

Abstract
Background
Impaired brain oxygen delivery can trigger and exacerbate migraine attacks. Normoxic hypercapnia increases brain oxygen delivery markedly by vasodilation of the cerebral vasculature, and hypercapnia has been shown to abort migraine attacks. Stable normoxic hypercapnia can be induced by a compact partial rebreathing device. This pilot study aimed to provide initial data on the device’s efficacy and safety.
Methods
Using a double-blinded, randomized, cross-over study design, adult migraine-with-aura patients self-administered the partial rebreathing device or a sham device for 20 minutes at the onset of aura symptoms.
Results
Eleven participants (mean age 35.5, three men) self-treated 41 migraine attacks (20 with the partial rebreathing device, 21 with sham). The partial rebreathing device increased mean End Tidal CO2 by 24%, while retaining mean oxygen saturation above 97%. The primary end point (headache intensity difference between first aura symptoms and two hours after treatment (0–3 scale) – active/sham difference) did not reach statistical significance (−0.55 (95% CI: −1.13–0.04), p = 0.096), whereas the difference in percentage of attacks with pain relief at two hours was significant (p = 0.043), as was user satisfaction (p = 0.022). A marked efficacy increase was seen from first to second time use of the partial rebreathing device. No adverse events occurred, and side effects were absent or mild.
Conclusion
Normoxic hypercapnia shows promise as an adjunctive/alternative migraine treatment, meriting further investigation in a larger population.
Clinical study registered at ClinicalTrials.gov with identifier NCT03472417
Keywords: Migraine, headache, hypercapnia, CO2 therapy, rebreathing
Introduction
Studies measuring cerebral blood flow (CBF) have shown that the early stages of migraine attacks are characterized by cerebral vasoconstriction (1–6), implicating hypoperfusion as a migraine trigger (7,8). Olesen et al. (1) were the first to demonstrate that hypoperfusion is often present more than an hour into or throughout the pain phase of the migraine attack – a finding which has been reported by several studies since then (3,4,9), refuting the previous theory that migraine pain is caused by cerebral vasodilation.
Rather than the hypoperfusion in itself, the actual attack trigger may be the resulting decrease in oxygen brain delivery (DO2,brain), since recent studies have demonstrated that hypoxia reliably triggers migraine attacks in migraine patients (10,11) and headache in healthy individuals (12), possibly because of hypoxia’s propensity for inducing and perpetuating cortical spreading depression (CSD) (13) – a migraine trigger phenomenon implicated in migraine with aura (MA) and possibly in migraine without aura (MO) as well (4,14–17).
Past clinical studies have shown that hypercapnia is efficacious in aborting migraine attacks (18–21) and post-spinal headache (22). Two of the studies (18,22) used CO2-rich gas mixtures administered from pressure bottles. However, pressure bottles are impractical, heavy and require refilling between treatments – a likely reason that this treatment option never came into clinical practice in spite of Marcussen and Wolff’s early demonstration of its efficacy in migraine (18).
Two other migraine studies used closed rebreathing bags to induce hypercapnia (19,20). However, in such devices, oxygen is quickly depleted, causing continually worsening – and potentially life-threatening – hypoxia if continued for too long.
Notably, stable normoxic hypercapnia can be achieved by a partial rebreathing device (PRD) (23) which works by capturing a controlled fraction of the expired air, which is then rebreathed together with a controlled amount of atmospheric air. The net effect is a moderate reduction of alveolar ventilation, in spite of the increase in minute ventilation elicited by raising the arterial CO2 tension (PaCO2). By nature of its particular design, the PRD is able to induce a steady state of moderate hypercapnia while retaining normal arterial oxygen saturation (SaO2) (23).
In contrast to gas bottles, the PRD used in this study is compact and very lightweight since no gas or power supply is needed. Because it does not incur hypoxemia, the PRD is inherently much safer than closed rebreathing bags.
Using a double-blinded, randomized, controlled, cross-over design, we performed a first clinical pilot study on the efficacy and safety of the PRD in treatment of migraine with aura (MA), with the additional aim to obtain sample-size estimates for a future large-scale clinical trial. MA was chosen as the patient group because of the comparative ease of standardizing the time point of treatment onset.
We aimed to control for any placebo effect and regression towards the mean by using a sham device as control and randomizing the device cross-over sequence.
It was our hypothesis that by using the PRD to increase PaCO2 and DO2,brain early in MA attacks, the attack severity could be reduced, without adverse effects.
Methods
The clinical trial was approved by the Danish Medicines Agency (ref. no. 2016092009) and the Committee for Ethics in Science of Central Denmark Region (ref. no. 1-10-72-245-16) and registered at ClinicalTrials.gov (NCT03472417). The study protocol is available from the authors by request.
All participant visits took place at the Headache Clinic at Aarhus University Hospital.
Partial rebreathing device
Figure 1 shows the elements of the PRD:
A flexible-volume rebreathing reservoir with laser perforations in its walls. The diameter and spacing of the perforations were carefully chosen to provide a measured gas flow between the atmosphere and the inside of the reservoir, so that even if the bypass valve (see below) was completely closed, gas exchange was still sufficient to prevent hypoxemia.
A mouthpiece connecting the reservoir with the mouth of the user.
A bypass valve in the wall of the reservoir, with a flow-through rate which can be adjusted by the use of a slider. The inspired CO2 concentration can thereby be up- or down-regulated between set limits, according to the baseline End-Tidal CO2 (ETCO2) and ventilatory response of the individual.
