

Abstract
Injury to the supraspinal motor systems, especially the corticospinal tract, leads to movement impairments. In addition to direct disruption of descending motor pathways, spinal motor circuits that are distant to and not directly damaged by the lesion undergo remodeling that contributes significantly to the impairments. Knowing which spinal circuits are remodeled and the underlying mechanisms are critical for understanding the functional changes in the motor pathway and for developing repair strategies. Here, we target spinal premotor cholinergic interneurons (IN) that directly modulate motoneuron excitability via their cholinergic C-bouton terminals. Using a model of unilateral medullary corticospinal tract lesion in male rats, we found transneuronal downregulation of the premotor cholinergic pathway. Phagocytic microglial cells were upregulated in parallel with cholinergic pathway downregulation and both were blocked by minocycline, a microglia activation inhibitor. Additionally, we found a transient increase in interneuronal complement protein C1q expression that preceded cell loss. 3D reconstructions showed ongoing phagocytosis of C1q-expressing cholinergic INs by microglia 3 d after injury, which was complete by 10 d after injury. Unilateral motor cortex inactivation using the GABAA receptor agonist muscimol replicated the changes detected at 3 d after lesion, indicating activity dependence. The neuronal loss after the lesion was rescued by increasing spinal activity using cathodal trans-spinal direct current stimulation. Our finding of activity-dependent modulation of cholinergic premotor INs after CST injury provides the mechanistic insight that maintaining activity, possibly during a critical period, helps to protect distant motor circuits from further damage and, as a result, may improve motor functional recovery and rehabilitation.
SIGNIFICANCE STATEMENT Supraspinal injury to the motor system disrupts descending motor pathways, leading to movement impairments. Whether and how intrinsic spinal circuits are remodeled after a brain injury is unclear. Using a rat model of unilateral corticospinal tract lesion in the medulla, we show activity-dependent, transneuronal downregulation of the spinal premotor cholinergic system, which is mediated by microglial phagocytosis, possibly involving a rapid and transient increase in neuronal C1q before neuronal loss. Spinal cord neuromodulation after injury to augment spinal activity rescued the premotor cholinergic system. Our findings provide the mechanistic insight that maintaining activity, possibly during an early critical period, could protect distant motor circuits from further damage mediated by microglia and interneuronal complement protein and improve motor functional outcomes.
Keywords: activity dependence, cholinergic interneuron, complement protein C1q, corticospinal tract, phagocytic microglia
Introduction
Brain and spinal cord injury cause an immediate loss of motor function due to the interruption of movement control signals by descending axon axotomy. Motor pathway lesion also produces longer-term circuit remodeling from days to years after the trauma (Yamada et al., 1998; Zhang et al., 2012), which leads to both adaptive and maladaptive changes. The goal of our study is to examine how spinal circuits are remodeled after loss of descending inputs. To repair the damaged motor systems after injury, a better understanding of the response that the nervous system mounts after injury is needed.
Our study targets the corticospinal tract (CST) because it is an excellent model system for producing selective axon loss after brain lesion and it is clinically highly relevant, producing skilled movement impairment after stroke or spinal cord injury (Papadopoulos et al., 2002; Carmel and Martin, 2014). Whereas axon loss contributes to development of impairments after injury, much less is known about the role of downstream circuit changes, such as in the spinal cord, in response to axon loss. Learning how spinal circuits remodel after injury, especially neuronal loss, is critical for understanding the mechanisms of motor impairment and for establishing standards for circuit repair for recovery and rehabilitation.
Circuit remodeling in the brain during development (Frost and Schafer, 2016) and neurodegenerative disease (Kreutzberg, 1996) is known to be mediated by the major phagocyte in the CNS, microglia, and to involve key phagocytosis-associated immune proteins (Veerhuis et al., 2011; Morgan and Harris, 2015). Microglial complement protein C1q is one protein that has been strongly suggested to mediate developmental synaptic pruning (Stevens et al., 2007; Bialas and Stevens, 2013) and neurodegeneration (Stephan et al., 2013; Hong et al., 2016). Both microglial activation (Shapiro et al., 2008) and phagocytosis-related immune molecules (Corriveau et al., 1998) can be directly modulated by neuronal activity. Importantly, CST circuit activity critically controls spinal motor circuit refinement during development and in adults (Martin et al., 2007; Jiang et al., 2013; Smith et al., 2017). How microglia affect transneuronal remodeling and the interplay with neural activity requires further investigation.
To address these questions, we use a rat model with a unilateral pyramidal tract lesion (PTx), in which one-half of the corticospinal projection is completely lesioned in the caudal medulla. This deprives the contralateral spinal cord of most of its corticospinal input. In this way, we isolate remote trans-synaptic changes in the spinal cord from a primary brain injury. We focused on a group of spinal premotor cholinergic interneurons (INs) as the CST neuronal target to investigate anterograde transneuronal changes. Premotor cholinergic INs (Barber et al., 1984) are located in the medial intermediate zone (IZ) and express the Pitx2 gene (Zagoraiou et al., 2009). These INs receive direct CST projections in mature rodents (Jiang et al., 2016), give rise to the cholinergic innervation (C bouton) on motoneurons (Zagoraiou et al., 2009), and modulate motoneuronal excitability through muscarinic type 2 receptors (Huang et al., 2000; Miles et al., 2007).
In this study, we show for the first time that selective CST loss after PTx triggers transneuronal downregulation of the premotor cholinergic innervation in the contralateral spinal cord in adult rats. This neuronal loss is mediated by microglial engulfment and likely requires interneuronal C1q overexpression. The microglia- and complement-associated loss of the cholinergic motoneuronal innervation is replicated by decreasing corticospinal pathway activity. Importantly, spinal cord neuromodulation after injury to augment activation rescued the premotor cholinergic system. Our findings suggest that activity changes critically contribute to anterograde trans-synaptic neuronal loss after CST injury. Manipulating the activity of spared spinal connections may prevent neuronal loss and help to restore restoration function after injury.
Materials and Methods
Animals
Experiments were performed in accordance with the National Institutes of Health's Guidelines for the Care and Use of Laboratory Animals. All animal protocols were approved by the City College of New York institutional animal care and use committee. Adult male Sprague Dawley rats (250–350 g) were housed under a 12 h light/dark cycle in a pathogen-free area with ad libitum water and food. All surgeries were performed under general anesthesia (70 mg/kg ketamine, 6 mg/kg xylazine, i.p.) and aseptic conditions.
Animal models
PTx.
Animals were anesthetized and a small craniotomy was made in the ventral surface of occipital bone to expose pyramid tract at the caudal medullary level. The entire right pyramid was transected as previously reported (Brus-Ramer et al., 2007). The completeness of transection was later confirmed using PKCγ immunofluorescence for CST axons, which shows unilateral loss of dorsal column staining contralateral to the PTx (see Fig. 1A, inset). Animals were studied 3 and 10 d after the lesion in two parallel groups.
Figure 1.

Downregulation of the cholinergic system in the spinal cord after unilateral pyramidal tract lesion. A, Schematic figure showing the location of the pyramidal tract lesion and the correspondence between spinal cord laterality and lesion side. The contralateral side of spinal cord is the denervated side. Inset image shows PKCγ staining of the ventral portion of the spinal cord dorsal column. Note complete loss of staining on the contralateral side. B, Representative staining of ChAT neurons in the control cervical spinal cord showing the location of cholinergic INs and motoneurons. Bottom image shows the region within the dotted rectangle. C, Density heat maps of cholinergic INs for the population of animals examined and representative C bouton images in control and the PTx3 and PTx10 spinal cord. A denser distribution of cholinergic INs can be seen in the medial IZ in both sides of the spinal cord (control), which is substantially less on the lesion side (contra side) at 10 d after PTx (PTx10). The color scale represents 0–4 cells/104 μm2. D, Numbers of cholinergic INs in the medial IZ, as shown in dotted boxes in Figure 1C, were quantified and compared between sides for each groups. The number of neurons in the lesion side of PTx10, but not PTx3, group significantly decreased. The ipsilateral to contralateral ratios indicated a significant decrease at PTx10 (neuronal count, **p = 0.0039; ratio comparison, p = 0.0225; n = 6–8). E, C bouton density significantly decreased in the contralateral spinal cord 3 d after PTx and further downregulated 10 d after PTx. Ipsilateral/contralateral ratios indicated significant decreases at both PTx3 and PTx10 (density, *p = 0.0313; ratio comparison, p = 0.0021. n = 5–6). F, Representative image of NeuN staining in PTx10 cervical spinal cord showing a loss of total neurons. G, NeuN+ neuron counts in the IZ showed a significant decrease in the spinal cord only at 10 d after PTx (*p = 0.0488, n = 4). H, Representative image of PV staining in control cervical spinal cord showing localization of PV+ INs. I, J, Density heat map and neuronal counting of PV+ INs did not show a change in the spinal cord 10 d after PTx. p = 0.375, n = 4. The color scale represents 0–8 cells/104 μm2. K, Numbers of motoneurons did not change 10 d after PTx. n = 8, p = 0.5. Scale in A, B, F, H: 0.2 mm, in C: 20 μm. See Table 1 for mean ± SEM values and Figure 1-1, for raw data. For all other statistical values, see Table 2. Wilcoxon matched-pairs test for within-group comparisons for D, E, G, J, and K, Kruskal–Wallis with Dunn's multiple-comparisons test for ratio comparison in D and E.
Chronic M1 muscimol infusion.
To block motor cortex activity chronically, GABAA receptor agonist muscimol (10 mM in normal saline; Sigma-Aldrich) was continuously infused using an implanted osmotic minipump (Alzet, model 2002, 0.5 μl/h, 10 d; see Fig. 8A) (Martin and Ghez, 1999). The infusion cannula was centered in the M1 forelimb representation ∼2.5 mm lateral to midline and 1 mm anterior to bregma and at a depth of 1.5 mm below the pial surface to target the pyramidal cell layer. The cannula was fixed to the skull with screws and dental acrylic cement and the pump was secured within a subcutaneous pocket between the shoulder blades.
Figure 8.
