

Abstract
The dissemination of multi-resistant bacteria represents an enormous burden on modern healthcare. Plasmid-borne conjugative transfer is the most prevalent mechanism, requiring a type IV secretion system that enables bacteria to spread beneficial traits, such as resistance to last-line antibiotics, among different genera. Inc18 plasmids, like the Gram-positive broad host-range plasmid pIP501, are substantially involved in propagation of vancomycin resistance from Enterococci to methicillin-resistant strains of Staphylococcus aureus. Here, we identified the small cytosolic protein TraN as a repressor of the pIP501-encoded conjugative transfer system, since deletion of traN resulted in upregulation of transfer factors, leading to highly enhanced conjugative transfer. Furthermore, we report the complex structure of TraN with DNA and define the exact sequence of its binding motif. Targeting this protein–DNA interaction might represent a novel therapeutic approach against the spreading of antibiotic resistances.
INTRODUCTION
The perpetual increase of antibiotic resistant strains among bacterial pathogens represents one of the most imminent challenges to human healthcare in the 21st century, annually accounting for estimated 700 000 deaths worldwide (1). Horizontal gene transfer (HGT) drives the spread of antibiotic resistances even among unrelated bacteria and conjugative plasmids are major players in this dissemination process. During conjugative transfer, single-stranded (ss) plasmid DNA, coding for antibiotic resistance genes, is conveyed from a donor to a recipient cell via direct cell-to-cell contact. The transferred DNA also encodes the conjugation machinery that in turn enables the recipient to further spread the plasmid DNA. The passage of ss-plasmid DNA across bacterial borders is carried out by a type IV secretion (T4S) conjugative machinery, consisting of three major parts: the relaxosome, the coupling protein and the mating pair formation (MPF) complex, a membrane-spanning multi-protein channel (2).
Despite Gram-positive (G+) bacterial species belonging to the most health-threatening antibiotic-resistant bacteria worldwide (1,3), their respective T4S mechanisms are not completely understood to date (4). Plasmids that have been frequently isolated in bacterial strains associated with nosocomial infections often belong to the incompatibility group 18 (Inc18) represented by G+ plasmids with an exceptional broad host-range, such as pRE25, pSM19035, pAMß1 and pIP501 (5,6). Inc18 plasmids confer resistance to numerous classes of antibiotics such as the macrolide-lincosamide-streptogramin (MLS) group of antibiotics and pIP501 further codes for chloramphenicol resistance. These broad host-range plasmids are important key players in mediating transfer of resistance genes from Enterococci to several multi-drug resistant bacterial strains. The ability of Enterococcus faecalis and Enterococcus faecium strains to acquire and disseminate mobile genetic elements encoding beneficial traits, among them resistance to last-line antibiotics, further contributed to their manifestation as leading hospital pathogens (5,7). Broad host-range Inc18 family plasmids were observed to confer resistance to the last-line antibiotic drug vancomycin to strains of methicillin-resistant Staphylococcus aureus (MRSA) (8,9). Nonetheless, research is just beginning to understand determinants facilitating the transfer of Inc18 plasmids.
Due to its small size and simplicity, pIP501 has become a paradigm to study conjugation of Inc18-like broad host-range plasmids in G+ bacteria. This plasmid was originally isolated from Streptococcus agalactiae (10) and the region responsible for conjugative transfer was identified as a single operon of ∼14 kb, encoding 15 transfer (tra) genes, traA to traO (11,12). The tra-operon was observed to be co-transcribed and the mRNA levels remained unchanged during growth (13). The relaxase TraA is encoded by the first gene of the tra-operon and was demonstrated to negatively autoregulate tra-gene transcription upon binding to the Ptra promoter (13,14). Such sophisticated autoregulation systems have been discovered in many plasmids and represent efficient mechanisms to ensure the optimal balance between the lowest effort for the cell and the maximum transfer capacity. Nevertheless, producing these multi-component transfer systems consumes a lot of energy and thus, the expression and assembly of the multi-protein conjugation machineries need to be regulated (15). To that end, additional regulatory circuits involving accessory factors might be required to ensure the highest possible adaptation to an ever-changing environment.
In this work, we establish the small cytosolic protein TraN as a novel repressor of the pIP501 encoded type IV secretion system (T4SS). We present the 1.9 Å co-crystal structure of TraN bound to its cognate 34 bp binding site DNA, enabling a detailed analysis to identify the precise binding motif. The protein–DNA co-crystal structure confirmed the postulated binding mode of TraN as a variation of the classic helix-turn-helix (HTH) motif. We could demonstrate that binding of TraN to its cognate binding site leads to a significant reduction of promoter activity. Furthermore, we observed elevated mRNA and steady-state protein levels of all tested tra-operon components, including the proposed MPF complex member TraM, in an E. faecalis traN deletion strain. These transcriptional changes were accompanied by highly increased transfer frequencies, suggesting that TraN is not only an essential factor for the pIP501 encoded T4SS, but also a novel repressor of this conjugative system. All observed phenotypical effects of the knockout mutant could be reversed by overexpression of TraN in trans. Moreover, we identified a second specific TraN binding site on the pIP501 plasmid, regulating an active promoter upstream of the traN gene. Additional potential TraN binding sites are present on several Inc18-like and other related multi-resistance plasmids, suggesting TraN as a potential pharmacological target to combat the problem of rising antibiotic resistances among bacteria.
MATERIALS AND METHODS
Strains and cloning procedures
All bacterial strains and plasmids used in this work are listed in Supplementary Table S1. If not stated otherwise, all E. faecalis strains were grown in brain heart infusion (BHI, Sigma-Aldrich) medium and used Escherichia coli strains were grown in Luria-Bertani (LB, Roth) medium at 37°C and 180 rpm, supplemented with appropriate antibiotics. For all experiments with E. faecalis, cells were grown overnight at 37°C and the initial cultures were inoculated to an optical density of OD600 = 0.01. Cultures were harvested at mid-exponential phase (reaching an OD600 of ∼1.5) and harvested by centrifugation (1 min; 16 100 g; 4°C). Oligonucleotides used in this study are listed in Supplementary Table S2.
Co-crystallization of TraN with DNA
Expression and purification of TraN was performed as described in (16). TraN was set up for co-crystallization with double-stranded (ds) oligonucleotides. The ds-oligonucleotides were ordered as desalted ss-oligonucleotides and annealed by heating up equimolar amounts to 95°C for 10 min, followed by slow cooling down to room temperature (RT). The DNA represents the 34 bp binding site as described in (16) with blunt ends. In all setups, TraN was used with a protein stock concentration of 1.85 mg/ml and premixed with the DNA at a molar ratio of 1.1:1. The plates had a drop volume of 1 μl with a drop ratio of 50% (v/v). Crystals from TraN set up with DNA were harvested from the Index screen (Hampton research) condition 88 (0.2 M ammonium citrate tribasic pH 7.0; 20% (w/v) PEG-3350).
Data collection, processing and refinement
Data collection was performed at 100 K on the synchrotron beamline ID29 (ESRF, Grenoble, France) (17) with a Pilatus 6M detector and a wavelength of 0.97625. All details regarding data collection, phasing and refinement statistics are given in Table 1. A crystal-to-detector distance of 389 mm, an oscillation range of 0.1° and an exposure time of 0.0371 s per image were chosen. In total, 3000 images were collected. The data set was processed and scaled using XDS (18) and AIMLESS (19). The 3D-DART webserver (20) was utilized to generate a search model of the oligonucleotide for molecular replacement. Molecular replacement was first performed with the program PHASER (21) of the PHENIX software suite (22), using the recently published structure of TraN (4P0Z; (16)) as a template. A first model with a very limited DNA fragment was refined with REFMAC5 (23) and used as a template for a second run with PHASER. The resulting model was subjected to the PHENIX program AutoBuild (24). The model was completed manually in Coot (25), refined with phenix.refine (26) and validated by the MolProbity server (27), resulting in a score of 1.49 (96th percentile). The Ramachandran-plot statistics showed 98.23% favored and no Ramachandran-outliers. 2.83% rotamer outliers occur in the structure. The final coordinates and structure factor amplitudes of the TraN-DNA co-crystal structures have been deposited in the PDB: 6G1T. The PDBePISA server (28) was used to calculate the interaction surfaces and the solvation energy of TraN and the two oligonucleotides forming the ds-binding site DNA. The CURVES+ server was utilized to analyze the axis bend and the displacement of the DNA molecule (29).
Table 1.
Data collection and refinement statistics. Values in parentheses are for the highest-resolution shell
| TraN-DNA structure (6G1T) | |
|---|---|
| Data collection | |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 122.77, 44.17, 56.46 |
| α, β, γ (°) | 90, 90.86, 90 |
| Resolution (Å) | 41.87–1.93 (2.00–1.93) |
| R merge | 0.08 (0.675) |
| I / σI | 12.9 (2.4) |
| Completeness (%) | 99.6 (99.4) |
| Redundancy | 5.6 (5.6) |
| Refinement | |
| Resolution (Å) | 1.93 |
| No. reflections | 22 268 |
| R work/Rfree | 0.1809/0.2125 |
| No. atoms | |
| Protein | 976 |
| Ligand/ion | 15 |
| Water | 121 |
| B-factors | |
| Protein | 23.90 |
| Ligand/ion | 29.12 |
| Water | 31.63 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.195 |
Circular dichroism measurements
Circular dichroism (CD) measurements were conducted on a Jasco J715 spectropolarimeter (Jasco). Temperature scans ranging from 25°C to 95°C with a temperature slope of 1°C/min were performed in a 0.02 cm cuvette with a protein concentration of 0.3 mg/ml mixed with ds-DNA oligonucleotides (sequences are given in Supplementary Table S3) in a 1:1 molar ratio when required. The upscan was performed with a starting temperature of 25°C and an end temperature of 95°C and the downscan was performed from 95°C back to 25°C. Full spectra from 190 to 260 nm were recorded every 5°C and calculations were performed utilizing the SpectraManager Software (Version 1.08.01). The secondary structure composition of selected spectra from the temperature scans (25°C, upscan; 95°C; 25°C, downscan) were analyzed with the DicroWeb server (30,31) with the same parameters as used in (16).
