


Abstract
The circadian protein Nocturnin (NOCT) belongs to the exonuclease, endonuclease and phosphatase superfamily and is most similar to the CCR4-class of deadenylases that degrade the poly-adenosine tails of mRNAs. NOCT-deficient mice are resistant to high-fat diet induced weight gain, and exhibit dysregulation of bone formation. However, the mechanisms by which NOCT regulates these processes remain to be determined. Here, we describe a pair of high-resolution crystal structures of the human NOCT catalytic domain. The active site of NOCT is highly conserved with other exoribonucleases, and when directed to a transcript in cells, NOCT can reduce translation and abundance of that mRNA in a manner dependent on key active site residues. In contrast to the related deadenylase CNOT6L, purified recombinant NOCT lacks in vitro ribonuclease activity, suggesting that unidentified factors are necessary for enzymatic activity. We also find the ability of NOCT to repress reporter mRNAs in cells depends upon the 3′ end of the mRNA, as reporters terminating with a 3′ MALAT1 structure cannot be repressed by NOCT. Together, these data demonstrate that NOCT is an exoribonuclease that can degrade mRNAs to inhibit protein expression, suggesting a molecular mechanism for its regulatory role in lipid metabolism and bone development.
INTRODUCTION
Messenger RNA (mRNA) degradation plays a fundamental role in regulating gene expression in diverse biological contexts (1). Characterizing the pathways and proteins involved in RNA decay and discerning their regulatory activities are crucial for understanding translational control. A key early step in degradation of many mRNAs is deadenylation, the enzymatic removal of the 3′ poly-adenosine (poly(A)) tail, which is catalyzed by deadenylases (2). The poly(A) tail promotes translation and stabilizes mRNA transcripts by binding the Poly(A) Binding Protein (PABP). Deadenylation reduces translation and promotes subsequent mRNA degradation. Once the tail is sufficiently shortened, the transcript can be rapidly destroyed by either of two pathways: (i) 3′-5′ decay catalyzed by exoribonucleases such as the exosome complex or (ii) decay in the 5′-3′ direction by exoribonucleases such as XRN1 following removal of the 5′ 7-methyl guanosine cap by decapping enzymes (3,4). The rates of deadenylation and decay vary considerably by transcript and can change in response to stimuli, stress or developmental programs, indicative of the importance of these processes in gene regulation (1,2).
Multiple deadenylases have been identified in eukaryotes and are classified into two families: CCR4-like and POP2-like. The CCR4 family shares a catalytic domain that is related to a superfamily of exonucleases, endonucleases and phosphatases (EEP). These deadenylases include the founding member, yeast CCR4, the human CCR4 orthologs CNOT6 and CNOT6L, and the divergent PDE12 enzyme, for which structures were recently reported (5,6). POP2 family members are related to the DEDD family of RNases, named for the active site aspartate and glutamate-containing motif conserved among family members, including POP2, PARN and PAN2 (7–9). Though unrelated in sequence and structure, these CCR4-like and POP2-like families share the characteristics of being hydrolytic 3′-5′ exoribonuclease enzymes with a general preference for poly(A) substrates and requiring divalent magnesium ions (2).
The presence of multiple deadenylases in eukaryotes suggests that these enzymes have overlapping functions; however, in certain contexts, individual deadenylases participate in the post-transcriptional regulation of unique sets of mRNAs. Deadenylases also have unique intracellular localization patterns (e.g. PDE12 is located in mitochondria and PARN is predominantly nuclear) (10,11). Moreover, genetic analysis of several deadenylases has revealed specific phenotypes, indicating that these enzymes can serve unique regulatory roles (12–20). A remaining challenge for the deadenylase field is to characterize the catalytic activities and substrate specificities of these enzymes and determine the mechanisms that control their regulatory functions. Compelling evidence supports the hypothesis that specific deadenylases are directed to specific target mRNAs by sequence-specific RNA-binding factors, including RNA-binding proteins and microRNAs (21–24).
In this study, we focus on human Nocturnin (NOCT), a protein containing an EEP domain that is most similar to CCR4 deadenylases (18% amino acid sequence identity and 33% sequence similarity with human CNOT6L). NOCT is widely conserved, with orthologs present from insects to mammals (13,16,25,26). Xenopus and mouse orthologs displayed weak deadenylase activity in in vitro assays using partially purified recombinant protein; however, characterization of the molecular function of NOCT remains incomplete (13,27). These findings suggested that NOCT may regulate mRNA stability and translation, although direct action of NOCT on target mRNAs has yet to be demonstrated in vivo and much remains to be learned about NOCT’s structure, function and enzymatic activities. In particular, natural NOCT substrates remain unknown, and NOCT’s regulatory effects on mRNAs in vivo remain unclear.
The potential importance of NOCT in controlling gene regulation is emphasized by the biological functions that it affects. NOCT was first identified as a transcript whose expression oscillates with circadian rhythms in the retina of Xenopus laevis, and subsequent analysis in mice showed NOCT is widely expressed and oscillates in multiple tissues (13,19,28). Major insights into NOCT’s physiological functions were obtained through phenotypic analysis of NOCT knockout mice, which have normal circadian phenotypes but exhibit metabolic defects (16). These data suggest that NOCT expression is a downstream output of circadian gene expression, and not a master regulator of the circadian regulatory pathway. Strikingly, Noct −/− mice are resistant to obesity and hepatic steatosis when fed a high-fat diet, whereas wild-type (WT) mice become obese (16). Further studies suggested that NOCT is essential for the regulation of lipid trafficking across the intestinal epithelium, as Noct −/− mice exhibited reduced levels of circulating triglycerides and increased accumulation of lipid droplets in intestinal enterocytes (29).
Characterization of NOCT function has also implied that the enzyme plays a regulatory role in adipogenesis and osteoblastogenesis. Noct −/− mice exhibit increased bone density and reduced adiposity, including in bone marrow. In the pre-adipocyte 3T3-L1 cell line, overexpression of NOCT enhanced adipogenesis when the cells were cultured in adipogenic media. Conversely, depletion of NOCT in 3T3-L1 cells reduced their adipogenic potential (30,31). Together, these results indicate a role for NOCT in promoting lipid uptake, trafficking and adipogenesis, potentially through mechanisms involving post-transcriptional control of gene expression. However, the potential role of NOCT ribonuclease activity and the target mRNAs that it may regulate remain unknown.
To better understand NOCT’s role in regulating fat metabolism and developmental processes, we sought to characterize its structure, biochemical functions, enzymatic activity and regulatory effects on mRNAs in cells. Here, we report the first crystal structure of the catalytic domain of human NOCT determined to 1.48 Å resolution, illustrating its strong structural homology to other CCR4-related EEP deadenylases. We devised cell-based assays to measure the effect of NOCT on reporter mRNAs. Using these assays, we assessed the contributions of the conserved active site residues to NOCT-mediated repression of a reporter mRNA, finding that residues in the NOCT active site contribute in part to its repressive activity. Furthermore, we use reporter mRNAs with varying 3′ end structures in our functional assays to demonstrate that NOCT repression activity is dependent on the accessibility of the 3′ end. Our findings provide new insight into the catalytic activity of human NOCT, providing a basis for characterization of the mechanism by which NOCT regulates mRNA targets to control lipid metabolism, body mass and skeletal development.
MATERIALS AND METHODS
Protein expression and purification
Strep(II)-Sumo-Nocturnin120–431 was expressed in a Strep(II) pSumo vector in BL21 Rosetta2 (DE3) Escherichia coli cells(MilliporeSigma) grown in 2xYT medium (Fisher Scientific) overnight at 18°C. Cells were lysed in 50 mM phosphate buffer (pH 7.5), 150 mM NaCl, 1 mM MgCl2 and 10% glycerol. Buffer was supplemented with 0.5 g lysozyme in 35 ml of lysate, Thermo Scientific Halt Protease Inhibitor Cocktail (0.5×), NEB Micrococcal Nuclease (100U) and 5.0 mM CaCl2. Lysate was incubated in the supplemented lysis buffer for 90 min at 4°C and then sonicated. Clarified lysate was purified using Strep-Tactin Superflow Plus resin (Qiagen) in 50 mM phosphate buffer (pH 7.5), 300 mM NaCl, 1 mM MgCl2 and 10% glycerol and eluted with 2.5 mM desthiobiotin. The Strep (II) tag was cleaved overnight at 4°C with Ulp-1 Sumo protease and NOCT was then purified further using Q Sepharose Fast Flow resin (GE Healthcare) in 10 mM phosphate buffer (pH 7.5), 20 mM NaCl, 1 mM MgCl2, 10% glycerol and 5 mM βME, and eluted with a 20 mM to 1.0 M NaCl gradient. The protein used to produce the 2.41 Å crystal structure was subsequently purified using a Superdex 75 gel filtration column (GE Healthcare) in 50 mM Tris (pH 8.0), 50 mM Na2SO4, 1 mM MgCl2, 5% glycerol and 1 mM TCEP. Similarly, the protein used to produce the 1.48 Å crystal structure was subsequently purified in 50 mM Tris (pH 8.0), 75 mM Na2SO4, 4 mM MgCl2, 10% glycerol and 1 mM TCEP.