Figure 1.
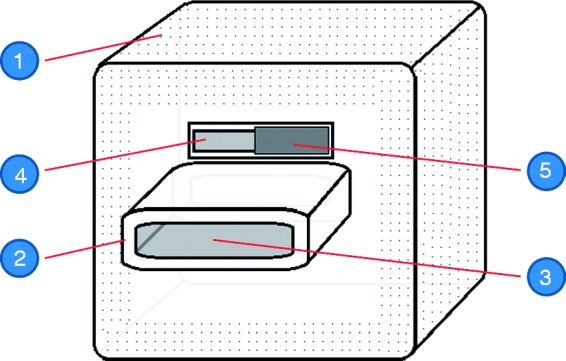
Schematic of the partial rebreathing device. 1: Flexible-volume rebreathing reservoir, having in its walls laser perforations; 2: Mouthpiece; 3: Mouthpiece opening, connecting the inside of the reservoir with the mouth of the user; 4: Bypass valve opening; 5: Adjustable slider, regulating the flow through the bypass valve.
Before use, the PRD is folded compactly and can be carried in a pocket or purse.
The participants used the device by breathing through the mouth piece, with the bypass valve previously fixed at the individual-specific setting determined in the Initial Device Test (see below). The participants wore nose clips while using the device and were instructed to breathe according to their spontaneous breathing drive.
The sham device only had a minimal effect on PaCO2 because the air flow had been redirected by means of a one-way check valve so that minimal rebreathing occurred (Figure 2).
Figure 2.

Photo of the active device (left) and the sham device (right), showing (a) the folded rebreathing reservoirs; (b) bypass valves; (c) reservoir connectors, shown dismounted from mouthpiece boxes (d); (e) sham device airflow redirection holes, with protruding structure for preventing accidental blocking by fingers, (f) sham device one-way check valve.
By necessity, the active and sham devices could not be completely identical. In order to preserve the placebo effect of the sham device in spite of this difference, the two devices were described to the test subjects as providing two different levels of inspired CO2, and not as a “real” versus a “fake” device.
Participants
Participants were recruited from a total group of 190 individuals who responded to an online post advertising the study. Individuals were enrolled and provided written and verbal informed consent if they conformed to the in/exclusion criteria (formulated according to the guidelines of the International Headache Society (24)):
Inclusion criteria were: Between one and six monthly attacks of migraine with typical aura, defined according to the ICHD-3 beta criteria (25); age 18 to 60 years; migraine onset before age 50; and, if in preventive migraine treatment, dose had been stable for three months.
Exclusion criteria were: More than 14 days with headache per month, or more than six days with non-migraine headache per month; medication overuse; pulmonary, cardiovascular, metabolic or psychiatric disease, cancer or anemia; and being pregnant or breastfeeding.
Study procedures and outcome assessment
For the individuals enrolled in the study, general and migraine-specific medical histories were recorded, and a neurological examination was performed.
Subsequently, an initial device test was performed, in which the participant used the active device for a minimum of 15 minutes while being guided by the study personnel and monitored with a capnograph and pulse oximeter (CapnoStream 20, Medtronic, Minneapolis, MN, USA), in order to ensure that the PRD elicited the target increase in ETCO2 and did not incur either hypoxemia (defined as SpO2 < 90%) or any side effects that were not pre-defined as acceptable (see below). The target ETCO2 increase was set according to a sliding scale so that participants with a high baseline ETCO2 were increased less than individuals with a low baseline ETCO2 (increases ranged from 8 mmHg for the highest ETCO2 baselines and 10 mmHg for the lowest).
After being instructed in using the device and study diary, each participant was randomized 1:1 to use either active or sham in the first treatment period. The informing/including study staff member performed the randomization by randomly choosing a sealed envelope containing an alpha-numeric code corresponding to a specific device; these codes were randomly generated, and the envelopes and devices packed by a different study staff member. The study staff member (CH Fuglsang) who enrolled and randomized the participants, provided device instructions and recorded all data was, by this procedure, blinded to which device (active or sham) the participant would use or had used in a given period.
The participant then received the corresponding home treatment kit containing the device, five disposable rebreathing reservoirs, a pulse oximeter (with an audio alarm preset at an SpO2 of 90%), instructions for use and the study diary for recording migraine symptoms.
The participants were instructed to use the device for 20 minutes at the onset of first aura symptoms, followed after 40 minutes by a further 20 minutes of device use. The severities of four symptoms were to be recorded at five time points: At first aura symptoms, immediately after the first device use, and after one, two and 24 hours. The four symptoms were headache, nausea, light/sound sensitivity and functional disability, all recorded on a scale from 0 to 3 (e.g. for headache: 0 = no headache, 1 = mild headache, 2 = moderate headache, 3 = severe headache). The participants were instructed not to use any other migraine treatment for the first two hours.
Moderate hypercapnia can produce mild side effects, including hyperpnea, feeling moderately colder or warmer, mild restlessness or increased perspiration (26). For the purposes of the study, these side effects had in advance been defined as acceptable and test subjects were informed that these side effects could occur but that they were natural and harmless. In some individuals, moderate hypercapnia can cause more unpleasant, though not dangerous, side effects such as dizziness, headache, and visual disturbances (26). In case of such non-acceptable side effects or a drop in SpO2 to below 90%, test subjects were instructed to pause the treatment for one minute, decrease the inspired CO2 fraction by means of the bypass valve slider, and then continue the treatment. If the unacceptable side effects persisted, they were instructed to abort the treatment completely.