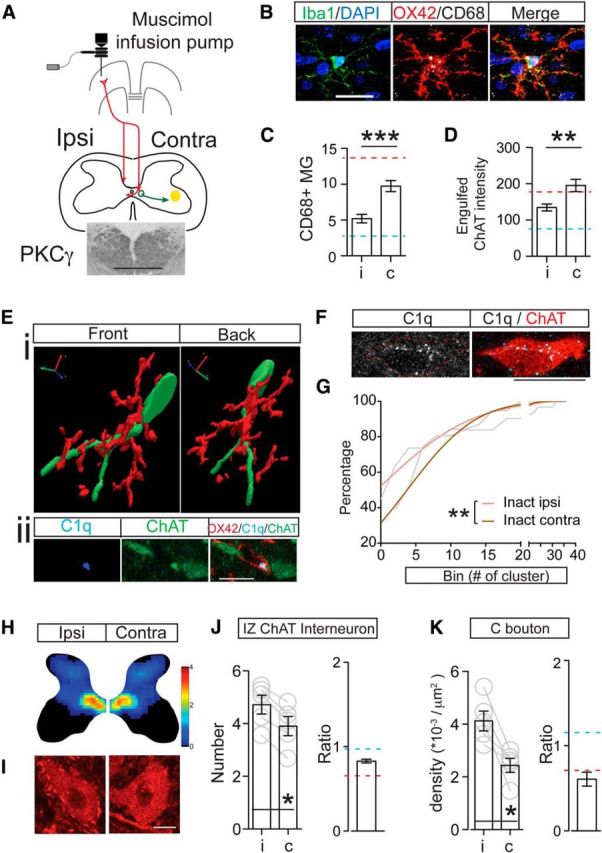
Unilateral motor cortical inactivation induced phagocytic microglial activation, increased neuronal C1q expression, and downregulated the premotor cholinergic innervation. A, Schematic figure showing the method for chronic infusion of the GABAA receptor agonist muscimol into the motor cortex using an implanted osmotic pump and intracortical cannula. Inset image shows symmetric PKCγ staining in the dorsal columns bilaterally, indicating no axon loss due to cortical inactivation. B, Images of individual Iba-1+ microglia showing increased OX42 and CD68 intensity after cortical inactivation. C, D, Numbers of CD68+ phagocytic microglia and optical intensity of engulfed ChAT clusters in microglial cells increased in medial IZ in animals receiving motor cortex inactivation. Mann–Whitney U test, **p < 0.01, ***p < 0.001. n = 10–11 sections for C, n = 30–33 cells for D. E, 3D reconstruction shows wrapping of a cholinergic IN by a microglial cell (Ei). Single optical images in Eii indicate ongoing engulfment as shown by the presence of a C1q+/ChAT+ cluster in the microglial cell that is contacting the neuron. F, Representative images showing increased C1q expression in a cholinergic IN. G, Cumulative distribution curves of C1q cluster numbers within cholinergic INs showing a significant rightward shift in cluster number on the inactivated side. Gray lines are original curves and colored lines are after fitting to a cumulative Gaussian frequency distribution (two-way ANOVA, **p < 0.01, n = 31–37). H, J, Density heat map and cell counts of cholinergic INs in the spinal cord medial IZ in muscimol inactivation animals showing a significant decrease. The color scale represents 0–4 cells/104 μm2. I, K, C bouton density also showed a significant contralateral loss after inactivation. Contralateral to ipsilateral ratios are much lower than those of control. Wilcoxon matched-pairs test, p = 0.0313, n = 5. The red dotted line indicates the mean value of PTx10 animals and the blue dotted line indicates the mean value of controls. Scale bars: A:, 0.2 mm; B, Eii, F, 10 μm; I, 20 μm. See Table 1 for mean ± SEM and Figure 8-1, for raw data. For all other statistical values, see Table 2.
Systemic minocycline administration.
Minocycline hydrochloride, a relatively specific inhibitor of microglia activation (Garrido-Mesa et al., 2013), was administered to PTx rats daily for 10 d (45 mg/kg, pH adjusted to 7.4, i.p.; Sigma-Aldrich). The first dose was given immediately after PTx.
Cathodal trans-spinal direct current stimulation (c-tsDCS).
c-tsDCS was delivered through hydrogel electrodes on the skin surface with the cathode over the C4–T2 vertebrae and the anode over the chest (0.5 × 1.5 inches; StimTent; see Fig. 9A) so that the maximal current density was induced within the cervical enlargement as predicted by finite element method modeling (Song et al., 2015). The stimulation was delivered using a programmable waveform/pulse generator (A-M Systems, model 3800) and stimulus isolation units (A-M Systems, model 3820). The current was ramped from 0 to −1.5 mA (3 × 10−3 mA/mm2) over 2.5 s and returned to 0 over 2.5 s after 27 min of stimulation. C-tsDCS was started the day after PTx surgery and continued daily for 10 d. Rats were stimulated in their home cages and showed no signs of distress during the experiments.
Figure 9.

Promoting spinal activity through c-tsDCS rescues downregulation of cholinergic pathway. A, Schematic figure showing c-tsDCS waveform and placement of the electrodes. Note that the cathode is placed on the dorsal neck skin and the anode is on the ventral surface. B, Density heat map of cholinergic INs and C boutons on motoneurons showing no difference after c-tsDCS in PTx animals. Scale bar, 20 μm. Color scale represents 0–4 cells/104 μm2. C, D, Number of IZ cholinergic INs (C) and C bouton density on motoneurons (D) did not show a significant difference bilaterally (Wilcoxon matched-pairs test: C, p = 0.1563; D, p = 0.5; n = 5). Furthermore, the contralateral to ipsilateral ratios for neurons (C) and C boutons (D) are close to 1. See Table 1 for mean ± SEM values and Figure 9-1, for raw data. For all other statistical values, see Table 2.
Tissue preparation and staining
Animals were deeply anesthetized and perfused with saline followed by 4% paraformaldehyde. Frozen sections were cut transversely (40 μm). Primary antibodies that were used for immunofluorescence staining include the following: goat anti-choline acetyltransferase (ChAT, 1:100; Millipore catalog #AB144P, RRID:AB_2079751), mouse anti-NeuN (1:100; Millipore catalog #MAB377, RRID:AB_2298772), rabbit anti-NeuN (1:500; Abcam catalog #ab104225, RRID:AB_10711153), mouse anti-parvalbumin (PV, 1:5000; Swant catalog #235, RRID:AB_10000343), rabbit anti-Iba-1 (1:1000, Wako catalog #019-19471, RRID:AB_2665520), mouse anti-OX42 (1:1000; Bio-Rad/AbD Serotec catalog #MCA275FA, RRID:AB_2129486), rat anti-CD68 (1:1000; Bio-Rad/AbD Serotec catalog #MCA1957, RRID:AB_322219), rabbit anti-C1q (1:1000; Abcam catalog #ab182451, RRID:AB_2732849), mouse anti-PKCγ (1:1000; Santa Cruz Biotechnology catalog #sc-166451, RRID:AB_2168997), rabbit anti-cleaved Caspase 3 (1:400; Abcam catalog #ab52294, RRID:AB_868672). Secondary antibodies are as follows: donkey anti-goat, donkey anti-mouse, donkey anti-rat, and donkey anti-rabbit IgG conjugated to FITC, Cy3, or Cy5 depending on the combination of colabeling. Sections were visualized and digitally imaged with a Zeiss LSM 880 meta confocal microscope using the standard and Airyscan modes. Whenever possible, we coprocessed all tissues together and performed microscopic analyses using the same confocal imaging parameters. All measurements were made by laboratory personnel blinded to the animal groups.
TUNEL assay
Floating spinal cord sections (40 μm) from PTx3 and PTx10 animals were divided into control and experiment groups. Control sections were incubated in DNase I recombinant (20 μg/ml, 0.01% Triton X-100, 0.01 m PBS) and experimental sections were incubated in buffer solution (0.01% Triton X-100, 0.01 m PBS) for 10 min at room temperature. Sections were washed with PBS twice and then all sections were incubated in TUNEL reaction solution (Roche) for 1 h at 37°C following the manufacturer's instructions. After this, sections were washed three times in PBS and mounted for confocal imaging.
Anatomical data acquisition and analysis
Spinal neuron counting and density heat maps.
Cholinergic INs, PV INs, and all spinal cord neurons were identified using ChAT, PV, and NeuN antibodies, respectively, for immunostaining (see Fig 1B,F,H). Positively stained ChAT and PV INs were marked using Neurolucida (MBF, RRID:SCR_001775) at the corresponding gray matter location to create TIFF files with each neuron marked by a single pixel, which were then quantified using custom programs written in MATLAB (The MathWorks, RRID:SCR_001622) to construct heat map representations of ChAT and PV IN density. These representations are intended to provide a topographic representation of the relative changes in cell density, in particular the control versus the affected side. Quantification and statistical determinations of changes in IN numbers are based on the absolute counts within the region of interest (ROI) where the cells are present. Output files with neuron markers were divided into 52 × 52 μm square ROIs. The distribution pattern, which depicts local cell density, is represented according to a color scale from the lowest (blue, represents close to 0) to the highest (dark red/maroon, represents ∼4 cells/104 μm2) neuron density. Black codes for no labeling. NeuN+ neurons were only counted within the area of medial IZ (212.5 × 212.5 μm), where the majority of ChAT INs are located (Fig. 1F). For neuronal counting, we analyzed eight to 15 sections for each animal and four to eight animals for each group.
C bouton density analysis.
Cholinergic immunolabeling associated with C boutons was captured at 40× magnification as z stacks (1 μm optical section thickness) containing individual ChAT+ motoneurons using confocal microscopy. The z-stack images were transferred to Neurolucida (RRID:SCR_001775), outlines were drawn around motoneuron somas, and C boutons were marked at each plane along the z stack. The tracing files were then analyzed in Neurolucida explorer to obtain estimates of soma surface area and total bouton numbers. The numbers of C boutons on individual motoneurons were divided by the surface areas of the corresponding motoneurons to obtain C bouton density. Four to six animals for each group and six to 10 motoneurons from each side of each animal were randomly chosen for the C bouton analysis.
Phagocytic microglia counting.
Phagocytic microglia were identified by immunolabeling with OX42. Colabeling of OX42 and the lysosomal marker CD68 was used to image the successful progress from microglial phagocytosis to degradation. Due to weak OX42 signal in control tissue, the counting was verified in a subset of four animals by Iba1, OX42, and CD68 triple labeling. Quantitative assessment indicated that the number of triple-labeled microglia corresponded to OX42 single-labeled microglia (data not shown). Images of 212.5 × 212.5 μm dimensions within the medial IZ were taken at 40× using the confocal microscope. Cells were counted within the same ROI using consistent thresholding for all images. Images were taken from three to four animals in each group and two to three sections from each animal for microglia counting. The total numbers of microglial cells that were analyzed ranged from 16 to 25 for each animal.
Microglia engulfment quantification.
Intensity of CD68+ and ChAT+ clusters within microglia was obtained using ImageJ (RRID:SCR_003070). Images were thresholded using consistent values and only clusters with ≥3 pixels were circled for optical intensity analysis. For clusters occupying several optical slices, we only analyzed the slice containing the largest cluster area. If multiple clusters appeared in one microglia, intensity of all clusters was summed to represent the value of the corresponding microglial cell. Images were taken from three animals in each group and eight to 10 microglia from each animal were analyzed. The total numbers of microglia cells that were analyzed ranged from 18 to 30 for each animal.
C1q quantification.
C1q signal was thresholded the same way across all images and the number of clusters with ≥3 pixels within IZ cholinergic INs were counted. C1q signal within microglia was compared using the optical intensity value obtained using ImageJ. Images were taken from three animals in each group and five to six sections from each animal were examined. The total neuronal numbers that were analyzed ranged from 17 to 59 for each animal.
3D reconstruction.
z-stack images of neuron–glia interactions were captured using the Airyscan function. Contours of cholinergic INs and microglial cells were drawn and reconstructed using Neurolucida (MBF, RRID:SCR_001775).
