

Macrophages and dendritic cells are usually the first point of contact with pathogens, including lentiviruses. Host restriction factors, including SAMHD1, mediate the innate immune response against these viruses. However, HIV-1 has evolved to circumvent the innate immune response and establishes disseminated infection. The cyclin-dependent kinase inhibitor p21, which is involved in differentiation and maturation of monocytes, blocks HIV-1 replication at the reverse transcription step. p21 is thought to suppress key enzymes involved in dNTP biosynthesis and activates SAMHD1 antiviral function. We report here that the human USP18 protein is a novel factor potentially contributing to HIV replication by blocking the antiviral function of p21 in differentiated human myeloid cells. USP18 downregulates p21 protein expression, which correlates with upregulated intracellular dNTP levels and the antiviral inactive form of SAMHD1. Depletion of USP18 stabilizes p21 protein expression, which correlates with dephosphorylated SAMHD1 and a block to HIV-1 replication.
KEYWORDS: THP-1, USP18, human immunodeficiency virus 1 (HIV-1), p21, restriction factor
ABSTRACT
The host intrinsic innate immune system drives antiviral defenses and viral restriction, which includes the production of soluble factors, such as type I and III interferon (IFN), and activation of restriction factors, including SAMHD1, a deoxynucleoside triphosphohydrolase. Interferon-stimulated gene 15 (ISG15)-specific ubiquitin-like protease 43 (USP18) abrogates IFN signaling pathways. The cyclin-dependent kinase inhibitor p21 (CIP1/WAF1), which is involved in the differentiation and maturation of monocytes, inhibits human immunodeficiency virus type 1 (HIV-1) in macrophages and dendritic cells. p21 inhibition of HIV-1 replication is thought to occur at the reverse transcription step, likely by suppressing cellular deoxynucleoside triphosphate (dNTP) biosynthesis and increasing the amount of antivirally active form of SAMHD1. SAMHD1 strongly inhibits HIV-1 replication in myeloid and resting CD4+ T cells. Here, we studied how USP18 influences HIV-1 replication in human myeloid THP-1 cells. We found that USP18 has the novel ability to inhibit the antiviral function of p21 in differentiated THP-1 cells. USP18 enhanced reverse transcription of HIV-1 by downregulating p21 expression and upregulating intracellular dNTP levels. p21 downregulation by USP18 was associated with the active form of SAMHD1, phosphorylated at T592. USP18 formed a complex with the E3 ubiquitin ligase recognition factor SKP2 (S-phase kinase associated protein 2) and SAMHD1. CRISPR-Cas9 knockout of USP18 increased p21 protein expression and blocked HIV-1 replication. Overall, we propose USP18 as a regulator of p21 antiviral function in differentiated myeloid THP-1 cells.
IMPORTANCE Macrophages and dendritic cells are usually the first point of contact with pathogens, including lentiviruses. Host restriction factors, including SAMHD1, mediate the innate immune response against these viruses. However, HIV-1 has evolved to circumvent the innate immune response and establishes disseminated infection. The cyclin-dependent kinase inhibitor p21, which is involved in differentiation and maturation of monocytes, blocks HIV-1 replication at the reverse transcription step. p21 is thought to suppress key enzymes involved in dNTP biosynthesis and activates SAMHD1 antiviral function. We report here that the human USP18 protein is a novel factor potentially contributing to HIV replication by blocking the antiviral function of p21 in differentiated human myeloid cells. USP18 downregulates p21 protein expression, which correlates with upregulated intracellular dNTP levels and the antiviral inactive form of SAMHD1. Depletion of USP18 stabilizes p21 protein expression, which correlates with dephosphorylated SAMHD1 and a block to HIV-1 replication.
INTRODUCTION
Cellular antiretroviral restriction factors are an important component of the innate immune response against HIV-1 (1–3). Many of these proteins are induced or activated by interferons (IFNs) (4–6). These proteins can inhibit retroviral replication at different stages of the viral life cycle. However, viruses have evolved mechanisms that antagonize the restriction abilities of these factors (1, 2). For example, viral proteins such as HIV-1 Vif target APOBEC3 proteins for proteasomal degradation (1, 2), and VPX of the human immunodeficiency virus type 2 (HIV-2)/simian immunodeficiency virus (SIV) can induce depletion of SAMHD1 (7–9). VPX loads SAMHD1 onto an E3 ubiquitin ligase complex, cullin ring-finger ligase 4 (CRL4)-DDB1, via the substrate adapter cullin-associated factor 1 (DCAF1) to initiate its proteasomal degradation (7, 10–13). This results in a drastic increase in the intracellular deoxynucleoside triphosphate (dNTP) pool to a level required for efficient reverse transcription (14, 15).
In vitro, quiescent CD4+ T cells, myeloid cells (including macrophages and dendritic cells), and phorbol 12-myristate 13-acetate (PMA)-differentiated THP-1 cells, express relatively high levels of SAMHD1 and restrict HIV-1 replication, likely because the deoxynucleoside triphosphohydrolase (dNTPase) activity of SAMHD1 leads to ablation of viral reverse transcription (14, 16–20). However, proliferating cells are highly permissive for infection and replication of HIV-1 (21–23). Together with upregulation of ribonucleotide reductase (RNR), downregulation of SAMHD1 expression and dNTPase activity during the S phase was considered a characteristic feature of proliferating cells (24, 25) until recently; Schott et al. (in 2018) and Yan et al. (in 2015) reported that SAMHD1 levels remain relatively unchanged during the cell cycle (26, 27).
The restriction ability of SAMHD1 is thought to be modulated in proliferating cells by phosphorylation at residue T592 by the cyclin A2/CDK1/2 complex and by dephopshorylation by the phosphatase PP2A-B55α during mitotic exit (21, 27–30). Intriguingly, the T592E mutant of SAMHD1, which mimics the phosphorylated form, is unable to restrict HIV-1 despite maintaining its ability to deplete the intracellular dNTP pool (28, 31). This finding suggests that SAMHD1 dNTPase activity is not sufficient to confer inhibition to retroviral replication. Thus, beyond its dNTPase activity, additional unidentified cellular regulatory mechanisms of SAMHD1 may be involved in its restriction of retroviruses.
Indeed, efficient replication of HIV-1 requires a sufficiently large cellular dNTP pool (32, 33), which is likely sustained by various cellular pathways (34, 35). De novo pathways synthesize new dNTPs, while salvage pathways recover nucleotides and their components from extracellular media and intracellular DNA degradation (34, 35). As a result, cycling cells exhibit higher dNTP levels compared to noncycling cells (15, 27, 34, 35). The CDK inhibitor p21 is one of the cellular factors regulating dNTP biosynthesis and is itself regulated during cell cycle. In cycling cells, p21 can be targeted for proteasomal degradation by the S-phase-associated protein 2 (SKP2) in a complex with cyclin A/E and CDK2 (36, 37). p21 blocks dNTP biosynthesis in monocyte-derived macrophages and dendritic cells by downmodulating the expression of the RNR2 subunit of ribonucleotide reductase, which is essential for the reduction of ribonucleotides to deoxynucleotides (23, 25, 38). On the other hand, the ability of p21 to inhibit cyclin/CDK activities likely regulates SAMHD1 antiviral activity by prohibiting the cyclinA/CDK1/2 phosphorylation of SAMHD1 at T592 (21, 22, 30, 39), suggesting that p21 regulates both de novo dNTP synthesis and the antiviral function of SAMHD1 (23, 39, 40).
Ubiquitin-like specific protease 18 (USP18, UBP43) is a cysteine protease that cleaves ISG15 (interferon-stimulated gene 15, a 17-kDa protein) from its conjugated targets. USP18 exerts both protease-dependent and -independent functions to balance immune responses in disease and nondisease states (41–46). USP18 is induced by IFNs, lipopolysaccharide, and viral infections and can modulate type I IFN responses (42). It acts as a negative regulator of NF-κB (nuclear factor “kappa-light-chain-enhancer” of activated B cells) activation by inhibiting ubiquitination of TAK1 (TGF-β-activated kinase 1) and NEMO (NF-κB essential modulator) (47, 48). USP18 binds to IFNAR2 (IFN receptor 2) and, in an isopeptidase-independent manner, blocks IFN signaling by disrupting IFNAR2-JAK (Janus-activated kinase) binding (42, 49). In the absence of free ISG15, SKP2 promotes USP18 ubiquitination and degradation by the proteasome (50, 51).
Experimental knockout of USP18 enhances JAK/STAT (signal transducer and activator of transcription) signaling and increases ISGs with elevated levels of protein ISGylation, thus providing resistance to viral infections (3, 42, 43, 52). Recent work by Honke et al. (49) showed in 2012 that high expression of USP18 in murine CD169+ macrophages in the splenic marginal zone was required to enforce a local replication of vesicular stomatitis virus (VSV), a negative-sense, single-stranded enveloped RNA virus belonging to the family Rhabdoviridae. The enforced viral replication was essential to provide adequate antigens for stimulation of a robust adaptive immunity to control the cytopathic virus infection (49). CD169+ macrophages and dendritic cells are known targets of HIV-1 (53); we therefore sought to determine whether human USP18 might be a factor that influences HIV-1 replication in macrophages by using the monocyte-derived macrophage cell line THP-1 as a model.
RESULTS
USP18 is HIV-1 inducible, and its expression enhances viral replication in differentiated THP-1 cells.
To evaluate the role of USP18 in HIV-1 infection, we generated THP-1 cells expressing USP18 at levels similar to those induced by IFN-β (Fig. 1A). HIV-1 infection upregulated USP18 expression, and this upregulation was even more robust in the presence of copackaged VPX (Fig. 1B). Expression of USP18 in undifferentiated THP-1 cells increased HIV-1 infection by up to 11-fold in the absence of type I IFN compared to the THP-1 control cells (Fig. 1C and D). The dose-dependent repression of HIV-1 by IFN-α (Fig. 1C) and IFN-β (Fig. 1D) in USP18-expressing THP-1 cells (THP-1.USP18) was significantly reduced compared to that of control cells (THP-1.Control). After PMA-induced differentiation, THP-1.USP18 and THP-1.Controls were transduced with a VPX-containing HIV-1 luciferase reporter virus produced in HEK293T cells, which had the ability to degrade SAMHD1 (Fig. 1E). In the presence of VPX, differentiated THP-1.USP18 cells showed significantly increased HIV-1 infection compared to THP-1.Control cells (Fig. 1F). Surprisingly, in the absence of VPX, USP18 overcame the SAMHD1 restriction, resulting in a >40-fold increase in HIV-1 infection (Fig. 1F). Thus, USP18 expression in the differentiated THP-1 cells mimicked the viral VPX function and allowed for higher infection even in the presence of increasing concentrations of IFN-α (Fig. 1G) and IFN-β (Fig. 1H) in the absence of VPX compared to the THP-1.Control cells.
