

Discovery of disease age-of-onset modifiers is important for clinical trials and drug design. Zhang et al. perform a genome-wide analysis of epigenetic functional polymorphisms and identify an association between the C6orf10/LOC101929163 locus and age of FTD/ALS onset. The risk allele may be associated with a pro-inflammatory state in the brain.
Keywords: C9orf72, genetic association, age of onset, amyotrophic lateral sclerosis, frontotemporal dementia
Abstract
The G4C2-repeat expansion in C9orf72 is the most common known cause of amyotrophic lateral sclerosis and frontotemporal dementia. The high phenotypic heterogeneity of C9orf72 patients includes a wide range in age of onset, modifiers of which are largely unknown. Age of onset could be influenced by environmental and genetic factors both of which may trigger DNA methylation changes at CpG sites. We tested the hypothesis that age of onset in C9orf72 patients is associated with some common single nucleotide polymorphisms causing a gain or loss of CpG sites and thus resulting in DNA methylation alterations. Combined analyses of epigenetic and genetic data have the advantage of detecting functional variants with reduced likelihood of false negative results due to excessive correction for multiple testing in genome-wide association studies. First, we estimated the association between age of onset in C9orf72 patients (n = 46) and the DNA methylation levels at all 7603 CpG sites available on the 450 k BeadChip that are mapped to common single nucleotide polymorphisms. This was followed by a genetic association study of the discovery (n = 144) and replication (n = 187) C9orf72 cohorts. We found that age of onset was reproducibly associated with polymorphisms within a 124.7 kb linkage disequilibrium block tagged by top-significant variation, rs9357140, and containing two overlapping genes (LOC101929163 and C6orf10). A meta-analysis of all 331 C9orf72 carriers revealed that every A-allele of rs9357140 reduced hazard by 30% (P = 0.0002); and the median age of onset in AA-carriers was 6 years later than GG-carriers. In addition, we investigated a cohort of C9orf72 negative patients (n = 2634) affected by frontotemporal dementia and/or amyotrophic lateral sclerosis; and also found that the AA-genotype of rs9357140 was associated with a later age of onset (adjusted P = 0.007 for recessive model). Phenotype analyses detected significant association only in the largest subgroup of patients with frontotemporal dementia (n = 2142, adjusted P = 0.01 for recessive model). Gene expression studies of frontal cortex tissues from 25 autopsy cases affected by amyotrophic lateral sclerosis revealed that the G-allele of rs9357140 is associated with increased brain expression of LOC101929163 (a non-coding RNA) and HLA-DRB1 (involved in initiating immune responses), while the A-allele is associated with their reduced expression. Our findings suggest that carriers of the rs9357140 GG-genotype (linked to an earlier age of onset) might be more prone to be in a pro-inflammatory state (e.g. by microglia) than AA-carriers. Further, investigating the functional links within the C6orf10/LOC101929163/HLA-DRB1 pathway will be critical to better define age-dependent pathogenesis of frontotemporal dementia and amyotrophic lateral sclerosis.
Introduction
The G4C2-repeat expansion in C9orf72 is the most common known cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Gijselinck et al., 2012) in Caucasians. It accounts for about 37% familial and 7% sporadic ALS patients; as well as 25% familial and 6% sporadic FTD patients (Rademakers, 2012) with age and sex dependent disease penetrance (Murphy et al., 2017). High phenotypic heterogeneity of C9orf72 patients also includes a wide range in disease age of onset (27–74 years) and duration (0.5–22 years) (Gijselinck et al., 2016). Yet, genetic modifiers of age of onset in C9orf72 patients are largely unknown [only the T-allele of rs1990622 in TMEM106B was associated with a later age of onset of FTD, but not ALS (Gallagher et al., 2014; van Blitterswijk et al., 2014)]. Detection of the age of onset modifier(s) might increase the accuracy of predicting age of onset in asymptomatic mutation carriers, which is important for clinical trials focused on early intervention.
Age of onset could be influenced by genetic and environmental modifiers, both of which may trigger epigenetic changes, such as DNA methylation at CpG sites (Zhang et al., 2016). Indeed, there is no a strict dichotomy between action of genetic and epigenetic factors; they often work in concert. Genome-wide DNA methylation profiles of identical twins are much more similar than between fraternal siblings (Zhang et al., 2016), demonstrating that many epigenetic changes are genetically controlled (e.g. the repeat expansion causes hypermethylation of the C9orf72 locus leading to downregulation of C9orf72 expression) (Xi et al., 2015b; Gijselinck et al., 2016). The DNA methylation levels of some CpGs are age-related allowing the estimation of DNA methylation age based on the cumulative assessment of 353 CpGs included on the genome-wide 450K BeadChip. Currently, DNA methylation age is the most accurate predictor of chronological age across multiple tissues (Horvath, 2013), but may in fact reflect biological age better than chronological age. Indeed, we recently reported that increased DNA methylation age acceleration (DNA methylation age minus chronological age) is associated with earlier age of onset in C9orf72 patients analysed on the 450 K BeadChip after exclusion of CpGs mapped to common single nucleotide polymorphisms (SNPs) (Zhang et al., 2017).
CpGs are the most mutable sites in the human genome because methyl-C can spontaneously deaminate to T (e.g. 35% of all coding mutations occur at CpG sites) (Lek et al., 2016). Hence, in the current study we tested the hypothesis that age of onset in C9orf72 patients is associated with some common SNPs causing a gain or loss of CpG sites (CpG-SNPs) and thus resulting in DNA methylation changes. Allele-specific DNA methylation is largely attributed to CpG-SNPs (Shoemaker et al., 2010), which have often been detected within promoter regions, transcription factor binding sites and DNase I hypersensitive sites (Gagliano et al., 2016), thus regulating the level of gene expression. CpG-SNPs belong to a group of methylation quantitative trait loci (Hannon et al., 2016), which are linked to some mental disorders (Gagliano et al., 2016).
We combined epigenetic and genetic approaches to map functional variants (CpG-SNPs) associated with age of onset in C9orf72 carriers. Such a study design reduces the likelihood of false negative results due to excessive correction for multiple testing in genome-wide association studies (GWASs). Our study also includes suggestions on how the significant SNPs exert their effects (e.g. by influencing gene expression).
Materials and methods
Participants
Informed consent was obtained from all participants in accordance with the respective ethics review boards. Sample characteristics are presented in Table 1 and Supplementary Table 1 for C9orf72 carriers, and Supplementary Table 2 for C9orf72 negative patients. Briefly, our study included patients diagnosed with bulbar or limb onset ALS, behavioural FTD (bvFTD), semantic dementia, progressive non-fluent aphasia (PNFA), and FTD-ALS. All patients were of Caucasian origin and diagnosed at hospitals specializing in neurodegenerative disorders using established clinical criteria for ALS (Brooks et al., 2000) and FTD (Neary et al., 1998), including the revised diagnostic criteria for bvFTD (Rascovsky et al., 2011) and language variants of FTD (Gorno-Tempini et al., 2011). Age of onset was defined as the age at which the first disease symptoms appeared, including initial bulbar or limb symptoms in ALS, and cognitive dysfunction in judgement, language, memory, or changes in behaviour or personality in FTD. Age of onset was either self-reported (for ALS) or obtained from unaffected family members (for FTD).
