

Abstract
DExD/H-box helicase 9 (DHX9), or RNA helicase A (RHA), is an abundant multifunctional nuclear protein. Although it was previously reported to act as a cytosolic DNA sensor in plasmacytoid dendritic cells (pDCs), the role and molecular mechanisms of action of DHX9 in cells that are not pDCs during DNA virus infection are not clear. Here, a macrophage-specific knockout and a fibroblast-specific knockdown of DHX9 impaired antiviral innate immunity against DNA viruses, leading to increased virus replication. DHX9 enhanced NF-κB–mediated transactivation in the nucleus, which required its ATPase-dependent helicase (ATPase/helicase) domain, but not the cytosolic DNA-sensing domain. In addition, DNA virus infection did not induce cytoplasmic translocation of nuclear DHX9 in macrophages and fibroblasts. Nuclear DHX9 was associated with a multiprotein complex including both NF-κB p65 and RNA polymerase II (RNAPII) in chromatin containing NF-κB–binding sites. DHX9 was essential for the recruitment of RNAPII rather than NF-κB p65, to the corresponding promoters; this function also required its ATPase/helicase activity. Taken together, our results show a critical role of nuclear DHX9 (as a transcription coactivator) in the stimulation of NF-κB–mediated innate immunity against DNA virus infection, independently of DHX9’s DNA-sensing function.
INTRODUCTION
An innate antiviral response depends on the sensing of viral nucleic acids by pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG-I)-like RNA helicases (RLHs). Three members of RLHs—RIG-I (DDX58), MDA5 and LGP2—are key mediators of antiviral immunity and belong to the family of DExD/H-box helicases (1,2). In addition to these RLHs, other DExD/H-box helicases, such as DHX9, DHX36, DDX41, DDX3, DHX15 and DDX60, have lately been identified as sensors of viral nucleic acids, as signaling adaptors, or transcriptional regulators of antiviral signaling pathways (3–9). Especially, aspartate-glutamate-x-aspartate/histidine (DExD/H)-box helicase 9, DHX9 (also known as DDX9, NDHII or RNA helicase A), has been identified as a CpG DNA–binding protein in plasmacytoid dendritic cells (pDCs) and alters cytokine responses via interaction with a protein called myeloid differentiation primary response 88 (MyD88) after its cytoplasmic translocation (3). DHX9 also binds to viral RNA in myeloid DCs, by acting through mitochondrial antiviral signaling (MAVS) (8). DHX9 is a human homolog of Drosophila melanogaster Maleless (Mle) (10), a transcriptional activator that increases transcription of male X-linked genes essential for dosage compensation (11). In mammals, DHX9 is an abundant nuclear protein with active involvement in transcriptional regulation and RNA metabolism (12–14).
Herpesviruses are double-stranded DNA viruses with an envelope, and eight human herpesviruses can be subdivided into α-, β- and γ-subfamilies, based on their biological functions and sequence similarities (15). Herpesviruses undergo productive lytic replication in permissive cells such as fibroblasts and epithelial cells or establish dormant latency in specific cell types of the hosts, thus leading to life-long persistent infection, and may cause severe diseases in humans, especially in children and immunocompromised individuals (16). Herpes simplex virus type 1 (HSV-1) is a prototypic α-herpesvirus and a causative agent of cold sores and encephalitis (17). Murine gammaherpesvirus 68 (MHV-68 or also known as γHV-68), is phylogenetically related two oncogenic human γ-herpesviruses, Epstein–Barr virus, and Kaposi's sarcoma–associated herpesvirus (18). Both HSV-1 and MHV-68 show robust lytic replication in various cell types of both humans and mice; these properties make them ideal DNA viruses for lab research.
Although increasing evidence suggests that DExD/H-box helicases including DHX9 perform significant functions in innate immunity by sensing viral nucleic acids, this cytosolic DNA sensing by DHX9 has not been examined during DNA virus infection; it is unclear whether cytosolic DNA sensing of DHX9 plays a critical role in the infected cells or is limited to pDCs only (3,8). Given the ability of a DNA virus to efficiently infect a wide range of permissive cells, especially fibroblasts and epithelial cells as primary target cells, understanding the function of DHX9 in these cell types is important (19). In this study, we demonstrated that DHX9 is intimately involved in innate immune responses against DNA virus infection. We found that a macrophage-specific knockout and a fibroblast-specific knockdown of DHX9 impaired antiviral innate immunity. DHX9 promoted nuclear factor kappa B (NF-κB)-mediated transcriptional activation of antiviral cytokines, via its physical association with p65 and RNA polymerase II (RNAPII) in the context of chromatin, by functioning as a bridging factor for RNAPII recruitment to NF-κB p65. Notably, DHX9 is unlikely to act as a cytosolic DNA sensor in cells that are not pDCs because DHX9 remained in the nucleus and DHX9 deficiency did not prevent NF-κB p65 from nuclear translocation following DNA virus infection. More importantly, the known cytosolic DNA-sensing domain of DHX9 was also found to be dispensable for the antiviral innate immune activity of DHX9 during DNA virus infection. Taken together, our results indicate that transcriptional regulation by nuclear DHX9 plays a critical role in antiviral responses of permissive cells with DNA virus infection, independently from DHX9’s cytosolic DNA-sensing activity.
MATERIALS AND METHODS
Mice
LysM (Lyz2)-cre (Lyz2tm1(cre)Ifo) mice were purchased from Jackson Laboratories (Bar Harbor, Maine, USA). The embryonic stem (ES) cell clone with gene targeting for Dhx9, called Dhx9tm1a(EUCOMM)Hmgu, was purchased from EUCOMM resource (Helmholtz Zentrum Munchen, Neurenberg, Germany) and used to generate Dhx9-floxed mice (Dhx9f/f). Gender-matched mice (6–8 weeks old) were used. All the procedures were approved by the Institutional Animal Care Committee of KAIST.
Primary cultures of bone marrow (BM)–derived macrophages (BMDMs) and BM–derived dendritic cells (BMDCs)
BM cells were isolated from the femurs and tibias of mice (20). BMDMs were generated by plating BM cells in a macrophage growth medium consisting of Dulbecco's modified Eagle's medium (DMEM, HyClone) supplemented with 10% of heat-inactivated FBS (HyClone), penicillin and streptomycin (10 U/ml) (HyClone), 10 mM HEPES (SH30237.01, HyClone), and 30% L929 cell–conditioned medium as a source of macrophage colony-stimulating factor (M-CSF) at 2 × 106 cells/ml in 10 ml per 10 cm petri dish. On day 3, 5 ml of the macrophage growth medium was added, and macrophages were harvested on day 5 with ice-cold culture medium. The L929 cell–conditioned medium (M-CSF source) was prepared by plating 4.7 × 105 L929 cells in a 75 cm2 flask containing 55 ml of the L929 medium and grown in a humidified incubator with 5% CO2 at 37°C for 7 days (20).
To obtain Fms-related tyrosine kinase 3 ligand (Flt3L)-induced BMDCs, BM cells were cultured in the DC growth medium consisting of Roswell Park Memorial Institute (RPMI) 1640 with 2 mM l-glutamine supplemented with 10% of heat-inactivated FBS (HyClone), penicillin and streptomycin (10 U/ml) (HyClone), 10 mM HEPES (SH30237.01, HyClone), 50 μM 2-mercaptoethanol (M6250, Sigma-Aldrich), 100 ng/ml murine Fms-related tyrosine kinase 3 ligand (Flt3L, 250-31L, Peprotech) for 7 days at 2 × 106 cells/ml in a 10 cm culture dish. Every 2 days of culture, half of the medium was removed and 7.5 ml of the DC growth medium was added. Nonadherent DCs were harvested from the cultures by gentle pipetting.
Cells culture, virus and plaque assays
Cell lines HEK293T (human embryonic kidney cells), HeLa (human epithelial cells) and Vero (green monkey kidney cells) were cultured in complete Dulbecco's modified Eagle's medium (DMEM, HyClone) containing 10% of fetal bovine serum (FBS, HyClone) supplemented with penicillin and streptomycin (10 U/ml) (HyClone), while NIH3T3 cells (mouse embryonic fibroblasts) were cultured with 10% of bovine calf serum (BCS, HyClone). MEFs (murine embryonic fibroblasts) were obtained from a pregnant BALB/c mouse (13–14 days postcoitum) as previously described (21) and cultured in DMEM with 10% of FBS. Cultures were incubated in a humidified incubator with 5% CO2 at 37°C. The wild-type MHV-68 (ATCC VR1465) and HSV-1 (ATCC VR-1493) were originally obtained from the American Type Culture Collection. Viral stocks of recombinant EGFP/MHV-68 (tw25) were prepared as previously described (22). The amplified viruses were titrated with plaque assays using Vero cells overlaid with 1% methylcellulose (Sigma-Aldrich) in normal growth media. After 2 or 5 days of infection, the cells were fixed and stained with 2% crystal violet (Merck Millipore) in 20% ethanol for HSV-1 or MHV-68, respectively. The plaques were then counted to determine the titers (23).