FIG 1.

HIV-1 induces USP18 and USP18 expression enhances HIV-1 infection in PMA-differentiated THP-1 cells. (A) Immunoblot analysis of basal, IFN-β-induced, and stably expressing USP18 levels in THP-1 cells. (B) PMA-differentiated THP-1 cells were mock transduced (media) or HIV-1 transduced with or without copackaged VPX. At 72 h postransduction, cells were harvested and immunoblotted for endogenous total and phosphorylated SAMHD1, USP18, and GAPDH as a loading control. THP-1.USP18 and THP-1.Control cells were treated with different concentrations of IFN-α (C) and IFN-β (D) for 4 h and subsequently transduced with single-round HIV-1 luciferase reporter virus (E). At 72 h postransduction, the luciferase activity was measured. (F) In a related experiment, these cells were PMA differentiated for 48 h and subsequently transduced with HIV-1 luciferase reporter virus with or without the copackaged VPX produced in panel E, which had the ability to deplete SAMHD1. The same experiment was reproduced in the presence of different concentrations of IFN-α (G) and IFN-β (H). Mean differences of three replicates for each group in a single experiment was analyzed and compared between the groups by using a Student t test, with results expressed as means ± the SD. A P value of <0.05 was considered statistically significant (*). The higher the number of asterisks, the lower the P value. Each panel is representative of at least three independent experiments.
USP18-mediated increase in HIV-1 infection is independent of USP18 isopeptidase activity.
To test whether the USP18-dependent enhanced HIV-1 replication was a result of USP18 isopeptidase activity, we mutated the catalytic site residue, cysteine 64 to either alanine (A) or serine (S) (Fig. 2A). We tested the reactivity of USP18 and its mutants toward the catalytic core of ISG15 in an ISG15-vinyl sulfone (VS) probe. The wild-type (WT) USP18 reacted strongly with the catalytic core of ISG15, as indicated by an upward shift in the USP18 39-kDa band toward a size of about 72 kDa (Fig. 2B). As expected, the catalytic site mutants lost this enzymatic activity (Fig. 2B). However, like the WT USP18, the two active site mutants retained the capacity to enhance permissiveness to HIV-1 in undifferentiated (Fig. 2C) and differentiated (Fig. 2D) THP-1 cells. To test whether this effect of USP18 was specific to HIV-1 or general to lentiviruses, we transduced undifferentiated and PMA-differentiated THP-1.Control, WT USP18, and mutant USP18 cells with HIV-2-WT and HIV-2Δvpx luciferase reporter viruses. We observed significantly higher HIV-2 infection in the undifferentiated WT and mutant USP18 cells compared to the THP-1.Control cells (Fig. 2E). As with HIV-1, both USP18 WT and mutant proteins increased HIV-2 infection in PMA-differentiated THP-1 cells by >7-fold (Fig. 2F) and additionally increased cell permissiveness in the presence of VPX (Fig. 2G).
FIG 2.

USP18 enhancement of HIV-1 infection is independent of USP18's isopeptidase activity. (A) Immunoblot of endogenous USP18 in THP-1 cells stably expressing pLOC empty vector control (CTL), WT USP18, and active site mutants (C64A, C64S), where the cysteine (C) at position 64 was mutated to alanine (A) or serine (S). (B) HA-tagged WT USP18 and its mutants were overexpressed in HEK293T cells, and protein lysates were incubated with ISG15-VS probe. Isopeptidase activity of USP18 toward ISG15 was revealed by the ISG15-VS adduct formation, which was detected by an upward shift in the USP18 band after immunoblotting with anti-USP18 antibody. THP-1.Control, THP-1.USP18 and its mutants (C64A, C64S) were transduced with a single round of HIV-1 luciferase reporter virus in an undifferentiated state (C) and in a PMA-differentiated state (D). The luciferase activity was measured at 72 h postransduction. In a related experiment, these cells were transduced with HIV-2Δvpx in an undifferentiated state (E) and a differentiated state (F). (G) Similarly, the PMA-differentiated cells were transduced with HIV-2 luciferase reporter virus, which had an active VPX to degrade SAMHD1. Mean differences of three replicates for each group in a single experiment was analyzed and compared between the groups by Student t test, expressed as means ± the SD. A P value of <0.05 was considered statistically significant (*). The higher the number of asterisks, the lower the P value. Each panel is representative of at least three independent experiments.
USP18 downregulates p21, which induces SAMHD1 phosphorylation in PMA-differentiated THP-1 cells.
Recent evidence suggests that the antiviral activity of SAMHD1 is positively regulated by p21 via inhibition of the cyclin and CDK complex that inactivates SAMHD1 by phosphorylation (22, 23, 39, 40). To understand the molecular mechanism behind the USP18-mediated enhancement of HIV-1 replication, we tested the expression levels of p21 in undifferentiated and PMA-differentiated THP-1 cells in the presence or absence of type I IFN, which is a known inducer of p21 (21, 54, 55). Remarkably, USP18 induced a downregulation of the p21 protein in PMA-differentiated THP-1 cells (Fig. 3A), which could not be rescued by IFN (Fig. 3A and B). The expression levels of cyclin D1 (Fig. 3A) and cyclin D2 (Fig. 3B) appeared elevated in the THP-1.USP18 cells. IFN-β induced significantly higher levels of both cyclins in the PMA-differentiated THP-1.Control cells but no further increase in the THP-1.USP18 cells (Fig. 3A and B). We further tested the phosphorylation status of SAMHD1 in cycling and noncycling THP-1 cell lines. In the absence of PMA treatment, THP-1.Control, WT USP18, and mutant USP18 cells expressed similar levels of total and phosphorylated SAMHD1 (Fig. 3C and D). However, the phosphorylation signal of SAMHD1 almost disappeared in PMA-differentiated THP-1.Control cells (Fig. 3C and D) but was retained in the THP-1.USP18 cells (Fig. 3C and D), although the level was lower than in the nondifferentiated cells (Fig. 3C). The increase in phosphorylated SAMHD1 in the differentiated THP-1 cells was independent of the USP18 isopeptidase activity, as phosphorylated SAMHD1 was also increased in the THP-1.USP18.C64A and THP-1.USP18.C64S cells (Fig. 3C). A subsequent probe for interaction partners of p21 and SAMHD1, including cyclin A, SKP2, CDK2, and CDK4 (21, 29, 56), demonstrated similar levels of CDK2 (Fig. 3C) and CDK4 (Fig. 3D) but upregulated levels of cyclin A and SKP2 in the differentiated WT USP18 (Fig. 3C and D) and mutant USP18-expressing (Fig. 3C) THP-1 cells compared to the THP-1.Control cells. In contrast, the cycling cell lines expressed similar levels of these proteins (Fig. 3C and D). SAMHD1 activity is tightly controlled during the cell cycle (24, 27). Considering that p21 is regulated in a cell cycle-dependent manner and in turn regulates the SAMHD1 antiviral function, we analyzed the cell cycle status of our cell lines expressing USP18 by staining the DNA with propidium iodide. As expected, a significant population of the nondifferentiated cells was in the S phase (Fig. 3E) (24). However, treatment of the cells with PMA shifted the population toward the G0/G1 and G2/M phases, with a conspicuously reduced S-phase population. No differences in the cell cycle populations were detected between the THP-1.USP18, mutant USP18, and control cells regardless of their differentiation status (Fig. 3E).
FIG 3.

USP18 downregulates p21 expression, which induces phosphorylated SAMHD1 in PMA-differentiated THP-1 cells. PMA-differentiated and undifferentiated THP-1 control and THP-1.USP18 cells were stimulated with 1,000 U/ml of IFN-β. At 48 h posttreatment, the cells were lysed and immunoblotted for p21, cyclin D1 (A), and cyclin D2 (B) with tubulin or GAPDH as a loading control. Romidepsin (FK288)-induced p21 in a cancer cell line (UM-UC-3) was included in the experiment as a positive control. (C) Immunoblots of total and phosphorylated SAMHD1, USP18, cyclin A, SKP2, and CDK2 in PMA undifferentiated and differentiated THP-1.Control cells (CTL), WT cells, and mutant THP-1.USP18 cells with β-actin as a loading control. (D) An immunoblot of CDK4 was included in subsequent experiments. For clarity and conciseness of data presentation, blots of different proteins on the same membranes detected with different antibodies were cropped and juxtaposed. Each panel is representative of at least three independent experiments. (E) Cycling and noncycling THP-1.Control, THP-1.USP18, and active-site mutant (C64A and C64S) cells were fixed, treated with RNase A, and then stained thoroughly with propidium iodide and analyzed with flow cytometry. The percentages of cells in the G1, S, and G2/M phases were quantified by FlowJo. Each panel is representative of at least three independent experiments.
USP18 complexes with SKP2, cyclin A, CDK1, CDK2, and SAMHD1.
To determine whether USP18 binds to SKP2 and other interacting partners of p21 (57), we tested the binding of SKP2 and SAMHD1 to USP18. HEK293T cells were either singly transfected or cotransfected with plasmids expressing hemagglutinin (HA)-tagged USP18, MYC-tagged SKP2, and FLAG-tagged SAMHD1. Cell lysates were immunoprecipitated with anti-HA beads and subsequently immunoblotted with anti-HA, anti-MYC, and anti-FLAG antibodies. The pulldown of USP18 precipitated SAMHD1 (Fig. 4A, lane 2), SKP2 (Fig. 4A, lane 3), and all three proteins complexed together (Fig. 4A, lane 4). Immunoprecipitation of SAMHD1 with anti-FLAG beads also pulled down SKP2 (Fig. 4A, lane 7), consistent with the observation of St Gelais et al. in 2014 (56). In a subsequent cotransfection of FLAG-SAMHD1 and pLOC-USP18, the cell lysates were immunoprecipitated with anti-FLAG beads and immunoblotted to detect the presence of endogenous cellular cyclin A2, CDK1, and CDK2 (Fig. 4B and C). The pulldown of SAMHD1 also precipitated USP18 and endogenous cyclin A2, CDK1 (Fig. 4B), and CDK2 (Fig. 4C), suggesting a possible complex comprising all five proteins.