After having treated two separate migraine attacks with the device, the participants visited the Headache Clinic and switched to the other device (active or sham). At the end of each treatment period, the participants answered questions assessing their: a) overall satisfaction with the device, b) subjectively experienced treatment effect, c) preference for the device treatment compared with their habitual migraine treatment, d) likelihood of using the device for treating their migraine attacks in the future if they had access to it, and e) side effects using the device, if any.
Participant enrollment took place from November 2016 to January 2017 and the last participant completed the study in October 2017.
The study protocol specified that each treatment period was limited to three months, and that subjects that did not have at least one attack within a treatment period would be excluded from the study. However, the overall attack frequency turned out to be lower than expected and initially reported by the test subjects, so to preserve the power of the study it was decided to extend the maximum duration of all test periods to five months. No other study procedures or end points were changed during the course or after completion of the study.
Study end points
The pre-specified primary end point of the study was Headache Intensity Difference between the moment of the first aura symptoms and two hours later (HID2).
Secondary end points were:
A: Comparing time point 0, and two hours’ post-treatment:
Nausea Intensity Difference (NID2)
Light/Sound Sensitivity Difference (LSSD2)
Functional Disability Difference (FDD2)
Pain relief (PR2); that is, the percentage of attacks in which there was no or mild pain two hours after first using the device
Pain freedom (PF2); that is, the percentage of attacks in which there was no pain two hours after first using the device
B: Recorded at the end of each treatment period:
Overall satisfaction with the device, −2 to +2 scale (−2 = wholly unsatisfied, +2 = wholly satisfied)
Subjective treatment effect, 0–4 scale (0 = no effect, 1 = small effect, 2 = moderate effect, 3 = good effect, 4 = very good effect)
Treatment preference vs. participant’s normal treatment, −2 to +2 scale (−2 = much worse than normal treatment,+2 = much better)
The likelihood that the participant would use this device to treat their migraine attacks in the future if it was available to them, −2 to +2 scale (−2 = would certainly not use the device, +2 = would certainly use the device)
Occurrence, type and severity of side effects
Adverse events
Statistical analysis
End point values for a device were averaged over the two attacks with that device and analyzed in a pairwise manner comparing the active and sham devices.
Wilcoxon signed rank tests were used for hypothesis testing of the primary end point as well as for the secondary end points concerning headache, nausea, light/sound sensitivity, functional disability, device satisfaction, treatment effect, preference and likelihood of use.
None of the end point distributions showed marked non-normality or outliers (see Figure 3, also confirmed by Shapiro-Wilk tests of non-normality), leading to the choice of means and standard deviations as the best descriptive parameters of the data.
Figure 3.

Box/scatter plots of the continuous-variable end points. (a) Active-sham differences of symptom scores of headache, nausea, light/sound sensitivity and functional disability, comparing zero and two hours. (b) Active-sham differences of end-of-period scores of subjective treatment effect, treatment preference, satisfaction with device and likelihood to use in future (for the subjective treatment effect and treatment preference, some data points are missing). Circles are individual patient data points, horizontal black bars indicate means, dark grey boxes indicate confidence intervals (mean ± 1.96 standard errors of the mean), light grey boxes indicate the interval of the mean ± one standard deviation, and the red horizontal line indicates the zero-line (corresponding to no difference between active and sham device). For the parameters in Figure 3(a), values below zero correspond to a better effect of the active over the sham. For the parameters in Figure 3(b), values above zero correspond to a better effect of the active over the sham.
For the secondary end points concerning pain relief and pain freedom, odds ratios and Pearson’s χ2 tests were used for comparing the proportions of attacks in which there was respectively pain relief and pain freedom. No interim analyses were performed. The statistical analyses did not adjust for multiple comparisons or include multivariable analysis. The 5% significance level was used, and all reported p values are two-tailed. Stata version 11 was used for all analyses (the signrank and cc commands respectively).
Results
In total, 18 MA patients were recruited and enrolled from a total group of 190 individuals responding to an online post advertising the study. The remaining 172 individuals did not conform to the in/exclusion criteria.
Three of the enrolled participants were subsequently excluded because they did not have at least one attack within the first or second five-month treatment period. One participant left the study due to illness unrelated to the study, one participant was lost to follow-up and two participants withdrew their consent before treating an attack. Characteristics of the 11 participants who completed the study are shown in Table 1 and 2.
Table 1.
Participant characteristics.
| Variable | Value |
|---|---|
| n | 11 |
| Age (mean ± SD, median) | 35.5 ± 12.0, 31.0 |
| Female/male | 8/3 |
| Caucasian (%) | 100% |
| Height in cm (mean ± SD, median) | 174.0 ± 8.5, 170.0 |
| Weight in kg (mean ± SD, median) | 70.0 ± 11.0, 67.0 |
| Age at migraine onset (mean ± SD, median) | 21.0 ± 11.5, 15.0 |
| Family history of migraine (%) | 73% |
| Current migraine medications: | Triptans: 55% (three additional participants had used triptans earlier but no longer did) NSAIDs or paracetamol: 73% Prophylactic medications: 9% |
| Baseline ETCO2 in mmHg (mean ± SD, median) | 37.1 ± 4.5, 38.0 |
| Baseline SpO2% (mean ± SD, median) | 98.8 ± 1.5, 99.0 |
| Average no. of days between attacks in study (mean ± SD, median) | 43.7 ± 22.6, 35.0 |
Table 2.
