

Abstract
Interstitial lung and liver disease (ILLD) is caused by biallelic mutations in the methionyl-tRNA synthetase (MARS) gene. To date, no genetic changes other than missense variants were reported in the literature. Here, we report a five-month old female infant with typical ILLD (failure to thrive, developmental delay, jaundice, diffuse interstitial lung disease, hepatomegaly with severe steatosis, anemia, and thrombocytosis) showing novel phenotypes such as kidney stones, acetabular dysplasia, prolonged fever, and extreme leukocytosis. Whole exome sequencing revealed a novel truncating variant (c.2158C>T/p.Gln720Stop) together with a novel tri-nucleotide insertion (c.893_894insTCG that caused the insertion of an arginine at amino acid position 299) in the MARS gene.
Keywords: Methionyl-tRNA synthetase, Infant, Kidney stone, Hip dysplasia, Leukocytosis, Interstitial lung and liver disease, Methionyl-tRNA synthetase gene
Core tip: Previously reported cases of interstitial lung and liver disease (ILLD) were associated with biallelic missense mutations in the methionyl-tRNA synthetase (MARS) gene. Here, we report a Chinese infant with typical ILLD (failure to thrive, developmental delay, interstitial lung disease, cholestasis, hepatomegaly, steatosis, anemia, and thrombocytosis) with novel phenotypes, such as kidney stones, acetabular dysplasia, prolonged fever, and extreme leukocytosis. Whole exome sequencing revealed a novel truncating variant (c.2158C>T/p.Gln720Stop), and a novel tri-nucleotide insertion (c.893_894insTCG) in the MARS gene. Despite the resolution of cholestasis, this patient died of respiratory failure at the age of 11 mo.
INTRODUCTION
The methionyl-tRNA synthetase (MARS) gene encodes cytoplasmic methionyl-tRNA synthetase (MetRS) responsible for catalyzing the ligation of methionine to tRNA[1]. MetRS belongs to a family of aminoacyl-tRNA synthetases that play critical roles in protein biosynthesis by charging tRNAs with their cognate amino acids[2]. Interstitial lung and liver disease (ILLD) (OMIM#615486) is caused by homozygous or compound heterozygous mutations in the MARS gene (156560) on chromosome 12q13[3-5]. Heterozygous MARS mutations have been reported to be associated with autosomal dominant Charcot-Marie-Tooth disease (CMT)[6-9]. The same MARS mutation may cause both ILLD and CMT[10]. MARS is also a candidate gene for hereditary spastic paraplegias (HSPs), a neuro-degenerative motor neuron disorder[11]. To date, no genetic changes other than missense variants have been reported in the literature. Here, we report a Chinese infant with lethal ILLD showing novel phenotypes such as kidney stones, acetabular dysplasia, prolonged fever, and extreme leukocytosis. Whole exome sequencing revealed a novel truncating variant together with a novel tri-nucleotide insertion in the MARS gene.
CASE REPORT
A five-month old female infant was presented with a failure to thrive, developmental delay, jaundice, and dark urine. She was born full-term with a normal birth weight (3100 g) after an uncomplicated first pregnancy and vaginal delivery. Weight gain and developmental milestones were normal until three months of age (weighted 6000 g), when she failed to thrive with a body weight of 5700 g at the age of 5 mo without the ability of rolling over.
At in-patient admission, this patient was 5.2 mo old with a body weight of 5500 g (2nd percentile by WHO standards), length of 55 cm (lower than the 1st percentile), and head circumference of 39 cm (2nd percentile). This infant had prolonged low-grade fever, pulmonary effusion, diffuse interstitial lung disease, significant leukocytosis, high procalcitonin (PCT)/CRP levels, and required nasal oxygen therapy. Serial chest X-rays showed some improvement in pulmonary effusion, but no improvement in interstitial lung involvement (Figure 1A). After serial antibiotic treatments (ceftriaxone, cefoperazone + slubactam, meropenem, norvancomycin, and fluconazole), body temperature was normalized, oxygen therapy was no longer needed, and leukocytosis improved, however the interstitial lung disease stayed the same. After treatment with ursodeoxycholic acid and fat-soluble vitamins, cholestasis improved significantly (Table 1).
Figure 1.
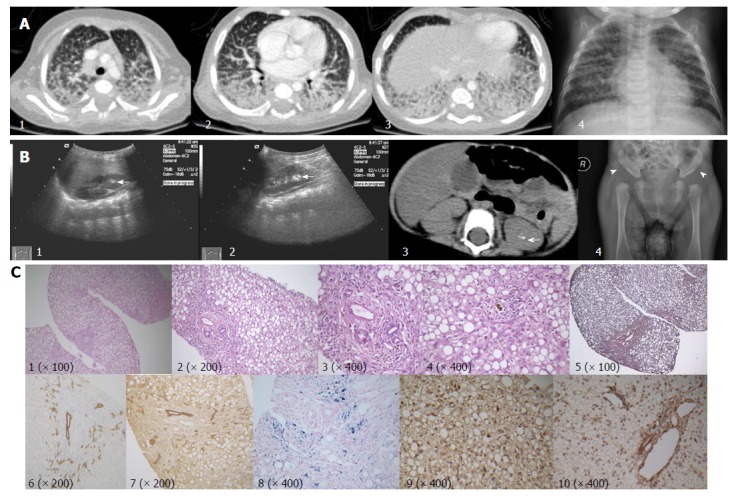
Imaging and histopathological features. A: Contrast enhanced pulmonary CT scan (1-3), and chest X-ray (4) showing pulmonary effusion with marked interstitial lung involvement; B: Hyper-echoic lesions consistent with stone formation on ultrasonography (arrows; 1, right kidney; 2, left kidney) and non-contrast abdominal computed tomography scan (arrow, 3). Acetabular dysplasia (4, arrowhead showing abnormally shallow hip socket); C: Liver biopsy (all originally magnified principal images): severe steatosis of hepatic cells with ballooning, lobular disarray, and cholestasis (1-4), mild fibrosis (5), mild lymphocyte infiltration (4), bile duct proliferation (6 CK-7, 7 CK-19), and hepatic iron deposition (8). MARS immunohistochemistry staining, coarsely granular pigments within the cytoplasm in the index patient (9), but not in samples of a healthy control (10). MARS: Methionyl-tRNA synthetase gene.