Table 1.
Sample characteristics of the discovery and replication C9orf72 datasets
| Discovery cohort | Replication cohort | |||
|---|---|---|---|---|
| Unrelated carriers | Symptomatic carriers from 16 families | Asymptomatic carriers from 16 families | Unrelated carriers | |
| Number of cases | 101 | 21 | 22 | 187 |
| Sex, male, n (%) | 55 (54.4) | 10 (47.6) | 12 (45.5) | 104 (55.6) |
| Age of onset, years, median (IQR) | 59 (54–66) | 55 (48–60) | NA | 58 (51–63) |
| Age of onset, years, mean (range) | 59.82 (37–78) | 54.86 (38–73) | NA | 57.2 (34–80) |
NA = not applicable.
The discovery cohort was recruited from Canada, Italy, Spain, UK, USA or Argentina and consisted of 144 C9orf72 carriers, including 21 symptomatic and 22 asymptomatic carriers from 16 pedigrees. The independent replication cohort was obtained from centres (different from those that collected the discovery cohort) participating in the International FTD-Genomics Consortium (IFGC; https://ifgcsite.wordpress.com/) (Ferrari et al., 2014). It consisted of 187 unrelated FTD or FTD-ALS C9orf72 carriers from the USA, Canada, UK, France, Belgium, Italy, Germany, Spain, Sweden, the Netherlands and Australia. Information about family relatedness was obtained from the clinical notes of the neurologists who collected the samples. In addition, the presence of relatedness in the replication cohort was previously assessed as part of a GWAS that identified and excluded all first-degree relatives (through identity by descent for any pair with an estimate <0.125) (Ferrari et al., 2014).
For a follow-up study of unrelated C9orf72 negative patients, we investigated 2142 FTD and 164 FTD-ALS patients from the IFGC (Ferrari et al., 2014), as well as 328 sporadic ALS patients from the ALS clinic at Sunnybrook Health Sciences Centre, Toronto (Supplementary Table 2), which also provided frontal cortex from 25 unrelated ALS autopsy cases without an expansion in C9orf72 (<30 repeats) for the gene expression studies (Supplementary Table 3).
Procedures
Blood genomic DNA was extracted using a QIAGEN kit. First, we analysed the genome-wide DNA methylation data from the 450K BeadChip (Illumina) that was previously generated using bisulfite converted DNA of 46 Canadian C9orf72 carriers (Zhang et al., 2017) to discover common CpG-SNPs with minor allele frequencies >5% that are associated with age of onset. The raw data were preprocessed and analysed using the minfi package in R-project (Aryee et al., 2014). The β-value was used to estimate the DNA methylation level of each CpG-site (β-value of 0: non-methylated; β-value of 1: completely methylated).
All participants of the discovery and replication cohorts (n = 331) were carriers of an expansion in C9orf72 (>30 repeats) based on previous analysis by repeat-primed PCR (Ferrari et al., 2014; Xi et al., 2015b). Genotypes for rs9357140, rs2143466 and rs1990622 were obtained by Sanger sequencing in the discovery cohort (Supplementary Table 4). For the replication dataset, these SNPs together with eight SNPs in strong linkage disequilibrium (LD) with rs9357140 and rs2143466 (R2 > 0.9) were either genotyped or imputed using the latest data from the Haplotype Reference Consortium (McCarthy et al., 2016) (Supplementary Table 5).
Genotypes for rs9357140 in a follow-up cohort of 2634 unrelated C9orf72 negative patients with ALS, FTD or FTD-ALS, were obtained by TaqManTM assay (C___9782529_10, ThermoFisher Scientific) for 328 ALS patients, or imputed from IFGC-GWAS for 2306 FTD and FTD-ALS patients (Ferrari et al., 2014) using the latest data from the Haplotype Reference Consortium (McCarthy et al., 2016).
To measure the degree of LD, we extracted R2 values (range from 0 to 1 with higher values indicating a higher LD) from the LDlink tool (https://analysistools.nci.nih.gov/LDlink) using the 1000 Genomes European population data. We searched for known variants within the boundaries of the LD block (R2 > 0.8) tagged by the top significant SNP (rs9357140) using the ‘proxy search’ in LDlink. Functional predictions for the missense SNPs were based on the PolyPhen-2 and SIFT data available from the Exome Aggregation Consortium database (Lek et al., 2016). Using the UCSC genome browser, the LD-block was also analysed for transcriptional factor binding sites and DNase I hypersensitivity.
To detect genes whose expression is associated with rs9357140, we searched for expression quantitative trait loci (eQTL) using Genotype-Tissue Expression (GTEx v7) data from 48 types of human tissues (GTEx Consortium et al., 2017). The GTEx portal (https://www.gtexportal.org/) was used to analyse the association between rs9357140 genotypes and gene expression by a linear regression method. Normalized effect size (NES) was defined as the slope of the linear regression.
To quantify gene expression, total RNA was extracted from human frontal cortex of ALS cases without C9orf72 expansions using the QIAzol plus RNeasy® Mini Kit (QIAGEN) and reverse transcribed to cDNA using oligo dT primers and the AffinityScript Multiple Temperature cDNA Synthesis Kit (Agilent Technologies). Quantitative RT-PCR was conducted for 25 samples (Supplementary Table 3) with an RNA integrity number >6.5 (based on an Agilent 2100 Bioanalyzer). To select endogenous control genes for the frontal cortex, we assessed four housekeeping genes including HPRT1 (MIM: 308000; Hs99999909_m1), UBC (MIM: 191340; Hs00824723_m1), B2M (MIM: 109700; Hs99999907_m1), and RPLP0 (MIM: 180510; Hs00420895_gH) (ThermoFisher Scientific) in nine samples (n = 3 per each rs9357140 genotype). We used Normfinder (Andersen et al., 2004) to identify the least variable housekeeping genes (B2M and RPLP0) in our samples (Supplementary Table 6). We measured expression of HLA-DRB1 transcript variant 1 (MIM:142857; Hs04192464_mH) and all C9orf72 transcripts (MIM:614260; Hs00376619_m1) (ThermoFisher Scientific) in triplicate for 25 samples with different rs9357140 genotypes: AA (n = 9), AG (n = 8) and GG (n = 8). Relative quantification was calculated with the ddCt method by geometric mean of housekeeping gene expression (B2M and RPLP0).