For biochemical assays, Strep(II)-Sumo-NOCT64–431, Strep(II)-Sumo-mNOCT62–429, Strep(II)-Sumo-CNOT6L158–555 and Strep(II)-Sumo-CNOT6L158–555 E240A were expressed using a Strep(II) pSumo vector in BL21 STAR (DE3) E. coli cells (ThermoFisher) expressing the pRare2 plasmid (MilliporeSigma) and purified as described above, with the addition of a 1.0 M NaCl wash (50 mM phosphate buffer (pH 7.5), 1.0 M NaCl, 1 mM MgCl2 and 10% glycerol) after the addition of lysate onto the Strep-Tactin Superflow Plus resin. The single step-purified mNOCT was concentrated and stored after the Strep-Tactin purification step. For all other proteins, the purifications were completed as described above. The ion exchange step was performed using Source 15Q resin (GE Healthcare) and the CNOT6L WT and E240A mutant proteins were purified in the phosphate and Tris buffers at pH 8.0.
Protein crystallization and structure determination
NOCT crystals were obtained using the hanging-drop vapor diffusion method. For the 2.41 Å resolution structure, a final concentration of 25 mM AMP and 10 mM MgCl2 was added to NOCT120–431 solutions before protein was concentrated to 9 mg/ml. Concentrated protein and well solution were combined at a 1:1 ratio. Crystals were obtained in a well solution of 100 mM HEPES buffer (pH 7.5), 5% isopropanol, 200 mM sodium acetate and 10% PEG 4000. For the 1.48 Å resolution structure, NOCT120–431 solutions at 8 mg/ml were prepared as described above with 10 mM 3′, 5′ adenosine diphosphate and 6.0 mM MgCl2. Concentrated protein, well solution and distilled water were combined at a 1:1:1 ratio. Crystals were obtained in 100 mM Tris (pH 8.5), 100 mM MgCl2, 100 mM sodium citrate and 14% PEG 4000. Diffraction data were collected at the LS-CAT beamline 21 ID-G at the Advanced Photon Source at Argonne National Laboratories. Data were indexed, integrated and scaled using HKL-2000 (32). Both structures were solved by molecular replacement (MR) using Phenix and a previously determined structure of NOCT120–431 as the search template. The initial NOCT structure was determined by MR utilizing a homology model of the enzyme as a search model, which was generated using I-TASSER and the crystal structure of CNOT6L (PDB ID: 3NGQ) as the template (data not shown) (5,33). Autobuilding in Phenix was followed with iterative rounds of refinement using Phenix Refine and model building using Coot (Supplementary Table S1) (34). The presence of one or two metal ions in the active sites of these structures was determined by generating an omit Fo-Fc map lacking Mg2+ ions and coordinated waters and contouring the map to 5σ. Here, we confirmed strong positive density peaks associated with the Mg2+ ions bound in the active site. Although crystallization was conducted in the presence of AMP and 3′, 5′ adenosine diphosphate, respectively, electron density corresponding to these nucleotides was not observed in the structures. X-ray data collection and refinement statistics are reported in Supplementary Table S1.
Surface electrostatic potentials were calculated using the APBS Tools 2.1 Plugin for MacPyMOLx11Hybrid. PDB2PQR was used to generate input files using AMBER charges (ff99) and radii, excluding both the bound Mg2+ and solvation shell during analysis (35–37). The surface potential was contoured to ±3 kT/e for NOCT (1.48 Å resolution dataset), PDE12 (PDB ID: 4Z0V) and CNOT6L (3NGQ). The surface rendering of NOCT colored by conservation was generated using the ConSurf server (http://consurf.tau.ac.il/2016/) and the 1.48 Å resolution dataset for the NOCT structure. HMMER was used for the homolog search algorithm and UniProt was used as the protein database. Minimal ID was set to 15% as NOCT has high structural but low sequence homology to the closely related human proteins CNOT6L and PDE12 (18 and 16% identical, respectively). The multiple sequence alignment was generated using MAFFT-L-INS-I (38–41).
Biochemical assays
Biochemical mRNA degradation reactions were performed in CNOT6L buffer (50 mM HEPES (pH 7.4), 150 mM NaCl, 10% glycerol, 2.0 mM MgCl2 and 1.0 mM dithiothreitol (DTT)). NOCT64–431, CNOT6L158–555 or CNOT6L158–555 E240A (1 μM) were mixed with 10 nM 5′ Cy3 labeled p(A)20 (Dharmacon), and RNasin RNase A inhibitor (Promega) and incubated over a 30-min time course at 37°C. In addition, biochemical assays were performed in the assay buffers described by Baggs and Green and in Garbarino-Pico et al. (13,27,42). Briefly, mRNA degradation assays were performed in (i) xNOCT buffer (5 mM HEPES (pH 7.5), 2.0 mM MgCl2, 0.5 mM DTT, 10% glycerol, 0.2 mg/ml bovine serum albumin, 0.02% (v/v) NP-40 and 0.3% (w/v) spermidine); and (ii) mNOCT buffer (50 mM HEPES (pH 8.0), 0.01 mM Mg(OAc)2 and 20 mM KOAc). NOCT64–431, mNOCT62–429, CNOT6L158–555 or CNOT6L158–555 E240A (1 μM) were mixed with 10 nM 5′ Cy3 labeled p(A)20 (Dharmacon), and RNasin RNase A inhibitor (Promega) and incubated over a 30-min time course at 37°C. Reaction products were resolved on a 15% polyacrylamide sequencing gel and detected using a Typhoon Trio + (GE Healthcare).
To titrate the ionic strength or Mg2+ concentrations, individual buffers were made with the indicated NaCl or MgCl2 concentrations in the buffer described above. To test various divalent metals, NOCT64–431 or CNOT6L158–555 were treated with 15 mM ethylenediaminetetraacetic acid (EDTA) and then dialyzed in assay buffer lacking MgCl2. The divalent metal ions tested (Mg2+, Mn2+, Ca2+ and Zn2+) were added to assay buffer at a final concentration of 2.0 mM and combined with NOCT64–431 or CNOT6L158–555. To control for the effect of dialysis on activity, CNOT6L158–555 activity was compared pre- and post-dialysis in buffer containing Mg2+. To measure NOCT activity in a range of substrate RNA concentrations, 10 nM 5′ Cy3 labeled p(A)20 (Dharmacon) was held constant and unlabeled p(A)19 (Dharmacon) was titrated into reactions at concentrations ranging from 0 nM to 10 μM unlabeled RNA. To measure NOCT activity against a long mRNA substrate analog, RNA substrate template was generated by polymerase chain reaction (PCR) using primers listed in Supplementary Table S2 and a plasmid containing the Renilla Luciferase ORF. RNA substrate was produced using the HiScribe T7 High Yield RNA Synthesis Kit (NEB) and purified using an RNA Clean and Concentrator -25 Kit (Zymo Research). Reactions were incubated with the long RNA substrate and RNasin at 37°C for a 30-min time course. As a positive control, RNase III (NEB) was incubated with the long substrate RNA in 1× NEB RNaseIII buffer at 37°C for a 30-min time course. Products were resolved on a 2.5% agarose/ 1× MOPS (200 mM 3-N-Morpholino propanesulphonic acid, 50 mM NaOAc, 10 mM EDTA)/4% (v/v) formaldehyde denaturing gel and detected by ethidium bromide (EtBr) staining and UV detection on a Syngene Gene Genius Bio Imaging System.
NOCT64–431 activity against ssDNA and dsDNA was tested using 5′ FAM-labeled DNA 25-mers. The dsDNA substrate contained the ssDNA substrate forward strand sequence annealed to the reverse complement sequence (Supplementary Table S2). NOCT64–431, Micrococcal Nuclease (NEB), or DNase I (Sigma) were incubated with ssDNA or dsDNA in assay buffer for 30 min and resolved on a polyacrylamide gel as described above.
Phosphatase activity assays against a panel of compounds (Supplementary Table S3) were conducted using 500 nM NOCT64–431 and 100 μM of each phosphorylated compound incubated in 50 mM HEPES (pH 7.4), 150 mM NaCl, 10% glycerol, 2.0 mM MgCl2 and 1.0 mM DTT for 30 min, and the resulting concentrations of phosphate were measured using a Malachite Green Assay (Cayman Chemical) with reactions performed in duplicate. To account for differences in the rate of phosphate production via spontaneous hydrolysis, each compound was tested in assay buffer lacking protein. Background phosphate production was then subtracted individually for each compound to account for non-enzymatic phosphate formation. Calf intestinal alkaline phosphatase (NEB) was used as a positive control.
