIN85 ligands and thereby recruiting CIN85 to the active RTK (43). While CIN85 has only an augmenting role in EGFR endocytosis (63), phosphorylated CIN85 in complex with Cbl licenses endocytic cargo trafficking from early (Rab5+) to late (Rab7+) endosomes (35, 64). Phosphorylated CIN85 also recruits dynamin to the EGFR-Cbl-CIN85 complex, which is necessary for trafficking of EGFR in Rab7+ late endosomes toward lysosomal degradation (65). Finally, the complex of EGFR-Cbl-CIN85 promotes the sorting of EGFR from the external membrane of a multivesicular body (MVB) into the intraluminal vesicles, a process necessary to attenuate EGFR signaling and promote EGFR degradation (35). In addition to the regulation of RTK trafficking throughout the endocytic pathway, CIN85 directly interacts with the PI3K regulatory subunit p85α to partially inhibit its catalytic activity (66). CIN85 also interacts with components of the mitogen-activated protein kinase (MAPK) signaling cascade, biasing signaling toward the ERK pathway (67).
Like CIN85, Abi-1 is also a ubiquitously expressed adaptor protein (27) with roles in Cbl E3 ubiquitin ligase-mediated endocytosis, signal transduction, and regulation of the actin cytoskeleton (reviewed in reference 32). Structurally, Abi-1 contains an N-terminal homeodomain homology region, several polyproline motifs, and a single C-terminal SH3 domain, which recruits Cbl to the plasma membrane for EGFR endocytosis (29). Additionally, Abi-1 also interacts with dynamin and synaptojanin at the plasma membrane to assist in endocytosis of RTKs, including EGFR (68). EGFR endocytosis and actin-based vesicular transport are regulated by Abi-1 in complex with N-WASP, while macropinocytosis-type endocytosis of EGFR requires an interaction between Abi-1 and the WAVE complex (29, 69). Abi-1 can enter a variety of protein complexes, including Sos/Eps8 or Sos/Grb2, which differentially regulates the activation of downstream signaling pathways (30, 31, 33). Mechanistically, pUL135 interactions with multiple adapter proteins involved in the regulation of EGFR signaling presents an intriguing means by which HCMV infection controls EGFR signaling, trafficking, and degradation during infection.
Our study demonstrates a requirement for interaction between pUL135 and both Abi-1 and CIN85 in regulating the trafficking and turnover of EGFR. Mutant viruses containing individual disruptions to SH3 ligands in pUL135 required for interaction with either Abi-1 or CIN85 did not significantly affect total or surface EGFR levels in fibroblasts. However, the combined disruption of both CIN85 and Abi-1 interactions with pUL135 significantly increased levels of total EGFR in fibroblasts (Fig. 5B). The increased total levels of EGFR in fibroblasts infected with ΔSH3cI/CIN85 correlated with enhanced cell surface levels of EGFR (Fig. 5C). This suggests that the interaction of pUL135 with both Abi-1 and CIN85 cooperatively modulates EGFR trafficking or turnover in infection. Despite the increased levels of EGFR at the cell surface, the efflux of EGFR from Rab5+ endosomes was slowed when UL135 or pUL135-host interactions were disrupted (Fig. 6). In this case, the loss of the pUL135-CIN85 interaction was sufficient to induce the retention of EGFR in Rab5+ endosomes to a level equivalent to that of the full UL135 disruption (UL135STOP) or disruption of both CIN85 and Abi-1 interactions (ΔSH3cI/CIN85). From these results, we infer a model whereby pUL135 recruits Abi-1 and CIN85 to facilitate distinct steps in the trafficking of EGFR from the early endosome toward the lysosome for degradation. When these interactions are lost, EGFR accumulates in Rab5+ early endosomes and is available for recycling back to the cell surface, which results in enhanced surface levels of EGFR in ΔSH3cI/CIN85 infection.
The interactions between pUL135 and CIN85 and Abi-1, and therefore their modulation of EGFR, played an important role in reactivation of HCMV from latency. In contrast to infection in fibroblasts, EGFR surface levels are increased by WT HCMV infection in CD34+ HPCs relative to levels in uninfected cells (18). Disruption of the interactions of pUL135 with both Abi-1 and CIN85 further enhanced EGFR levels on the surface of CD34+ HPCs to the level of UL135STOP virus infection (Fig. 7A). Enhanced surface levels required disruption of both Abi-1 and CIN85 and mimicked the phenotypes observed in fibroblasts (Fig. 5C). These results suggest that the interactions between pUL135 and Abi-1 and CIN85 function similarly in CD34+ HPCs but with greater significance for infection. Disruption of these host interactions, individually or in combination, abrogated the ability of HCMV to reactivate from latency in CD34+ HPCs (Fig. 7). Therefore, these studies mechanistically link the modulation of EGFR trafficking to reactivation.
Stanton et al. previously demonstrated that the interaction between pUL135 and Abi-1 recruited the WAVE complex, including Wave2, CYFIP1/Sra1, and Nap1, to facilitate the depolymerization of actin filaments and actin stress fibers (25). This prevented the formation of a functional immune synapse and protected infected cells from both NK and CD8+ T cell recognition and cytolysis. The roles of pUL135 in mediating the actin cytoskeleton and trafficking and turnover of EGFR are undoubtedly linked. Stanton et al. also detected an interaction between the C terminus of pUL135 and talin, which was shown to play a role in diminishing cell adhesion (25). While we also identified talin as a candidate interaction by IP/MS experiment, it was excluded from further analysis because it was also detected in our negative control (FLAG pulldown from infected cells where pUL135 lacked the FLAG epitope tag).
Chimpanzee CMV (ChCMV) UL135 is the only known ortholog of HCMV UL135 (17, 70). The amino acid sequences of HCMV and ChCMV pUL135 proteins are 35% identical and 46% similar. Interestingly, ChCMV also contains putative SH3cI and CIN85 ligands at approximately the same locations as in HCMV pUL135 (Fig. 8), suggesting that ChCMV pUL135 may functional similarly to its human homologue. While rhesus CMV (RhCMV) has a positionally conserved region corresponding to the HCMV and ChCMV ULb′ region, these RhCMV genes share little identity with HCMV ULb′ genes. There is no ULb′-like region or genes in lower orders, such as murine or rat CMV. This suggests that pUL135 and its function in downregulating EGFR are conserved adaptations to high-order primates.
FIG 8.

SH3cI and CIN85 ligands of pUL135 are conserved between human and chimpanzee CMV strains. A schematic of HCMV pUL135 and ChCMV pUL135 is shown to scale. M1, M21, and M97 indicate the methionine codons where translation of UL135 initiates for HCMV pUL135. SH3cI ligands are shown in light gray; CIN85 ligands are shown in dark gray. Amino acid sequences are shown for ChCMV, and underlined residues indicate anchor residues essential for binding to Abi-1 or CIN85. TMD, transmembrane domains.