Differential scanning fluorimetry assays
ss-DNA oligonucleotides with a concentration of 100 pmol/μl were treated as described in the crystallization section (sequences are given in Supplementary Table S3). 5 μl of TraN (1.9 mg/ml) was mixed with ds-DNA oligonucleotides in a 1:1 molar ratio. 5 μl of buffer (25 mM HEPES; 75 mM Na2SO4; pH 7.6) was added, the reaction volume was stocked up to 20 μl with distilled water and 5 μl of 50× SYPRO Orange (Sigma-Aldrich) was admixed. After incubation at RT for 5 min, the melting curve was measured from 25°C to 95°C in increments of 0.1°C and with 6 s incubation time at every step with a CFX96 Real-Time PCR Detection System (Bio-Rad).
Analysis of promoter activity
To assess the activity of the original pIP501 Ptra promoter in E. coli, the β-galactosidase gene from the promoter-probe plasmid pQF120 was set under the control of the Ptra promoter with (pQF120_oBS_Ptra) and without the original TraN binding site, o BS, (pQF120_Ptra) present, as described elsewhere (13). For activity determination of the newly identified promoter PtraNO, the promoter sequence with (pQF120_BS3_PtraNO) or without (pQF120_PtraNO) the alternative binding site BS 3 was cloned into pQF120. A second plasmid either used as empty vector control (pET28a_p15) or harboring TraN (pET28a_p15_traN) or the control protein T25 (used in Figure 4A), a fragment of the adenylate cyclase (pET28a_p15_T25), was co-expressed. E. coli JM109 carrying variants of the two plasmids pQF120 and pET28a_p15 was grown at 30°C. Two hours after induction with 1 mM IPTG, a β-galactosidase assay, adapted from (32), was performed. The absorption at 405 and 600 nm was measured with a TECAN GENios Pro plate reader and relative Miller units were calculated.
Figure 4.

TraN acts as a negative regulator of tra-operon components. (A) β-galactosidase assay to monitor Ptra promoter activity in E. coli. The β-galactosidase gene was set under the control of the P promoter with or without the original TraN binding site (o BS) and TraN or two controls (empty vector and T25, a fragment of the bacterial adenylate cyclase) were supplied in trans. Enzymatic activity was assessed in E. coli with a β-galactosidase assay and Miller units are given as fold of empty vector control. n = 24. (B) Analysis of tra-gene mRNA levels in E. faecalis JH2-2 wild type (pIP501) and mutant (pIP501ΔtraN) cells. Pyrroline-5-carboxylate reductase (proC) and glyceraldehyde-3-phosphate dehydrogenase (gapdh) were used as housekeeping genes. Relative gene expression was calculated applying the comparative CT method (ΔΔCT Method (37)) using proC as housekeeping gene and values are given as fold of the respective wild type gene expression. n = 4. (C, D) Immunoblot analysis of steady-state protein levels from E. faecalis JH2-2 (negative control), E. faecalis JH2-2 wild type (pIP501) and mutant (pIP501ΔtraN) cells. Representative immunoblots (C) and densitometric quantification (D) are shown. Blots were probed with antibodies directed against distinct Tra-proteins and EF-Tu and GroEL as loading controls. Values are given as fold of the respective wild type protein levels. n = 12. Mean (square) and median (center line) are depicted. For box plots, whiskers show minima and maxima within 2.2 interquartile range (IQR), for dot plots standard error of mean (s.e.m.) is given. n.s. not significant, *P< 0.05, **P< 0.01, ***P< 0.001.
Construction of a markerless traN knockout mutant and a TraN overexpression plasmid for complementation
A pIP501 traN in-frame deletion mutant in E. faecalis JH2–2 (pIP501ΔtraN) was generated using a method consisting of two homologous recombination steps as described in (33). 95% of traN was deleted, maintaining the two N-terminal and the four C-terminal codons intact to avoid polar effects on downstream genes in the pIP501 tra-operon. The knockout was confirmed by sequencing. To complement the knockout phenotype, the traN gene with an optimized ribosomal binding site was cloned into pEU327 (34) and the overexpression plasmid was transformed into the traN deletion strain.
Biparental mating assays
Filter mating experiments with E. faecalis OG1X as recipient were employed as described previously (35). Transfer rates of three independent experiments are given.
Assessment of growth and cellular viability
To analyze and compare growth of E. faecalis JH2–2 (negative control) with the E. faecalis JH2–2 wild type (pIP501) and mutant (pIP501ΔtraN) strain, cells were inoculated to OD600 = 0.01 and OD600 was assessed every 30 min for 7.5 h on a Beckman coulter DU 730 Life Science UV/VIS Spectrophotometer.
Loss of membrane integrity indicated via propidium iodide (PI) staining was utilized to analyze cellular viability at distinct time points. 100–200 μl (depending on optical density) of culture was harvested by centrifugation (1 min; 16 100 g; 4°C) and resuspended in 250 μl phosphate buffered saline (PBS; 25 mM potassium phosphate; 0.9% (w/v) NaCl; pH 7.2) containing 100 μg/l PI. After 10 min incubation in the dark at RT, cells were washed once with 250 μl PBS. 30 000 cells per sample were analyzed with flow cytometry (BD LSR Fortessa; BD FACSDivia software).
SDS-PAGE and immunoblotting
For determination of steady-state protein levels and comparison between negative control, wild type (pIP501), knockout (pIP501ΔtraN) and complementation (pIP501ΔtraN, pEU327-traN) strain, E. faecalis JH2-2 samples were resuspended in lysis buffer (1× PBS pH 7.2; 1 mM PMSF; 1 mM benzamidine) and sonicated for 30 s (Sonopuls, HD 2070, Bandelin; 60% amplitude, 0.5 s pulses). After addition of SDS sample buffer and incubation for 10 min at 95°C, samples were centrifuged (1 min; 16 100 g; RT). The supernatant was loaded onto a SDS-polyacrylamide-gel, separated by electrophoresis and blotted onto a PVDF membrane (ROTH) using wet electro-transfer. Blots were probed with primary polyclonal rabbit antibodies against Tra-proteins (dilution 1:10 000; Biogenes GmbH, Berlin) or monoclonal antibodies against EF-Tu (dilution 1:10 000; mouse, Hycult Biotech, HM6010) and GroEL (dilution 1:10 000; rabbit, Sigma-Aldrich, G6532) and the respective secondary antibodies (goat anti-mouse, Thermo Scientific, 31430; goat anti-rabbit, Promega, W4011; both horseradish-peroxidase conjugated; dilution 1:20 000). Immunodetection was performed using an ECL kit from GE Healthcare on a Chemidoc™ Touch Imaging System (Bio-Rad). For analysis of steady-state protein expression levels, the blots were stripped and re-probed with GroEL and EF-Tu used as loading controls. Densitometric quantification was performed utilizing Image Lab 5.2 software (Bio-Rad).
Quantitative real-time PCR
For expression analysis on transcriptional level of the wild type (pIP501), knockout (pIP501ΔtraN) and complementation (pIP501ΔtraN, pEU327-traN) strain, samples of OD600 = 1 were harvested (1 min; 16 100 g; 4°C) and total RNA was isolated using TRIzol reagent (Thermo Scientific). Briefly, the cell pellets were resuspended in 500 μl TRIzol plus approximately 200 μl glass beads (500 μm diameter) and mechanical lysis was performed in three cycles of 1 min with 4000 g (PowerLyzer® 24 Bench Top Bead-Based Homogenizer, Qiagen). The following steps were employed as described in the manufacturer's manual. Integrity of the isolated RNA was validated by visualizing rRNAs using an agarose gel-electrophoresis protocol adapted from (36). Contaminating DNA was removed with the DNA-free Kit (Ambion) and the RNA concentration was determined spectrometrically with a NanoDrop (ND 1000 Spectrophotometer). 2 μg of total RNA were reverse transcribed using the M-MLV Reverse Transcriptase RNase H- (Solis Biodyne) and random hexamer primers (Thermo Scientific). The actual q-RT-PCR was performed with the 5× HOT FIREPol EvaGreen qPCR Supermix (Solis Biodyne) as described in the manual. First, standard curve experiments were performed to ensure primer efficiency lying between 90% and 110%. The actual runs were accomplished in quadruplicate. Pyrroline-5-carboxylate reductase (proC) and glyceraldehyde-3-phosphate dehydrogenase (gapdh) were used as housekeeping genes and gene expression of proC was utilized to calculate the relative gene expression levels with the comparative CT method (ΔΔCT method (37)).
Immunolabeling of TraM in E. faecalis
For visualization of TraM in E. faecalis JH2-2 wild type (pIP501) and the knockout (pIP501ΔtraN) strain with E. faecalis JH2-2 as negative control, we applied a recently established immunostaining method (38), using a TraM specific primary antibody (dilution 1:50; Biogenes GmbH, Berlin) and an Alexa-594 conjugated secondary antibody (dilution 1:100; goat anti-rabbit, Thermo Scientific, R37117), combined with MDY-64 (Thermo Scientific) counterstaining of the cytosol, enabling us to discriminate between bacterial cells and background. To only visualize correctly localized TraM molecules at the cell membrane, the fixing and the digestion time were reduced (fixation for 30 min followed by enzymatic digestion of the cell wall for 10 min), no permeabilization step was included and no detergent was added to the buffers.