Table 1.
Changes in complete blood count, procalcitonin, serum biochemistry, and blood coagulation profiles
| Age (mo) (1in-patient admission; 2discharge to out-patient follow-up) | 5.1 | 5.61 | 6 | 6.2 | 6.5 | 6.8 | 7 | 7.22 | 9.5 | |
| Complete blood count (reference range) | White blood cell (4-10 × 109/L) | 16.9 | 21.1 | 71.7 | 26.4 | 33.7 | 45.8 | 30.3 | 24.3 | 14.3 |
| Neutrophil (20%-50%) | 58.1 | 39.8 | 58.0 | 63.1 | 62.0 | 63.0 | 62.7 | 64.4 | 38.9 | |
| Lymphocyte (45%-75%) | 36.2 | 53.1 | 31.1 | 28.7 | 28.9 | 15.0 | 29.5 | 27.8 | 51.4 | |
| Abnormal lymphocytes (0%) | NA | 0.0 | 0.0 | NA | 0.0 | 17.0 | 0.0 | 0.0 | NA | |
| Platelet count (100-300 × 109/L) | 764.0 | 513.0 | 993.0 | 464 | 387.0 | 494.0 | 279.0 | 397 | 386.0 | |
| Hemoglobin (110-160 g/L) | 78.0 | 85.2 | 78.2 | 60.1 | 64.0 | 65.2 | 90.0 | 88.0 | 122.0 | |
| Red blood cell count (4.0-5.5 × 1012 /L) | 3.5 | 3.1 | 2.8 | 2.0 | 2.0 | 2.2 | 2.9 | 2.9 | 4.3 | |
| Reticulocyte (0.5%-1.5%) | NA | 2.9 | 6.7 | NA | 6.3 | 7.8 | 3.3 | 6.8 | 1.0 | |
| C-reactive protein (< 8 mg/L) | 1.0 | 8.0 | 90.0 | 32.0 | 43.0 | 37.0 | 45.0 | 8.0 | 8.0 | |
| Procalcitonin (< 0.05 ng/mL) | NA | 4.6 | 17.4 | 7.7 | 13.4 | NA | NA | NA | NA | |
| Serum biochemistry (reference range) | Albumin (35-55 g/L) | 29.0 | 34.6 | 27.3 | 30.8 | 32.3 | 28.7 | 38.5 | 39.1 | 43.0 |
| Alanine aminotransferase (0-40 IU/L) | 41.0 | 45.0 | 17.0 | 13.0 | 4.0 | 50.0 | 49.0 | 38.0 | 29.0 | |
| Aspartate aminotransferase (0-40 IU/L) | 100.0 | 104.0 | 46.0 | 37.0 | 66.0 | 98.0 | 70.0 | 62.0 | 41.0 | |
| Total bilirubin (5.1-17.1 μmol/L) | 68.0 | 120.4 | 133.0 | 132.9 | 126.8 | 110.6 | 90.1 | 42.9 | 8.1 | |
| Direct bilirubin (0-6 μmol/L) | 53.0 | 76.9 | 93.7 | 96.1 | 86.6 | 70.4 | 61.8 | 29.8 | 4.4 | |
| γ-glutamyl transferase (7-50 IU/L) | 73.0 | 61.0 | 76.0 | 58.0 | 54.0 | 57.0 | 107.0 | 230.0 | 122.0 | |
| Total bile acid (0-10 μmol/L) | NA | 182.8 | 123.3 | 152.4 | 137.2 | 157.4 | 311.7 | 282.3 | 34.6 | |
| Alkaline phosphatase (42-383 IU/L) | 307.0 | 137.0 | 149.0 | 119.0 | 122.0 | 148.0 | 178.0 | 214.0 | 378.0 | |
| Blood glucose (3.9-5.8 mmol/L) | NA | 1.2 | 1.6 | 8.4 | 1.1 | NA | NA | 3.6 | NA | |
| Lactic acid (0-2 mmol/L) | NA | 3.9 | NA | 3.6 | 3.6 | NA | NA | NA | NA | |
| Ammonia (10-47 μmol/L) | NA | 88.0 | NA | NA | NA | NA | NA | 55.0 | NA | |
| Total cholesterol (3.1-5.2 mmol/L) | 3.1 | 2.0 | NA | 2.3 | 2.5 | NA | 2.8 | 4.4 | 3.1 | |
| LDL-cholesterol (1.30-3.90 mmol/L) | NA | NA | NA | 1.0 | NA | NA | NA | NA | NA | |
| HDL-cholesterol (0.91-2.05 mmol/L) | NA | NA | NA | 0.3 | NA | NA | NA | NA | NA | |
| Triglyceride (0.56-1.70 mmol/L) | NA | 2.0 | NA | 2.7 | 2.1 | NA | 2.1 | 1.8 | 1.5 | |
| Blood coagulation profiles (reference range) | Activated partial thromboplastin time (28.0-44.5 s) | NA | 48.1 | NA | 57.5 | 56.4 | 53.9 | 47.7 | 42.3 | 43.8 |
| D-dimer (0-0.3 mg/L) | NA | 0.94 | NA | 2.06 | 1.15 | 0.97 | 0.7 | 0.51 | NA | |
| Fibrinogen (2-4 g/L) | NA | 1.45 | NA | 1.82 | 2.29 | 2.54 | 3.03 | 3.46 | 3.44 | |
| Fibrinogen degradation products (0-5 μg/ML) | NA | 1.31 | NA | 5.22 | 2.35 | 2.78 | 1.47 | 1.16 | NA | |