Statistical analyses
We used the linear regression model of the R minfi package to assess the genome-wide association between the DNA methylation status of CpG-SNPs and age of onset in C9orf72 patients, as well as to evaluate the false discovery rate to generate adjusted q-values (Zhang et al., 2017). We used a Manhattan plot to prioritize significant variants (P < 0.01 and q < 0.05) for further genetic study, and a Q-Q plot to highlight potential confounders using the R qqman package (Turner, 2018).
To assess if genotypes affect age of onset, we used a Cox proportional hazard regression model (R survival and survminer packages) (Grambsch, 2000) adjusting for sex, rs1990622 genotypes, disease phenotypes, and censoring age of last follow-up for the 22 currently asymptomatic C9orf72 carriers. To adjust for relatedness in the Cox proportional hazard regression analysis of the discovery cohort, we created an indicator number for each family; then used the coxph function of the R coxme package with a frailty approach (Ripatti and Palmgren, 2000). The hazard ratio (HR) with 95% confidence interval (CI) is presented. To analyse the association between genotypes and age of onset in the C9orf72 disease subgroups, we used multivariate linear regression with an additive, dominant or recessive model adjusting for sex, rs1990622 genotypes, or DNA methylation age-acceleration. We also used multivariate linear regression to analyse the association between genotypes and age of onset in C9orf72 negative disease subgroups (adjusting for sex). We present the linear regression coefficient (B) with standard error (SE) and percentage of response variance explained by the linear regression model (r2). Results of additive model were presented, unless otherwise specified.
We used a meta-analysis (R metafor package) with a fixed-effect model to assess the pooled effect size of the Cox regression coefficient (logHR) from the discovery and replication stages (Trinh et al., 2016). We performed a trend analysis using the Cochran–Armitage test to analyse if rs9357140 genotypes are associated with C9orf72 disease subgroups. A non-parametric Mann-Whitney U-test or Kruskal-Wallis test was used to assess differences in age of onset or gene expression among two or more groups where appropriate. Sex and rs1990622 genotype adjusted P-values are shown, unless otherwise specified. The results with P < 0.05 were accepted as statistically significant.
Data availability
The data that support the findings of this study are available on request from the corresponding authors (E.R., M.Z.). The data are not publicly available because of information that could compromise the privacy of the research participants.
Results
Epigenetic analysis suggested CpG-SNPs associated with age of onset
The study design is presented in Fig. 1. First, we estimated the association between age of onset in a Canadian cohort of 46 unrelated C9orf72 patients and DNA methylation levels at 7603 common CpG-SNPs available on the 450K BeadChip. Age of onset was significantly associated with DNA methylation levels (q < 0.05) at three CpG-SNPs (rs12763379 on 10q24.2; rs9357140 and rs2143466 on 6p21.3): P = 9.6 × 10−6, P = 6.0 × 10−6 and P = 1.8 × 10−5, respectively (Fig. 2A and Supplementary Table 7). However, rs12763379 in PYROXD2 was removed from follow-up study because of its overlap with insertion/deletion variations and single tandem repeats precluding reliable genotyping.
Figure 1.

Flow chart of the study design. AO = age of onset.
Figure 2.

Genome-wide DNA methylation analysis of the CpG-SNPs in C9orf72 patients. (A) Manhattan plot presenting the association between DNA methylation status of CpG-SNPs and age of onset, including a locus on chr6:32160000–32580000 with two age of onset-associated CpG-SNPs (rs9357140 and rs2143466 indicated by the box). Arrows indicate the transcriptional direction of each gene (5′ to 3′). ‘Me’ in red represent methylation sites controlled by rs9357140 and rs2143466. The LD block tagged by rs9357140 (R2 > 0.8) is highlighted in green. (B) Genotypes of rs9357140 are significantly associated with DNA methylation status: P < 1.0 × 10−6, B = −0.39 (SE: 0.01); and age of onset: P = 2.2 × 10−5 adjusted for sex and rs1990622 genotypes, B = 7.01 (SE: 1.47). The dashed line represents the linear regression trend.
Genetic association study confirmed the association between rs9357140/rs2143466 and age of onset
Multivariate linear regression suggested that rs9357140 genotypes control the gain or loss of DNA methylation at CpG-site cg18698799 (P < 1.0 × 10−6), thereby underlying the association with age of onset: adjusted P = 2.2 × 10−5, B = 7.01 (SE: 1.47) (Fig. 2B). The association remained significant after adjusting for DNA methylation age-acceleration: P = 2.7 × 10−4, B = 6.72 (SE: 1.68). AA-carriers have significantly lower DNA methylation levels compared to AG-carriers (P = 2.2 × 10−5, Mann-Whitney U-test) or GG-carriers (P = 4.7 × 10−5, Mann-Whitney U-test); mean β-value: 0.04 (AA-carriers) versus 0.54 (AG-carriers) versus 0.88 (GG-carriers) (Fig. 2B). Similar results were observed for rs2143466 (Supplementary Fig. 1). The Q-Q plot suggested that there are no other confounders for the association (Supplementary Fig. 2). Both SNPs belong to a strong 124.7 kb LD-block (R2 > 0.8) on chr6:32213638–32338386 containing two overlapping genes: a long non-coding RNA (LOC101929163) and C6orf10—an uncharacterized testes-specific gene with rs9357140 mapped to intron 9 and rs2143466 mapped to intron 14 (Fig. 2).
Next, we enlarged our discovery dataset to 144 carriers by genotyping rs9357140 and rs2143466 in 98 recently collected C9orf72 carriers, including 101 unrelated symptomatic carriers and 16 families with 21 symptomatic and 22 asymptomatic C9orf72 carriers (Fig. 1 and Table 1). To obtain the median age of onset for different SNP genotypes, we used the Kaplan-Meier estimate, censoring age of last follow-up for asymptomatic carriers. The median age of onset difference between rs9357140 AA- and GG-carriers was 12 years: 67 years for AA (95% CI: 60–71), 59 years for AG (95% CI: 56–64) and 55 years for GG genotype (95% CI: 54–60) (Fig. 3A). Cox proportional hazard regression analysis also revealed that age of onset in C9orf72 carriers is significantly associated with rs9357140 genotypes: adjusted P = 1.1 × 10−4, HR = 0.43 (95% CI: 0.28-0.66), suggesting that every A-allele could reduce hazard by 57% (Fig. 3B and Table 2). A similar association with age of onset was also observed for rs2143466: adjusted P = 1.1 × 10−4, HR = 0.43 (95% CI: 0.28–0.68) (Supplementary Fig. 3).
Figure 3.

The association between rs9357140 genotypes and age of onset in C9orf72 carriers. (A) Kaplan-Meier curve of cumulative incidence of disease onset in the discovery cohort (n = 144) stratified by rs9357140 genotypes. (B) Meta-analysis of the Cox regression coefficient from the discovery cohort (n = 144) and the replication cohort (n = 187). The regression coefficient equals logHR.
