


Abstract
With the rapid development of biotechnology, multi-dimensional genomic data are available for us to study the regulatory associations among multiple levels. Thus, it is essential to develop a tool to identify not only the modular patterns from multiple levels, but also the relationships among these modules. In this study, we adopt a novel non-negative matrix factorization framework (NetNMF) to integrate pairwise genomic data in a network manner. NetNMF could reveal the modules of each dimension and the connections within and between both types of modules. We first demonstrated the effectiveness of NetNMF using a set of simulated data and compared it with two typical NMF methods. Further, we applied it to two different types of pairwise genomic datasets including microRNA (miRNA) and gene expression data from The Cancer Genome Atlas and gene expression and pharmacological data from the Cancer Genome Project. We respectively identified a two-level miRNA–gene module network and a two-level gene–drug module network. Not only have the majority of identified modules significantly functional implications, but also the three types of module pairs have closely biological associations. This module discovery tool provides us comprehensive insights into the mechanisms of how the two levels of molecules cooperate with each other.
INTRODUCTION
Cellular system is complicatedly organized and cellular functions are mainly carried out in a highly modular manner (1,2). Thus, module discovery is helpful to investigate the complex regulation mechanisms of how different elements interact with each other in biological systems. Previous studies have proposed a number of methods to identify modular structure. One class is network topology-based methods, which identify highly connected sub-graphs in biological networks as modules (3–5). For example, MINE developed by Rhrissorrakrai and Gunsalus (4) performs an iterative cluster discovery procedure to find subnetworks in which nodes have high edge degree and local neighborhood density. OCG clustering method (5) focuses on decomposing a human protein–protein interaction network into overlapping modules based on the extension of Newman’s modularity function (3) to correctly assign multifunctional proteins. Another class is expression-based methods, which capture groups of genes with similar expression patterns in multiple samples. Existing studies have proposed many clustering approaches (6,7) such as hierarchical clustering, k-means, self-organizing map and matrix decomposition techniques (8–10) to analyze gene expression data to capture the global clusters, where a subset of genes exhibit highly correlated activities under all or a set of samples. For example, Zhang et al. (9) developed a singular value decomposition (SVD) tool svdPPCS to identify the conserved and divergent co-expression modules of two time series microarray datasets. Kim and Tidor (8) applied non-negative matrix factorization (NMF) to identify local patterns in gene expression data. In addition, gene expression profiles could also be used to detect modules by constructing a co-expression network. For example, the widely used tool WGCNA applies hierarchical clustering to the weighted gene co-expression network, creates a tree with branches and identifies modules by cutting the branches at a certain height (11).
Biological molecules also demonstrate multi-layer interaction and modular organization at multiple levels. The advance of genomic technologies makes it possible to simultaneously perform multi-platform genomic profiling and provides us the opportunities to integrate multi-dimensional genomic data to study the coordinate regulatory mechanisms. For example, in order to investigate the roles of microRNAs (miRNAs) in post-transcriptional gene regulation, several studies have proposed computational methods to discover miRNA–gene regulatory co-modules (12–15). Such co-module discovery methods have also been adopted for gene–drug co-modules from pairwise gene expression data and drug response data (16,17). In addition, there exist some methods for identifying modules from more than two levels of molecules (18,19). For instance, joint NMF (18) was adopted to discover multi-dimensional modules by integrating DNA methylation, gene and miRNA expression data across the same set of ovarian cancer samples. Moreover, network alignment and conserved module discovery across pairwise or multiple species are also popular paradigms (20–22). For example, Yang et al. (21) developed OrthoClust to simultaneously detect conserved and species-specific modules across multiple species. ModuleAlign (22) is a module-based global alignment of protein–protein interaction networks from two species.
Exploring the complex biological systems from the perspective of molecular modules rather than individual ones is very helpful for us to understand biological network design and systems behavior. Previous module discovery has made great progresses in many aspects, but most of them did not reveal module interactions from the same molecular level and different levels from a systematic view. Here, we not only aim to identify modules in one regulatory layer and co-modules in two different layers, but also the relationships among these identified modules. Moreover, these results could be demonstrated by a multi-layer module network, where each node represents a module and each edge represents existing interaction relationship between the two modules.
To this end, we develop a novel non-negative matrix factorization method NetNMF to construct the module network by integrating large-scale pairwise datasets in a network manner (Figure 1). We applied NetNMF to a set of simulated data and compared it with two typical NMF methods to demonstrate its effectiveness. We further applied it to the expression profiles of 12 106 genes and 804 miRNAs across the same set of 748 breast cancers from TCGA (23) and identified a two-layer miRNA–gene module network consisting of 69 miRNA–gene co-modules, 99 miRNA module links and 88 gene module links, which aids us to understand the mechanisms of how the miRNAs and genes cooperate with each other to perform certain functions. We also applied it to the expression profiles of 17 419 genes and drug response data of 205 drugs across 901 diverse types of cancer cell lines from the Cancer Genome Project (CGP) (24), and identified a two-layer gene–drug module networks consisting of 88 gene–drug co-modules, 113 gene module links and 122 drug module links. We found that not only the majority of identified modules have significantly functional implications, but also the three types of module pairs (gene–gene, drug–drug, gene–drug). The discovery of gene–drug module network here provides us a new tool to learn the drug action mechanisms from the gene regulation level and also predict the drug–target relationships and potential drug combinations for early clinical trials.
Figure 1.

Overview of the NetNMF for discovering a two-level module network by integrating pairwise genomic data. Three matrices R11, R12, R22 are computed via Pearson correlation, representing the similarities within and between two types of features in the pairwise input data matrices X1 and X2. NetNMF simultaneously decomposes R11, R12, R22 to get the underlying co-modules and their associations. The ith co-module is identified based on the ith column vector in factored matrices G1 and G2; the association degree between the ith and jth modules is determined by S11(i, j) (or S22(i, j)), where S(i, j) represents the element of the ith row and jth column in this matrix. Thus, a two-layer module network could be constructed in which a node represents a module.