The coevolution of viruses, especially herpesviruses, with their hosts has allowed deep integration of viral mechanisms into host pathways, including the EGFR-PI3K pathways. EGFR and it is downstream signaling effects are key regulators of cellular homeostasis, growth, adhesion, and survival (reviewed in references 71 and 72). Many viruses stimulate EGFR and PI3K/Akt signaling pathways during lytic replication to induce antiapoptotic responses, stimulate transcription factors required for viral replication, and stimulate cell transformation for continued viral survival (reviewed in reference 73). Like most herpesviruses, HCMV glycoprotein binding and viral entry potently stimulate EGFR/PI3K (reviewed in reference 74), activating downstream signaling pathways to change cellular transcription, morphology, motility, and survival (75–78). After HCMV entry and viral gene expression, HCMV uses multiple methods to negatively regulate EGFR, including transcriptional repression of EGFR synthesis via induction of the transcription factor WT1 during replication in fibroblasts (59). Interestingly, despite the virus-induced reduction in EGFR total and cell surface levels, HCMV infection retains active EGFR in the viral assembly compartment (VAC) independently of EGF ligand, isolating the cell from its environment or the need to receive exogenous EGF ligand stimulation (18). This work mechanistically defines the means by which HCMV regulates EGFR trafficking and turnover as another aspect of HCMV's ability to manipulate and isolate the host cell, creating or maintaining conditions suitable for viral reactivation and replication. The ability of HCMV to isolate the cell from perturbations to homeostasis has important implications for host signaling, stress responses, and metabolism (reviewed in references 79 and 80).
As a general theme, herpesvirus latency requires PI3K/Akt signaling to maintain latency and downregulates it to promote reactivation. Herpes simplex virus 1 (HSV-1) latency requires sustained PI3K-Akt signaling provided by nerve growth factor to maintain latency; interruption of nerve growth factor signaling or disruption of sustained PI3K-Akt signaling leads to an epigenetic derepression of lytic gene promoters and increases the frequency of reactivation (81, 82). Similarly, Epstein-Barr virus (EBV) latent membrane protein 1 (LMP-1) increases EGFR expression (83), while both LMP-1 and LMP-2a stimulate sustained PI3K signaling (84). Kaposi's sarcoma-associated herpesvirus (KSHV) protein K1 is expressed during lytic and latent infections and activates PI3K signaling (85, 86). KSHV with a K1 deletion reactivates poorly from latency (86), and inhibition of the PI3K pathway stimulates KSHV reactivation (87). Consistent with these studies, inhibition of EGFR or PI3K stimulates reactivation of latent HCMV (18, 88). Further, the induction of EGFR expression in CD34+ HPCs observed after HCMV infection is an important aspect of creating conditions supportive of latency (88), and HCMV latency requires sustained PI3K signaling (18).
EGFR adaptor proteins are both the stoichiometric and functional limiting factors in the EGFR/PI3K signaling cascade due to their low expression levels (89). Due to their low levels, targeting EGFR signal transduction adaptor proteins, including Abi-1 and CIN85, represents an efficient means to control EGFR signaling and exert maximal influence on the cellular environment. The multifunctional HSV-1 immediate early protein ICP0 antagonizes EGFR and PI3K-Akt signaling through an interaction with CIN85 (90). Further, it was recently shown that interaction between CIN85 and ICP0 recruits Cbl to downregulate Nectin-1 at the surface of infected cells and prevents superinfection (91). Because EGFR/PI3K signaling promotes the establishment of latency and because its attenuation enhances reactivation, we postulate that UL135-mediated control of EGFR is to primarily regulate exit from latency. However, because EGFR is a receptor for HCMV entry (92, 93), it is also possible that the decrease of surface EGFR may prevent HCMV superinfection.
In addition to the regulation of EGFR trafficking, we have recently demonstrated that HCMV infection induces the retention of a number surface proteins in EEA1+ early endosomes following their endocytosis (60). HCMV-mediated retention of cargoes in the early endosome and prevention of recycling back to the cell surface may be a ubiquitination-dependent phenomenon as overexpression of the deubiquitinase TRE17/USP6 restores surface levels of some clathrin-independent endocytosis (CIE) cargo in the context of HCMV infection (60). These studies together suggest that regulating endocytic trafficking is a critical point of host control in virus infection that significantly impacts outcomes of infection. Going forward, it will be important to determine how pUL135 and its recruitment of CIN85 and Abi-1 affect the retention of cargos other than EGFR.
MATERIALS AND METHODS
Viruses and cells.
Virus strain TB40/E-GFP was grown and purified, and titers were determined, following which the virus was used for infection as previously described (16, 57). MRC-5 (ATCC CCL-1171), MRC-9 (ATCC CCL-212), and HEK 293-T/17 (ATCC CRL-11268) cells were cultured as previously described (23). Bone marrow stromal cells Sl/Sl and M2-10B4 (MG3) cells expressing granulocyte colony-stimulating factor (G-CSF) and interleukin-3 (IL-3) were obtained courtesy of Donna Hogge (Terry Fox Laboratories, Vancouver, British Columbia) via Stemcell Technologies; cells were maintained and used for long-term culture of human CD34+ cells as previously described (23). For latency assays, CD34+ human hematopoietic cells (HPCs) were isolated from deidentified medical waste following bone marrow harvest from healthy donors for clinical procedures at the University of Arizona Medical Center. Cytomegalovirus latency assays in primary CD34+ cells were performed as previously described (18, 23).
Plasmids.
For the creation of UL135 mutants, we used the previously described plasmid pCIG-UL135myc (23) as the template for site-directed Phusion mutagenesis. We created construct pCIG-UL135mycΔAtypSH3 using Phusion mutagenesis primers UL135ΔAtypSH3-FWD and -REV. Similarly, we created vectors pCIG-UL135mycΔSH3cI and pCIG-UL135mycΔSH3cII using the primer pair UL135ΔSH3cI-FWD and -REV and the pair UL135ΔSH3cII-FWD and -REV, respectively. We created plasmid pCIG-UL135mycΔSH3x3 by serial mutagenesis of the ΔAtypSH3, ΔSH3cI, and ΔSH3cII putative ligands, using the primers listed in Table 2. pCIG-UL135mycΔCIN85 was created by mutagenesis of the first predicted CIN85 ligand with UL135ΔCIN85#1-FWD and -REV, followed by en bloc mutagenesis of the second through fifth predicted CIN85 ligands using the primer pair UL135ΔCIN85#2-5-FWD and -REV. We created the construct pCIG-UL135mycΔSH3cI/CIN85 by mutagenizing the SH3cI putative ligand in pCIG-UL135mycΔCIN85.
TABLE 2.