Participant migraine characteristics.
| Gender | Age | Age at migraine onset | Typical migraine symptoms | Aura symptoms, normal onset time before pain |
|---|---|---|---|---|
| F | 26 | 15 | P, Pu, WPA, N, V, Pnp, Ptp, VD, CD | VD, 20–25 min. |
| F | 26 | 10 | P, WPA, N, V, Pnp, Ptp, VD, CD | VD, 30–45 min. |
| F | 23 | 25 | P, Pu, WPA, N, Pnp, Ptp, VD, CD | VD, SpD, 15 min. |
| F | 29 | 27 | P, Pu, N, Pnp, Ptp, CD | VD, 30–60 min. |
| F | 35 | 13 | P, Pu, N, Pnp, Ptp, VD, CD | VD, SeD, 60–180 min. |
| M | 49 | 12 | P, Pu, WPA, N, V, PtP, VD, CD | VD, SpD, 30–60 min. |
| F | 20 | 8 | P, Pu, WPA, N, V, PnP, PtP, VD, CD | VD, 45 min. |
| M | 31 | 13 | P, Pu, V, Ptp, VD | VD, 60 min. |
| M | 47 | 35 | P, N, Pnp, Ptp, VD, CD | VD, SpD, 120–180 min. |
| F | 59 | 46 | P, Pu, N, Pnp, Ptp, CD | SeD, 60–120 min. |
| F | 45 | 27 | P, Pu, WPA, N, V, Pnp, Ptp, VD, CD | VD, SpD, 30–60 min. |
Abbreviations of migraine symptoms: P: Moderate/severe pain; Pu: Pain is pulsating/throbbing; WPA: Pain worsening with physical activity; N: nausea; V: vomiting; Pnp: phonophobia; Ptp: photophobia; VD: visual disturbances; CD: cognitive disturbances.
Abbreviations of aura symptoms: VD: visual disturbances; SeD: sensory disturbances; SpD: speech disturbances. Gender: F, Age: 45, Age at migraine onset: 27, Typical migraine symptoms: P, Pu,WPA, N, V, Pnp, Ptp, VD, CD. Aura symptoms, normal onset time: VD, 30-60 min.
Three of the test subjects had one treatment period in which they only had one attack. For this reason, the total number of attacks in the study was 41, of which 20 were with the PRD and 21 with the sham device.
Device effect on ETCO2 and SpO2
Among the 18 enrolled participants that completed the initial device test, mean baseline ETCO2 was 36.8 mmHg (range 26–41 mmHg). Using the active device, ETCO2 increased to a mean of 45.8 mmHg (range 39 to 51); that is, a mean increase of 9.0 mmHg (equal to a 24% increase compared to baseline).
The average SpO2 at baseline was 98.7% (range 95 to 100%), which decreased to 97.3% (likewise range 95 to 100%) during the active device use.
The baseline values and increases of ETCO2 and SpO2 among the 11 participants who completed the study were very close to the values in the group of 18 (Table 1).
Migraine outcomes
Figure 3 and Table 3 show the results for the continuous-variable end points.
Table 3.
Trial end points I, continuous-variable data.
| Active |
Sham |
Active-sham difference |
p-value (Wilcoxon signed | ||||
|---|---|---|---|---|---|---|---|
| Mean | 95% CI | Mean | 95% CI | Mean | 95% CI | rank test) | |
| Headache intensity difference, 0–2 hours | 0.77 | 0.24–1.31 | 1.32 | 0.68–1.96 | −0.55 | −1.13–0.04 | 0.096 |
| Headache intensity difference, 0–1 hours | 0.80 | 0.19–1.40 | 1.11 | 0.46–1.77 | −0.32 | −0.96–0.32 | 0.26 |
| Nausea intensity difference, 0–2 hours | 0.09 | −0.34–0.53 | 0.55 | −0.08–1.17 | −0.45 | −1.08–0.17 | 0.28 |
| Nausea intensity difference, 0–1 hours | 0.00 | −0.40–0.40 | 0.48 | −0.05–1.01 | −0.48 | −0.97–0.02 | 0.072 |
| Light/sound sensitivity difference, 0–2 hours | −0.16 | −0.83–0.52 | 0.30 | −0.37–0.96 | −0.45 | −1.24–0.33 | 0.34 |
| Light/sound sensitivity difference, 0–1 hours | −0.20 | −0.75–0.34 | 0.59 | −0.05–1.24 | −0.80 | −1.43– −0.17 | 0.043 + |
| Functional disability difference, 0–2 hours | 0.14 | −0.34–0.61 | 0.43 | −0.24–1.10 | −0.30 | −0.98–0.39 | 0.50 |
| Functional disability difference, 0–1 hours | 0.14 | −0.26–0.53 | 0.43 | −0.15–1.01 | −0.30 | -0.96–0.36 | 0.39 |
| Satisfaction with device’s effect | 0.59 | −0.09–1.28 | −1.00 | −1.92– −0.08 | 1.59 | 0.53–2.65 | 0.022 + |
| Subjective treatment effect | 1.40 | 0.56–2.24 | 0.50 | −0.17–1.17 | 1.00 | −0.08–2.08 | 0.099 |
| Treatment preference vs. normal treatment | −0.17 | −0.78–0.44 | −0.75 | −1.54–0.04 | 0.78 | −0.23–1.79 | 0.19 |
| Likelihood to use device in future | −0.09 | −1.06–0.88 | −1.18 | −1.92– −0.44 | 1.09 | −0.10–2.29 | 0.081 |
Statistically significant at the 5% level.
