

The results of this study demonstrate that the gene-editing complex Cas9/sgRNA can be programmed to target and cleave HIV-1 PBS DNA, and thus, inhibit HIV-1 infection. Given that almost all HIV-1 strains have the same PBS, which is copied from the cellular tRNA3Lys during reverse transcription, PBS-targeting sgRNA can be used to inactivate HIV-1 DNA of different strains. We also discovered that HIV-1 uses different mechanisms to resist Cas9/sgRNAs, depending on whether they target the plus or the minus strand of PBS DNA. These findings allow us to predict that a Cas9 variant that uses the CCA sequence as the protospacer adjacent motif (PAM) should more strongly and persistently suppress HIV-1 replication. Together, these data have identified the PBS as the target DNA of Cas9/sgRNA and have predicted how to improve Cas9/sgRNA to achieve more efficient and sustainable suppression of HIV-1 infection, therefore improving the capacity of Cas9/sgRNA in curing HIV-1 infection.
KEYWORDS: CRISPR-Cas9, human immunodeficiency virus, primer binding site
ABSTRACT
The clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated protein 9 (Cas9) gene-editing technology has been used to inactivate viral DNA as a new strategy to eliminate chronic viral infections, including HIV-1. This utility of CRISPR-Cas9 is challenged by the high heterogeneity of HIV-1 sequences, which requires the design of the single guide RNA (sgRNA; utilized by the CRISPR-Cas9 system to recognize the target DNA) to match a specific HIV-1 strain in an HIV patient. One solution to this challenge is to target the viral primer binding site (PBS), which HIV-1 copies from cellular tRNA3Lys in each round of reverse transcription and is thus conserved in almost all HIV-1 strains. In this study, we demonstrate that PBS-targeting sgRNA directs Cas9 to cleave the PBS DNA, which evokes deletions or insertions (indels) and strongly diminishes the production of infectious HIV-1. While HIV-1 escapes from PBS-targeting Cas9/sgRNA, unique resistance mechanisms are observed that are dependent on whether the plus or the minus strand of the PBS DNA is bound by sgRNA. Characterization of these viral escape mechanisms will inform future engineering of Cas9 variants that can more potently and persistently inhibit HIV-1 infection.
IMPORTANCE The results of this study demonstrate that the gene-editing complex Cas9/sgRNA can be programmed to target and cleave HIV-1 PBS DNA, and thus, inhibit HIV-1 infection. Given that almost all HIV-1 strains have the same PBS, which is copied from the cellular tRNA3Lys during reverse transcription, PBS-targeting sgRNA can be used to inactivate HIV-1 DNA of different strains. We also discovered that HIV-1 uses different mechanisms to resist Cas9/sgRNAs, depending on whether they target the plus or the minus strand of PBS DNA. These findings allow us to predict that a Cas9 variant that uses the CCA sequence as the protospacer adjacent motif (PAM) should more strongly and persistently suppress HIV-1 replication. Together, these data have identified the PBS as the target DNA of Cas9/sgRNA and have predicted how to improve Cas9/sgRNA to achieve more efficient and sustainable suppression of HIV-1 infection, therefore improving the capacity of Cas9/sgRNA in curing HIV-1 infection.
INTRODUCTION
Highly active antiretroviral therapy (HAART) has transformed HIV-1 infection from a deadly disease to a manageable chronic infection. However, HAART represents a lifelong treatment, because it suppresses HIV-1 infection but does not clear the latent HIV-1 reservoir. The root of HIV-1 latency is the quiescent proviral DNA that persists in a transcriptionally silent state but maintains the ability to produce infectious virions upon viral activation (1). In theory, eliminating this latent proviral DNA should lead to the cure of HIV-1 infection. This view has inspired the development and application of several molecular tools to attack and destroy HIV-1 DNA, including recombinases (2), zinc finger nucleases (ZFNs) (3, 4), transcription activator-like endonucleases (TALENs) (5, 6), and clustered regularly interspersed short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) (7, 8). Compared to recombinases, ZFNs, and TALENs, which achieve recognition of specific DNA sequences by protein engineering (9), the CRISPR-Cas9 system utilizes a single guide RNA (sgRNA) to recognize the target DNA, which makes any DNA sequence editable through sgRNA programming (10). Akin to its role in bacteria, which is to restrict foreign DNA, including bacteriophages, CRISPR-Cas9 has been tested for inactivation of the DNA genome of human viruses, including HIV-1 (11–15), herpesviruses (16–18), hepatitis B virus (19–21), adenovirus (22), and others (23).
Cas9 cleavage at a specific DNA site leads to a double-stranded DNA break, which is repaired by the nonhomology end-joining (NHEJ) complex when a homologous DNA template is unavailable. NHEJ repair creates genomic deletions or insertions (indels). These aberrations may eliminate the infectious viral genome when found within essential viral genes. However, some of the indels, although in low abundance, give rise to viable virions. Because they alter the sequence of the Cas9 cleavage site, such genomic alterations confer resistance to attack by the same Cas9/sgRNA (24, 25). This mechanism of viral escape is surmountable, as recently reported in two studies showing that HIV-1 infection can be halted using two carefully selected sgRNAs that target two conserved sites in HIV-1 DNA (26, 27). Similar observations were reported for herpesviruses (18).
In addition to viral resistance, another challenge of applying Cas9/sgRNA to eliminate infectious HIV-1 DNA in patients stems from the high diversity of circulating HIV-1 strains. This high diversity necessitates tailoring the design of effective sgRNAs to the specific strain in each patient (28). Ideally, a functionally important DNA sequence that is highly conserved across HIV-1 strains, once validated, could facilitate effective CRISPR-Cas9-mediated eradication of HIV-1 infection, regardless of viral subtypes. A viral DNA region with such potential is the primer binding site (PBS). The PBS is generated during each cycle of HIV-1 reverse transcription by copying the 3′-terminal 18 nucleotides of the cellular tRNA3Lys (29). Since tRNA3Lys needs to bind to the PBS to initiate viral reverse transcription, and the PBS mediates the second template switch of reverse transcription (Fig. 1A), we hypothesize that by targeting and cleaving the PBS DNA, Cas9/sgRNA causes indels in the PBS, which impair HIV-1 reverse transcription and inhibit the replication of all HIV-1 strains. Not surprisingly, PBS-targeting small interfering RNA (siRNA) has been tested for suppressing HIV-1 infection (30). This antiretrovirus strategy has been exploited by cells to control the mobility of long terminal repeat (LTR) retrotransposons, using tRNA-derived small RNA to target the PBS (31). It is expected that HIV-1 will evolve resistant mutations in the PBS as a result of NHEJ repair (Fig. 1A). However, since the PBS sequence is copied from the tRNA3Lys in each round of reverse transcription, we predict that the newly synthesized wild-type (wt) PBS will again be subjected to Cas9/sgRNA cleavage (Fig. 1A), which leads to prolonged suppression of HIV-1 replication by the same PBS-targeting sgRNA.
FIG 1.

Inhibition of HIV-1 infection by Cas9/sgRNA targeting viral PBS. (A) Illustration of the potential mechanisms by which the PBS-targeting Cas9/sgRNA inhibits HIV-1 infection. When the viral RNA carrying the resistant mutation in the PBS is reverse transcribed into viral DNA, the plus strand has the wild-type PBS sequence, thus potentially becoming susceptible to cleavage by Cas9/sgRNA again. vRNA, viral RNA; vDNA, viral DNA. (B) Location of the PBS-targeting sgRNA. The PBS sequence is highlighted in green letters. The PAM is indicated with blue lines and letters. (C) HIV-1 strain NL4-3 was used to infect SupT1 cells that stably express Cas9 and PBS-targeting sgRNA. Expression of Cas9 and viral p24 was examined by immunostaining and flow cytometry. Results of one representative infection experiment are shown. Results of three independent experiments are summarized in the bar graph, with the values of the control set at 100. ***, P < 0.001. (D) Levels of HIV-1 in the supernatants of infected SupT1 cells were determined either by measuring viral reverse transcriptase activity (representing total viral particles) or by infecting the TZM-bl indicator cells (representing infectious viral particles). Results of three independent infection experiments were averaged and are shown in the bar graph, with values of the control set at 100. ***, P < 0.001. (E, F, G) Indels caused by the PBS-targeting Cas9/sgRNA. Total cellular DNA was extracted from the infected SupT1 cells that expressed the sgPBS1, sgPBS2, or sgPBS3 guide RNA. Viral DNA was amplified by PCR and sequenced. Indels are shown by red letters for sgPBS1 (E), sgPBS2 (F), and sgPBS3 (G). The PAM is highlighted in blue letters. Red arrow indicates Cas9 cleavage site. The frequency of each indel among the sequenced DNA clones is presented. Error bars show standard deviations.
RESULTS
Cas9/sgRNA targeting the HIV-1 PBS inhibits infection.