Primer sequences
| Primer function and name | Sequencea |
|---|---|
| Y2H screen | |
| EcoRI-UL135Y2HF | 5′-GGGAATTCCATATGTCCTTATTTACCCAGCGCCGAGGCCGCAAGCGATC-3′ |
| UL135R-BamHI | 5′-CGCGGATCCTCAGGTCATCTGCATTGACTCGGCGTCCTTCATGAC-3′ |
| pGAD-FWD | 5′-TAATACGACTCACTATAGGGCGA-3′ |
| pGAD-REV | 5′-AGATGGTGCACGATGCACAG-3′ |
| Mutagenesis | |
| UL135ΔSH3cI-FWD | 5′-PHOS-CGCCTACGCCGGCGGTCCG-3′ |
| UL135ΔSH3c1-REV | 5′-GCCGCCTCTTGCGCCCGG-3′ |
| UL135ΔSH3cII-FWD | 5′-PHOS-CCGGGGCCAAGAAGCGGCCGCCTACG-3′ |
| UL135ΔSH3cII-REV | 5′-GCGCCGGAGGTGGCTTCCAGGA-3′ |
| UL135ΔAtypSH3-FWD | 5′-PHOS-CCGCTGCAGTCGGCTGGACACCG-3′ |
| UL135ΔAtypSH3-REV | 5′-GTTTCGTGGGCGCCGGTGTTTTCT-3′ |
| UL135ΔCIN85#1-FWD | 5′-GCCGCTAAGAACCTGAGCACGCCGCCCA-3′ |
| UL135ΔCIN85#1-REV | 5′-PHOS-CGCGGGTATCGGCGTCGGGG-3′ |
| UL135ΔCIN85#2-5-FWD | 5′-GCCGCTCTGCCGCGCACCGTCGGGCTGGAGAATCTCTCGAAGGTGGGACTCTCGTGTCCCTGTCCCCGAGCCGCTACGCCGACGGAGCCGACCA-3′ |
| UL135ΔCIN85#2-5-REV | 5′-PHOS-ATTAGCGGCTGGCTTTTGTGGCGTCGGCGTTTTCGGGAAGGGAGCGGCCGTCACCGGCGGTGTCCAG-3′ |
| Subcloning | |
| NheI-HA-GGGG-Abi1-FWD | 5′-GGGGGCTAGCACCATGTACCCATACGATGTTCCAGATTACGCGGGCGGTGGCGGTGCAGAGCTGCAGATGTTACTAGAGG-3′ |
| Abi1-REV-XhoI | 5′-GGGGCTCGAGTTAATCAGTATAGTGCATGATTGATTCAAC-3′ |
| XbaI-CIN85-FWD | 5′-ATGGTGGAGGCCATAGTGGAGTTT-3′ |
| CIN85-HA-REV-BamHI | 5′-ATGGGATCCTCAGGCGTAGTCGGGCACGTCGTAGGGGTATTTTGATTGTAGAGCTTTCTTTATG-3′ |
| Recombinant virus construction | |
| UL135-Fwd-63nt | 5′-GTGTTTGGACAATAAACACATTCCTTGCCAAAAAATGACGTTTCCAGAAATCCAAGGCATAAATGTCCGTACACCGGCC-3′ |
| Myc-REV-UL135 | 5′-GAGGGAAGGCGTGTGCTGCTATACAACTGTACAACGGACGCGCTCGCTGTTTCGGTCTCACAGATCCTCTTCTGAGATGA-3′ |
| UL135-GalK-ins-FWD | 5′-AAAAGGCGGTGCAGAGCGTCATGAAGGACGCCGAGTCAATGCAGATGACCCCTGTTGACAATTAATCATCGGCA-3′ |
| UL135-GalK-ins-REV | 5′-GTGTGCTGCTATACAACTGTACAACGGACGCGCTCGCTGTTTCGGTCTCATCAGCACTGTCCTGCTCCTT-3′ |
| UL135-3xFLAG-FWD | 5′-AAAAGGCGGTGCAGAGCGTCATGAAGGACGCCGAGTCAATGCAGATGACCGATTATAAAGATGATGATGATAAA-3′ |
| UL135-3xFLAG-REV | 5′-GTGTGCTGCTATACAACTGTACAACGGACGCGCTCGCTGTTTCGGTCTCACTTGTCGTCGTCGTCCTTGTAGTC-3′ |
PHOS, 5′ phosphorylation of oligonucleotide.
To create a CIN85 expression vector with a C-terminal HA tag, we amplified the CIN85 CDS from pCMV6-XL4-CIN85 (SC108004; Origene) (GenBank accession number NM_031892.1; CIN85 transcript variant 1) with primers CIN85-FWD and CIN85-HA-REV and subcloned the amplicon into vector pCIG. Similarly, we created an Abi-1 expression vector with an N-terminal HA tag by amplifying the Abi-1 CDS from pOTB7-Abi1 (94) (Mammalian Gene Collection cDNA clone 4024, IMAGE consortium ID 3531592; obtained via Open Biosystems) using primers NheI-HA-GGGG-Abi1-FWD and Abi1-REV-XhoI and then subcloning the amplicon into vector pCIG. These cDNA clones of both Abi-1 and CIN85 represent the most complete isoforms of each protein.
Transfection of plasmids into 293/T cells was conduced with polyethyleneimine ([PEI] molecular weight, 25,000) (Polysciences). The inserts of all plasmids were confirmed by Sanger sequencing.
Creation of mutant viruses.
Recombinant viruses were created using the bacterial artificial chromosome (BAC) clone of the TB40/E genome, wherein a SV40-GFP cassette was introduced between US34 and TRS1 as a marker of infection, as previously described (17, 57). All BAC recombination was conducted as previously described (20, 95), using a two-step GalK selection/counterselection method that leaves no excision scars or extraneous sequence in the final construct. All TB40/E UL135myc recombinant BACs were created by first replacing the entire UL135 open reading frame with the GalK gene via homologous recombination, as previously described (23). To generate TB40/E UL135myc ΔSH3cI, UL135myc ΔCIN85, and UL135myc ΔSH3cI/CIN85 mutant viruses, we PCR amplified alanine-substituted constructs of UL135myc from the respective pCIG vectors, using the primers UL135-FWD-63nt and Myc-Rev-UL135, followed by DpnI digestion, gel purification, and recombination of the amplicon into the TB40/E ΔUL135-GalK BAC using 2-deoxygalactose counterselection.
TB40/E UL135myc contains a C-terminal myc tag and was created in the same manner as Fix-UL135myc (96). The recombination cassette used to create TB40/E UL135myc contains a short amino acid linker sequence (LEGFPR; ∼0.7 kDa) between UL135 and the C-terminal myc tag. The UL135 SH3 mutant viruses do not contain any additional linker sequence between UL135 and the C-terminal myc tag.
TB40/E UL1353×FLAG was generated by amplifying a GalK cassette with primers UL135-GalK-Ins-FWD and UL135-GalK-Ins-REV (Table 2), and the product was inserted between the UL135 CDS and stop codon. A 3×FLAG sequence was amplified from plasmid pGTE-3xFLAG with primers UL135-3xFLAG-FWD and UL135-3xFLAG-REV, which was then inserted in place of the GalK gene to create TB40/E UL1353×FLAG. pGTE-3xFLAG was a gift from Caroline Kulesza and encodes the 3×FLAG peptide DYKDDDDKGADYKDDDDKEFDYKDDDDK. The UL135STOP virus contains stop codons in place of UL135 M1, M21, and M97 and has been described previously (23).
The inserts of all recombinant BACs were confirmed by Sanger sequencing.
Immunoprecipitation and mass spectrometry.
Approximately 1 × 108 MRC-5 fibroblasts were infected with TB40/E UL1353×FLAG virus at an MOI of 3 and then harvested 72 h postinfection, as previously described (28). Lysates were collected, immunoprecipitated with an anti-FLAG antibody covalently conjugated to Dynabeads (Fisher Scientific), and prepared for mass spectrometry as previously described (28). Tryptic digests of the immunoprecipitated proteins were subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) using an electrospray ionization-linear trap quadrupole (ESI-LTQ) XL mass spectrometer (Thermo Scientific). Peptide and protein identification was performed with SEQUEST software, with a global false discovery set to 5%. To generate a negative-control data set, the same immunoprecipitation was performed on cells infected with WT HCMV, which contains no 3×FLAG tag. Any protein identified in both the WT (no tag) negative control sample and the 3×FLAG sample was excluded.
Yeast two-hybrid screen.