MATERIALS AND METHODS
Data
We downloaded the gene expression data and miRNA expression data across the same set of 845 samples (748 breast cancer samples and 97 normal samples) from TCGA (23). We first removed the genes and miRNAs whose symbols could not be mapped to HGNC symbols. Then, we filtered the genes and miRNAs whose expression values are zeros across more than 90% of samples. Next, we did differential expression analysis for genes using the limma package in R (25) with adjusted P-value < 0.01, and log2 (fold change) > 0.5 in order to pre-filter genes less related to breast cancer. We obtained the breast cancer dataset including gene expression data 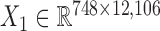 and miRNA expression data
and miRNA expression data 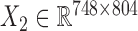 . At last, we calculated the gene co-expression matrix (network)
. At last, we calculated the gene co-expression matrix (network) 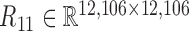 , gene-miRNA co-expression matrix (network)
, gene-miRNA co-expression matrix (network) 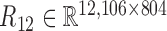 and miRNA co-expression matrix (network)
and miRNA co-expression matrix (network) 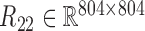 by means of Pearson correlation based on matrices X1 and X2.
by means of Pearson correlation based on matrices X1 and X2.
We also downloaded the pharmacogenomic data including gene expression data (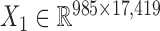 ) and drug response data (
) and drug response data (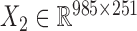 ) across the same set of 985 cell lines of various cancer types from CGP (24). For the drug response data, we first removed the drugs (or samples) with missing values across more than 30% of samples (or drugs) and then imputed the missing data with R package mice (26). At last, we obtained the pharmacogenomic dataset with
) across the same set of 985 cell lines of various cancer types from CGP (24). For the drug response data, we first removed the drugs (or samples) with missing values across more than 30% of samples (or drugs) and then imputed the missing data with R package mice (26). At last, we obtained the pharmacogenomic dataset with 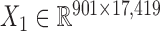 and
and 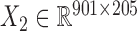 and calculated three correlation matrices
and calculated three correlation matrices 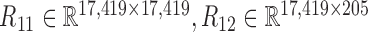 and
and 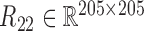 as done for the breast cancer data. Moreover, we replaced the correlation matrices R11, R12 and R22 by their corresponding absolute values respectively.
as done for the breast cancer data. Moreover, we replaced the correlation matrices R11, R12 and R22 by their corresponding absolute values respectively.
The NetNMF model
NMF and its variants have been increasingly applied to diverse fields including bioinformatics (8,13,18,27). The typical NMF decomposes a non-negative matrix X of size m × n into two non-negative matrices including the basis matrix 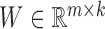 and the loading matrix
and the loading matrix 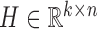 , such that X ≈ WH, where k < min{m, n}. That is, data X is explained as a positive linear combinations of basis vectors. We could obtain such a factorization by solving the following optimization problem:
, such that X ≈ WH, where k < min{m, n}. That is, data X is explained as a positive linear combinations of basis vectors. We could obtain such a factorization by solving the following optimization problem:
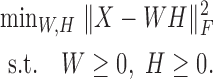 |
Besides the two-factor NMF, three-factor NMF (that is, X ≈ FSG) is also an important class of matrix factorization technique (28,29). Such format provides a framework to perform biclustering of data matrix X by matrices F and G, respectively. Factored matrix S not only provides an additional degree of freedom to make the approximation tight, but also indicates the relations between the identified clusters. Particularly, for the symmetric similarity matrix R, it could be factored into GSGT. The similarity matrix captures the intrinsic module or cluster structure within its original feature matrix (30,31). Here, we propose NetNMF to simultaneously decompose three similarity matrices calculated from X1 and X2. It combines the idea of two-factor and three-factor NMF and is formulated as follows:
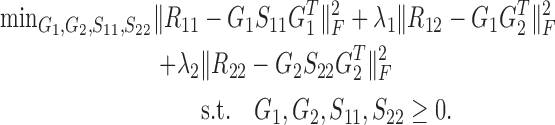 |
(1) |
where 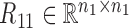 ,
, 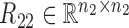 are the symmetric similarity matrices corresponding to two types of features, respectively and
are the symmetric similarity matrices corresponding to two types of features, respectively and 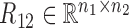 is for the similarities between them, which are all non-negative.
is for the similarities between them, which are all non-negative. 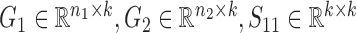 and
and 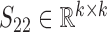 are the non-negative factored matrices. Here, k is a pre-determined parameter, and λ1, λ2 are the parameters to balance the scales of three terms in Equation (1). In the objective function Equation (1), the term of
are the non-negative factored matrices. Here, k is a pre-determined parameter, and λ1, λ2 are the parameters to balance the scales of three terms in Equation (1). In the objective function Equation (1), the term of 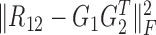 identifies the one-to-one relationships between the two types of modules, and it could also be regarded as a three-factor NMF version
identifies the one-to-one relationships between the two types of modules, and it could also be regarded as a three-factor NMF version 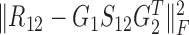 under the constraint S12 = I, which is used to enforce the ith module identified by G1 is only related to the ith module by G2; the other two terms respectively aim at identifying one type of modules as well as exploring the relationships within them via matrices S11 and S22.
under the constraint S12 = I, which is used to enforce the ith module identified by G1 is only related to the ith module by G2; the other two terms respectively aim at identifying one type of modules as well as exploring the relationships within them via matrices S11 and S22.