Construction of DHX9 knockdown cells
The empty pLKO.1 TRC cloning vector (plasmid #10878) and control shRNA construct (plasmid #1864) (24) were purchased from Addgene. The target sequence of the mouse DHX9 shRNA construct (TRCN0000071116) was obtained from the Broad TRC RNAi shRNA library (The RNAi Consortium). The sense strand of the mouse DHX9 short hairpin RNA (shRNA) sequence was 5′-ccggCGCAAAGTGTTTGATCCAGTActcgagTACTGGATCAAACACTTTGCGtttttg-3′ (based on positions 2242–2262 of mouse DHX9 coding DNA sequence [CDS]). The 21-nucleotide DHX9 target sequences are indicated in uppercase letters, whereas the hairpin and the sequences necessary for the directional cloning are depicted in lowercase letters. The forward sequence was 5′-ccggCGCAAAGTGTTTGATCCAGTActcgagTACTGGATCAAACACTTTGCGtttttg-3′ and the reverse sequence was 5′-aattcaaaaaCGCAAAGTGTTTGATCCAGTActcgagTACTGGATCAAACACTTTGCG-3′. The sense and antisense strands were annealed, and the annealed oligonucleotide was cloned into the AgeI and EcoRI sites downstream of the U6 promoter in the pLKO.1 TRC vector according to TRC protocols available on their website (https://www.addgene.org/tools/protocols/plko/). The cloned shRNA expression cassette was verified by sequencing. The human DHX9 shRNA construct was a kind gift from Dr. Mien-Chie Hung (The University of Texas MD Anderson Cancer Center, Houston, TX, USA) (25). To produce lentiviruses, HEK293T cells were transfected with lentiviral vectors together with the packaging and envelope plasmids psPAX2 and pMD2.G from Dr Seungmin Hwang (The University of Chicago, Chicago, IL, USA) (26). Two days after transfection, the culture media containing the lentiviruses were harvested and spun down at 1300 × g for 5 min to remove cell debris. HeLa and NIH3T3 cells were transduced with the lentivirus supernatant supplemented with 8 μg/ml polybrene (Sigma-Aldrich). The cells were selected beginning at 48 h for ∼2 weeks in DMEM containing 10% of FBS, penicillin and streptomycin (10 U/ml) and 2 μg/ml puromycin (Sigma-Aldrich). The knockdown efficiency was determined by real-time reverse transcription-PCR (RT-PCR) and western blot analysis.
Plasmids
FLAG-DHX9 and DHX9-K417R were kind gifts from Dr Kathleen Boris-Lawrie (University of Minnesota, St. Paul, MN, USA). FLAG-DHX9(ΔDUF) (1-920) was generated from FLAG-DHX9 by standard PCR subcloning, using the primers F: 5′-tatgcagaattcccgggctagcaccatggattacaaggatgatga-3′ and R: 5′- tattgcggccgcttataaaagggctacgtgatcagaaaatctgtttcc-3′. FLAG-DHX9(ΔDUF+NTD) was constructed by standard PCR subcloning in a separate reaction to generate two fragments. The ΔDUF fragment was generated using the primers F: 5′-tatgcagaattcccgggctagcaccatggattacaaggatgatga-3′ and R: 5′-acgtggaccatctccataccgtaaaagggctacgtgatcaga-3′, and the NTD fragment via primers F: 5′- gatcacgtagcccttttacggtatggagatggtccacgtcct-3′ and R: 5′-tgccttagcggccgcttactgtctacacacaggaactga-3′. The two fragments were then overlap-fused using the following PCR conditions: 94°C 10 min; 94°C 30 s, 50°C for 25 min and 72°C for 5 min for 10 cycles; and finally 72°C for 10 min. The overlap extension PCR product was amplified again using a standard PCR protocol. All the constructs were verified by sequencing. FLAG-DHX9(ΔDUF+NLSSV40) was constructed by standard PCR subcloning (primers F: 5′-tatgcagaattcccgggctagcaccatggattacaaggatgatga-3′, R: 5′-tattgcggccgcttatacctttctcttcttttttggtaaaagggctacgtgatcaga-3′); the reverse primer introduced the fused NLS, a stop codon and the NotI restriction sites. The DHX9 cDNA without the 3′ untranslated region (UTR) was amplified and subcloned into EcoRI and NotI sites of pcDNA3.1 vector (27).
Antibodies and western blotting
For western blotting, cell extracts were prepared in 5 × SDS sample buffer (60 mM Tris–Cl pH 6.8, 2% sodium dodecyl sulfate, 5% β-mercaptoethanol, 10% glycerol and 0.01% bromophenol blue), subjected to electrophoresis on SDS polyacrylamide gels and analyzed with the following primary antibodies: a rabbit polyclonal antibody to DHX9 (ab26271, 1:1000) from Abcam, mouse monoclonal antibody to NF-κB p65 (F-6, sc-8008, 1:500), mouse monoclonal antibody to RNAPII (F-12, sc-55492, 1:500) from Santa Cruz Biotechnology, rabbit polyclonal antibody to MyD88 (IMG-178, 1:500) from Imgenex, mouse monoclonal antibody to α-tubulin (DM1A, 1:2000) from Sigma-Aldrich, mouse monoclonal antibody to PARP-1 (4C10-5, 1:1000) from BD Pharmingen, rabbit polyclonal antibody to ORF45 (1:500), rabbit polyclonal antibody to ORF65 (1:1000) (28), mouse monoclonal antibody to FLAG-M2 (1:2000) from Sigma-Aldrich, rabbit anti-HA polyclonal antibody (HA-probe, Y-11, sc-805, 1:1000) from Santa Cruz Biotechnology, and a mouse monoclonal antibody to HSV-1 + HSV-2 ICP27 (ab31631, 1:1000) from Abcam. Anti-rabbit or anti-mouse immunoglobulin G (IgG) antibody conjugated with horseradish peroxidase (1:5000) from Santa Cruz Biotechnology served as a secondary antibody. The proteins in the western blots were detected with an EPD western blot detection kit (ELPIS, Republic of Korea), and the signals were detected and analyzed using LAS-4000, a chemiluminescent image analyzer (Fujifilm).
An immunofluorescence assay and confocal microscopy
The cells on coverslips were washed with 1 × PBS, then fixed with 4% paraformaldehyde at room temperature for 10 min, and permeabilized and blocked in 1 × PBS containing 0.1% of bovine serum albumin, 10% of normal goat serum, and 0.3% of Triton X-100. The cells were then incubated with a rabbit polyclonal anti-DHX9 antibody (ab26271, 1:500) or a rabbit polyclonal antibody to RNA polymerase II CTD repeat YSPTSPS (phospho S5, ab5131, 1:500) from Abcam and a mouse monoclonal anti-p65 antibody (F-6, sc-8008, 1:200) or mouse monoclonal antibody to RNAPII (F-12, sc-55492, 1:500) from Santa Cruz Biotechnology at 4°C overnight followed by incubation with a FITC-conjugated goat anti-rabbit IgG antibody (Jackson ImmunoResearch) and rhodamine-conjugated goat anti-mouse IgG antibody (Jackson ImmunoResearch) at room temperature for 1 h. The cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Inc.), and the coverslips were mounted onto glass slides using mounting medium Immu-Mount (Thermo Scientific). The slides were examined, and representative images were photographed under a confocal laser scanning microscope (LSM 700, Carl-Zeiss) with the LSM analysis software.
RNA isolation and reverse transcription
The total RNA was extracted from cells using the TRI Reagent (Molecular Research Center, Inc.) and chloroform extraction methods (29). The cDNA was synthesized with random hexamer primers using RevertAid First strand cDNA synthesis (Fermentas, USA) as previously described (29). The cellular transcripts were quantified with the following primers: human DHX9 (F: 5′-tccaactggaatccttggac-3′; R: 5′-ttttcccacatccagtagcc-3′), human IL6 (F: 5′-agccctgagaaaggagacatgta-3′; R: 5′-tctgccagtgcctctttgc-3′), human IFN-β (F: 5′-tgacatccctgaggagattaagc-3′; R: 5′-gcgtcctccttctggaactg-3′), murine Dhx9 (F: 5′-cccccacctgttgatccttc-3′; R: 5′-gaccaaggaaccactcccac-3′), murine Il-6 (F: 5′-tccatccagttgccttcttg-3′; R: 5′-ggtctgttgggagtggtatc-3′), murine Ifn-β (F: 5′-aaactcatgaccagtctgca-3′; R: 5′-aggagatcttcagtttcggagc-3′), murine Tnf (F: 5′-cgtcagccgatttgctatct-3′; R: 5′-cggactccgcaaagtctaag-3′), murine Il-1β (F: 5′-caaccaacaagtgatattctccatg-3′; R: 5′-gatccacactctccagctgca-3′), ICP0 (F: 5′-aagcttggatccgagccccgccc-3′; R: 5′-aagcggtgcatgcacgggaaggt-3′), RTA (F: 5′-gcctggaggcgcttagg-3′; R: 5′-aacacattgcgcccaaatg-3′). The transcripts were normalized to actin (F: 5′-gtatcctgaccctgaagtacc-3′; R: 5′-tgaaggtctcaaacatgatct-3′). The experiment was performed on an iCycler iQ multicolor Real-time PCR detection system and analyzed in the Optical system software (Bio-Rad). Real-time PCR was carried out in triplicate and run at 50°C for 2 min and followed by 45 cycles at 95°C for 10 s, 55°C for 15 s and 72°C for 20 s with a melting curve analysis. The results were analyzed in the Optical system software (Bio-Rad).