Yeast two-hybrid (Y2H) analysis was performed using a Matchmaker Gold Y2H system (Clontech). The cytosolic domain of UL135 (encoding residues 44 to 328) was amplified with primers EcoRI-UL135Y2HF and UL135R-BamHI and then cloned into pGBKT7 to create the bait plasmid pGBKT7-UL135. This plasmid was transformed into Y2HGold Saccharomyces cerevisiae and tested for autoactivation and toxicity; neither was observed. Yeast strain Y2HGold containing bait plasmid pGBKT7-UL135 was mated with the universal human cDNA Mate and Plate library in yeast strain Y187 (Clontech), using the manufacturer's recommended protocols. After mating, diploid yeast cells were plated onto double-dropout medium with 5-bromo-4-chloro-3-indolyl-α-galactopyranoside (DDO/X-α-Gal) with 125 ng/ml aureobasidin A. A total of 2.8 × 106 diploid mates were screened. After three rounds of outgrowth under stringent selection (quadruple-dropout medium with X-α-Gal and aureobasidin A [QDO/X/A]), prey plasmids were recovered by transformation into DH10B Escherichia coli and selection on LB agar with 100 μg/ml ampicillin. Prey sequences were amplified from each clone with primers PGAD-FWD and PGAD-REV, Sanger sequenced from both 5′ and 3′ ends of the insert, assembled into contigs (when possible) with Macvector software, and then identified by NCBI BLAST alignment to human mRNAs. After sequencing, prey inserts that did not map to known human CDS exons (n = 9) were excluded.
Coimmunoprecipitations.
Immunoprecipitations from transfected 293 cells were performed as previously described (18). MRC-5 cells were infected with recombinant HCMV at an MOI of 2. Forty-eight hours after infection, cells were washed twice with phosphate-buffered saline (PBS), lysed in situ with TNEN lysis buffer (50 mM NaCl, 50 mM Tris-HCl pH 7.5 at 4C, 10 mM EDTA, 10% glycerol, 2 mM phenylmethylsulfonyl fluoride [PMSF], 1× HALT protease, phosphatase inhibitors [Thermo Fisher], 10 mM N-ethylmaleimide, and 0.5% Nonidet P-40 substitute) as previously described (18), and insoluble debris was pelleted at 10,000 × g for 10 min at 4°C. Soluble protein was quantified with a bicinchoninic acid (BCA) assay, and equal quantities of protein were used for immunoprecipitation. Myc epitope-tagged variants of pUL135 were precipitated with 20 μl of EZView anti-c-myc affinity gel (E6654; Sigma) for 90 min at 4°C. CIN85HA and HAAbi-1 were immunoprecipitated using 5 μl of rabbit (Rb) anti-HA antibody per 300 μg of protein lysate for 3 h at 4C, followed by capture of antigen-antibody complexes with 25 μl of Protein G Plus resin (Pierce) for 45 min at 4°C (Table 3). The resin beads were then rapidly washed five times with TNEN buffer containing 0.1% Nonidet P-40. Immunoprecipitates were eluted from the affinity resin in 1× lithium dodecyl sulfate (LDS)-PAGE sample buffer (Invitrogen) with 50 mM Tris-(2-carboxyethyl) phosphine (TCEP) for 10 min at 80°C and then subjected to SDS-PAGE and Western blotting as previously described (18).
TABLE 3.
Ligand and antibodies used in this work
| Antigen or ligand | Identification no. | Hosta | Source | Concn or amt (application)b |
|---|---|---|---|---|
| Antigens | ||||
| α-Tubulin | DM1A | M | Sigma | 300 ng/ml (WB) |
| Myc epitope tag | 71D10 | R | Cell Signaling | 1:1,000 (WB), 1:200 (IF) |
| 9E10 | M | BioXCell | 5 μg (IP) | |
| HA epitope tag | C29F4 | R | Cell Signaling | 1:1,000 (WB) |
| 12CA5 | M | BioXCell | 5 μg (IP) | |
| CIN85 | 84 | M | Upstate/Millipore | 2 μg/ml (WB) |
| Abi1 | A5106 | R | Sigma | 200 ng/ml (WB) |
| EGFR | D38B1 | R | Cell Signaling | 1:1,000 (WB) |
| Rab5 | C8B1 | R | Cell Signaling | 1:200 (IF) |
| Myc agarose beads | E6654 | R | Sigma | 25 μl (IP) |
| GM130 | Clone 35 | M | BD Biosciences | 1:100 (IF) |
| CD34, PE conjugate | 581 | M | BD Biosciences | 20 μl/million cells (FACS) |
| EGFR, BV421 conjugate | AY13 | M | BioLegend | 5 μl/million cells (FCA) |
| Ligand | ||||
| EGF647 | MSG | Molecular Probes | 200 ng/ml (IF), 4 μg/ml (FCA) |
M, mouse; R, rabbit.
WB, Western blotting; IF, immunofluorescence; IP, immunoprecipitation; FACS, fluorescence-activated cell sorting; FCA, flow cytometry analysis.
Quantitation of EGFR by flow cytometry.
EGFR surface levels were quantified as previously described (18). Briefly, cells were infected at an MOI of 1, and infection was allowed to proceed for 48 h. Cells were washed with ice-cold PBS to stop membrane trafficking, trypsinized on ice, labeled with Brilliant Violet 421 (BV421) mouse (Ms) anti-EGFR antibody at 4°C, washed with FACS buffer, and fixed with 2% formaldehyde prior to flow cytometry analysis. Surface EGFR was quantitated from the population of GFP-positive (GFP+; infected) cells. All samples were analyzed on a BD LSRII instrument, and data were analyzed using FlowJo software.
Immunofluorescence.
Preparation of cells, fixation, and immunofluorescence staining were performed as previously described (23). For the experiments measuring trafficking of EGF, cells on coverslips were infected for 24 h and then serum starved for 24 h. Coverslips with cells were washed with PBS–0.1% bovine serum albumin (PBS+B) and then labeled with Alexa-Fluor 647-conjugated EGF (EGF647) on ice for 15 min. Coverslips were washed with cold PBS+B and then transferred to prewarmed medium containing fetal bovine serum (FBS). At indicated time points (Fig. 6B), cells on coverslips were rapidly washed with PBS+B, and fixed with 2% formaldehyde. After cells were stained, images were acquired using a DeltaVision RT inverted deconvolution microscope. Only infected cells expressing GFP were imaged (GFP channel not shown). When necessary, images were cropped to the single cell being analyzed. Colocalization analysis was conducted on z-stacked images using Mosaic Suite's Squassh workflow for ImageJ (97), as previously described (60). The area of EGF647 pixels that is also Rab5 positive divided by the total area of EGF647 positive pixels is used as the metric of colocalization. Representative single planar images were adjusted for brightness and contrast prior to publication.
Immunoblotting.
Infected cells were washed twice with PBS and lysed in situ with radioimmunoprecipitation assay (RIPA) buffer or TNEN lysis buffer with 0.5% Nonidet P-40 substitute, and insoluble debris was pelleted at 15,000 × g for 10 min at 4°C. Protein concentration was quantitated by BCA assay (Pierce/ThermoFisher). Forty micrograms of each lysate was separated on 4 to 12% or 4 to 20% Bis-Tris acrylamide gels (Genscript) with MOPS-SDS buffer, followed by transfer onto polyvinylidene difluoride membrane for fluorescence applications (PVDL-FL; Millipore) and Western blotting as previously described (18). Membranes were scanned with a Li-Cor Odyssey CLx scanner, and proteins were quantified using Image Studio software according to the manufacturer's guidelines.
Infectious centers assay.