Table 2.
The Cox proportional hazard regression results for the association between the rs9357140 genotypes and age of onset in the discovery and replication cohorts
| Discovery (n = 144) | Replication (n = 187) | |
|---|---|---|
| HR (95% CI) | 0.59 (0.45–0.77) | 0.79 (0.64–0.98) |
| P-value for AA versus AG versus GG | 0.0001 | 0.029 |
| Adjusted HR (95% CI)* | 0.43 (0.28–0.68) | 0.79 (0.64–0.98) |
| Adjusted P-value for AA versus AG versus GG* | 0.00011 | 0.03 |
*The hazard ratio (HR) and P-value was adjusted for sex, rs1990622 genotypes, disease phenotypes and family relationship in the discovery stage. HR was adjusted for sex, rs1990622 genotypes and disease phenotypes in the replication stage.
The replication study validated the association between rs9357140/rs2143466 and age of onset
In the replication stage (Fig. 1 and Table 1), we obtained genotypes from the IFGC-GWAS (Ferrari et al., 2014) for 10 SNPs tagged by rs9357140 (R2 > 0.9) (Supplementary Table 5) for 187 C9orf72 patients with a median age of onset of 58 years and interquartile range (IQR) of 51–80 years. Cox proportional hazard regression analysis showed that age of onset was significantly associated with rs9357140: adjusted P = 0.03, HR = 0.79 (95% CI: 0.64–0.98), suggesting that every A-allele reduced hazard by 21% (Table 2 and Fig. 3). As expected, similar associations with age of onset were observed for rs2143466: adjusted P = 0.025, HR = 0.79 (95% CI: 0.64–0.97) (Supplementary Fig. 3) and for the other eight SNPs within the LD block listed in Supplementary Table 5 (data not shown).
Meta-analysis revealed overall effect of rs9357140/rs2143466 on age of onset
We conducted a meta-analysis of logHR in all 331 C9orf72 carriers using a fixed-effects model and observed that every A-allele of rs9357140 reduced hazard by 30% (pooled HR = 0.70, P = 0.0003) (Fig. 3B). Again, a similar effect was observed for rs2143466 (pooled HR = 0.70, P = 0.0002) (Supplementary Fig. 3). The association between age of onset and rs9357140 was also significant in 304 unrelated C9orf72 patients: adjusted P = 2.3 × 10−6, B = 3.2 (SE: 0.67) (Supplementary Table 1). The median age of onset of rs9357140 AA-carriers was 6 years later than GG-carriers: 62 years (IQR: 57–68) versus 56 years (IQR: 50–62).
Subgroup analyses of the association between rs9357140 and age of onset
The association between age of onset and rs9357140 was evident in unrelated C9orf72 patients with either pure ALS (n = 59; adjusted P = 0.002, B = 4.97, SE: 1.53) or pure FTD (n = 174; adjusted P = 0.0008, B = 2.82, SE: 0.83), but not in patients with FTD-ALS (n = 71; adjusted P = 0.125, B = 2.63, SE: 1.69) (Supplementary Table 1 and Supplementary Fig. 4A–C). A similar result was observed for rs2143466 (Supplementary Fig. 4D–F). Notably, we found no significant difference in age of onset among patients affected by pure ALS, pure FTD or FTD-ALS; or ALS/FTD subtypes (bulbar ALS, limb ALS, unspecified ALS, bvFTD, semantic dementia, PNFA, unspecified FTD): P > 0.05, Kruskal-Wallis test (Supplementary Fig. 5). Multivariate linear regression analysis in different disease subtypes revealed that age of onset was associated with rs9357140 genotypes in limb ALS (n = 35) and bulbar ALS (n = 23) under a dominant model (adjusted P < 0.05), and bvFTD (n = 157) under a recessive model (adjusted P < 0.05), but not in FTD-ALS patients (n = 71) (Supplementary Table 1).
To evaluate if rs9357140 genotypes modify disease phenotypes, we performed a trend analysis using the Cochran–Armitage test to analyse the association between rs9357140 genotypes and C9orf72 disease phenotypes (ALS versus FTD, ALS versus FTD-ALS, or FTD versus FTD-ALS) under an additive model (AA versus AG versus GG) (Supplementary Table 8); and found no statistically significant results (P > 0.05).
Age of onset in C9orf72 negative patients is associated with rs9357140
We analysed 2634 C9orf72-negative patients with ALS, FTD or FTD-ALS (Supplementary Table 2), and found a significant association between rs9357140 genotypes and age of onset (adjusted P = 0.007 for recessive model) (Supplementary Table 2). Subgroup analysis detected a significant association only in the largest subgroup of FTD patients (n = 2142, adjusted P = 0.01 for recessive model) with a small effect size (B = 1.44, SE: 0.55) (Supplementary Table 2). The association is evident in the bvFTD patients (n = 1364, adjusted P = 0.035 for recessive model), but not the other smaller FTD subtypes (Supplementary Table 2). We also observed that age of onset differed significantly among the FTD subtypes (P < 0.01, Kruskal-Wallis test). Cox proportional hazard regression analysis revealed that the AA-genotype is associated with age of onset [P = 0.036, adjusted for sex and FTD subtype; HR = 0.94 (95% CI: 0.88–0.99)], suggesting that the AA-genotype could reduce hazard by 6% relative to the GG- and AG-genotypes. Kaplan-Meier estimate analysis revealed that AA-carriers have a slightly later median age of onset (63 years in AA-carriers versus 62 years in GG + AG-carriers) (Fig. 4). A similar association with age of onset was also observed for rs2143466 in the C9orf72 negative FTD patients: P = 0.036, adjusted for sex and FTD subtype; HR = 0.94 (95% CI: 0.88–0.99).
Figure 4.
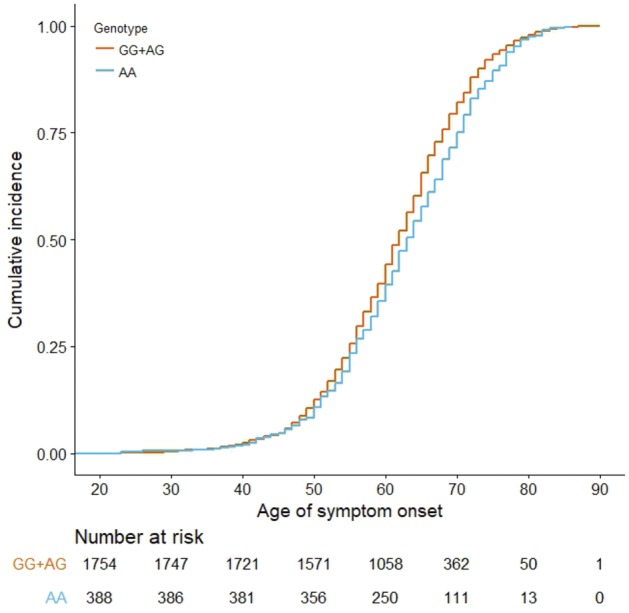
Kaplan-Meier curve of cumulative incidence of disease age of onset in 2142 C9orf72-negative patients with FTD stratified by rs9357140 genotype (AA versus GG+AG).