For all end points, using the active device resulted in lower average symptom scores (Table 3, Figure 3(a)) and higher average end-of-period test subject scores (Table 3, Figure 3(b)) respectively, though variances were large.
For HID2 (the pre-specified primary end point), headache increased by 0.77 points (0–3 scale) from first aura symptoms to two hours, as compared to an increase of 1.32 with the sham device (mean difference −0.55 (95% CI: −1.13–0.04)), a result which was not statistically significant (p = 0.096). Interestingly, a post hoc analysis showed that the three participants with greatest effect on HID2 were the three men.
The mean severity of all symptoms increased from first aura symptoms to two hours, except for light/sound sensitivity, which decreased slightly with the active device (but increased with the sham device).
The higher satisfaction with the active over the sham device was statistically significant (0.59 vs. −1.00 on the −2/+2 scale (95% CI: 0.53–2.65), p = 0.022).
In 60% of attacks with the active device there was pain relief at two hours, compared to 29% with the sham device (Table 4), a difference which was statistically significant (odds ratio = 3.75 (95% CI, exact: 0.86–16.97), p = 0.043). Total pain freedom at two hours was only achieved in 15% of attacks with the active device and 14% with the sham device.
Table 4.
Trial end points II, binary data.
| Active % | Sham % | Odds ratio | 95% CI (exact) | p-value (χ2 test) | |
|---|---|---|---|---|---|
| Pain relief % at two hours, average of both attacks | 60 | 29 | 3.75 | 0.86–16.97 | 0.043 + |
| Pain relief % at two hours, first attack | 45 | 27 | 2.22 | 0.27–19.76 | 0.38 |
| Pain relief % at two hours, second attack | 78 | 30 | 8.17 | 0.75–113.44 | 0.037 + |
| Pain freedom % at two hours, average of both attacks | 15 | 14 | 1.06 | 0.12–9.03 | 0.95 |
| Pain freedom % at two hours, first attack | 9 | 18 | 0.45 | 0.01–10.42 | 0.53 |
| Pain freedom % at two hours, second attack | 22 | 10 | 2.57 | 0.11–168.26 | 0.47 |
Statistically significant at the 5% level.
A post hoc comparison of the first and second attack with a given device indicated a training benefit when using the active device, PR2 being 45% in the first attack with the active device and 78% in the second attack (Figure 4, Table 4), PF2 also increasing (from 9% to 22%). This training effect was not seen with the sham device (Figure 4, Table 4).
Figure 4.

Comparison of active vs. sham device effect on pain relief at two hours (left) and pain freedom at two hours (right), showing differences between first use and second use of device.
Figure 5 shows the changes in mean migraine symptoms over the first two hours, for the active and sham devices respectively.
Figure 5.

Changes in symptom severity during the first two hours, for headache (top left), nausea (top right), light/sound sensitivity (bottom left) and functional disability (bottom right). Mean values and corresponding 95% confidence intervals are shown.
Values at 24 hours were not analyzed due to a high incidence of missing study diary data at this time point.
Side effects
During the initial device tests, two out of 18 participants experienced transient side effects (dizziness and mild nausea respectively), which disappeared after a break of two minutes and a one-step increase in the bypass valve opening and did not reoccur after device use was resumed.
In the course of the treatments at home, the following side effects were recorded:
Sham device: Warm sensation (two participants, one attack each), dry mouth (one participant, two attacks), increased perspiration (one participant, two attacks), irritated sensation (one participant, one attack), unpleasant taste (one participant, one attack).
Active device: Hyperpnea (three participants: One, one and two attacks), dyspnea (two participants: One and two attacks), warm sensation (two participants, one attack each), mild claustrophobia (one participant, one attack), increased salivation (one participant, one attack), anxiety (one participant, one attack), restlessness (one participant, one attack).
In total, eight of the test subjects experienced a side effect during one or both of the attacks with the active device, while five experienced a side effect during one or both of the attacks with the sham device. Side effects occurred more frequently in the first than in the second attack in a treatment period.
The participants had been instructed to stop using the device in the event of excessive side effects, but at no point during the study did a participant feel that the side effects were strong enough for them to stop the treatment.
No test subject reported any occurrence of arterial desaturation to below the pulse oximeter alarm limit.
No adverse events occurred in the course of the study.
Discussion
This small-scale randomized and controlled trial constitutes the first clinical test of partial rebreathing for migraine treatment. The primary end point, measuring difference in headache at time of first aura symptoms and after two hours, did not reach statistical significance (p = 0.096). This could be because of the small number of participants and large variance in this pilot study, but it cannot of course be ruled out that the trial would be negative even if sufficiently powered. However, a full-scale trial of the device seems warranted by the fact that the secondary end points measuring device satisfaction and pain relief both reached significance (p = 0.022 and p = 0.043 respectively). For the remaining secondary end points, the mean active/sham differences were likewise in favor of the active device over the sham (i.e. symptom scores were on average lower and end-of-period test subject scores were higher), though these differences were not statistically significant.