Cell-based assays
HEK293 cells (ATCC) were cultured in DMEM with glucose, 10% (v/v) Fetal Bovine Serum (FBS), 100 U/ml penicillin, 100 U/ml streptomycin and 292 μg/ml glutamine (penicillin/streptomycin/glutamine from Gibco), at 37°C and 5% CO2. The full-length NOCT coding region was cloned into the pF5K vector (Promega) to express MS2-NOCT in HEK293 cells. All protein constructs contained a V5 epitope tag for western blot detection. As a negative control, HaloTag (HT) fused to MS2 coat protein in the pFN21A vector was used (Promega). Point mutations were generated using the oligonucleotide mediated site-directed mutagenesis method (Quikchange, Agilent). The reporter plasmid, pNLP 4xMS2BS p(A), was derived from the plasmid psiCheck1 (Promega) and contained the SV40 promoter driving NanoLuc Luciferase (NLuc) with a 3′ UTR containing four stem-loop binding sites for phage MS2 coat protein. The NLuc 4xMS2BS p(A) and NLuc 4xMS2BS MALAT1 reporters contained either a minimal synthetic 3′ cleavage and polyadenylation signal or a 3′ MALAT1 triple helix, respectively. The Firefly Luciferase internal control plasmid was pGL4.13 (Promega).
For transfections, cells were plated at 600 000 cells per well in 6-well culturing plates. Twenty-four hours post plating, DNA was transfected into cells using a 3 μl:1 μg ratio of ViaFect (Promega) to DNA including 1.25 μg of effector plasmid, 500 ng of Firefly plasmid and 1.25 μg of the NLuc reporter plasmid. Forty-eight hours post-transfection, cells were harvested in 1× phosphate buffered saline (PBS) (pH 7.5). Cell suspensions were then plated into white-walled 96-well plates and assayed for luminescence using the NanoGlo Dual Luciferase Assay System and a GloMax Multi+ luminometer according to the manufacturer’s instructions (Promega).
Tethered function assay data were analyzed using a hierarchical Bayesian model in order to account for the differences in variation between technical and biological replicates. The input data were the log2 ratios of NLuc activity to that of the Firefly Luciferase internal control (ffLuc). We modeled the average value for each biological replicate as arising from a t-distribution centered on the overall average value μprot for the particular protein variant of interest; a separately inferred average value for the ffLuc intensity on each particular day (μffluc,day, shared across all proteins considered on that day) was applied as an additive offset to the day-wise values. The values for the technical replicates were further modeled with t-distributions centered on the appropriate daily average value μprot,day - μffluc,day. The protein-wise scale parameters σprot were modeled as arising from a common gamma distribution with uninformative hyperpriors, whereas the other scale parameters, and the degree of freedom parameters for the t distributions were simply assigned uninformative priors. We fitted the models using JAGS via the rjags interface, running four independent Monte Carlo chains for 250 000 iterations with 25 000 steps of burn-in; we ensured convergence by verifying that the Gelman-Rubin shrinkage statistic for all parameters of interest was <1.1 (43,44). Reported credible intervals were calculated using the highest posterior density approach with the R boa package; reported probabilities were calculated directly from the posterior distributions (45). We performed separate fits for each reporter construct. The main topic of interest in all cases is the central log ratio value μprot for each protein, which reflects the relative Luciferase level observed when that protein is present in the assay. We most frequently report the differences observed for the μprot value of a protein of interest relative to either that of a HT negative control or of WT NOCT. All raw data, number of replicates, and statistics from the fitted models are also shown in Supplementary Table S4.
Western blotting
Cells harvested from the transfected cells used in the tethered function assays were harvested in 1× PBS, pelleted, and resuspended in radioactive immunoprecipitation assay buffer (RIPA buffer, 50 mM Tris (pH 8.0), 150 mM NaCl, 1% (v/v) IGEPAL CA-630, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) sodium dodecyl sulphate) for lysis. Lysates were homogenized using QIAShredder Columns (Qiagen) and sample concentrations were determined using the BioRad DC Lowry assay. Each sample (8 μg total protein) was resolved on 4–20% TGX gradient gels (BioRad) and then transferred to Millipore EMD Immobilon membrane. Transfected NOCT proteins were detected using anti-V5 antibody (Invitrogen, R960–25) and HT was detected using anti-HT antibody (Promega, G9211). Blots were probed using HRP-conjugated Secondary Antibody (Sigma, A1047) and Pierce ECL Western Blotting substrate before exposure to autoradiography film. As a loading control, Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was detected on the same blots using anti-GAPDH antibody (Ambion, AM4300).
Northern blotting
Cells, transfected as described in the cell-based assay method above, were harvested in 1× PBS and total cellular RNA was extracted using the ReliaPrep RNA Cell Miniprep System (Promega). For each experiment, triplicate RNA samples were pooled. RNA from each sample (3–5 μg total RNA) was prepared for electrophoresis in 1× MOPS, 3.7% (v/v) formaldehyde, 25% (v/v) formamide, 5% (v/v) glycerol, 1 mM EDTA, 0.025% (w/v) bromophenol blue and 0.025% (w/v) xylene cyanol FF. Samples were resolved on a 1% MOPS agarose gel containing 4% formaldehyde before transfer to Millipore EMD Ny+ membrane using 20× Saline Sodium Citrate (SSC). The membrane was UV-crosslinked and prehybridized with Ultrahyb buffer (Invitrogen).
Antisense Northern probe templates for NLuc and Firefly were PCR-amplified with primers containing the T7 promoter sequence (Supplementary Table S2). Probes were transcribed using the MAXIscript T7 Transcription Kit (Ambion) with αP32 UTP (20 μCi at 3 μCi/mmol) and cold UTP (8 μM). Radiolabeled probes were purified using Sephadex G-25 resin (GE Healthcare). The final activity of the probes ranged from 1.3 × 106 – 2.5 × 106 counts per million/μg probe. Blots were probed overnight at 68°C before washing with 2× SSC + 0.1% (w/v) sodium dodecyl sulphate (SDS) and then 0.1x SSC + 0.1% (w/v) SDS. Blots were exposed to phosphoimager screens for 3 days and then imaged using a Typhoon FLA 9500. Image Quant (GE Healthcare) was used to quantify the intensity of the bands for NLuc and Firefly for each sample. Background correction was performed using the rolling ball method with a radius set to 200 to determine the baseline before peak integration.
The quantitation data were analyzed using a Bayesian hierarchical model similar to that described above for the tethered function assays, using the log2 ratio of NLuc to ffLuc peak areas as a key statistic for each sample. As before, we modeled the log2 ratio values as arising from a t-distribution, centered on the difference between day-wise values for NLuc (independently inferred for each target) and ffLuc (shared for all targets on that day). All such t-distributions used shared values for the scale and degree of freedom parameters, using an uninformative prior for the scale and the Gamma(2,0.1) prior suggested for the degrees of freedom (46). Day-wise means were, in turn, modeled as arising from a normal distribution centered on μprot,day - μffluc,day with a shared variance parameter representing technical noise in the measurements themselves. The prior for the square root of the variance was set to be a Gamma(1,2) distribution, amounting to a weak assumption that the technical noise would make no more than 1–2 log2 units of difference in the abundance measurements. The model was fitted in JAGS (43,47) using eight independent Monte Carlo chains, with 500 000 iterations per chain and a 25 000 step burn-in period and convergence assessed as described above. All data, number of replicates, and statistics are shown in Supplementary Table S4.
RESULTS
Crystal structure of NOCT reveals a conserved structure, active site and basic cleft
As the structure of NOCT had not been previously characterized, we sought to purify the protein and determine its crystal structure to gain insight into its molecular functions. Our attempts to purify the full-length human protein (residues 1–431) were hindered by its relative insolubility and degradation. We overcame this problem by truncating the N-terminus, which is predicted to be disordered (48). Through optimization of expression and purification conditions, we successfully purified human NOCT120–431, encompassing the entire EEP domain and NOCT64–431, which includes half of the N-terminus of the longest annotated form of NOCT. During the course of optimization, we found that bacterial nucleic acid and ribonuclease contaminants co-purified with NOCT, a common issue that can lead to mischaracterization of ribonuclease activity (49). Consequently, we utilized a number of strategies to eliminate contaminants, including the use of a commercial E. coli strain with a mutation in the RNaseE gene that diminishes ribonuclease activity, and implementation of a multistep purification scheme that included affinity, ion exchange and gel filtration chromatography coupled with stringent, high-salt washing conditions. Through these efforts, we obtained highly purified recombinant NOCT (Supplementary Figure S1).
We then determined crystal structures of the human NOCT catalytic domain (residues 120–431) in two different space groups to 2.41 Å and 1.48 Å resolution (Supplementary Table S1 and Figure 1A). The NOCT catalytic domain adopts the overall structure of an α/β-sandwich (Figure 1A and B). The core of the domain comprises two anti-parallel β sheet that face one another, with one sheet formed by β4- β5- β3- β2- β15- β14 and β1 strands and the other composed of β6- β7- β8- β9- β13 and β10 strands. Several α and 310 helices surround the two central β sheets, effectively burying a substantial portion of the β sheet core. An unusual π helix immediately precedes α3 (Supplementary Figure S2). This π helix contains Pro205, which disrupts the canonical i, i+4 hydrogen bonding pattern observed in α-helices. The π helix is surface exposed and could potentially serve a functional role, as has been described for other proteins (50,51).