Experimental design and statistics
Statistical analyses were performed using Prism 5 (GraphPad, RRID:SCR_002798). The comparisons of neuron counts and C bouton density were conducted between the two sides of spinal cord within individual groups using Wilcoxon matched-pairs test; the ratios were compared between groups using Kruskal–Wallis test with Dunn's multiple comparisons. The properties of individual cells for a single group were compared between two sides of spinal cord primarily using the Mann–Whitney U test. Microglial C1q intensity data were normally distributed and were compared with an unpaired t test and one-way ANOVA with Bonferroni's multiple-comparisons test. Comparison of cumulative distributions of C1q cluster numbers within cholinergic INs was performed using two-way repeated-measures ANOVA. All data are presented as mean ± SEM, as shown in Table 1, and the significance was determined at p < 0.05. Statistical values are shown in Table 2 or in the corresponding text. All analyses were conducted blinded to the treatment group.
Table 1.
Data presented as mean ± SEM
| Control |
PTx3 |
PTx10 |
PTx10 + minocycline |
Inactivation |
PTx10 + tsDCs |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ipsilateral | Contralateral | Ipsilateral | Contralateral | Ipsilateral | Contralateral | Ipsilateral | Contralateral | Ipsilateral | Contralateral | Ipsilateral | Contralateral | |
| No. of IZ ChAT+ INs | 4.8 ± 0.3 | 4.5 ± 0.4 | 4.0 ± 0.3 | 4.0 ± 0.6 | 5.1 ± 0.2 | 3.4 ± 0.2 | 3.7 ± 0.7 | 3.8 ± 0.7 | 4.7 ± 0.4 | 3.9 ± 0.4 | 5.7 ± 0.3 | 5.1 ± 0.2 |
| Ratio of contralateral to ipsilateral INs (no.) | 0.96 ± 0.09 | 1.01 ± 0.14 | 0.65 ± 0.04 | 1.07 ± 0.15 | 0.82 ± 0.02 | 0.93 ± 0.06 | ||||||
| C bouton density (no. *10−3/μm2 ) | 3.9 ± 0.5 | 4.4 ± 0.3 | 4.8 ± 0.4 | 4.3 ± 0.4 | 4.5 ± 0.2 | 3.2 ± 0.2 | 3.7 ± 0.3 | 3.7 ± 0.3 | 4.1 ± 0.4 | 2.4 ± 0.3 | 5.2 ± 0.4 | 5.2 ± 0.4 |
| Ratio of contralateral to ipsilateral bouton density | 1.15 ± 0.08 | 0.90 ± 0.02 | 0.71 ± 0.01 | 1.03 ± 0.09 | 0.61 ± 0.08 | 1.03 ± 0.12 | ||||||
| No. of IZ NeuN+ neurons | 48.2 ± 2.2 | 49.5 ± 1.8 | 45.5 ± 1.9 | 37.6 ± 2.6 | ||||||||
| No. of motoneurons | 22.8 ± 1.0 | 22.7 ± 0.9 | ||||||||||
| No. of PV+ INs | 22.3 ± 1.1 | 22.7 ± 1.4 | ||||||||||
| No. of CD68+ microglia in IZ | 3.3 ± 0.6 | 2.7 ± 0.6 | 5.4 ± 0.9 | 5.4 ± 0.8 | 8.7 ± 0.6 | 13.7 ± 1.7 | 5.0 ± 1.0 | 5.0 ± 1.1 | 5.2 ± 0.6 | 9.7 ± 0.8 | ||
| Intensity of microglia CD68 cluster | 260.6 ± 21.9 | 225.8 ± 31.2 | 162.9 ± 21.3 | 195.5 ± 29.0 | 387.4 ± 34.8 | 529.7 ± 53.9 | 184.8 ± 22.0 | 211.3 ± 25.3 | 267.2 ± 26.4 | 371.4 ± 52.9 | ||
| Intensity of engulfed ChAT | 83.22 ± 8.9 | 76.0 ± 7.1 | 95.0 ± 8.2 | 119.7 ± 15.1 | 150.2 ± 15.4 | 177.6 ± 15.1 | 132.5 ± 15.2 | 150.2 ± 13.9 | 134.6 ± 9.5 | 195.4 ± 17.0 | ||
| Intensity of microglial C1q | 18.7 ± 3.6 | 21.5 ± 3.3 | 34.7 ± 5.2 | 38.1 ± 4.9 | 48.4 ± 4.8 | 65.8 ± 5.9 | 26.0 ± 3.2 | 28.9 ± 3.0 | 43.1 ± 5.0 | 48.0 ± 5.7 | ||
Table 2.
Wilcoxon matched-pairs test within each group
| Control | PTx3 | PTx10 | PTx10 + minocycline | Inactivation | PTx10 + tsDCs | |
|---|---|---|---|---|---|---|
| No. of IZ ChAT+ INs | W = 5.00, p = 0.3125 | W = 5.00, p = 0.5 | W = 0.00, p = 0.0039** | W = 3.00, p = 0.5 | W = 0.00, p = 0.0313* | W = 3.00, p = 0.1563 |
| C bouton density | W = −2.00, p = 0.0938 | W = 0.00, p = 0.0625 | W = 0.00, p = 0.0313* | W = 5.00, p = 0.5 | W = 0.00, p = 0.0313* | W = −7.00, p = 0.5 |
| No. of IZ NeuN+ neurons | W = −0.00, p = 0.1346 | W = 1.5, p = 0.0488* | ||||
| No. of motoneurons | W = 18.00, p = 0.5 | |||||
| No. of PV+ INs | W = −2.00, p = 0.375 | |||||
| Mann–Whitney U test within each group | ||||||
| No. of CD68+ microglia | U = 17.5, p = 0.5 | U = 32, p = 0.4789 | U = 3.0, p = 0.005** | U = 17.0, p = 0.4676 | U = 6.5, p = 0.0003*** | |
| Intensity of microglial CD68 clusters | U = 168.0, p = 009545 | U = 123.0,p = 0.2397 | U = 124.0, p = 0.0229* | U = 202.0, p = 0.2443 | ||
| Intensity of engulfed ChAT | U = 164.0, p = 0.3252 | U = 159.0, p = 0.203 | U = 228.0, p = 0.0426* | U = 245.0, p = 0.1029 | U = 295.0, p = 0.003** | |
| Unpaired t test within each group | ||||||
| Intensity of microglial C1q | t(50) = 0.56, p = 0.5725 | t(43) = 0.45, p = 0.6499 | t(112) = 2.246, p = 0.0266* | t(89) = 0.66, p = 0.5111 | t(88) = 0.65, p = 0.5147 |
*p < 0.05;
**p < 0.01;
***p < 0.001.
Results
Downregulation of spinal premotor cholinergic innervation after unilateral corticospinal tract lesion
Typical ChAT staining at the seventh cervical level (C7) shows, in addition to motoneurons in the motor pools, a cluster of ChAT+ INs in the medial IZ, adjacent to central canal (Fig. 1B, dotted rectangle and enlargement in below; Barber et al., 1984). These INs in the medial IZ are likely Pitx2+ premotor cholinergic INs that were identified in mice (Zagoraiou et al., 2009), which give rise to C boutons that synapse on motoneuron cell bodies and proximal dendrites (Miles et al., 2007). Additional ChAT+ INs are sparsely scattered throughout the gray matter. We counted all cholinergic INs in the C6–C7 spinal cord of control, 3 d postinjury (PTx3), and 10 d postinjury (PTx10) rats and reconstructed regional cell density heat maps (Fig. 1C). Spinal cords from both control and PTx3 rats showed a symmetrical distribution of cholinergic INs in the medial IZ. In PTx10 rats, there was a remarkable decrease in neuron density in the medial IZ on the denervated side; ChAT IN density in the dorsal and ventral horns did not seem to change. We also examined C boutons on motoneurons in the lateral motor pool (Fig. 1C, insets). C boutons can be easily identified as bright and large ChAT+ clusters that contact motoneuron soma and proximal dendrites. The micrographs show that the number of C boutons is slightly fewer in contralateral PTx3 spinal cord and is further decreased in PTx10 spinal cord. We quantified and plotted the number of cholinergic INs in the medial IZ. The area of interest is shown in Figure 1C as a dotted rectangle, and the results of the counting analysis is shown in Figure 1D. The mean raw values are shown in Table 1 and raw values in Figure 1-1. We chose this area based on the known location of the Pitx2+ premotor cholinergic IN class (Zagoraiou et al., 2009). We also quantified the C bouton density (Fig. 1E) of all groups (data values are shown in Table 1 and Fig. 1-1). Compared with the normal variation of both ChAT+ IN numbers and C bouton densities in individual control animals, PTx3 animals showed no significant changes in either IN numbers or C bouton density. By contrast, PTx10 animals showed consistent reductions in both IZ ChAT+ IN numbers (∼35% decrease, Wilcoxon matched-pairs test, p = 0.0039) and C bouton density (∼29% decrease, Wilcoxon matched-pairs test, p = 0.0313) on the contralateral (CST-denervated) side compared with the ipsilateral side. The ratio of contralateral to ipsilateral values in Figure 1, D and E, demonstrate the gradual decrease of cholinergic innervation from 3 to 10 d after PTx (Kruskal–Wallis test with Dunn's multiple-comparisons test; Fig. 1D: H = 7.592, #p = 0.0225; Fig. 1E: H = 12.38, p = 0.0021, ctrl-PTx10, ##p < 0.01). We then quantified total neuronal numbers in the IZ using NeuN as pan-neuronal marker. We found significantly fewer (∼17%) NeuN+ neurons in the contralateral side at 10 d, but not at 3 d, after PTx (Wilcoxon matched-pairs test, PTx10: p = 0.0488; Fig. 1F,G; mean values are in Table 1 and the raw values are in Fig. 1-1). The decrease in number of total neurons in the IZ with NeuN staining is consistent with the reduction in cholinergic IN number, which implies a true neuronal loss induced by the remote injury or possibly severe neuronal stress (Duan et al., 2016).
Raw data for bar graphs in Figure 1. Fig. 1D. Cholinergic interneuron counts and ratios. Fig. 1E. C bouton density and ratios. Fig. 1G. NeuN+ neuronal counts. Fig. 1J. Parvalbumin interneuron counts. Fig. 1K. Motoneuron counts. Download Figure 1-1, XLSX file (43.1KB, xlsx)
To determine the specificity of this neuronal loss, we also looked at two other spinal neuron classes: PV+ INs and motoneurons. The distribution of PV+ INs (Fig. 1H) shows a relatively higher density of neurons in the medial dorsal horn, with others scattered ventrally close to the motor pools, including Ia inhibitory INs and Renshaw cells (Alvarez et al., 2005). PV+ IN density (Fig. 1I) and total number (Fig. 1J, Table 1) were not significantly different in PTx10 rats (Wilcoxon matched-pairs test, p = 0.375), which is when we see significant ChAT+ IN loss. We also did not find a significant change in cervical motoneuron numbers of PTx10 animals (Wilcoxon matched-pairs test, p = 0.5; Fig. 1K, Table 1; raw values are in Fig. 1-1). These findings indicate that a unilateral medullary lesion of the corticospinal projection produces a progressive decrease in the premotor cholinergic system. The lack of an effect of the lesion on motoneurons and PV+ INs suggests specificity of the downregulation. IN specificity may be due to a paucity of monosynaptic CST projections on PV+ INs (Brownstone and Bui, 2010) and motoneurons in rodents (Liang et al., 1991; Yang and Lemon, 2003).