Primary CD34+ HPCs were used to assess the latency and reactivation of HCMV in vitro as previously described (17). CD34+ HPCs were isolated from bone marrow and then infected with HCMV at an MOI of 2 for 20 h. Cells were labeled with phycoerythrin (PE)-conjugated CD34 antibody (BD Biosciences) and FACS sorted (FACSAria; BD Bioscience) to obtain a >97% pure population of CD34+, GFP+ (infected) cells. These cells were cultured for 10 days in collagen-coated transwells (Corning) above an irradiated (4,000 rads; 137Cs Gammacell-40 irradiator) mix of Sl/Sl and MG3 stromal cells. The cells were then divided for limiting dilution assay, as described previously (62); one half was directly lysed and seeded in serial 2-fold dilutions on MRC-5 fibroblasts to quantitate infectious centers formed during the establishment of latency, while the other half were seeded intact in serial 2-fold dilutions onto MRC-5 fibroblasts in a cytokine-rich medium to promote differentiation and reactivation. Fourteen days after seeding onto fibroblasts, viral plaques were counted, and the frequency of infectious centers was calculated using ELDA software (http://bioinf.wehi.edu.au/software/elda/) (98).
Accession number(s).
Where strains, protein names, and CDS nucleotide or amino acid numbers are given in this paper, they correspond to the following reference sequences in GenBank: HCMV TB40/E (57), accession number EF999921; chimpanzee CMV, Heberling strain (70), NC_003521; rhesus CMV strain 68-1, NC_006150.1; KF011492; pUL135, ABV71656.1, Abi-1 mRNA, NM_005470.3 (UniProt accession number Q8IZP0); CIN85, NM_031892.2 (UniProt accession number Q5JPT6). The sequence for CD2AP corresponds to UniProt accession number Q9Y5K6.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Mahadevaiah Umashankar for assistance with the yeast two-hybrid assay and Katie Caviness for cloning the HAAbi-1 plasmid. We acknowledge Paula Campbell and the Arizona Cancer Center/Arizona Research Laboratories Division of Biotechnology Cytometry Core Facility for expertise and assistance in flow cytometry and Patricia Jansma of the Molecular and Cellular Biology Imaging Facility for microscopy expertise and assistance. We especially thank the Terry Fox Laboratory for providing the M2-10B4 and Sl/Sl cells.
This work was supported by Public Health Service grants AI079059 (to F.G.) and AI083281 (to S.T.) from the National Institute of Allergy and Infectious Diseases. This work was also supported in part by the Cytometry Shared Resource and the University of Arizona Cancer Center (P30CA023074). J.B. is supported by an American Cancer Society Fellowship.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00919-18.
REFERENCES
- 1.Lumbreras C, Manuel O, Len O, Berge ten IJM, Sgarabotto D, Hirsch HH. 2014. Cytomegalovirus infection in solid organ transplant recipients. Clin Microbiol Infect 20(Suppl 7):19–26. doi: 10.1111/1469-0691.12594. [DOI] [PubMed] [Google Scholar]
- 2.Teira P, Battiwalla M, Ramanathan M, Barrett AJ, Ahn KW, Chen M, Green JS, Saad A, Antin JH, Savani BN, Lazarus HM, Seftel M, Saber W, Marks D, Aljurf M, Norkin M, Wingard JR, Lindemans CA, Boeckh M, Riches ML, Auletta JJ. 2016. Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: a CIBMTR analysis. Blood 127:2427–2438. doi: 10.1182/blood-2015-11-679639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cannon MJ, Davis KF. 2005. Washing our hands of the congenital cytomegalovirus disease epidemic. BMC Public Health 5:70. doi: 10.1186/1471-2458-5-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendelson M, Monard S, Sissons P, Sinclair J. 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J Gen Virol 77:3099–3102. doi: 10.1099/0022-1317-77-12-3099. [DOI] [PubMed] [Google Scholar]
- 6.Maciejewski JP, St Jeor SC. 1999. Human cytomegalovirus infection of human hematopoietic progenitor cells. Leuk Lymphoma 33:1–13. doi: 10.3109/10428199909093720. [DOI] [PubMed] [Google Scholar]
- 7.Söderberg-Nauclér C, Streblow DN, Fish KN, Allan-Yorke J, Smith P, Nelson JA. 2001. Reactivation of latent human cytomegalovirus in CD14+ monocytes is differentiation dependent. J Virol 75:7543–7554. doi: 10.1128/JVI.75.16.7543-7554.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Söderberg-Nauclér C, Fish KN, Nelson JA. 1997. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 91:119–126. doi: 10.1016/S0092-8674(01)80014-3. [DOI] [PubMed] [Google Scholar]
- 9.Cheng S, Caviness K, Buehler J, Smithey M, Nikolich-Zugich J, Goodrum F. 2017. Transcriptome-wide characterization of human cytomegalovirus in natural infection and experimental latency. Proc Natl Acad Sci U S A 114:E10586–E10595. doi: 10.1073/pnas.1710522114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rossetto CC, Tarrant-Elorza M, Pari GS. 2013. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14+ monocytes and CD34+ cells. PLoS Pathog 9:e1003366. doi: 10.1371/journal.ppat.1003366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Damme E, Thys K, Tuefferd M, Van Hove C, Aerssens J, Van Loock M. 2016. HCMV displays a unique transcriptome of immunomodulatory genes in primary monocyte-derived cell types. PLoS One 11:e0164843. doi: 10.1371/journal.pone.0164843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slobedman B, Mocarski ES. 1999. Quantitative analysis of latent human cytomegalovirus. J Virol 73:4806–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goodrum F. 2016. Human cytomegalovirus latency: approaching the Gordian knot. Annu Rev Virol 3:333–357. doi: 10.1146/annurev-virology-110615-042422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinclair J, Reeves MB. 2013. Human cytomegalovirus manipulation of latently infected cells. Viruses 5:2803–2824. doi: 10.3390/v5112803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cha TA, Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR. 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol 70:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petrucelli A, Rak M, Grainger L, Goodrum F. 2009. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J Virol 83:5615–5629. doi: 10.1128/JVI.01989-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umashankar M, Petrucelli A, Cicchini L, Caposio P, Kreklywich CN, Rak M, Rak M, Goldman DC, Hamlin KL, Nelson JA, Fleming WH, Streblow DN, Goodrum F. 2011. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog 7:e1002444. doi: 10.1371/journal.ppat.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buehler J, Zeltzer S, Reitsma JM, Petrucelli A, Umashankar M, Rak M, Zagallo P, Schroeder J, Terhune S, Goodrum F. 2016. Opposing regulation of the EGF receptor: a molecular switch controlling cytomegalovirus latency and replication. PLoS Pathog 12:e1005655. doi: 10.1371/journal.ppat.1005655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caviness K, Cicchini L, Rak M, Umashankar M, Goodrum F. 2014. Complex expression of the UL136 gene of human cytomegalovirus results in multiple protein isoforms with unique roles in replication. J Virol 88:14412–14425. doi: 10.1128/JVI.02711-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caviness K, Rak M, Crawford LB, Streblow DN, Nelson JA, Caposio P, Goodrum F. 2016. Complex interplay of the UL136 isoforms balances cytomegalovirus replication and latency. mBio 7:e01986-. doi: 10.1128/mBio.01986-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodrum F, Reeves MB, Sinclair J, High K, Shenk T. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–945. doi: 10.1182/blood-2007-01-070078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SH, Caviness K, Albright ER, Lee J-H, Gelbmann CB, Rak M, Goodrum F, Kalejta RF. 2016. Long and short isoforms of the human cytomegalovirus UL138 protein silence IE transcription and promote latency. J Virol 90:9483–9494. doi: 10.1128/JVI.01547-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Umashankar M, Rak M, Bughio F, Zagallo P, Caviness K, Goodrum FD. 2014. Antagonistic determinants controlling replicative and latent states of human cytomegalovirus infection. J Virol 88:5987–6002. doi: 10.1128/JVI.03506-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bughio F, Umashankar M, Wilson J, Goodrum F. 2015. Human cytomegalovirus UL135 and UL136 genes are required for postentry tropism in endothelial cells. J Virol 89:6536–6550. doi: 10.1128/JVI.00284-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stanton RJ, Prod'homme V, Purbhoo MA, Moore M, Aicheler RJ, Heinzmann M, Bailer SM, Haas J, Antrobus R, Weekes MP, Lehner PJ, Vojtesek B, Miners KL, Man S, Wilkie GS, Davison AJ, Wang ECY, Tomasec P, Wilkinson GWG. 2014. HCMV pUL135 remodels the actin cytoskeleton to impair immune recognition of infected cells. Cell Host Microbe 16:201–214. doi: 10.1016/j.chom.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tirosh O, Cohen Y, Shitrit A, Shani O, Le-Trilling VTK, Trilling M, Friedlander G, Tanenbaum M, Stern-Ginossar N. 2015. The transcription and translation landscapes during human cytomegalovirus infection reveal novel host-pathogen interactions. PLoS Pathog 11:e1005288. doi: 10.1371/journal.ppat.1005288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biesova Z, Piccoli C, Wong WT. 1997. Isolation and characterization of e3B1, an eps8 binding protein that regulates cell growth. Oncogene 14:233–241. doi: 10.1038/sj.onc.1200822. [DOI] [PubMed] [Google Scholar]
- 28.Reitsma JM, Savaryn JP, Faust K, Sato H, Halligan BD, Terhune SS. 2011. Antiviral inhibition targeting the HCMV kinase pUL97 requires pUL27-dependent degradation of Tip60 acetyltransferase and cell-cycle arrest. Cell Host Microbe 9:103–114. doi: 10.1016/j.chom.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Tanos BE, Pendergast AM. 2007. Abi-1 forms an epidermal growth factor-inducible complex with Cbl: role in receptor endocytosis. Cell Signal 19:1602–1609. doi: 10.1016/j.cellsig.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan PD, Goff SP. 2000. Abl interactor 1 binds to Sos and inhibits epidermal growth factor- and v-Abl-induced activation of extracellular signal-regulated kinases. Mol Cell Biol 20:7591–7601. doi: 10.1128/MCB.20.20.7591-7601.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Innocenti M, Frittoli E, Ponzanelli I, Falck JR, Brachmann SM, Di Fiore PP, Scita G. 2003. Phosphoinositide 3-kinase activates Rac by entering in a complex with Eps8, Abi1, and Sos-1. J Cell Biol 160:17–23. doi: 10.1083/jcb.200206079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kotula L. 2012. Abi1, a critical molecule coordinating actin cytoskeleton reorganization with PI-3 kinase and growth signaling. FEBS Lett 586:2790–2794. doi: 10.1016/j.febslet.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Innocenti M, Tenca P, Frittoli E, Faretta M, Tocchetti A, Di Fiore PP, Scita G. 2002. Mechanisms through which Sos-1 coordinates the activation of Ras and Rac. J Cell Biol 156:125–136. doi: 10.1083/jcb.200108035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rønning SB, Pedersen NM, Madshus IH, Stang E. 2011. CIN85 regulates ubiquitination and degradative endosomal sorting of the EGF receptor. Exp Cell Res 317:1804–1816. doi: 10.1016/j.yexcr.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 35.Schroeder B, Srivatsan S, Shaw AS, Billadeau D, McNiven MA. 2012. CIN85 phosphorylation is essential for EGFR ubiquitination and sorting into multivesicular bodies. Mol Biol Cell 23:3602–3611. doi: 10.1091/mbc.e11-08-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kowanetz K. 2004. CIN85 associates with multiple effectors controlling intracellular trafficking of epidermal growth factor receptors. Mol Biol Cell 15:3155–3166. doi: 10.1091/mbc.e03-09-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cormont M, Metón I, Mari M, Monzo P, Keslair F, Gaskin C, McGraw TE, Le Marchand-Brustel Y. 2003. CD2AP/CMS regulates endosome morphology and traffic to the degradative pathway through its interaction with Rab4 and c-Cbl. Traffic 4:97–112. doi: 10.1034/j.1600-0854.2003.40205.x. [DOI] [PubMed] [Google Scholar]
- 38.Havrylov S, Jolanta Redowicz M, Buchman VL. 2010. Emerging roles of Ruk/CIN85 in vesicle-mediated transport, adhesion, migration and malignancy. Traffic 11:721–731. doi: 10.1111/j.1600-0854.2010.01061.x. [DOI] [PubMed] [Google Scholar]
- 39.Minegishi Y, Shibagaki Y, Mizutani A, Fujita K, Tezuka T, Kinoshita M, Kuroda M, Hattori S, Gotoh N. 2013. Adaptor protein complex of FRS2β and CIN85/CD2AP provides a novel mechanism for ErbB2/HER2 protein downregulation. Cancer Sci 104:345–352. doi: 10.1111/cas.12086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt MHH, Hoeller D, Yu J, Furnari FB, Cavenee WK, Dikic I, Bogler O. 2004. Alix/AIP1 antagonizes epidermal growth factor receptor downregulation by the Cbl-SETA/CIN85 complex. Mol Cell Biol 24:8981–8993. doi: 10.1128/MCB.24.20.8981-8993.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kowanetz K, Terzic J, Dikic I. 2003. Dab2 links CIN85 with clathrin-mediated receptor internalization. FEBS Lett 554:81–87. doi: 10.1016/S0014-5793(03)01111-6. [DOI] [PubMed] [Google Scholar]
- 42.Brett TJ, Traub LM, Fremont DH. 2002. Accessory protein recruitment motifs in clathrin-mediated endocytosis. Structure 10:797–809. doi: 10.1016/S0969-2126(02)00784-0. [DOI] [PubMed] [Google Scholar]
- 43.Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I. 2002. Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature 416:183–187. doi: 10.1038/416183a. [DOI] [PubMed] [Google Scholar]
- 44.Petrelli A, Gilestro GF, Lanzardo S, Comoglio PM, Migone N, Giordano S. 2002. The endophilin-CIN85-Cbl complex mediates ligand-dependent downregulation of c-Met. Nature 416:187–190. doi: 10.1038/416187a. [DOI] [PubMed] [Google Scholar]
- 45.Ceregido MA, Garcia-Pino A, Ortega Roldan JL, Casares S, López Mayorga O, Bravo J, van Nuland NAJ, Azuaga AI. 2013. Multimeric and differential binding of CIN85/CD2AP with two atypical proline-rich sequences from CD2 and Cbl-b*. FEBS J 280:3399–3415. doi: 10.1111/febs.12333. [DOI] [PubMed] [Google Scholar]