The NetNMF algorithm
Obviously, the optimization problem Equation (1) is not convex. Thus, it is unrealistic to find a global minimal solution. The idea of multiplicative update rules is one of the mostly used to solve NMF problems (28). By adopting this strategy, we develop the following algorithm to find a local minimal solution by updating matrices S11, S22, G1 and G2 alternately (Supplementary Materials).

In addition, parameters λ1 and λ2 are used to balance the three terms in the objective function Equation (1) and the elements of R11, R12, R22 are in [0,1]. Thus, an intuitive way is to set  ,
, 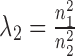 . To validate this setting, we compared the performance of NetNMF under it with several other settings when applied to the simulated datasets (Supplementary Materials). It indicates that there is no significantly difference between this setting and the optimal one for (λ1, λ2). Thus, such a setting could be as the default in NetNMF. When applying NMF-based methods to real data, we need to pre-determine the reduced dimension of the matrix factorization k, which is also the expected number of identified modules. Here, considering the dimensions of each dataset, we selected k = 70 from {50, 60, 70, 80, 90} for breast cancer dataset, and k = 90 from {80, 90, 100, 110, 120} for pharmacogenomic data. Under such setting, we found that the frequency of identified gene modules with significantly enriched GO terms is highest, indicating that we could discover the biologically meaningful modules as much as possible by classifying these features into this number of modules. Since this algorithm could not guarantee a global optimal solution, we repeated it for several times with different initializations and chose the best one with minimal objective value as the final decomposition.
. To validate this setting, we compared the performance of NetNMF under it with several other settings when applied to the simulated datasets (Supplementary Materials). It indicates that there is no significantly difference between this setting and the optimal one for (λ1, λ2). Thus, such a setting could be as the default in NetNMF. When applying NMF-based methods to real data, we need to pre-determine the reduced dimension of the matrix factorization k, which is also the expected number of identified modules. Here, considering the dimensions of each dataset, we selected k = 70 from {50, 60, 70, 80, 90} for breast cancer dataset, and k = 90 from {80, 90, 100, 110, 120} for pharmacogenomic data. Under such setting, we found that the frequency of identified gene modules with significantly enriched GO terms is highest, indicating that we could discover the biologically meaningful modules as much as possible by classifying these features into this number of modules. Since this algorithm could not guarantee a global optimal solution, we repeated it for several times with different initializations and chose the best one with minimal objective value as the final decomposition.
Determination of modules
The factored matrices G1 and G2 guide us to identify two types of modules, respectively. The main idea is to select the features with relatively large values of each column of G1 (or G2) as the members of each module. Specifically, we calculated z-scores of each column vector 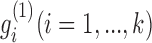 of G1 (or
of G1 (or 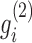 of G2) as below:
of G2) as below:
 |
(2) |
where  . Based on this transformation, we determined the ith module members if
. Based on this transformation, we determined the ith module members if 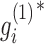 and
and 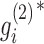 are larger than a given threshold T. Here, we set T = 3.5 for breast cancer dataset from TCGA and T = 3.7 for pharmacogenomic dataset from CGP to identify modules with proper resolution (Supplementary Figure S1). Too small T leads to big size module containing much redundant information, whereas too large T makes modules small leaving key molecules out.
are larger than a given threshold T. Here, we set T = 3.5 for breast cancer dataset from TCGA and T = 3.7 for pharmacogenomic dataset from CGP to identify modules with proper resolution (Supplementary Figure S1). Too small T leads to big size module containing much redundant information, whereas too large T makes modules small leaving key molecules out.
Determination of module links
Given the factorization 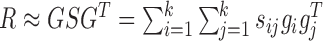 where gi is the ith column vector of G and sij is the ith row and jth column element of S, the decomposed latent vectors gis could reconstruct the original relationship matrix R, and sij could be regarded as the weight of
where gi is the ith column vector of G and sij is the ith row and jth column element of S, the decomposed latent vectors gis could reconstruct the original relationship matrix R, and sij could be regarded as the weight of 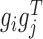 in the reconstruction of R. That is, under the normalization of gi (i = 1, ..., k), the larger sij is, the larger the elements of R for all the combinations of the selected features based on gi and gj are, which indicates high similarity between modules determined by gi and gj. Thus, we could utilize the diagonal elements in S to evaluate the quality of identified modules, and use the non-diagonal elements to determine the possible links between distinct modules (Supplementary Materials).
in the reconstruction of R. That is, under the normalization of gi (i = 1, ..., k), the larger sij is, the larger the elements of R for all the combinations of the selected features based on gi and gj are, which indicates high similarity between modules determined by gi and gj. Thus, we could utilize the diagonal elements in S to evaluate the quality of identified modules, and use the non-diagonal elements to determine the possible links between distinct modules (Supplementary Materials).
Functional enrichment analysis for co-modules
We utilized the gProfileR package in R (32) to conduct functional enrichment analysis for gene modules. For miRNA modules, we firstly extracted the target genes of each miRNA supported by more than two databases of miRTarBase (33), TarBase (34) and miRecords (35), and then we performed the enrichment analysis for target gene set of each miRNA module. We selected the significantly enriched GO biological process (BP) and KEGG pathway terms with less than 500 genes for each identified gene or miRNA module if Bonferroni-corrected P-value < 0.05.
RESULTS
Simulation study and comparison
We compared NetNMF with NMF and TriNMF by applying them to a set of simulated data (Supplementary Materials). NetNMF simultaneously decomposes three similarity matrices R11, R12 and R22. NMF factors only one matrix R12 into G1 and G2 to identify co-modules; TriNMF decomposes R11, R22 into three matrices to obtain G1, G2, respectively.