Viral DNA isolation and analysis by quantitative PCR (qPCR)
The cells were harvested and washed once with 1 × PBS. The cell pellet was resuspended in lysis buffer (40 mM Tris pH 7.7, 20 mM EDTA pH 8.0, 0.2 M NaCl and 1% sodium dodecyl sulfate). Proteinase K was added to a final concentration of 0.5 mg/ml, and the suspension was incubated overnight at 56°C. Genomic DNA samples, including viral DNA samples, were isolated via a standard phenol:chloroform:isoamyl alcohol (25:24:1) extraction and ethanol precipitation method and resuspended in TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0). The whole genomic DNA (50 ng) was used for real-time PCR. The copy number of viral genomic DNA from virus-infected cells was quantitated with the iCycler iQ multicolor Real-time PCR detection system (Bio-Rad) using SYBR green I dye (Invitrogen) with ORF56 locus specific primers in a 20 μl reaction mixture (30). Real-time PCR was carried out in triplicate and run at 50°C for 2 min followed by 45 cycles at 95°C for 10 s, 55°C for 15 s, and 72°C for 20 s with a melting curve analysis. The standard DNA samples were prepared in triplicate with 10-fold serial dilutions of pBAC/MHV-68 ranging from 2 to 2 × 107 copies and used to generate a standard curve for each real-time PCR assay. Isolated viral DNA samples from pBAC/MHV-68-transfected cells were treated with DpnI to remove the input plasmid DNAs of the virus genome (23,28). Real-time PCR was performed using SYBR green PCR and specific primers in a 20 μl reaction mixture for M1 (F: 5′-cctggccatggttacatactc-3′; R: 5′-ggaacataatccataagcagggt-3′), the replicon of which contains the DpnI site (23,28). Real-time PCR was conducted in triplicate and was run at 50°C for 2 min followed by 45 cycles at 95°C for 10 s, 58°C for 15 s and 72°C for 20 s, and a melting curve analysis.
Cell transfection
Plasmid DNAs and BAC DNAs were prepared by standard methods using a Qiagen Plasmid Maxi or Midi kit (Qiagen). To transfect NIH3T3 cells, approximately 2.5 × 105 cells per well were seeded in a 12-well culture plate 1 day prior to the transfection. BAC DNA was transfected into the cells using Lipofectamine Plus (Invitrogen). To transfect HeLa cells, the cells were seeded in 12-well and 24-well culture plates at approximately 2 × 105 cells or 1 × 105 cells per well, respectively. The plasmid DNA was transfected into cells using Lipofectamine 2000 (Invitrogen). To transfect HEK293T cells, approximately 4 × 105 cells per well were seeded in 12-well culture plate 1 day prior to the transfection. Plasmid DNA was transfected into cells using polyethylenimine (1 mg/ml) (Sigma-Aldrich). For the luciferase reporter assays, 2.5 × 105 HEK293T cells per well in a 24-well culture plate were transfected with 50 ng of a reporter plasmid via polyethylenimine. Each transfection for the reporter assays was performed in triplicate and involved 10 ng of the β-galactosidase expression plasmid as an internal control. Twenty-eight hours post-transfection, the cells were harvested and analyzed by a Luciferase Assay System (Promega).
Coimmunoprecipitation
Approximately 6 × 106 NIH3T3 or 1 × 107 HeLa cells were seeded on a 10 cm culture dish overnight, infected with MHV-68 or HSV-1 virus and incubated for 12 h. The cells were scraped and resuspended in immunoprecipitation (IP) buffer (20 mM HEPES (pH 7.4), 100 mM NaCl, 0.5% Nonidet P-40 and 1% Triton X-100) supplemented with 1/100 volume of a protease inhibitor cocktail (Sigma-Aldrich) and 1 mM phenylmethanesulfonylfluoride (PMSF). The cells were lysed at 4°C for 1 h, and the cell debris were removed by centrifugation (13 500 × g, 4°C, 10 min) using a refrigerated micro centrifuge. The cell lysates were incubated at 4°C with an appropriate antibody for 1 h and next incubated with protein A/G agarose beads (Thermo Fisher Scientific) overnight at 4°C. The beads were washed three to five times with IP buffer and subjected to a western blot analysis.
Cytosolic and nuclear fractional assay
NIH3T3 cells or HeLa cells were washed twice with ice-cold 1× PBS and scraped in 500 μl of PBS supplemented with 1/100 volume of the protease inhibitor cocktail (Sigma-Aldrich) and 1 mM phenylmethanesulfonylfluoride (PMSF). The cells were spun down at 850 × g for 4 min at 4°C. The cells were resuspended in 300 μl of hypotonic lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 1 mM DTT, 1/100 volume of protease inhibitor cocktail, and 0.5 mM PMSF) and incubated on ice for 10 min prior to the addition of 1/20 volume of 10% NP-40; the resultant mixture was briefly vortexed. The cell lysate was centrifuged at 3500 × g at 4°C for 4 min to pellet the cell nuclei. The nuclei were then resuspended in high-salt buffer (25% glycerol, 20 mM HEPES pH 7.9, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF and 1/100 volume of protease inhibitor cocktail) with vigorous shaking. The nuclear extract was collected after centrifugation at 18 000 × g for 10 min at 4°C.
A chromatin immunoprecipitation (ChIP) assay
The ChIP assays were based on the SimpleChIP® enzymatic chromatin IP kit (magnetic beads) (Cell Signaling Upstate Biotechnology) and were performed according to the manufacturer's protocol. Briefly, 1.5 × 107 shCtrl (control) or DHX9 knockdown (shDHX9) HeLa cells were mock-infected or infected with the MHV-68 virus at MOI 2 for 12 h. The cells were fixed with 1% formaldehyde, cell extracts were prepared, and chromatin was sheared by sonication. The ChIP assays involved antibodies to p65 (C-20, sc-372, Santa Cruz Biotechnology), to RNAPII (N20, sc-899, Santa Cruz Biotechnology), DHX9 (ab26271, Abcam), and negative control IgG. The bound DNA was quantified by qPCR and expressed as the fold enrichment relative to normal IgG. For re-ChIP experiments, protein–DNA complexes after the first antibody immunoprecipitation were eluted by incubation of the protein G beads with equal volume of 10 mM DTT for 30 min at 37°C. The eluted samples were diluted 40-fold with 1 × ChIP buffer and subjected to immunoprecipitation with the second antibody (31). The following promoter-specific primers were used: IL6 (F: 5′-cctcaccctccaacaaagat-3′; R: 5′-agttcatagctgggctcctg-3′), IFN-β (F: 5′-gggagaagt gaaagtgggaa-3′; R: 5′-caggagagcaatttggagga-3′), and GAPDH (F: 5′-tactagcggttttacgggcg-3′; R: 5′-tcgaacagga ggagcagagagcga-3′).
RESULTS
BMDMs from macrophage-specific DHX9-knockout mice manifest impaired innate immunity after DNA virus infection
To gain insights into the impact of the DHX9 knockout in macrophages after virus infection, the ES cell clone with gene targeting of Dhx9, Dhx9tm1a(EUCOMM)Hmgu, was used to generate Dhx9-floxed mice (Dhx9f/f), and macrophage-specific DHX9 knockout mice were generated by crossing Dhx9f/f with LysM-cre (referred to as LysM-cre Dhx9f/f). BMDMs were generated from BM cells isolated from LysM-cre Dhx9f/f mice (where DHX9 was specifically deleted) and from Dhx9f/f mice as a control. We confirmed the loss of DHX9 protein expression in BMDMs but not in BMDCs from LysM-cre Dhx9f/f mice by western blotting (Figure 1A) and either in mock-infected BMDMs in serum-free media without a virus or in BMDMs infected with a DNA virus (MHV-68 or HSV-1) in serum-free media at MOI 5 for 9 and 18 h. The levels of proinflammatory cytokine transcripts, such as Il-6, Ifn-β, Tnf and Il-1β, were significantly attenuated in virus-infected DHX9 knockout BMDMs (Figure 1B–E, F–I), compared to the control. These data indicated that the DHX9 knockout reduces the innate immune response in macrophages after DNA virus infection.
Figure 1.

BMDMs from macrophage-specific DHX9 knockout mice display impaired innate immunity after DNA virus infection. (A) Loss of DHX9 protein in BMDMs, but not in BMDCs from LysM-cre Dhx9f/f mice was confirmed by western blotting. (B–I) BMDMs from Dhx9f/f and LysM-cre Dhx9f/f mice were mock-infected or infected with MHV-68 or HSV-1 at MOI 5 for 9 and 18 h. Total cellular RNA was extracted and subjected to RT-qPCR analysis of Il-6 (B, F), Ifn-β (C, G), Tnf (D, H), and Il-1β (E, I) mRNA levels. The data represent the mean ± SD of triplicate assays. *P < 0.05; **P < 0.01; ***P < 0.001 versus control, ns = nonsignificant (Student's t test).
The DHX9 knockdown enhances MHV-68 virus replication in fibroblasts and epithelial cells
To study the role of endogenous DHX9 during DNA virus infection, DHX9 knockdown NIH3T3 fibroblasts (shDHX9/3T3) were created by lentiviral transduction; they expressed shRNAs against DHX9 (shDHX9) or control sequences (shCtrl) (32). The knockdown of endogenous DHX9 was confirmed to yield ∼80% downregulation at both RNA and protein levels as quantified by RT-qPCR and western blot analysis, respectively (Figure 2A and B). The shDHX9/3T3 cells were infected with MHV-68 at MOI 0.5 or 2.0 for 24, 36 or 48 h and were harvested to analyze viral protein expression and viral genome replication (Figure 2C–F). The DHX9 knockdown enhanced viral protein expression and viral genome replication, when compared with the shCtrl/3T3 cells in both low- and high-MOI infection groups. As shown in Figure 2 G and H, the virus titers were consistently higher in DHX9 knockdown cells than in shCtrl/3T3 cells. We also examined the effect of the DHX9 knockdown on HeLa epithelial cells (shDHX9/HeLa) infected with either MHV-68 or HSV-1 and obtained the same results (Supplementary Figure S1).