Analysis of TraM fluorescence signal with confocal microscopy and flow cytometry
In all experiments, immunolabeled cells were immobilized on agar slides and visualized with a Leica SP5 confocal microscope, with spectral detection and a HCX PL APO 63× NA 1.4 oil immersion objective. MDY-64 was excited at 488 nm and emission was detected between 500 and 530 nm. For Alexa-594 detection, excitation was performed at 561 nm and emission was detected between 580 and 630 nm. Pictures were subsequently processed and analyzed using the open-source software FIJI (39). In all experiments Z-stacks using 63 × 63 × 125 (x/y/z) nm sampling were acquired and subsequently Z-projections with the maximum-intensity projection method were performed for all channels. To measure the fraction area of imaged TraM structures per cell, MDY-64 stained cells in intensity projections were segmented using the Otsu-method, while excluding not fully acquired objects at the image borders. Pictures were subsequently transformed to binary images and the watershed method was applied to separate cells in close proximity. Individual segmented cells were afterwards added to the region of interest (ROI) manager with the analyze particles function. These ROIs were used to measure the fraction area of structures in respective TraM-fluorescence channels. Six independent measurements (n = 6) were conducted and 527 (negative), 317 (pIP501) and 306 (pIP501ΔtraN) cells in total were analyzed. To quantify the fluorescence signal intensity of TraM, the measurements were conducted with a single-photon detector. For computation of the intensity of fluorescently labeled TraM structures, pictures were segmented as described above. The mean grey value of each TraM particle was measured using the analyze particles function and the fluorescence intensity per pixel is presented as fold of wild type (pIP501). Five independent measurements (n = 5) with 1576 (pIP501) and 1250 (pIP501ΔtraN) cells in total were analyzed. In addition to greyscale images, a spectrum look-up table was applied to easier appreciate intensity differences. Brightness and contrast were adjusted in representative pictures. All pictures within an experiment were processed equally.
For flow cytometry, samples were immunostained as described above and each strain was analyzed in quadruplicate with a flow cytometer (BD LSR Fortessa) and the mean fluorescent intensity of 30 000 cells per sample was determined with BD FACSDivia software. To avoid artefacts from background signals (e.g. autofluorescence), the values for fluorescence intensity of the negative strain were subtracted from wild type and mutant strain prior to calculation.
Search for similar binding sites and promoter motifs
To analyze whether related plasmids or bacterial strains also harbor a TraN binding motif, we initially conducted a BLAST search (40) for potential traN-gene homologs and further tested if those also carry homologs coding for the relaxase TraA and the motor proteins TraE and TraJ. To search for similar binding sites, the respective sequences were analyzed with the SnapGene viewer software (BSL Biotech LLC, version 4.1.4) and the search function, where individual bases can be set as ambiguous, was applied. Three different search terms were used: (i) Original binding site, o BS (A6T7G8T9C10A11G12G13T14T15A16A17A18C19A20T21A22 T23); (ii) Binding site involving class (I) interactions (N6T7G8N9C10N11N12N13N14N15A16N17A18C19A20T21 N22N23); (iii) Binding site with nucleotides required for TraN specificity (N6T7G8T9C10A/T11N12N13T14N15A16A17A18C19A20T21N22T23). The identified potential binding sites were further characterized by investigating adjacent stretches for the presence of a promoter. Where annotated, the sequence of the origin of transfer (oriT) is given. Otherwise, in silico prediction of the oriT according to the consensus sequence identified in (41) was made. The protein identity of Tra-protein homologs was calculated with Clustal Omega (42).
Statistics
Results are visualized either as line graphs showing mean ± standard error of mean (s.e.m.), dot plots (if n < 8) where mean (square), median (center line) and s.e.m are depicted, or as box plots (if n ≥ 8) with mean (square) and median (center line) as well as whiskers presenting minima and maxima within 2.2 interquartile range (IQR). Student's t-test (applied for Figures 5A, C, 7C, Supplementary Figure S5B) and analysis of variance (ANOVA) with a Bonferroni post hoc test (applied for Figure 4A) were used to compare between two or multiple groups, respectively, and displayed significances are two-sided (calculated with Origin Pro 2016, OriginLab). Normal distribution of data was examined with a Shapiro-Wilk's test and homogeneity of variances was confirmed with a Levene's test (both analyzed with Origin Pro 2016). In case of heterogeneous variances, a Welch's t-test (applied for Figure 4B, D, calculated with Origin Pro 2016) or a Welch's ANOVA with a Games-Howell post hoc test (applied for Figures 6A, B, D, 7A, B, Supplementary Figure S6B, calculated with IBM SPSS Statistics, Version 24) was used. Outliers were identified using the 2.2-fold IQR labeling rule and were only detected in Figure 3B (indicated with magenta diamonds). Outlier containing data was analyzed using a nonparametric Kruskal–Wallis test, calculated with IBM SPSS Statistics, Version 24. For the assessment of viability of different strains (negative, pIP501 and pIP501ΔtraN; = between-subject factor) over time (within-subject factor), a two-way ANOVA mixed design with a Bonferroni post hoc test was applied (Supplementary Figure S4B; conducted with Origin Pro 2016). Significances are indicated with asterisks: ***P < 0.001, **P < 0.01, *P < 0.05, n.s. not significant. All figures were processed with Origin Pro 2016 and Adobe Illustrator CS6 (Adobe).
Figure 5.

A significant quantity of overproduced TraM localizes to the cell periphery in the E. faecalis traN deletion mutant. (A) Flow cytometric analysis of E. faecalis JH2-2 wild type (pIP501) and mutant (pIP501ΔtraN) cells to analyze TraM signal intensities with E. faecalis JH2-2 as negative control. Fixed cells were probed with primary TraM-antibodies and corresponding secondary antibodies conjugated with Alexa-594. 30,000 cells per sample were analyzed in quadruplicate with flow cytometry. The signal of the negative strain was subtracted from the TraM-signal of wild type and knockout strain as a background control. The mean fluorescence intensity of the TraM signal is depicted as fold of wild type. n = 4. For quantification of TraM fluorescence signal intensity using confocal microscopy techniques, see Supplementary Figure S5A, B. (B, C) Immunolabeling of E. faecalis JH2-2 wild type (pIP501) and the mutant strain E. faecalis JH2-2 (pIP501ΔtraN) with E. faecalis JH2-2 as negative control to analyze TraM signal area. Cells were prepared as described in (A). Z-projection of three-dimensional representative micrographs (B) and quantification of the area of TraM spots (C) are displayed. Values are given as fold of the wild type. No fraction area of the negative strain was detectable. Six independent measurements (n = 6) with 527 (negative), 317 (pIP501) and 306 (pIP501ΔtraN) cells in total were analyzed. Scale bar represents 2 μm. Mean (square) and median (center line) are depicted. For dot plots standard error of mean (s.e.m.) is given. ***P< 0.001.
Figure 7.

An alternative TraN binding site on pIP501 regulates a promoter upstream of the traN gene. (A) Differential scanning fluorimetry (DSF) assay of TraN without DNA, with random DNA and the original binding site (o BS) as well as with alternative binding sites identified on the pIP501 plasmid (m BS, BS 2, BS 3, BS 4). For ‘no DNA’ n = 9, for all others n = 10. For a detailed sequence alignment, please see Supplementary Figure S6A. (B) β-galactosidase assay to monitor promoter activity in E. coli. The β-galactosidase gene was set under the control of either the Ptra promoter with / without the original TraN binding site (o BS) or the PtraNO promoter with / without the alternative specific binding site (BS 3). TraN or the empty vector control were supplied in trans. Enzymatic activity was assessed in E. coli with a β-galactosidase assay and Miller units are given as fold of the respective empty vector control. n = 6. (C) Ratio of traO mRNA levels to selected tra-gene mRNA levels (traJ, traK and traM). Wild type (pIP501) and mutant cells (pIP501ΔtraN) were compared to assess the activity of the potential PtraNO promoter and its regulation by TraN by qPCR. mRNA levels were normalized to pyrroline-5-carboxylate reductase (proC) as housekeeping gene and the ratio of gene expression is given. n = 3. Mean (square) and median (center line) are depicted. For box plots, whiskers show minima and maxima within 2.2 interquartile range (IQR), for dot plots standard error of mean (s.e.m.) is given. n.s. not significant, *P< 0.05, **P< 0.01, ***P< 0.001.
Figure 6.

traN knockout mutant shows increased transfer rates in E. faecalis and phenotypic effects of traN deletion can be compensated by complementation. (A) Results of biparental mating assays monitoring the transfer rate of the E. faecalis JH2-2 wild type (pIP501), the mutant strain E. faecalis JH2-2 (pIP501ΔtraN) and the complementation strain, where traN is supplied in trans (pIP501ΔtraN, pEU327-traN). n = 3. (B) Analysis of tra-gene mRNA levels of cells as described in (A). Relative gene expression was calculated applying the comparative CT method (ΔΔCT Method (37)) using pyrroline-5-carboxylate reductase (proC) as housekeeping gene and values are given as fold of the respective wild type gene expression. n = 3. (C, D) Immunoblot analysis of steady-state protein levels from E. faecalis JH2-2 wild type (pIP501), mutant (pIP501ΔtraN) and complementation (pIP501ΔtraN, pEU327-traN) strain with E. faecalis JH2-2 as a negative control. Representative immunoblots (C) and densitometric quantification (D) are shown. Blots were probed with antibodies directed against distinct Tra-proteins and GroEL as loading control. Values are given as fold of the respective wild type protein levels. n = 12. Mean (square) and median (center line) are depicted. For dot plots standard error of mean (s.e.m.) is given. n.s. not significant, *P< 0.05, **P< 0.01.
Figure 3.