We adopted purity and the area under receiver operating characteristic curves (AUC) measures to evaluate the performance of different methods; purity measures the accuracy of modules identified relative to the real embedded modules in simulated data. Here, to exclude the impact of threshold T, we demonstrated the comparison results of three methods under two different threshold values (i.e. T = 1 and 1.5). Both of them show that NetNMF always performs better than TriNMF and NMF under the increasing noise levels (Figure 2A and B). Moreover, the AUC scores without any pre-defined thresholds of NetNMF are also higher than those of TriNMF and NMF especially for the data with high noise (Figure 2C). These results suggest that NetNMF performs better in identifying pairwise modules, which proves that incorporating much relationships within and between different types of data is indeed helpful to discover the hidden co-modular patterns in complex pairwise datasets.
Figure 2.

Performance comparison of NetNMF, NMF and TriNMF in terms of purity as well as AUC in simulated datasets. (A and B) The boxplots of purity scores for identified co-modules in 30 realizations on the simulated data with respect to different noise levels. Different thresholds (T = 1 for (A) and T = 1.5 for (B)) are used for selecting features from both factored matrices G1 and G2. (C) The boxplots of AUC scores without any pre-defined parameters in the same 30 realizations.
TCGA breast cancer data
First, we applied NetNMF to miRNA and gene expression data across the breast cancer samples derived from TCGA (23) (‘Materials and Methods’ section) and identified 69 matched miRNA–gene modules after removing one empty miRNA module. The 69 miRNA–gene co-modules cover 179 genes and 11 miRNAs on average (Supplementary Table S1 and Figure S1A and B). We also applied hypergeometric test to evaluate the degree of overlap of any two gene (or miRNA) modules. Only 169 out of 2346 (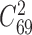 ) pairs of gene modules and 16 pairs of miRNA modules have significant overlap (FDR < 0.05). Besides, the co-module member genes and miRNAs exhibit highly co-expression patterns compared with randomly selected features (e.g. co-module 48 in Figure 3D).
) pairs of gene modules and 16 pairs of miRNA modules have significant overlap (FDR < 0.05). Besides, the co-module member genes and miRNAs exhibit highly co-expression patterns compared with randomly selected features (e.g. co-module 48 in Figure 3D).
Figure 3.

Illustration of the two-layer module network using TCGA breast cancer dataset. (A) The miRNA–gene module network consists of 69 miRNA modules in the top layer, 69 gene modules in the bottom layer, 69 edges (dash lines with equal weights) of one-to-one matching miRNA–gene co-modules and 99 edges between gene modules and 88 edges between miRNA modules weighted by the corresponding values in factored matrices S11 and S22, respectively. gMx (or mMx) indicate a gene (or miRNA) module with index x. (B) The module 48-centered subnetwork. (C) The detailed network for each module in (B). Some pairs of miRNAs in one miRNA module are linked if the two miRNAs share at least one target. The gene network for one gene module is constructed based on GeneMANIA (41). (D) Heat map of co-module 48 consisting of 171 genes and 11 miRNAs (squared boxes) based on the input similarity matrices of NetNMF. We extended the heat map to cover more variables by randomly selecting 171 genes and 11 miRNAs for contrasting. (E and F) Top biological terms enriched in the gene modules (E) and miRNA modules (F) in (B). The enrichment ratio indicates the functional significance of a module with −log10 (P-value) (Bonferroni-corrected P-value). Similar setting is used in Figure 5.
Gene and miRNA modules have significant functions
In total, 43 (62%) gene modules and 52 (75%) miRNA modules are enriched in at least one BP term or KEGG pathway (Supplementary Table S1). These modules are enriched in 752 distinct GO BP terms and 75 KEGG pathways. For gene modules, the most frequent enriched BP terms are microtubule-based process and microtubule cytoskeleton organization, which are closely related to cancer cells. Microtubules are dynamic filamentous cytoskeletal proteins (36). They are usually present in interphase cells and dividing cells constituting the mitotic spindle. Changes in microtubule stability have been reported for a range of cancers. Moreover, microtubules binding agents are a key class of anticancer agents with high activity in patients. For miRNA modules, the most frequent BP term is cell cycle G1/S phase transition, which is a key stage in cell cycle. The most frequently enriched KEGG pathways are cytokine–cytokine receptor interaction for gene modules and miRNAs in cancer for miRNA modules. Cytokines are secretory proteins that mediate intercellular communication in the immune system. Previous studies have showed that several cytokines, such as Interleukin (IL) -1, -6 and transforming growth factor beta (TGF-β), regulate the inflammatory tumor micro-environment, and thus stimulate cancer cell proliferation and invasion (37).
MiRNA–gene co-modules demonstrate regulatory relationships
In 37 of 69 identified miRNA–gene co-modules, the target genes of miRNAs also exist in their matched gene modules. Gene and miRNA members in 33 co-modules are both enriched in at least one BP term or KEGG pathway. The genes and miRNAs in five co-modules share the same enriched biological functions. For example, in co-module 41, two miRNA target genes (GIMAP4 for hsa-mir-146a and CARD11 for hsa-mir-155) are in this gene module. Meanwhile, this gene module and miRNA module are enriched in the same BP—endocytosis.
Besides, although the genes and miRNAs in other 28 co-modules do not share any biological functions, most of them are enriched in the highly related BPs. For example, both genes and miRNAs in co-module 22 are enriched in blood vessel development related functions, that is, gene module is enriched in endothelium development and sprouting angiogenesis; miRNA module is enriched in the terms of positive regulation of vascular endothelial growth factor receptor signaling pathway and positive regulation of sprouting angiogenesis. This analysis shows obvious regulatory relationships from miRNA level to gene level.