Neuronal loss typically is preceded by different kinds of neuronal death, especially apoptosis (Fricker et al., 2018). We therefore aimed to determine whether spinal cholinergic INs could undergo apoptosis after PTx. We first used the early apoptotic marker cleaved caspase 3 (c-casp 3). Importantly, we did not detect any positive signal with c-casp 3 (Fig. 2A). To verify that our assay was working, we stained the motor cortex that is ipsilateral to the PTx for c-casp3 and NeuN+ 10 d after injury (Fig. 2B), which is when we would expect abundant cell death after pyramidal axotomy (Hains et al., 2003). As expected, we saw many double-labeled neurons, consistent with retrograde neuronal apoptosis induced by axotomy (Hains et al., 2003). This indicates that our c-casp3 assay was working and the negative finding in the spinal cord suggests the absence of apoptosis. We further investigated apoptosis in the spinal cord using TUNEL staining in both PTx3 (Fig. 2A) and PTx10 (data not shown) animals. Again, we did not observe any staining. To verify efficacy of the TUNEL assay, we performed a positive control (DNase I recombinant pretreated) in the spinal cord of PTx animals, which revealed staining (Fig. 2A). Together, our findings show that eliminating the CST induced a substantial decrease in the premotor cholinergic innervation. This cell loss is not preceded by neuronal apoptosis, similar to the findings in other in vivo and in vitro study (Cunningham et al., 2013; Neher et al., 2014).
Figure 2.

Neuronal apoptosis is not detected in the spinal cord after PTx. A, Spinal cords of PTx3 animals had no detectable signal with either active caspase 3 or TUNEL staining. The TUNEL assay was effective in revealing DNA degradation in tissue pretreated with DNase recombinase. Scale bar, 0.2 mm. B, Neurons in ipsilateral motor cortex showed apoptosis at 10 d after PTx using cleaved (active) caspase 3 staining, validating the immunohistochemical assay. Scale bar, 20 μm.
Activation of phagocytic microglia in the denervated spinal cord
Microglia are the only resident immune cells in the CNS and they show rapid activation and phagocytic function, mostly at the primary injury site after CNS insult (Kumar and Loane, 2012). To investigate how cervical microglia respond to remote CST injury (PTx), we stained the spinal cord with Iba1, a constitutively expressed calcium-binding adapter molecule and general microglial marker, and OX42, which recognizes the complement iC3b receptor. iC3b binding is known to lead to phagocytosis by C3R/OX42-expressing cells (Underhill and Ozinsky, 2002). OX42 can thus indicate the potential of microglia phagocytosis. The contralateral ventral dorsal column showed increased Iba1 and OX42 signal (Fig. 3A), which obviously directly results from CST axotomy and axonal degeneration. In the gray matter, Iba-1 showed a similar expression pattern and intensity across all conditions (Fig. 3A, left column), thus suggesting the lack of microglial proliferation. In contrast, the level of OX42 showed a strong increase throughout the spinal gray and white matter of PTx3 rats (Fig. 3A, right column, middle) compared with very weak expression in control animals (Fig. 3A, right column, top). The contralateral and ipsilateral sides at PTx3 are not particularly different, which is likely due to the bilateral projection of CST axons. PTx10 rats (Fig. 3A, right column bottom) still show stronger OX42 signal than controls, especially on the contralateral side, which is, however, weaker than that of PTx3. The upregulation of OX42 expression in microglia therefore strongly implies increased microglial phagocytic potential in the spinal cord. Interestingly, OX42 expression is strongly bilateral, suggesting a sensitive response to the bilateral loss of CST axons throughout the spinal cord despite the stronger contralateral projection.
Figure 3.

Microglia activated in response to unilateral PTx. A, Overview of Iba-1- and OX42-labeled microglial cell expression in the spinal cord of control, PTx3, and PTx10 animals. Note the much higher OX42, but not Iba-1, intensity in the affected dorsal column at both 3 and 10 d after injury. B, Images of individual Iba-1+ microglial cells showing increased OX42 and CD68 intensity after PTx. Arrows point to CD68+ clusters within microglial cells. C, Numbers of CD68+ microglia in medial IZ. **p = 0.005, n = 6–8 sections. D, Optical intensity of CD68+ clusters in microglia. *p = 0.0229, n = 16–22 cells. Mann–Whitney U test for within-group comparison. Scale bars: A, 0.2 mm; B, 10 μm. See Table 1 for mean ± SEM values and Figure 3-1, for raw data. For all other statistical values, see Table 2.
Raw data for bar graphs in Figure 3. Fig. 3C. CD68+ microglia counts. Fig. 3D. Optical intensity of microglial CD68+ clusters. Download Figure 3-1, XLSX file (42KB, xlsx)
Phagocytosis led by iC3b binding of OX42/C3R is typically followed and completed by lysosome degradation of phagocytosed targets. The level of lysosome expression thus implies the successful progress of microglial phagocytosis. We next examined expression of the lysosome marker CD68 in the spinal cord. CD68 signal is quite weak in the IZ of the control spinal cord (Fig. 3B, top). At PTx3, it started to appear as clusters (Fig. 3B, middle, arrow). The clusters are commonly detected within the microglia soma with both Iba1+ and OX42+ staining. At 10 d after injury, the intensity of CD68 clusters showed a larger increase in microglial cells (Fig. 3B, bottom, arrows). We counted CD68+ microglial cells in the IZ of the spinal cord and analyzed the intensity of CD68 clusters in individual microglia. We found a significant contralateral (i.e., denervated side) increase at PTx10 for both values (Mann–Whitney U test within groups, Fig. 3C, PTx10: p = 0.005; and Fig. 3D, PTx10: p = 0.0229; values in Table 1; raw values are in Fig. 3-1. The presence of significantly more CD68+ microglial cells and stronger cluster intensities on the contralateral side reflects that microglial phagocytosis has gradually and successfully progressed in the spinal cord, especially the contralateral side, which has the greatest extent of CST axonal denervation.
Progressive microglial phagocytosis of cholinergic IN fragments in the denervated spinal cord
Loss of cholinergic INs and the absence of apoptosis led us to determine whether the INs are targeted by phagocytic microglial cells. Double labeling of ChAT with OX42 in the spinal cord reveals very little spatial interaction between microglia and cholinergic INs (Fig. 4A, top). By contrast, at 3 d after injury, we frequently detected close contact between microglia and cholinergic INs. As shown in the representative example (Fig. 4A, middle, arrowheads), the processes of activated microglial cells typically wrap around the soma (stars) and dendrites (rectangle and enlarged in Fig. 4Aa) of nearly 54.2 ± 6.8% of IZ cholinergic INs analyzed in contralateral side of PTx3 spinal cord. Recall that, at this time point, there is no significant cell loss. Ten days after PTx, the close contact between ChAT INs and microglial cells are not detected as frequently as at 3 d (Fig. 4A, bottom) because there is now abundant cell loss. Instead, large ChAT+ clusters appeared within microglia (Fig. 4A, bottom, arrows, and enlarged in Fig. 4Ab). Because of their large size and colocalization within OX42 microglia, we interpret these as engulfed fragments of cholinergic IN. Typically, in the PTx10 spinal cord, these internalized ChAT+ clusters also were positively labeled by immunostaining for CD68+ and located beside DAPI+ nuclei within microglia soma (Fig. 4B, arrows), which indicates the start of lysosomal degradation of cholinergic tissue fragments. We analyzed the intensity of ChAT+ clusters inside microglia in the IZ to assay microglial phagocytosis at different stages after PTx. Similar to the CD68 cluster analysis, the mean intensities of internalized ChAT+ clusters did not show a significant upregulation until 10 d. The contralateral side showed a significantly higher mean value compared with the ipsilateral side at PTx10 and with control rats, which suggests extensive internalization/engulfment of cholinergic neuronal fragments in the contralateral spinal cord (Mann–Whitney U test, PTx10: p = 0.0426; Fig. 4C, mean values in Table 1; raw values are in Fig. 4-1). Engulfed ChAT clusters also showed an increase on the ipsilateral side of the PTx10 spinal cord, which may reflect the bilateral projection of premotor cholinergic INs (Stepien et al., 2010). These data together suggest that active engulfment of cholinergic neurons by phagocytic microglia may lead to downregulation of the spinal cholinergic system (Fig. 4D, red arrow).
Figure 4.
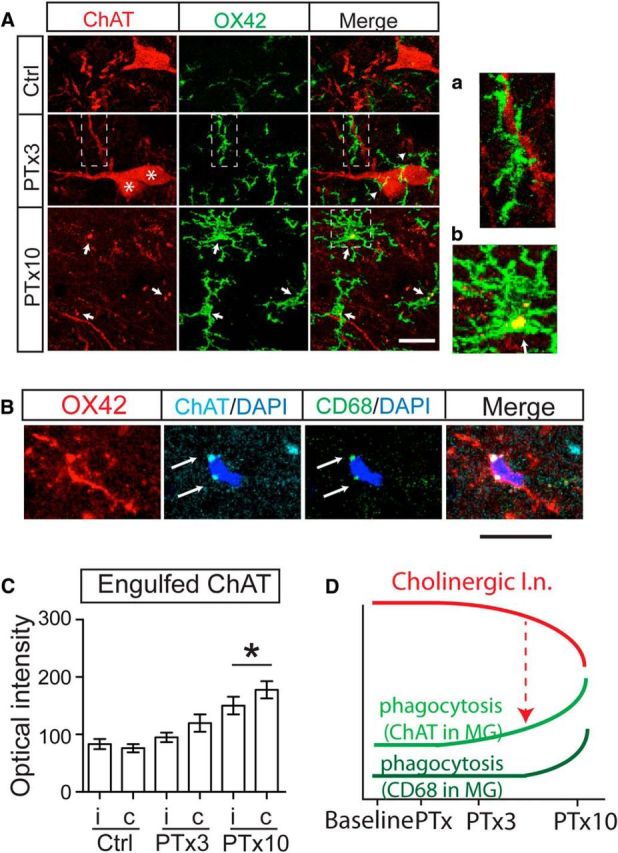
Microglial phagocytosis of cholinergic IN fragments increased in response to unilateral PTx. A, Images show microglia wrapping (arrowheads) cholinergic IN somata (stars) and dendrites (rectangle) at PTx3, and internalization of ChAT fragments at PTx10 (arrows). a. Enlarged image of rectangular area in PTx3 images. b. Enlarged image of square area in PTx10 images. B, Representative images showing ChAT+/ CD68+ clusters within one IZ microglial cell in the PTx10 spinal cord. C, Optical intensity of engulfed ChAT clusters in microglia in the medial IZ. Mann–Whitney U test for within group comparison, *p = 0.0426, n = 18–30 cells. D, Schematic summary of changes in the cholinergic pathway and phagocytic microglia. The parallel but opposite changes in the cholinergic pathway (red curve; i.e., ChAT INs and C boutons) compared with phagocytosis associated with microglial ChAT (light green curve) and CD68 (dark green curve), suggesting phagocytosis of ChAT neurons (red dotted arrow). Scale in A and B: 10 μm. See Table 1 for mean ± SEM values and Figure 4-1, for raw data. For all other statistical values, see Table 2.