TraN is stabilized by binding to its specific DNA site. (A) Circular dichroism (CD) temperature scans of TraN without DNA, with the original binding site (o BS) DNA as well as with random DNA. Temperature scans were performed with a temperature slope of 1°C per min. The upscan started from 25°C to an end temperature of 95°C and the downscan began at 95°C and cooled down back to 25°C. Full spectra between 190 nm and 260 nm at 25°C before heating to 95°C (black line; 25°C, upscan), at the maximum temperature of 95°C (blue line) and after cooling down to 25°C (gray line; 25°C, downscan) are depicted. θmre = Molar ellipticity. For secondary structure analysis using the DicroWeb server (30,31), please see Supplementary Table S5. (B) Differential scanning fluorimetry (DSF) assay of TraN without DNA, with random DNA and the original binding site DNA (o BS) as well as with reverse charge (RC) or different spacing (DS) mutations in the upstream (left), downstream (right) and both interaction sites with the melting temperature depicted. Outliers are denoted as magenta diamonds. For ‘no DNA’ n = 16, for all others n = 10. Mean (square) and median (center line) are depicted. Whiskers within box plots show minima and maxima within 2.2 interquartile range (IQR). ***P< 0.001.
RESULTS
The TraN-DNA co-crystal structure gives detailed insights into the binding mode
Recently, we solved the atomic structure of TraN and experimentally identified its approximate binding site location on the pIP501 plasmid (16). To get detailed insights into TraN’s mode of action on the molecular level, oligonucleotides comprising the proposed binding site were set up with TraN in protein–DNA co-crystallization trials. We obtained crystals of a 1:1 complex of TraN with a 34-mer double-stranded (ds) oligonucleotide, which contained one complex per asymmetric unit and diffracted to 1.93 Å (Table 1).
The structure is well ordered except for the N-terminal methionine and six residues at the C-terminal end of TraN, which were not observed in the final electron density map. In addition, five nucleotides at the 3′-end of the forward strand (D-strand) and two nucleotides at the 5′-end of the reverse strand (E-strand) exhibited low electron density due to apparent flexibility. TraN shows an internal dimer fold with an α–β composition comprising of 9 α-helices (h1–h9) and 6 β-strands (s1–s6) (Figure 1A, for details on the secondary structure elements, see Figure 2E). Helices 3 and 7 represent the recognition helices of two helix-turn-helix (HTH) motifs, constituting the N- and the C-terminal domains of TraN. The β-strands in the center of the protein form a β-barrel-like motif at the internal dimer axis. The co-crystal structure presented in this study confirms the postulated binding mode of TraN: A winged-helix (WH) protein–DNA interaction, a common variation of the classic HTH-motif (43–46) (Figure 1A). The recognition helices h3 and h7 of the TraN internal dimer HTH-domains indeed reach into two adjacent major grooves of the binding site of the ds-oligonucleotide (Figure 1A). Both wings of the WH-motif consisting of the β-strands s2 and s3 of the N-terminal domain and s5 and s6 of the C-terminal domain, respectively, protrude into the central minor groove of the binding site and are involved in numerous direct or indirect DNA interactions. This arrangement is in significant contrast to the DNA binding mode of homo-dimeric WH transcription factors, such as the LysR-type transcriptional regulators, where the wing of each WH-motif contacts the distal minor groove (47).
Figure 1.

TraN shows highly specific interaction with its cognate binding site. (A) Cartoon representation of TraN bound to the double-stranded (ds) oligonucleotide comprising the specific binding site (forward strand/D-strand; reverse strand/E-strand). Front view and rotated by 180° around the vertical axis are depicted. Amino acids involved in specific interaction with the DNA are shown as cyan sticks. (B, C) Calculation of the axis bend by the CURVES+ server (29). (B) The trajectory of the DNA helix is drawn as a black line over the ds-oligonucleotide and the bend of the DNA is given. (C) The axis bend of the forward/D-strand from C1 to T29 is depicted with the TraN binding region shaded in grey. (D) Surface representation of the electrostatic potential of TraN with the binding site DNA slightly elevated from the protein. Only nucleotides involved in the direct interaction are colored. Front view and rotated by 180° around the vertical axis are depicted.
Figure 2.

Interactions of TraN with the DNA lead to highly specific binding. (A–C) Class (I) interactions between amino acid side chains and DNA bases are depicted. Interactions of the N-terminal half of TraN (A), the C-terminal half of TraN (B) and the second loop protruding into the minor groove of the DNA (C) are shown with important amino acids and bases highlighted in color. Coordinated water molecules are depicted as spheres. The forward / D-strand and/or the reverse/E-strand are labeled. (D) Sequence of the double-stranded (ds) oligonucleotides used in the crystallization trials, the nucleotides visible in the X-ray structure are marked with square brackets. A dashed line indicates the pseudo-2-fold axis of the imperfect inverted binding site repeats. All nucleotides interacting with TraN are shaded in grey. Direct base to side chain interactions (class (I)) are highlighted in magenta and bases which are part of class (II) interactions (between either amino acid side chains or peptide backbone with the DNA backbone) are marked with an asterisk. Class (III) water-mediated interactions are indicated with a blue dot and van-der-Waals interactions with a circle in magenta. An alternative labeling according to the base pair position of L39769.1 is given. (E) Sequence and secondary structure-based alignment of the two TraN domains. Secondary-structure elements are labeled, identical amino acids are shaded in grey. Class (I) interactions are highlighted in magenta, class (II) interactions with an asterisk, class (III) interactions are marked with a blue dot and van-der-Waals interactions with a circle in magenta.
When comparing the complex structure of TraN with the previously published apo-form, no significant differences regarding the fold of the protein were observed (RMSD of 0.6 Å). However, the complex formation leads to a significant deformation of the B-form DNA, introducing an overall bend of 32.4° (Figure 1B) that is localized at the contact positions with the two recognition helices h3 and h7 (Figure 1C). This causes a displacement of the base pairs in these areas away from the helical axis (Supplementary Figure S1A, B) and consequently a significant widening of the major groove at the binding sites, resulting in a compression of the adjacent minor grooves. DNA bending assays using an electrophoretic mobility shift assay (EMSA) approach did not reveal significant bending of the DNA under the conditions tested (data not shown), which might be caused by a compensation of the local bends in the neighboring regions.
Due to the positive charge of the TraN surface facing the ds-DNA, a close interplay with the phosphate groups of the DNA backbone can be observed (Figure 1D). An exception is the surface area of the loop connecting the α-helices h6 and h7 of the C-terminal HTH-motif (Figure 1D), where two negatively charged residues (Asp-77 and Glu-78) are replacing a positively charged and a neutral residue (Arg-23 and Asn-24) when compared to the N-terminal domain (Figure 1D and Supplementary Figure S1C; for details on the alignment, see Figure 2E). The buried interaction surface of TraN with the ds-DNA amounts to 883 and 850 Å2 for forward (D-strand) and reverse (E-strand) strand, respectively. Due to overlapping areas, the total buried interaction surface is slightly smaller with 1660 Å2. These values are similar to the protein–DNA interaction surface of the MerR family of transcriptional regulators (48). MerR-like proteins share a similar WH-fold with TraN and bind to their designated binding sites as dimers, reminiscent of the internal dimer composition of TraN (e.g. MerR-like protein MtaN from Bacillus subtilis, PDB: 1R8D; 1679 Å2 of protein–DNA interaction surface). Due to this structural similarity, a potential role of TraN as a regulator of the pIP501 encoded T4SS was proposed (16).
The specificity of TraN DNA-binding depends on a distinct motif
The polar interactions fall into three classes: (I) interactions between distinct amino acid side chains and the bases of the DNA, (II) interactions concerning either the amino acid side chains (IIa) or the protein backbone (IIb) with the backbone of the DNA and (III) water-mediated interactions (Supplementary Table S4). Sequence specific interactions (class (I)) are mediated by Arg-23, Asn-24 and Gln-28 of the N-terminal domain forming hydrogen bonds (H-bonds) to the bases in the right half site (Figure 2A) and by Glu-78, His-82 and Arg-86 of the C-terminal domain forming H-bonds to bases in the left half site (Figure 2B). In addition to the major groove contacts, the β-hairpins reach into the central minor groove where Lys-101 forms a direct base contact (Figure 2C). These interactions are important for the recognition and the strong specific binding of TraN to its binding site (Figure 2D, E). Interestingly, van-der-Waals interactions of the C5M-methyl groups of thymine bases with hydrophobic patches of TraN are also contributing to the sequence specificity (Figure 2D, E and Supplementary Figure S2A–C). This concerns T15 of the E-chain forming contacts with the N-terminal domain (especially with Asn-22, Arg-23 and Asn-24; Supplementary Figure S2A) and T7 (interacting with Phe-87), T9 (interacting with Asn-76) of the D-chain (Supplementary Figure S2B) and T24 of the E-chain (forming contacts with Glu-78, Ala-81 and His-82; Supplementary Figure S2C), respectively, interacting with the C-terminal domain. Here, a clear difference between the binding modes of the N- and the C-terminal domains is evident. The asymmetry of the two half sites results in only one hydrophobic patch being in contact with a thymine base in the N-terminal domain (Supplementary Figure S2A), whereas three such patches exist in the C-terminal domain (Supplementary Figure S2B, C), indicating a much better complementary fit to the left half site (Figure 2D, E). The importance of such hydrophobic patches for the sequence specific DNA recognition has been noted for the structurally related LysR-type transcription regulators (47).