Linked gene (or miRNA) modules perform related functions
Based on the factored matrices S11 and S22, we determined 88 links between gene modules and 99 links between miRNA modules (‘Materials and Methods’ section). Among them, 49 pairs of gene modules and 5 pairs of miRNA modules have significant overlap (hypergeometric test with FDR < 0.05). A total of 14 of 44 pairs of gene modules and 25 of 54 pairs of miRNA modules, both of which are enriched in at least one BP term or KEGG pathway, possess the common enriched biological functions. For example, gene modules 38 and 41 share the inflammatory response function, but focus on different aspects. In gene module 38, the inflammatory response is mainly related with the processes of lipid metabolism in blood and foam cell differentiation. Foam cell is formed when macrophage tries to destroy the lipid deposit on the blood vessel walls and this process correlates to inflammatory responses (38). Another gene module 41 highly involves in the inflammatory response induced by lymphocyte and leukocyte activation processes. For the pair of miRNA modules 59 and 60 consisting of 12 and 8 miRNAs, in which there is only one common miRNA. But they are both enriched in several KEGG pathways including miRNAs in cancer, cell cycle and so on.
On the other hand, for those gene (or miRNA) module pairs without any common enriched biological functions, they also have closely functional associations. For example, for the pair of gene modules 9 and 15, which are respectively enriched in 28 and 13 functional terms significantly, they both involve in cellular metabolic processes. The top significantly enriched BPs in gene module 9 are mainly cyclic nucleotide metabolic process-related categories, including regulation of cyclic adenosine monophosphate (cAMP) metabolic process and cyclic nucleotide metabolic process. The cyclic nucleotides and cAMP are both important intracellular signal transduction molecules, acting as the second messengers between an extracellular signal and the elicited intracellular response (39). Interestingly, gene module 15 is enriched in extracellular matrix (ECM)-related metabolic terms such as ECM organization and collagen metabolic process. Collagen is the main structural element of ECM, playing a key role in cell adhesion and cell-to-cell communication (40).
In summary, for the detected module pairs by NetNMF, some of them share certain biological functions, but also have their own specific roles. Meanwhile, other module pairs without common enriched functional terms also have distinct coordinating relationships. Thus, NetNMF not only identifies gene modules and miRNA modules with highly co-expression patterns and significant functions, but also detects the associations between gene modules, miRNA modules and miRNA–gene co-modules.
A two-level modular network reveals the regulatory relationships between genes and miRNAs
Based on these identified relationships, we could construct a two-layer module network, in which each node represents a gene module or a miRNA module at different levels (Figure 3A). Such a network provides us a new way to explore the regulatory mechanisms between miRNAs and genes. For example, module 48-centered subnetwork (Figure 3B) contains three miRNA modules and four gene modules. The detailed network constructed for each module (based on GeneMANIA (41)) is very dense (Figure 3C). Moreover, these modules all involve in angiogenesis related functions (Figure 3E and F). Angiogenesis is a hallmark of wound healing, cancer and inflammatory diseases (42). From the gene level, the centered gene module 48 is enriched in blood vessel morphogenesis and endothelial cell proliferation. Endothelial cells form the inner lining of blood vessels. Based on the computed matrix S11, gene module 43 and 63 link to gene module 48 with high weights. Gene module 63 is enriched in the positive regulation of cell differentiation process. In the initialization of vascular growth, angioblasts migrate to discrete locations, differentiate in situ and assemble into solid endothelial cords and then form a plexus with endocardial tubes (43). The most four enriched BPs with gene module 43 are respectively inflammatory response, leukocyte activation, lymphocyte activation and cytokine-mediated signaling pathway. This module likely function during organismal injury recovery such as wound healing, in which the inflammatory response is activated to produce a number of immune cells and new blood vessels occur (42). In such a process, cytokines and small proteins play important roles in cell signaling transition. Majority of them act as stimulus for cell proliferation and differentiation, especially for immune cells. Besides, they could also induce vascular cell growth and migration (44). Another gene module 62 with weak link to gene module 48, has no significantly enriched BPs, but has overlap with gene module 63, which indicates that this module is likely to involve in the regulation of angiogenesis. From the miRNA level, the target gene set of centered miRNA module 48 is also enriched in vasculature development. It links to another two miRNA modules—1 and 49. These two modules are both enriched in the pathway of miRNAs in cancer. They target several genes including BRCA1, GATA3 and NOTCH1, which are closely related to cancers such as breast or ovarian cancers. It is well known that angiogenesis plays an important role in tumor development, growth and metastasis (45). New blood vessels could supply adequate nutrients, oxygen and remove waste products for cancer cells. There have been some antiangiogenic therapy for cancer patients. Therefore, the module 48-centered subnetwork demonstrates highly cooperative biological functions. It enables us to have a comprehensive understanding for the angiogenesis process.
Comparison with other methods
To demonstrate the effectiveness of NetNMF, we also compared it with TriNMF and NMF when applying to the TCGA breast cancer data (Figure 4) as well as the CGP data (Supplementary Figure S3). We compared the modules identified by the three methods in terms of biologically functional enrichment when applied to TCGA breast cancer dataset (Figure 4) and CGP dataset (Supplementary Figure S3). The enriched BP terms by NetNMF have more significant P-value than those of TriNMF and NMF: for gene modules, 59% (NetNMF versus TriNMF, P < 0.001, one-sided Wilcoxon signed rank tests) and 66% (NetNMF versus NMF, P < 9.09e −7) BP terms are above the diagonal line, respectively (Figure 4A); for miRNA modules, 60% (NetNMF versus TriNMF, P < 5.88e −10) and 57% (NetNMF versus NMF, P < 9.44e −4) BP terms are above the diagonal line, respectively (Figure 4B). The advantages of NetNMF indicate that NetNMF is superior to TriNMF and NMF in identifying more biologically relevant gene or miRNA modules.
Figure 4.