Figure 1.

The crystal structure of NOCT reveals a conserved structure, active site and basic cleft. (A) A cartoon representation of the 1.48 Å resolution structure of the NOCT catalytic domain (residues 120–431). (B) Cartoon representation of the NOCT120–431 1.48 Å resolution structure denoting the secondary structural elements of the NOCT catalytic domain colored using the PyMOL chainbow settings. (C) A detailed view of the active site of the 2.41 Å resolution structure. Two Mg2+ ions are bound in the active site (one coordinated to Glu195 and the other to Asp324) and a sulphate anion is bound between Lys219 and Lys288 adjacent to the active site. (D) A detailed view of the active site of the 1.48 Å resolution structure. A single Mg2+ ion is bound in the active site, coordinated to Glu195. Hydrogen bonds are depicted with dashed lines. Hydrogen bond distance cutoffs were assigned an upper limit of 3.3 Å. (E) The active site of the 2.41 Å resolution structure and the adjacent basic residues are shown with the enzyme surface corresponding to the active site and basic patch superimposed. (F) A surface electrostatic rendering of the NOCT catalytic domain with surface potential contoured to ±3 kT/e. NOCT has a prominent basic cleft adjacent to the active site. The bound Mg2+ ion is shown as a sphere. (G) A surface representation of the NOCT 1.48 Å resolution structure. Residues are colored by conservation based on a multiple sequence alignment generated by ConSurf. The residues that are conserved are colored in violet, and the most variable residues are colored in teal. Neutral residues that are neither highly variable or conserved are white. The most conserved residues are found in the active site and adjacent basic cleft.
The predicted active site of NOCT possesses residues that are highly conserved in NOCT and other CCR4 family members, including residues Asn149 on β2, Glu195 on β5, His286 on β8, Asp324 on β9 and Asp377 on β13 (Figure 1C and D). The remaining active site residues include His414, located in the β15–3105 loop and Asn326 in the β9–α4 loop. The crystal structure of NOCT reveals two variations of the bound ligands in the active site of the enzyme. In the 2.41 Å resolution structure, two Mg2+ ions occupy the active site and are directly coordinated via an octahedral geometry to Glu195 (Mg2+1) and Asp324 (Mg2+2), respectively, with water molecules occupying the remaining metal coordination sites (Figure 1C and Supplementary Figure S3A). In the 1.48 Å resolution structure, only a single Mg2+ ion is bound in the active site and is directly coordinated by Glu195 (Figure 1D and Supplementary Figure S3A). We aligned the two structures of the NOCT catalytic domain (RMSD of 0.268 Å over 1619 atoms) and found no changes in conformation overall or locally in the active site (Supplementary Figure S3B and C). Correlating with the observation of different Mg2+ ion occupancy, crystal structures of the related CCR4-family enzymes human CNOT6L and human PDE12 have been reported with one or two Mg2+ ions bound in their respective active sites (5,6). The overall architecture of NOCT is most similar to the catalytic domains of the CNOT6L (RMSD 1.154 Å over 1203 atoms for all atoms) and PDE12 (RMSD 1.170 Å over 1122 atoms for all atoms), in addition to the arrangement and identity of the active site residues (Supplementary Figure S3D). For comparison, an alignment with apurinic/apyrimidinic endonuclease-1 (APE1; RMSD 8.692 Å over 1032 atoms for all atoms), a more distantly related EEP family member, is shown (Supplementary Figure S3D).
We examined the electrostatic surface of NOCT, finding a pronounced basic patch ∼13 Å long and 7.5 Å wide along the surfaces adjacent to the putative active site, suggestive of an mRNA binding cleft (Figure 1E and F). Similar, albeit less prominent, basic clefts are observed on CNOT6L and PDE12 structures (Supplementary Figure S3E). We also observed a sulphate anion from the buffer bound in the basic cleft of NOCT, adjacent to the active site, via a hydrogen bond to Lys219, potentially mimicking binding of the phosphodiester backbone of RNA (Figure 1C and E). ConSurf analysis indicated that the most conserved residues cluster in the predicted active site and extend into the basic cleft, indicating that the residues located in these regions likely serve important functional roles (Figure 1G and Supplementary Figure S3F) (38–41). In summary, the NOCT structure illustrates a conserved globular domain with predicted active site residues and divalent metal ion cofactors located adjacent to a basic cleft, consistent with its proposed function as a ribonuclease.
Biochemical analysis of NOCT catalytic activity
We next sought to characterize the putative RNA degradation activity of NOCT. To obtain highly purified recombinant enzyme, we isolated NOCT64–431 using a stringent, multistep protocol (described in the ‘Materials and Methods’ section). As positive and negative controls for RNA degradation, we purified the well-characterized human deadenylase CNOT6L158–555 and a catalytically inactive CNOT6L158–555 E240A mutant using the same expression and purification procedure (5). Because mouse and Xenopus NOCT proteins were previously reported to degrade poly(A), we first measured the activities of WT NOCT64–431 against a poly(A)20 RNA substrate in CNOT6L buffer (13,27). Our data show that WT NOCT64–431 did not degrade the poly(A)20 RNA, whereas WT CNOT6L readily degraded this substrate (Figure 2). As expected, the CNOT6L E240A catalytic mutant was inactive toward this RNA, and chelation of Mg2+ with EDTA inhibited WT CNOT6L, consistent with prior studies (5).
Figure 2.

NOCT is not active against poly(A)20 substrates in vitro. Recombinant NOCT64–431, CNOT6L158–555 and CNOT6L158–555 E240A were incubated with 5′ Cy3 labeled poly(A)20 RNA substrate. A representative gel is shown here. Where indicated, the chelator EDTA was added to stop the ribonuclease reaction.
We considered that the reaction conditions may be suboptimal for human NOCT, and thus explored the effects of divalent and monovalent ions. First, we titrated Mg2+and NaCl concentrations, spanning concentrations necessary for activities of other deadenylases (49). Additionally, we titrated the amount of poly(A) RNA substrate over a broad range of concentrations, ranging from concentrations below that of the enzyme to concentrations of RNA in excess of NOCT. We also screened other biologically relevant divalent metals including Mn2+, Ca2+ and Zn2+ individually and in combination with Mg2+. None of these conditions enabled NOCT to degrade the poly(A) substrate (Supplementary Figure S4A–D). To examine whether NOCT requires longer RNA substrates, or if NOCT has RNA endonuclease activity, we tested a long ∼1100 nt RNA substrate containing the Renilla Luciferase ORF with a short 3′ UTR and a 60 nt poly(A) tail; NOCT was unable to degrade this RNA (Supplementary Figure S4E). Because NOCT is a member of the EEP superfamily which includes enzymes that hydrolyze a wide range of phosphorylated substrates, we also screened a diverse collection of potential alternative substrates including dsDNA, ssDNA and an array of phosphorylated signaling molecules, nucleotides, lipids and carbohydrates to test for potential deoxyribonuclease and phosphatase activities (52). NOCT did not display activity toward any of these nucleic acids or phosphorylated molecules (Supplementary Figure S4F–G and Table S3).
These results seemingly contradict previous reports that recombinant mouse and Xenopus NOCT proteins could degrade poly(A) substrates in vitro (13,27). It is noteworthy that those proteins were partially purified using a single affinity step under low-stringency purification conditions. In our experience, such strategies are prone to bacterial ribonuclease contamination. Indeed, we found that single-step purified NOCT had such contaminants under some purification conditions (data not shown). We also considered the potential for species-specific differences in NOCT activity; therefore, we compared the activities of human NOCT (NOCT) and murine NOCT (mNOCT) purified under our multi-step, stringent conditions. Mouse NOCT62–429 was additionally purified using a single step with stringent conditions that include a high salt wash, which can reduce or eliminate contaminating activity (Supplementary Figures S1E and S4H). After testing a broad range of conditions, neither human NOCT nor mouse NOCT had detectible enzymatic activity in comparison to CNOT6L (Supplementary Figure S4H). We additionally assayed NOCT and mNOCT activity in two reaction buffers (mNOCT and xNOCT buffers) that were previously reported to support activity of mNOCT or Xenopus NOCT (13,27). It is noteworthy that the reaction conditions previously employed for measuring mouse or Xenopus NOCT activity contained either very low ionic strength or sub-physiological Mg2+ concentrations, in contrast to the conditions used in Figure 2 and Supplementary Figure S4A–F, or previously used to characterize catalytic activity of human CNOT6L and PDE12 (5,6,53). We did not observe deadenylase activity of the stringently purified NOCT or mNOCT in these conditions, whereas CNOT6L, but not mutant CNOT6L, could degrade the poly(A) RNA (Supplementary Figures S1E and S4H). We conclude that purified recombinant human or mouse NOCT do not exhibit deadenylase activity under these conditions.