All interactions are listed in Supplementary Table S4 and an overview of the amino acids and bases involved in the interplay of TraN with its binding site are depicted in Figure 2D, E. Investigating the binding interfaces of the two domains separately using the PDBePISA server (28) yielded a significant difference in the free energy of assembly dissociation with 7.2 kcal/mol for the N-terminal domain and 10.7 kcal/mol for the C-terminal domain. Together with the observed differences in the van der Waals contacts, these results indicate a stronger interaction of the C-terminal domain of TraN with the DNA compared to the N-terminal domain.
The thermal stability of TraN in the presence or absence of DNA was assessed with circular dichroism (CD) measurements. Without DNA, the full TraN spectrum showed signs of significant unfolding at 95°C, but the protein refolded almost completely when cooled down to 25°C. In comparison the protein is stabilized by complexation with DNA, leading to only a slight conformational change upon heating to 95°C. A similar effect was observed for both the specific binding site and the random sequence, pointing towards a general stabilization of the protein by DNA binding (Figure 3A). The secondary structure composition of TraN and its change upon temperature variation was assessed by the DicroWeb server (30,31) and can be found in Supplementary Table S5.
To examine the significance of the directly interacting bases, we conducted differential scanning fluorimetry (DSF) assays in the presence of mutated oligonucleotides (details on the mutated bases can be found in Supplementary Table S3 and Supplementary Figure S3A, B). The complex with the original binding site (o BS) showed an increase in the melting temperature (TM) of 16°C, whereas with random DNA it was 10°C, respectively (Figure 3B). Mutations in the individual halves diminished the thermal stability of TraN with a drop in the TM of up to 5°C, being more prominent when the left half binding site (contacted by the C-terminal domain of TraN) was affected (Figure 3B). The melting temperature dropped even further when both half sites were mutated simultaneously (Figure 3B). Together, these experiments confirm the identified bases as crucial for the specificity of TraN binding and thus represent the actual TraN binding motif.
TraN acts as negative regulator of tra-operon components
Due to TraN’s structural similarity with transcriptional regulators from the MerR family, a potential role of TraN as a regulator of the pIP501 encoded T4SS was proposed (16). To test this hypothesis, the promoter activity was evaluated in E. coli using a reporter plasmid expressing β-galactosidase under the control of the Ptra promoter, with or without the o BS of TraN. A second plasmid encoding either traN or selected controls (empty vector control; T25, adenylate cyclase fragment) was supplied in trans. Whereas neither the empty vector, nor T25 expression changed β-galactosidase activity when comparing plasmids with or without the binding site, a significant reduction of β-galactosidase activity was observed upon TraN in trans expression when the test plasmid harbored o BS (Figure 4A). These results clearly indicate that TraN binding to its cognate binding site leads to repression of the Ptra promoter.
To confirm that TraN also represses the Ptra promoter in the native T4SS environment in E. faecalis, a knockout mutant of traN was constructed. First, growth and viability of the constructed knockout (pIP501ΔtraN) was assessed. When measuring optical density, the knockout mutant E. faecalis JH2-2 pIP501ΔtraN displayed delayed growth during exponential phase compared to wild type (pIP501) cells and the negative control E. faecalis JH2.2, while still reaching a comparable optical density in stationary phase (Supplementary Figure S4A). Flow cytometric analysis of membrane integrity using propidium iodide (PI) showed a significantly increased portion of dead cells in the traN knockout mutant when compared to wild type and negative control in both exponential and stationary phase (Supplementary Figure S4B). Thus, the lack of traN seems to pose an additional burden to the cells, resulting in slightly impeded growth and decreased viability.
To further characterize the deletion mutant, gene expression levels, normalized to pyrroline-5-carboxylate reductase (proC) as housekeeping gene using the ΔΔCT Method (37), were compared between the E. faecalis JH2-2 knockout mutant and the wild type strain. mRNA levels of all tested tra-genes were significantly upregulated upon deletion of traN (Figure 4B). When assessing steady-state protein levels using quantitative immunoblotting, all tested Tra-proteins were elevated in the E. faecalis traN deletion mutant compared to wild type cells harboring pIP501 (Figure 4C, D). Taken together, these results suggest a negative regulation of tra-component expression by TraN.
A significant proportion of upregulated TraM localizes correctly in the traN deletion mutant
Due to the high homology of TraM to VirB8-like proteins (49) and the fact that TraM was shown to interact with the peptidoglycan-digesting protein TraG—a central key player in the assembly of the MPF complex—(38,50), the expression levels of this essential Tra-protein were further assessed with complementary approaches. Analysis of steady-state protein levels gives insights into the whole expression load at a certain time point but does not discriminate between different protein subpopulations ranging from correctly localized to mislocalized, unfolded and maybe even aggregated protein. To that end, we performed immunostaining of E. faecalis JH2-2, either harboring wild type pIP501 or the deletion variant pIP501ΔtraN with the plasmid-free strain as negative control, using primary anti-TraM specific antibodies and corresponding Alexa-594-labeled secondary antibodies. With this approach, only correctly localized TraM molecules at the cell's periphery should show a fluorescence signal since mild conditions without permeabilization steps were applied. 30 000 cells per sample were analyzed with flow cytometry, confirming the upregulation of TraM in the E. faecalis traN deletion mutant (Figure 5A). To verify this result with a visual approach, the samples were further analyzed with confocal microscopy. Using a single-photon detector, the mean fluorescence signal intensity of >1200 cells per strain was quantified, confirming the flow cytometric data (Supplementary Figure S5A, B). Interestingly, while steady-state protein levels revealed an upregulation of ∼5-fold in the E. faecalis knockout strain (pIP501ΔtraN), the elevation of the fluorescence signal was ∼1.5-fold. Due to the proposed essential role of TraM in MPF complex assembly (38,49,50), we next investigated potential changes in the subcellular distribution of TraM upon traN deletion. The wild type strain harboring pIP501 and the traN knockout mutant revealed a similar localization pattern of TraM in distinct foci at the cell membrane, while the number of foci appeared to be elevated in mutant cells (Figure 5B). Aiming to quantify the number of distinct TraM-positive dots, we observed a variable size of these structures, potentially due to their close spatial proximity and the resolution limit of confocal microscopy. To provide quantitative data, we measured the area captured by the TraM signal in individual cells instead. When analyzing at least 300 cells of each strain, we observed an average 1.7-fold increase in the TraM signal area in the E. faecalis knockout strain (pIP501ΔtraN) compared to cells harboring wild type pIP501 (Figure 5C). Combining flow cytometry with confocal imaging techniques further confirmed the observed upregulation of TraM in the E. faecalis knockout mutant pIP501ΔtraN. While indicating that not the whole protein load detected with immunoblotting seems to be correctly localized at the cell membrane, still a significant increase of the TraM protein at the cell periphery is detected in the traN knockout mutant.
Deletion of traN increases the frequency of conjugative transfer
Finally, we investigated whether the elevated expression level of tra-components results in an upregulation of conjugative transfer. Utilizing biparental mating assays, transfer rates of the traN knockout strain were determined and compared to that of the pIP501 wild type. The transfer rate of the E. faecalis mutant strain (pIP501ΔtraN) increased by two orders of magnitude (Figure 6A). To complement the observed phenotype of the traN deletion strain, a plasmid constitutively overexpressing TraN was supplied in trans. Interestingly, the elevated transfer frequency observed in the traN deletion mutant was not only reverted but overcompensated such that the conjugative transfer was more than two orders of magnitude below wild type levels (Figure 6A). Conclusively, traN complementation reverted effects of traN deletion on gene expression (Figure 6B) and steady-state protein levels (Figure 6C, D) and also resulted in a significant overcompensation of all analyzed protein levels and most assessed mRNA quantities in respect to wild type cells harboring pIP501.
Taken together, our results identify TraN as a negative regulator of conjugative transfer, by repressing transcription of pIP501 T4S components, resulting in decreased levels of Tra-proteins, ultimately leading to significantly reduced transfer rates.
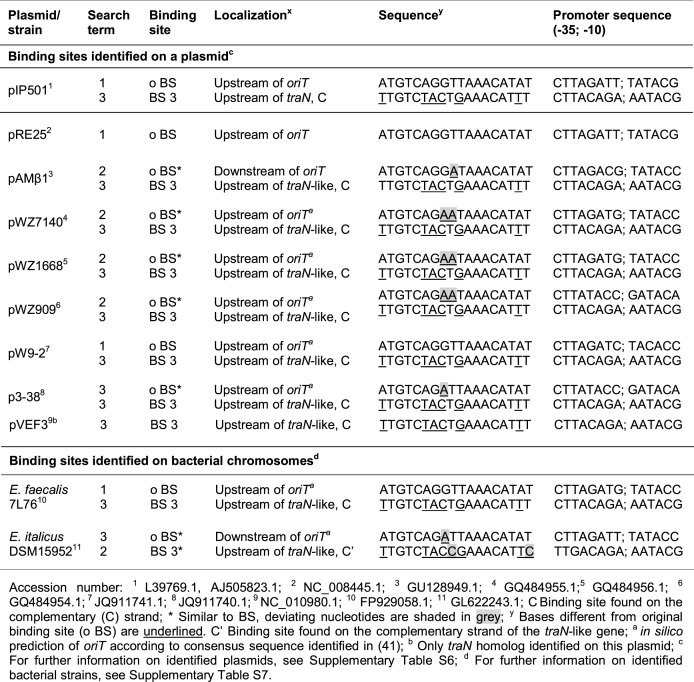
A second specific TraN binding site identified on the pIP501 plasmid regulates a promoter directly upstream of the traN gene
In a sequence search for equivalent TraN binding motifs on the pIP501 plasmid, we fixed the identified bases of class (I) interactions in the query sequence. Three additional potential binding sites within the pIP501 plasmid were found and termed BS 2, BS 3 and BS 4. By setting position 8 of the query sequence as variable, we observed another mutated potential binding site (m BS; Supplementary Table S3). Conducting further DSF assays, we observed that the thermal stability of TraN was significantly decreased when oligonucleotides comprising the BS 2, BS 4 and m BS sequences were provided, compared to the o BS (Figure 7A). Interestingly, providing BS 3 oligonucleotides led to a thermal stability equal to the original site (Figure 7A). Furthermore, a potential promoter, termed PtraNO, was found in close proximity to the BS 3 site (Table 2), directly upstream of the traN gene. Taken together, these results suggest that the identified BS 3 might represent a potential second binding site for TraN on the pIP501 plasmid. An alignment of all sequences (Supplementary Figure S6A) revealed that the observed difference in thermal stability between BS 3 and the other alternative binding sites might be due to the variation of hydrophobic interactions via the methyl groups of thymine bases and by unspecific interactions (class (II) and (III)). Thus, these variations might be caused by the thymine bases at positions 9 and 14 and adenine at position 17. Furthermore, based on the observed van der Waals interactions, we propose that an adenine or a thymine at position 11 is required for the binding specificity (Supplementary Figure S6A).