Comparison of all the enriched GO BPs of modules detected by NetNMF, TriNMF and NMF using TCGA breast cancer dataset. NetNMF performs better than TriNMF and NMF via one-sided Wilcoxon signed rank tests. (A) Comparison for gene modules. For each GO BP, we compute enrichment scores (−log10(P-value)) and the highest score among all modules is taken as the final score of this GO BP for each method. The scores for NetNMF are plotted against those of TriNMF and NMF. Majority of terms are above the central diagonal line, that is, they are more significantly enriched using NetNMF than TriNMF (59%) or NMF (66%). (B) Comparison for the target gene set of miRNA modules. Similar setting with (A). About 60 and 57% of terms are above the central diagonal line comparing NetNMF with TriNMF and NMF, respectively.
CGP dataset
We also applied NetNMF to the gene expression data and drug response data from the same set of cancer cell lines (24) (‘Materials and Methods’ section), and extracted 88 matched gene–drug modules (after removing two empty ones) consisting of 200 genes and 3 drugs in each one on average (Supplementary Table S2 and Figure S1C and D). Only 298 out of 3828 (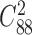 ) pairs of gene modules and no pair of drug modules have significant overlap with hypergeometric test (FDR < 0.05), indicating the identified modules tend to be distinct with each other. We found 66 out of 88 (75%) gene modules are enriched in at least one BP term or KEGG pathway. In total, they cover 984 different BP terms and 110 KEGG pathways respectively, in which the most frequent ones are leukocyte activation and lymphocyte activation.
) pairs of gene modules and no pair of drug modules have significant overlap with hypergeometric test (FDR < 0.05), indicating the identified modules tend to be distinct with each other. We found 66 out of 88 (75%) gene modules are enriched in at least one BP term or KEGG pathway. In total, they cover 984 different BP terms and 110 KEGG pathways respectively, in which the most frequent ones are leukocyte activation and lymphocyte activation.
For each drug module, we summarized their targets or target pathways. For 68 drug modules including more than one drug, the drugs in 33 modules (49%) share the same targets or pathways. For example, GSK690693 and MK-2206 in the 10th drug module, both have effects on PI3K signaling pathway; the five drugs (RDEA119, CI-1040, PD-0325901, Selumetinib and Trametinib) in the 72th drug module are all MEK inhibitors and target ERK MAPK signaling pathway.
Matched gene–drug modules demonstrate close associations
The enriched BPs in gene modules and the signaling pathway targeted by matched drug modules demonstrate strong relevance. In 10 gene–drug co-modules, the drug targets appear in the corresponding gene module. For example, in the 18th gene–drug co-module, the gene module and drug module are both related to cell-cycle arrest. The 197 genes in this module are significantly enriched in the negative regulation of G1/S transition of mitotic cell cycle, preventing the commitment of a cell from G1 to S phase of the mitotic cell cycle; positive regulation of cell cycle arrest by p53-mediated DNA damage response, resulting in the stopping or reduction in rate of the cell cycle; and some other negatively regulation of cell cycle phase transition. Nutlin-3a, one of the two drugs in this module, targets genes MDM2 and tumor suppressor p53, which are included in this gene module. Nutlin-3a inhibits the interaction between MDM2 and p53, which stabilizes p53 and then selectively induces senescence in cancer cells. Another drug, XMD15-27, targets CAMK2, which was reported as regulators of the cell cycle machinery. CAMK2 involves in the cell cycle associating with multiple cell signaling pathways. Its inhibition has various effects (promotion or suppression) on cell-cycle progression in various cancers (46).
For another example, the 60th gene–drug module includes 211 genes and four drugs. The genes in this module have significantly functional relevance with pigmentation such as development pigmentation, melanin metabolic and biosynthetic process. The four drugs are respectively PLX4720, SB590885, selumetinib and dabrafenib, where PLX4720, SB590885 and dabrafenib target BRAF, and selumetinib targets MEK1 and MEK2. These drugs all target ERK MAPK signaling pathway. BRAF has been an attractive target for melanoma drug development (47); MEK1 and MEK2 are key components in the MAPK signaling pathway. Moreover, a V600E mutation of the BRAF serine/threonine kinase (S/T kinase) is found occurred in more than 50% of all melanoma (48). Combination of BRAF and MEK inhibition in melanoma with BRAF V600 mutation, compared with BRAF inhibition alone, can delay the emergence of resistance and reduce toxic effects in patients, thereby improves the rate of progression-free survival (49).
Linked gene (or drug) modules have similar functions
We identified 113 links between gene modules, where 58 pairs have significant overlap (hypergeometric test, FDR < 0.05) and 122 links between drug modules. Among 113 pairs of gene modules, the two gene modules in 65 pairs are both enriched in at least one GO BP term or KEGG pathway and 28 of these 65 pairs share the same biological functions; 14 pairs of drug modules have the common targets or target pathways. For example, gene modules 11 and 29 are both enriched in cell cycle phase transition. However, there is little difference: gene module 11 involves in G2/M phase transition, whereas gene module 29 focuses on G1/S phase transition.
In addition, for the gene module pairs with no common enriched GO terms, we also found they tend to involve in the related BPs, such as the gene modules 10 and 36. They are respectively enriched in B-cell receptor (BCR) signaling pathway and lipid raft assembly. Recent studies have reported that lipid rafts participate in many of the cell surface events involved in B cell activation, including BCR signaling. Lipid rafts act as platforms for BCR signaling and might facilitate amplification of the BCR signaling after ligand binding (50).
For the drug modules, drug modules 10 and 14, respectively, target AKT1/AKT2 and mTOR, all of which are the components of their common target pathway—PI3K signaling pathway. Drug modules 1 and 15 affect distinct signaling pathways, which are ERK MAPK pathway and RTK pathway, respectively, but these two pathways are highly related. RTK and ERK MAPK signaling pathways both function in cell proliferation and differentiation regulation (51). Cross-talk occurs between these two pathways. The stimulation of RTKs triggers the activation of MAPKs in a multi-step process (52). All these analysis has suggested that NetNMF can reveal biologically meaningful links between modules, which provides deep insights into their organization.