Figure 2.

DHX9 knockdown fibroblasts are more susceptible to MHV-68 infection. (A and B) DHX9 knockdown NIH3T3 cells were generated by transducing the cells with lentiviruses expressing shRNAs against DHX9 (shDHX9/3T3) or control shRNA (shCtrl/3T3). The magnitude of the DHX9 knockdown was determined by RT-qPCR (A) and western blot analysis of 3 independent experiments (B). (C–H) The shDHX9/3T3 cells and shCtrl/3T3 cells were infected with MHV-68 at MOI 0.5 (C, E and G) or 2 (D, F, and H) for 24, 36 and 48 h. (C, D) Viral protein expression levels in the infected cells were determined with antibodies against ORF45 (a viral tegument protein) and ORF65 (a small capsid protein, M9). α-Tubulin served as a loading control. (E, F) Total genomic DNAs were extracted from infected cells, and real-time PCR was conducted using primers against MHV-68 ORF56 to quantitate the copy number of viral DNAs. (G, H) Cell-free supernatants were collected and virion production was determined by plaque assays. The data represent the mean ± SD of triplicate assays. *P < 0.05; **P < 0.01; ***P < 0.001 versus control, ns = nonsignificant (Student's t test).
To test whether transfection with viral DNA results in a similar phenotype in DHX9 knockdown cells, we transfected shDHX9/3T3 cells with the viral genomic DNA from an infectious BAC clone of wild-type (WT) MHV-68 (pBAC/MHV-68) (33) for up to 72 h (Supplementary Figure S2). Representative microscopic images showed that shDHX9/3T3 clearly manifested major viral cytopathic effects (CPEs) at 60 h post-transfection, whereas shCtrl/3T3 cells did not show CPEs until 72 h (Supplementary Figure S2A). Viral replication was upregulated in shDHX9/3T3 cells relative to shCtrl/3T3 cells (Supplementary Figure S2B–D). Taken together, our results indicated that the DHX9 knockdown facilitated MHV-68 replication during both viral infection and viral genomic DNA transfection.
DHX9 is necessary for an antiviral response to MHV-68 infection
We found that overexpression of DHX9 decreased MHV-68 and HSV-1 replication and potentiated virus-triggered induction of IL6 and IFN-β transcriptions in infected HeLa and HEK293T cells in a dose-dependent manner (Supplementary Figure S3 and S4). Next, we determined whether endogenous DHX9 is required for the host innate immune responses induced by MHV-68 in fibroblasts and epithelial cells. First, the cytokine responses were measured after MHV-68 infection at MOI 2 in shDHX9/3T3 cells (Figure 3A and B). The induction of Il-6 and Ifn-β transcripts in response to MHV-68 infection was impaired in shDHX9/3T3 cells. Similar results were also obtained in DHX9 knockdown HeLa cells (shDHX9/HeLa) where we saw a significant reduction in the IL6 and IFN-β transcript levels in shDHX9/HeLa cells, when compared with shCtrl/HeLa cells (Figure 3C and D). HSV-1 infection in shDHX9/HEK293T cells yielded a similar phenotype (Supplementary Figure S5). We also performed 2 × кB-luc and IFN-β-luc reporter assays in shDHX9/HeLa cells. Cells devoid of DHX9 expression showed marked attenuation of 2 × кB-luc and IFN-β-luc reporter activities induced by MHV-68 infection (Figure 3E and F). Likewise, the deficient expression of DHX9 inhibited p65-mediated NF-κB activation (Figure 3G). These results suggested that regulation of NF-κB activation by endogenous DHX9 may be necessary for the antiviral responses to DNA virus infection. In agreement with this hypothesis, our preliminary transcriptome analysis (by RNA-seq) of shCtrl/HeLa and shDHX9/HeLa cells with or without HSV-1 infection revealed that most of the known NF-κB–dependent genes (66 out of 68 genes; ∼97%) were dramatically downregulated in shDHX9/HeLa cells (Supplementary Figure S6).
Figure 3.

DHX9 is essential for induction of IL6 and type I IFN upon MHV-68 infection. (A–D) shDHX9/3T3 (A, B) and shDHX9/HeLa (C, D) cells were mock-infected or infected with MHV-68 at MOI 2 for 6 and 12 h. Total cellular RNA was extracted and subjected to RT-qPCR for IL6 (A, C) and IFN-β (B, D) mRNA levels. (E, F) shDHX9/HeLa and shCtrl cells were transfected with 2 × кB-luc (E) or IFN-β-luc (F) reporter plasmid (100 ng) for 20 h, then infected with MHV-68 at MOI 2 for 12 h, and harvested for luciferase assays. (G) shDHX9/HeLa and shCtrl cells were transfected with 2 × кB-luc (100 ng) and HA-p65 (50 ng) for 28 h and harvested for luciferase assays. The relative luciferase activity (RLA) of each sample was determined by normalizing the activity to the sample's β-galactosidase activity. The data represent the mean ± SD of triplicate assays. ***P < 0.001 versus control, ns = nonsignificant (Student's t test).
The ATPase/helicase activity, but not the DNA-sensing domain of DHX9, is essential for its antiviral activity during replication of a DNA virus
DHX9 belongs to the DExH family of ATPase-dependent helicase (ATPase/helicase) with the ability to bind to and unwind DNA and RNA (34). DHX9 consists of two double-stranded RNA-binding domains (dsRBDs) at the N-terminus, an ATP-binding site in the DEIH helicase core, helicase-associated domain 2 (HA2), an oligonucleotide- or oligosaccharide-binding region (OB-fold) formerly known as the domain of unknown function (DUF), and a nuclear transport domain (NTD) at the C-terminus that overlaps with the arginine–glycine–glycine (RGG) repeats (35) (Figure 4A). To determine the domain required for the antiviral function of DHX9 in nonimmune cells during DNA virus infection, we used various mutant constructs of DHX9, which encode an ATPase/helicase mutant (K417R) with a point mutation in the conserved ATP-binding motif (Gly-Lys-Thr) in the catalytic domain (35), a DNA-sensing mutant (ΔDUF; amino acid [aa] positions 1–920) (3), or a DNA-sensing mutant with the addition of NTD (ΔDUF+NTDDHX9; aa 1–920 and 1171–1270). The ΔDUF+NTDDHX9 mutant construct was generated by inserting the aa 1171–1270 region into the ΔDUF mutant, because the C terminus of DHX9 (NTD) is required for the nuclear localization of DHX9 (36). The expression and the subcellular localization of the constructs were examined along with the WT DHX9 in HEK293T cells (Supplementary Figure S7). All mutants were expressed at the levels comparable to the expression of WT DHX9 and were predominantly localized to the nucleus, except for the ΔDUF mutant. We cotransfected the FLAG-tagged DHX9 mutant constructs with reporter plasmids into HeLa cells for 20 h and infected the cells with MHV-68 at MOI 2 for another 12 h (Figure 4B–E). Unlike the WT DHX9, the ATPase/helicase mutant of K417R failed to enhance the activation of 2 × кB-luc and IFN-β-luc promoters during viral infection (Figure 4B and C). In contrast, ΔDUF+NTDDHX9 further increased the MHV-68-induced activation of 2 × кB-luc and IFN-β-luc promoters, whereas the ΔDUF mutant did not, presumably because of its cytoplasmic localization. Similar results were obtained when the abundance of IL6 and IFN-β transcripts was analyzed during MHV-68 infection in HeLa cells transfected with DHX9 mutant constructs (Figure 4D and E). In these cells, the quantities of the RTA transcript from a viral genome inversely correlated with the antiviral activity of DHX9 mutants (Figure 4F). The ΔDUF+NTDDHX9 mutant, but not the K417R and ΔDUF mutants, had active antiviral function like WT DHX9 did (Figure 4F). Moreover, the reconstitution of DHX9 knockdown cells (shDHX9/HeLa) with the WT DHX9 by transfection significantly restored the MHV-68-induced activation of the 2 × кB promoters to the levels similar to those in the shCtrl cells; this effect was not recapitulated with the ATPase/helicase mutant, K417R (34) (Figure 4G). In the same experiment, ΔDUF+NTDDHX9 also regained the virus-induced activation of the 2 × кB promoters in DHX9 knockdown cells, whereas ΔDUF (aa 1–920) did not.
Figure 4.