The present findings are consistent with previous studies having shown efficacy of systemic or local hypercapnia in migraine treatment (18–21).
The difference between PR2 at first PRD treatment (45%) and second PRD treatment (78%) indicated an increased efficacy with training and/or repeated use.
No adverse events occurred in the study, and side effects were in general mild or absent. In the few cases where non-acceptable side effects occurred (see Study procedures and outcome assessment), the initial device tests demonstrated that these could be effectively and quickly addressed by briefly interrupting the treatment, and their recurrence prevented by decreasing the rebreathing level.
The small scale of this study naturally restricts the strength of the conclusions that can be drawn, as does the effect that cognitive impairment during MA attacks could have on self-reported symptom scores. Additionally, since attacks were treated at home, we cannot confirm that treatment was performed correctly or for the intended duration.
As is unavoidable in many device trials, subject blinding was not perfect, specifically because of the necessary physical differences between the two devices and due to the higher incidence of CO2-induced hyperpnea with the active device. We sought to mitigate this problem by describing the devices to the test subjects as having different levels of CO2 increase (as opposed to one being “real” and the other “fake”) and stating that it was not known which CO2 level would have the best effect. Even so, there may still have been some residual bias due to imperfect blinding.
The migraine patients in the study were recruited from the general population via a post on a social media profile of Aarhus University Hospital. Our aim with this approach was to recruit more typical migraine patients than the patients normally referred to the highly specialized Headache Clinic at the hospital, of which many have chronic and/or very severe or atypical migraine. It is possible that there may have been a recruitment bias in the type of patients that responded to this type of online post – a bias which would reduce the generalizability of the results.
Even in the light of the trial’s limitations and small size, the data indicate that the PRD treatment may be effective in a non-trivial percentage of MA patients. On the other hand, it is also clear that some test subjects only experienced a negligible treatment effect or none at all. For these patients, it is promising that the study indicated an increase in efficacy with each subsequent treatment.
Several physiological mechanisms may underpin the migraine treatment efficacy of normoxic hypercapnia, indicated by this and earlier studies. A number of studies have indicated that transient increases of the extracellular K+ concentration play an important role in the triggering and propagation of CSD (17,28–32), which may be an underlying attack trigger not only in MA but MO as well (4,14–17). Clearance of K+ from the extracellular space depends critically on proper functioning of the Na+/K+-ATPase transporter, which in turn requires an adequate supply of oxygen and glucose to function (33,34). Indeed, inhibition of Na+/K+-ATPase directly triggers spreading depression in hippocampal slices (35).
Inadequate Na+/K+-ATPase function may thus be the underlying mechanism behind the propensity of ischemia for triggering spreading depolarizations in animal models (36), and of hypoxia for triggering migraine (10–12).
It is well established that hypercapnia has a strong vasodilatory effect in the brain (37,38), increasing total cerebral blood flow by as much as 7.4% for each mmHg increase in ETCO2 (27), in turn increasing total DO2,brain by the same percentage if normal SaO2 is preserved. The efficacy of normoxic hypercapnia in migraine may thus be a function of bolstering Na+/K+-ATPase activity by reversing the hypoperfusion (and any resulting tissue hypoxia) responsible for triggering and propagating the spread of CSD. Indeed, hypercapnic acidosis has in animal models shown a marked inhibitory effect on the triggering, propagation and duration of CSD (39–41), as well as a general inhibition of neural excitability (42,43).
For the purpose of increasing DO2,brain, an alternative to normoxic hypercapnia is to increase the inspired oxygen fraction, and a recent study showed some efficacy of high-flow oxygen for acute migraine relief (44). However, due to the sigmoidal shape of the oxygen dissociation curve, such increases in PaO2 only increase the arterial oxygen concentration slightly compared to normoxia, and very high inspired oxygen fractions may even incur vasoconstriction and hypoperfusion (45). Being the mathematical product of CBF and CaO2, DO2,brain can be increased significantly by the strong increase in CBF achievable by normoxic hypercapnia, but comparatively little by the small increase in CaO2 elicited by hyperoxic normocapnia (though hyperoxia could have other relevant effects apart from its impact on DO2,brain).
Compared to pharmaceutical relief medications, the PRD could potentially have a number of advantages, among them:
CO2-induced increases in CBF happen within 10 seconds of starting to breathe an increased CO2 fraction (46) and wash out of the body within a few minutes of ending the treatment. This fast onset of action enables early intervention, possibly aborting or containing the progress of the CSD wave – a hypothesis that could be tested in an imaging study.
As a drug-free treatment, PRDs could potentially be combined with pharmaceuticals or replace them in groups for whom standard medications are contraindicated, provided that sufficient PRD safety data is obtained for the patient groups in question.
PRDs avoid the common problem of oral medications being expelled by emesis before uptake through the gastric tract is complete.
The effects of moderate hypercapnia are well known and generally mild, and any excessive CO2 increases can be directly sensed by the user, allowing him/her to immediately decrease the device’s rebreathing level and its bodily effects.
In light of such potential advantages and the promising results of this small-scale pilot study, we believe that a large-scale clinical trial of the PRD treatment in migraine is warranted.