Raw data for bar graphs in Figure 4C. Optical intensity of engulfed ChAT+ clusters. Download Figure 4-1, XLSX file (72.7KB, xlsx)
Minocycline inhibition of microglial function prevents microglial engulfment of INs and rescues premotor cholinergic system downregulation
The findings we have presented so far show that phagocytic microglial cells are associated with cholinergic neuronal loss in the spinal cord after PTx. To determine whether phagocytic microglia are directly involved in this transneuronal remodeling, we next applied minocycline, a commonly used tetracycline antibiotic and a relatively specific microglial inhibitor, and determined CST lesion-dependent changes to the spinal cholinergic system. We first verified microglial activity in control and PTx10 animals using Iba-1 and OX42 as indices (Fig. 5A, top two rows). Minocycline treatment (Fig. 5A, bottom row), as we expected, markedly reduced both Iba-1 and OX42 intensity in the contralateral IZ compared with that of the untreated PTx10 spinal cord (Fig. 5A, middle row). Importantly, the increase in the number of CD68+ microglia (Fig. 5C), the elevation in the intracellular CD68 level (Fig. 5D), and engulfed ChAT in individual microglia (Fig. 5E) were largely prevented in the IZ (see Table 1; Mann–Whitney U test, Fig. 5C: p = 0.4676; Fig. 5D: p = 0.2443; Fig. 5E: p = 0.1029). The values are similar to those of controls and are substantially lower than that of contralateral spinal cord at PTx10 (Fig. 5C–E, blue dotted line for control, red dotted lines for PTx10; mean values are in Table 1 and raw values are in Fig. 5-1). These findings strongly suggest that chronic minocycline effectively inhibited microglial phagocytosis, including cholinergic fragments, induced by remote injury.
Figure 5.

Systemic minocycline prevents microglial phagocytosis and rescues the downregulation of the cholinergic system after PTx. A, OX42, but not Iba-1, signal in microglia was substantially depressed in the IZ of the minocycline-treated spinal cord compared with that of untreated PTx10 spinal cord. B, Images of individual Iba-1+ microglial cells showed suppressed expression of OX42 and CD68 after minocycline treatment. C, Numbers of CD68+ phagocytic microglia in the medial IZ of minocycline-treated animals showing no increase. D, E, Optical intensity of CD68 and engulfed ChAT clusters in microglial cells in the medial IZ did not show the expected increase after injury. C–E, Mann–Whitney U test, C; p = 0.4676, n = 6 sections, D: p = 0.2443, E: p = 0.1029, n = 21–27 cells. The red dotted line indicates the mean value of PTx10 contralateral and the blue dotted line indicates the mean value of controls. F–H, Cholinergic IN distribution heat map and neuronal counting in the medial IZ showing a symmetric distribution pattern in the spinal cord of minocycline-treated animals, similar to that of control animals. The characteristic decrease in C bouton density after PTx also was blocked with minocycline treatment. G, H, Wilcoxon matched-pairs test, p = 0.5, n = 4. The color scale represents 0–4 cells/104 μm2. I, Schematic summary of changes in the cholinergic pathway and phagocytosis after minocycline treatment. With minocycline prevention of microglial phagocytosis (microglial ChAT/CD68), the cholinergic pathway (including INs and C boutons) was rescued from downregulation (broken red dotted arrow). Scale bars: A, 0.2 mm; B, F, 10 μm. See Table 1 for mean ± SEM values and Figure 5-1, for raw data. For all other statistical values, see Table 2.
Raw data for bar graphs in Figure 5. Fig. 5C. CD68+ microglia counts. Fig. 5D. Optical intensity of microglial CD68+ clusters. Fig. 5G. Cholinergic interneuron counts and ratios. Fig. 5H. C bouton density and ratios. Download Figure 5-1, XLSX file (36.4KB, xlsx)
We then examined cholinergic INs in the medial IZ and C boutons in PTx10 animals with chronic minocycline treatment. Indeed, minocycline abrogated the decrease in both ChAT+ IN numbers and C bouton density in the contralateral compared with the ipsilateral sides (paired t test, n = 4, p > 0.05; Fig. 5F–H, Table 1; raw values are in Fig. 5-1). The rescue of the cholinergic system by minocycline strongly suggests that phagocytic microglial cells contribute to the elimination of the premotor cholinergic pathway after remote corticospinal axon lesion. Without microglial phagocytosis, the premotor cholinergic system was protected from the lesion-dependent transneuronal downregulation (Fig. 5I, interrupted red arrow).
Neuronal C1q showed early and unilateral upregulation in CST denervated spinal cord
We next investigated whether the complement protein C1q changed in association with transneuronal remodeling of the cholinergic pathway. Figure 6A shows representative Airyscan high-resolution images with colabeling of C1q, ChAT, and OX42 in the contralateral medial IZ from all animal groups. In control tissue (Fig. 6A, top row), the C1q signal is barely differentiable from background staining. This is consistent with a low basal expression level in the adult brain (Stephan et al., 2013). In the PTx3 spinal cord (Fig. 6A, second row), however, C1q signal showed a remarkable increase. Dense C1q clusters were especially prominent in ChAT+ INs (enlarged in Fig. 6Aa). In contrast, the C1q signal is lower at PTx10 (Fig. 6A, third row) than in PTx3 and is comparable to controls.
Figure 6.

C1q is upregulated in the contralateral spinal cord after PTx. A, Colabeling of C1q, OX42, and ChAT in the contralateral medial IZ in control, PTx3, PTx10, and PTx10 + minocycline spinal cords. a and b are enlarged images of two cholinergic INs from the PTx3 and minocycline images. Scale bar, 10 μm. B, Cumulative distribution curves of C1q cluster numbers within individual cholinergic INs. Gray lines are original curves, colored lines are curves fitted to a cumulative Gaussian frequency distribution. Bi, Comparison of C1q cumulative curves of the contralateral side of PTx animals with controls. Note the rightward shift of the PTx3 curve and return of PTx10 curve to the control distribution. Minocycline treatment partially rescued the C1q increase in INs. Bii, Comparison of C1q cumulative curves between the two sides within each group. Note the unilateral rightward shift in PTx3 and PTx10 + minocycline groups. Two-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001). C, Representative images of microglial C1q in different groups and optical intensity of microglial C1q clusters (top). C1q expression in microglia (bar graphs; bottom) is significantly increased 10 d after PTx, which is completely prevented by minocycline treatment. Unpaired t test within groups, *p = 0.0266. One-way ANOVA with Bonferroni's posttest between groups, ##p < 0.01, ###p < 0.001, n = 17–59 cells). D, Schematic illustration of changes in neuronal C1q (blue), microglial phagocytosis (C1q and ChAT, green), and cholinergic pathways (red) in different groups. Di, In addition to the opposite changes in the cholinergic pathway and microglial ChAT and C1q expression, neuronal C1q showed an early increase that subsequently decreased. Dii, Minocycline treatment completely prevents microglial phagocytosis. In contrast, the increase in neuronal C1q is only partially prevented. The preserved increase in neuronal C1q is indicated in the dotted box in both Di and Dii. See Table 1 for mean ± SEM values and Figure 6-1, and Figure 6-2, for raw data. For all other statistical values, see Table 2.
Raw data for line plots in Figure 6B. Number of C1q clusters in ChAT interneurons. Download Figure 6-1, XLSX file (10.8KB, xlsx)
Raw data for bar graphs in Figure 6C. Optical intensity of microglial C1q+ clusters. Download Figure 6-2, XLSX file (12.5KB, xlsx)
We counted the numbers of C1q clusters within ChAT+ INs in images taken from the medial IZ of all animals and plotted this as cumulative distribution curves (raw data, gray curves; Gaussian-frequency curves, colored curves; Fig. 6B, raw values in Fig. 6-1). In the control spinal cord, <20% of INs have intracellular C1q expression. This percentage increased to >40% in the CST-denervated contralateral side of PTx3 rats, whereas there was, as yet, no loss of cholinergic INs (Fig. 1C,D) and a substantial number of cells containing much higher cluster numbers (up to 36). This leads to a significant rightward shift in the cumulative distribution. Interestingly, at PTx10, when there was an ∼35% loss specifically in the medial premotor class (Fig. 1D and our estimate), the cumulative curve plotted for spared INs shifted back to the control level (two-way ANOVA, Ctrl-PTx3: F(1,18) = 17.1, p = 0.0006; PTx3-PTx10: F(1,18) = 26.1, p < 0.0001, Ctrl-PTx10: F(1,18) = 4.06, p = 0.059; Fig. 6Bi). We hypothesize that only neurons with a low C1q expression survived. Comparison of bilateral cumulative distributions within each group (Fig. 6Bii, same scale as Fig. 6Bi) show a significant contralateral increase of C1q expression at PTx3 (two-way ANOVA, PTx3: F(1,18) = 26.1, p < 0.0001).
By contrast, microglial cells, which are the major source of C1q, did not show a significant increase in the average intensity of intracellular C1q until 10 d after PTx and minocycline fully suppressed the microglial C1q increase (unpaired t test within groups, PTx10: t(112) = 2.246, p = 0.0266; one-way ANOVA between groups: F(93,157) = 16.43, p < 0.0001, Ctrl-PTx10: ###p < 0.01, PTx3-PTx10: ##p < 0.01. PTx10-minocycline: ###p < 0.001; Fig. 6C, Table 1; raw values are in Fig. 6-2), which verified the effective inhibition of microglia function by minocycline. Intriguingly, interneuronal C1q expression in minocycline-treated-PTx10 animals also significantly decreased (Fig. 6A, fourth row), as reflected in a leftward shift in the cumulative distribution compared with PTx3 (two-way ANOVA, F(1,18) = 47.51, p < 0.0001; Fig. 6Bi). The leftward shift with minocycline may have resulted indirectly from suppression of the microglia source of neuronal C1q or by acting directly on cholinergic INs.
However, it is important to recognize that there are still C1q clusters preserved in some neurons at PTx10 with minocycline treatment (Fig. 6A, fourth row, and enlarged in Fig. 6Ab). The cumulative distribution curve after minocycline treatment remains intermediate between PTx3 and control/PTx10 levels and significantly more than PTx10 (two-way ANOVA, F(1,18) = 4.54, p = 0.0473; Fig. 6Bi). Moreover, the increase in microglial C1q occurs well after the early interneuronal C1q upregulation at PTx3. Both the delayed increase in microglial C1q and preserved neuronal C1q clusters after minocycline treatment suggest that microglia are unlikely to be the only source of the early, transient increase of neuronal C1q. Endogenous C1q synthesis by cholinergic INs may also contribute to the neuronal C1q overexpression (Fig. 6Di,Dii, dotted box). Together, these findings suggest the close association of an early increase in contralateral neuronal C1q, possibly of neuronal origin (Stevens et al., 2007; Stephan et al., 2013), with the net transneuronal downregulation of cholinergic INs and C bouton loss. The increased neuronal C1q level, which is limited to the contralateral side, likely serves as an “eat me” signal that specifies the targets of the bilateral increased microglial phagocytosis.