FIG 4.

USP18 interacts with SKP2 and SAMHD1 and complexes with cyclin A2, CDK1, and CDK2. (A) Immunoprecipitation of HA-USP18, coexpressed with FLAG-SAMHD1 (lane 2), MYC-SKP2 (lane 3) or all three expression plasmids in a ratio of 1:1:1 (lane 4) using anti-HA affinity matrix beads. In parallel, FLAG-SAMHD1 was coexpressed with MYC-SKP2 and immunoprecipitated with anti-FLAG affinity matrix beads (lanes 7). Single and double transfections were supplemented with pcDNA3.1(+). Cell lysates were immunoblotted for USP18, SKP2, and SAMHD1, using their respective epitopped tags. (B) Immunoprecititation of FLAG-SAMHD1 coexpressed with pLOC-USP18 or without pLOC-USP18 and replaced by pcDNA3.1+. Proteins of HEK293T cells were immunoprecipitated with anti-FLAG affinity matrix beads and immunoblotted with FLAG and USP18 and endogenous expression of cyclin A2 and CDK1, cyclin A2 (B), and CDK2 (C). Each panel is representative of at least three independent experiments.
Presence or absence of USP18 regulates the level of p21.
To further evaluate the mechanism of the USP18-mediated increase in HIV-1 replication, we knocked out USP18 in THP-1 cells by the CRISPR-Cas9 system (see Materials and Methods) and immunoblotted the PMA-differentiated cells for p21, total and phosphorylated SAMHD1, USP18, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) as the loading control (Fig. 5A). USP18KO cells contained significantly upregulated p21 protein levels, which could be further enhanced by IFN-β treatment (Fig. 5A, lanes 3 and 4, and Fig. 5C). The highly induced p21 protein correlated significantly with reduced phosphorylated form of SAMHD1 to a level similar to exogenous IFN-β-induced dephosphorylated SAMHD1 (Fig. 5A, compare lanes 2 and 3) (21). The increased p21 protein levels also correlated significantly with diminished HIV-1 infection in the USP18KO THP-1 cells (Fig. 5B). In contrast, SKP2 protein levels were unchanged in the USP18KO THP-1 cells (Fig. 5C).
FIG 5.

The presence or absence of USP18 regulates the level of p21. (A) PMA-differentiated USP18KO THP-1 cells and vector control cells were stimulated with 1,000 U/ml of IFN-β. At 24 h after stimulation, the cells were lysed and immunoblotted for the endogenous expression of p21, total and phosphorylated SAMHD1, USP18, and GAPDH as a loading control. (B) After 48 h of PMA treatment, the cells were also tested for HIV-1 replication and transduced with HIV-1 reporter virus in the presence or absence of copackaged VPX. (C) In a related experiment, PMA-differentiated cells USP18KO and control cells were treated with PMA and subsequently immunoblotted for p21, total, and phosphorylated SAMHD1, SKP2, USP18, and GAPDH as a loading control. (D) PMA-differentiated SAMHD1KO THP-1 cells expressing the vector control and USP18 were immunoblotted for p21, USP18, SAMHD1, and GAPDH as a loading control. These cells were treated with 25 ng/ml PMA. (E) At 48 h posttreatment, the cells were transduced with a single round of HIV-1 reporter virus, and the luciferase activity was measured after 72 h. (F) In a related experiment, the cells were tested for SKP2 protein expression levels by immunoblotting the PMA-differentiated vector control and USP18 expressing SAMHD1KO THP-1 cells for p21, SAMHD1, SKP2, USP18, and GAPDH as a loading control. Each panel is representative of at least two independent experiments.
To understand how p21 downregulation by USP18 might affect HIV-1 replication in the absence of SAMHD1, we obtained SAMHD1KO THP-1 cells (58), and stably expressed USP18 or a vector control in these cells (Fig. 5D). The PMA-differentiated SAMHD1KO THP-1 cell lines were then immunoblotted for p21, total SAMHD1, USP18, and GAPDH as the loading control. USP18 overexpression significantly reduced p21 protein levels as seen before. The cells were further tested for HIV-1 infection following PMA differentiation. SAMHD1KO THP-1 cells demonstrated significantly enhanced levels of HIV-1 replication compared to wild-type THP-1 cells (Fig. 5E, compare white and black bars). The infection was further enhanced by USP18 in the absence of SAMHD1 by more than 100-fold (Fig. 5E, compare green and red bars). The significantly reduced p21 protein levels in the USP18 overexpressed SAMHD1KO THP-1 cells correlated strongly with upregulated SKP2 protein levels (Fig. 5F).
USP18 enhances HIV-1 replication at the reverse transcription step.
To estimate the frequencies of HIV-1-infected THP-1.USP18 and control cells in a PMA-differentiated state, we generated USP18-expressing cells that lacked GFP expression by retroviral transduction compared to the lentiviral pLOC vector (Fig. 6A). This allowed us to infect the cells with an HIV-1.IRES-GFP reporter virus and to quantify the percentage of green fluorescent protein (GFP)-expressing cells (Fig. 6B). Flow cytometric analysis showed significantly increased frequencies of GFP+ cells (6-fold) in the THP-1.USP18 cells compared to the vector control cells (Fig. 6B and C). To rule out a block at the membrane fusion step, we tested the HIV-1 VSV-G pseudotype fusion using a virion-based fusion assay as described previously (59, 60). Flow cytometric analysis allowed us to quantify the proportion of cells whose membranes were successfully fused by the virus (Fig. 6D). No significant difference in virion fusion was observed between the THP-1.USP18 and control cells in both PMA-undifferentiated (Fig. 6E) and differentiated (Fig. 6F) states. In parallel, we tested these cells for luciferase activity 48 h postinfection with HIV-1 NL-LucR− E− reporter viruses. As expected, THP-1.USP18 cells demonstrated higher infection than the control cells (Fig. 6G). To test whether USP18 could relieve the block at the reverse transcription step in PMA-differentiated cells, we quantified the early and late reverse transcription products 12 h postinfection in the presence and absence of the antiretroviral drug nevirapine, a nonnucleoside reverse transcriptase inhibitor. Interestingly, the THP-1.USP18 cells contained significantly increased levels of early (Fig. 6H) and late (Fig. 6I) reverse transcription products compared to the vector controls.
FIG 6.

USP18 enhancement of HIV-1 infection occurs at the reverse transcription step. (A) Immunoblot of total and phosphorylated SAMHD1, USP18, and GAPDH as a loading control in retrovirus-based USP18 expression in THP-1 cells and controls. (B) Representative gating strategy for the frequencies of GFP-expressing, PMA-differentiated THP-1.USP18 and control cells, transduced with HIV-1.IRES.GFP reporter virus at an MOI of 2 for 72 h. (C) The mean (n = 2) difference in the percentage of GFP+ cells between the groups was analyzed by a Student t test. (D) Representative gating strategy for the frequencies of cleaved CCF2+ (blue) in PMA-undifferentiated and differentiated THP-1.Control and USP18-expressing cells were transduced with HIV-1 pseudotyped virus with copackaged β-lactamase-Vpr with or without VSV-G. The mean (n = 3) difference in percentage cleaved CCF2+ cells between the groups in an undifferentiated state (E) and a differentiated state (F) was analyzed by using a Student t test. (G) PMA-differentiated THP-1.USP18 and vector controls were transduced with HIV-1 NL-LucR−E−. At 48 h postransduction, the cells were measured for luciferase activity. Values are means ± the SD for three independent infections. Asterisks represent statistically significant differences (***, P < 0.0001). Similarly, cells were infected or uninfected (mock = media) in the presence or absence of nevirapine (NVP) for the quantification of early (H) and late (I) RT products by qPCR. At 12 h postransduction, the DNA of infected cells was isolated, and HIV-1 reverse transcripts were quantified by real-time qPCR. Each panel is representative of at least two independent experiments.
USP18 upregulates intracellular dNTPs.
p21 induction is known to reduce the intracellular dNTP pool by repressing key enzymes involved in de novo dNTP biosynthesis (23, 25, 34, 35, 39, 40). Because USP18 significantly downregulated p21 expression (Fig. 3A and 5D), we evaluated the impact of USP18 expression on the intracellular dNTP pool. Interestingly, dATP and dGTP levels were significantly upregulated in USP18-expressing THP-1 cells compared to controls (Fig. 7A) and, even more interestingly, USP18 significantly upregulated all four intracellular dNTPs in USP18-overexpressing SAMHD1KO THP-1 cells (Fig. 7B) compared to SAMHD1KO THP-1 cells without overexpression (Fig. 7B). Indeed, we could rescue the SAMHD1 block to HIV-1 replication by supplementing the differentiated THP-1 cells with deoxynucleosides (Fig. 7C).
FIG 7.