A two-layer module network predicts the potential relationships between genes and drugs
Similarly, for the CGP dataset, we also constructed a two-layer module network (Figure 5A), which enables us to comprehensively explore not only the associations between gene modules with specific biological functions from the gene level (or drug modules with distinct drug targets and target pathways from the drug level), but also the multi-to-multi relationships between drugs and genes. For example, the module 37-centered subnetwork (Figure 5B and C) includes three gene modules and three drug modules, where the centered co-module 37 member genes and drug exhibit distinct co-expression patterns (Figure 5D). These gene modules are all involved in mRNA transcription-related BPs (Figure 5E). Module 37 including 44 genes are significantly enriched in regulation of histone H3K4 methylation. Classically, H3K4 methylation is implicated in activation of transcription (53) such as H3K4-me1, -me2 and -me3. This module links to two gene modules—87 and 28. Gene module 87 mainly participates in the process of spliceosomal complex assembly, which could catalyze nuclear mRNA splicing (54). The spliceosome is composed of small nuclear RNAs and protein factors. It plays the role of scissor to remove introns from a transcribed pre-mRNA. For another gene module 28, its top two enriched functions are respectively mRNA processing and mRNA splicing via spliceosomem (54). Besides, it is significantly enriched in mRNA surveillance pathway, which is a quality control mechanism that detects and degrades abnormal mRNAs (51). In short, these three gene modules show significant functional associations, participating in different aspects of mRNA transcription process.
Figure 5.

Illustration of the two-layer module network using the CGP dataset. (A) This gene–drug module network includes 88 gene–drug co-modules, 122 edges between drug modules in the top layer and 113 edges between gene modules in the bottom layer. (B) The module 37-centered subnetwork. (C) Both drug module 37 and 84 contain only one drug; drug module 10 includes two drugs targeting the same pathway (F). (D) Heat map of co-module 37 consisting of 44 genes and one drug (squared boxes). (E) Top enriched biological terms in gene modules in (B). (F) The details about drug modules in (B).
For the drug level, the centered drug module 37 contains only one drug—Navitoclax, targeting apoptosis suppressor proteins BCL-2, BCL-XL and BCL-W (Figure 5F). Thus, Navitoclax could trigger apoptosis in tumor cells, especially for the cancers with overexpressed BCL-2, BCL-XL and BCL-W. The drug FMK in module 84 targets RSK protein family, which is a group of highly conserved Ser/Thr kinases. As the downstream effectors of the ERK MAPK signaling cascade, RSKs play the roles of translational control in various stages (55). Another drug module 10 includes two drugs—MK-2206 and GSK690693, both of which are Akt inhibitors, but with different mode of action. MK-2206 is a kind of allosteric Akt inhibitor whereas GSK690693 is an adenosine triphosphate-competitive Akt inhibitor. Their combination displays a synergistic and cytotoxic effect, affecting PI3K-Akt signaling pathway at much lower concentration than using single drug (56). Akt lies at a critical signaling node downstream of PI3K-Akt pathway, and is important in regulating fundamental cellular functions such as transcription and translation (51). Moreover, ERK MAPK pathway and PI3K-Akt pathway are functionally correlated in tumorigenesis, and extensive cross-talk between these two pathways has been reported (56,57). Thus, all the four drugs have effects on the transcription activities, which are the main functions enriched by the corresponding gene modules (Figure 5E). The analysis above concludes that the members in module 37-centered subnetwork have distinct biological relevance. We could further make use of such a two-level subnetwork to predict new drug target candidates or potential drug combinations for clinical cancer therapy.
DISCUSSION
Module detection in complex biological networks is a crucial problem, which simplifies a complex system into several small parts with specific functions and thus aids us to study the mechanisms of molecular actions. With the dramatic advance of biotechnologies, large-scale genomic data from multiple dimensions are available, providing us the opportunities to detect modules from different levels together. Meanwhile, since a biological process is accomplished successfully by the cooperation of individual modules, thus identifying the relationships between different modules are also essential. Using the identified individual modules from different levels and their associations, we are able to construct multi-layer module networks to understand how the biological system functions. In this study, we present a method NetNMF to construct a two-layer module network via integrating three similarity matrices within and between two types of biological features. Compared with other two NMF-based methods, NetNMF can simultaneously discover the modular patterns and their relationships in a more accurate manner. This model could also be extended to integrate more than two types of features. Besides, prior interaction knowledge between molecules could be incorporated into the NetNMF framework in the form of network-based penalty terms (13) to make the linked features in the network more likely to be placed into the same module, which will improve the accuracy of module discovery and biological interpretability of modules.