| Thrombin time (14-21 s) | NA | 20.4 | NA | 19.1 | 19.9 | 19.9 | 15.8 | 18.4 | 15.2 | |
| International normalized ratio (0.8-1.2) | NA | NA | NA | 1.29 | 1.26 | 1.35 | 1.3 | 1.03 | 0.99 | |
| Prothrombin time (12.0-14.8 s) | NA | NA | NA | 16 | 15.7 | 16.5 | 16.1 | 13.5 | 13.1 | |
| Prothrombin time activity (80%-100%) | NA | NA | NA | 67 | 69 | 63 | 66 | 95 | 103 | |
NA: Not available.
The patient was discharged with normal oxygen saturation on room air without apparent respiratory distress or cough. A liver function test and complete blood count were normal at a 9.5 mo follow-up. However, the infant was admitted to a provincial level pediatric intensive care unit for acute respiratory distress at 11 mo of age and received mechanical ventilation. Despite treatment, she died of respiratory failure and hypoxic encephalopathy.
A genetic cause was suspected due to multiple system involvement, although a liver panel consisting of 41 genes (Table 2) related to liver diseases came back negative. Lysosomal storage disease was considered, but an enzyme panel for the screening of common lysosomal storage diseases was normal, as was the urine acidoglycoprotein level. This patient was enrolled for the undiagnosed disease patient program in our hospital, and whole exome sequencing was ordered. Compound heterozygous MARS gene variants, c.2158C>T/p.Gln720Stop and c.893_894insTCG/p.Arg299dup, were detected. Presence of these mutations was confirmed with Sanger sequencing, and parental origins were ascertained. Both variants were not reported in the dbSNP137 (http://www. ncbi.nlm.nih.gov/snp/), 1000 Genome Database (http://www.1000genomes.org/), and Exome Variant Server (http://evs.gs.washington.edu/EVS/). The c.2158C>T mutation was inherited from the healthy mother, which caused the change of a glutamine amino acid at position 720 to a stop codon, which was predicted to be disease-causing by MutationTaster (http://www.mutationtaster.org). The tri-nucleotide insertion (c.893_894insTCG) inherited from her healthy father caused the insertion of a single amino acid (arginine) at position 299, which was predicted to be disease-causing by MutationTaster (Figure 2A). The detailed genetic testing results and secondary findings are provided in Table 2.
Table 2.
Genetic testing results
| Genetic Tests | Gene | Transcript ID | Associated conditions (Inheritance patterns) in OMIM | Variant | Amino-acid change | Hom/Het | Parental origin |
Prediction of pathogenicity |
|||
| Mutation taster | SIFT | Provean | Polyphen2 | ||||||||
| Liver Panel1 | ATP8B1 | NM _005603 | Cholestasis, benign recurrent, intrahepatic (AR); cholestasis, intrahepatic, of pregnancy, 1 (AD); cholestasis, progressive familial intrahepatic 1 (AR) | c.234C> G | p.His78Gln | Het | NA | Polymorphism | Tolerated | Neutral | Benign |
| c.1729A>G | p.Ile577Val | Het | NA | Polymorphism | Tolerated | Neutral | Possibly damaging | ||||
| c.2021T>C | p. Met674Thr | Het | NA | Polymorphism | Tolerated | Neutral | Benign | ||||
| c.3477C>T | Synonymous | Het | NA | Polymorphism | Tolerated | Neutral | NA | ||||
| c.3744C>A | Synonymous | Het | NA | Polymorphism | Tolerated | Neutral | NA | ||||