The ATPase/helicase activity, not the cytosolic DNA-sensing domain of DHX9 is required for induction of IL6 and type I IFN upon MHV-68 infection. (A) Schematic diagrams of DHX9 domains and its deletion mutants. The numbers represent the positions of DHX9 amino acid residues that delineate the domains. dsRBDs, double-stranded RNA-binding domains; HA2, helicase-associated domain 2; OB-fold, oligonucleotide/oligosaccharide-binding; NTD, nuclear transport domain; and RGG-box, glycine-rich domain. K417R represents a substitution mutant of DHX9 Lys 417 to Arg that abrogates the ATP-dependent helicase activity, while DUF is a domain responsible for cytosolic DNA sensing. (B, C) HeLa cells in 24-well plates were transfected with 2 × кB-luc (B) or IFN-β-luc (C) reporter plasmid (100 ng) and various DHX9 mutants (200 or 400 ng) for 20 h, followed by infection with MHV-68 at MOI 2 for another 12 h, and harvested for a luciferase assay. (D–F) HeLa cells in 12-well plates were transfected with various DHX9 mutants (1 or 2 μg) for 24 h, and either mock-infected or infected with MHV-68 at MOI 2 for 12 h, total RNAs were extracted for RT-qPCR. The relative levels of IL6 (D), IFN-β (E) and MHV-68 RTA (F) mRNAs were determined using ACTIN as a control. (G) shCtrl/HeLa and shDHX9/HeLa cells were transfected with 2 × кB-luc (100 ng) and various DHX9 mutants (200 or 400 ng) for 20 h, followed by infection with MHV-68 at MOI 2 for another 12 h, and the cells were harvested for a luciferase assay. The data represent mean ± SD of triplicate assays.
To rule out the possibility that NTDDHX9 (aa 1170–1270) still contains a part of the DNA-sensing domain, a new mutant, DHX9(ΔDUF+NLSSV40) was generated by replacing the NTD domain of DHX9(ΔDUF+NTDDHX9) with a classical nuclear localization signal (NLS) from the SV40 large T-antigen (PKKKRKV) (27). The new construct showed an antiviral activity similar to that of WT DHX9 (Supplementary Figure S8). These results confirmed the antiviral activity of DHX9 against DNA virus infection, which is shown to be dependent on its ATPase/helicase domain in the nucleus rather than the DNA-sensing domain in the cytoplasm, in stimulation of NF-κB–dependent transcription of antiviral genes.
DNA virus infection did not induce cytoplasmic translocation of DHX9 in cells that are not pDCs
DHX9 was previously reported to induce NF-κB–dependent transcription in pDCs via interaction with MyD88 after its cytoplasmic translocation upon the exposure to CpG DNA or DNA virus (3). To delineate the molecular mechanisms of DHX9-induced NF-κB activation in cells that are not pDCs, we investigated the subcellular localization of DHX9 in BMDMs and MEFs after infection with recombinant EGFP/MHV-68 or GFP-HSV-1 (Figure 5). Virus-infected BMDMs showed no cytosolic translocation of DHX9, as indicated in GFP-positive cells (Figure 5A). Likewise, the subcellular localization of DHX9 was restricted to the nucleus of MEFs, indicating a critical role of nuclear DHX9 in permissive fibroblasts after virus infection (Figure 5B). It was also noted that endogenous DHX9 underwent changes in nuclear distribution (from a diffuse to punctate pattern) after virus infection. To examine the effects of DHX9 domain mutations on nuclear distribution, DHX9 knockdown (shDHX9/HeLa) cells were transfected with FLAG-DHX9 WT, K417R or ΔDUF+NLSSV40 followed by infection with EGFP/MHV-68 (Supplementary Figure S9). Upon infection, punctate distribution of nuclear DHX9 was seen in shDHX9/HeLa cells transfected with any of the constructs, regardless of their antiviral activity, suggesting that defects of K417R in antiviral activity are unlikely to be due to aberrant nuclear distribution.
Figure 5.

Nuclear DHX9 is not translocated to the cytosol after DNA virus infection in BMDMs and MEFs. (A) Two representative confocal-microscopy images of BMDMs that were either mock-infected (top) or infected with EGFP/MHV-68 (middle) or HSV-GFP (bottom) at MOI 5 for 12 h. (B) Two representative confocal-microscopy images of MEFs that were either mock-infected (top) or infected with EGFP/MHV-68 (middle) or HSV-GFP (bottom) at MOI 2 for 12 h. The cells were fixed and stained for endogenous DHX9 (Cy3-red), while DAPI staining (blue) was performed to visualize nuclei. Images were obtained by means of an LSM 700 confocal microscope from Carl Zeiss. All panels show × 1,000 magnification (scale bar: 20 μm).
DHX9 interacts with NF-κB p65 and RNAPII in the nucleus during DNA virus infection
Next, we studied the molecular mechanisms by which DHX9 facilitates NF-κB–mediated transactivation during DNA virus infection. Although DHX9 reportedly interacts with NF-κB p65 in an overexpression system (14), the functional significance of this specific protein association during viral infection has never been validated. Our coimmunoprecipitation (co-IP) experiments with HEK293T cells transfected with FLAG-DHX9 and HA-p65 confirmed this interaction (Figure 6A), and the interaction was stronger in MHV-68-infected cells. To further elucidate the kinetics of NF-κB activation and DHX9 localization during MHV-68 infection, we conducted biochemical subcellular fractionation assays (Figure 6B). Endogenous NF-κB p65 was detected entirely in the cytosol of the mock-infected cells, whereas endogenous DHX9 was predominantly found in the nucleus. During MHV-68 infection, NF-κB p65 began to relocate to the nucleus as early as within 6 h and continued until 24 h after the infection. These results were confirmed by the immunofluorescence assays, showing that NF-κB p65 was translocated to the nucleus and colocalized with DHX9 during virus infection (Figure 6C).
Figure 6.

Endogenous DHX9 interacts with NF-κB p65 in MHV-68–infected fibroblasts and epithelial cells. (A) HEK293T cells were transfected with FLAG-DHX9 (3 μg) and HA-p65 (3 μg) for 36 h and infected with MHV-68 at MOI 2 for 12 h. Cell lysates were subjected to immunoprecipitation (IP) with an anti-FLAG antibody and western blot analysis with an anti-HA antibody. (B) Subcellular fractions of mock-infected and MHV-68-infected HeLa cells at MOI 2 were prepared at the indicated time points. PARP-1 and α-tubulin were used to confirm the integrity of nuclear and cytosolic extracts, respectively. (C) Two representative confocal microscopy images of HeLa cells that were either mock-infected or infected with MHV-68 at MOI 2 for 12 h. The cells were fixed and stained for endogenous DHX9 (FITC-green) and p65 (Cy3-red). DAPI staining (blue) was used to visualize nuclei. (D and E) Interactions of DHX9 with NF-κB p65 and RNAPII in fibroblasts and epithelial cells during viral infection. NIH3T3 cells (D) and HeLa cells (E) were mock-infected or infected with MHV-68 at MOI 2 for 12 h. The cytosolic and the nuclear extracts from the cells were subjected to immunoprecipitation with the anti-DHX9 antibody and immunoblotted with the indicated antibodies. The input lysates represent 10% of the extracts by volume used for each immunoprecipitation. (F) The cells were prepared as described in (C) and stained for endogenous DHX9 (FITC-green) and RNAPII (Cy3-red). All panels show ×1000 magnification (scale bar: 20 μm).
To validate the subcellular compartment where endogenous DHX9 physically associates with NF-κB p65 during viral infection, we carried out endogenous co-IP with an anti-DHX9 antibody in the nuclear and the cytosolic extracts of NIH3T3 cells and HeLa cells with mock or MHV-68 infection (Figure 6D and E). DHX9 strongly interacted with NF-κB p65 in the nucleus of both NIH3T3 and HeLa cells during MHV-68 infection. DHX9 was found to weakly interact with RNAPII during mock infection, and this interaction was intensified during viral infection. In contrast, an abundant nuclear protein, poly(ADP-ribose) polymerase-1 (PARP-1), did not interact with DHX9 (Figure 6D and E, lanes 3 and 4). We also noticed that DHX9 did not interact with NF-κB p65 or MyD88 in the cytosol (Figure 6D and E, lane 2). Similar results were obtained for HSV-1 infection (Supplementary Figure S10A).
Immunofluorescence assays confirmed that the punctate pattern of DHX9 colocalized in part with the RNAPII in the nucleus during MHV-68 infection (Figure 6F). Additionally, we confirmed the colocalization of endogenous DHX9 and initiating RNAPII using anti-RNA Pol II CTD repeat YSPTSPS (phospho S5) antibody as well as colocalization of NF-κB p65 and RNAPII (phospho S5) after virus infection (Supplementary Figure S11). Taken together, these data suggested that endogenous DHX9 was specifically associated with NF-κB p65 and RNAPII in the nucleus of DNA virus–infected fibroblasts and epithelial cells.
DHX9 is required for the recruitment of RNAPII to the NF-κB p65–dependent promoters of antiviral cytokines during DNA virus infection
To test whether DHX9 is necessary for the interactions between NF-κB and RNAPII during viral infection, we performed reciprocal co-IP with an antibody against endogenous NF-κB p65 in shCtrl/HeLa or shDHX9/HeLa cells with or without MHV-68 infection (Figure 7A). Although endogenous DHX9 and RNAPII were readily detectable in the immunoprecipitates of the nuclear extracts from control cells during MHV-68 infection, nuclear p65 failed to interact with RNAPII in DHX9 knockdown cells. Similar results were obtained for HSV-1 infection (Supplementary Figure S10B). This finding suggested that DHX9 may contribute to the association between NF-κB p65 and RNAPII during DNA virus infection as part of the innate immune response. We then carried out chromatin IP (ChIP) assays to test whether DHX9 is required for the recruitment of NF-κB p65 and RNAPII to the specific antiviral promoters, such as IL6 or IFN-β, during MHV-68 infection (Figure 7B–D). DHX9 knockdown HeLa cells and control cells were either mock-infected or infected with MHV-68 for 12 h. The cross-linked DNA was immunoprecipitated with an anti-p65, anti-RNAPII, or nonspecific antibody (anti-IgG). Quantitated ChIP data with or without MHV-68 infection in shDHX9/HeLa cells revealed compromised RNAPII’s occupancy of IL6 and IFN-β promoters in these cells, whereas NF-κB p65 binding was barely affected (Figure 7B and C). In the same experiment, equivalent amounts of RNAPII were detected on the GAPDH promoter in either shCtrl/HeLa or shDHX9/HeLa cells regardless of virus infection (Figure 7D). These results are in agreement with the reduced activation of IL6 and IFN-β expression observed in DHX9 knockdown cells during viral infection (Figure 3C and D) and indicate that DHX9 is necessary to recruit RNAPII for the activation of the antiviral gene promoters after nuclear translocation of p65.