Importantly, our purification strategy is fully compatible with deadenylase activity, as shown by the activity of CNOT6L enzyme purified in an identical manner (Figure 2). A caveat of our approach is that in order to obtain soluble NOCT protein, we had to truncate the N-terminal 63 amino acids. It is noteworthy that similar NOCT protein isoforms starting at the equivalent position to Met67, are annotated in the GenBank database. The N-terminal 120 amino acids of NOCT are predicted to be disordered according to GlobPlot analysis and are unlikely to contribute to catalytic function (48). Moreover, NOCT64–431 contains the entire EEP domain, is functional in cells (see below) and similar truncations in other CCR4-family members, such as CNOT6L, remain catalytically active (5,6). Thus, our results indicate that NOCT protein does not display RNA degrading activity on its own. We speculated that NOCT activity might require other currently unidentified cofactors, such as protein partners and/or certain post-translational modifications. Although the enzymatic function of NOCT in cells remained largely unexplored, we hypothesized that such cofactors and/or modifications would be present in vivo and therefore devised an assay to measure NOCT function in this context.
NOCT represses translation and causes degradation of reporter mRNAs in vivo
We examined whether NOCT could repress gene expression in cells. Natural direct target mRNAs of NOCT have not been identified to date; therefore, we employed a tethered function assay to measure NOCT’s ability to regulate translation and stability of a NanoLuc Luciferase (NLuc) reporter mRNA in cells (54–56). This reporter, NLuc 4xMS2BS p(A), has a 3′UTR bearing four binding sites for the MS2 coat protein followed by a cleavage and polyadenylation site. We used HEK293 cells, which do not express detectable endogenous NOCT protein (data not shown). A Firefly Luciferase plasmid (ffLuc) was co-transfected and served as an internal control in all assays. A plasmid that expressed WT NOCT fused to MS2 coat protein (MS2-NOCT) was co-transfected with the Luciferase constructs into the cells and its effect on reporter expression was measured relative to a negative control protein, MS2 fused to HaloTag (MS2-HT). MS2-NOCT reproducibly repressed reporter expression by 2.7- to 3.0-fold (Figures 3 and 4). Likewise, a positive control fusion of MS2-HT to the deadenylase CNOT7 (MS2-HT-CNOT7) repressed the reporter, consistent with its well-documented ability to repress translation and deadenylate mRNA (57–59). We then compared the activities of the truncated MS2-NOCT64–431 to MS2-NOCT1–431 in the tethered function assay. Repression of reporter expression by NOCT64–431 and NOCT1–431 was comparable, indicating that the truncation of the N-terminus does not negatively affect NOCT activity (Figure 3). This direct assay provides evidence that NOCT can repress gene expression when directed to an mRNA in vivo.
Figure 3.
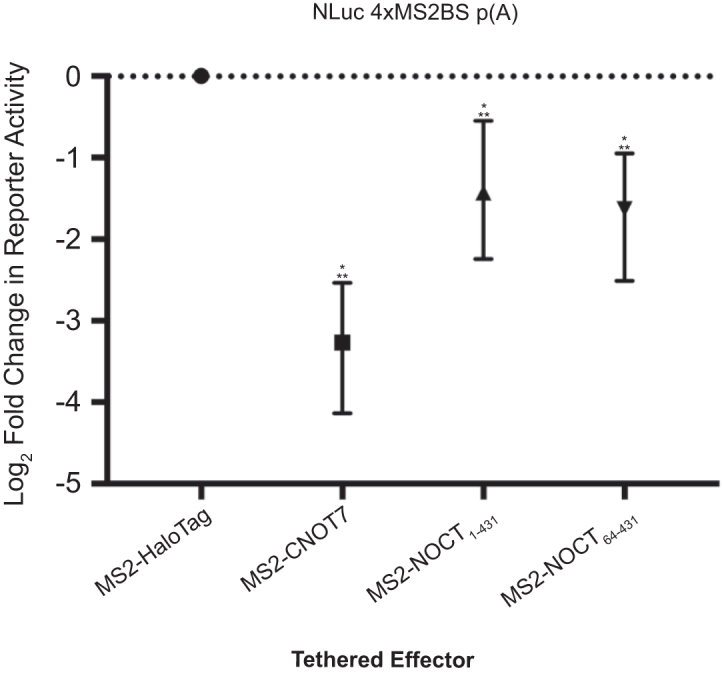
NOCT translationally represses a reporter mRNA in a cell-based tethered function assay. The effect of NOCT or control CNOT7 on normalized expression of NLuc from the NLuc 4xMS2BS mRNA p(A) reporter was measured in HEK293 cells, and log2 fold change in reporter activity is plotted relative to negative control MS2-HaloTag. Tethered effector proteins included MS2 coat protein fusions to WT NOCT1–431, NOCT64–431, CNOT7 and HaloTag. Error bars show 95% credible intervals; any comparisons where the 95% credible intervals do not overlap zero are considered significant and are denoted by a single star. Two stars denote a 95% posterior probability that the difference measured is >1.3-fold.
Figure 4.

NOCT repression of protein expression depends on conserved active site residues and the identity of the mRNA 3′ end. (A) Mutation of NOCT the active site reduced but did not eliminate translational repression of the NLuc 4xMS2BS p(A) reporter. Tethered effectors fused to MS2 coat protein are listed on the bottom of the graph. Log2 fold change in reporter protein expression calculated relative to negative control MS2-HaloTag (MS2-HT) is plotted for each tethered effector. (B) As in (A), but with activity shown relative to WT NOCT. (C) Western blot detection of tethered effectors for three replicate samples using anti-V5 monoclonal antibody (WT and mutant MS2-NOCT effectors) or anti-HaloTag monoclonal antibody (MS2-HT and MS2-HT-CNOT7). (D) Tethered function assays (as in A and B) comparing repression of NLuc targets with the indicated 3′ ends by MS2-tethered effectors relative to that of MS2-HT. Error bars and significance stars are as in Figure 3.
Next, we assessed the regulatory role of NOCT active site residues in translational repression by individually mutating Glu195, Asn149, His286, Asp324, Asn326, Asp377 and His414 to alanine in the MS2-NOCT construct. We also created a His414 to asparagine mutant, which is more likely to preserve hydrogen-bonding interactions of His414 within the active site. Mutation of His414 (H414A and H414N), as well as N149A and E377A reduced NOCT-mediated repression of the reporter (Figure 4A and B). In contrast, the H286A, E324A and N326A substitutions did not appreciably affect NOCT-mediated repression, whereas the E195A mutation slightly enhanced repression activity. Interestingly, the E195A mutation is analogous to the murine E193A mutant that was previously reported to inactivate partially purified, recombinant mouse NOCT (27). The differences in the ability of NOCT to repress reporter mRNAs following mutagenesis of various active site residues cannot be attributed to differences in expression of the NOCT fusion proteins, as all constructs were well-expressed, and slight differences among expression levels of the mutant proteins do not correlate with differences in reporter expression (Figure 4C). Together, our observations indicate that NOCT possesses the ability to repress protein expression when directed to a specific mRNA, and that this repressive activity is dependent at least in part on specific conserved active site residues.
Given the relationship of NOCT to CCR4-type 3′ exoribonucleases, we next explored the effect of the 3′ end of the reporter mRNA on repression. To facilitate this analysis, we compared NOCT-mediated repression of a NLuc 4xMS2BS reporter terminating with a poly(A) tail to a similar reporter terminating with a MALAT1 triple-helical structure. The MALAT1 3′ end is processed by RNase P, and does not undergo canonical cleavage and polyadenylation (60). Instead, a short, encoded poly(A) sequence is incorporated into a triple helix structure (61). As expected, WT NOCT repressed the poly(A) reporter, as did the positive control CNOT7 (Figure 4D). In contrast, the NOCT H414A mutant exhibited reduced activity toward the poly(A) reporter compared to the WT enzyme. Strikingly, NOCT did not have any substantial activity toward the MALAT1 reporter, suggesting that the mechanism of repression is dependent on either poly(A) and/or the accessibility of the 3′ end (Figure 4D). Tethered CNOT7 retained partial inhibitory function on the MALAT1 reporter, consistent with the ability of the CNOT complex to cause translational repression and promote 5′ decapping and decay (62–64). Taken together, these observations indicate that NOCT acts at the 3′ end of the mRNA, consistent with CCR4-type exoribonucleases, and unlike CNOT7, does not appear to recruit 5′-directed decay factors or endonucleases to target mRNAs.