Close spatial interactions between microglial cells and cholinergic INs at PTx3
We observed close spatial interactions between microglia and cholinergic INs (Fig. 4A, middle row) and a transient C1q increase in cholinergic INs at 3 d, but not 10 d, after injury. This led us to explore how neuronal C1q could be involved in the glia–IN interaction and transneuronal modification of the spinal cholinergic system. Using 3D reconstructions, we were able to achieve a better view of this interaction, as shown in Figure 7A. In this example, the cholinergic IN is tightly wrapped by one microglial cell (different views shown in Fig. 7Ai). The microglia soma (*) and at least 2 of its processes (1 and 2) appear to cling to part of the cholinergic IN soma or dendrites when observed from different views. In addition, numerous C1q clusters can be detected inside this IN (Fig. 7Ai, transparent view). We then chose two optical slices from the Airyscan z-stack image to better demonstrate the contacts (Fig. 7Aii,Aiii). In such thin optical slice images (0.3 μm thickness), we verified the contacts at multiple locations (Fig. 7Aii,Aiii, far right panel, arrows). Interestingly, we also noticed that ChAT+/C1q+/OX42+ clusters were located where the microglia and the neuron contact each other (Fig. 7Aii, Aiii, arrows). These ChAT clusters are double labeled for OX42, suggesting that they are partially inside the neuron and microglia. This shows the ongoing engulfment of cholinergic IN fragments. There is another group of C1q+ clusters in the microglial cell that shows no ChAT labeling (Fig. 7Aiii, arrowhead), which may be endogenous to this microglial cell. Moreover, two unlabeled regions were captured in this OX42+ microglia soma, as shown in Figure 7Aiii (stars, also Fig. 3B, middle). We commonly observed this morphology on the contralateral side of PTx3 IZ. This morphology is similar to the multinucleated giant microglial cells described in other studies reporting a higher potential of phagocytosis, especially targeting large neurons (Beyer et al., 2000; Abiega et al., 2016).
Figure 7.

3D reconstruction of the interactions between microglial cells and cholinergic INs in the contralateral medial IZ. A, One ChAT+ IN and one microglial cell from a PTx3 animal. Ai, 3D reconstruction images show tight wrapping of the IN by a microglial cell from different angles. The semitransparent image shows numerous C1q clusters inside the cholinergic IN and microglia. Representative single optical images in Aii and Aiii show C1q+/ChAT+ clusters at the contact point of the neuron and microglial cell (arrows), indicating ongoing engulfment, endogenously expressed C1q in microglial cells (arrowhead), and the two chambers of the same microglial cell (*). B, Cholinergic INs and a microglial cell from a PTx10 animal. Bi, 3D reconstruction images showing little contact between the IN and the microglial cell. Transparent view shows dense C1q clusters inside the microglia, but none in neurons. Bii, Single optical images showing several C1q+/ChAT+ clusters inside microglia (1–3), indicating completed engulfment. Scale bar, 10 μm.
At PTx10, when we observed significant contralateral IN loss, we did not observe close contacts between microglia and INs as shown in Figure 7Bi. Instead, dense C1q+/ChAT+ clusters were now accumulated within microglial cells (Fig. 7Bi, transparent view, Bii, 1, 2, 3), implying completed engulfment of C1q+/ChAT+ fragments.
To summarize, ongoing engulfment begins to take place as early as 3 d after PTx, although without detectable loss of INs, and the engulfment or phagocytosis seems to be completed by 10 d after PTx. Importantly, the systematic timing of changes in C1q levels and cell loss, as well as the colocalization of C1q and engulfed ChAT fragments, suggests the critical involvement of C1q in the process of microglial engulfment of cholinergic INs.
Downregulation of the premotor cholinergic innervation of motoneurons is regulated by CST activity
Injury to the descending CST not only produces Wallerian degeneration, it also rapidly reduces excitatory inputs to its postsynaptic cholinergic spinal IN targets. To determine how activity loss contributes to the anterograde remodeling of the cholinergic system, we inactivated one hemisphere of the motor cortex in adult rats by chronic infusion of muscimol, a GABAA receptor agonist, using an implanted osmotic pump (Fig. 8A). Ten days after muscimol infusion, PKCγ showed the normal symmetrical dense staining intensity in both sides of dorsal column (Fig. 8A, inset), indicating that muscimol inactivation did not lesion the CST. However, localized CD68+ phagocytic microglial cells remained significantly increased (Mann–Whitney U test, p = 0.0003; Fig. 8B,C, Table 1; raw values are in Fig. 8-1). Similar to PTx, the intensity of microglial engulfed ChAT+ clusters also significantly increased in the contralateral compared with the ipsilateral gray matter (Mann–Whitney U test, p = 0.003; Fig. 8D, Table 1; raw values are in Fig. 8-1). These data suggest increased microglial phagocytosis of cholinergic neurons in response to CS inactivation. In addition, and as with PTx, we were also able to capture microglia wrapping cholinergic INs in the medial IZ in the contralateral spinal cord (Fig. 8Ei, 3D reconstruction) and C1q-associated engulfment (Fig. 8Eii, single optical plane from the cell in Fig. 8Ei showing ChAT and C1q internalized within an OX42+ microglial cell). During inactivation, cholinergic INs in the contralateral spinal cord express more C1q clusters intracellularly (Fig. 8F) and show a significant rightward shift in the cumulative distribution of neuronal C1q cluster numbers (two-way ANOVA, F(1,18) = 10.65, p = 0.0043; Fig. 8G; raw values are in Fig. 8-1). Importantly, on quantification, we detected significantly decreased numbers of ChAT+ INs in the medial IZ and decreased C bouton density (Fig. 8H,J for cholinergic INs, ∼19% decrease, Wilcoxon matched-pairs test, p = 0.0313, n = 5; Fig. 8I,K for C bouton, ∼40% decrease, Wilcoxon matched-pairs test, p = 0.0313, n = 5; mean values in Table 1; raw values are in Fig. 8-1). Our findings suggest that motor cortex inactivation initiates the process of spinal cholinergic circuit remodeling that is remarkably similar to that of PTx3. The relatively mild CST manipulation produced by inactivation compared with the complete loss of the CST from one hemisphere after PT lesion helps to explain the quantitatively smaller effect of inactivation on IN numbers.
Raw data for bar graphs and line plots in Figure 8. Fig. 8C. CD68+ microglia counts. Fig. 8D. Optical intensity of engulfed ChAT+ clusters. Fig. 8G. Number of C1q clusters in ChAT+ interneurons. Fig. 8J. Cholinergic interneuron counts and ratios. Fig. 8K. C bouton density and ratio. Download Figure 8-1, XLSX file (42KB, xlsx)
Promoting spinal cord activity through tsDCS rescues the cholinergic innervation after PTx
The activity-dependent transneuronal modification suggested by the inactivation experiment led us to test whether we could rescue the premotor cholinergic system by increasing spinal cord activity. We applied c-tsDCS beginning 1 d after PTx (see Materials and Methods and Fig. 9A). c-tsDCS enhances the excitability of spinal circuits to afferent fiber and descending corticospinal inputs (Song et al., 2015) at both spinal INs in the IZ and motoneurons in the ventral horn (Song and Martin, 2017). Ten days of c-tsDCS resulted in a symmetrical distribution of cholinergic INs and C bouton density on motor neurons (Fig. 9B). Quantification revealed no significant difference bilaterally (Wilcoxon matched-pairs test, INs: p = 0.1563; and C boutons: p = 0.5, n = 5; Fig. 9C,D, mean values are in Table 1 and raw values are in Fig. 9-1), with the contralateral to ipsilateral ratios close to 1, and significantly higher than those of the PTx10 group (Fig. 9C,D, right). These findings provide further support activity dependence in the transneuronal modulation of the cholinergic premotor system and further suggest the potential for c-tsDCS as a noninvasive and efficient method of repair after spinal cord injury.
Raw data for bar graphs in Figure 9. Fig. 9C. Cholinergic interneuron counts and ratios. Fig. 9D. C bouton density and ratios. Download Figure 9-1, XLSX file (38KB, xlsx)
Discussion
In this study, we demonstrated transneuronal downregulation of the spinal premotor cholinergic system in response to PTx. IN loss is preceded by overexpression of the complement molecule C1q and is produced by increased microglial phagocytosis on the affected side. Interestingly, IN loss was not associated with neuronal apoptosis, suggesting phagocytosis of live ChAT+ INs, a process termed phagoptosis (Brown and Neher, 2012). Depression of CST activity after 10 d of localized motor cortex inactivation replicates the cholinergic system downregulation by contralateral microglial activation and C1q overexpression. We propose that deprival of excitatory CST inputs to the spinal cord, either by injury or inactivation, triggers complement molecule upregulation in INs that receive a robust CST projection (Chakrabarty et al., 2009; Jiang et al., 2016). This, in turn, leads to microglial engulfment of the complement-tagged cholinergic INs and downregulation of the cholinergic innervation of motoneurons (Figure 10). We hypothesize that activity critically regulates the fate of CST target neurons at locations distant from the site of injury. Consistent with this, we successfully rescued neuronal loss and C bouton reduction using c-tsDCS neuromodulation of spinal activity. This does not necessarily preclude additional direct effects of the loss of inputs in mediating transneuronal changes. Downregulation of the cholinergic premotor system is potentially an important mechanism underlying motor impairment after stroke or spinal injury. This activity regulation has important implications for therapeutic intervention.
Figure 10.

Schematic illustration of the anterograde trans-synaptic downregulation hypothesis of the spinal cholinergic pathway after CST loss or inactivation. Each panel shows the hypothesized changes in the spinal cord after loss of the CST (orange axons; action potential size represents the level of CST activity). A, In the normal condition, the cholinergic INs (green) convey corticospinal signals to motoneurons (green action potential) and, in turn, CS activation maintains the survival and function of the cholinergic INs. The microglia remain silent (not activated) as surveillance cells. B, Shortly after pyramidal tract lesion (PTx3) or 10 d of cortical inactivation, denervated cholinergic INs are less active due to the loss of CST signaling and intrinsically express more C1q (red dots). Microglia become activated and are attracted to the site and wrap and then engulf the denervated cholinergic INs, which leads to the disconnection of distal cholinergic axon terminals with the IN soma. These disconnected axons and corresponding terminal C boutons undergo Wallerian degeneration and thus show a decrease earlier than the INs. The INs that are not directly affected by PTx and their C boutons would not be expected to show C1q overexpression and would not be targets for microglial phagocytosis. C, Ten days after PTx (PTx10) or anticipated after a longer period of CST inactivation, denervated cholinergic INs and all of their terminal C boutons are fully cleared by phagocytic microglial cells. This manifests as a progressive decrease in IN numbers and C bouton density. Surviving INs are expected to maintain their C boutons or possibly to sprout more C boutons to replace those lost by the lesion/inactivation (data not shown).