In future studies, it will also be relevant to investigate the background for (and correlates of) the PRD efficacy variance, for at least two reasons. Firstly, it could help identify predictors for a patient’s likely response to the treatment, allowing more targeted studies into, and targeted clinical use of, PRD treatment. Secondly, it could make it possible to develop improvements to the device, its timing/dosage or duration of use, in order to increase the percentage of migraine patients deriving benefit from the treatment. Some MA patients may be further in the attack evolution than others before experiencing the migraine aura, meaning that some of the participants in this study had possibly passed a pathophysiological “point of no return” at the time of their cue to start using the device (in this study the cue was the aura symptoms). For this reason, it may be better to start the treatment at the appearance of the very first prodromes (which occur in the majority of both MA and MO patients (6)), and not wait until the appearance of aura.
This study specifically studied MA, so its results may not be generalizable to other types of migraine such as MO. However, several studies indicate that CSD and/or cerebral hypoperfusion play important roles in MO as well (4,14–17), and hypoxia has been shown to trigger migraine attacks in both MO and MA patients (10). This seems to indicate that a clinical study of PRD treatment in MO would be warranted.
In future clinical studies, the efficacy of PRD treatment as an add-on to pharmaceuticals would be another interesting focus.
Clinical implications
Normoxic hypercapnia induced by a partial rebreathing device showed promise as an adjunctive or alternative, non-pharmacological treatment of episodic migraine with aura.
The device efficacy increased with each subsequent use.
Further studies should be undertaken to investigate the efficacy in a larger population and in migraine without aura.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: T. Johansen is a co-founder, shareholder and employee of BalancAir.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by BalancAir.
References
- 1.Olesen J, Friberg L, Skyhoj Olsen T, et al. Timing and topography of cerebral blood flow, aura, and headache during migraine attacks. Ann Neurol 1990; 28: 791–798. [DOI] [PubMed] [Google Scholar]
- 2.De Benedittis G. CBF changes during headache-free periods and spontaneous/induced attacks in migraine with and without aura: A TCD and SPECT comparison study. J Neurosurg Sci 1999; 43: 141–147. [PubMed] [Google Scholar]
- 3.Sanchez Del Rio M, Bakker D, Wu O, et al. Perfusion weighted imaging during migraine: Spontaneous visual aura and headache. Cephalalgia 1999; 19: 701–707. [DOI] [PubMed] [Google Scholar]
- 4.Denuelle M, Fabre N, Payoux P, et al. Posterior cerebral hypoperfusion in migraine without aura. Cephalalgia 2008; 28: 856–862. [DOI] [PubMed] [Google Scholar]
- 5.Hansen JM, Schytz HW, Larsen VA, et al. Hemiplegic migraine aura begins with cerebral hypoperfusion: Imaging in the acute phase. Headache 2011; 51: 1289–1296. [DOI] [PubMed] [Google Scholar]
- 6.Charles A. The evolution of a migraine attack – a review of recent evidence. Headache 2013; 53: 413–419. [DOI] [PubMed] [Google Scholar]
- 7.Brennan KC, Charles A. An update on the blood vessel in migraine. Curr Opin Neurol 2010; 23: 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelman L. The biological basis of headache. Expert Rev Neurother 2011; 11: 363–378. [DOI] [PubMed] [Google Scholar]
- 9.Bednarczyk EM, Remler B, Weikart C, et al. Global cerebral blood flow, blood volume, and oxygen metabolism in patients with migraine headache. Neurology 1998; 50: 1736–1740. [DOI] [PubMed] [Google Scholar]
- 10.Schoonman GG, Sándor PS, Agosti RM, et al. Normobaric hypoxia and nitroglycerin as trigger factors for migraine. Cephalalgia 2006; 26: 816–819. [DOI] [PubMed] [Google Scholar]
- 11.Arngrim N, Schytz HW, Britze J, et al. Migraine induced by hypoxia: An MRI spectroscopy and angiography study. Brain 2016; 139: 723–737. [DOI] [PubMed] [Google Scholar]
- 12.Broessner G, Rohregger J, Wille M, et al. Hypoxia triggers high-altitude headache with migraine features: A prospective trial. Cephalalgia 2016; 36: 765–771. [DOI] [PubMed] [Google Scholar]
- 13.Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 2015; 95: 953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woods RP, Iacoboni M, Mazziotta JC. Brief report: Bilateral spreading cerebral hypoperfusion during spontaneous migraine headache. New Engl J Med 1994; 331: 1689–1692. [DOI] [PubMed] [Google Scholar]
- 15.Cavestri R, Arreghini M, Longhini M, et al. Interictal abnormalities of regional cerebral blood flow in migraine with and without aura. Minerva Med 1995; 86: 257–264. [PubMed] [Google Scholar]
- 16.Chalaupka FD. Reversible imaging abnormalities consistent with CSD during migraine without aura attack. Headache 2008; 48: 1229–1232. [DOI] [PubMed] [Google Scholar]
- 17.Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Ann Rev Physiol 2013; 75: 365–391. [DOI] [PubMed] [Google Scholar]