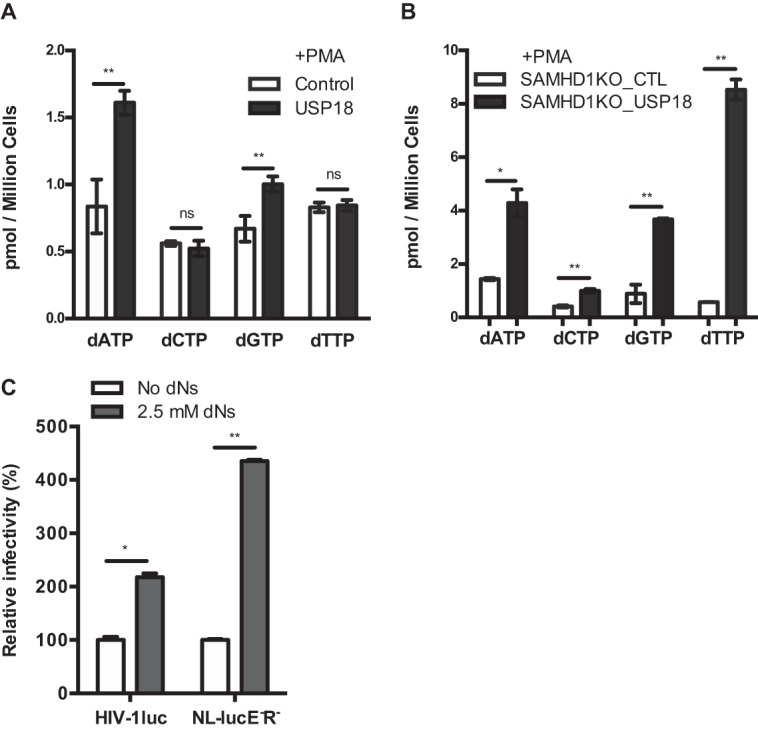
USP18 enhances HIV-1 replication by upregulating intracellular dNTP concentrations. (A) The intracellular levels of dATP, dCTP, dGTP, and dTTP were quantified by a single nucleotide primer extension assay in differentiated THP-1.Control, THP-1.USP18, SAMHD1KO_CTL, and SAMHD1KO_USP18 cells. Mean differences of three replicates for each dNTP level in a single experiment were analyzed and compared between THP-1.Control and THP-1.USP18 (A) cells, as well as between SAMHD1KO_CTL and SAMHD1KO_USP18 (B) cells, by a Student t test, and are expressed as means ± the SD. A P value of <0.05 was considered statistically significant (*). The higher the number of asterisks, the lower the P value. Each panel is representative of at least three independent experiments. (C) PMA-differentiated THP-1 cells were treated with 2.5 mM dNs and transduced with HIV-1 luciferase reporter virus (HIV-1luc) from the three-plasmid system (see Materials and Methods) and from the NL-LucE−R− construct. At 48 h postinfection, the intracellular luciferase activity was measured. The values (i.e., for the luciferase activity) obtained in cells untreated with dNs were set as 100%, and the viral gene expression from cells treated with dNs was calculated relative to the untreated cells. The data are representative of two independent experiments.
DISCUSSION
USP18 is expressed at different levels in many mammalian tissues, including the liver, spleen, and thymus (41, 61). In addition, high expression is found in several innate immune cells, such as murine CD169+ macrophages, bone marrow-derived dendritic cells, peritoneal macrophages, monocyte-derived macrophages, and white matter microglia, and it is differentially regulated during T-cell activation (41, 45, 47, 49, 61, 62). USP18 has been implicated in the innate immunity against bacteria and viruses (43, 44, 49, 52, 62). Our findings show that USP18 can be induced by HIV-1, an observation that has recently been independently confirmed in HIV-1-infected monocyte-derived macrophages (3). The expression of USP18 allowed for enhanced replication of HIV-1, HIV-2, and SIVmac (data not shown), enabling infection in noncycling THP-1 cells even in the presence of the potent restriction factor SAMHD1. The observed positive effect of USP18 is associated with the downregulation of the CDK inhibitor p21. USP18 overcame the inhibitory effect of p21 at the HIV-1 reverse transcription step likely by relieving the p21 block of SAMHD1 phosphorylation and by rescuing the p21-dependent repression of key enzymes of de novo dNTP biosynthesis (23, 25, 40). Thus, USP18 increased the supply of dNTPs for reverse transcription to take place. Moreover, the enhancement of HIV-1 infection by USP18 appeared to be independent of its isopeptidase activity, suggesting that de-ISGylation is not involved in overcoming the inhibitory effect of p21.
p21 is known as an important factor regulating cell growth, monocyte differentiation, survival, and maturation (63–65). Notably, its dysregulation is common in many cancers (57, 66). Many recent reports have highlighted HIV-1 inhibition by p21, which is likely achieved by its ability to regulate de novo dNTP biosynthesis and the cyclin-dependent kinases (CDKs) required for cell cycle progression (22, 23, 25, 34, 38–40, 54, 55, 63–65, 67–69). Despite the importance and the physiological relevance of the p21 protein, the mechanisms and the cellular factors required for the regulation of p21 in vivo are only partly understood. It is thought that p21 is regulated transcriptionally both in a p53-dependent and -independent manner; the p53-dependent manner is mediated by phosphorylation and also likely by ISG15 modification (36, 38, 57, 66, 70). p21 also undergoes extensive posttranslational regulation (36, 37, 57). In actively dividing cells, p21 is a highly unstable protein with a half-life of about 20 to 60 min (37, 57). G1/S- and S-phase transition of the cell cycle requires ubiquitin-dependent degradation of p21, mediated by the E3 ubiquitin ligase complex substrate recognition factor SKP2, which promotes polyubiquitylation of p21 in a complex with CDK2 and cyclin E or A to initiate its proteasomal degradation (36, 57). However, another ubiquitin-independent proteolysis of p21 has been postulated, which may occur in a cell type-dependent manner (57, 71–73).
Here, we demonstrate that USP18 might be involved in the regulation of p21 protein expression. The expression of USP18 dramatically diminishes p21 protein levels, which cannot be rescued by either PMA or type I IFN. On the other hand, the absence of USP18 stabilizes p21 protein and enhances its anti-HIV activity. How USP18 mediates p21 downregulation is currently not clear. Our data appear to exclude the involvement of ISG15 and rather support a mechanism involving SKP2-dependent regulation of p21 protein. USP18 likely recruits or retains SKP2 in an environment in which USP18 cannot be degraded by the proteasome due to the presence of free ISG15 (50, 51). By retaining SKP2, USP18 likely primes p21 ubiquitylation by SKP2, facilitating its degradation by the proteasome. The degradation of p21 thus retains cyclin A/CDK2 to phosphorylate SAMHD1 at residue T592 and potentially activates a de novo dNTP biosynthesis pathway in the differentiated THP-1 cells.
Overall, the identification of a novel function of USP18 in abrogating the antiviral activites of p21 underscores the importance of USP18 in the innate immune cells. In vivo, constitutive and HIV-1-induced expression of USP18 in innate target cells could potentially facilitate the replication of the virus and help it to escape the innate immune restriction mediated by p21. Further investigation to understand the mechanism of USP18-mediated downregulation of p21 and how this likely affects de novo dNTP biosynthesis is warranted and should be helpful for the design of better therapeutics for the control of HIV-1 replication in innate immune cells.
MATERIALS AND METHODS
Plasmids.
The human USP18 open reading frame (ORF) was cloned into the pLOC lentiviral vector containing turbo GFP and blasticidin S resistance (Thermo Fisher Scientific, Inc., Darmstadt, Germany), using the NheI and SpeI restriction sites. An empty vector control was obtained by excising the ORF using the same sites and religating. The USP18 cDNA was alternatively cloned into the retroviral vector pMSCVneo (74) using the HpaI and Xhol restriction sites. All vector constructs were verified by sequencing and tested for USP18 protein expression. HIV-1 vector, pSIN.PPT.CMV.Luc.IRES.GFP (75, 76), HIV-2 virus containing HIV-2 construct (pHIV-2D4), and pHIV-2 Luc SV40 (previously called pHIV-2 SEW Luc SV40) have been described before (76). The HIV-1 construct psPAX2 was obtained from the NIH, AIDS Reagent Program repository. pRSV-Rev (77) and pMDLg/pRRE and pMD.G (77) have been described previously. HIV-1 NL-LucR− E−, pMDLx g/pRRE, HIV-2ROD, and SIVmac239 VPX, cloned into pcDNA6/myc-His (Invitrogen/Life Technologies, Germany), were obtained from Nathaniel R. Landau (78). pHIT60 was kindly provided by Jonathan Stoye (79). The active site of human USP18 was mutated by site-directed mutagenesis from cysteine (C) 64 to alanine (A) or serine (S) to obtain pLOC-USP18-C64A or pLOC-USP18-C64S plasmids. HA- and V5-epitope-tagged-human USP18 were cloned into pcDNA3.1(+) at the HindIII and NotI restriction sites to obtain pcDNA3.1-HA-USP18 and pcDNA3.1-V5-USP18. C-terminal HA-tagged USP18 or V5-tagged USP18 was cloned into pLOC-empty vector at the NheI and SpeI restriction sites to obtain pLOC-HA-USP18 and pLOC-V5-USP18 plasmids.
Cell culture.
THP-1 cells (ATCC TIB-202) (80) were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (PAN-Biotech, Aidenbach, Germany) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, and 100 U/ml penicillin-streptomycin at 37°C in a humidified atmosphere with 5% CO2. HEK293T cells (ATCC CRL-3216) (81, 82), HOS cells (ATCC CRL-1543) (83), and TZM-bl cells (NIH, AIDS Reagent Program) (84–88) were maintained in Dulbecco modified Eagle complete medium (Biochrom, Berlin, Germany) supplemented with 10% FBS, 2 mM l-glutamine, and 100 U/ml penicillin-streptomycin at 37°C in a humidified atmosphere with 5% CO2. Cell lines were generated by transduction of THP-1 cells with lentiviral vectors made by cotransfection of pLOC-USP18, pLOC-USP18-C64A, pLOC-USP18-C64S, or pEV, together with psPAX2, pRSV-Rev, and pMD.G in HEK293T cells. Viral particles were layered on 2 ml 20% (wt/vol) sucrose, concentrated by ultracentrifugation at 284,061 × g (SW-41 rotor; Beckman Coulter, Krefeld Germany) for 2 h at 4°C and resuspended in RPMI. The cells were spinoculated at 1200 × g for 2 h at 30°C and selected using blasticidin S hydrochloride (Sigma-Aldrich, Taufkirchen, Germany). Blasticidin S-resistant cell pools were tested for GFP protein expression by fluorescence microscopy and for USP18 expression by using immunoblots. Alternatively, the cell lines were generated by retroviral transduction of THP-1 cells with particles generated from cotransfection of HEK293T with plasmid pMSCV-USP18 or pMSCV empty vector, together with pHIT60 for packaging and VSV-G (pMD.G) for entry. The retrovirus-based USP18 expressing THP-1 cells were selected using neomycin (G418-BC liquid, A 2912; Biochrom GmbH, Berlin, Germany). Neomycin-resistant cell pools were tested for USP18 expression by immunoblotting.
USP18 knockout by LentiCRISPRv2.