The HIV-1 PBS has three potential protospacer adjacent motifs (PAMs), which allows the design of three PBS-targeting sgRNAs (sgPBS1, sgPBS2, and sgPBS3) (Fig. 1B). These three sgRNAs were cloned into the plentiCRISPR-v2 vector and stably transduced into CD4+ SupT1 T cells. The expression of Cas9 (bearing a FLAG tag) was detected with flow cytometry after immunostaining for FLAG (Fig. 1C, y axis). When these cell lines were challenged with the HIV-1 strain NL4-3, the numbers of infected cells staining positive for HIV-1 p24 protein (Fig. 1C, x axis) decreased 3-fold in the cells expressing sgPBS1, sgPBS2, and sgPBS3 compared to the level in the control cells expressing Cas9 alone (Fig. 1C). As a result, Cas9/sgPBS led to a 5-fold decrease in the production of HIV-1 particles, as determined by the level of viral reverse transcriptase (RT) activity in the culture supernatants, and a 10-fold decrease in the level of infectious HIV-1, as measured by infecting TZM-bl indicator cells (Fig. 1D).
To determine whether the impaired viral infection and viral production resulted from Cas9/sgPBS-induced indels, specifically in the PBS region, we amplified viral DNA from the infected SupT1 cells and sequenced the PBS DNA. A wide range of indels of various lengths were detected in the sgPBS1-, sgPBS2-, and sgPBS3-expressing SupT1 cells (Fig. 1E to G). These data demonstrate that Cas9/sgRNA strongly impairs HIV-1 infection and production when targeted to the PBS DNA.
Since 15 of the 18 nucleotides of the PBS are present in the tRNA3Lys genes (Fig. 2A), we tested whether any of the PBS-targeting sgRNAs sgPBS1, sgPBS2, and sgPBS3 target tRNA3Lys DNA (Fig. 2B). Accordingly, we designed primers to amplify tRNA3Lys DNA from SupT1 cells that stably express Cas9/sgPBS1, Cas9/sgPBS2, or Cas9/sgPBS3. Among the 6 tRNA3Lys genes that we tried to amplify, PCR products were obtained for 4 genes (Fig. 2C). We then sequenced these PCR products from either control or sgPBS-expressing SupT1 cells and observed no indels that were expected from Cas9/sgRNA cleavage (Fig. 2D), highlighting the specificity of the targeting mechanism. We also cloned the PCR products of the tRNALys-3-5 gene and sequenced 24 DNA clones from the control or sgPBS-expressing SupT1 cells. Again, no indels were detected (Fig. 2E). These data suggest that Cas9/sgPBS does not cause detectable mutations in cellular tRNA3Lys genes.
FIG 2.

Sequencing of tRNA3Lys genes in SupT1 cells that express Cas9 and sgPBS1, sgPBS2, or sgPBS3. Control cells express Cas9 only. (A) Illustration of tRNA3Lys binding to the HIV-1 PBS region (highlighted in green). (B) Incomplete complementation of sgPBS1, sgPBS2, or sgPBS3 to the tRNALys-TTT-3-5 gene. (C) PCR products of amplified tRNA3Lys genes from control SupT1 cells or SupT1 cells expressing Cas9/sgPBS1, Cas9/sgPBS2, or Cas9/sgPBS3. Sizes of DNA markers are shown on the left of the agarose gels. (D) Sanger sequencing data of the PCR products. The reference sequence for each tRNA3Lys gene was obtained from GenBank. (E) PCR products for gene tRNALys-TTT-3-5 in the control or Cas9/sgPBS-expressing SupT1 cells were cloned. About 24 DNA clones for each PCR product were sequenced. The frequency of each variant is presented.
HIV-1 escapes from Cas9/sgPBS.
We next investigated whether HIV-1 escapes from Cas9/sgPBS inhibition. Control and Cas9/sgPBS-expressing SupT1 cells were infected with HIV-1 for prolonged culture periods. The levels of viral RT activity in culture supernatants were determined at various time intervals to monitor HIV-1 replication until a severe cytopathic effect was observed. Compared to HIV-1 replication in the control cells, a significant delay was observed in SupT1 cells that expressed Cas9 together with sgPBS1, sgPBS2, or sgPBS3 (Fig. 3A). And yet, viral replication eventually reached peak levels in the Cas9/sgPBS-expressing SupT1 cells, similar to that in the control cells, indicating viral escape from Cas9/sgPBS inhibition (Fig. 3A). In order to verify viral escape, we performed a second round of viral replication by infecting the SupT1 cell line with HIV-1 that was collected at the peak of the first round of viral replication from the same SupT1 cell line. As expected, HIV-1 had escaped from Cas9/sgPBS3; however, it was still suppressed by Cas9/sgPBS1 and Cas9/PBS2 in the second round of replication (Fig. 3B). Only at the third round of replication did HIV-1 exhibit similar growth kinetics in the control and all three Cas9/sgPBS-expressing SupT1 cell lines (Fig. 3C). We observed this delayed escape from Cas9/sgPBS1 and Cas9/sgPBS2 in another, independently performed HIV-1 evolution experiment. These data demonstrate that HIV-1 can escape from the inhibition by Cas9/sgPBS but also indicate that it takes two passages for HIV-1 to acquire complete resistance to Cas9/sgPBS1 and Cas9/sgPBS2.
FIG 3.
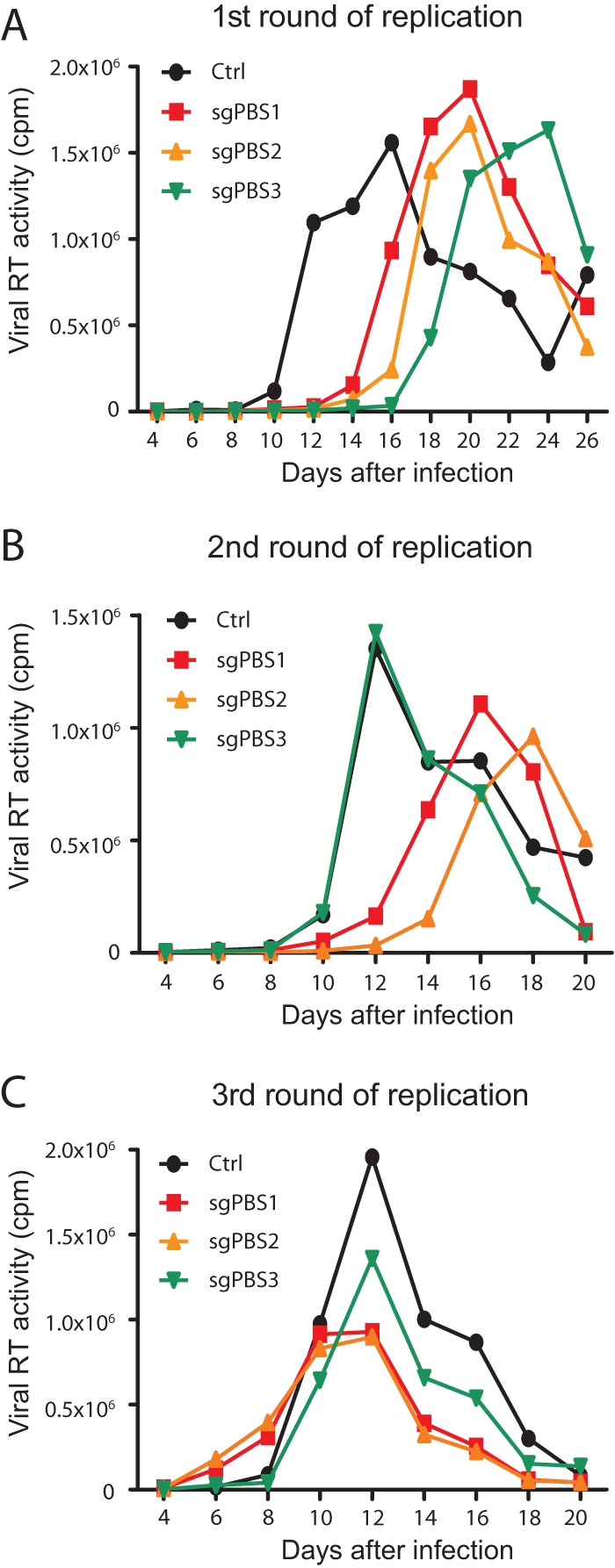
Escape of HIV-1 from inhibition by Cas9/sgPBS1, Cas9/sgPBS2, or Cas9/sgPBS3. (A) HIV-1 that was produced by transfecting HEK293T cells was used to infect SupT1 cells expressing Cas9 and sgPBS1, sgPBS2, or sgPBS3. Viral replication was monitored for a prolonged period of time by measuring viral RT activity in the culture supernatants at various time intervals. Control (ctrl) SupT1 cells expressed Cas9 only. This is called the first (1st) round of viral replication. (B) Viruses at the peak of the 1st round of viral replication in each cell line were collected and used to infect the same cell line using viruses with the same amounts of viral RT activity. This is called the second (2nd) round of viral replication. (C) The third (3rd) round of viral replication was performed using viruses collected at the peak of the second round to infect the same cell line. Results shown represent two independent replication experiments.