- 46.Kirsch KH, Georgescu M-M, Shishido T, Langdon WY, Birge RB, Hanafusa H. 2001. The adapter type protein CMS/CD2AP binds to the proto-oncogenic protein c-Cbl through a tyrosine phosphorylation-regulated Src homology 3 domain interaction. J Biol Chem 276:4957–4963. doi: 10.1074/jbc.M005784200. [DOI] [PubMed] [Google Scholar]
- 47.Dikic I. 2002. CIN85/CMS family of adaptor molecules. FEBS Lett 529:110–115. doi: 10.1016/S0014-5793(02)03188-5. [DOI] [PubMed] [Google Scholar]
- 48.Take H, Watanabe S, Takeda K, Yu Z-X, Iwata N, Kajigaya S. 2000. Cloning and characterization of a novel adaptor protein, CIN85, that interacts with c-Cbl. Biochem Biophys Res Commun 268:321–328. doi: 10.1006/bbrc.2000.2147. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe S, Take H, Takeda K, Yu Z-X, Iwata N, Kajigaya S. 2000. Characterization of the CIN85 adaptor protein and identification of components involved in CIN85 complexes. Biochem Biophys Res Commun 278:167–174. doi: 10.1006/bbrc.2000.3760. [DOI] [PubMed] [Google Scholar]
- 50.Dustin ML, Olszowy MW, Holdorf AD, Li J, Bromley S, Desai N, Widder P, Rosenberger F, van der Merwe PA, Allen PM, Shaw AS. 1998. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell 94:667–677. doi: 10.1016/S0092-8674(00)81608-6. [DOI] [PubMed] [Google Scholar]
- 51.Kirsch KH, Georgescu MM, Ishimaru S, Hanafusa H. 1999. CMS: an adapter molecule involved in cytoskeletal rearrangements. Proc Natl Acad Sci U S A 96:6211–6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kühn J, Wong LE, Pirkuliyeva S, Schulz K, Schwiegk C, Fünfgeld KG, Keppler S, Batista FD, Urlaub H, Habeck M, Becker S, Griesinger C, Wienands J. 2016. The adaptor protein CIN85 assembles intracellular signaling clusters for B cell activation. Sci Signal 9:ra66. doi: 10.1126/scisignal.aad6275. [DOI] [PubMed] [Google Scholar]
- 53.Mi T, Merlin JC, Deverasetty S, Gryk MR, Bill TJ, Brooks AW, Lee LY, Rathnayake V, Ross CA, Sargeant DP, Strong CL, Watts P, Rajasekaran S, Schiller MR. 2012. Minimotif Miner 3.0: database expansion and significantly improved reduction of false-positive predictions from consensus sequences. Nucleic Acids Res 40:D252–D260. doi: 10.1093/nar/gkr1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Proepper C, Johannsen S, Liebau S, Dahl J, Vaida B, Bockmann J, Kreutz MR, Gundelfinger ED, Boeckers TM. 2007. Abelson interacting protein 1 (Abi-1) is essential for dendrite morphogenesis and synapse formation. EMBO J 26:1397–1409. doi: 10.1038/sj.emboj.7601569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kurakin AV, Wu S, Bredesen DE. 2003. Atypical recognition consensus of CIN85/SETA/Ruk SH3 domains revealed by target-assisted iterative screening. J Biol Chem 278:34102–34109. doi: 10.1074/jbc.M305264200. [DOI] [PubMed] [Google Scholar]
- 56.Kowanetz K, Szymkiewicz I, Haglund K, Kowanetz M, Husnjak K, Taylor JD, Soubeyran P, Engstrom U, Ladbury JE, Dikic I. 2003. Identification of a novel proline-arginine motif involved in CIN85-dependent clustering of Cbl and down-regulation of epidermal growth factor receptors. J Biol Chem 278:39735–39746. doi: 10.1074/jbc.M304541200. [DOI] [PubMed] [Google Scholar]
- 57.Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J Gen Virol 89:359–368. doi: 10.1099/vir.0.83286-0. [DOI] [PubMed] [Google Scholar]
- 58.Fairley JA, Baillie J, Bain M, Sinclair J. 2002. Human cytomegalovirus infection inhibits epidermal growth factor (EGF) signalling by targeting EGF receptors. J Gen Virol 83:2803–2810. doi: 10.1099/0022-1317-83-11-2803. [DOI] [PubMed] [Google Scholar]
- 59.Jafferji I, Bain M, King C, Sinclair JH. 2009. Inhibition of epidermal growth factor receptor (EGFR) expression by human cytomegalovirus correlates with an increase in the expression and binding of Wilms' tumour 1 protein to the EGFR promoter. J Gen Virol 90:1569–1574. doi: 10.1099/vir.0.009670-0. [DOI] [PubMed] [Google Scholar]
- 60.Zeltzer S, Zeltzer CA, Wilson J, Donaldson JG, Goodrum F. 2018. Virus control of trafficking from sorting endosomes. mBio 9:e00683-. doi: 10.1128/mBio.00683-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller CL, Eaves CJ. 2001. Long-term culture-initiating cell assays for human and murine cells, p 123–141. In Klug CA, Jordan CT (ed), Hematopoietic stem cell protocols. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 62.Umashankar M, Goodrum F. 2014. Hematopoietic long-term culture (hLTC) for human cytomegalovirus latency and reactivation. Methods Mol Biol 1119:99–112. doi: 10.1007/978-1-62703-788-4_7. [DOI] [PubMed] [Google Scholar]
- 63.Ahmad G, Mohapatra BC, Schulte NA, Nadeau SA, Luan H, Zutshi N, Tom E, Ortega-Cava C, Tu C, Sanada M, Ogawa S, Toews ML, Band V, Band H. 2014. Cbl-family ubiquitin ligases and their recruitment of CIN85 are largely dispensable for epidermal growth factor receptor endocytosis. Int J Biochem Cell Biol 57:123–134. doi: 10.1016/j.biocel.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ravid T, Heidinger JM, Gee P, Khan EM, Goldkorn T. 2004. c-Cbl-mediated ubiquitinylation is required for epidermal growth factor receptor exit from the early endosomes. J Biol Chem 279:37153–37162. doi: 10.1074/jbc.M403210200. [DOI] [PubMed] [Google Scholar]
- 65.Schroeder B, Weller SG, Chen J, Billadeau D, McNiven MA. 2010. A Dyn2-CIN85 complex mediates degradative traffic of the EGFR by regulation of late endosomal budding. EMBO J 29:3039–3053. doi: 10.1038/emboj.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gout I, Middleton G, Adu J, Ninkina NN, Drobot LB, Filonenko V, Matsuka G, Davies AM, Waterfield M, Buchman VL. 2000. Negative regulation of PI 3-kinase by Ruk, a novel adaptor protein. EMBO J 19:4015–4025. doi: 10.1093/emboj/19.15.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aissouni Y, Zapart G, Iovanna JL, Dikic I, Soubeyran P. 2005. CIN85 regulates the ability of MEKK4 to activate the p38 MAP kinase pathway. Biochem Biophys Res Commun 338:808–814. doi: 10.1016/j.bbrc.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 68.So CW, So CK, Cheung N, Chew SL, Sham MH, Chan LC. 2000. The interaction between EEN and Abi-1, two MLL fusion partners, and synaptojanin and dynamin: implications for leukaemogenesis. Leukemia 14:594–601. doi: 10.1038/sj.leu.2401692. [DOI] [PubMed] [Google Scholar]
- 69.Innocenti M, Gerboth S, Rottner K, Lai FP, Hertzog M, Stradal TE, Frittoli E, Didry D, Polo S, Disanza A, Benesch S, Di Fiore PP, Carlier M-F, Scita G. 2005. Abi1 regulates the activity of N-WASP and WAVE in distinct actin-based processes. Nat Cell Biol 7:969–976. doi: 10.1038/ncb1304. [DOI] [PubMed] [Google Scholar]
- 70.Davison AJ, Dolan A, Akter P, Addison C, Dargan DJ, Alcendor DJ, McGeoch DJ, Hayward GS. 2003. The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. J Gen Virol 84:17–28. doi: 10.1099/vir.0.18606-0. [DOI] [PubMed] [Google Scholar]