The expression of HLA-DRB1 and LOC101929163 is associated with rs9357140
Since rs9357140 and rs2143466 control the loss or gain of CpG-sites and therefore DNA methylation levels (Fig. 2), we hypothesized that CpG-SNPs at the C6orf10/LOC101929163 locus may modulate age of onset by regulating gene expression. We used the public eQTL dataset of 48 types of human tissue (GTEx portal) to analyse if genotypes of the top-significant tagging SNP (rs9357140) are associated with the expression of nearby genes (10 top-significant hits are shown in Supplementary Table 9). Among brain tissues, the A-allele of rs9357140 was associated with reduced expression of the LOC101929163 (P = 7.6 × 10−6, NES = −0.66 in the nucleus accumbens, part of the basal ganglia; Supplementary Fig. 6A); and HLA-DRB1, encoding major histocompatibility complex, class II, DR beta 1 (P = 4.1 × 10−6, NES = −0.42 in the frontal cortex; Supplementary Fig. 6B).
To validate the link between rs9357140 genotypes and HLA-DRB1 expression, we conducted quantitative RT-PCR using frontal cortex from 25 unrelated ALS cases (Supplementary Fig. 6C). Mann-Whitney U-test confirmed that AA-carriers had significantly lower HLA-DRB1 expression compared to AG-carriers (P = 0.001) or GG-carriers (P = 0.000003). Of note, C9orf72 expression did not differ among the rs9357140 genotypes (P > 0.05, Mann-Whitney U-test, Supplementary Fig. 7) and was not correlated with HLA-DRB1 expression (adjusted P = 0.23, linear regression).
Bioinformatics analysis predicted multiple DNase I hypersensitivity sites within the LD-block associated with age of onset
The LD-block associated with age of onset contains 196 known variants tagged by rs9357140 (R2 > 0.8), including five missense substitutions with minor allele frequencies of 0.36–0.38 and conflicting functional predictions by PolyPhen-2 and SIFT (Supplementary Table 10). Since C6orf10 is expressed mainly in testes, its coding variability is likely not relevant to C9orf72 pathology. We found no transcriptional factor binding sites mapped to any of the 196 SNPs; however, 12 of these SNPs are located at DNase I hypersensitivity sites; including rs9268000 (78 kb upstream of rs9357140) with a high cluster score of 1000 (Supplementary Table 10). These results suggest that age of onset modifiers of C9orf72 disease could be associated with DNase I hypersensitivity sites and HLA-DRB1 expression (250 kb away from the investigated LD-block, Fig. 2).
Discussion
The current study combined epigenetic and genetic data to detect functional variants associated with age of onset in a large dataset of 331 C9orf72 carriers. A DNA methylation study of CpG-SNPs in the discovery stage enabled prioritizing age of onset modifiers linked to DNA methylation status for further genetic investigation. Such a novel strategy has the advantage of reducing noise from GWAS signals. Indeed, CpG-SNPs could help rank GWAS hits (Gagliano et al., 2016), as they are important elements of methylation quantitative trait loci (Hannon et al., 2016). Our genome-wide DNA methylation analysis of CpG-SNPs followed by a genetic association study of the discovery and replication C9orf72 cohorts revealed that age of onset is associated with SNPs within a 124.7 kb LD-block tagged by rs9357140. Overall, every A-allele of rs9357140 may reduce hazard by 30% (the median age of onset of AA-carriers was 6 years later than GG-carriers). The genotypes of rs9357140 were also moderately associated with age of onset in C9orf72-negative patients although the effect size was small (e.g. the median age of onset in AA-carriers affected by FTD was 1 year later than GG-carriers).
Recently, a key tool for connecting phenotypes to genetic variations has emerged from gene expression studies. Since the locus with significant SNPs may not be the actual disease-related target, cis-acting eQTLs can provide a mechanistic link between SNPs and the biological processes they affect (GTEx Consortium et al., 2017). In our study, the minor A-allele of rs9357140 (top-significant SNP within the C6orf10/LOC101929163 locus) is associated with reduced brain expression of LOC101929163 (in nucleus accumbens) and HLA-DRB1 (in frontal cortex), while the major G-allele is associated with their increased expression (Supplementary Fig. 6). Future functional studies have to investigate if the non-coding RNA LOC101929163 is a modulator of HLA-DRB1 expression (e.g. affecting transcriptional factors relevant to HLA-DRB1). The major histocompatibility complex class II protein HLA-DR is implicated in neurodegenerative diseases as a marker of activated microglia (Walker and Lue, 2015) and is important in initiating immune responses by presenting peptides derived not only from exogenous but also endogenous proteins, such as peptides resulting from autophagy of intracellular proteins by lysosomes (Dengjel et al., 2005).
Our results support the notion that microglial/autophagy pathways play key roles in modulating C9orf72 disease, the pathogenesis of which might involve both gain and loss of function mechanisms (Hardy and Rogaeva, 2014). Normal function of C9orf72 is essential for the lysosome/autophagosome pathway and immune responses in macrophages or microglia (O’Rourke et al., 2016; Shi et al., 2018). For instance, transcriptome and histologic analyses of C9orf72 carriers support the idea that decreased C9orf72 expression leads to altered microglial function and neuroinflammation (O’Rourke et al., 2016), while increased C9orf72 levels could be neuroprotective (McGoldrick et al., 2018; Shi et al., 2018). It is important to investigate if rs9357140 GG-carriers, which have an earlier age of onset and upregulated HLA-DRB1, are in a more pro-inflammatory state (e.g. by microglia) than AA-carriers.
Our survey of the literature and the GWAS catalogue database (https://www.ebi.ac.uk/gwas/) revealed that SNPs within or close to the C6orf10/LOC101929163 locus (Supplementary Fig. 8) are associated with autoimmune disorders (multiple sclerosis, rheumatoid arthritis, systemic sclerosis, Grave’s disease and asthma), as well as neurodegenerative diseases (FTD, Parkinson’s disease and Alzheimer’s disease) (Lambert et al., 2013; Ferrari et al., 2014; Lu et al., 2017), highlighting the role of the immune system in neurodegeneration (Supplementary Table 11). Notably, several dementia genes are linked to microglia/immune function (e.g. TREM2 and CD33) (Lambert et al., 2013). Our study of C9orf72-negative patients suggests that the C6orf10/LOC101929163 locus could be a modest age of onset modifier for the general population of FTD patients. Intriguingly, another two SNPs (rs9268877 and rs9268856) near this locus have been reported in a case-control GWAS as modifiers of FTD risk (Ferrari et al., 2014). Of note, rs9357140 is not in LD with rs9268877 and rs9268856 representing an independent association signal (Supplementary Table 11), yet the mechanism behind the association with age of onset or disease risk could be similarly pointing to the functional significance of the HLA-DRA/HLA-DRB5 locus.