Plasmids of LentiCRISPRv2 targeting USP18 were constructed according to previously described protocols (89, 90). Briefly, complementary oligonucleotides containing the specific USP18 sgRNA sequences, including sequence TAATGAATGTGGACTTCACC, and overhangs complementary to the overhangs generated by BsmBI digestion of LentiCRISPRv2 were ligated into the BsmBI-digested LentiCRISPRv2 plasmid to generate the functional transfer vector. LentiCRISPRv2 plasmid lacking sgRNA sequence was used as an empty vector control. HIV-1 pseudotype virus containing the pLentiCRISPRv2 transfer vector, packaging plasmid psPAX2, and VSV-G were cotransfected into HEK293T cells. At 48 h posttransfection, viral supernatants were harvested, concentrated, and purified over 20% (wt/vol) sucrose and resuspended in fresh RPMI media. THP-1 cells were transduced with the HIV-1 pseudovirus and, at 72 h postransduction, the cells were subcultivated in fresh media containing 2 μg/ml puromycin for selection over a period of 14 days. The selected cells were single cell cloned by serial dilution in a 96-well plate. Single cells were clonally expanded and tested for USP18 expression by immunoblot analysis. Cells that tested negative for USP18 expression were further analyzed by PCR amplification of genomic DNA flanking the CRISPR-targeted region. The forward primer 5′-CTGGTTGGTTTACACAACATTGGACAG-3′ and the reverse primer 5′-GATATTGAAGAGGTAAGACTGTTCTTCAGG-3′ were used to amplify exon 3. The gDNA amplicon was subsequently cloned into pJET1.2/blunt cloning vector and sequenced (CloneJET PCR cloning kit, K1232; Thermo Fisher). Multiple sequence alignments were performed using Vector NTI (91). SAMHD1 knockout THP-1 cells were obtained as a gift from Veit Hornung (58).
Virus production and transduction.
HIV-1 luciferase reporter viruses were generated by transfecting HEK293T cells with 600 ng of either pNL-LucR− E− and 150 ng of pMD.G or alternatively, using 600 ng of pMDLg/pRRE or pMDLx g/pRRE, together with 250 ng of pRSV-Rev, 600 ng of pSIN.PPT.CMV.Luc.IRES.GFP, and 150 ng of pMD.G with or without pcDNA6/myc-His-VPX or HIV-2rod VPX using Lipofectamine LTX (Thermo Fisher Scientific, Schwerte, Germany), according to manufacturer's recommendations in a 6-well plates. For HIV-2, transfection consisted of 850 ng of pHIV-2D4, 150 ng of pMD.G, and 600 ng of HIV-2.Luc.SV40 with or without pcDNA6/myc-His-VPX or HIV-2rod VPX. Viral supernatants were collected at 48 h after transfection, concentrated (see above), treated with DNase I (ENO521; Thermo Fisher Scientific), and then titrated using TZM-bl or HEK293T cells. Cell lysates of transfected HEK293T cells were immunoblotted for endogenous SAMHD1 to confirm the degradation of SAMHD1 by VPX. PMA-differentiated (25 ng PMA/ml; Calbiochem, Darmstadt, Germany) and undifferentiated USP18-expressing and control THP-1 cells were treated with or without human type I IFNs: IFN-αA/D (Sigma-Aldrich) and IFN-β-1a (PBL Assay Science, Piscataway Township, NJ) at concentrations of 10, 100, and 1,000 U/ml for 4 h and subsequently transduced with HIV-1 or HIV-2 luciferase reporter viruses. The luciferase activity was measured 3 days later. All experiments were independently repeated at least three times in triplicates.
β-Lactamase-based virion fusion assay.
A β-lactamase-containing HIV-1 fusion assay was performed as previously described (92). Briefly, β-lactamase containing HIV-1 pseudotype virus was generated by cotransfection of HEK293T cells with pMDLg/pRRE, pRSV-Rev, pSIN.PPT.CMV.Luc.iresGFP, and pMD.G for pseudotyping and pMM310 (59) for β-lactamase-Vpr chimeric protein expression. At 48 h posttransfection, viral supernatant was collected and concentrated by centrifugation over 20% (wt/vol) sucrose. For a negative-control viral particle, pMD.G (VSV-G) was replaced with pcDNA3.1+ to exclude background effect of the fluorescence substrate. Titers of the concentrated viral particles were determined on HEK293T cells. The viral particles were used to infect 106 PMA-differentiated and undifferentiated THP-1.USP18 and vector control at a multiplicity of infection (MOI) of 0.5 via spinoculation at 1,200 × g for 2 h at 30°C. The cells were incubated for additional 3 h at 37°C and then washed with serum-free media. To allow for fluorescence substrate uptake, the cells were resuspended in 1 ml of freshly prepared loading solution consisting of serum-free media, 10 mM HEPES, 1% probenecid, 0.015% solution A (CCF2-AM), and 0.08% solution B (100 mg/ml Pluronic-F127R, 0.1% acetic acid; GeneBLAzer detection kit; Invitrogen, Germany). The cells were subsequently incubated at 25°C in a 5% CO2 incubator. At 16 h after incubation, the cells were washed thoroughly with phosphate-buffered saline (PBS), and the fluorescence was measured using BD FACSCanto II (BD Biosciences). Analysis was subsequently done using the FlowJo software version 9.9.6 (FlowJo LLC, Ashland, OR).
qPCR quantification of HIV-1 reverse transcripts.
HIV-1 NL-LucR− E−-pseudotyped viral particles were produced and titered in HEK293T cells. The particles were DNase I treated at 37°C for 1 h. PMA-differentiated THP1.USP18 or control cells were transduced with NL-LucR−E− virus at an MOI of 0.5 in the presence or absence of 10 μM nevirapine, a gift from Henning Hofmann. Cells were harvested at 12 h postransduction (93) and stored at −80°C until processed for DNA isolation. 250 ng of total DNA was used for qPCR amplification of early and late HIV-1 reverse transcripts (early RT: forward, 5′-GTGCCCGTCTGTTGTGTGAC, and reverse, 5′-GGCGCCACTGCTAGAGATTT; late RT: forward, 5′-TGTGTGCCCGTCTGTTGTGT, and reverse, 5′-GAGTCCTGCGTCGAGAGAGC) (93). Assays were performed on a Roche LightCycler 96 (Hoffmann-La Roche, Ltd., Basel, Switzerland) using SYBR green (Applied Biosciences/Thermo Fisher, Inc.). All PCR data were adjusted to genomic GAPDH levels (GAPDH: forward, 5′-CATCATCCCTGCCTCTACTGG, and reverse, 5′-GGTCCACCACTGACACGTT). The data were normalized to a standard curve generated with proviral plasmid DNA serially diluted in HEK293T cell genomic DNA (94).
Transfection.
For USP18, SKP2, and SAMHD1 interaction experiments, 106 HEK293T cells were cotransfected with 0.8 μg of HA-USP18, 0.8 μg of MYC-SKP2, and 0.8 μg of FLAG-SAMHD1-expressing plasmids in 6-well plates for 48 h. In a related experiment, 1.25 μg of FLAG-SAMHD1 was cotransfected with 1.25 μg of pLOC-USP18. Single and double transfections were supplemented with pcDNA 3.1+.
ISG15-VS probe reaction.
HA-tagged wild-type human USP18 and its mutants (C64A and C64S) were transfected in HEK293T cells. At 48 h posttransfection, cells were lysed in 50 mM Tris (pH 7.4), 5 mM MgCl2, 250 mM sucrose, 1 mM dithiothreitol (DTT) using glass beads (44, 95). Lysates (20 μg) were incubated with 1 μg of HA-ISG15-VS probe (Boston Biochem) for 1 h at 37°C. ISG15 and USP18 and its mutant proteins were separated on a sodium dodecyl sulfate (SDS) gel and immunoblotted for ISG15, USP18, and tubulin as a control using rabbit anti-ISG15 (ab 36765; Abcam, Germany) and rabbit anti-USP18 (D4E7; Cell Signaling, Frankfurt am Main, Germany) respective antibodies and mouse anti-tubulin (1:8,000, dilution, clone B5-1-2; Sigma-Aldrich).
Immunoblot analysis.
PMA-differentiated WT, SAMHD1KO, and USP18KO THP-1 cells, as well as PMA-differentiated and undifferentiated THP-1.USP18, mutant and control cells were lysed in radioimmunoprecipitation assay buffer (25 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, protease inhibitor cocktail set III [Calbiochem], and phosphatase inhibitor cocktail [Roche, Mannheim, Germany]). USP18 expression cells were detected by rabbit anti-human USP18 at a 1:1,000 dilution (D4E7; Cell Signaling). SAMHD1 expression was detected by rabbit anti-human SAMHD1 at a 1:1000 dilution (12586-1-AP; Proteintech, Manchester, United Kingdom). Phosphorylated SAMHD1 was detected by rabbit anti-human phosphorylated SAMHD1 (T592, 8005; ProSci, Biocat, Heidelberg, Germany) at a 1:1,000 dilution. Cell cycle regulators were detected by rabbit anti-human cyclin A (sc-751, H-432; Santa Cruz Biotechnology, Inc., Heidelberg, Germany), cyclin D1 (sc-753, H-295; Santa Cruz Biotechnology), goat anti-human cyclin D2 (sc-181-G, C-17; Santa Cruz Biotechnology), CDK2 (sc-163-G, M2; Santa Cruz Biotechnology), or CDK4 (sc-260-G, C-22; Santa Cruz Biotechnology) using a 1:500 dilution. Endogenous and overexpressed SKP2 was detected using rabbit anti-human SKP2 (4358; Cell Signaling) at a 1:500 dilution and human endogenous p21CIP1 was detected using mouse anti-human p21CIP1 (556430, SX118; BD Pharmingen, Heidelberg, Germany) at a 1:500 dilution. Overexpressed FLAG-tagged SAMHD1 and HA-tagged USP18 in transfected HEK293T were detected by using mouse anti-FLAG at a 1:1,000 dilution (M2; Sigma-Aldrich) and mouse anti-HA at a 1:7,500 dilution (MMS-101P; Covance, Münster, Germany), respectively. Overexpressed c-MYC-tagged SKP2 in HEK293T was detected by mouse anti-c-MYC at a 1:500 dilution (9E10, MCA2200; Bio-Rad Abd Serotec, Puchheim, Germany). Tubulin and β-actin were detected using mouse anti-tubulin antibody (1:8,000, dilution, clone B5-1-2; Sigma-Aldrich) and rabbit anti-β-actin-linked HRP (5125, 13E5; Cell Signaling), respectively. GAPDH was detected with goat anti-GAPDH at a 1:10,000 dilution (EB06377; Everest Biotech, VWR, Darmstadt, Germany).