HIV-1 mutates the viral PBS to escape from inhibition by Cas9/sgPBS1 and Cas9/sgPBS2.
To understand the genetic mechanism behind the delayed HIV-1 escape from Cas9/sgPBS1 and Cas9/sgPBS2 compared to the escape from Cas9/sgPBS3, we sequenced the HIV-1 genome at the peak of replication for each of the three rounds of infection. No mutation was detected in the PBS of HIV-1 that had replicated in the control SupT1 cells. For viruses that were recovered from the first round of replication in Cas9/sgPBS1-expressing cells, short indels were detected at the Cas9 cleavage site within the PBS (Fig. 4). Of the viruses that were sequenced, 11% contained single-nucleotide (1nt) mutations (including deletion, insertion, or substitution), 48% contained double-nucleotide (2nt) mutations, and 41% had triple-nucleotide (3nt) mutations, as well as insertions of more than 3 nucleotides. When viruses from the second round of replication in the Cas9/sgPBS1-expressing SupT1 cells were sequenced, the percentage of viruses containing single-nucleotide mutations in the PBS increased to 29% (Fig. 4). At the third round of HIV-1 replication, only one virus had a 3nt insertion in the PBS, with the rest having either 1nt mutations (38%) or 2nt mutations (57%) (Fig. 4). These sequencing data reveal a gradual evolution of escape mutations toward minimal change in the PBS at the Cas9 cleavage site. This conclusion is further supported by the sequencing data of HIV-1 that escaped from Cas9/sgPBS2 inhibition after multiple rounds of replication, showing enrichment of viruses containing single-nucleotide mutations at the cleavage site of Cas9 in the PBS (Fig. 5).
FIG 4.

Development of HIV-1 resistance to Cas9/sgPBS1. Viruses at the peaks of the first, second, and third rounds of replication in the Cas9/sgPBS1-expressing SupT1 cells were harvested. Viral RNA was extracted and subjected to RT-PCR and sequencing. Each indel in the PBS was registered, and its frequency in the sequenced DNA clones was calculated. The bar graph shows the frequencies of mutations for single-nucleotide (1nt) changes, double-nucleotide (2nt) changes, or changes involving three or more nucleotides (3nt).
FIG 5.

Development of HIV-1 resistance to Cas9/sgPBS2. Viruses at the peaks of the first, second, and third rounds of replication in the Cas9/sgPBS2-expressing SupT1 cells were collected and sequenced for the PBS DNA. The detected indels are shown, and their frequencies are presented. The bar graphs summarize the percentages of indels involving changes of one nucleotide (1nt), two nucleotides (2nt), or three or more nucleotides (3nt).
We then inserted these PBS mutations that were identified in the escaped viruses into the parental HIV-1 NL4-3 and examined whether these mutated viruses can resist Cas9 inhibition. We first tested the two most frequent mutations, a G insertion (named PBS-M1) and a TT insertion (named PBS-M2), which were detected at the cleavage site of Cas9 guided by sgPBS1 (Fig. 6A). These two mutations are expected to disrupt the binding of sgPBS1 RNA to its target DNA and, thus, resist Cas9 attack. In the short-term infection experiments, the M1 and M2 mutated viruses produced similar levels of viruses in both the control cells and cells that expressed Cas9/sgPBS1 (Fig. 6B), indicating that the M1 and M2 mutations in the PBS confer resistance to sgPBS1. This finding is supported by the data from long-term replication of the wild-type and mutated HIV-1 in the control and sgPBS1-expressing SupT1 cells, which showed that the M1 and M2 mutated viruses replicated equally well in the control and the sgPBS1-expressing SupT1 cells (Fig. 6C).
FIG 6.

The PBS-M1 and PBS-M2 mutations allow HIV-1 to resist Cas9/sgPBS1. (A) Depiction of the M1 and M2 mutations (in red letters) in the PBS. (B) M1 and M2 mutations resist Cas9/sgPBS1 inhibition in short-term infection. Wild-type HIV-1 or the M1 or M2 mutant, with the same p24 amounts, was used to infect SupT1 cells expressing the Cas9 clone (control) or Cas9/sgPBS1. Forty hours after infection, the levels of HIV-1 in the culture supernatants were determined by infecting the TZM-bl indicator cells. Results shown are the average values from three independent infection experiments. Levels of each virus from the control cells are set at 100. ***, P < 0.001. (C) Replication of wild-type HIV-1 and the M1 and M2 mutants in control or Cas9/sgPBS1-expressing SupT1 cells. Levels of HIV-1 in the culture supernatants were determined by measuring viral RT activity over various time intervals. Results represent two independent viral replication experiments. (D) Persistence of the M1 and M2 mutations in HIV-1 during long-term replication in the control SupT1 cells or cells expressing Cas9/sgPBS1. Viruses at the peak of replication shown in panel C were collected, and the PBS DNA was amplified and sequenced. The frequencies of the wild-type and mutated sequences are presented. (E) Prevalences of wild-type and mutated PBS sequences in HIV-1-infected cells and in viruses. M1 or M2 mutated virus was used to infect control or Cas9/sgPBS1-expressing SupT1 cells for 40 h. The PBS region was amplified from HIV-1 DNA in the infected cells or from HIV-1 RNA in virus particles. The PCR products were analyzed by MiSeq to register the wild-type and mutated PBS sequences. Results shown are the MiSeq data from three independent infections. (F) Viral DNA was extracted from the M2 virus-infected control SupT1 cells and then treated with HpaI to remove the M2/M2 viral DNA, followed by PCR and MiSeq to determine the percentages of wild-type and M2 PBS sequences. Equal amounts of wild-type HIV-1 plasmid DNA and the M2 HIV-1 plasmid DNA were mixed, treated with HpaI, and then subjected to PCR and MiSeq analysis. The results serve as the control for effective removal of M2 DNA by HpaI digestion. The percentages of wt/wt, wt/M2, and M2/M2 double-stranded PBS DNA in the M2-infected control SupT1 cells were calculated and are presented in the table. Error bars show standard deviations.
Persistence of PBS escape mutations during long-term HIV-1 replication.
Given that each replication cycle of HIV-1 regenerates the wild-type PBS sequence by copying cellular tRNA3Lys, we expected that, during long-term replication of the M1 and M2 mutants in control SupT1 cells, the wild-type PBS viruses would become the prevalent viral population in the absence of the selection pressure of sgPBS1. This mechanism has been reported for various PBS mutations that revert to the wild-type sequence after prolonged replication (32–35). We thus sequenced the viruses that were harvested at the peak of long-term replication of M1 and M2 viruses in control SupT1 cells. Unexpectedly, 30% of the sequenced viruses had the M1 sequence, and 74% had the M2 sequence (Fig. 6D). We further sequenced the viruses from long-term culture of M1 and M2 viruses in the Cas9/sgPBS1-expressing cells. Above 90% of the viruses had the M1 or M2 PBS sequence (Fig. 6D). These data demonstrate that even in the absence of Cas9/sgPBS1 pressure, the M1 or M2 PBS mutation can be preserved during HIV-1 replication, instead of simply reverting to the wild-type PBS.
The M1 and M2 mutated viruses produce more mutated than wild-type viruses in short-term infection of control SupT1 cells.
In order to understand the mechanism behind the persistence of M1 and M2 mutations in the long-term viral replication, we used the MiSeq method to sequence HIV-1 DNA within the control or Cas9/sgPBS1-expressing SupT1 cells that were infected by either M1 or M2 viruses for 40 h. We performed Alu-PCR to amplify the integrated HIV-1 DNA (57), which expresses viral genes and produces viruses. The results showed that, in M1-infected control SupT1 cells, 41% of viral DNA had wild-type PBS and 54% had the M1 mutation (Fig. 6E). This is largely in agreement with the fact that one strand of PBS DNA is copied from tRNA3Lys, whereas the other strand is copied from the M1 viral RNA. Interestingly, in M2-infected control SupT1 cells, 31% of viral DNA had wild-type PBS, while 65% had the M2 mutation, which indicates that when cells repair the mismatch at the M2 mutation site in the PBS, the repair outcome is skewed to retain the M2 mutation; this is also seen in the case of M1 (41% wild type), albeit to a lesser extent (Fig. 6E). In contrast, above 90% of viral DNA had the M1 or M2 mutation in the infected Cas9/sgPBS1-expressing cells (Fig. 6E), which may have resulted from the cleavage and subsequent removal of the wild-type PBS by Cas9/sgPBS1.
We next sequenced the viruses that were produced from these infected SupT1 cells. In agreement with the dominance of mutated PBS DNA in the infected Cas9/sgPBS1-expressing cells, close to 90% of the viruses had the M1 or M2 mutation (Fig. 6E). Surprisingly, in the M1- or M2-infected control SupT1 cells, 69% of viruses had the M1 PBS and 78% had the M2 PBS, respectively (Fig. 6E), which is significantly higher than the percentages of these two mutations within the viral DNA of the infected cells (54% M1 and 65% M2). One possible explanation of this discrepancy is that significant portions of viral DNAs are wt/M1 or wt/M2 hybrids at the PBS, which are the result of reverse transcription and remain unrepaired even after integration. Since the minus strand (i.e., containing the M1 or M2 mutation) serves as the template for transcription, only M1 or M2 mutated viral RNA is synthesized.