| Whole exome sequencing | MARS | NM_004990 | Charcot-Marie-Tooth disease, axonal, type 2U (AD); Interstitial lung and liver disease (AR) | c.2158C>T | p.Gln720Stop | Het | Maternal | Disease causing | NA | NA | NA |
| c.893_894insTCG | p.Arg299dup | Het | Paternal | Disease causing | NA | Deleterious | NA | ||||
| ATP8B1 | NM_005603 | Cholestasis, benign recurrent, intrahepatic (AR); cholestasis, intrahepatic, of pregnancy, 1 (AD); cholestasis, progressive familial intrahepatic 1 (AR) | c.2021T>C | p. Met674Thr | Het | Paternal | polymorphism | Tolerated | Neutral | Benign | |
| CPT1A | NM_001876 | CPT deficiency, hepatic, type IA (AR) | c.1163+5G>A | - | Het | Maternal | Disease causing | NA | NA | NA | |
| LRPPRC | NM_133259 | Leigh syndrome, French-Canadian type (AR) | c.2965C>T | p.Arg989Cys | Het | Maternal | Disease causing | Damaging | Deleterious | Probably damaging | |
| FLG | NM_002106 | Ichthyosis vulgaris (AD); (Dermatitis, atopic, susceptibility to, 2) | c.5841G>A | p.Trp1947Stop | Het | Maternal | Disease causing | NA | NA | NA | |
| G6PD | NM_00104251 | Hemolytic anemia, G6PD deficient (favism) (XLD); (Resistance to malaria due to G6PD deficiency) | c.241C>T | p.Arg81Cys | Het | Maternal | Disease causing | Damaging | Deleterious | Benign | |
| POMGNT1 | NM_017739 | Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type A, 3 (AR); Muscular dystrophy-dystroglycanopathy (congenital with mental retardation), type B, 3 (AR); Muscular dystrophy-dystroglycanopathy (limb-girdle), type C, 3 (AR); Retinitis pigmentosa 76 (AR) | c.794G>A | p.Arg265His | Het | Maternal | Disease causing | Damaging | Deleterious | Probably damaging | |
| SERPINC1 | NM_000488 | Thrombophilia due to antithrombin III deficiency (AD/AR) | c.719A>G | p.Asn240Ser | Het | Maternal | Polymorphism | Tolerated | Neutral | Benign | |
| TG | NM_003235 | Thyroid dyshormonogenesis 3 (AR); (autoimmune thyroid disease, susceptibility to, 3) | c.5791A>G | p.Ile1931Val | Het | Paternal | Polymorphism | Tolerated | Neutral | Benign | |
| USH2A | NM_206933 | Retinitis pigmentosa 39; Usher syndrome type 2A (AR) | c.8559-2A>G | - | Het | Paternal | Disease causing | NA | NA | NA | |
Genes included in liver panel: ATP8B1, ABCB11, ABCB4, TJP2, BAAT, CLDN1, HSD3B7, AKR1D1, CYP7B1, AMACR, CYP27A1, DHCR7, JAG1, NOTCH2, SLC25A13, DGUOK, MPV17, FAH, ABCC2, UGT1A1, NPC1, NPC2, GALT, GALE, ALDOA, ALDOB, KRT18, KRT8, CIRH1A, CFTR, GFDM1, EARS2, HSD17B4, LIPA, PEX1, PEX5, POU1F1, HESX1, SERPINA1, VIPAS39, and VPS33B. NA: Not available.
Figure 2.
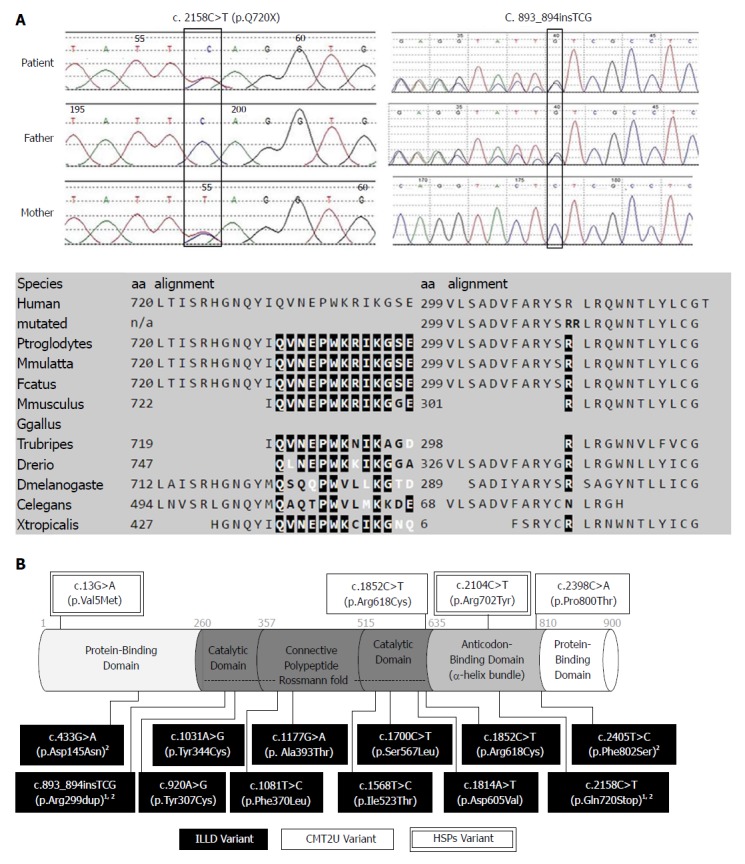
Genetic testing results, protein features, and distribution of reported variants within the methionyl-tRNA synthetase protein. A: Sanger sequencing confirmation of the index case and parents, both variants affect highly conserved amino acid residues of the MetRS protein; B: Illustration of MetRS protein domains, location of amino acid changes of the reported variants so far. 1Variants from our report; 2Variants from Chinese ILLD cases. MetRS: Methionyl-tRNA synthetase; ILLD: Interstitial lung and liver disease.