- 71.Wee P, Wang Z. 2017. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel) 9:52. doi: 10.3390/cancers9050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tomas A, Futter CE, Eden ER. 2014. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol 24:26–34. doi: 10.1016/j.tcb.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cooray S. 2004. The pivotal role of phosphatidylinositol 3-kinase–Akt signal transduction in virus survival. J Gen Virol 85:1065–1076. doi: 10.1099/vir.0.19771-0. [DOI] [PubMed] [Google Scholar]
- 74.Liu X, Cohen JI. 2015. The role of PI3K/Akt in human herpesvirus infection: from the bench to the bedside. Virology 479–480:568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chan G, Nogalski MT, Stevenson EV, Yurochko AD. 2012. Human cytomegalovirus induction of a unique signalsome during viral entry into monocytes mediates distinct functional changes: a strategy for viral dissemination. J Leukoc Biol 92:743–752. doi: 10.1189/jlb.0112040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chan G, Nogalski MT, Bentz GL, Smith MS, Parmater A, Yurochko AD. 2010. PI3K-dependent upregulation of Mcl-1 by human cytomegalovirus is mediated by epidermal growth factor receptor and inhibits apoptosis in short-lived monocytes. J Immunol 184:3213–3222. doi: 10.4049/jimmunol.0903025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Collins-McMillen D, Kim JH, Nogalski MT, Stevenson EV, Chan GC, Caskey JR, Cieply SJ, Yurochko AD. 2015. Human cytomegalovirus promotes survival of infected monocytes via a distinct temporal regulation of cellular Bcl-2 family proteins. J Virol 90:2356–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith MS, Bivins-Smith ER, Tilley AM, Bentz GL, Chan G, Minard J, Yurochko AD. 2007. Roles of phosphatidylinositol 3-kinase and NF-κB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: strategy for viral persistence. J Virol 81:7683–7694. doi: 10.1128/JVI.02839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shenk T, Alwine JC. 2014. Human cytomegalovirus: coordinating cellular stress, signaling, and metabolic pathways. Annu Rev Virol 1:355–374. doi: 10.1146/annurev-virology-031413-085425. [DOI] [PubMed] [Google Scholar]
- 80.Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. 2008. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat Rev Microbiol 6:266–275. doi: 10.1038/nrmicro1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. 2010. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M. 2015. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 18:649–658. doi: 10.1016/j.chom.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miller WE, Earp HS, Raab-Traub N. 1995. The Epstein-Barr virus latent membrane protein 1 induces expression of the epidermal growth factor receptor. J Virol 69:4390–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kung CP, Meckes DG, Raab-Traub N. 2011. Epstein-Barr virus LMP1 activates EGFR, STAT3, and ERK through effects on PKC. J Virol 85:4399–4408. doi: 10.1128/JVI.01703-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang L, Dittmer DP, Tomlinson CC, Fakhari FD, Damania B. 2006. Immortalization of primary endothelial cells by the K1 protein of Kaposi's sarcoma-associated herpesvirus. Cancer Res 66:3658–3666. doi: 10.1158/0008-5472.CAN-05-3680. [DOI] [PubMed] [Google Scholar]
- 86.Zhang Z, Chen W, Sanders MK, Brulois KF, Dittmer DP, Damania B. 2016. The K1 protein of Kaposi's sarcoma-associated herpesvirus augments viral lytic replication. J Virol 90:7657–7666. doi: 10.1128/JVI.03102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Peng L, Wu T-T, Tchieu JH, Feng J, Brown HJ, Feng J, Li X, Qi J, Deng H, Vivanco I, Mellinghoff IK, Jamieson C, Sun R. 2010. Inhibition of the phosphatidylinositol 3-kinase-Akt pathway enhances gamma-2 herpesvirus lytic replication and facilitates reactivation from latency. J Gen Virol 91:463–469. doi: 10.1099/vir.0.015073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim JH, Collins-McMillen D, Buehler JC, Goodrum F, Yurochko AD. 2017. Human cytomegalovirus requires epidermal growth factor receptor signaling to enter and initiate the early steps in the establishment of latency in CD34+ human progenitor cells. J Virol 91:e01206-. doi: 10.1128/JVI.01206-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shi T, Niepel M, McDermott JE, Gao Y, Nicora CD, Chrisler WB, Markillie LM, Petyuk VA, Smith RD, Rodland KD, Sorger PK, Qian W-J, Wiley HS. 2016. Conservation of protein abundance patterns reveals the regulatory architecture of the EGFR-MAPK pathway. Sci Signal 9:rs6. doi: 10.1126/scisignal.aaf0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liang Y, Kurakin A, Roizman B. 2005. Herpes simplex virus 1 infected cell protein 0 forms a complex with CIN85 and Cbl and mediates the degradation of EGF receptor from cell surfaces. Proc Natl Acad Sci U S A 102:5838–5843. doi: 10.1073/pnas.0501253102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Deschamps T, Dogrammatzis C, Mullick R, Kalamvoki M. 2017. Cbl E3 ligase mediates the removal of nectin-1 from the surface of herpes simplex virus 1-infected cells. J Virol 91:e00393-. doi: 10.1128/JVI.00393-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang X, Huong S-M, Chiu ML, Raab-Traub N, Huang E-S. 2003. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 424:456–461. doi: 10.1038/nature01818. [DOI] [PubMed] [Google Scholar]
- 93.Chan G, Nogalski MT, Yurochko AD. 2009. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc Natl Acad Sci U S A 106:22369–22374. doi: 10.1073/pnas.0908787106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen DM, Schuler GD, Altschul SF, Zeeberg B, Buetow KH, Schaefer CF, Bhat NK, Hopkins RF, Jordan H, Moore T, Max SI, Wang J, Hsieh F, Diatchenko L, Marusina K, Farmer AA, Rubin GM, Hong L, Stapleton M, Soares MB, Bonaldo MF, Casavant TL, Scheetz TE, Brownstein MJ, Usdin TB, Toshiyuki S, Caminci P, Prage C, Raha SS, Loquellano NA, Peters GJ, Abramsom RD, Mullahy SJ, Bosak SA, McEwan PJ, McKernan KJ, Malek JA, Gunaratne PH, Richards S, Worley KC, Hale S, Garcia AM, Gay LJ, et al. 2002. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci U S A 99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Grainger L, Cicchini L, Rak M, Petrucelli A, Fitzgerald KD, Semler BL, Goodrum F. 2010. Stress-inducible alternative translation initiation of human cytomegalovirus latency protein pUL138. J Virol 84:9472–9486. doi: 10.1128/JVI.00855-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rizk A, Paul G, Incardona P, Bugarski M, Mansouri M, Niemann A, Ziegler U, Berger P, Sbalzarini IF. 2014. Segmentation and quantification of subcellular structures in fluorescence microscopy images using Squassh. Nat Protoc 9:586–596. doi: 10.1038/nprot.2014.037. [DOI] [PubMed] [Google Scholar]
- 98.Hu Y, Smyth GK. 2009. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.