Notably, age of onset estimation is more objective for ALS (self-reported) than FTD (reported by family members) (Pottier et al., 2018). Hence, the less significant result in the replication C9orf72 cohort enriched in FTD patients (72.7% versus 27.8% in the discovery stage) may be explained by a less accurate age of onset estimation (Supplementary Table 12). In addition, the subgroup analysis could be further complicated by the less accurate estimation of age of onset for the complex FTD-ALS phenotype and reduced statistical power for the smaller subgroup.
One of the limitations of our study is the lack of unified deep phenotyping for each patient and healthy control data, however our findings set the basis for future research (e.g. aimed at investigating the link between CpG-SNPs and disease phenotype, risk, progression or severity). Another limitation is the absence of information on the expansions size in our study participants, because C9orf72 genotyping was done by repeat-primed PCR. This is of note, since repeat length examined by Southern blot was inversely correlated with age of onset (van Blitterswijk et al., 2013), and the clinical data support disease anticipation in C9orf72 families, which is evident by an earlier age of onset across successive generations (van Blitterswijk et al., 2013; Xi et al., 2015a; Van Mossevelde et al., 2017). However, C9orf72 repeat expansions are difficult to size accurately by Southern blot because of their large size (up to several thousand repeats) and somatic mosaicism masking the true length of the expansion (Xi et al., 2015a; McGoldrick et al., 2018). It would also be important to understand the genetic-epigenetic links across human tissues relevant to neurodegenerative disorders, since DNA methylation changes reflect the complex interactions between genes, environmental factors, and ageing (Zhang et al., 2016).
Our findings suggest that CpG-SNPs at the C6orf10/LOC101929163 locus might modify age of onset in C9orf72 carriers belonging to the entire ALS-FTD spectrum by controlling DNA methylation and gene expression (e.g. HLA-DRB1). CpG-SNPs at the C6orf10/LOC101929163 locus might also be age of onset modifiers for general FTD patients to a lesser extent. Understanding the functional mechanisms of the C6orf10/LOC101929163/HLA-DRB1 pathway (e.g. to investigate if the non-coding RNA LOC101929163 is a modulator of HLA-DRB1 expression) might prove critical for identifying biomarkers and/or designing drugs to modify age of onset in C9orf72 driven disease. Finally, the detected CpG-SNPs could be used to better predict age of onset in C9orf72 asymptomatic carriers in preventive clinical trials (e.g. based on the Genetic Frontotemporal dementia Initiative study) (Rohrer et al., 2015), for designing conditional and/or modifiers studies in the sporadic FTLD spectrum, such as based on IFGC related projects (https://ifgcsite.wordpress.com/) and for genetic counselling.
Supplementary Material
Acknowledgements
We would like to thank the patients who participated in this study.
Glossary
Abbreviations
- ALS
amyotrophic lateral sclerosis
- FTD
frontotemporal dementia
- GWAS
genome wide association study
- LD
linkage disequilibrium
- SNP
single nucleotide polymorphism
Appendix 1
We acknowledge the following members of the International FTD-Genomics Consortium (https://ifgcsite.wordpress.com/) for their contribution of genotyping and clinical data of FTD/FTD-ALS patients (funding, as well as full location and affiliations details can be found in the Supplementary material): Raffaele Ferrari, Dena G. Hernandez, Michael A. Nalls, Jonathan D. Rohrer, Adaikalavan Ramasamy, John B. J. Kwok, Carol Dobson-Stone, William S. Brooks, Peter R. Schofield, Glenda M. Halliday, John R. Hodges, Olivier Piguet, Lauren Bartley, Elizabeth Thompson, Isabel Hernández, Agustín Ruiz, Mercè Boada, Barbara Borroni, Alessandro Padovani, Carlos Cruchaga, Nigel J. Cairns, Luisa Benussi, Giuliano Binetti, Roberta Ghidoni, Gianluigi Forloni, Diego Albani, Daniela Galimberti, Chiara Fenoglio, Maria Serpente, Elio Scarpini, Jordi Clarimón, Alberto Lleó, Rafael Blesa; Maria Landqvist Waldö, Karin Nilsson, Christer Nilsson, Ian R. A. Mackenzie, Ging-Yuek R. Hsiung, David M. A. Mann, Jordan Grafman, Christopher M. Morris, Johannes Attems, Timothy D. Griffiths, Ian G. McKeith, Alan J. Thomas, Pietro Pietrini, Edward D. Huey, Eric M. Wassermann, Atik Baborie, Evelyn Jaros, Michael C. Tierney, Pau Pastor, Cristina Razquin, Sara Ortega-Cubero, Elena Alonso, Robert Perneczky, Janine Diehl-Schmid, Panagiotis Alexopoulos, Alexander Kurz, Innocenzo Rainero, Elisa Rubino, Lorenzo Pinessi, Ekaterina Rogaeva, Peter St George-Hyslop, Giacomina Rossi, Fabrizio Tagliavini, Giorgio Giaccone, James B. Rowe, Johannes C. M. Schlachetzki, James Uphill, John Collinge, Simon Mead, Adrian Danek, Vivianna M. Van Deerlin, Murray Grossman, John Q. Trojanowski, Julie van der Zee, Christine Van Broeckhoven, Stefano F. Cappa, Isabelle Leber, Didier Hannequin, Véronique Golfier, Martine Vercelletto, Alexis Brice, Benedetta Nacmias, Sandro Sorbi, Silvia Bagnoli, Irene Piaceri, Jørgen E. Nielsen, Lena E. Hjermind, Matthias Riemenschneider, Manuel Mayhaus, Bernd Ibach, Gilles Gasparoni, Sabrina Pichler, Wei Gu, Martin N Rossor, Nick C. Fox, Jason D. Warren, Maria Grazia Spillantini, Huw R. Morris, Patrizia Rizzu, Peter Heutink, Julie S. Snowden, Sara Rollinson, Anna Richardson, Alexander Gerhard, Amalia C. Bruni, Raffaele Maletta, Francesca Frangipane, Chiara Cupidi, Livia Bernardi, Maria Anfossi, Maura Gallo, Maria Elena Conidi, Nicoletta Smirne, Rosa Rademakers, Matt Baker, Dennis W. Dickson, Neill R. Graff-Radford, Ronald C. Petersen, David Knopman, Keith A. Josephs, Bradley F. Boeve, Joseph E. Parisi, William W. Seeley, Bruce L. Miller, Anna M. Karydas, Howard Rosen, John C. van Swieten, Elise G. P. Dopper, Harro Seelaar, Yolande A. L. Pijnenburg, Philip Scheltens, Giancarlo Logroscino, Rosa Capozzo, Valeria Novelli, Annibale A. Puca, Massimo Franceschi, Alfredo Postiglione, Graziella Milan, Paolo Sorrentino, Mark Kristiansen, Huei-Hsin Chiang, Caroline Graff, Florence Pasquier, Adeline Rollin, Vincent Deramecourt, Thibaud Lebouvier, Dimitrios Kapogiannis, Luigi Ferrucci, Stuart Pickering-Brown, Andrew B. Singleton, John Hardy, Parastoo Momeni.