Liver biopsy results showed severe steatosis of hepatic cells with ballooning, lobular disarray, and cholestasis. Mild changes, such as fibrosis, lymphocyte infiltration, and bile duct proliferation, were seen within the portal region. Hepatic iron deposition was seen after iron staining, but copper staining was negative (Figure 1C). Sinosoids and Kupffer cells seemed normal. Immunohistochemical staining for hepatitis B surface antigen, core antigen, Epstein-Barr virus, and langerin cells were negative. Immunohistochemical staining for cholestasis-related proteins, such as BSEP, MDR3, MRP2, TJP2, and MYO5B, were all normal. After genetic diagnosis, we used a rabbit anti-MARS monoclonal antibody (purchased from http://www.abcam.cn, product code: ab180497) to perform immunohistochemical staining on paraffin-embedded liver biopsy samples. When compared to a normal liver sample (donated for liver transplantation), coarsely granular pigments within the cytoplasm were seen in the index patient sample.
Ultrasound examination revealed marked hepatomegaly (liver 4 cm below the right costal margin, and 5 cm below the xiphoid process) and reduced hepatic echogenicity. Hyper-echoic lesions consistent with stone formation were seen on both kidneys. Abdominal computed tomography scans showed hepatic steatosis and hyper-echoic lesions suggestive of kidney stones in the left kidney but not in the right kidney (Figure 1B). X-ray imaging of the skull was normal, as were the long bones of both arms and legs. X-ray imaging also picked up abnormally shallow hip sockets on both sides, which is suggestive of acetabular dysplasia or congenital hip dysplasia (Figure 1B). Other diagnostic evaluations are provided in Table 3.
Table 3.
Diagnostic evaluation of the patient with a methionyl-tRNA synthetase mutation
| Etiological assessment | Investigations performed (normal unless otherwise indicated) |
| Infections | Serum procalcitonin levels (significantly elevated, Table 1); |
| Serology for Hepatitis B, C, HIV, syphilis, EBV, CMV, HSV, toxoplasmin, and rubella virus; | |
| PCR for CMV; beta-d-glucan assay; galactomannan assay; T-Spot.TB test; | |
| Cerebrospinal fluid analysis for white blood cell count, protein, and glucose level; | |
| Complete blood count: anemia, elevated WBC and C-reactive protein (Table 1); | |
| Culture for blood, urine, sputum, alveolar lavage fluid, and cerebrospinal fluid; | |
| Sputum and alveolar lavage fluid for mycoplasma/chlamydia DNA detection; | |
| Sputum and alveolar lavage fluid for detection of respiratory syncytial virus, adenovirus, influenza virus, and para-influenza virus antigens; | |
| Alveolar lavage fluid smear for fungus detection | |
| Radiology, endoscopy, and histopathology | Multiple chest X-rays and a contrast-enhanced computed tomography scan of the lung (alveolar effusions with severe interstitial lung disease) (Figure 1); |
| Abdominal ultrasonography and CT scan (hepatomegaly, liver steatosis, kidney stones) (Figure 1); | |
| Bronchoscopy (chronic inflammatory changes in bronchiolar mucosa); | |
| X-ray imaging of the skull; CT scan of adrenal gland; | |
| X-ray imaging of long bones: (abnormally shallow hip socket that is suggestive of acetabular dysplasia or congenital hip dysplasia) (Figure 1); | |
| Liver biopsy (severe steatosis of hepatic cells with ballooning, lobular disarrays; mild changes, such as cholestasis, fibrosis, lymphocyte infiltration, Iron deposition, and bile duct proliferation); | |
| Bone marrow aspirate (extreme proliferation of bone marrow cells with few hemophagocytic cells); peripheral blood smear | |
| Immunology | Immunoglobulin levels (after IVIG therapy at local hospital): elevated IgG (20.2 g/L, normal range 3.7-8.3 g/L), IgM (1.47 g/L, normal range 0.33-1.25 g/L), and IgA (0.63 g/L, normal range 0.14-0.5) levels; normal IgE, complement 4, and complement 3 levels; |
| Neutrophil oxidative burst activity, and lymphocyte subpopulations; | |
| Autoimmune antibodies | |
| Biochemical, metabolic and endocrine profiling | Glucose profiling (hypoglycemia); slightly elevated serum lactate (Table 1); |
| Liver function test: cholestasis, hypoalbuminemia, abnormal blood coagulation profiles (Table 1); | |
| Creatine kinase, lactate dehydrogenase; | |
| Serum amino acids (proline 1803 μmol/L, normal range: 165-700 μmol/L; threonine 171 μmol/L, normal range: 17-90 μmol/L) and acyl-carnitine profile; urine organic acids (including succinylacetone); Urine acidoglycoprotein (51.98 mg/mmol creatinine, normal range: 59.70-78.52 mg/mmol creatinine). | |
| Low levels of total serum cholesterol, HDL and LDL cholesterol (Table 1). | |
| Serum cortisol level; thyroid function test (total triiodothyronine 52.6 ng/dL, normal range: 70-220 ng/dL) | |
| Ophthalmology, electrocardiology, and echocardiogram (patent foramen ovale, 2.6 mm) | |
| Genetic disorders | White blood cell lysosomal enzyme screening for GM1 gangliosidosis, GM2 gangliosidosis, Sandhoff disease, Krabbe leukodystrophy, Gaucher disease, Fabry disease, Pompe disease, metachromatic leukodystrophy, Nieman-Pick disease, neuronal ceroid lipofuscinoses (1 and 2), mucopolysaccharidosis (type I-VII, IX), muculipidosis (type II and III). |
| Liver panel including 41 genes known to cause liver diseases, and trio whole exome sequencing (Table 2). |
DISCUSSION
MetRS is one of 20 ubiquitously expressed enzymes essential for protein biosynthesis, and covalently links methionine with its cognate tRNA. Since initial reports of MARS gene mutations causing ILLD[3] and CMT[6] in 2013, a total of 34 cases of ILLD[4,5,10] and eight cases of CMT[7-10] have been reported.