In other experiments, endogenous cyclin A2, CDK1, and CDK2 were detected using rabbit anti-human cyclin A2 (18202-1-AP; Proteintech; 1:500 dilution), rabbit anti-human Cdc2 p34/CDK1 (H-297, sc-747; Santa Cruz; 1:500), and rabbit anti-human CDK2 (78B2, 2546; Cell Signaling; 1:500), respectively, after immunoprecipitation of overexpressed SAMHD1-FLAG cotransfected with USP18. Secondary antibodies were horseradish peroxidase-conjugated sheep anti-mouse antibody (α-mouse-IgG-HRP; GE Healthcare, Munich, Germany), donkey anti-rabbit (α-rabbit-IgG-HRP; GE Healthcare), and rabbit anti-goat antibody (α-rabbit-IgG-HRP; Santa Cruz), and blots were developed with ECL reagents (GE Healthcare).
Immunoprecipitation.
To determine SKP2 and SAMHD1 binding to USP18, HEK293T cells were singly or cotransfected with expression plasmids of 0.8 μg of HA-USP18, SKP2-MYC, and FLAG-SAMHD1 in a ratio of 1:1:1. Single and double transfections were supplemented with pcDNA3.1(+). After 48 h, the cells were lysed in immunoprecipitation (IP) lysis buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 0.8% NP-40, 10% glycerol, 1 mM phenylmethanesulfonyl fluoride solution [Sigma-Aldrich], and protease inhibitor cocktail set III [Calbiochem]). The lysates were cleared by centrifugation. The supernatant were incubated with 20 μl of anti-HA affinity matrix beads and anti-HA affinity resin beads (Roche) at 4°C for 2 h. The samples were washed four times with IP buffer on ice. Bound proteins were eluted by boiling the beads for 5 min at 95°C in reducing sample buffer. Interaction between MYC-SKP2 and FLAG-SAMHD1 was determined by single or cotransfection of their respective plasmids in HEK293T cells at a concentration of 0.8 μg each. Cell lysates were immunoprecipitated with 10 μl anti-FLAG affinity resin beads (Biotool; Absource, Munich, Germany), followed by incubation for 2 h at 4°C and then washed six times. Immunoblot analysis and detection were performed as described previously. To analyze the interaction of USP18, cyclin A, CKD1/2, and SAMHD1, FLAG-SAMHD1 was cotransfected with pLOC-USP18. HEK293T cells were harvested at 48 h posttransfection and lysed in 200 μl of NET lysis buffer (50 mM Tris/HCl [pH 7.4], 150 mM NaCl, 15 mM EDTA [pH 7.4], 1% NP-40 containing protease and phosphatase inhibitors)/dish for 30 min on ice. Lysates were centrifuged at 17,000 × g for 15 min at 4°C. For preclearing, lysates were incubated with 25 μl Protein G Sepharose 4 Fast Flow (GE Healthcare) in 500 μl of Tris-buffered saline (TBS) plus 0.1% NP-40 (containing protease inhibitor) for 1.5 h at 4°C. After centrifugation, 200 μl of NENT100 (20 mM Tris [pH 7.4], 100 mM NaCl, 1 mM EDTA [pH 7.4], 0.1% NP-40, 25% glycerol) plus 1 mg/ml bovine serum albumin was added to precleared lysates, which were subsequently incubated with 25 μl of anti-FLAG M2 affinity gel (Sigma-Aldrich) for 1 h at 4°C. Beads were washed twice with 300 μl NENT300 (20 mM Tris [pH 7.4], 300 mM NaCl, 1 mM EDTA [pH 7.4], 0.1% NP-40, 25% glycerol) and twice with 300 μl of TBS plus 0.1% NP-40, each time for 2 min at 4°C under constant rotation. Bound immune complexes were released in 25 μl of 2× sample buffer through boiling (95°C, 5 min).
Cell cycle analysis.
THP-1.USP18, mutant cells, and controls were treated with 25 ng/ml PMA, and 3 days after differentiation, the cells were harvested and resuspended in fresh RPMI media, washed with PBS, and fixed with 70% ice-cold ethanol. At 24 h after fixation, the cells were washed thoroughly and treated with RNase A for 30 min at 37°C. The cells were then stained with propidium iodide for 30 min at 4°C in the dark and analyzed at a 488-nm excitation wavelength by flow cytometry (FACS Canto II; BD Biosciences, Heidelberg, Germany). The data were evaluated using FlowJo software version 9.8.2 (Tree Star, San Carlos, CA).
dNTP quantification.
Cellular dNTPs were extracted according to a previously described protocol (32). Briefly, 2 × 106 PMA-differentiated THP-1.USP18, THP-1.Control, and SAMHD1 knockout THP-1.USP18 and THP-1.Control cells were harvested and washed with cold PBS and lysed in ice-cold 0.2 ml of 65% (vol/vol) aqueous methanol. Lysates were heated at 95°C for 3 min and clarified by centrifugation at 18,800 × g for 3 min. Supernatants were transferred into new sterile tubes and dried using a SpeedVac (Eppendorf GmbH, Hamburg, Germany) at 30°C. The dried dNTPs were then resuspended and quantified using primer extension assay as described earlier (32). The required linear range of the dNTP assay was between 2 and 32% of the primer extension. The intracellular dNTP concentrations (pmol) were based on 106 cells.
Exogenous dN treatment.
Wild-type THP-1 cells were differentiated with 25 ng/ml PMA. At 48 h posttreatment, the cells were treated with or without deoxynucleosides containing 2.5 mM concentrations (each) of 2′-deoxyadenosine monohydrate (dA), dC (Sigma), dT, and dG (Abcam) 40 min prior to infection and subsequently transduced with HIV-1, generated from a three-plasmid system and NL-LucE−R− (with or without deoxynucleosides [dNs]). After 24 h, fresh medium was exchanged and, at 48 h postinfection, the intracellular luciferase activity was measured.
Statistical analysis.
Data were analyzed using GraphPad Prism version 6 (GraphPad Software, Inc., La Jolla, CA). The study groups were compared using a two-tailed, unpaired Student t test, and a P value of <0.05 was considered statistically significant. The data represent means ± the standard deviations (SD), as indicated in the figures.
ACKNOWLEDGMENTS
We thank Wioletta Hörschken for her excellent technical assistance. We thank Kate Bishop, Alexander Hölscher, Michele J. Hoffmann, Henning Hofmann, Veit Hornung, Neeltje Kootstra, Nathaniel R. Landau, David Looney, Mathias Schweizer, and Jonathan P. Stoye for reagents. The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: TZM-bl (catalog no. 8129) from John C. Kappes, Xiaoyun Wu, and Tranzyme, Inc., and psPAX2 (catalog no. 11348) from Didier Trono.
C.M. is supported by the Heinz-Ansmann Foundation for AIDS Research. This study was supported by the Deutsche Forschungsgemeinschaft (DFG; CRC1292 project TP04 to R.K. and SPP1923 project KO 4573/1-1 to R.K. and SPP1923 project MU 1608/9-1 to C.M.) and by National Institutes of Health grants GM104198 and R01 AI136581 to B.K.
E.O.K., C.M., K.S.L., and P.A.L. conceptualized the study. E.O.K. and C.M. designed the methodology. E.O.K., K.S., A.A.J.V., and J.H. performed experiments. E.O.K., C.M., R.K., K.S., J.H., B.K., W.A.S., and D.H. interpreted the results. E.O.K. wrote the original draft of the manuscript, and E.O.K., C.M., R.K., K.S., K.S.L., A.A.J.V., B.K., and W.A.S. reviewed and edited the manuscript. C.M. and E.O.K. acquired funding. D.H., K.S.L., R.K., P.A.L., and W.A.S. obtained resources. D.H. and C.M. supervised the study.