Contributor Information
International FTD-Genomics Consortium (IFGC):
Raffaele Ferrari, Dena G Hernandez, Michael A Nalls, Jonathan D Rohrer, Adaikalavan Ramasamy, John B J Kwok, Carol Dobson-Stone, William S Brooks, Peter R Schofield, Glenda M Halliday, John R Hodges, Olivier Piguet, Lauren Bartley, Elizabeth Thompson, Isabel Hernández, Agustín Ruiz, Mercè Boada, Barbara Borroni, Alessandro Padovani, Carlos Cruchaga, Nigel J Cairns, Luisa Benussi, Giuliano Binetti, Roberta Ghidoni, Gianluigi Forloni, Diego Albani, Daniela Galimberti, Chiara Fenoglio, Maria Serpente, Elio Scarpini, Jordi Clarimón, Alberto Lleó, Rafael Blesa, Maria Landqvist Waldö, Karin Nilsson, Christer Nilsson, Ian R A Mackenzie, Ging-Yuek R Hsiung, David M A Mann, Jordan Grafman, Christopher M Morris, Johannes Attems, Timothy D Griffiths, Ian G McKeith, Alan J Thomas, Pietro Pietrini, Edward D Huey, Eric M Wassermann, Atik Baborie, Evelyn Jaros, Michael C Tierney, Pau Pastor, Cristina Razquin, Sara Ortega-Cubero, Elena Alonso, Robert Perneczky, Janine Diehl-Schmid, Panagiotis Alexopoulos, Alexander Kurz, Innocenzo Rainero, Elisa Rubino, Lorenzo Pinessi, Ekaterina Rogaeva, Peter St George-Hyslop, Giacomina Rossi, Fabrizio Tagliavini, Giorgio Giaccone, James B Rowe, Johannes C M Schlachetzki, James Uphill, John Collinge, Simon Mead, Adrian Danek, Vivianna M Van Deerlin, Murray Grossman, John Q Trojanowski, Julie van der Zee, Christine Van Broeckhoven, Stefano F Cappa, Isabelle Leber, Didier Hannequin, Véronique Golfier, Martine Vercelletto, Alexis Brice, Benedetta Nacmias, Sandro Sorbi, Silvia Bagnoli, Irene Piaceri, Jørgen E Nielsen, Lena E Hjermind, Matthias Riemenschneider, Manuel Mayhaus, Bernd Ibach, Gilles Gasparoni, Sabrina Pichler, Wei Gu, Martin N Rossor, Nick C Fox, Jason D Warren, Maria Grazia Spillantini, Huw R Morris, Patrizia Rizzu, Peter Heutink, Julie S Snowden, Sara Rollinson, Anna Richardson, Alexander Gerhard, Amalia C Bruni, Raffaele Maletta, Francesca Frangipane, Chiara Cupidi, Livia Bernardi, Maria Anfossi, Maura Gallo, Maria Elena Conidi, Nicoletta Smirne, Rosa Rademakers, Matt Baker, Dennis W Dickson, Neill R Graff-Radford, Ronald C Petersen, David Knopman, Keith A Josephs, Bradley F Boeve, Joseph E Parisi, William W Seeley, Bruce L Miller, Anna M Karydas, Howard Rosen, John C van Swieten, Elise G P Dopper, Harro Seelaar, Yolande A L Pijnenburg, Philip Scheltens, Giancarlo Logroscino, Rosa Capozzo, Valeria Novelli, Annibale A Puca, Massimo Franceschi, Alfredo Postiglione, Graziella Milan, Paolo Sorrentino, Mark Kristiansen, Huei-Hsin Chiang, Caroline Graff, Florence Pasquier, Adeline Rollin, Vincent Deramecourt, Thibaud Lebouvier, Dimitrios Kapogiannis, Luigi Ferrucci, Stuart Pickering-Brown, Andrew B Singleton, John Hardy, and Parastoo Momeni
Funding
This work was in part supported by the Canadian Consortium on Neurodegeneration in Aging (E.R., M.Z.), the ALS Canada-Brain Canada Hudson Grant (J.R., E.R., L.Z.), James Hunter ALS Initiative and the Temerty Family Foundation (L.Z., J.R.), Alzheimer’s Society grant #284 (R.F.), Argentine National Research Council (CONICET) (EIS), ALS Canada Clinical Research Fellowship (R.S.), National Institutes of Health (NIH) R35 NS097261, P50 AG016574, P01 NS084974 (RR), P50 AG016574 (N.R.G., D.W.D., J.E.P., B.F.B., R.C.P.), NIH P01 NS084974 (D.W.D.), NIH P01 AG019724 (B.L.M., W.W.S.), JPND PreFrontALS (733051042), JPND RiMOD (733051024), Memorabel-FTD (733050103) (J.C.v-S), the Flemish Government initiated Impulse Program on Networks for Dementia Research (VIND), the Methusalem Excellence Program, the Research Foundation Flanders (FWO) and the University of Antwerp Research Fund (C.V.B., J.v-d-Z.), NIH P01-AG-017586 (V.V.D.), “Investissements d’avenir” ANR-10-IAIHU-06, Assistance Publique—Hôpitaux de Paris (Clinical Research and Development Department), Programme Hospitalier de Recherche Clinique, FTLD-exome RCAOM-12123, the ANR-PRTS PREV-DEMALS project (I.L.B.), an MRC Clinician Scientist Fellowship (MR/M008525/1), the NIHR Rare Disease Translational Research Collaboration (BRC149/NS/MH), the MRC UK GENFI grant (MR/M023664/1) (J.D.R.), Swedish Research Council (Dnr 521-2010-3134, 529-2014-7504, 2015-02926), Alzheimer foundation Sweden, Brain Foundation Sweden, Swedish FTD Initiative, Swedish Brain Power, Karolinska Institutet doctoral funding, Gamla tjänarinnor, Stohnes foundation, Dementia foundation Sweden and the Stockholm County Council (ALF project) (CG), Ricerca Corrente, Italian Ministry of Health (G.R., G.B., L.B.), a National Health & Medical Research Council of Australia (NHMRC) Boosting Dementia Research Leadership Fellowship (1138223) (C.D.S.), NHMRC Senior Principal Research Fellowship (1079679) (G.M.H.), NHMRC Senior Research Fellowship (1103258) (O.P.), Fondazione CRF Grant 2015.0722, Fondo di Ateneo ex 60% anno 2017 (B.N.), Italian Ministry of health, grant RF08900000 (E.S.), Ministero dell’Istruzione, dell’Università e della Ricerca – MIUR project “Dipartimenti di Eccellenza 2018 – 2022” to Dept. of Neuroscience “Rita Levi Montalcini”, University of Torino (I.R., E.R.), grants PI13/02434 and PI16/01861, Acción Estratégica en Salud, integrated in the Spanish National R + D + I Plan and financed by ISCIII (Instituto de Salud Carlos III)-Subdirección General de Evaluación and the Fondo Europeo de Desarrollo Regional (FEDER- “Una manera de Hacer Europa”), Innovative Medicines Initiative 2 Joint Undertaking which receives support from the European Union’s Horizon 2020 research and innovation programme (ADAPTED Grant No. 115975) (A.R.). Replication data for C9orf72 carriers and for C9orf72 negative FTD/FTD-ALS patients have been obtained from the International FTD-Genomics Consortium (https://ifgcsite.wordpress.com/) dataset, funding sources of which are listed in the Supplementary material).