We subsequently analyzed the effect of NOCT on NLuc 4xMS2BS p(A) reporter mRNA levels using northern blot analysis to determine whether NOCT-mediated repression occurs through inhibition of translation, enhanced RNA decay or a combination of both mechanisms (Supplementary Figure S5A and B). Relative to the MS2-HT control, tethered MS2-NOCT reduced the reporter mRNA levels by 2.2-fold (Figure 5A; Supplementary Figure S5A and B). In contrast, based on our statistical criteria, the NOCT active site mutants did not significantly reduce reporter mRNA levels relative to the MS2-HT negative control, suggesting that the mutations compromised their ability to degrade the mRNA (Figure 5A). However, when compared to WT NOCT, loss of activity could only be firmly established for the H414A mutant (Figure 5B and C). The other active site mutants showed some evidence of impaired activity relative to WT NOCT, but measurement noise precludes a firm conclusion on their relative activity compared with both HT and WT NOCT. Nevertheless, these data (particularly regarding the H414A mutant) indicate that the catalytic site of NOCT contributes to RNA decay in vivo. We also tested the ability of tethered NOCT to reduce levels of the NLuc 4xMS2BS 3′ MALAT1 mRNA and observed that NOCT was incapable of degrading the RNA, consistent with its inability to translationally repress the MALAT1 reporter (Figure 5C). The positive control MS2-HT-CNOT7 was capable of degrading the MALAT1 mRNA, albeit with reduced effectiveness compared to the poly(A) mRNA, consistent with CNOT-mediated RNA degradation and 5′ decay. These observations indicate that NOCT-mediated repression involves mRNA degradation and is dependent on either the sequence and/or structure of the 3′ end.
Figure 5.

WT NOCT degrades poly(A) RNA and mutating active site residues stabilizes NLuc RNA. (A) Fusion proteins of MS2 coat proteins with WT NOCT or NOCT active site mutants were transfected into HEK293 cells along with the NLuc 4xMS2 poly(A) reporter. Resulting levels of NLuc RNA relative to the MS2-HT control were measured by northern blot analysis. WT NOCT reduced the steady state levels of the NLuc reporter RNA. Mutating the NOCT active site residues to alanine appeared to reduce degredation; however, the width of the credible intervals precludes a firm conclusion as to whether or not there is residual activity relative to HT. (B) As in panel (A), but with data shown relative to WT NOCT. The data suggest that all tested mutations reduce activity relative to WT, although only the H414A and N326A mutations showed strong evidence of loss of activity. (C) As in A and B, showing mRNA levels of the indicated reporter. MS2-NOCT reduced the steady state levels of NLuc 4xMS2BS p(A) but not 4xMS2BS MALAT1. Statistical significance is denoted as previously described in Figure 3.
DISCUSSION
The results presented here contribute two major advances: (i) we describe the first crystal structure of the NOCT catalytic domain, illuminating a highly conserved active site and basic cleft that may mediate interactions with the phosphodiester backbone of RNA substrates. (ii) We demonstrate that NOCT can translationally repress and degrade an mRNA when directed to that transcript in vivo, providing initial evidence for post-transcriptional regulatory activity.
The NOCT catalytic domain exhibits sequence and structural homology with other CCR4-type EEP family members (e.g. the deadenylases CNOT6L and PDE12), including the identity and conformation of active site residues and the presence of magnesium ion cofactors. Accordingly, the structural homology supports a role for NOCT in RNA degradation, corroborated by the in vivo data demonstrating that NOCT reduces reporter levels. We also showed that this activity is dependent on key conserved active site residues (Figure 5). Moreover, NOCT activity is also dependent on the sequence and/or structure of the 3′ end of the mRNA, as introduction of the MALAT1 structure blocks NOCT-mediated decay and translational repression. If NOCT acted through an endonucleolytic mechanism, then a 3′ MALAT1 structure would not be expected to prevent RNA degradation and repression. While the structure and/or accessibility of the 3′ end is crucial, the potential for 3′ end sequence specificity of NOCT remains to be determined. Both the canonical poly-adenylated and MALAT1 reporter mRNAs terminate in adenosine residues, but the 3′ end of MALAT1 is sequestered within a stable triple helix, suggesting that the MALAT1 structure prevents NOCT from accessing the reporter's 3′ end (61). It is noteworthy that our Northern blot analysis did not detect RNA decay intermediates. At this time, we cannot rule out that NOCT works in concert with other ribonucleases to promote mRNA decay. For instance, NOCT may deadenylate mRNA targets before another enzyme, such as the exosome, degrades the remainder of the RNA message (65).
NOCT is most similar to the poly(A) specific CNOT6L, a well-documented CCR4-type deadenylase, but the precise determinants of deadenylase poly(A) specificity remain incompletely understood. Structural and functional studies of CNOT6L have provided some clues regarding this specificity and 3′-exoribonuclease mechanism (5). Mutational analysis of conserved actives site residues in CNOT6L demonstrated that alanine mutations of Glu240, Asp489 and His529 abolished deadenylase activity. In agreement with this finding, alanine substitutions of the NOCT residues corresponding to the latter two residues in NOCT, Asp377 and His414, to alanine diminished mRNA repression in the tethered function assay (Figure 4A and B). In addition, the NOCT H414A mutant reduced mRNA degradation in vivo relative to the WT enzyme, further illustrating the catalytic defects associated with this mutant (Figure 5). Alignments of NOCT with the EEP enzymes CNOT6L, PDE12 and APE1 illustrate that the aspartate and histidine form a conserved pair that interact through side chain-mediated hydrogen bonding (Supplementary Figure S3D) (5,6,66). The imidazolium cation of the histidine may promote active site binding of the reactive phosphate of the nucleic acid substrate through hydrogen bonding, as proposed for the catalytic mechanism of APE1 (66). Interestingly, substitution of NOCT Glu195 with alanine does not significantly alter mRNA repression or degradation in vivo (Figures 4 and 5), in contrast to the corresponding E240A mutant in CNOT6L that abolishes deadenylase activity (Figure 2) (5). In CNOT6L and NOCT structures, the carboxylate groups of these glutamates coordinate Mg2+ ions (Supplementary Figure S3A). Coordination of these Mg2+ ions mediates the binding of poly(A) DNA, a nonreactive substrate analog, to CNOT6L, consistent with the catalytic defect observed with the CNOT6L E240A mutant. The observation that the analogous E195A mutant does not significantly hinder NOCT activity or function in vivo suggests that the enzyme may employ an alternate RNA-binding mode. Further structural and functional studies of NOCT and related CCR4-type deadenylases are required to fully elucidate the functions of the conserved active site residues in RNA substrate binding and catalysis.
Our biochemical assays indicated that recombinant purified NOCT did not degrade RNA substrates in vitro. The truncation of NOCT’s N-terminus (residues 1–63) is unlikely to account for this lack of catalytic activity as the truncated protein has a fully formed catalytic domain that was equally active in repressing RNA in vivo (Figure 3). Moreover, similar truncations of yeast CCR4 and human CNOT6L and PDE12 are active in vitro (this study and (5,6,42)). To date, our attempts to detect binding of recombinant purified NOCT to poly(A) RNA have been negative. Based on these observations, we posit that NOCT requires one or more unidentified protein partners to be active. Such a partner may facilitate substrate binding, such as an RNA-binding protein that would direct NOCT to target mRNAs, thereby regulating its substrate specificity. Precedent for this idea is exemplified by the Pan2 deadenylase, which is inactive without its RNA-binding protein partner, Pan3 (67,68). The tethered function assay used in this study may bypass the necessity of such a partner. Additionally, it cannot be ruled out that NOCT binds to mRNAs by recognizing specific sequence motifs or three-dimensional RNA structures. At last, it is conceivable that NOCT activity may require post-translational modifications, which would be absent in the recombinant protein expressed in bacteria.
The CCR4-NOT deadenylase complex is a multiple subunit, multifunctional repressive complex that causes mRNA deadenylation, 5′ decay and translational repression (62,64,69–71). Our data suggest that NOCT may function in both translational repression and mRNA decay, functioning similarly to other described deadenylases. First, the H414A mutation diminishes NOCT-mediated decay of poly(A) reporter mRNA, indicating that its exoribonuclease function is important, yet this same mutant retains some translational repression activity (Figure 4). The translational repression activity of H414A is reduced relative to WT NOCT, suggesting that a combination of exonuclease activity and additional activity acts on polyadenylated target mRNAs. At this time, we cannot definitively discriminate the contributions of these mechanisms. Second, NOCT-mediated mRNA decay and translational repression activities are abrogated on the MALAT1 reporter, suggesting that both activities of NOCT are dependent on its ability to antagonize 3′ poly(A) and PABP function. The effect of NOCT on MALAT1 contrasts with that of CNOT7, which retains some of its inhibitory activity toward the MALAT1 mRNA, reflecting its previously described 5′ cap-dependent translational repression and mRNA decay mechanisms (62–64).