REFERENCES
- 1.Harris RS, Hultquist JF, Evans DT. 2012. The restriction factors of human immunodeficiency virus. J Biol Chem 287:40875–40883. doi: 10.1074/jbc.R112.416925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duggal NK, Emerman M. 2012. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol 12:687–695. doi: 10.1038/nri3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor JP, Cash MN, Santostefano KE, Nakanishi M, Terada N, Wallet MA. 2018. CRISPR/Cas9 knockout of USP18 enhances type I IFN responsiveness and restricts HIV-1 infection in macrophages. J Leukoc Biol doi: 10.1002/JLB.3MIA0917-352R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fensterl V, Sen GC. 2009. Interferons and viral infections. Biofactors 35:14–20. doi: 10.1002/biof.6. [DOI] [PubMed] [Google Scholar]
- 5.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A 95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berger A, Sommer AF, Zwarg J, Hamdorf M, Welzel K, Esly N, Panitz S, Reuter A, Ramos I, Jatiani A, Mulder LC, Fernandez-Sesma A, Rutsch F, Simon V, König R, Flory E. 2011. SAMHD1-deficient CD14+ cells from individuals with Aicardi-Goutieres syndrome are highly susceptible to HIV-1 infection. PLoS Pathog 7:e1002425. doi: 10.1371/journal.ppat.1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahn J, Hao C, Yan J, DeLucia M, Mehrens J, Wang C, Gronenborn AM, Skowronski J. 2012. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J Biol Chem 287:12550–12558. doi: 10.1074/jbc.M112.340711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwefel D, Groom HC, Boucherit VC, Christodoulou E, Walker PA, Stoye JP, Bishop KN, Taylor IA. 2014. Structural basis of lentiviral subversion of a cellular protein degradation pathway. Nature 505:234–238. doi: 10.1038/nature12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwefel D, Boucherit VC, Christodoulou E, Walker PA, Stoye JP, Bishop KN, Taylor IA. 2015. Molecular determinants for recognition of divergent SAMHD1 proteins by the lentiviral accessory protein Vpx. Cell Host Microbe 17:489–499. doi: 10.1016/j.chom.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hofmann H, Logue EC, Bloch N, Daddacha W, Polsky SB, Schultz ML, Kim B, Landau NR. 2012. The Vpx lentiviral accessory protein targets SAMHD1 for degradation in the nucleus. J Virol 86:12552–12560. doi: 10.1128/JVI.01657-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, Bloch N, Maudet C, Bertrand M, Gramberg T, Pancino G, Priet S, Canard B, Laguette N, Benkirane M, Transy C, Landau NR, Kim B, Margottin-Goguet F. 2012. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol 13:223–228. doi: 10.1038/ni.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amie SM, Noble E, Kim B. 2013. Intracellular nucleotide levels and the control of retroviral infections. Virology 436:247–254. doi: 10.1016/j.virol.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, de Carvalho LP, Stoye JP, Crow YJ, Taylor IA, Webb M. 2011. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480:379–382. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- 17.Kim B, Nguyen LA, Daddacha W, Hollenbaugh JA. 2012. Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J Biol Chem 287:21570–21574. doi: 10.1074/jbc.C112.374843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, Yatim A, Schwartz O, Laguette N, Benkirane M. 2012. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4+ T cells. Retrovirology 9:87. doi: 10.1186/1742-4690-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnold LH, Groom HC, Kunzelmann S, Schwefel D, Caswell SJ, Ordonez P, Mann MC, Rueschenbaum S, Goldstone DC, Pennell S, Howell SA, Stoye JP, Webb M, Taylor IA, Bishop KN. 2015. Phospho-dependent regulation of SAMHD1 oligomerisation couples catalysis and restriction. PLoS Pathog 11:e1005194. doi: 10.1371/journal.ppat.1005194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofmann H, Norton TD, Schultz ML, Polsky SB, Sunseri N, Landau NR. 2013. Inhibition of CUL4A Neddylation causes a reversible block to SAMHD1-mediated restriction of HIV-1. J Virol 87:11741–11750. doi: 10.1128/JVI.02002-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cribier A, Descours B, Valadao AL, Laguette N, Benkirane M. 2013. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep 3:1036–1043. doi: 10.1016/j.celrep.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 22.Badia R, Pujantell M, Riveira-Munoz E, Puig T, Torres-Torronteras J, Marti R, Clotet B, Ampudia RM, Vives-Pi M, Este JA, Ballana E. 2016. The G1/S specific cyclin D2 is a regulator of HIV-1 restriction in non-proliferating cells. PLoS Pathog 12:e1005829. doi: 10.1371/journal.ppat.1005829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valle-Casuso JC, Allouch A, David A, Lenzi GM, Studdard L, Barre-Sinoussi F, Muller-Trutwin M, Kim B, Pancino G, Saez-Cirion A. 2017. p21 restricts HIV-1 in monocyte-derived dendritic cells through the reduction of dNTP biosynthesis and regulation of SAMHD1 antiviral activity. J Virol 91:e01324-17. doi: 10.1128/JVI.01324-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franzolin E, Pontarin G, Rampazzo C, Miazzi C, Ferraro P, Palumbo E, Reichard P, Bianchi V. 2013. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc Natl Acad Sci U S A 110:14272–14277. doi: 10.1073/pnas.1312033110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allouch A, David A, Amie SM, Lahouassa H, Chartier L, Margottin-Goguet F, Barre-Sinoussi F, Kim B, Saez-Cirion A, Pancino G. 2013. p21-mediated RNR2 repression restricts HIV-1 replication in macrophages by inhibiting dNTP biosynthesis pathway. Proc Natl Acad Sci U S A 110:E3997–E4006. doi: 10.1073/pnas.1306719110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan J, Hao C, DeLucia M, Swanson S, Florens L, Washburn MP, Ahn J, Skowronski J. 2015. CyclinA2-cyclin-dependent kinase regulates SAMHD1 protein phosphohydrolase domain. J Biol Chem 290:13279–13292. doi: 10.1074/jbc.M115.646588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schott K, Fuchs NV, Derua R, Mahboubi B, Schnellbacher E, Seifried J, Tondera C, Schmitz H, Shepard C, Brandariz-Nunez A, Diaz-Griffero F, Reuter A, Kim B, Janssens V, König R. 2018. Dephosphorylation of the HIV-1 restriction factor SAMHD1 is mediated by PP2A-B55α holoenzymes during mitotic exit. Nat Commun 9:2227. doi: 10.1038/s41467-018-04671-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White TE, Brandariz-Nunez A, Valle-Casuso JC, Amie S, Nguyen LA, Kim B, Tuzova M, Diaz-Griffero F. 2013. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 13:441–451. doi: 10.1016/j.chom.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.St Gelais C, Kim SH, Ding L, Yount J, Ivanov D, Spearman P, Wu L. 2016. A putative cyclin-binding motif in human SAMHD1 contributes to protein phosphorylation, localization and stability. J Biol Chem doi: 10.1074/jbc.M116.753947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welbourn S, Dutta SM, Semmes OJ, Strebel K. 2013. Restriction of virus infection but not catalytic dNTPase activity is regulated by phosphorylation of SAMHD1. J Virol 87:11516–11524. doi: 10.1128/JVI.01642-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhattacharya A, Wang Z, White T, Buffone C, Nguyen LA, Shepard CN, Kim B, Demeler B, Diaz-Griffero F, Ivanov DN. 2016. Effects of T592 phosphomimetic mutations on tetramer stability and dNTPase activity of SAMHD1 cannot explain the retroviral restriction defect. Sci Rep 6:31353. doi: 10.1038/srep31353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, Balakrishnan M, Bambara RA, Planelles V, Dewhurst S, Kim B. 2004. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem 279:51545–51553. doi: 10.1074/jbc.M408573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kennedy EM, Amie SM, Bambara RA, Kim B. 2012. Frequent incorporation of ribonucleotides during HIV-1 reverse transcription and their attenuated repair in macrophages. J Biol Chem 287:14280–14288. doi: 10.1074/jbc.M112.348482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathews CK. 2015. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat Rev Cancer 15:528–539. doi: 10.1038/nrc3981. [DOI] [PubMed] [Google Scholar]
- 35.Kohnken R, Kodigepalli KM, Wu L. 2015. Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol Cancer 14:176. doi: 10.1186/s12943-015-0446-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A. 2003. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem 278:25752–25757. doi: 10.1074/jbc.M301774200. [DOI] [PubMed] [Google Scholar]
- 37.Sheaff RJ, Singer JD, Swanger J, Smitherman M, Roberts JM, Clurman BE. 2000. Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Mol Cell 5:403–410. doi: 10.1016/S1097-2765(00)80435-9. [DOI] [PubMed] [Google Scholar]
- 38.Shi B, Sharifi HJ, DiGrigoli S, Kinnetz M, Mellon K, Hu W, de Noronha CMC. 2018. Inhibition of HIV early replication by the p53 and its downstream gene p21. Virol J 15:53. doi: 10.1186/s12985-018-0959-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pauls E, Ruiz A, Riveira-Munoz E, Permanyer M, Badia R, Clotet B, Keppler OT, Ballana E, Este JA. 2014. p21 regulates the HIV-1 restriction factor SAMHD1. Proc Natl Acad Sci U S A 111:E1322–E1324. doi: 10.1073/pnas.1322059111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allouch A, David A, Amie SM, Lahouassa H, Chartier L, Margottin-Goguet F, Barre-Sinoussi F, Kim B, Saez-Cirion A, Pancino G. 2014. Reply to Pauls et al.: p21 is a master regulator of HIV replication in macrophages through dNTP synthesis block. Proc Natl Acad Sci U S A 111:E1325–E1326. doi: 10.1073/pnas.1322699111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. 2002. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem 277:9976–9981. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- 42.Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, Fuchs SY, Shuai K, Zhang DE. 2006. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J 25:2358–2367. doi: 10.1038/sj.emboj.7601149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, de la Torre JC, Zhang DE. 2004. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat Med 10:1374–1378. doi: 10.1038/nm1133. [DOI] [PubMed] [Google Scholar]
- 44.Ketscher L, Hannss R, Morales DJ, Basters A, Guerra S, Goldmann T, Hausmann A, Prinz M, Naumann R, Pekosz A, Utermohlen O, Lenschow DJ, Knobeloch KP. 2015. Selective inactivation of USP18 isopeptidase activity in vivo enhances ISG15 conjugation and viral resistance. Proc Natl Acad Sci U S A 112:1577–1582. doi: 10.1073/pnas.1412881112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goldmann T, Zeller N, Raasch J, Kierdorf K, Frenzel K, Ketscher L, Basters A, Staszewski O, Brendecke SM, Spiess A, Tay TL, Kreutz C, Timmer J, Mancini GM, Blank T, Fritz G, Biber K, Lang R, Malo D, Merkler D, Heikenwalder M, Knobeloch KP, Prinz M. 2015. USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J 34:1612–1629. doi: 10.15252/embj.201490791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Honke N, Shaabani N, Zhang DE, Hardt C, Lang KS. 2016. Multiple functions of USP18. Cell Death Dis 7:e2444. doi: 10.1038/cddis.2016.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Li H, Zhong B, Blonska M, Gorjestani S, Yan M, Tian Q, Zhang DE, Lin X, Dong C. 2013. USP18 inhibits NF-κB and NFAT activation during Th17 differentiation by deubiquitinating the TAK1-TAB1 complex. J Exp Med 210:1575–1590. doi: 10.1084/jem.20122327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Z, Xian H, Hu J, Tian S, Qin Y, Wang RF, Cui J. 2015. USP18 negatively regulates NF-κB signaling by targeting TAK1 and NEMO for deubiquitination through distinct mechanisms. Sci Rep 5:12738. doi: 10.1038/srep12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Honke N, Shaabani N, Cadeddu G, Sorg UR, Zhang DE, Trilling M, Klingel K, Sauter M, Kandolf R, Gailus N, van Rooijen N, Burkart C, Baldus SE, Grusdat M, Lohning M, Hengel H, Pfeffer K, Tanaka M, Häussinger D, Recher M, Lang PA, Lang KS. 2012. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat Immunol 13:51–57. doi: 10.1038/ni.2169. [DOI] [PubMed] [Google Scholar]