Table 2.
TraN binding motifs on Inc18-like and related multi-resistance plasmids and on bacterial chromosomes. Three different search terms were used: 1: Original binding site (A6T7G8T9C10A11G12G13T14T15A16A17A18C19A20T21A22T23); 2: Binding site involving class (I) interactions (N6T7G8N9C10N11N12N13N14N15A16N17A18C19A20T21N22N23); 3: Binding site with nucleotides required for TraN specificity (N6T7G8T9C10A/T11N12N13T14N15A16A17A18C19A20T21N22T23)
|
To assess the activity of the potential PtraNO promoter in close proximity to the alternative TraN binding site BS 3 just upstream of the traN gene, we generated a reporter plasmid carrying the β-galactosidase gene under the control of the potential PtraNO promoter with and without the BS 3 sequence and performed a β-galactosidase assay in E. coli (Figure 7B). In trans expression of TraN revealed a significant and comparable downregulation of β-galactosidase activity when either o BS or BS 3 were available (Figure 7B), pointing toward a role of TraN in regulating the identified promoter PtraNO upstream of the traN coding region, therefore indicating a potential autoregulation. To analyze the activity of the identified PtraNO promoter in its native environment in E. faecalis, the ratios of relative mRNA levels of traO to other tra-genes were assessed. As traO levels were overrepresented compared to the other tested tra-genes in the wild type harboring pIP501, as indicated by a ratio higher than one (Figure 7C), the PtraNO promoter seems to be active in its native environment under the conditions tested. Interestingly, all calculated ratios were increased upon deletion of traN, suggesting that our approach led to the identification of a new promoter in E. faecalis and a role of TraN in its regulation.
TraN binding motifs are common in Inc18-like and related multi-resistance plasmids
To identify potential TraN binding sites in related T4SSs, a search for the o BS site as well as for sequences comprising either only class (I) interactions or all nucleotides apparently responsible for specific binding was performed (Table 2). First, the transfer region of the Inc18 family plasmid pRE25, being almost identical to that of pIP501 (51), was analyzed. Interestingly, while the o BS was present upstream of the origin of transfer (oriT), pRE25 lacked BS 3 upstream of the traN-like gene (Table 2). Even though we were able to identify a set of potential binding sites (PS 1–5) upstream of the traN-homolog (Supplementary Table S3), none of these sites revealed increased thermal stability in DSF assays (Supplementary Figure S6B). The alignment of the potential binding sites (PS 1–5) with the original site can be found in Supplementary Figure S6C.
Additionally, we analyzed other Inc18-like and related multi-resistance plasmids (Supplementary Table S6) as well as bacterial strains, where chromosomally encoded homologs of traN have been found (Supplementary Table S7). Intriguingly, in most cases an o BS-like site near the oriT on the leading strand and a BS 3-like site upstream of the traN-like gene on the complementary strand - just as on the pIP501 plasmid - was present (Table 2 and Supplementary Tables S6, S7). Potential promoter sequences were found in close proximity to each binding site (Table 2). On most of these plasmids and bacterial chromosomes, homologs of the relaxase TraA and the motor proteins TraE and TraJ were present (Supplementary Tables S6, S7).
DISCUSSION
The efficient dissemination of conjugative plasmids encoding antibiotic resistances and virulence factors via T4SSs within bacterial communities and across species boundaries represents a major threat to modern healthcare. As the production of the conjugative multi-component machinery responsible for these processes comes with significant energetic costs for bacteria, sophisticated control systems must be in place to minimize these expenses by accurate regulation of tra-component expression (52).
In this study, we establish the small cytosolic protein TraN as an additional repressor of the Inc18-plasmid pIP501. Using X-ray crystallography, we solved the complex structure of TraN bound to its cognate binding site DNA and identified the distinct binding motif. TraN belongs to the family of winged-helix (WH) proteins that can be classified into three types according to their binding modes (53). The first class of WH proteins comprises the canonical (HNF3-like) DNA interaction profile (54), where the respective recognition helix contacts the DNA via the major groove. In the second binding mode, represented by the RFX1 transcription factor (55), the wing recognizes the major groove of the DNA and the recognition helix interacts with the minor groove. The third class, exemplified by PCG2-DBD, involves contacts between the wing and the minor groove, as well as between the recognition helix and the major groove of the target DNA (53). TraN clearly belongs to the class 1 binding mode and exists as an internal dimer that mostly resembles dimeric transcription factors of the MerR family (16,48). The wings of TraN are involved in the formation of the hydrophobic interface between the two WH domains of the internal dimer and insert into the central minor groove. A clear difference of TraN to the dimeric transcription regulators of the MerR family that bind palindromic sequences, is the pronounced asymmetry of the two half sites, which is most notable in the DNA contacting residues, thus demanding different DNA recognition motifs. This then leads to an asymmetric DNA binding, as reflected in the observed different binding strengths of the two half sites. The proposed binding asymmetry was verified with differential scanning fluorimetry (DSF) assays, where changing distinct bases in the left binding site (contacted by the C-terminal domain of TraN) led to more pronounced alterations in temperature stability. This asymmetric DNA binding might have functional implications in the regulation of the observed tra-component expression or during conjugative DNA transfer, which will be discussed later. Further, a general stabilization of TraN by binding to DNA was observed.
In this study we experimentally validated TraN’s proposed role as a repressor of the tra-operon. Comparison of steady-state protein levels with data from immunofluorescence analyses in E. faecalis revealed a discrepancy in terms of upregulated TraM protein load in the traN knockout mutant (pIP501ΔtraN). This is most likely due to the fact that only correctly localized TraM on the cell periphery can be detected with the immunofluorescence approach, suggesting that not all of the upregulated protein quantity in the traN knockout is properly localized and functional. We selected the VirB8-like protein TraM for immunofluorescence experiments, as it was not only shown to be indispensable for conjugative transfer (56) but also postulated to be a central member of the MPF complex (49,50,57). Furthermore, it was demonstrated that localization of TraM was highly dependent on the peptidoglycan-degrading protein TraG, playing an initiatory role in the process of MPF complex assembly (35,38). Although we demonstrated correct localization of TraM only, it can be assumed that a significant portion of the other upregulated T4S components is also functional in the E. faecalis traN knockout, as the transfer rate of the mutant massively increased compared to the wild type strain, clearly demonstrating TraN as a repressor of the pIP501 tra-operon.
The higher transfer frequency upon traN deletion is most likely caused by the enhanced production of transfer components, as indicated by the elevated Tra-protein load and the increase of TraM-positive foci at the cell periphery. Nevertheless, a role of TraN in regulating conjugative processes directly, potentially decreasing the efficiency of transfer, cannot be excluded at the moment. Furthermore, it is feasible that TraN might exert repression of transfer frequency by downregulation of TraO, a proposed surface adhesin (50,57,58). As G+ T4SSs lack conjugative pili, they rely on surface adhesins to establish the tight contact between donor and recipient cells (4). The observed elevated transfer frequencies in the deletion mutant E. faecalis pIP501ΔtraN might thus be caused by an increased number of TraO molecules, giving rise to a better contact to potential recipient cells and/or a contact to more recipients. Indeed, the observed transcriptional activity from the second promoter PtraNO, upstream of the traN gene, increased the abundance of traO transcripts compared to other tra-genes. We could further demonstrate that PtraNO is also regulated by TraN, most probably by TraN binding to the alternative binding site BS 3. The possibility of transcriptional repression via BS 3 might prevent an overproduction of TraN, which would result in a marked downregulation of tra-operon production, as observed when overexpressing TraN in trans for complementation experiments. Thus, TraN might at least partially autoregulate its own synthesis. Furthermore, the fine-tuning of the transcription of the last gene of the operon, traO, might be essential to avoid toxicity by overproduction of surface adhesins, as demonstrated for other systems. (59). Until now, effects of TraO overexpression on bacterial viability have not been directly investigated, whereas overproduction of PrgB, a surface adhesin from the E. faecalis pheromone-responsive plasmid pCF10, was demonstrated to lead to cellular lysis, thus regulation of its synthesis was shown to be necessary (4,59,60). The observed growth delay and reduced viability of the E. faecalis traN deletion mutant might be attributable to overexpression of TraO, leading to toxic effects similar to those seen for PrgB. Thus, the repression of the PtraNO promoter by TraN binding to BS 3 might not only be important for regulation of TraN expression levels but also to maintain TraO levels in a range that ensures optimal transfer activity while minimizing the risk of toxic effects due to overproduction.