Figure 7.
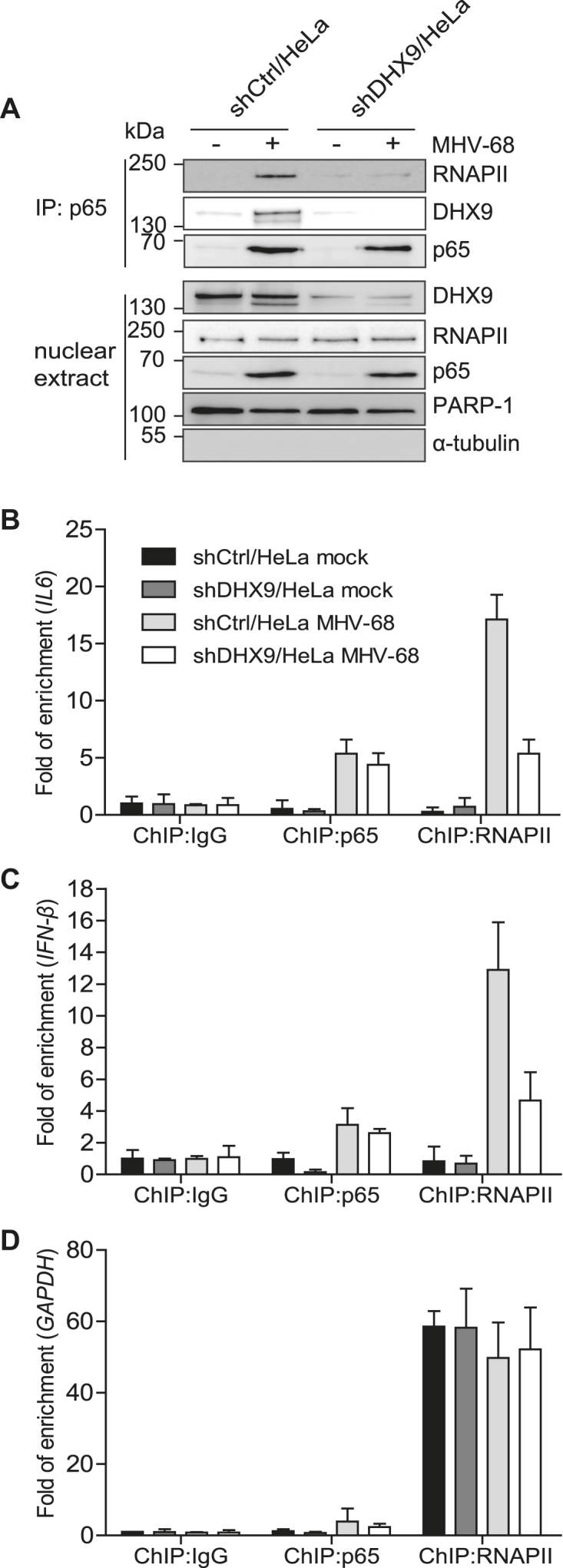
Endogenous DHX9 is required for RNAPII recruitment to NF-κB–dependent antiviral promoters. (A) shCtrl and shDHX9/HeLa cells were infected with MHV-68 at MOI 2 for 12 h. The nuclear extract was subjected to immunoprecipitation with the anti-p65 antibody and immunoblotted with the indicated antibodies. The input lysates represent 10% of nuclear extracts by volume used for each immunoprecipitation. (B–D) shCtrl/HeLa and shDHX9/HeLa cells were infected with MHV-68 at MOI 2 for 12 h. Cell lysates were prepared and analyzed by a ChIP assay with an anti-p65 or anti-RNAPII antibody. Normal rabbit IgG was included as a control for nonspecific immunoprecipitation. The amounts of precipitated DNAs were measured by qPCR using primers flanking the promoter regions of IL6 (B), IFN-β (C), and GAPDH (D). The data represent mean ± SD of triplicate assays.
The ATPase activity of DHX9 is required for RNAPII recruitment to the NF-κB p65–dependent promoters during DNA virus infection
To further delineate the mechanisms of action of DHX9, we determined which domain of DHX9 is required for recruitment of RNAPII to the promoter in nonimmune cells. For this purpose, HEK293T cells stably expressing the shRNA targeting the region within the 3′ UTR of DHX9 mRNA were generated by means of a lentiviral system. Cells lacking DHX9 were transfected with an siRNA-resistant construct encoding WT DHX9, K417R, ΔDUF, ΔDUF+NTDDHX9 or ΔDUF+NLSSV40 for 36 h, followed by mock- or virus infection for another 12 h (Figure 8). DHX9 proteins were all similarly expressed (Figure 8A). Consistent with our previous results (Figure 7), DHX9 depletion correlated with reduced RNAPII recruitment to IL6 and IFN-β promoters, while the occupancy by p65 was affected only marginally (Figure 8B and C). The loss of RNAPII recruitment in DHX9-depleted cells was reversed by transfection with either WT DHX9 or two other independent DNA-sensing mutants (ΔDUF+NTDDHX9 and ΔDUF+NLSSV40) that were localized to the nucleus. The ATPase/helicase mutant (K417R) and the DNA-sensing mutant (ΔDUF) with cytoplasmic localization failed to reconstitute this activity or abrogated the RNAPII recruitment to the antiviral promoters (Figure 8B and C) (14). In contrast, RNAPII recruitment to the GAPDH promoter was not affected in any cases (Figure 8D). Together with our results on these mutants in reporter assays (Figure 4), these trans-complementation results for ChIP assays revealed that the ATPase/helicase activity was necessary for DHX9 to drive proper transcription of antiviral genes by recruiting RNAPII to NF-κB–responsive promoters during DNA virus infection.
Figure 8.

The ATPase/helicase activity of DHX9 is required for RNAPII recruitment to NF-κB–dependent antiviral promoters. Stable cells expressing 3′ UTR shDHX9 were reconstituted with FLAG-DHX9-WT (lacking the 3′ UTR), K417R, ΔDUF, ΔDUF+NTDDHX9, or ΔDUF+NLSSV40 (18 μg) for 36 h. The transfected cells were either mock-infected or infected with MHV-68 at MOI 2 for 12 h. (A) Cell lysates were subjected to western blot analysis with anti-DHX9 and anti-α-tubulin antibodies to assess the expression levels. (B–D) Cell lysates were analyzed by ChIP assays involving the anti-p65 or anti-RNAPII antibody. Normal rabbit IgG was included as a control for nonspecific immunoprecipitation. The amounts of input and precipitated DNAs were measured by qPCR with primers flanking the promoter regions of IL6 (B), IFN-β (C), and ‘GAPDH (D). The data represent mean ± SD of triplicate assays.
DHX9 itself is recruited to the NF-κB p65–dependent promoters during viral infection
Our data suggested that DHX9 may act as a bridging factor for NF-κB p65 and RNAPII during virus infection because DHX9 was required for RNAPII recruitment to the NF-κB–dependent promoters. To determine whether DHX9 directly binds to the NF-κB–dependent promoters in the context of chromatin, cross-linked DNA was immunoprecipitated with an anti-p65, anti-DHX9, anti-RNAPII, or nonspecific antibody (anti-IgG). Quantitated ChIP data from HeLa cells with MHV-68 infection showed the occupancy of IL6 and IFN-β promoters by DHX9 in addition to p65 and RNAPII, suggesting that DHX9 itself is recruited to NF-κB–dependent promoters together with the NF-κB subunit p65 (Figure 9 A and B). Next, we performed a re-ChIP experiment to determine whether DHX9 is associated with the multiprotein complex involving NF-κB p65 in the chromatin containing NF-κB–binding sites after virus infection. Virus-infected cells were used to immunoprecipitate chromatin with the anti-p65 antibody, followed by the second immunoprecipitation with either the anti-RNAPII or anti-DHX9 antibody (Figure 9C). The re-ChIP experiment revealed that DHX9 was recruited to IL6 and IFN-β promoters upon interaction with p65 after virus infection, thus indicating p65-associated DHX9 and RNAPII at the genomic sites containing NF-κB recognition sites in the specific promoters (Figure 9C, lanes 3–6). Reciprocal re-ChIP using the anti-RNAPII or anti-DHX9 antibody confirmed the co-occupancy of the NF-κB target gene promoters by p65 and DHX9 together with RNAPII (Figure 9C, lanes 7–14). These results suggested that DHX9 may directly bind to the NF-κB target gene promoters and recruit RNAPII, forming the multiprotein transcription activator complex to induce the NF-κB–dependent antiviral genes during DNA virus infection.
Figure 9.