Similar to previous reports, the patient in our case showed a failure to thrive, developmental delay, interstitial lung disease, liver involvement (hepatomegaly, cholestasis, hepatic steatosis, fibrosis, and iron deposition), anemia, and thrombocytosis. An active proliferation of bone marrow cells has been reported by Sun et al[5]. Our patient had marked leukocytosis (white blood cell count up to 71.7 × 109/L), and a bone marrow biopsy showed extreme proliferation of bone marrow cells with few hemophagocytic cells. MetRS is also a component of a cytoplasmic multiaminoacyl-tRNA synthetase complex with multiple roles in immune response, inflammation, and tumorigenesis[12,13]. Prolonged low-grade fever, leukocytosis, thrombocytosis, and elevated c-reactive protein in this patient responded to intensive antibiotic treatment, and could be viewed as an exaggerated inflammatory or immune response to infection. Unlike previous reports of an arrest in red blood cell maturity[3,5], a bone marrow biopsy from this patient showed marked proliferation of normal erythrocyte precursors.
While aminoaciduria has been reported[3], kidney stones have never been reported to be associated with a MARS mutation. No evidence of urinary tract infection, proteinuria, or organic aciduria was found in our case, and serum electrolytes with urea and creatinine were essentially normal. An evaluation of urinary citrate, calcium, and 24 h urine output in future ILLD cases might be necessary in order to rule out factors that promote renal stone formation[14]. Mutations in genes encoding mitochondrial seryl-tRNA synthetases have been reported to cause renal damage[15,16], but no association of cytoplasmic aminoacyl-tRNA synthetases, including MARS, have been reported. Since previously reported mutations were all non-synonymous in nature, severe mutations (such as a truncation or single amino acid insertion as in our case) may have caused some renal impairment leading to stone formation.
No skeletal abnormality has been reported, with the exception of two ILLD cases with delayed bone age[5]. Our case had marked acetabular dysplasia consistent with developmental hip dysplasia. Other than being female, this infant did not have other risk factors[17], such as breach presentation upon delivery, local infection, or trauma. Whole exome sequencing did not reveal abnormalities in previously reported susceptible genes such as GDF5, TBX4, ASPN, IL-6, TGF-b1, and PAPPA2[18]. Hip dysplasia is associated with CMT[19], and the rate of hip dysplasia among children with CMT ranges from 6% to 8.1%[20]. Novarino et al[11] reported four cases of HSPs with compound heterozygous variants of the MARS gene in a family with infantile onset delayed motor milestones and disabilities upon crawling/walking. Two cases had bilateral Achilles contracture, one had scoliosis, but none had hip-joint abnormalities. A recent report of an ILLD case[10] with a p.Arg618Cys variant was also associated with CMT in a previous report[6], indicating ILLD and CMT may share a similar disease-causing mechanism. All reported cases of CMT, ILLD, and HSPs associated with the MARS gene had missense mutations. Our case had a truncating mutation and an insertion of a single amino acid. Severe mutations may have been responsible for the hip dysplasia, which could be an early manifestation of CMT in this patient.
The c.2158C>T/p.Gln720Stop, which was inherited from the mother, caused the glutamine amino acid change at position 720, leading to a stop codon at a well-conserved α-helix bundle domain (anti-codon binding domain) of the methionyl-tRNA synthetase protein.
The tri-nucleotide insertion (c.893_894insTCG) with paternal origin caused the insertion of a single amino acid (arginine) at position 299 in the Rossmann fold domain (catalysis center). Nine out of 12 ILLD variants reported so far affected an amino acid in the Rossmann fold domain (Figure 2B). Arg299 is adjacent to the active methionine-binding site of human MetRS, which is surrounded by the amino acid residues Arg12, Leu13, Pro14, Thr257, Gly259, Tyr260, Asn297, and His301[21].
All eight mutations from European ILLD cases were located in the Rossmann fold of the MARS protein. However, only one out of four mutations from Chinese cases carried mutations in the Rossmann fold domain, and the location of mutations among Chinese ILLD cases was significantly different from that of European ILLD cases (Fisher's exact = 0.018) (Figure 2B). Our case also suggested that severe mutations may lead to more organ/system involvement and severe outcomes.
In vivo yeast complementation assays were used to predict the effects of MARS variants, including 1852C>T/p.Arg618Cys[6], c.920A>G/p.Tyr307Cys[10] and 1852C>T/p.Arg618Cys[10]. The in vitro aminoacylation assay with HEK293 cells was used to confirm the effects of c.1108T>C/p.Phe370Leu, and c.1568T>C/p.Ile523Thr MARS variants[3]. The effects of c.1031A>G/p.Tyr344Cys, c.1177G>A/p.Ala393Thr, c.1700C>T/p.Ser567Leu and c.1814A>T/p.Asp605Val were studied using the in vitro yeast aminoacylation assay[4], and later by Comisso et al[22] using the E. Coli-based aminoacylation assay. Further functional studies are needed to confirm the effects of variants in our case, as well as variants reported by others (c.2398C>A/p.Pro800Thr[7], c.433G>A/p.Asp145Asn and c.2405T>C/p.Phe802Ser[5]). Besides previously used methods, one may consider the use of animal models such as Drosophila and C. elegans to predict the pathogenicity of other aminoacyl-tRNA synthetase mutations[23].