We expect that the insights into NOCT structure and function reported here will enhance understanding of its regulatory roles in lipid metabolism, adipogenesis, osteogenesis and circadian biology. Currently, all physiological NOCT studies have been performed in mice, and the regulatory roles of human NOCT remain to be explored. The high degree of conservation between human and murine NOCT (91% sequence identity) suggests that the enzymes are likely to have comparable regulatory functions. Murine NOCT is expressed in multiple tissues and its expression is influenced by feeding behavior, dietary fat intake and the circadian clock (16,19,27,29,72). How human NOCT is expressed and regulated remains unexplored. A crucial challenge is to identify the natural mRNAs that are bound and regulated by NOCT. Our findings indicate that NOCT-regulated mRNAs will have reduced levels and shorter half-lives. Consequently, these RNAs are unlikely to accumulate as deadenylated intermediates, which could explain difficulties in identifying direct NOCT targets by surveying increased poly(A) tail length in NOCT-deficient mice (73). NOCT repression is manifested in reduced protein expression from a target mRNA; therefore, quantitative proteomics approaches may also facilitate identification of its regulatory impact on natural target mRNAs. Such approaches, conducted in physiologically relevant tissues and cell types, will be necessary to discover the post-transcriptional regulatory networks that underlie NOCT’s important physiological functions.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank under accession numbers 6BT1 and 6BT2.
Supplementary Material
ACKNOWLEDGEMENTS
We thank R. Arvola for assistance with the Northern Blotting experiments; K. Hughes for critical reading of the manuscript; J. Wilusz for the MALAT1 sequences; R. Fick for assistance in structure determination; and the scientific staff at the LS-CAT beamlines at Argonne National Laboratories for data collection technical support.
Contributor Information
Elizabeth T Abshire, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
Jennifer Chasseur, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
Jennifer A Bohn, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
Paul A Del Rizzo, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
Lydia Freddolino, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA; Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA.
Aaron C Goldstrohm, Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN 55455, USA.
Raymond C Trievel, Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Edward Mallinckrodt Jr. Foundation Grant (to A.C.G.); University of Michigan Nutrition and Obesity Research Center Pilot Grant (supported by the National Institutes of Health (NIH) grant [DK089503 to R.C.T.]; American Heart Association Predoctoral Fellowship [16PRE26700002 to E.T.A.]; NIH Chemistry-Biology Training Program Fellowship [5T32GM008597 to E.T.A.]; NIH Cellular Biotechnology Training Program Fellowship [5T32GM008353 to J.B.]; National Institute of General Medical Sciences, NIH grant [R01GM105707 to A.C.G.]; U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory [DE-AC02-06CH11357]; Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor [085P1000817]. Funding for open access charge: University of Minnesota, Institutional Funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Garneau N.L., Wilusz J., Wilusz C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007; 8:113–126. [DOI] [PubMed] [Google Scholar]
- 2. Goldstrohm A.C., Wickens M. Multifunctional deadenylase complexes diversify mRNA control. Nat. Rev. Mol. Cell Biol. 2008; 9:337–344. [DOI] [PubMed] [Google Scholar]
- 3. Liu Q., Greimann J.C., Lima C.D. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell. 2006; 127:1223–1237. [DOI] [PubMed] [Google Scholar]
- 4. Geisler S., Coller J. XRN1: a Major 5′ to 3′ exoribonuclease in eukaryotic cells. Enzymes. 2012; 31:97–114. [DOI] [PubMed] [Google Scholar]
- 5. Wang H., Morita M., Yang X., Suzuki T., Yang W., Wang J., Ito K., Wang Q., Zhao C., Bartlam M.et al. Crystal structure of the human CNOT6L nuclease domain reveals strict poly(A) substrate specificity. EMBO J. 2010; 29:2566–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wood E.R., Bledsoe R., Chai J., Daka P., Deng H., Ding Y., Harris-Gurley S., Kryn L.H., Nartey E., Nichols J.et al. The role of phosphodiesterase 12 (PDE12) as a negative regulator of the innate immune response and the discovery of antiviral inhibitors. J. Biol. Chem. 2015; 290:19681–19696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schafer I.B., Rode M., Bonneau F., Schussler S., Conti E. The structure of the Pan2-Pan3 core complex reveals cross-talk between deadenylase and pseudokinase. Nat. Struct. Mol. Biol. 2014; 21:591–598. [DOI] [PubMed] [Google Scholar]
- 8. Virtanen A., Henriksson N., Nilsson P., Nissbeck M. Poly(A)-specific ribonuclease (PARN): an allosterically regulated, processive and mRNA cap-interacting deadenylase. Crit. Rev. Biochem. Mol. Biol. 2013; 48:192–209. [DOI] [PubMed] [Google Scholar]
- 9. Thore S., Mauxion F., Seraphin B., Suck D. X-ray structure and activity of the yeast Pop2 protein: a nuclease subunit of the mRNA deadenylase complex. EMBO Rep. 2003; 4:1150–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rorbach J., Nicholls T.J., Minczuk M. PDE12 removes mitochondrial RNA poly(A) tails and controls translation in human mitochondria. Nucleic Acids Res. 2011; 39:7750–7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Montellese C., Montel-Lehry N., Henras A.K., Kutay U., Gleizes P.E., O’Donohue M.F. Poly(A)-specific ribonuclease is a nuclear ribosome biogenesis factor involved in human 18S rRNA maturation. Nucleic Acids Res. 2017; 45:6822–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Albrecht U. The circadian clock, metabolism and obesity. Obes. Rev. 2017; 18(Suppl. 1):25–33. [DOI] [PubMed] [Google Scholar]
- 13. Baggs J.E., Green C.B. Nocturnin, a deadenylase in Xenopus laevis retina: a mechanism for posttranscriptional control of circadian-related mRNA. Curr. Biol. 2003; 13:189–198. [DOI] [PubMed] [Google Scholar]
- 14. Berthet C., Morera A.M., Asensio M.J., Chauvin M.A., Morel A.P., Dijoud F., Magaud J.P., Durand P., Rouault J.P. CCR4-associated factor CAF1 is an essential factor for spermatogenesis. Mol. Cell. Biol. 2004; 24:5808–5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dietrich P., Hellerbrand C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract. Res. Clin. Gastroenterol. 2014; 28:637–653. [DOI] [PubMed] [Google Scholar]
- 16. Green C.B., Douris N., Kojima S., Strayer C.A., Fogerty J., Lourim D., Keller S.R., Besharse J.C. Loss of Nocturnin, a circadian deadenylase, confers resistance to hepatic steatosis and diet-induced obesity. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:9888–9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakamura T., Yao R., Ogawa T., Suzuki T., Ito C., Tsunekawa N., Inoue K., Ajima R., Miyasaka T., Yoshida Y.et al. Oligo-astheno-teratozoospermia in mice lacking Cnot7, a regulator of retinoid X receptor beta. Nat. Genet. 2004; 36:528–533. [DOI] [PubMed] [Google Scholar]
- 18. Reverdatto S.V., Dutko J.A., Chekanova J.A., Hamilton D.A., Belostotsky D.A. mRNA deadenylation by PARN is essential for embryogenesis in higher plants. RNA. 2004; 10:1200–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Y., Osterbur D.L., Megaw P.L., Tosini G., Fukuhara C., Green C.B., Besharse J.C. Rhythmic expression of Nocturnin mRNA in multiple tissues of the mouse. BMC Dev. Biol. 2001; 1:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Washio-Oikawa K., Nakamura T., Usui M., Yoneda M., Ezura Y., Ishikawa I., Nakashima K., Noda T., Yamamoto T., Noda M. Cnot7-null mice exhibit high bone mass phenotype and modulation of BMP actions. J. Bone Miner. Res. 2007; 22:1217–1223. [DOI] [PubMed] [Google Scholar]
- 21. Christie M., Boland A., Huntzinger E., Weichenrieder O., Izaurralde E. Structure of the PAN3 pseudokinase reveals the basis for interactions with the PAN2 deadenylase and the GW182 proteins. Mol. Cell. 2013; 51:360–373. [DOI] [PubMed] [Google Scholar]
- 22. Zhang X., Devany E., Murphy M.R., Glazman G., Persaud M., Kleiman F.E. PARN deadenylase is involved in miRNA-dependent degradation of TP53 mRNA in mammalian cells. Nucleic Acids Res. 2015; 43:10925–10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van Etten J., Schagat T.L., Hrit J., Weidmann C.A., Brumbaugh J., Coon J.J., Goldstrohm A.C. Human Pumilio proteins recruit multiple deadenylases to efficiently repress messenger RNAs. J. Biol. Chem. 2012; 287:36370–36383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Raisch T., Bhandari D., Sabath K., Helms S., Valkov E., Weichenrieder O., Izaurralde E. Distinct modes of recruitment of the CCR4-NOT complex by Drosophila and vertebrate Nanos. EMBO J. 2016; 35:974–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blanco A.M., Gomez-Boronat M., Madera D., Valenciano A.I., Alonso-Gomez A.L., Delgado M.J. First evidences of nocturnin in fish: two isoforms in goldfish differentially regulated by feeding. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017; 314:R304–R312. [DOI] [PubMed] [Google Scholar]