Anterograde transneuronal remodeling critically contributes to impaired motor function after spinal cord injury
The transneuronal downregulation of the cholinergic system in the cervical spinal cord after unilateral PTx is distinguishable from typical secondary injury (Kapitza et al., 2012). The C6–C7 spinal cord segments are too far from the lesion site to be directly affected (Profyris et al., 2004). The finding that there is no change in the number of PV+ INs or motoneurons strongly suggests that the transneuronal remodeling is not a global response. Instead, it is conferred by being targeted by the CST. Cholinergic INs in the medial IZ are one of the groups robustly innervated by CST axons (Jiang et al., 2016) and an association between the fate of cholinergic INs with the activity of corticospinal axons has also been shown during development (Chakrabarty et al., 2009). We thus consider that the specific downregulation in the cholinergic pathway is largely due to loss of CST inputs as part of anterograde transneuronal remodeling. However, it cannot be ruled out that the cholinergic INs are more vulnerable to CST inputs/activity than other spinal cord neurons.
Premotor cholinergic INs in the spinal cord have been suggested to be critically involved in locomotion (Huang et al., 2000; Miles et al., 2007), motor attention (Zagoraiou et al., 2009), and task-dependent coordination and posture control (Stepien et al., 2010). We found ∼35% loss of premotor cholinergic INs 10 d after PTx and a single premotor cholinergic IN is estimated to form ∼1000 C boutons onto motoneurons (Zagoraiou et al., 2009). Therefore, an enormous loss of cholinergic inputs would be expected after a corticospinal system injury. Such a loss of cholinergic INs and C boutons could contribute significantly to the rapid exhaustion of the central drive required for locomotion—and, more important, for skilled movements under cortical control—after a stroke or spinal cord injury (Kapitza et al., 2012). Rescuing the cholinergic INs and their C boutons from this novel form of anterograde circuit remodeling that we show would no doubt improve the capacity for functional compensation.
Early induction of neuronal C1q may mediate microglia phagocytosis of cholinergic INs
The complement cascade is critically involved in the phagocytic pathway for neuronal loss in normal aging and neurodegenerative disease and synaptic pruning during development (Veerhuis et al., 2011; Stephan et al., 2013). Neuronal C1q protein was found to be significantly increased in the brains of Alzheimer's disease patients postmortem (Shen et al., 1997) and in developing retinal ganglion cells (Bialas and Stevens, 2013). This increase precedes neuronal or synaptic loss in animal models (Schafer et al., 2012; Hong et al., 2016). We saw the same pattern, in which a complement protein is expressed in cholinergic INs targeted by injured CST (Fig. 6B). Importantly, in both PTx3 and inactivation animals, we identified cholinergic INs with high intracellular C1q expression that were tightly wrapped by activated microglia. Some were undergoing engulfment and the engulfed neuronal fragments colabeled with C1q. This strong linkage implies that neuronal C1q expression not only contributes to neuronal elimination by microglial cells, but may also serve as a contralateral (and active) “eat me” marker for a more general bilateral microglia phagocytosis. Therapeutic targeting of neuronal C1q early after injury might block the subsequent progress of phagocytosis.
C1q is considered to be predominantly generated by microglial cells (Fonseca et al., 2017) and the microglial C1q increase after PTx was completely prevented by minocycline treatment in our study. However, the neuronal C1q increase was not completely prevented compared with that of microglial C1q expression (Garrido-Mesa et al., 2013). Moreover, neuronal C1q after PTx increased much earlier than that of microglia C1q (Fig. 6D). These findings point additionally to microglial-independent C1q production, which could derive from cholinergic INs denervated by CST lesion. Indeed, C1q mRNAs are detected in rodent cortical INs (Shen et al., 1997; Stephan et al., 2013), suggesting that neurons are capable of generating C1q. It is plausible that there are neuronal-microglial interactions, akin to those discussed by Stephens and colleagues for the developing and mature nervous system (Schafer et al., 2013), whereby premotor cholinergic INs sense and react to the loss of presynaptic CS input earlier than microglial cells. This could manifest as early synthesis of neuronal C1q that, in turn, marks itself for elimination.
Neuronal activity contributes to transneuronal downregulation of cholinergic INs and circuit remodeling
Neuronal activity is a key regulator of synaptic pruning and circuit refinement during motor (Martin et al., 2007) and sensory (Jamann et al., 2018) development. Refinement of the entire circuit by activity manipulation is not limited to the directly manipulated neurons/axons via competition (Martin and Ghez, 1999; Martin et al., 2005; Friel et al., 2007; Schafer et al., 2012; Jiang et al., 2016), but also involves transneuronal remodeling (Chakrabarty et al., 2009; Jiang et al., 2016). In our study, we found remarkably similar downregulation of cholinergic INs in animals with motor cortex inactivation and 3 d after PTx. tsDCS of cervical segments early after injury effectively increased spinal cord activity and rescued cholinergic INs from downregulation. These findings together strongly suggest that loss of CS activity, and subsequent interneuronal activity, may be the underlying regulator of anterograde transneuronal modulation early after PTx.
The robust activity dependence implies that intervention of neuronal activity early after injury with neuromodulatory approaches could have potential for rescuing this premotor system and help functional restoration. Indeed, our tsDCS-mediated rescue supports this idea and further raises the question of dissecting the different sources of activity in repair. Many postinjury rehabilitation approaches rely on direct activity manipulations or increasing activity through motor performance. For example, forced use of the impaired forelimb by constraint of the nonaffected limb (Maier et al., 2008; Friel et al., 2012) and motor cortex stimulation, alone or in combination with tsDCS (Carmel et al., 2010; Song et al., 2015, 2016), promote sprouting of spared ipsilateral CST and improve motor function after PTx. These manipulations may also be rescuing spinal neurons from anterograde trans-synaptic neuronal loss, possibly mediated by microglia and complement proteins.
What exactly bridges presynaptic activity changes with postsynaptic remodeling is not well understood. It could be due to loss of anterograde neurotrophic support (von Bartheld et al., 1996; Altar and DiStefano, 1998), which is strongly modulated by presynaptic axonal activity (Bruno and Cuello, 2006). In addition, the expression of phagocytic-related immune proteins such as C1q (Simonetti et al., 2013) and MHC I (Corriveau et al., 1998) are also sensitive to neuronal activity manipulation. Furthermore, microglia–synapse interactions as a basis for microglia surveillance and engulfment of weak synapses (Wake et al., 2013) can be regulated by blocking of visual inputs by tetrodotoxin (Wake et al., 2009). Further investigation is needed to explore whether there is causality to the chronological order of activity interference/axon loss, the change in complement protein expression, and phagocytic microglia activation.
Implication for neural repair
The primary objectives for motor rehabilitation after spinal cord injury are to limit damage inflicted by the injury and to promote spared pathways to compensate for lost functions due to damaged neuronal circuits. In contrast to the suddenness of primary damage to neuronal circuits after stroke or spinal injury, transneuronal remodeling produced by remote injury can occur over days to weeks. Therapeutic interventions to prevent this relatively late and protracted remodeling may offer a better opportunity for functional restoration. Our study shows the critical involvement of phagocytic microglia, early interneuronal C1q, and presynaptic activity in trans-synaptic neuronal loss after supraspinal injury. A better understanding of this process is necessary for developing target- and location-specific rehabilitation strategies that depend on the stage and severity of motor injury.
Footnotes
This work was supported by The National Institutes of Health (Grant 2R01NS064004 to J.H.M.), the Craig H. Neilsen Foundation (Grant 261214 to J.H.M.), and the New York State Department of Health Spinal Cord Injury Research Board (Grant C030606GG to J.H.M. and Grant C30860GG to A.A.). We thank Xiuli Wu for histology and immunohistochemistry and the staff of the City University of New York Graduate Center Quantitative Research Consulting Center for statistics consultation. We thank the Andreas Kottman lab at the CUNY School of Medicine for kindly providing the DNase I recombinant used in TUNEL positive control staining.
The authors declare no competing financial interests.
References
- Abiega OS, et al. (2016) Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol 14:e1002466. 10.1371/journal.pbio.1002466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altar CA, DiStefano PS (1998) Neurotrophin trafficking by anterograde transport. Trends Neurosci 21:433–437. 10.1016/S0166-2236(98)01273-9 [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Jonas PC, Sapir T, Hartley R, Berrocal MC, Geiman EJ, Todd AJ, Goulding M (2005) Postnatal phenotype and localization of spinal cord V1 derived interneurons. J Comp Neurol 493:177–192. 10.1002/cne.20711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber RP, Phelps PE, Houser CR, Crawford GD, Salvaterra PM, Vaughn JE (1984) The morphology and distribution of neurons containing choline acetyltransferase in the adult rat spinal cord: an immunocytochemical study. J Comp Neurol 229:329–346. 10.1002/cne.902290305 [DOI] [PubMed] [Google Scholar]
- Beyer M, Gimsa U, Eyüpoglu IY, Hailer NP, Nitsch R (2000) Phagocytosis of neuronal or glial debris by microglial cells: upregulation of MHC class II expression and multinuclear giant cell formation in vitro. Glia 31:262–266. 10.1002/1098-1136(200009)31:3%3C262::AID-GLIA70%3E3.0.CO;2-2 [DOI] [PubMed] [Google Scholar]
- Bialas AR, Stevens B (2013) TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci 16:1773–1782. 10.1038/nn.3560 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Brown GC, Neher JJ (2012) Eaten alive! cell death by primary phagocytosis: ‘phagoptosis’. Trends Biochem Sci 37:325–332. 10.1016/j.tibs.2012.05.002 [DOI] [PubMed] [Google Scholar]
- Brownstone RM, Bui TV (2010) Spinal interneurons providing input to the final common path during locomotion. Prog Brain Res 187:81–95. 10.1016/B978-0-444-53613-6.00006-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno MA, Cuello AC (2006) Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc Natl Acad Sci U S A 103:6735–6740. 10.1073/pnas.0510645103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brus-Ramer M, Carmel JB, Chakrabarty S, Martin JH (2007) Electrical stimulation of spared corticospinal axons augments connections with ipsilateral spinal motor circuits after injury. J Neurosci 27:13793–13801. 10.1523/JNEUROSCI.3489-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmel JB, Martin JH (2014) Motor cortex electrical stimulation augments sprouting of the corticospinal tract and promotes recovery of motor function. Front Integr Neurosci 8:51. 10.3389/fnint.2014.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmel JB, Berrol LJ, Brus-Ramer M, Martin JH (2010) Chronic electrical stimulation of the intact corticospinal system after unilateral injury restores skilled locomotor control and promotes spinal axon outgrowth. J Neurosci 30:10918–10926. 10.1523/JNEUROSCI.1435-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty S, Shulman B, Martin JH (2009) Activity-dependent codevelopment of the corticospinal system and target interneurons in the cervical spinal cord. J Neurosci 29:8816–8827. 10.1523/JNEUROSCI.0735-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corriveau RA, Huh GS, Shatz CJ (1998) Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron 21:505–520. 10.1016/S0896-6273(00)80562-0 [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Martínez-Cerdeño V, Noctor SC (2013) Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci 33:4216–4233. 10.1523/JNEUROSCI.3441-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Zhang YP, Hou Z, Huang C, Zhu H, Zhang CQ, Yin Q (2016) Novel insights into NeuN: from neuronal marker to splicing regulator. Mol Neurobiol 53:1637–1647. 10.1007/s12035-015-9122-5 [DOI] [PubMed] [Google Scholar]
- Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, MacGregor GR, Tenner AJ (2017) Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation 14:48. 10.1186/s12974-017-0814-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC (2018) Neuronal cell death. Physiol Rev 98:813–880. 10.1152/physrev.00011.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel KM, Drew T, Martin JH (2007) Differential activity-dependent development of corticospinal control of movement and final limb position during visually guided locomotion. J Neurophysiol 97:3396–3406. 10.1152/jn.00750.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel K, Chakrabarty S, Kuo HC, Martin J (2012) Using motor behavior during an early critical period to restore skilled limb movement after damage to the corticospinal system during development. J Neurosci 32:9265–9276. 10.1523/JNEUROSCI.1198-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost JL, Schafer DP (2016) Microglia: architects of the developing nervous system. Trends in cell biology 26:587–597. 10.1016/j.tcb.2016.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido-Mesa N, Zarzuelo A, Gálvez Jz (2013) Minocycline: far beyond an antibiotic. Br J Pharmacol 169:337–352. 10.1111/bph.12139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Black JA, Waxman SG (2003) Primary cortical motor neurons undergo apoptosis after axotomizing spinal cord injury. J Comp Neurol 462:328–341. 10.1002/cne.10733 [DOI] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in alzheimer mouse models. Science 352:712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Noga BR, Carr PA, Fedirchuk B, Jordan LM (2000) Spinal cholinergic neurons activated during locomotion: localization and electrophysiological characterization. J Neurophysiol 83:3537–3547. 10.1152/jn.2000.83.6.3537 [DOI] [PubMed] [Google Scholar]
- Jamann N, Jordan M, Engelhardt M (2018) Activity-dependent axonal plasticity in sensory systems. Neuroscience 368:268–282. 10.1016/j.neuroscience.2017.07.035 [DOI] [PubMed] [Google Scholar]
- Jiang YQ, Williams PT, Martin JH (2013) Rapid and persistent impairments of the forelimb motor representations following cervical deafferentation in rats. Eur J Neurosci 38:3702–3711. 10.1111/ejn.12372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YQ, Zaaimi B, Martin JH (2016) Competition with primary sensory afferents drives remodeling of corticospinal axons in mature spinal motor circuits. J Neurosci 36:193–203. 10.1523/JNEUROSCI.3441-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitza S, Zörner B, Weinmann O, Bolliger M, Filli L, Dietz V, Schwab ME (2012) Tail spasms in rat spinal cord injury: changes in interneuronal connectivity. Exp Neurol 236:179–189. 10.1016/j.expneurol.2012.04.023 [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–318. 10.1016/0166-2236(96)10049-7 [DOI] [PubMed] [Google Scholar]
- Kumar A, Loane DJ (2012) Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun 26:1191–1201. 10.1016/j.bbi.2012.06.008 [DOI] [PubMed] [Google Scholar]
- Liang FY, Moret V, Wiesendanger M, Rouiller EM (1991) Corticomotoneuronal connections in the rat: evidence from double-labeling of motoneurons and corticospinal axon arborizations. J Comp Neurol 311:356–366. 10.1002/cne.903110306 [DOI] [PubMed] [Google Scholar]
- Maier IC, Baumann K, Thallmair M, Weinmann O, Scholl J, Schwab ME (2008) Constraint-induced movement therapy in the adult rat after unilateral corticospinal tract injury. J Neurosci 28:9386–9403. 10.1523/JNEUROSCI.1697-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JH, Ghez C (1999) Pharmacological inactivation in the analysis of the central control of movement. J Neurosci Methods 86:145–159. 10.1016/S0165-0270(98)00163-0 [DOI] [PubMed] [Google Scholar]
- Martin JH, Engber D, Meng Z (2005) Effect of forelimb use on postnatal development of the forelimb motor representation in primary motor cortex of the cat. J Neurophysiol 93:2822–2831. 10.1152/jn.01060.2004 [DOI] [PubMed] [Google Scholar]
- Martin JH, Friel KM, Salimi I, Chakrabarty S (2007) Activity- and use-dependent plasticity of the developing corticospinal system. Neurosci Biobehav Rev 31:1125–1135. 10.1016/j.neubiorev.2007.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles GB, Hartley R, Todd AJ, Brownstone RM (2007) Spinal cholinergic interneurons regulate the excitability of motoneurons during locomotion. Proc Natl Acad Sci U S A 104:2448–2453. 10.1073/pnas.0611134104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan BP, Harris CL (2015) Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov 14:857–877. 10.1038/nrd4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher JJ, Neniskyte U, Hornik T, Brown GC (2014) Inhibition of UDP/P2Y6 purinergic signaling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia 62:1463–1475. 10.1002/glia.22693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos CM, Tsai SY, Alsbiei T, O'Brien TE, Schwab ME, Kartje GL (2002) Functional recovery and neuroanatomical plasticity following middle cerebral artery occlusion and IN-1 antibody treatment in the adult rat. Ann Neurol 51:433–441. 10.1002/ana.10144 [DOI] [PubMed] [Google Scholar]
- Profyris C, Cheema SS, Zang D, Azari MF, Boyle K, Petratos S (2004) Degenerative and regenerative mechanisms governing spinal cord injury. Neurobiol Dis 15:415–436. 10.1016/j.nbd.2003.11.015 [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74:691–705. 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Stevens B (2013) The “quad-partite” synapse: microglia-synapse interactions in the developing and mature CNS. Glia 61:24–36. 10.1002/glia.22389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro LA, Wang L, Ribak CE (2008) Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia 49:33–41. 10.1111/j.1528-1167.2008.01491.x [DOI] [PubMed] [Google Scholar]
- Shen Y, Li R, McGeer EG, McGeer PL (1997) Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Res 769:391–395. 10.1016/S0006-8993(97)00850-0 [DOI] [PubMed] [Google Scholar]
- Simonetti M, Hagenston AM, Vardeh D, Freitag HE, Mauceri D, Lu J, Satagopam VP, Schneider R, Costigan M, Bading H, Kuner R (2013) Nuclear calcium signaling in spinal neurons drives a genomic program required for persistent inflammatory pain. Neuron 77:43–57. 10.1016/j.neuron.2012.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, Paton JFR, Chakrabarty S, Ichiyama RM (2017) Descending systems direct development of key spinal motor circuits. J Neurosci 37:6372–6387. 10.1523/JNEUROSCI.0149-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Martin JH (2017) Spinal cord direct current stimulation differentially modulates neuronal activity in the dorsal and ventral spinal cord. J Neurophysiol 117:1143–1155. 10.1152/jn.00584.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Truong DQ, Bikson M, Martin JH (2015) Transspinal direct current stimulation immediately modifies motor cortex sensorimotor maps. J Neurophysiol 113:2801–2811. 10.1152/jn.00784.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Amer A, Ryan D, Martin JH (2016) Combined motor cortex and spinal cord neuromodulation promotes corticospinal system functional and structural plasticity and motor function after injury. Exp Neurol 277:46–57. 10.1016/j.expneurol.2015.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai HH, Huang EJ, Rowitch DH, Berns DS, Tenner AJ, Shamloo M, Barres BA (2013) A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci 33:13460–13474. 10.1523/JNEUROSCI.1333-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepien AE, Tripodi M, Arber S (2010) Monosynaptic rabies virus reveals premotor network organization and synaptic specificity of cholinergic partition cells. Neuron 68:456–472. 10.1016/j.neuron.2010.10.019 [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178. 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- Underhill DM, Ozinsky A (2002) Phagocytosis of microbes: complexity in action. Annu Rev Immunol 20:825–852. 10.1146/annurev.immunol.20.103001.114744 [DOI] [PubMed] [Google Scholar]
- Veerhuis R, Nielsen HM, Tenner AJ (2011) Complement in the brain. Mol Immunol 48:1592–1603. 10.1016/j.molimm.2011.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld CS, Byers MR, Williams R, Bothwell M (1996) Anterograde transport of neurotrophins and axodendritic transfer in the developing visual system. Nature 379:830–833. 10.1038/379830a0 [DOI] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29:3974–3980. 10.1523/JNEUROSCI.4363-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Miyamoto A, Nabekura J (2013) Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci 36:209–217. 10.1016/j.tins.2012.11.007 [DOI] [PubMed] [Google Scholar]
- Yamada K, Patel U, Shrier DA, Tanaka H, Chang JK, Numaguchi Y (1998) MR imaging of CNS tractopathy: wallerian and transneuronal degeneration. AJR Am J Roentgenol 171:813–818. 10.2214/ajr.171.3.9725322 [DOI] [PubMed] [Google Scholar]
- Yang HW, Lemon RN (2003) An electron microscopic examination of the corticospinal projection to the cervical spinal cord in the rat: lack of evidence for cortico-motoneuronal synapses. Exp Brain Res 149:458–469. 10.1007/s00221-003-1393-9 [DOI] [PubMed] [Google Scholar]
- Zagoraiou L, Akay T, Martin JF, Brownstone RM, Jessell TM, Miles GB (2009) A cluster of cholinergic premotor interneurons modulates mouse locomotor activity. Neuron 64:645–662. 10.1016/j.neuron.2009.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang Y, Xing S, Liang Z, Zeng J (2012) Secondary neurodegeneration in remote regions after focal cerebral infarction: a new target for stroke management? Stroke 43:1700–1705. 10.1161/STROKEAHA.111.632448 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Raw data for bar graphs in Figure 1. Fig. 1D. Cholinergic interneuron counts and ratios. Fig. 1E. C bouton density and ratios. Fig. 1G. NeuN+ neuronal counts. Fig. 1J. Parvalbumin interneuron counts. Fig. 1K. Motoneuron counts. Download Figure 1-1, XLSX file (43.1KB, xlsx)
Raw data for bar graphs in Figure 3. Fig. 3C. CD68+ microglia counts. Fig. 3D. Optical intensity of microglial CD68+ clusters. Download Figure 3-1, XLSX file (42KB, xlsx)
Raw data for bar graphs in Figure 4C. Optical intensity of engulfed ChAT+ clusters. Download Figure 4-1, XLSX file (72.7KB, xlsx)
Raw data for bar graphs in Figure 5. Fig. 5C. CD68+ microglia counts. Fig. 5D. Optical intensity of microglial CD68+ clusters. Fig. 5G. Cholinergic interneuron counts and ratios. Fig. 5H. C bouton density and ratios. Download Figure 5-1, XLSX file (36.4KB, xlsx)
Raw data for line plots in Figure 6B. Number of C1q clusters in ChAT interneurons. Download Figure 6-1, XLSX file (10.8KB, xlsx)
Raw data for bar graphs in Figure 6C. Optical intensity of microglial C1q+ clusters. Download Figure 6-2, XLSX file (12.5KB, xlsx)
Raw data for bar graphs and line plots in Figure 8. Fig. 8C. CD68+ microglia counts. Fig. 8D. Optical intensity of engulfed ChAT+ clusters. Fig. 8G. Number of C1q clusters in ChAT+ interneurons. Fig. 8J. Cholinergic interneuron counts and ratios. Fig. 8K. C bouton density and ratio. Download Figure 8-1, XLSX file (42KB, xlsx)
Raw data for bar graphs in Figure 9. Fig. 9C. Cholinergic interneuron counts and ratios. Fig. 9D. C bouton density and ratios. Download Figure 9-1, XLSX file (38KB, xlsx)