We were able to test this possibility by determining the percentages of the wt/wt, wt/M2, and M2/M2 PBS DNA variants in the infected cells, because the M2 mutation creates a digestion site for the restriction enzyme HpaI, which recognizes and cleaves the 5′-GTTAAC-3′ DNA sequence (Fig. 6F). This method was first tested by mixing equal amounts of wild-type viral plasmid DNA and the M2 viral plasmid DNA in the presence of DNA that was extracted from uninfected control SupT1 cells and subsequently digested with HpaI. The digested DNA samples were analyzed by MiSeq. The results showed that almost all M2 viral DNA was digested and removed (Fig. 6F). We then incubated HpaI with viral DNA that was extracted from control SupT1 cells that were infected with the M2 virus. Compared to the untreated samples, which had 35% wild-type PBS and 62% M2 PBS, HpaI digestion changed these percentages to 76% wild-type PBS and 23% M2 PBS because the M2/M2 PBS DNA was digested by HpaI. The detected M2 sequence (23%) indicates the existence of wt/M2 hybrid PBS that resists HpaI digestion. Using these data, we were able to calculate the percentage of wt/wt PBS as 46% and wt/M2 PBS as 54% in the HpaI-treated samples. Knowing the relative ratio of wt/wt and wt/M2 (46:54), together with the percentages of wt (35%) and M2 (62%), in the M2-infected control cells, we calculated the percentages of wt/wt PBS as 23%, wt/M2 PBS as 27%, and M2/M2 PBS as 50%. These data explain why close to 40% of viral DNA in the infected cells has the wild-type PBS sequence, yet only about 20% of the viruses have wild-type PBS, because both wt/M2 (27%) and M2/M2 (50%) viral DNA express the M2 viral RNA.
The M3 and M4 PBS mutants resist Cas9/sgPBS2.
Two PBS mutations, M3 and M4, were the most frequently identified in HIV-1 that had escaped from Cas9/sgPBS2 (Fig. 7A). The results of both short-term and long-term viral-infection experiments showed that M3 and M4 were refractory to Cas9/sgPBS2 inhibition (Fig. 7B and C). When we sequenced the entire genome of the viruses from the second round of replication in the Cas9/sgPBS2-expressing cells, we also found two mutations, a change of G to A at position 2557 (G2557A) in reverse transcriptase and G4405A in integrase. G2557A causes the amino acid substitution S3N at the beginning of RT, and G4405A leads to the mutation G59E in integrase (Fig. 7A). However, neither of these two mutations conferred resistance to Cas9/sgPBS2 (Fig. 7B and C). The MiSeq data showed that in the infected control SupT1 cells, similar levels of wild-type and M3 PBS sequences were detected following M3 virus infection, whereas 34% wild-type PBS and 61% M4 PBS were detected in M4 virus-infected control SupT1 cells (Fig. 7D). Similar to the results for M1 and M2 virus infection of control SupT1 cells, the percentages of viruses containing the wild-type PBS diminished significantly compared to the levels of wild-type PBS DNA in the cells that were infected by M3 or M4 viruses (Fig. 7D). Also, wild-type PBS DNA was greatly reduced in the Cas9/sgPBS2-expressing SupT1 cells that were infected by M3 or M4 virus (Fig. 7D).
FIG 7.

The M3 and M4 PBS mutations resist inhibition by Cas9/sgPBS2. (A) Illustration of the M3 and M4 mutations in the PBS. Also shown are mutations G2557A (S3N in RT) and G4405A (G59E in integrase [IN]), which were detected in viruses that escaped from Cas9/sgPBS2. (B) The M3 and M4 mutants were refractory to Cas9/sgPBS2 in short-term infections. Wild-type and mutated HIV-1 were used to infect control or Cas9/sgPBS2-expressing SupT1 cells. Forty hours later, the levels of infectious HIV-1 in the culture supernatants were determined by infecting the TZM-bl cells. Results shown are the average values from three independent infections. Levels of each virus from the control cells are set at 100. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) Replication of wild-type and mutated HIV-1 in SupT1 cells expressing Cas9/sgPBS2. Levels of viral reverse transcriptase activity were measured to monitor virus growth over time. Results shown represent two independent long-term infection experiments. (D) Percentages of wild-type and mutated viral PBS DNA sequences in the SupT1 cells that were infected with either the M3 or M4 mutant and in viruses thus produced, as determined by MiSeq analysis. Error bars show standard deviations.
HIV-1 evades Cas9/sgPBS3 by selecting for specific insertion sequences.
Finally, we sequenced HIV-1 that escaped from Cas9/sgPBS3 and found short insertions of 6 to 9 nucleotides at the cleavage site of Cas9 (Fig. 8). Viruses containing single-nucleotide changes in the PBS were not detected. Strikingly, these short insertions are not random sequences; they all restore the wild-type PBS, leaving a repeat of a partial PBS sequence immediately downstream, and thus disrupt the binding of sgPBS3 to its target DNA (Fig. 9A). These short insertions were not detected in the 46 clones of viral DNA that were collected from the short-term infection (Fig. 1G), indicating their rarity following NHEJ repair of the double-stranded DNA break caused by Cas9/sgPBS3. This also explains the relatively longer delay of HIV-1 growth under Cas9/sgPBS3 inhibition compared to the viral recovery from sgPBS1 or sgPBS2 inhibition (Fig. 3A), because it may have taken longer for the virus to capture these specific rare insertions to resist Cas9/sgPBS3 than to acquire the single- or double-nucleotide mutations that occur at relatively higher frequencies and are exploited by HIV-1 to resist Cas9/sgPBS1 and Cas9/sgPBS2. In addition to the NHEJ repair mechanism, these specific short insertions might have been generated by viral reverse transcriptase when copying the 3′-terminal 18 nucleotides of tRNA3Lys. Instead of continuously copying this tRNA sequence, reverse transcriptase may pause due to a local RNA structure and recopy a portion of the PBS sequence.
FIG 8.

Development of HIV-1 resistance to Cas9/sgPBS3. Viruses at the peaks of replication in Cas9/sgPBS3-expressing SupT1 cells were collected and sequenced. Indels in the PBS DNA were detected and are shown for the first, second, and third rounds of replication. The frequency of each indel in the sequenced DNA clones is also presented.
FIG 9.

The M5 and M6 PBS mutations resist Cas9/sgPBS3 inhibition. (A) Illustration of the M5 and M6 mutations (in red letters) in HIV-1 PBS. (B) Resistance of the M5 and M6 mutants to Cas9/sgPBS3 in short-term infections of SupT1 cells. Results shown are the average values from three independent infection experiments. ***, P < 0.001. (C) The M5 and M6 viruses replicate as efficiently in control SupT1 cells as in SupT1 cells expressing Cas9/sgPBS3. (D) Infection of control and Cas9/sgPBS3-expressing SupT1 cells by wild-type or M1 mutated viruses. Levels of viruses in the culture supernatants were measured by infecting the TZM-bl indicator cells. Levels of wild-type HIV-1 from the control cells are set at 100. Results shown are the average values from three independent infection experiments. ***, P < 0.001. (E) Indels in HIV-1 PBS DNA from the Cas9/sgPBS3-expressing SupT1 cells that were infected by the M1 mutated virus. Frequency of each indel is shown. (F) Cleavage of the reverse transcription products from the M1 RNA by Cas9/sgPBS3. The plus strand of the PBS DNA (shown as the top strand) is copied from the cellular tRNA3Lys, does not have the M1 mutation, is therefore recognized by sgPBS3, and is consequently cleaved by Cas9. Error bars show standard deviations.
We then tested two of these insertions, named M5 and M6, for their resistance to Cas9/sgPBS3 (Fig. 9A). The results of both short-term viral infection and long-term viral replication showed that M5 and M6 viruses infected the control and Cas9/sgPBS3-expressing SupT1 cells equally well (Fig. 9B and C). Given the greater difficulty of selecting the specific 6- to 9-nucleotide sequences compared to a single-nucleotide mutation at the Cas9 cleavage site, we speculated that such single-nucleotide mutations may not resist Cas9/sgPBS3 attack. To test this possibility, we used the M1 virus to infect the Cas9/sgPBS3-expressing SupT1 cells, since the M1 mutation changes the target DNA sequence for both sgPBS1 and sgPBS3. In contrast to the resistance of M1 virus to Cas9/sgPBS1, this virus mutant was inhibited by Cas9/sgPBS3 as much as the wild-type virus was (Fig. 9D). We further sequenced the M1 viral DNA in the infected Cas9/sgPBS3-expressing SupT1 cells and observed an array of indels that are characteristic of Cas9 cleavage (Fig. 9E). Therefore, single-nucleotide mutations at the Cas9 cleavage site do not render HIV-1 resistant to Cas9/sgPBS3.