While TraN seems to independently regulate this feedback loop at the PtraNO promoter site, transcriptional regulation at the Ptra promoter might be conducted by both the relaxase TraA and TraN. TraA has been shown to autoregulate its own synthesis by binding to an area overlapping with the −10 region of the Ptra promoter (13) and thus the protein is hypothesized to tune early steps of tra-component expression. We further propose that upon reaching a certain threshold level, TraN will occupy its cognate binding sites. This then represses the transcription from the Ptra as well as the PtraNO promoter, accomplished either by spatial hindrance of the transcription machinery or by bending of the DNA. Indeed, such a bend in the DNA can be observed in the TraN-DNA co-crystal structure.
Responding to yet unknown signals from the environment and/or potential recipients, conjugative transfer is initiated by the relaxase TraA, introducing a single strand cut at the oriT. Concomitant with conjugative transfer, replication of the plasmid is initiated. Due to TraN’s strong interaction with its binding motif, it is conceivable that the protein is only released from the o BS site upstream of the oriT when the first steps of conjugation are initiated. In this scenario, TraN might remain bound to the second site upstream of the traN gene (BS 3) and is only released upon unwinding of the ds-DNA during the DNA transfer and the concomitant DNA-replication process. Once the ds-DNA has been re-established through replication, TraN might again bind to its cognate binding sites, repressing both the Ptra as well as the PtraNO promoter. This might occur in a way already observed for distinct transcription factors (61), namely by interaction with unspecific DNA sequences, thus sliding along the ds-DNA until the specific site is reached. This hypothesis is strengthened by the observation that TraN shows relatively strong binding affinity to random DNA, although weaker than to the cognate binding site sequence (16). When the specific binding site is reached, the C-terminal domain of TraN might contribute substantially to the strong protein–DNA interaction.
In sum, we propose the following mechanism of concerted tra-operon regulation by the repressor protein TraN and the relaxase TraA (Figure 8A): First, the transcription of the tra-operon is initiated. The relaxase TraA, encoded as the very first tra-gene, is expressed, binds to its binding site at the Ptra promoter and tunes the level of tra-operon transcription. Simultaneously, the other Tra-proteins are produced and the MPF complex assembles at the bacterial membrane. The PtraNO promoter leads to enhanced production of TraN and the surface adhesin TraO. Once TraN protein levels reach a certain threshold, both high affinity binding sites on the pIP501 plasmid will be occupied and transcription repressed. This results in a feedback loop and minimizes the risk of cytotoxicity mediated by overproduction of TraO and/or other Tra-proteins. Responding to yet unknown signals from the recipient or the environment (Figure 8B), the first steps of conjugation are initiated. The relaxase TraA nicks the plasmid at the oriT, and the unwinding and replication processes of the pIP501 plasmid are started. At this time point, TraN has to be released from its binding site upstream of the oriT (o BS) and the plasmid translocation process takes place. TraN might remain bound to its second binding site (BS 3) except for a short time interval during the replication process. Once replication of the plasmid is completed in the donor, TraN again binds to its binding sites at the Ptra and PtraNO promoter and thus further represses transcriptional processes, regulating tra-component production. While the exact sequence of tra-operon regulation and conjugative processes has not been revealed yet, it can be assumed that those processes are intertwined and well-matched. Thus, conjugative efficiency is optimized, while overshooting production of Tra-proteins is reduced to a minimum.
Figure 8.

Scheme of the proposed regulation of pIP501 by TraN and the relaxase TraA. (A) Regulation of tra-operon expression by TraN and TraA. (1) The expression of Tra-proteins from the tra-operon is initiated and mRNA is produced from the Ptra and the PtraNO promoters. (2) The relaxase TraA binds to its binding site at the Ptra promoter and controls the level of tra-operon transcription, while the PtraNO promoter remains active. Simultaneously, Tra-proteins are produced and the mating pair formation (MPF) complex assembles at the bacterial membrane. (3) TraN binding to its binding sites upstream of the Ptra and the PtraNO promoters leads to repression of transcription. (B) Initiation of conjugative transfer by the relaxase TraA. Responding to yet unknown signals, conjugative processes are initiated by the relaxase TraA introducing a single-strand (ss) cut at the origin of transfer (oriT). TraN is released from its binding site at the Ptrapromoter and replication takes place in the donor, while the ss-DNA is transferred to the recipient.
Intriguingly, we could identify further potential TraN binding sites on several Inc18 or related multi-resistance plasmids as well as on bacterial chromosomes. Inc18 plasmids have been demonstrated to be major key players in the dissemination of vanA-encoded vancomycin resistance and other determinants facilitating non-susceptibility of MRSA strains to last-line antibiotics (7,50,62,63). Interestingly, most investigated bacterial plasmids and bacterial chromosomes containing a potential TraN binding site not only encoded a TraN-homolog but also a putative TraA-like relaxase and putative homologs of the motor proteins TraE and TraJ. As all studied plasmids of the Inc18 and related incompatibility groups encoding a traN homolog also possess at least one specific putative TraN binding site, we speculate that the regulatory mechanism established for TraN applies to a wide range of systems.
Taken together, we identified TraN as a repressor of conjugative transfer and demonstrate its mode of action, which most probably is conserved across a wide range of Inc18-like and related multi-resistance plasmids responsible for dissemination of antibiotic resistances. We postulate that stimulating the expression of TraN-like proteins or synthetic TraN mimetics might result in diminished horizontal gene transfer not only of pIP501, but rather of a large group of Inc18 or related multi-resistance plasmids. Thus, further studies on TraN and similar T4S regulators might pave the way to efficient interventions decelerating the spreading of antibiotic resistances.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structure have been deposited with the Protein Data bank under accession number 6G1T.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Austrian Science Foundation [P27383 to W.K., P27183-B24 to S.B.]; Deutsches Zentrum für Luft- und Raumfahrt [50WB1466 to E.G.]; Swedish Research Council Vetenskapsrådet [2015-05468 to SB]. Funding for open access charge: Austrian Science Foundation [P27383].
Conflict of interest statement. None declared.
REFERENCES
- 1. Willyard C. The drug-resistant bacteria that pose the greatest health threats. Nature. 2017; 543:15–15. [DOI] [PubMed] [Google Scholar]
- 2. Christie P.J. The mosaic type IV secretion systems. EcoSal Plus. 2016; 7:doi:10.1128/ecosalplus.ESP-0020-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tacconelli E., Carrara E., Savoldi A., Harbarth S., Mendelson M., Monnet D.L., Pulcini C., Kahlmeter G., Kluytmans J., Carmeli Y. et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2017; 18:318–327. [DOI] [PubMed] [Google Scholar]
- 4. Grohmann E., Christie P.J., Waksman G., Backert S.. Type IV secretion in Gram-negative and Gram-positive bacteria. Mol. Microbiol. 2018; 107:455–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mikalsen T., Pedersen T., Willems R., Coque T.M., Werner G., Sadowy E., van Schaik W., Jensen L.B., Sundsfjord A., Hegstad K.. Investigating the mobilome in clinically important lineages of Enterococcus faecium and Enterococcus faecalis. BMC Genomics. 2015; 16:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Palmer K.L., Kos V.N., Gilmore M.S.. Horizontal gene transfer and the genomics of enterococcal antibiotic resistance. Curr. Opin. Microbiol. 2010; 13:632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saheed M., Rothman R.. Update on emerging Infections: news from the centers for disease control and prevention. Ann. Emerg. Med. 2016; 67:386–387. [PubMed] [Google Scholar]
- 8. Jensen L.B., Garcia-Migura L., Valenzuela A.J.S., Løhr M., Hasman H., Aarestrup F.M.. A classification system for plasmids from enterococci and other Gram-positive bacteria. J. Microbiol. Methods. 2010; 80:25–43. [DOI] [PubMed] [Google Scholar]
- 9. Zhu W., Clark N.C., McDougal L.K., Hageman J., McDonald L.C., Patel J.B.. Vancomycin-resistant Staphylococcus aureus isolates associated with Inc18-like vanA plasmids in Michigan. Antimicrob. Agents Chemother. 2008; 52:452–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Evans R.P., Macrina F.L.. Streptococcal R plasmid pIP501: Endonuclease site map, resistance determinant location, and construction of novel derivatives. J. Bacteriol. 1983; 154:1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang A., Macrina F.L.. Characterization of six linked open reading frames necessery for pIP501-mediated conjugation. Plasmid. 1995; 34:206–210. [DOI] [PubMed] [Google Scholar]
- 12. Abajy M.Y., Kopeć J., Schiwon K., Burzynski M., Döring M., Bohn C., Grohmann E.. A type IV-secretion-like system is required for conjugative DNA transport of broad-host-range plasmid pIP501 in gram-positive bacteria. J. Bacteriol. 2007; 189:2487–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kurenbach B., Kopeć J., Mägdefrau M., Andreas K., Keller W., Bohn C., Abajy M.Y., Grohmann E.. The TraA relaxase autoregulates the putative type IV secretion-like system encoded by the broad-host-range Streptococcus agalactiae plasmid plP501. Microbiology. 2006; 152:637–645. [DOI] [PubMed] [Google Scholar]