- 18.Marcussen RM, Wolff HG. Effects of carbon dioxide-oxygen mixtures given during preheadache phase of the migraine attack; further analysis of the pain mechanisms in headache. Arch Neurol Psychiatry 1950; 63: 42–51. [PubMed] [Google Scholar]
- 19.Dexter SL. Rebreathing aborts migraine attacks. Br Med J (Clin Res Ed) 1982; 284: 312–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pradalier A, Baron JF, Dry J, et al. Trial treatment of migraine attack by rebreathing of expired air. Presse Med 1984; 13: 1901–1901. [PubMed] [Google Scholar]
- 21.Spierings ELH. Non-inhaled, intranasal carbon dioxide for the abortive treatment of migraine headache: Efficacy, tolerability, and safety. Annals of Neurology 2005; 58: S17. DOI: 10.1002/ana.11300. [DOI]
- 22.Sikh SS, Agarwal G. Post spinal headache. A preliminary report on the effect of inhaled carbon dioxide. Anaesthesia 1974; 29: 297–300. [DOI] [PubMed] [Google Scholar]
- 23.Johansen T, Jack S, Dahl R. Normalizing CO2 in chronic hyperventilation by means of a novel breathing mask: A pilot study. Clin Respir J 2013; 7: 359–366. [DOI] [PubMed] [Google Scholar]
- 24.Tfelt-Hansen P, Pascual J, Ramadan N, et al. Guidelines for controlled trials of drugs in migraine: 3rd edn. A guide for investigators. Cephalalgia 2012; 32: 6–38. [DOI] [PubMed] [Google Scholar]
- 25.Bes A, Kunkel R, Lance JW, et al. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 2013; 33: 629–808. [DOI] [PubMed]
- 26.Schaefer KE. Respiratory pattern and respiratory response to CO2. J Appl Physiol 1958; 13: 1–14. [DOI] [PubMed] [Google Scholar]
- 27.Pollock JM, Deibler AR, Whitlow CT, et al. Hypercapnia-induced cerebral hyperperfusion: An underrecognized clinical entity. Am J Neuroradiol 2009; 30: 378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grafstein B. Mechanism of spreading cortical depression. J Neurophysiol 1956; 19: 154–171. [DOI] [PubMed] [Google Scholar]
- 29.Hansen A, Zeuthen T. Extracellular ion concentrations during spreading depression and ischemia in the rat-brain cortex. Acta Physiol Scand 1981; 113: 437–445. [DOI] [PubMed] [Google Scholar]
- 30.Lothman E, Lamanna J, Cordingley G, et al. Responses of electrical potential, potassium levels, and oxidative metabolic-activity of cerebral neocortex of cats. Brain Res 1975; 88: 15–36. [DOI] [PubMed] [Google Scholar]
- 31.Kager H, Wadman W, Somjen G. Conditions for the triggering of spreading depression studied with computer simulations. J Neurophysiol 2002; 88: 2700–2712. [DOI] [PubMed] [Google Scholar]
- 32.Matsuura T, Bures J. Minimum volume of depolarized neural tissue required for triggering cortical spreading depression in rat. Exper Brain Res 1971; 12: 238–249– 238–249. [DOI] [PubMed] [Google Scholar]
- 33.Lipton P, Whittingham T. Reduced ATP concentration as a basis for synaptic transmission failure during hypoxia in the invitro guinea-pig hippocampus. J Physiol (Lond) 1982; 325: 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang JC, Brennan KC, He D, et al. A mathematical model of the metabolic and perfusion effects on cortical spreading depression. PLOS One 2013; 8: e70469–e70469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Balestrino M, Young J, Aitken P. Block of (Na+,K+)ATPase with ouabain induces spreading depression-like depolarization in hippocampal slices. Brain Res 1999; 838: 37–44. [DOI] [PubMed] [Google Scholar]
- 36.von Bornstädt D, Houben T, Seidel JL, et al. Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron 2015; 85: 1117–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madden JA. The effect of carbon dioxide on cerebral arteries. Pharmacol Ther 1993; 59: 229–250. [DOI] [PubMed] [Google Scholar]
- 38.Claassen JA, Zhang R, Fu Q, et al. Transcranial Doppler estimation of cerebral blood flow and cerebrovascular conductance during modified rebreathing. J Appl Physiol 2007; 102: 870–877. [DOI] [PubMed] [Google Scholar]
- 39.Gardner-Medwin AR. Possible roles of vertebrate neuroglia in potassium dynamics, spreading depression and migraine. J Exp Biol 1981; 95: 111–127. [DOI] [PubMed] [Google Scholar]
- 40.Tombaugh GC. Mild acidosis delays hypoxic spreading depression and improves neuronal recovery in hippocampal slices. J Neurosci 1994; 14: 5635–5643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tong CK, Chesler M. Modulation of spreading depression by changes in extracellular pH. J Neurophysiol 2000; 84: 2449–2457. [DOI] [PubMed] [Google Scholar]
- 42.Kaila K and Ransom BR. pH and brain function, 1st edn. New York, NY: Wiley-Liss, 1998, pp.371–506.
- 43.Ruusuvuori E, Kaila K. Carbonic anhydrases and brain pH in the control of neuronal excitability. Subcell Biochem 2014; 75: 271–290. [DOI] [PubMed] [Google Scholar]
- 44.Singhal AB, Maas MB, Goldstein JN, et al. High-flow oxygen therapy for treatment of acute migraine: A randomized crossover trial. Cephalalgia 2017; 37: 730–736. [DOI] [PubMed] [Google Scholar]
- 45.Ashkanian M, Borghammer P, Gjedde A, et al. Improvement of brain tissue oxygenation by inhalation of carbogen. Neuroscience 2008; 156: 932–938. [DOI] [PubMed] [Google Scholar]
- 46.Poulin MJ, Liang PJ, Robbins PA. Dynamics of the cerebral blood flow response to step changes in end-tidal PCO2 and PO2 in humans. J Appl Physiol 1996; 81: 1084–1095. [DOI] [PubMed] [Google Scholar]