The different responses of the M1 mutation to sgPBS1 and sgPBS3 are a result of the locations of their PAMs on different strands of viral DNA. The PAM of sgPBS1 is on the plus-strand DNA, whereas the PAM of sgPBS3 is on the minus strand (Fig. 9F). Therefore, sgPBS1 binds to the minus-strand DNA and sgPBS3 binds to the plus-strand DNA. During reverse transcription, the plus strand is copied from cellular tRNA3Lys and is always available for binding by sgPBS3, which supports Cas9 cleavage of viral DNA (Fig. 9F). In contrast, the minus strand is copied from the M1 viral RNA and does not support functional binding by sgPBS1. As a result, the M1 virus resists sgPBS1 but not sgPBS3. These data support the use of sgRNA that binds to the plus strand of the PBS DNA and, thus, guides Cas9 to cleave reverse transcription products even from viral RNA containing resistant indels.
DISCUSSION
In this study, we demonstrate that CRISPR-Cas9 can be programmed to target and cleave the HIV-1 PBS, thereby strongly inhibiting HIV-1 infection. Since more than 95% of the circulating HIV-1 strains use tRNA3Lys as the primer for reverse transcription (36, 37), the PBS-targeting sgRNA can guide Cas9 to inhibit the vast majority of HIV-1 strains. This provides one solution to the difficulty of designing sgRNAs to target highly divergent HIV-1 sequences. We noticed that, compared to targeting viral gene coding sequences, such as Gag, Tat, or Rev (24–27), targeting the PBS with Cas9/sgRNA caused a relatively shorter delay of HIV-1 replication, which necessitates a combinational use of PBS-targeting sgRNA with sgRNA targeting conserved viral genes to achieve long-term suppression of HIV-1 infection. It is somewhat expected that HIV-1 escapes from PBS-targeting Cas9/sgRNA, but it is intriguing that HIV-1 uses unique mechanisms to resist different PBS-targeting Cas9/sgRNAs. The mode of resistance depends on which strand of viral DNA is bound by the sgRNA; in other words, on which strand the PAM is located.
When the PAM is held within the plus strand of the PBS DNA, or the sgRNA binds to the minus-strand DNA, HIV-1 selects single- or dual-nucleotide mutations that are generated by the NHEJ complex when repairing the Cas9-cleaved PBS DNA (Fig. 10). These PBS mutations, for example, M1, M2, M3, and M4 in this study, do not markedly affect HIV-1 infection of SupT1 cells, likely because viral reverse transcriptase is able to tolerate the minimal mismatch between viral RNA and tRNA3Lys and start reverse transcription. In theory, PBS mutations should be quickly lost in HIV-1 replication due to the regeneration of the wild-type PBS sequence by copying the first 18 nucleotides of cellular tRNA3Lys (32–35). However, our study shows that HIV-1 takes advantage of several mechanisms to maintain these escape mutations in PBS and achieve wild-type-like replication in SupT1 cells that express Cas9 and sgPBS1 or sgPBS2.
FIG 10.
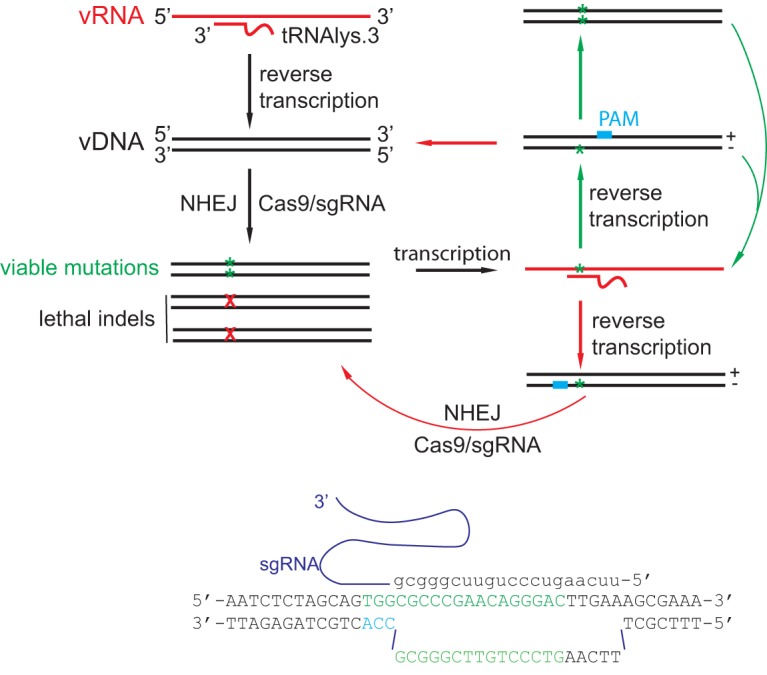
Illustration of the effect of PAM position in the PBS DNA on Cas9/sgRNA restriction of HIV-1 infection. If PAM is positioned on the plus strand of the PBS, HIV-1 is able to escape by acquiring and maintaining single- or double-nucleotide mutations in the PBS. If PAM is located on the minus strand of the PBS DNA, Cas9/sgRNA is able to target and cleave viral cDNA that is reverse transcribed from viral RNA that bears mutated PBS sequences. Based on these mechanisms, it can be predicted that a greater and more persistent suppression of HIV-1 infection can be achieved with a Cas9 variant using the CCA PAM that is on the minus strand of the PBS DNA and also allows the design of sgRNA with its seed region (14 to 15 nt) included in the PBS.
The primary target for these viral mechanisms is the reverse transcription product from the viral RNA carrying PBS mutations. The newly synthesized viral PBS DNA has the wild-type plus strand and the mutated minus strand (Fig. 10). This wt/mutant (mut) PBS hybrid DNA may be processed via three pathways. First, the mismatched nucleotides can be repaired by the cellular mismatch repair (MMR) mechanism (38), which may lead to an increase in the ratio of mut/mut PBS sequences compared to wt/wt PBS sequences, as was seen for the PBS mutations M1, M2, and M4 (Fig. 7 and 8). This biased outcome of MMR likely results from the transient presence of a nick at the 5′ end of viral DNA during integration (39), which guides the MMR machinery to use the strand without the nick (in this case, the minus strand) as the template for repair in eukaryotic cells (38). This nick-guided repair allows the inheritance of the genetic information held in the intact DNA strand, which acts as the template strand during cellular DNA replication. Second, the wt/mut PBS hybrid DNA may segregate into wt/wt and mut/mut DNA in a 50:50 fashion during cellular DNA duplication and cell division. Finally, some wt/mut PBS hybrids may not resolve and may remain as hybrid DNA. Alternatively, if transcription occurs before the wt/mut hybrid DNA is repaired, only the mut viral RNA will be synthesized. In the sgPBS-expressing cells, the wt/wt PBS DNA is cleaved by the Cas9/sgPBS complex, while the wt/mt and mut/mut PBS DNA resist and survive, and both are transcribed to produce viral RNA bearing the mutation, which gives rise to the resistant viruses.
However, when the PAM is located on the minus strand of the PBS DNA, HIV-1 cannot resist Cas9/sgPBS by mutating the PBS, because the newly synthesized wt/mut PBS DNA is recognized and cleaved by the Cas9/sgRNA as a result of the sgRNA binding to the wild-type plus-strand DNA (Fig. 10). The virus has two potential pathways to escape. One is to mutate the sequences adjacent to the PBS when a significant part of the target DNA is located outside the PBS, as was seen for sgPBS3 (Fig. 9). This weakness of sgPBS3 can be overcome by identifying a Cas9 variant that uses a CCA PAM, by which means 15 nucleotides of the target DNA, which covers the seed region of an sgRNA, can be included in the PBS (Fig. 10). Therefore, the resistance effect is expected to be minimal if HIV-1 mutates the target DNA sequence adjacent to the 3′ end of the PBS (Fig. 10). In addition, this Cas9 variant is expected to cut at the site which is 3 nucleotides from the 5′ end of the PBS. Indels at this site should inhibit HIV-1 infection much more strongly than indels at the downstream sites associated with Cas9/sgPBS that were tested in this study (Fig. 1B), because of the greater importance of the 5′ end of the PBS in tRNA annealing (40). We have examined the Cas9 variants that have been engineered to use alternative PAMs (41–46), and unfortunately, none uses CCA as the PAM. The second escape pathway is to use a cellular tRNA other than tRNA3Lys to prime viral reverse transcription. The genetic barrier for the latter escape pathway might be high, because HIV-1 reverse transcriptase and the viral 5′ untranslated region (UTR) also need to change in order to use a new tRNA primer (40, 47–51), which has rarely occurred in studies where the PBS was changed to match the sequences of different tRNAs (32–34).
We noticed a change of CCC to CCT in the viral PBS in the infected SupT1 cells that express Cas9/sgPBS (Fig. 1E and F, 8, and 9E). This nucleotide change could have been the result of virus using tRNA5Lys to initiate reverse transcription, as reported by Das et al. (36, 37). However, this C-to-T change was not selected by the virus to evade sgPBS1 or sgPBS2, likely because of the infrequent use of tRNA5Lys as the primer and/or the low level of resistance to sgPBS1 or sgPBS2 conferred by this mutation.