There is currently no cure for ILLD, and thus treatment is only supportive. Provided that in vitro enzyme activity may partly be restored by increasing methionine[22], methionine supplementation could be considered in studies of animal models, or possibly even in humans. However, plasma levels of methionine and its toxic product homocysteine should be closely monitored.
In conclusion, truncation and insertion variants in the MARS gene may cause ILLD, and phenotypes of ILLD may also include kidney stones, acetabular dysplasia, prolonged fever, and extreme leukocytosis.
ARTICLE HIGHLIGHTS
Case characteristics
A five-month old female infant presented with failure to thrive, developmental delay, jaundice, and dark urine.
Clinical diagnosis
Typical clinical findings and whole exome sequencing results led to a diagnosis of interstitial lung and liver disease (ILLD).
Differential diagnosis
Genetic cause was suspected due to multiple system involvement, but a liver panel consisting of 41 genes related to liver diseases came back negative. Lysosomal storage disease was considered, but an enzyme panel for screening common lysosomal storage diseases was normal, as was the urine acidoglycoprotein level.
Laboratory diagnosis
Laboratory findings were Cholestasis, anemia, abnormal blood coagulation profiled, thrombocytosis, and extreme leukocytosis. Whole exome sequencing revealed a novel truncating variant (c.2158C>T/p.Gln720Stop) and a novel tri-nucleotide insertion (c.893_894insTCG) in the methionyl-tRNA synthetase (MARS) gene.
Imaging diagnosis
X-ray, computed tomography scan, and ultrasound imaging revealed interstitial lung disease, hepatomegaly, kidney stones, and acetabular dysplasia.
Pathological diagnosis
Liver biopsy results showed severe hepatic steatosis, hepatic cells ballooning, lobular disarray, cholestasis, iron deposition, and mild fibrosis/lymphocyte infiltration/bile duct proliferation within the portal region.
Treatment
Ursodeoxycholic acid, fat-soluble vitamins, antibiotics, oxygen therapy, and supportive treatment.
Related reports
Previous reports of ILLD were associated with biallelic missense mutations in the MARS gene. Phenotypes, such as kidney stones, acetabular dysplasia, prolonged fever, and extreme leukocytosis have never been reported to be associated with ILLD.
Term explanation
ILLD is interstitial lung and liver disease caused by homozygous or compound heterozygous mutations in the MARS gene. Typical findings in ILLD include failure to thrive, developmental delay, interstitial lung disease, liver involvement (hepatomegaly, cholestasis, hepatic steatosis, fibrosis, and iron deposition), anemia, and thrombocytosis.
Experiences and lessons
Regardless of race or ethnicity, ILLD should be considered in all patients with chronic liver diseases showing progressive interstitial lung involvement. Severe mutations may lead to more organ/system involvement and severe outcomes.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): D
Grade E (Poor): 0
Informed consent statement: Informed consent was obtained from the parents.
Conflict-of-interest statement: The authors declare that they have no conflicts of interest.
CARE Checklist (2013) statement: The authors have read the CARE Checklist (2013), and the manuscript was prepared and revised according to the CARE Checklist (2013).
Peer-review started: June 22, 2018
First decision: July 31, 2018
Article in press: August 24, 2018
P- Reviewer: Al-Haggar M, Arslan N, Hamaguchi M S- Editor: Wang XJ L- Editor: Filipodia E- Editor: Huang Y
Contributor Information
Kuerbanjiang Abuduxikuer, Department of Hepatology, Children’s Hospital of Fudan University, Shanghai 201102, China.
Jia-Yan Feng, Department of Pathology, Children’s Hospital of Fudan University, Shanghai 201102, China.
Yi Lu, Department of Hepatology, Children’s Hospital of Fudan University, Shanghai 201102, China.
Xin-Bao Xie, Department of Hepatology, Children’s Hospital of Fudan University, Shanghai 201102, China.
Lian Chen, Department of Pathology, Children’s Hospital of Fudan University, Shanghai 201102, China.
Jian-She Wang, Department of Hepatology, Children’s Hospital of Fudan University, Shanghai 201102, China; Department of Pediatrics, Jinshan Hospital of Fudan University, Shanghai 201508, China. jshwang@shmu.edu.cn.