DHX9 is recruited to the NF-κB p65–dependent promoters during viral infection. HeLa cells were either mock-infected or infected with MHV-68 at MOI of 2 for 12 h. (A and B) Cell lysates were prepared and analyzed by ChIP assays with an anti-p65, anti-RNAPII, or anti-DHX9 antibody. Normal rabbit IgG was included as a control for nonspecific immunoprecipitation. The amounts of precipitated DNA were measured by quantitative PCR (qPCR) involving primers flanking the promoter regions of IL6 (A) and IFN-β (B). The data represent mean ± SD of triplicate assays. (C) The cell lysates were prepared for ChIP, first subjected to immunoprecipitation with the anti-p65 antibody and then subjected to re-ChIP with anti-RNAPII and anti-DHX9 antibodies. Genomic DNA samples sequentially enriched in the NF-κB target gene promoters (IL6 and IFN-β promoters) were amplified by PCR and analyzed by agarose gel electrophoresis (lanes 3–6). Reciprocal re-ChIP using the anti-RNAPII or anti-DHX9 antibody was also conducted with the first antibody followed by incubation with the second antibody as indicated (lanes 7–14). Normal IgG was included as a control for nonspecific immunoprecipitation and input lanes indicate the chromatin complexes prior to immunoprecipitation. A set of representative agarose gel images is shown from two independent experiments.
DISCUSSION
DHX9, a member of the DExD/H subclass (also known as RNA helicase A) is an abundant nuclear protein and performs an active function in transcription and RNA metabolism (37). However, how DHX9 participates in the regulation of a transcriptional response to viral infection has not been studied. In this report, we demonstrated a novel mechanism of action of DHX9 as a transcription regulator in the control of DNA virus infection in fibroblasts and epithelial cells, aside from its role as a cytosolic nucleic-acid sensor in pDCs (3). It is essential to understand the function and mechanisms of action of DHX9 in permissive cells in addition to pDCs during DNA virus infection because most DNA viruses deposit and replicate their genomes within the nucleus (19).
NF-κB is reported to be critical for antiviral immune responses to DNA viruses. For instance, loss of NF-κB p50 in mice causes an inability to efficiently control MHV-68 infection, thereby leading to persistent virus replication in the lungs and high levels of latently infected B cells in the spleen (38). NF-κB activation in monocytes plays a key role in the inhibition of HSV-1 replication (39). Besides, NF-κB inhibitors abrogated the expression of antiviral genes in response to HSV-2 infection in human cervical epithelial cells (40). Here, our results provided several lines of evidence that DHX9 performs a critical function as a nuclear transcriptional coactivator for NF-κB–mediated transactivation rather than as a cytosolic DNA sensor in the antiviral response of permissive cells during DNA virus infection. Both the macrophage-specific knockout and fibroblast-specific knockdown of DHX9 attenuated NF-κB–mediated induction of antiviral genes and suppressed the concomitantly increased viral replication (Figures 1–3 and Supplementary Figures S1, S2 and S5), whereas DHX9 overexpression promoted NF-κB–dependent antiviral cytokine expression and suppressed viral replication (Figure 4 and Supplementary Figures S3 and S4). In contrast to pDCs, in the cytosol of macrophages and fibroblasts, DHX9 neither was present (via translocation) nor interacted with MyD88 during DNA virus infection, but appeared as a punctate pattern in the nucleus, colocalizing with RNAPII (Figures 5 and 6). Moreover, DHX9 deficiency abrogated the interaction between RNAPII and NF-κB and the occupancy of IL-6 and IFN-β promoters by RNAPII but not by NF-κB p65 (Figures 6–8). In the context of chromatin, DHX9 itself effectively bound to these promoters in the form of a multiprotein complex together with RNAPII and NF-κB p65 (Figure 9). To our knowledge, this is the first demonstration of DHX9 binding and recruitment to the promoters in the context of chromatin. Taken together, these results highlight the importance of nuclear DHX9 as a co–transcription factor for expression of antiviral genes for defense against DNA viruses. Of note, the influence of DHX9 on the promoters of antiviral cytokines seems to be specific to NF-κB because DHX9 failed to affect the IRF3-mediated transactivation of IFN-β-luc promoters (Supplementary Figure S12).
DHX9 has been shown to interact with various nuclear transcription regulatory factors, including the CBP–p300 complex (12), BRCA1 (41), SMN complex (42), topoisomerase II (37), NF-κB p65 (14), the STAT6–p100 complex (43) and EGFR (25). These interactions are often linked to the RNAPII holoenzyme and are intended to facilitate the transcriptional regulation of specific gene expression (13). Nevertheless, the detailed mechanisms by which DHX9 can facilitate the transcription-regulatory activity of these factors are poorly understood. In our mutant study, the antiviral activity of DHX9 required its ATPase/helicase activity in the nucleus, rather than the DUF domain, an important DNA-sensing domain in the cytosol. Consistent with these results, reduced RNAPII’s occupancy of the antiviral promoters in DHX9-depleted cells was restored by complementation with WT DHX9 or a nucleus-localized DNA-sensing mutant (ΔDUF+NTDDHX9 and ΔDUF+NLSSV40), whereas complementation with the ATPase/helicase mutant (K417R) or the cytosolic ΔDUF mutant (ΔDUF) alone failed to reconstitute this activity (Figure 8). In addition, all DHX9 domain mutants were able to interact with p65 and RNAPII in the transfected cells and underwent a punctate distribution in the nucleus during DNA virus infection (Supplementary Figures S9 and S13). These results reinforced the notion that the ATPase/helicase activity of DHX9 may be critical for NF-κB–mediated antiviral activity even in the presence of an interaction of DHX9 with p65 and RNAPII (Supplementary Figures S11 and S13). In line with our findings, DHX9-K417R has been shown to decrease CREB-dependent transcription in mammals (12) and male X-linked gene transcription in Drosophila (44). Given that our ChIP and re-ChIP results indicate that DHX9 binds to the promoters and constitutes a multiprotein complex together with RNAPII and NF-κB (Figure 9), it is plausible that unwinding of the promoter regions by DHX9 may serve as a platform for RNAPII recruitment to the promoters. Similarly, TFIIH exerts its helicase action to help RNAPII to gain access to the template (45). As seen in the case with Akirin2, which bridges NF-κB and the chromatin-remodeling SWI–SNF complex and forms a complex with the NF-κB subunit p50 (46), it will be interesting to investigate whether DHX9 can interact with additional chromatin-remodeling complexes during DNA virus infection.
We also noticed that complementation with the cytosolic ΔDUF mutant in DHX9 knockdown cells abrogated NF-κB recruitment, while other mutants as well as the WT only marginally affected the occupancy by NF-κB (Figure 8). Because the DHX9 knockdown itself did not alter NF-κB recruitment significantly, it will be interesting to test whether the cytosolic ΔDUF mutant can interfere with nuclear translocation of NF-κB. Consistent with this notion, the ΔDUF mutant also abrogated RNAPII recruitment; this effect may be due to the possible defects in nuclear translocation of NF-κB under the influence of ΔDUF, affecting the recruitment of RNAPII to the NF-κB–dependent promoters. In conclusion, our results show that nuclear DHX9 is crucial for innate immunity against DNA virus infection in fibroblasts and epithelial cells and may serve as a bridging factor to recruit RNAPII to the NF-κB–specific promoters of antiviral genes with its own ability to bind to and unwind the promoter DNA. Because many DNA viruses including herpesviruses inevitably target important host antiviral factors (47–49), it will be worthwhile to identify the molecular strategy that a virus may develop to evade the antiviral action of DHX9.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Kathleen Boris-Lawrie (University of Minnesota, St. Paul, MN, USA) for the FLAG-DHX9 and DHX9 (K417R) constructs and Dr. Mien-Chie Hung (The University of Texas, MD Anderson Cancer Center, Houston, TX, USA) for the human DHX9 shRNA constructs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Korea University Grant and the National Research Foundation of Korea (NRF) funded by the Ministry of Education [2018R1A2B6001363 to M.J.S.]. Funding for open access charge: National Research Foundation of Korea (NRF) funded by the Ministry of Education [2018R1A2B6001363].
Conflict of interest statement. None declared.