- 26. Gronke S., Bickmeyer I., Wunderlich R., Jackle H., Kuhnlein R.P. Curled encodes the Drosophila homolog of the vertebrate circadian deadenylase Nocturnin. Genetics. 2009; 183:219–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garbarino-Pico E., Niu S., Rollag M.D., Strayer C.A., Besharse J.C., Green C.B. Immediate early response of the circadian polyA ribonuclease nocturnin to two extracellular stimuli. RNA. 2007; 13:745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baggs J.E., Green C.B. Functional analysis of nocturnin: a circadian clock-regulated gene identified by differential display. Methods Mol. Biol. 2006; 317:243–254. [DOI] [PubMed] [Google Scholar]
- 29. Douris N., Kojima S., Pan X., Lerch-Gaggl A.F., Duong S.Q., Hussain M.M., Green C.B. Nocturnin regulates circadian trafficking of dietary lipid in intestinal enterocytes. Curr. Biol. 2011; 21:1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kawai M., Green C.B., Lecka-Czernik B., Douris N., Gilbert M.R., Kojima S., Ackert-Bicknell C., Garg N., Horowitz M.C., Adamo M.L.et al. A circadian-regulated gene, Nocturnin, promotes adipogenesis by stimulating PPAR-gamma nuclear translocation. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:10508–10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guntur A.R., Kawai M., Le P., Bouxsein M.L., Bornstein S., Green C.B., Rosen C.J. An essential role for the circadian-regulated gene nocturnin in osteogenesis: the importance of local timekeeping in skeletal homeostasis. Ann. N.Y. Acad. Sci. 2011; 1237:58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 33. Roy A., Kucukural A., Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010; 5:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dolinsky T.J., Czodrowski P., Li H., Nielsen J.E., Jensen J.H., Klebe G., Baker N.A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007; 35:W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dolinsky T.J., Nielsen J.E., McCammon J.A., Baker N.A. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004; 32:W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Celniker G., Nimrod G., Ashkenazy H., Glaser F., Martz E., Mayrose I., Pupko T., Ben-Tal N. ConSurf: Using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 2013; 53:199–206. [Google Scholar]
- 39. Glaser F., Pupko T., Paz I., Bell R.E., Bechor-Shental D., Martz E., Ben-Tal N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003; 19:163–164. [DOI] [PubMed] [Google Scholar]
- 40. Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005; 33:W299–W302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., Ben-Tal N. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016; 44:W344–W350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Viswanathan P., Chen J., Chiang Y.C., Denis C.L. Identification of multiple RNA features that influence CCR4 deadenylation activity. J. Biol. Chem. 2003; 278:14949–14955. [DOI] [PubMed] [Google Scholar]
- 43. Plummer M. JAGS: a program for analysis of Bayesian graphical models using Gibbs sampling. Proceedings of the 3rd International Workshop on Distributed Statistical Computing. 2003; Vienna: Technische Universität Wien; 1–10. [Google Scholar]
- 44. Gelman A., Rubin D.B. Inference from iterative simulation using multiple sequences. Stat. Sci. 1992; 7:457–472. [Google Scholar]
- 45. Smith B.J. boa: an R Package for MCMC output convergence assessment and posterior inference. J. Stat. Softw. 2007; 21:1–37. [Google Scholar]
- 46. Juárez M.A., Steel M.F.J. Model-based clustering of non-gaussian panel data based on Skew-t distributions. J. Bus. Econ. Stati. 2010; 28:52–66. [Google Scholar]
- 47. Plummer M. rjags: Bayesian graphical models using MCMC. 2016; https://cran.r-project.org/web/packages/rjag
- 48. Linding R., Russell R.B., Neduva V., Gibson T.J. GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003; 31:3701–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hrit J., Raynard N., Van Etten J., Sankar K., Petterson A., Goldstrohm A.C. In vitro analysis of RNA degradation catalyzed by deadenylase enzymes. Methods Mol. Biol. 2014; 1125:325–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weaver T.M. The pi-helix translates structure into function. Protein Sci. 2000; 9:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kumar P., Bansal M. Identification of local variations within secondary structures of proteins. Acta Crystallogr. D Biol. Crystallogr. 2015; 71:1077–1086. [DOI] [PubMed] [Google Scholar]
- 52. Dlakic M. Functionally unrelated signalling proteins contain a fold similar to Mg2+-dependent endonucleases. Trends Biochem. Sci. 2000; 25:272–273. [DOI] [PubMed] [Google Scholar]
- 53. Romani A.M. Cellular magnesium homeostasis. Arch. Biochem. Biophys. 2011; 512:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bos T.J., Nussbacher J.K., Aigner S., Yeo G.W. Tethered function assays as tools to elucidate the molecular roles of RNA-Binding proteins. Adv. Exp. Med. Biol. 2016; 907:61–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Coller J., Wickens M. Tethered function assays: an adaptable approach to study RNA regulatory proteins. Methods Enzymol. 2007; 429:299–321. [DOI] [PubMed] [Google Scholar]
- 56. Coller J., Wickens M. Tethered function assays using 3′ untranslated regions. Methods. 2002; 26:142–150. [DOI] [PubMed] [Google Scholar]
- 57. Viswanathan P., Ohn T., Chiang Y.C., Chen J., Denis C.L. Mouse CAF1 can function as a processive deadenylase/3′-5′-exonuclease in vitro but in yeast the deadenylase function of CAF1 is not required for mRNA poly(A) removal. J. Biol. Chem. 2004; 279:23988–23995. [DOI] [PubMed] [Google Scholar]
- 58. Aslam A., Mittal S., Koch F., Andrau J.C., Winkler G.S. The Ccr4-NOT deadenylase subunits CNOT7 and CNOT8 have overlapping roles and modulate cell proliferation. Mol. Biol. Cell. 2009; 20:3840–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Doidge R., Mittal S., Aslam A., Winkler G.S. The anti-proliferative activity of BTG/TOB proteins is mediated via the Caf1a (CNOT7) and Caf1b (CNOT8) deadenylase subunits of the Ccr4-not complex. PLoS One. 2012; 7:e51331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wilusz J.E. Long noncoding RNAs: re-writing dogmas of RNA processing and stability. Biochim. Biophys. Acta. 2016; 1859:128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brown J.A., Bulkley D., Wang J., Valenstein M.L., Yario T.A., Steitz T.A., Steitz J.A. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat. Struct. Mol. Biol. 2014; 21:633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cooke A., Prigge A., Wickens M. Translational repression by deadenylases. J. Biol. Chem. 2010; 285:28506–28513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Behm-Ansmant I., Rehwinkel J., Doerks T., Stark A., Bork P., Izaurralde E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006; 20:1885–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Waghray S., Williams C., Coon J.J., Wickens M. Xenopus CAF1 requires NOT1-mediated interaction with 4E-T to repress translation in vivo. RNA. 2015; 21:1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zinder J.C., Lima C.D. Targeting RNA for processing or destruction by the eukaryotic RNA exosome and its cofactors. Genes Dev. 2017; 31:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Freudenthal B.D., Beard W.A., Cuneo M.J., Dyrkheeva N.S., Wilson S.H. Capturing snapshots of APE1 processing DNA damage. Nat. Struct. Mol. Biol. 2015; 22:924–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Brown C.E., Tarun S.Z. Jr., Boeck R., Sachs A.B. PAN3 encodes a subunit of the Pab1p-dependent poly(A) nuclease in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996; 16:5744–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wolf J., Valkov E., Allen M.D., Meineke B., Gordiyenko Y., McLaughlin S.H., Olsen T.M., Robinson C.V., Bycroft M., Stewart M.et al. Structural basis for Pan3 binding to Pan2 and its function in mRNA recruitment and deadenylation. EMBO J. 2014; 33:1514–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fabian M.R., Frank F., Rouya C., Siddiqui N., Lai W.S., Karetnikov A., Blackshear P.J., Nagar B., Sonenberg N. Structural basis for the recruitment of the human CCR4-NOT deadenylase complex by tristetraprolin. Nat. Struct. Mol. Biol. 2013; 20:735–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fabian M.R., Cieplak M.K., Frank F., Morita M., Green J., Srikumar T., Nagar B., Yamamoto T., Raught B., Duchaine T.F.et al. miRNA-mediated deadenylation is orchestrated by GW182 through two conserved motifs that interact with CCR4-NOT. Nat. Struct. Mol. Biol. 2011; 18:1211–1217. [DOI] [PubMed] [Google Scholar]
- 71. Fabian M.R., Mathonnet G., Sundermeier T., Mathys H., Zipprich J.T., Svitkin Y.V., Rivas F., Jinek M., Wohlschlegel J., Doudna J.A.et al. Mammalian miRNA RISC recruits CAF1 and PABP to affect PABP-dependent deadenylation. Mol. Cell. 2009; 35:868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gilbert M.R., Douris N., Tongjai S., Green C.B. Nocturnin expression is induced by fasting in the white adipose tissue of restricted fed mice. PLoS One. 2011; 6:e17051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kojima S., Gendreau K.L., Sher-Chen E.L., Gao P., Green C.B. Changes in poly(A) tail length dynamics from the loss of the circadian deadenylase Nocturnin. Sci. Rep. 2015; 5:17059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank under accession numbers 6BT1 and 6BT2.