- 14. Kopec J., Bergmann A., Fritz G., Grohmann E., Keller W.. TraA and its N-terminal relaxase domain of the Gram-positive plasmid pIP501 show specific oriT binding and behave as dimers in solution. Biochem. J. 2005; 387:401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. San Millan A., MacLean R.C.. Fitness costs of plasmids: a limit to plasmid transmission. Microbiol. Spectr. 2017; 5:doi:10.1128/microbiolspec.MTBP-0016-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goessweiner-Mohr N., Eder M., Hofer G., Fercher C., Arends K., Birner-Gruenberger R., Grohmann E., Keller W.. Structure of the double-stranded DNA-binding type IV secretion protein TraN from Enterococcus. Acta Crystallogr. D. Biol. Crystallogr. 2014; 70:2376–2389. [DOI] [PubMed] [Google Scholar]
- 17. De Sanctis D., Beteva A., Caserotto H., Dobias F., Gabadinho J., Giraud T., Gobbo A., Guijarro M., Lentini M., Lavault B. et al. ID29: a high-intensity highly automated ESRF beamline for macromolecular crystallography experiments exploiting anomalous scattering. J. Synchrotron Radiat. 2012; 19:455–461. [DOI] [PubMed] [Google Scholar]
- 18. Kabsch W. XDS. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Evans P.R., Murshudov G.N.. How good are my data and what is the resolution. Acta Crystallogr. D. Biol. Crystallogr. 2013; 69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Dijk M., Bonvin A.M.J.J.. 3D-DART: a DNA structure modelling server. Nucleic Acids Res. 2009; 37:235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adams P.D., Afonine P.V, Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Murshudov G.N., Vagin A.A., Dodson E.J.. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997; 53:240–255. [DOI] [PubMed] [Google Scholar]
- 24. Terwilliger T.C., Grosse-Kunstleve R.W., Afonine P. V, Moriarty N.W., Zwart P.H., Hung L.W., Read R.J., Adams P.D.. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D. Biol. Crystallogr. 2008; 64:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Afonine P. V, Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D.. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D. Biol. Crystallogr. 2012; 68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krissinel E., Henrick K.. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007; 372:774–797. [DOI] [PubMed] [Google Scholar]
- 29. Blanchet C., Pasi M., Zakrzewska K., Lavery R.. CURVES+ web server for analyzing and visualizing the helical, backbone and groove parameters of nucleic acid structures. Nucleic Acids Res. 2011; 39:68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Whitmore L., Wallace B.A.. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004; 32:668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Whitmore L., Wallace B.A.. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008; 89:392–400. [DOI] [PubMed] [Google Scholar]
- 32. Griffith K.L., Wolf R.E.. Measuring β-galactosidase activity in bacteria: cell growth, permeabilization, and enzyme assays in 96-well arrays. Biochem. Biophys. Res. Commun. 2002; 290:397–402. [DOI] [PubMed] [Google Scholar]
- 33. Fercher C., Probst I., Kohler V., Goessweiner-Mohr N., Arends K., Grohmann E., Zangger K., Meyer N.H., Keller W.. VirB8-like protein TraH is crucial for DNA transfer in Enterococcus faecalis. Sci. Rep. 2016; 6:24643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eichenbaum Z., Federle M.J., Marra D., De Vos W.M., Kuipers O.P., Kleerebezem M., Scott J.R.. Use of the lactococcal nisA promoter to regulate gene expression in gram-positive bacteria: Comparison of induction level and promoter strength. Appl. Environ. Microbiol. 1998; 64:2763–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arends K., Celik E.-K.K., Probst I., Goessweiner-Mohr N., Fercher C., Grumet L., Soellue C., Abajy M.Y., Sakinc T., Broszat M. et al. TraG encoded by the pIP501 type IV secretion system is a two-domain peptidoglycan-degrading enzyme essential for conjugative transfer. J. Bacteriol. 2013; 195:4436–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aranda P.S., Lajoie D.M., Jorcyk C.L.. Bleach gel: a simple agarose gel for analyzing RNA quality. Electrophoresis. 2012; 33:366–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and 2(-Delta Delta C(T)) method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 38. Kohler V., Probst I., Aufschnaiter A., Büttner S., Schaden L., Rechberger G.N., Koraimann G., Grohmann E., Keller W.. Conjugative type IV secretion in Gram-positive pathogens: TraG, a lytic transglycosylase and endopeptidase, interacts with translocation channel protein TraM. Plasmid. 2017; 91:9–18. [DOI] [PubMed] [Google Scholar]
- 39. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012; 9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boratyn G.M., Camacho C., Cooper P.S., Coulouris G., Fong A., Ma N., Madden T.L., Matten W.T., McGinnis S.D., Merezhuk Y. et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 2013; 41:W29–W33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grohmann E., Muth G., Espinosa M.. Conjugative plasmid transfer in Gram-positive bacteria. Microbiol Mol. Biol. Rev. 2003; 67:277–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011; 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aravind L., Anantharaman V., Balaji S., Babu M.M., Iyer L.M.. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol. Rev. 2005; 29:231–262. [DOI] [PubMed] [Google Scholar]
- 44. Gajiwala K.S., Burley S.K.. Winged helix proteins. Curr. Opin. Struct. Biol. 2000; 10:110–116. [DOI] [PubMed] [Google Scholar]
- 45. Kenney L.J. Structure/function relationships in OmpR and other winged-helix transcription factors. Curr. Opin. Microbiol. 2002; 5:135–141. [DOI] [PubMed] [Google Scholar]
- 46. Harami G.M., Gyimesi M., Kovács M.. From keys to bulldozers: expanding roles for winged helix domains in nucleic-acid-binding proteins. Trends Biochem. Sci. 2013; 38:364–371. [DOI] [PubMed] [Google Scholar]
- 47. Alanazi A.M., Neidle E.L., Momany C. IUCr . The DNA-binding domain of BenM reveals the structural basis for the recognition of a T-N11-A sequence motif by LysR-type transcriptional regulators. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013; 69:1995–2007. [DOI] [PubMed] [Google Scholar]
- 48. Brown N.L., Stoyanov J.V., Kidd S.P., Hobman J.L.. The MerR family of transcriptional regulators. FEMS Microbiol. Rev. 2003; 27:145–163. [DOI] [PubMed] [Google Scholar]
- 49. Goessweiner-Mohr N., Grumet L., Arends K., Pavkov-Keller T., Gruber C.C., Gruber K., Birner-Gruenberger R., Kropec-Huebner A., Huebner J., Grohmann E. et al. The 2.5 Å structure of the Enterococcus conjugation protein TraM resembles VirB8 type IV secretion proteins. J. Biol. Chem. 2013; 288:2018–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kohler V., Vaishampayan A., Grohmann E.. Broad-host-range Inc18 plasmids: occurrence, spread and transfer mechanisms. Plasmid. 2018; doi:10.1016/J.PLASMID.2018.06.001. [DOI] [PubMed] [Google Scholar]
- 51. Schwarz F.V., Perreten V., Teuber M.. Sequence of the 50-kb conjugative multiresistance plasmid pRE25 from Enterococcus faecalis RE25. Plasmid. 2001; 46:170–187. [DOI] [PubMed] [Google Scholar]
- 52. Bañuelos-Vazquez L.A., Torres Tejerizo G., Brom S.. Regulation of conjugative transfer of plasmids and integrative conjugative elements. Plasmid. 2017; 91:82–89. [DOI] [PubMed] [Google Scholar]
- 53. Liu J., Huang J., Zhao Y., Liu H., Wang D., Yang J., Zhao W., Taylor I.A., Peng Y.L.. Structural basis of DNA recognition by PCG2 reveals a novel DNA binding mode for winged helix-turn-helix domains. Nucleic Acids Res. 2015; 43:1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Clark K.L., Halay E.D., Lai E., Burley S.K.. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature. 1993; 364:412–420. [DOI] [PubMed] [Google Scholar]
- 55. Gajiwala K.S., Chen H., Cornille F., Roques B.P., Reith W., Mach B., Burley S.K.. Structure of the winged-helix protein hRFX1 reveals a new mode of DNA binding. Nature. 2000; 403:916–921. [DOI] [PubMed] [Google Scholar]
- 56. Laverde D., Probst I., Romero-Saavedra F., Kropec A., Wobser D., Keller W., Grohmann E., Huebner J.. Targeting type IV secretion system proteins to combat multidrug-resistant Gram-positive pathogens. J. Infect. Dis. 2017; 215:1836–1845. [DOI] [PubMed] [Google Scholar]
- 57. Bhatty M., Laverde Gomez J.A., Christie P.J.. The expanding bacterial type IV secretion lexicon. Res. Microbiol. 2013; 164:620–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Goessweiner-Mohr N., Arends K., Keller W., Grohmann E., Arends K., Grohmann E.. Conjugation in Gram-Positive Bacteria. Microbiol. Spectr. 2014; 2:PLAS-0004-2013. [DOI] [PubMed] [Google Scholar]
- 59. Bhatty M., Cruz M.R., Frank K.L., Laverde Gomez J.A., Andrade F., Garsin D.A., Dunny G.M., Kaplan H.B., Christie P.J.. Enterococcus faecalis pCF10-encoded surface proteins PrgA, PrgB (aggregation substance) and PrgC contribute to plasmid transfer, biofilm formation and virulence. Mol. Microbiol. 2015; 95:660–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bhatty M., Camacho M.I., Gonzalez-Rivera C., Frank K.L., Dale J.L., Manias D.A., Dunny G.M., Christie P.J.. PrgU: a suppressor of sex pheromone toxicity in Enterococcus faecalis. Mol. Microbiol. 2017; 103:398–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Elf J., Li G.W., Xie X.S.. Probing transcription factor dynamics at the single-molecule level in a living cell. Science. 2007; 316:1191–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Monaco M., Pimentel de Araujo F., Cruciani M., Coccia E.M., Pantosti A.. Worldwide epidemiology and antibiotic resistance of Staphylococcus aureus. Current Topics in Microbiology and Immunology. 2016; 409:Berlin, Heidelberg: Springer; 21–56. [DOI] [PubMed] [Google Scholar]
- 63. Freitas A.R., Tedim A.P., Francia M. V, Jensen L.B., Novais C., Peixe L., Sánchez-Valenzuela A., Sundsfjord A., Hegstad K., Werner G. et al. Multilevel population genetic analysis of vanA and vanB Enterococcus faecium causing nosocomial outbreaks in 27 countries (1986–2012). J. Antimicrob. Chemother. 2016; 71:3351–3366. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structure have been deposited with the Protein Data bank under accession number 6G1T.