In summary, our study has generated a useful system to help advance the application of CRISPR-Cas9 for HIV-1 cure. We expect that the PBS sgRNA can be combined with sgRNAs targeting other conserved HIV-1 sequences to achieve efficient and persistent inactivation and excision of HIV-1 proviral DNA, a concept which has been demonstrated by recent studies showing ablation of HIV-1 infection in tissue culture with two select sgRNAs (26, 27). Along this line, a recent study also showed that the HIV-1 TAR sequence appears to be vulnerable to Cas9/sgRNA attack, since HIV-1 was unable to escape from the suppression by Cas9/sgRNA(TAR1) that targets the TAR (52). With the promising effect of CRISPR-Cas9 in cleaving HIV-1 DNA in the HIV-1-transgenic and HIV-1-infected mouse models (53, 54), the utility of this gene-editing system in curing HIV-1 infection is worth exploring further.
MATERIALS AND METHODS
Plasmids.
Guide RNA sequences sgPBS1, sgPBS2, and sgPBS3 were cloned into the plentiCRISPRv2 vector (catalog number 52961; Addgene) using the restriction enzyme BsmBI. The sequences of these sgRNAs are as follows: sgPBS1, 5′-TCT AGC AGT GGC GCC CGA AC-3′; sgPBS2, 5′-CTA GCA GTG GCG CCC GAA CA-3′; and sgPBS3, 5′-TCG CTT TCA AGT CCC TGT TC-3′. HIV-1 proviral DNA construct NL4-3 was obtained from the NIH AIDS Reagent Program. psPAX2 DNA (catalog number 12260) was purchased from Addgene. Mutations in HIV-1 DNA were generated using a PCR-based mutagenesis method (Clontech). The primers for each mutation are as follows: PBS-M1, 5′-GTG GAA AAT CTC TAG CAG TGG CGC CCG GAA CAG GGA CTT GAA AGC GAA AGT AAA G-3′/5′-CTT TAC TTT CGC TTT CAA GTC CCT GTT CCG GGC GCC ACT GCT AGA GAT TTT CCA C-3′; PBS-M2, 5′-GTG GAA AAT CTC TAG CAG TGG CGC CCG TTA ACA GGG ACT TGA AAG CGA AAG TAA AG-3′/5′-CTT TAC TTT CGC TTT CAA GTC CCT GTT AAC GGG CGC CAC TGC TAG AGA TTT TCC AC-3′; PBS-M3, 5′-GAA AAT CTC TAG CAG TGG CGC CCG ACA GGG ACT TGA AAG CGA AAG TA AAG-3′/5′-CTT TAC TTT CGC TTT CAA GTC CCT GTC GGG CGC CAC TGC TAG AGA TTT TC-3′; PBS-M4, 5′-GGA AAA TCT CTA GCA GTG GCG CCC GAG ACA GGG ACT TGA AAG CGA AAG TAA AG-3′/5′-CTT TAC TTT CGC TTT CAA GTC CCT GTC TCG GGC GCC ACT GCT AGA GAT TTT CC-3′; G2557A, 5′-GGC TGC ACT TTA AAT TTT CCC ATT AAT CCT ATT GAG ACT GTA CCA GTA AA-3′/5′-TTT ACT GGT ACA GTC TCA ATA GG ATT AAT GGG AAA ATT TAA AGT GCA GCC-3′; G4405A, 5′-GGA CAA GTA GAC TGT AGC CCA GAA ATA TGG CAG CTA GAT TGT ACA C-3′/5′-GTG TAC AAT CTA GCT GCC ATA TTT CTG GGC TAC AGT CTA CTT GTC C-3′; PBS-M5, 5′-GAA AAT CTC TAG CAG TGG CGC CCG AAC AGG GAC GCC AGG GAC TTG AAA GCG AAA GTA AAG C-3′/5′-GCT TTA CTT TCG CTT TCA AGT CCC TGG CGT CCC TGT TCG GGC GCC ACT GCT AGA GAT TTT C-3′; and PBS-M6, 5′-GAA AAT CTC TAG CAG TGG CGC CCG AAC AGG GAC AGG GAC TTG AAA GCG AAA GTA AAG C-3′/5′-GCT TTA CTT TCG CTT TCA AGT CCC TGT CCC TGT TCG GGC GCC ACT GCT AGA GAT TTT C-3′.
Cells.
HEK293T and TZM-bl cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). TZM-bl cells contain the HIV-1 LTR-luciferase reporter DNA cassette that is activated by HIV-1 Tat following HIV-1 infection (55). The CD4+ SupT1 cells were maintained in RPMI 1640 medium supplemented with 10% FBS.
Generation of stable cell lines.
The plentiCRISPRv2 DNA, which expresses Cas9 and sgPBS, was transfected into HEK293T cells together with the psPAX2 and pVSV-G DNA to produce lentiviral particles, which were used to infect the CD4+ SupT1 cells. Forty hours after infection, puromycin (2 μg/ml) was added to select for stably transduced cells. The expression of Cas9-FLAG was detected by immunostaining with DyLight 649-conjugated anti-FLAG antibodies (catalog number 600-443-383, 1:1,000 dilution; Rockland), followed by analysis using a flow cytometer (BD FACSCalibur).
HIV-1 production and infection.
HIV-1 was produced by transfecting HEK293T cells with the HIV-1 DNA clone NL4-3. Transfections were performed with polyethyleneimine (PEI) (catalog number 23966; Polysciences, Inc.). Viruses in the culture supernatants were collected and clarified by centrifugation to remove cell debris. The level of HIV-1 was determined either by measuring virion-associated viral p24 with an enzyme-linked immunosorbent assay (ELISA) or by infecting the TZM-bl indicator cells as we previously described (25).
For short-term infection, SupT1 cells were infected with HIV-1 equivalent to 100 ng of p24. The inoculated viruses were washed off 16 h postinfection. After a further 24-h infection, the number of infected SupT1 cells was determined by immunostaining of viral p24 using fluorescein isothiocyanate (FITC)-conjugated anti-p24 antibodies (catalog number 6604665, 1:200 dilution; Beckman Coulter), followed by flow cytometry analysis. The levels of viruses that were produced in the supernatants were quantified by measuring viral reverse transcriptase activity or by infecting the TZM-bl indicator cells as we previously described (25). The levels of luciferase activity of the infected TZM-bl cells reflect the amounts of infectious HIV-1 particles.
For long-term infection, HIV-1 equivalent to 5 ng viral p24 was used to infect SupT1 cells. Viral growth was monitored by measuring the levels of viral reverse transcriptase activity in the culture supernatants at various time intervals. All HIV-1 infection experiments were performed in the certified biocontainment level 3 laboratory at the Lady Davis Institute.
Sequencing the HIV-1 genome.
RNA was extracted from HIV-1 particles that were harvested from either the short-term or long-term infections by ultracentrifugation at 10,000 × g. Viral DNA was amplified from viral RNA by RT-PCR using the OneTaq one-step RT-PCR kit (catalog number E5315S; NEB) and cloned using the NEB PCR cloning kit (catalog number E1203S; NEB). On average, 40 or more DNA clones for each RT-PCR product were prepared and sequenced by MCLAB (San Francisco, CA). The PBS region was amplified using primer pair 5′-GAG CCC TCA GAT GCT ACA TA-3′ and 5′-TAG CTC CCT GCT TGC CCA TA-3′.
To sequence HIV-1 DNA from the infected SupT1 cells, total DNA was first extracted using the DNeasy blood and tissue kit (catalog number 69504; Qiagen). To sequence the entire HIV-1 genomes, four pairs of primers were used to amplify four segments of viral DNA as we described previously (56). These four primer pairs are as follows: 5′-GCA GTG GCG CCC GAA CAG GGA C-3′/5′-CGC TGC CAA AGA GTG ATC T-3′ (to amplify nt positions 632 to 2275, referring to the genome sequence of strain NL4-3 with GenBank accession number AF324493); 5′-GCA GGG CCC CTA GGA AAA AGG-3′/5′-CCT ATT CTG CTA TGT CGA CAC C-3′ (to amplify nt positions 2003 to 5803); 5′-TAA AGC CAC CTT TGC CTA G-3′/5′-CTA AGG ATC CGT TCA CTA ATC GAA TGG-3′ (to amplify nt positions 5516 to 8474); and 5′-GGA GGC TTG GTA GGT TTA A-3′/5′-TGC TAG AGA TTT TCC ACA CTG AC-3′ (to amplify nt positions 8282 to 9709). The PCR products were cloned and sequenced.