REFERENCES
- 1. Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004; 5:730–737. [DOI] [PubMed] [Google Scholar]
- 2. Schmidt A., Endres S., Rothenfusser S. Pattern recognition of viral nucleic acids by RIG-I-like helicases. J. Mol. Med. 2011; 89:5–12. [DOI] [PubMed] [Google Scholar]
- 3. Kim T., Pazhoor S., Bao M., Zhang Z., Hanabuchi S., Facchinetti V., Bover L., Plumas J., Chaperot L., Qin J. et al. Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:15181–15186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miyashita M., Oshiumi H., Matsumoto M., Seya T. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell. Biol. 2011; 31:3802–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oshiumi H., Sakai K., Matsumoto M., Seya T.. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur. J. Immunol. 2010; 40:940–948. [DOI] [PubMed] [Google Scholar]
- 6. Soulat D., Burckstummer T., Westermayer S., Goncalves A., Bauch A., Stefanovic A., Hantschel O., Bennett K.L., Decker T., Superti-Furga G. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 2008; 27:2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang Z., Yuan B., Bao M., Lu N., Kim T., Liu Y.J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011; 12:959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Z., Yuan B., Lu N., Facchinetti V., Liu Y.J. DHX9 pairs with IPS-1 to sense double-stranded RNA in myeloid dendritic cells. J. Immunol. 2011; 187:4501–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mosallanejad K., Sekine Y., Ishikura-Kinoshita S., Kumagai K., Nagano T., Matsuzawa A., Takeda K., Naguro I., Ichijo H.. The DEAH-box RNA helicase DHX15 activates NF-κB and MAPK signaling downstream of MAVS during antiviral responses. Sci. Signal. 2014; 7:ra40. [DOI] [PubMed] [Google Scholar]
- 10. Lee C.G., Hurwitz J.. Human RNA helicase A is homologous to the maleless protein of Drosophila. J. Biol. Chem. 1993; 268:16822–16830. [PubMed] [Google Scholar]
- 11. Kuroda M.I., Kernan M.J., Kreber R., Ganetzky B., Baker B.S.. The maleless protein associates with the X chromosome to regulate dosage compensation in Drosophila. Cell. 1991; 66:935–947. [DOI] [PubMed] [Google Scholar]
- 12. Nakajima T., Uchida C., Anderson S.F., Lee C.G., Hurwitz J., Parvin J.D., Montminy M.. RNA helicase A mediates association of CBP with RNA polymerase II. Cell. 1997; 90:1107–1112. [DOI] [PubMed] [Google Scholar]
- 13. Aratani S., Fujii R., Oishi T., Fujita H., Amano T., Ohshima T., Hagiwara M., Fukamizu A., Nakajima T.. Dual roles of RNA helicase A in CREB-dependent transcription. Mol. Cell. Biol. 2001; 21:4460–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tetsuka T., Uranishi H., Sanda T., Asamitsu K., Yang J.P., Wong-Staal F., Okamoto T.. RNA helicase A interacts with nuclear factor kappaB p65 and functions as a transcriptional coactivator. Eur. J. Biochem. 2004; 271:3741–3751. [DOI] [PubMed] [Google Scholar]
- 15. Weir J.P. Genomic organization and evolution of the human herpesviruses. Virus Genes. 1998; 16:85–93. [DOI] [PubMed] [Google Scholar]
- 16. Grinde B. Herpesviruses: latency and reactivation–viral strategies and host response. J. Oral Microbiol. 2013; 5:22766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whitley R.J., Roizman B.. Herpes simplex virus infections. Lancet. 2001; 357:1513–1518. [DOI] [PubMed] [Google Scholar]
- 18. Efstathiou S., Ho Y.M., Hall S., Styles C.J., Scott S.D., Gompels U.A.. Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus saimiri. J. Gen. Virol. 1990; 71:1365–1372. [DOI] [PubMed] [Google Scholar]
- 19. Paludan S.R., Bowie A.G., Horan K.A., Fitzgerald K.A.. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 2011; 11:143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weischenfeldt J., Porse B.. Bone Marrow-Derived Macrophages (BMM): Isolation and applications. CSH Protoc. 2008; 2008:pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- 21. Kang H.R., Cheong W.C., Park J.E., Ryu S., Cho H.J., Youn H., Ahn J.H., Song M.J.. Murine gammaherpesvirus 68 encoding open reading frame 11 targets TANK binding kinase 1 to negatively regulate the host type I interferon response. J. Virol. 2014; 88:6832–6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu T.T., Tong L., Rickabaugh T., Speck S., Sun R.. Function of Rta is essential for lytic replication of murine gammaherpesvirus 68. J. Virol. 2001; 75:9262–9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee S., Cho H.J., Park J.J., Kim Y.S., Hwang S., Sun R., Song M.J.. The ORF49 protein of murine gammaherpesvirus 68 cooperates with RTA in regulating virus replication. J. Virol. 2007; 81:9870–9877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moffat J., Grueneberg D.A., Yang X., Kim S.Y., Kloepfer A.M., Hinkle G., Piqani B., Eisenhaure T.M., Luo B., Grenier J.K. et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006; 124:1283–1298. [DOI] [PubMed] [Google Scholar]
- 25. Huo L., Wang Y.N., Xia W., Hsu S.C., Lai C.C., Li L.Y., Chang W.C., Wang Y., Hsu M.C., Yu Y.L. et al. RNA helicase A is a DNA-binding partner for EGFR-mediated transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:16125–16130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hwang S., Maloney N.S., Bruinsma M.W., Goel G., Duan E., Zhang L., Shrestha B., Diamond M.S., Dani A., Sosnovtsev S.V. et al. Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe. 2012; 11:397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Z., Kenworthy R., Green C., Tang H.. Molecular determinants of nucleolar translocation of RNA helicase A. Exp. Cell Res. 2007; 313:3743–3754. [DOI] [PubMed] [Google Scholar]
- 28. Noh C.W., Cho H.J., Kang H.R., Jin H.Y., Lee S., Deng H., Wu T.T., Arumugaswami V., Sun R., Song M.J.. The virion-associated open reading frame 49 of murine gammaherpesvirus 68 promotes viral replication both in vitro and in vivo as a derepressor of RTA. J. Virol. 2012; 86:1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ng Y.C., Kim Y.W., Ryu S., Lee A., Lee J.-S., Song M.J.. Suppression of norovirus by natural phytochemicals from Aloe vera and Eriobotryae Folium. Food Control. 2017; 73:1362–1370. [Google Scholar]
- 30. Song M.J., Hwang S., Wong W.H., Wu T.T., Lee S., Liao H.I., Sun R.. Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:3805–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schumm K., Rocha S., Caamano J., Perkins N.D.. Regulation of p53 tumour suppressor target gene expression by the p52 NF-κB subunit. EMBO J. 2006; 25:4820–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M.. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005; 307:1098–1101. [DOI] [PubMed] [Google Scholar]
- 33. Wu T.T., Liao H.I., Tong L., Leang R.S., Smith G., Sun R.. Construction and characterization of an infectious murine gammaherpesivrus-68 bacterial artificial chromosome. J. Biomed. Biotechnol. 2011; 2011:926258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee C.G., Hurwitz J.. A new RNA helicase isolated from HeLa cells that catalytically translocates in the 3′ to 5′ direction. J. Biol. Chem. 1992; 267:4398–4407. [PubMed] [Google Scholar]
- 35. Zhang S., Grosse F.. Domain structure of human nuclear DNA helicase II (RNA helicase A). J. Biol. Chem. 1997; 272:11487–11494. [DOI] [PubMed] [Google Scholar]
- 36. Tang H., McDonald D., Middlesworth T., Hope T.J., Wong-Staal F.. The carboxyl terminus of RNA helicase A contains a bidirectional nuclear transport domain. Mol. Cell. Biol. 1999; 19:3540–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou K., Choe K.T., Zaidi Z., Wang Q., Mathews M.B., Lee C.G.. RNA helicase A interacts with dsDNA and topoisomerase IIalpha. Nucleic Acids Res. 2003; 31:2253–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krug L.T., Collins C.M., Gargano L.M., Speck S.H.. NF-κB p50 plays distinct roles in the establishment and control of murine gammaherpesvirus 68 latency. J. Virol. 2009; 83:4732–4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marino-Merlo F., Papaianni E., Medici M.A., Macchi B., Grelli S., Mosca C., Borner C., Mastino A.. HSV-1-induced activation of NF-κB protects U937 monocytic cells against both virus replication and apoptosis. Cell Death Dis. 2016; 7:e2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li H., Li X., Wei Y., Tan Y., Liu X., Wu X.. HSV-2 induces TLRs and NF-κB-dependent cytokines in cervical epithelial cells. Biochem. Biophys. Res. Commun. 2009; 379:686–690. [DOI] [PubMed] [Google Scholar]
- 41. Anderson S.F., Schlegel B.P., Nakajima T., Wolpin E.S., Parvin J.D.. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat. Genet. 1998; 19:254–256. [DOI] [PubMed] [Google Scholar]
- 42. Pellizzoni L., Charroux B., Rappsilber J., Mann M., Dreyfuss G.. A functional interaction between the survival motor neuron complex and RNA polymerase II. J. Cell Biol. 2001; 152:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Valineva T., Yang J., Silvennoinen O.. Characterization of RNA helicase A as component of STAT6-dependent enhanceosome. Nucleic Acids Res. 2006; 34:3938–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee C.G., Chang K.A., Kuroda M.I., Hurwitz J.. The NTPase/helicase activities of Drosophila maleless, an essential factor in dosage compensation. EMBO J. 1997; 16:2671–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guzmán E., Lis J.T.. Transcription factor TFIIH is required for promoter melting in vivo. Mol. Cell. Biol. 1999; 19:5652–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tartey S., Matsushita K., Vandenbon A., Ori D., Imamura T., Mino T., Standley D.M., Hoffmann J.A., Reichhart J.M., Akira S. et al. Akirin2 is critical for inducing inflammatory genes by bridging IkappaB-zeta and the SWI/SNF complex. EMBO J. 2014; 33:2332–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Christensen M.H., Paludan S.R.. Viral evasion of DNA-stimulated innate immune responses. Cell. Mol. Immunol. 2017; 14:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sun C., Schattgen S.A., Pisitkun P., Jorgensen J.P., Hilterbrand A.T., Wang L.J., West J.A., Hansen K., Horan K.A., Jakobsen M.R. et al. Evasion of innate cytosolic DNA sensing by a gammaherpesvirus facilitates establishment of latent infection. J. Immunol. 2015; 194:1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zheng C. Evasion of cytosolic DNA-Stimulated innate immune responses by herpes simplex virus 1. J. Virol. 2018; 92:e00099-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.