References
- 1.Deniziak MA, Barciszewski J. Methionyl-tRNA synthetase. Acta Biochim Pol. 2001;48:337–350. [PubMed] [Google Scholar]
- 2.Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 3.van Meel E, Wegner DJ, Cliften P, Willing MC, White FV, Kornfeld S, Cole FS. Rare recessive loss-of-function methionyl-tRNA synthetase mutations presenting as a multi-organ phenotype. BMC Med Genet. 2013;14:106. doi: 10.1186/1471-2350-14-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hadchouel A, Wieland T, Griese M, Baruffini E, Lorenz-Depiereux B, Enaud L, Graf E, Dubus JC, Halioui-Louhaichi S, Coulomb A, et al. Biallelic Mutations of Methionyl-tRNA Synthetase Cause a Specific Type of Pulmonary Alveolar Proteinosis Prevalent on Réunion Island. Am J Hum Genet. 2015;96:826–831. doi: 10.1016/j.ajhg.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun Y, Hu G, Luo J, Fang D, Yu Y, Wang X, Chen J, Qiu W. Mutations in methionyl-tRNA synthetase gene in a Chinese family with interstitial lung and liver disease, postnatal growth failure and anemia. J Hum Genet. 2017;62:647–651. doi: 10.1038/jhg.2017.10. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez M, McLaughlin H, Houlden H, Guo M, Yo-Tsen L, Hadjivassilious M, Speziani F, Yang XL, Antonellis A, Reilly MM, et al. Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J Neurol Neurosurg Psychiatry. 2013;84:1247–1249. doi: 10.1136/jnnp-2013-305049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyun YS, Park HJ, Heo SH, Yoon BR, Nam SH, Kim SB, Park CI, Choi BO, Chung KW. Rare variants in methionyl- and tyrosyl-tRNA synthetase genes in late-onset autosomal dominant Charcot-Marie-Tooth neuropathy. Clin Genet. 2014;86:592–594. doi: 10.1111/cge.12327. [DOI] [PubMed] [Google Scholar]
- 8.Nam SH, Hong YB, Hyun YS, Nam da E, Kwak G, Hwang SH, Choi BO, Chung KW. Identification of Genetic Causes of Inherited Peripheral Neuropathies by Targeted Gene Panel Sequencing. Mol Cells. 2016;39:382–388. doi: 10.14348/molcells.2016.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirano M, Oka N, Hashiguchi A, Ueno S, Sakamoto H, Takashima H, Higuchi Y, Kusunoki S, Nakamura Y. Histopathological features of a patient with Charcot-Marie-Tooth disease type 2U/AD-CMTax-MARS. J Peripher Nerv Syst. 2016;21:370–374. doi: 10.1111/jns.12193. [DOI] [PubMed] [Google Scholar]
- 10.Rips J, Meyer-Schuman R, Breuer O, Tsabari R, Shaag A, Revel-Vilk S, Reif S, Elpeleg O, Antonellis A, Harel T. MARS variant associated with both recessive interstitial lung and liver disease and dominant Charcot-Marie-Tooth disease. Eur J Med Genet. 2018;61:616–620. doi: 10.1016/j.ejmg.2018.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Novarino G, Fenstermaker AG, Zaki MS, Hofree M, Silhavy JL, Heiberg AD, Abdellateef M, Rosti B, Scott E, Mansour L, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343:506–511. doi: 10.1126/science.1247363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo M, Schimmel P. Essential nontranslational functions of tRNA synthetases. Nat Chem Biol. 2013;9:145–153. doi: 10.1038/nchembio.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao P, Fox PL. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol Med. 2013;5:332–343. doi: 10.1002/emmm.201100626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bevill M, Kattula A, Cooper CS, Storm DW. The Modern Metabolic Stone Evaluation in Children. Urology. 2017;101:15–20. doi: 10.1016/j.urology.2016.09.058. [DOI] [PubMed] [Google Scholar]
- 15.Rivera H, Martín-Hernández E, Delmiro A, García-Silva MT, Quijada-Fraile P, Muley R, Arenas J, Martín MA, Martínez-Azorín F. A new mutation in the gene encoding mitochondrial seryl-tRNA synthetase as a cause of HUPRA syndrome. BMC Nephrol. 2013;14:195. doi: 10.1186/1471-2369-14-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belostotsky R, Ben-Shalom E, Rinat C, Becker-Cohen R, Feinstein S, Zeligson S, Segel R, Elpeleg O, Nassar S, Frishberg Y. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet. 2011;88:193–200. doi: 10.1016/j.ajhg.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw BA, Segal LS; Section on orthopaedics. Evaluation and Referral for Developmental Dysplasia of the Hip in Infants. Pediatrics. 2016;138 doi: 10.1542/peds.2016-3107. [DOI] [PubMed] [Google Scholar]
- 18.Shi D, Dai J, Ikegawa S, Jiang Q. Genetic study on developmental dysplasia of the hip. Eur J Clin Invest. 2012;42:1121–1125. doi: 10.1111/j.1365-2362.2012.02682.x. [DOI] [PubMed] [Google Scholar]
- 19.Novais EN, Bixby SD, Rennick J, Carry PM, Kim YJ, Millis MB. Hip dysplasia is more severe in Charcot-Marie-Tooth disease than in developmental dysplasia of the hip. Clin Orthop Relat Res. 2014;472:665–673. doi: 10.1007/s11999-013-3127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker JL, Nelson KR, Heavilon JA, Stevens DB, Lubicky JP, Ogden JA, VandenBrink KA. Hip abnormalities in children with Charcot-Marie-Tooth disease. J Pediatr Orthop. 1994;14:54–59. doi: 10.1097/01241398-199401000-00012. [DOI] [PubMed] [Google Scholar]
- 21.Nadarajan SP, Mathew S, Deepankumar K, Yun H. An in silico approach to evaluate the polyspecificity of methionyl-tRNA synthetases. J Mol Graph Model. 2013;39:79–86. doi: 10.1016/j.jmgm.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 22.Comisso M, Hadchouel A, de Blic J, Mirande M. Mutations in MARS identified in a specific type of pulmonary alveolar proteinosis alter methionyl-tRNA synthetase activity. FEBS J. 2018 doi: 10.1111/febs.14510. [DOI] [PubMed] [Google Scholar]
- 23.Oprescu SN, Griffin LB, Beg AA, Antonellis A. Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods. 2017;113:139–151. doi: 10.1016/j.ymeth.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]