To amplify the integrated HIV-1 DNA, Alu-PCR was performed as described previously (57), starting with the first PCR using primers Alu forward (5′-GCC TCC CAA ACT GCT GGG ATT ACA G-3′) and HIV gag reverse (5′-GTT CCT GCT ATG TCA CTT CC-3′), followed by a second PCR with primers LTR (R) forward (5′-TTA AGC CTC AAT AAA GCT TGC C-3′) and LTR (U5) reverse (5′-GTT CGG GCG CCA CTG CTA GA-3′). To eliminate the plasmid DNA that may associate with HIV-1 particles that were used for long-term or short-term infections, virus stocks were incubated with DNase I (catalog number 18047-019, 200 U/μl; Invitrogen) in the presence of 2 mM Mg2Cl at 37°C for 30 min before infecting SupT1 cells. One infection was performed in the presence of the HIV-1 RT inhibitor nevirapine (used at 2 μM; NIH AIDS Reagent Program). The results controlled for the contamination of plasmid DNA in the subsequent PCR.
Sequencing tRNA3Lys genes.
Total DNA was extracted from SupT1 cells that expressed Cas9, Cas9/sgPBS1, Cas9/sgPBS2, or Cas9/sgPBS3 using the DNeasy blood and tissue kit (catalog number 69504; Qiagen). Primers were designed to amplify the following six tRNA3Lys genes: tRNALys-TTT-3-1 gene (GenBank accession numbers NC_000001.11 and HG983931.1), 5′-TGG CGA CAG AGT AAG ACA CCA T-3′/5′-TTA CGA TGT CCG CTC TTG GGA CTT-3′; tRNALys-TTT-3-2 gene (NC_000001.11 and HG983932.1), 5′-GGT ATG CGA GGT AAT GCA TAA CC-3′/5′-AGT CTC ACA GTG TGG ATG ATG C-3′; tRNALys-TTT-3-3 gene (NC_000006.12 and HG983935.1), 5′-CTC ACT AGA ATG TGC AGG AAG-3′/5′-GAA TCA GGT ACT AAA AGC CAC CAC TC-3′; tRNALys-TTT-3-4 gene (NC_000011.10 and HG983933.1), 5′-CGC AGT TTA CAC AAT CCA GG-3′/5′-CAC GGC ATA ATG GTA ACA GAG-3′; tRNALys-TTT-3-5 gene (NC_000017.11 and HG983934.1), 5′-ATC CTC TGT CGG TTC TAC AAG-3′/5′-TTA CCC TCC ACC GTC GTT TGC TGT A-3′; and tRNALys-TTT-5-1 gene (NC_000011.10 and HG983937.1), 5′-TAA TCG CGA AGG GAA AGA ATG CGG-3′/5′-CCC AAG CGG GTT TCA CAT AA-3′.
PCR products were recovered from agarose gels using a gel extraction kit (catalog number D2500-02; Omega) and sent to MCLAB (San Francisco, CA) for sequencing. The PCR products of the tRNALys-TTT-3-5 gene were also cloned using a PCR cloning kit (catalog number E1203S; NEB), and 24 DNA clones were prepared and sequenced by MCLAB.
Illumina MiSeq.
HIV-1 DNA or RNA was amplified using primer pair 5′-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG GTG GCG AGC CCT CAG ATG CTA C-3′/5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GTT GGC GTA CTC ACC AGT CGC CG-3′. The sequences in italics were used for a second round of PCR with primers containing barcode sequences. The PCR products were extracted from agarose gels using the gel extraction kit (catalog number D2500-02; Omega) and sent to the Genomics Platform at IRIC (Institute for Research in Immunology and Cancer, University of Montreal) for processing with MiSeq reagent nanokit v2 (500 cycles) (Illumina). The average number of reads per DNA sample was 8,000. The MiSeq results were analyzed as previously described (25). In brief, sequences were demultiplexed and isolated from PhiX using bcl2fastq. The program Trimmomatic (version 0.32) was used to remove sequencing adapters and low-quality bases from the 3′ end. Sequenced pairs of reads were combined into single overlapping fragments using FLASH version 1.2.11 and inspected to identify the various populations of variants in each sample. km software (https://www.biorxiv.org/content/early/2018/04/17/295808) was used to reconstruct all observed variants, as well as quantify their frequencies of occurrence with respect to the viral reference sequences. Each viral infection was performed in triplicate. The amplified viral DNA from three infections was all analyzed by MiSeq.
Statistical analysis.
P values were calculated using the unpaired two-tailed t test, based on data from three or more independent experiments. A P value of <0.05 was deemed statistically significant.
ACKNOWLEDGMENTS
This work was supported by CIHR operating grant IBC150405 to C.L.
We thank Lambert Raphaelle and Jennifer Huber for the Illumina MiSeq service. We are indebted to the late Mark A. Wainberg for his support of this work and for his lifelong dedication to HIV research.
REFERENCES
- 1.Dahabieh MS, Battivelli E, Verdin E. 2015. Understanding HIV latency: the road to an HIV cure. Annu Rev Med 66:407–421. doi: 10.1146/annurev-med-092112-152941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karpinski JHI, Chemnitz J, Schäfer C, Paszkowski-Rogacz M, Chakraborty D, Beschorner N, Hofmann-Sieber H, Lange UC, Grundhoff A, Hackmann K, Schrock E, Abi-Ghanem J, Pisabarro MT, Surendranath V, Schambach A, Lindner C, van Lunzen J, Hauber J, Buchholz F. 2016. Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity. Nat Biotechnol 34:401–409. doi: 10.1038/nbt.3467. [DOI] [PubMed] [Google Scholar]
- 3.Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, Guschin DY, Rupniewski I, Waite AJ, Carpenito C, Carroll RG, Orange JS, Urnov FD, Rebar EJ, Ando D, Gregory PD, Riley JL, Holmes MC, June CH. 2008. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, Crooks GM, Kohn DB, Gregory PD, Holmes MC, Cannon PM. 2010. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol 28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, Cost GJ, Zhang L, Santiago Y, Miller JC, Zeitler B, Cherone JM, Meng X, Hinkley SJ, Rebar EJ, Gregory PD, Urnov FD, Jaenisch R. 2011. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol 29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ. 2011. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 7.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schiffer JT, Aubert M, Weber ND, Mintzer E, Stone D, Jerome KR. 2012. Targeted DNA mutagenesis for the cure of chronic viral infections. J Virol 86:8920–8936. doi: 10.1128/JVI.00052-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wright AV, Nunez JK, Doudna JA. 2016. Biology and applications of CRISPR systems: harnessing nature's toolbox for genome engineering. Cell 164:29–44. doi: 10.1016/j.cell.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 11.Ebina H, Misawa N, Kanemura Y, Koyanagi Y. 2013. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep 3:2510. doi: 10.1038/srep02510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu W, Kaminski R, Yang F, Zhang Y, Cosentino L, Li F, Luo B, Alvarez-Carbonell D, Garcia-Mesa Y, Karn J, Mo X, Khalili K. 2014. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A 111:11461–11466. doi: 10.1073/pnas.1405186111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaminski R, Chen Y, Fischer T, Tedaldi E, Napoli A, Zhang Y, Karn J, Hu W, Khalili K. 2016. Elimination of HIV-1 genomes from human T-lymphoid cells by CRISPR/Cas9 gene editing. Sci Rep 6:22555. doi: 10.1038/srep22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao HK, Gu Y, Diaz A, Marlett J, Takahashi Y, Li M, Suzuki K, Xu R, Hishida T, Chang CJ, Esteban CR, Young J, Izpisua Belmonte JC. 2015. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat Commun 6:6413. doi: 10.1038/ncomms7413. [DOI] [PubMed] [Google Scholar]
- 15.Zhu W, Lei R, Le Duff Y, Li J, Guo F, Wainberg MA, Liang C. 2015. The CRISPR/Cas9 system inactivates latent HIV-1 proviral DNA. Retrovirology 12:22. doi: 10.1186/s12977-015-0150-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Quake SR. 2014. RNA-guided endonuclease provides a therapeutic strategy to cure latent herpesviridae infection. Proc Natl Acad Sci U S A 111:13157–13162. doi: 10.1073/pnas.1410785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuen KS, Chan CP, Wong NH, Ho CH, Ho TH, Lei T, Deng W, Tsao SW, Chen H, Kok KH, Jin DY. 2015. CRISPR/Cas9-mediated genome editing of Epstein-Barr virus in human cells. J Gen Virol 96:626–636. doi: 10.1099/jgv.0.000012. [DOI] [PubMed] [Google Scholar]