- 50.Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, Tezcan I, Rice GI, Chen C, Mansouri N, Mahdaviani SA, Itan Y, Boisson B, Okada S, Zeng L, Wang X, Jiang H, Liu W, Han T, Liu D, Ma T, Wang B, Liu M, Liu JY, Wang QK, Yalnizoglu D, Radoshevich L, Uze G, Gros P, Rozenberg F, Zhang SY, Jouanguy E, Bustamante J, Garcia-Sastre A, Abel L, Lebon P, Notarangelo LD, Crow YJ, Boisson-Dupuis S, Casanova JL, Pellegrini S. 2015. Human intracellular ISG15 prevents interferon-alpha/beta overamplification and auto-inflammation. Nature 517:89–93. doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tokarz S, Berset C, La Rue J, Friedman K, Nakayama K, Nakayama K, Zhang DE, Lanker S. 2004. The ISG15 isopeptidase UBP43 is regulated by proteolysis via the SCFSkp2 ubiquitin ligase. J Biol Chem 279:46424–46430. doi: 10.1074/jbc.M403189200. [DOI] [PubMed] [Google Scholar]
- 52.Kim KI, Malakhova OA, Hoebe K, Yan M, Beutler B, Zhang DE. 2005. Enhanced antibacterial potential in UBP43-deficient mice against Salmonella typhimurium infection by up-regulating type I IFN signaling. J Immunol 175:847–854. doi: 10.4049/jimmunol.175.2.847. [DOI] [PubMed] [Google Scholar]
- 53.Sewald X, Ladinsky MS, Uchil PD, Beloor J, Pi R, Herrmann C, Motamedi N, Murooka TT, Brehm MA, Greiner DL, Shultz LD, Mempel TR, Bjorkman PJ, Kumar P, Mothes W. 2015. Retroviruses use CD169-mediated trans-infection of permissive lymphocytes to establish infection. Science 350:563–567. doi: 10.1126/science.aab2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katayama T, Nakanishi K, Nishihara H, Kamiyama N, Nakagawa T, Kamiyama T, Iseki K, Tanaka S, Todo S. 2007. Type I interferon prolongs cell cycle progression via p21WAF1/CIP1 induction in human colon cancer cells. Int J Oncol 31:613–620. [PubMed] [Google Scholar]
- 55.Mandal M, Bandyopadhyay D, Goepfert TM, Kumar R. 1998. Interferon-induces expression of cyclin-dependent kinase-inhibitors p21WAF1 and p27Kip1 that prevent activation of cyclin-dependent kinase by CDK-activating kinase (CAK). Oncogene 16:217–225. doi: 10.1038/sj.onc.1201529. [DOI] [PubMed] [Google Scholar]
- 56.St Gelais C, de Silva S, Hach JC, White TE, Diaz-Griffero F, Yount JS, Wu L. 2014. Identification of cellular proteins interacting with the retroviral restriction factor SAMHD1. J Virol 88:5834–5844. doi: 10.1128/JVI.00155-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abbas T, Dutta A. 2009. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wittmann S, Behrendt R, Eissmann K, Volkmann B, Thomas D, Ebert T, Cribier A, Benkirane M, Hornung V, Bouzas NF, Gramberg T. 2015. Phosphorylation of murine SAMHD1 regulates its antiretroviral activity. Retrovirology 12:103. doi: 10.1186/s12977-015-0229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Münk C, Brandt SM, Lucero G, Landau NR. 2002. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc Natl Acad Sci U S A 99:13843–13848. doi: 10.1073/pnas.212400099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cavrois M, De Noronha C, Greene WC. 2002. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol 20:1151–1154. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- 61.Liu LQ, Ilaria R Jr, Kingsley PD, Iwama A, van Etten RA, Palis J, Zhang DE. 1999. A novel ubiquitin-specific protease, UBP43, cloned from leukemia fusion protein AML1-ETO-expressing mice, functions in hematopoietic cell differentiation. Mol Cell Biol 19:3029–3038. doi: 10.1128/MCB.19.4.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Honke N, Shaabani N, Zhang DE, Iliakis G, Xu HC, Häussinger D, Recher M, Lohning M, Lang PA, Lang KS. 2013. Usp18 driven enforced viral replication in dendritic cells contributes to break of immunological tolerance in autoimmune diabetes. PLoS Pathog 9:e1003650. doi: 10.1371/journal.ppat.1003650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Asada M, Yamada T, Ichijo H, Delia D, Miyazono K, Fukumuro K, Mizutani S. 1999. Apoptosis inhibitory activity of cytoplasmic p21Cip1/WAF1 in monocytic differentiation. EMBO J 18:1223–1234. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu M, Iavarone A, Freedman LP. 1996. Transcriptional activation of the human p21WAF1/CIP1 gene by retinoic acid receptor. Correlation with retinoid induction of U937 cell differentiation. J Biol Chem 271:31723–31728. [DOI] [PubMed] [Google Scholar]
- 65.Hobeika AC, Etienne W, Torres BA, Johnson HM, Subramaniam PS. 1999. IFN-gamma induction of p21WAF1 is required for cell cycle inhibition and suppression of apoptosis. J Interferon Cytokine Res 19:1351–1361. doi: 10.1089/107999099312812. [DOI] [PubMed] [Google Scholar]
- 66.El-Deiry WS. 2016. p21WAF1 mediates cell cycle inhibition, relevant to cancer suppression and therapy. Cancer Res 76:5189–5191. doi: 10.1158/1538-7445.AM2016-5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bergamaschi A, David A, Le Rouzic E, Nisole S, Barre-Sinoussi F, Pancino G. 2009. The CDK inhibitor p21Cip1/WAF1 is induced by FcγR activation and restricts the replication of human immunodeficiency virus type 1 and related primate lentiviruses in human macrophages. J Virol 83:12253–12265. doi: 10.1128/JVI.01395-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lloberas J, Celada A. 2009. p21waf1/CIP1, a CDK inhibitor and a negative feedback system that controls macrophage activation. Eur J Immunol 39:691–694. doi: 10.1002/eji.200939262. [DOI] [PubMed] [Google Scholar]
- 69.Lane AN, Fan TW. 2015. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res 43:2466–2485. doi: 10.1093/nar/gkv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang YF, Wee S, Gunaratne J, Lane DP, Bulavin DV. 2014. Isg15 controls p53 stability and functions. Cell Cycle 13:2200–2210. doi: 10.4161/cc.29209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Touitou R, Richardson J, Bose S, Nakanishi M, Rivett J, Allday MJ. 2001. A degradation signal located in the C terminus of p21WAF1/CIP1 is a binding site for the C8 alpha-subunit of the 20S proteasome. EMBO J 20:2367–2375. doi: 10.1093/emboj/20.10.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen X, Barton LF, Chi Y, Clurman BE, Roberts JM. 2007. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGγ proteasome. Mol Cell 26:843–852. doi: 10.1016/j.molcel.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li X, Amazit L, Long W, Lonard DM, Monaco JJ, O'Malley BW. 2007. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGγ-proteasome pathway. Mol Cell 26:831–842. doi: 10.1016/j.molcel.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 74.Grez M, Akgun E, Hilberg F, Ostertag W. 1990. Embryonic stem cell virus, a recombinant murine retrovirus with expression in embryonic stem cells. Proc Natl Acad Sci U S A 87:9202–9206. doi: 10.1073/pnas.87.23.9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kootstra NA, Münk C, Tonnu N, Landau NR, Verma IM. 2003. Abrogation of postentry restriction of HIV-1-based lentiviral vector transduction in simian cells. Proc Natl Acad Sci U S A 100:1298–1303. doi: 10.1073/pnas.0337541100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bahr A, Singer A, Hain A, Vasudevan AA, Schilling M, Reh J, Riess M, Panitz S, Serrano V, Schweizer M, König R, Chanda S, Häussinger D, Kochs G, Lindemann D, Münk C. 2016. Interferon but not MxB inhibits foamy retroviruses. Virology 488:51–60. doi: 10.1016/j.virol.2015.10.034. [DOI] [PubMed] [Google Scholar]
- 77.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. 1998. A third-generation lentivirus vector with a conditional packaging system. J Virol 72:8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sunseri N, O'Brien M, Bhardwaj N, Landau NR. 2011. Human immunodeficiency virus type 1 modified to package simian immunodeficiency virus Vpx efficiently infects macrophages and dendritic cells. J Virol 85:6263–6274. doi: 10.1128/JVI.00346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Soneoka Y, Cannon PM, Ramsdale EE, Griffiths JC, Romano G, Kingsman SM, Kingsman AJ. 1995. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res 23:628–633. doi: 10.1093/nar/23.4.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. 1980. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer 26:171–176. doi: 10.1002/ijc.2910260208. [DOI] [PubMed] [Google Scholar]
- 81.Pear WS, Nolan GP, Scott ML, Baltimore D. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A 90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. 1987. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol Cell Biol 7:379–387. doi: 10.1128/MCB.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McAllister RM, Gardner MB, Greene AE, Bradt C, Nichols WW, Landing BH. 1971. Cultivation in vitro of cells derived from a human osteosarcoma. Cancer 27:397–402. doi: 10.1002/1097-0142(197102)27:2<397::AID-CNCR2820270224>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 84.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol 72:2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Takeuchi Y, McClure MO, Pizzato M. 2008. Identification of gammaretroviruses constitutively released from cell lines used for human immunodeficiency virus research. J Virol 82:12585–12588. doi: 10.1128/JVI.01726-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O'Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. 2000. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol 74:8358–8367. doi: 10.1128/JVI.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Platt EJ, Bilska M, Kozak SL, Kabat D, Montefiori DC. 2009. Evidence that ecotropic murine leukemia virus contamination in TZM-bl cells does not affect the outcome of neutralizing antibody assays with human immunodeficiency virus type 1. J Virol 83:8289–8292. doi: 10.1128/JVI.00709-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu G, Moriyama EN. 2004. Vector NTI, a balanced all-in-one sequence analysis suite. Brief Bioinform 5:378–388. doi: 10.1093/bib/5.4.378. [DOI] [PubMed] [Google Scholar]
- 92.Hain A, Krämer M, Linka RM, Nakhaei-Rad S, Ahmadian MR, Häussinger D, Borkhardt A, Münk C. 2018. IL-2 inducible kinase ITK is critical for HIV-1 infection of Jurkat T cells. Sci Rep 8:3217. doi: 10.1038/s41598-018-21344-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat Med 7:631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 94.Hofmann H, Vanwalscappel B, Bloch N, Landau NR. 2016. TLR7/8 agonist induces a postentry SAMHD1-independent block to HIV-1 infection of monocytes. Retrovirology 13:83. doi: 10.1186/s12977-016-0316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Borodovsky A, Ovaa H, Kolli N, Gan-Erdene T, Wilkinson KD, Ploegh HL, Kessler BM. 2002. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem Biol 9:1149–1159. doi: 10.1016/S1074-5521(02)00248-X. [DOI] [PubMed] [Google Scholar]