Competing interests
The authors report no competing interests.
References
- Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 2004; 64: 5245–50. [DOI] [PubMed] [Google Scholar]
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014; 30: 1363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1: 293–9. [DOI] [PubMed] [Google Scholar]
- GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group; Statistical Methods groups—Analysis Working Group; Enhancing GTEx (eGTEx) groups; NIH Common Fund, et al . Genetic effects on gene expression across human tissues. Nature 2017; 550: 204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72: 245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci USA 2005; 102: 7922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari R, Hernandez DG, Nalls MA, Rohrer JD, Ramasamy A, Kwok JB et al. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol 2014; 13: 686–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliano SA, Ptak C, Mak DYF, Shamsi M, Oh G, Knight J et al. Allele-skewed DNA modification in the brain: relevance to a schizophrenia GWAS. Am J Hum Genet 2016; 98: 956–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC et al. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol 2014; 127: 407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 2012; 11: 54–65. [DOI] [PubMed] [Google Scholar]
- Gijselinck I, Van Mossevelde S, van der Zee J, Sieben A, Engelborghs S, De Bleecker J et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol Psychiatry 2016; 21: 1112–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF et al. Classification of primary progressive aphasia and its variants. Neurology 2011; 76: 1006–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grambsch TMTaPM. Modeling survival data: extending the cox model. New York, NY: Springer; 2000. [Google Scholar]
- Hannon E, Spiers H, Viana J, Pidsley R, Burrage J, Murphy TM et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci 2016; 19: 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Rogaeva E. Motor neuron disease and frontotemporal dementia: sometimes related, sometimes not. Exp Neurol 2014; 262 (Pt B): 75–83. [DOI] [PubMed] [Google Scholar]
- Horvath S. DNA methylation age of human tissues and cell types. Genome Biol 2013; 14: R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C et al. Meta-analysis of 74 046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013; 45: 1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T et al. Analysis of protein-coding genetic variation in 60 706 humans. Nature 2016; 536: 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu RC, Yang W, Tan L, Sun FR, Tan MS, Zhang W et al. Association of HLA-DRB1 polymorphism with Alzheimer’s disease: a replication and meta-analysis. Oncotarget 2017; 8: 93219–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A et al. A reference panel of 64 976 haplotypes for genotype imputation. Nat Genet 2016; 48: 1279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGoldrick P, Zhang M, van Blitterswijk M, Sato C, Moreno D, Xiao S et al. Unaffected mosaic C9orf72 case: RNA foci, dipeptide proteins, but upregulated C9orf72 expression. Neurology 2018; 90: e323–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy NA, Arthur KC, Tienari PJ, Houlden H, Chio A, Traynor BJ. Age-related penetrance of the C9orf72 repeat expansion. Sci Rep 2017; 7: 2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51: 1546–54. [DOI] [PubMed] [Google Scholar]
- O’Rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AK et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016; 351: 1324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C, Zhou X, Perkerson RB, 3rd, Baker M, Jenkins GD, Serie DJ et al. Potential genetic modifiers of disease risk and age at onset in patients with frontotemporal lobar degeneration and GRN mutations: a genome-wide association study. Lancet Neurol 2018; 17: 548–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R. C9orf72 repeat expansions in patients with ALS and FTD. Lancet Neurol 2012; 11: 297–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011; 134 (Pt 9): 2456–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011; 72: 257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripatti S, Palmgren J. Estimation of multivariate frailty models using penalized partial likelihood. Biometrics 2000; 56: 1016–22. [DOI] [PubMed] [Google Scholar]
- Rohrer JD, Nicholas JM, Cash DM, van Swieten J, Dopper E, Jiskoot L et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol 2015; 14: 253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 2018; 24: 313–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker R, Deng J, Wang W, Zhang K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res 2010; 20: 883–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh J, Gustavsson EK, Vilarino-Guell C, Bortnick S, Latourelle J, McKenzie MB et al. DNM3 and genetic modifiers of age of onset in LRRK2 Gly2019Ser parkinsonism: a genome-wide linkage and association study. Lancet Neurol 2016; 15: 1248–56. [DOI] [PubMed] [Google Scholar]
- Turner SD. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. Journal of Open Source Software 2018; 3: 731. [Google Scholar]
- van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, Murray ME, Heckman MG, Diehl NN et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol 2013; 12: 978–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC et al. TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol 2014; 127: 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Mossevelde S, van der Zee J, Gijselinck I, Sleegers K, De Bleecker J, Sieben A et al. Clinical evidence of disease anticipation in families segregating a c9orf72 repeat expansion. JAMA Neurol 2017; 74: 445–52. [DOI] [PubMed] [Google Scholar]
- Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimer’s Res Ther 2015; 7: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z, van Blitterswijk M, Zhang M, McGoldrick P, McLean JR, Yunusova Y et al. Jump from pre-mutation to pathologic expansion in C9orf72. Am J Hum Genet 2015a; 96: 962–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z, Zhang M, Bruni AC, Maletta RG, Colao R, Fratta P et al. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol 2015b; 129: 715–27. [DOI] [PubMed] [Google Scholar]
- Zhang M, Tartaglia MC, Moreno D, Sato C, McKeever P, Weichert A et al. DNA methylation age-acceleration is associated with disease duration and age at onset in C9orf72 patients. Acta Neuropathol 2017; 134: 271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Xi Z, Ghani M, Jia P, Pal M, Werynska K et al. Genetic and epigenetic study of ALS-discordant identical twins with double mutations in SOD1 and ARHGEF28. J Neurol Neurosurg Psychiatry 2016; 87: 1268–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding authors (E.R., M.Z.). The data are not publicly available because of information that could compromise the privacy of the research participants.