- 18.van Diemen FR, Kruse EM, Hooykaas MJ, Bruggeling CE, Schurch AC, van Ham PM, Imhof SM, Nijhuis M, Wiertz EJ, Lebbink RJ. 2016. CRISPR/Cas9-mediated genome editing of herpesviruses limits productive and latent infections. PLoS Pathog 12:e1005701. doi: 10.1371/journal.ppat.1005701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin SR, Yang HC, Kuo YT, Liu CJ, Yang TY, Sung KC, Lin YY, Wang HY, Wang CC, Shen YC, Wu FY, Kao JH, Chen DS, Chen PJ. 2014. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol Ther Nucleic Acids 3:e186. doi: 10.1038/mtna.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seeger C, Sohn JA. 2014. Targeting hepatitis B virus with CRISPR/Cas9. Mol Ther Nucleic Acids 3:e216. doi: 10.1038/mtna.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kennedy EM, Bassit LC, Mueller H, Kornepati AV, Bogerd HP, Nie T, Chatterjee P, Javanbakht H, Schinazi RF, Cullen BR. 2015. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology 476:196–205. doi: 10.1016/j.virol.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bi Y, Sun L, Gao D, Ding C, Li Z, Li Y, Cun W, Li Q. 2014. High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog 10:e1004090. doi: 10.1371/journal.ppat.1004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price AA, Grakoui A, Weiss DS. 2016. Harnessing the prokaryotic adaptive immune system as a eukaryotic antiviral defense. Trends Microbiol 24:294–306. doi: 10.1016/j.tim.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang G, Zhao N, Berkhout B, Das AT. 2016. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol Ther 24:522. doi: 10.1038/mt.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Pan Q, Gendron P, Zhu W, Guo F, Cen S, Wainberg MA, Liang C. 2016. CRISPR/Cas9-derived mutations both inhibit HIV-1 replication and accelerate viral escape. Cell Rep 15:481–489. doi: 10.1016/j.celrep.2016.03.042. [DOI] [PubMed] [Google Scholar]
- 26.Wang G, Zhao N, Berkhout B, Das AT. 2016. A combinatorial CRISPR-Cas9 Attack on HIV-1 DNA extinguishes all infectious provirus in infected T cell cultures. Cell Rep 17:2819–2826. doi: 10.1016/j.celrep.2016.11.057. [DOI] [PubMed] [Google Scholar]
- 27.Lebbink RJ, de Jong DC, Wolters F, Kruse EM, van Ham PM, Wiertz EJ, Nijhuis M. 2017. A combinational CRISPR/Cas9 gene-editing approach can halt HIV replication and prevent viral escape. Sci Rep 7:41968. doi: 10.1038/srep41968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dampier W, Sullivan NT, Chung CH, Mell JC, Nonnemacher MR, Wigdahl B. 2017. Designing broad-spectrum anti-HIV-1 gRNAs to target patient-derived variants. Sci Rep 7:14413. doi: 10.1038/s41598-017-12612-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu WS, Hughes SH. 2012. HIV-1 reverse transcription. Cold Spring Harb Perspect Med 2:a006882. doi: 10.1101/cshperspect.a006882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das AT, Brummelkamp TR, Westerhout EM, Vink M, Madiredjo M, Bernards R, Berkhout B. 2004. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J Virol 78:2601–2605. doi: 10.1128/JVI.78.5.2601-2605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schorn AJ, Gutbrod MJ, LeBlanc C, Martienssen R. 2017. LTR-retrotransposon control by tRNA-derived small RNAs. Cell 170:61–71e11. doi: 10.1016/j.cell.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das AT, Klaver B, Berkhout B. 1995. Reduced replication of human immunodeficiency virus type 1 mutants that use reverse transcription primers other than the natural tRNA(3Lys). J Virol 69:3090–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Mak J, Arts EJ, Gu Z, Kleiman L, Wainberg MA, Parniak MA. 1994. Effects of alterations of primer-binding site sequences on human immunodeficiency virus type 1 replication. J Virol 68:6198–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wakefield JK, Wolf AG, Morrow CD. 1995. Human immunodeficiency virus type 1 can use different tRNAs as primers for reverse transcription but selectively maintains a primer binding site complementary to tRNA(3Lys). J Virol 69:6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang C, Rong L, Morin N, Cherry E, Huang Y, Kleiman L, Wainberg MA. 1997. The roles of the human immunodeficiency virus type 1 Pol protein and the primer binding site in the placement of primer tRNA(3Lys) onto viral genomic RNA. J Virol 71:9075–9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das AT, Klaver B, Berkhout B. 1997. Sequence variation of the human immunodeficiency virus primer-binding site suggests the use of an alternative tRNA(Lys) molecule in reverse transcription. J Gen Virol 78(Pt 4):837–840. doi: 10.1099/0022-1317-78-4-837. [DOI] [PubMed] [Google Scholar]
- 37.Das AT, Vink M, Berkhout B. 2005. Alternative tRNA priming of human immunodeficiency virus type 1 reverse transcription explains sequence variation in the primer-binding site that has been attributed to APOBEC3G activity. J Virol 79:3179–3181. doi: 10.1128/JVI.79.5.3179-3181.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Modrich P. 2016. Mechanisms in E. coli and human mismatch repair (Nobel Lecture). Angew Chem Int Ed Engl 55:8490–8501. doi: 10.1002/anie.201601412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Craigie R, Bushman FD. 2012. HIV DNA integration. Cold Spring Harb Perspect Med 2:a006890. doi: 10.1101/cshperspect.a006890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oude Essink BB, Das AT, Berkhout B. 1995. Structural requirements for the binding of tRNA Lys3 to reverse transcriptase of the human immunodeficiency virus type 1. J Biol Chem 270:23867–23874. doi: 10.1074/jbc.270.40.23867. [DOI] [PubMed] [Google Scholar]
- 41.Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z, Liu DR. 2018. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, Joung JK. 2015. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol 33:1293–1298. doi: 10.1038/nbt.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales AP, Li Z, Peterson RT, Yeh JR, Aryee MJ, Joung JK. 2015. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishimasu H, Yamano T, Gao L, Zhang F, Ishitani R, Nureki O. 2017. Structural basis for the altered PAM recognition by engineered CRISPR-Cpf1. Mol Cell 67:139–147e132. doi: 10.1016/j.molcel.2017.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao L, Cox DBT, Yan WX, Manteiga JC, Schneider MW, Yamano T, Nishimasu H, Nureki O, Crosetto N, Zhang F. 2017. Engineered Cpf1 variants with altered PAM specificities. Nat Biotechnol 35:789–792. doi: 10.1038/nbt.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anders C, Bargsten K, Jinek M. 2016. Structural plasticity of PAM recognition by engineered variants of the RNA-guided endonuclease Cas9. Mol Cell 61:895–902. doi: 10.1016/j.molcel.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beerens N, Klaver B, Berkhout B. 2000. A structured RNA motif is involved in correct placement of the tRNA(3)(Lys) primer onto the human immunodeficiency virus genome. J Virol 74:2227–2238. doi: 10.1128/JVI.74.5.2227-2238.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beerens N, Groot F, Berkhout B. 2001. Initiation of HIV-1 reverse transcription is regulated by a primer activation signal. J Biol Chem 276:31247–31256. doi: 10.1074/jbc.M102441200. [DOI] [PubMed] [Google Scholar]
- 49.Liang C, Li X, Rong L, Inouye P, Quan Y, Kleiman L, Wainberg MA. 1997. The importance of the A-rich loop in human immunodeficiency virus type 1 reverse transcription and infectivity. J Virol 71:5750–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Isel C, Ehresmann C, Marquet R. 2010. Initiation of HIV reverse transcription. Viruses 2:213–243. doi: 10.3390/v2010213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abbink TE, Beerens N, Berkhout B. 2004. Forced selection of a human immunodeficiency virus type 1 variant that uses a non-self tRNA primer for reverse transcription: involvement of viral RNA sequences and the reverse transcriptase enzyme. J Virol 78:10706–10714. doi: 10.1128/JVI.78.19.10706-10714.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mefferd AL, Bogerd HP, Irwan ID, Cullen BR. 2018. Insights into the mechanisms underlying the inactivation of HIV-1 proviruses by CRISPR/Cas. Virology 520:116–126. doi: 10.1016/j.virol.2018.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yin C, Zhang T, Qu X, Zhang Y, Putatunda R, Xiao X, Li F, Xiao W, Zhao H, Dai S, Qin X, Mo X, Young WB, Khalili K, Hu W. 2017. In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol Ther 25:1168–1186. doi: 10.1016/j.ymthe.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaminski R, Bella R, Yin C, Otte J, Ferrante P, Gendelman HE, Li H, Booze R, Gordon J, Hu W, Khalili K. 2016. Excision of HIV-1 DNA by gene editing: a proof-of-concept in vivo study. Gene Ther 23:696. doi: 10.1038/gt.2016.45. [DOI] [PubMed] [Google Scholar]
- 55.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang C, Rong L, Russell RS, Wainberg MA. 2000. Deletion mutagenesis downstream of the 5′ long terminal repeat of human immunodeficiency virus type 1 is compensated for by point mutations in both the U5 region and gag gene. J Virol 74:6251–6261. doi: 10.1128/JVI.74.14.6251-6261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu JJ, Wu TL, Liszewski MK, Dai J, Swiggard WJ, Baytop C, Frank I, Levine BL, Yang W, Theodosopoulos T, O'Doherty U. 2008. A more precise HIV integration assay designed to detect small differences finds lower levels of integrated DNA in HAART treated patients. Virology 379:78–86. doi: 10.1016/j.virol.2008.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]