DATA AVAILABILITY
A MATLAB package NetNMF is available at http://page.amss.ac.cn/shihua.zhang/.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [11661141019, 61621003, 61422309, 61379092]; Strategic Priority Research Program of the Chinese Academy of Sciences (CAS) [XDB13040600]; National Ten Thousand Talent Program for Young Top-notch Talents; Key Research Program of the Chinese Academy of Sciences [KFZD-SW-219]; National Key Research and Development Program of China [2017YFC0908405]; CAS Frontier Science Research Key Project for Top Young Scientist [QYZDB-SSW-SYS008]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Barabasi A., Oltvai Z.. Network biology: understanding the cell’s functional organization. Nat. Rev. Genet. 2004; 5:101–113. [DOI] [PubMed] [Google Scholar]
- 2. Zhang S., Jin G., Zhang X., Chen L.. Discovering functions and revealing mechanisms at molecular level from biological networks. Proteomics. 2007; 7:2856–2869. [DOI] [PubMed] [Google Scholar]
- 3. Girvan M., Newman M.. Community structure in social and biological networks. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rhrissorrakrai K., Gunsalus K.. MINE: module identification in networks. BMC Bioinformatics. 2011; 12:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Becker E., Robisson B., Chapple C., Guénoche A., Brun C.. Multifunctional proteins revealed by overlapping clustering in protein interaction network. Bioinformatics. 2012; 28:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kerr G., Ruskin H., Crane M., Doolan P.. Techniques for clustering gene expression data. Comput. Biol. Med. 2008; 38:283–293. [DOI] [PubMed] [Google Scholar]
- 7. Madeira S., Oliveira A.. Biclustering algorithms for biological data analysis: a survey. IEEE/ACM Trans. Comput. Biol. Bioinform. 2004; 1:24–45. [DOI] [PubMed] [Google Scholar]
- 8. Kim P., Tidor B.. Subsystem identification through dimensionality reduction of large-scale gene expression data. Genome Res. 2003; 13:1706–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang W., Edwards A., Fan W., Zhu D., Zhang K.. svdPPCS: an effective singular value decomposition-based method for conserved and divergent co-expression gene module identification. BMC Bioinformatics. 2010; 11:338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ihmels J., Bergmann S., Barkai N.. Defining transcription modules using large-scale gene expression data. Bioinformatics. 2004; 20:1993–2003. [DOI] [PubMed] [Google Scholar]
- 11. Zhang B., Horvath S.. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005; 4:1128. [DOI] [PubMed] [Google Scholar]
- 12. Liu B., Li J., Tsykin A.. Discovery of functional miRNA–mRNA regulatory modules with computational methods. J. Biomed. Inform. 2009; 42:685–691. [DOI] [PubMed] [Google Scholar]
- 13. Zhang S., Li Q., Liu J., Zhou X.. A novel computational framework for simultaneous integration of multiple types of genomic data to identify microRNA-gene regulatory modules. Bioinformatics. 2011; 27:401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu Y., Guo M., Liu X., Wang C., Liu Y., Liu G.. Identify bilayer modules via pseudo-3D clustering: applications to miRNA-gene bilayer networks. Nucleic Acids Res. 2016; 44:e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y., Liu W., Xu Y., Li C., Wang Y., Yang H., Zhang C., Su F., Li Y., Li X.. Identification of subtype specific miRNA-mRNA functional regulatory modules in matched miRNA-mRNA expression data: multiple myeloma as a case. Biomed. Res. Int. 2015; 2015:501262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kutalik Z., Beckmann J., Bergmann S.. A modular approach for integrative analysis of large-scale gene-expression and drug-response data. Nat. Biotechnol. 2008; 26:531–539. [DOI] [PubMed] [Google Scholar]
- 17. Chen J., Zhang S.. Integrative analysis for identifying joint modular patterns of gene-expression and drug-response data. Bioinformatics. 2016; 32:1724–1732. [DOI] [PubMed] [Google Scholar]
- 18. Zhang S., Liu C., Li W., Shen H., Laird P., Zhou X.. Discovery of multi-dimensional modules by integrative analysis of cancer genomic data. Nucleic Acids Res. 2012; 40:9379–9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li W., Zhang S., Liu C., Zhou X.. Identifying multi-layer gene regulatory modules from multi-dimensional genomic data. Bioinformatics. 2012; 28:2458–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ali W., Deane C.. Functionally guided alignment of protein interaction networks for module detection. Bioinformatics. 2009; 25:3166–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan K., Wang D., Rozowsky J., Zheng H., Cheng C., Gerstein M.. OrthoClust: an orthology-based network framework for clustering data across multiple species. Genome Biol. 2014; 15:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hashemifar S., Ma J., Naveed H., Canzar S., Xu J.. ModuleAlign: module-based global alignment of protein–protein interaction networks. Bioinformatics. 2016; 32:658–664. [DOI] [PubMed] [Google Scholar]
- 23. Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garnett M., Edelman E., Heidorn S., Greenman C., Dastur A., Lau K., Greninger P., Thompson I., Luo X., Soares J. et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012; 483:570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ritchie M., Phipson B., Wu D., Hu Y., Law C., Shi W., Smyth G.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Buuren S., Groothuis-Oudshoorn K.. mice: multivariate imputation by chained equations in R. J. Stat. Softw. 2011; 45:1–67. [Google Scholar]
- 27. Brunet J., Tamayo P., Golub T., Mesirov J.. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ding C., Li T., Peng W., Park H.. Orthogonal nonnegative matrix t-factorizations for clustering. ACM SIGKDD. 2006; 2006:126–135. [Google Scholar]
- 29. Žitnik M., Zupan B.. Data fusion by matrix factorization. IEEE Trans. Pattern Anal. Mach. Intell. 2015; 37:41–53. [DOI] [PubMed] [Google Scholar]
- 30. Eisen M., Spellman P., Brown P., Botstein D.. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Dam S., Võsa U., van der Graaf A., Franke L., de Magalhães J.. Gene co-expression analysis for functional classification and gene–disease predictions. Brief. Bioinform. 2017; bbw139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reimand J., Kull M., Peterson H., Hansen J., Vilo J.. g:Profiler – a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007; 35:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hsu S., Lin F., Wu W., Liang C., Huang W., Chan W., Tsai W., Chen G., Lee C., Chiu C. et al. miRTarBase: a database curates experimentally validated microRNA–target interactions. Nucleic Acids Res. 2010; 39:D163–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sethupathy P., Corda B., Hatzigeorgiou A.. TarBase: a comprehensive database of experimentally supported animal microRNA targets. RNA. 2006; 12:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xiao F., Zuo Z., Cai G., Kang S., Gao X., Li T.. miRecords: an integrated resource for microRNA–target interactions. Nucleic Acids Res. 2009; 37:105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dumontet C., Jordan M.. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010; 9:790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Esquivel-Velázquez M., Ostoa-Saloma P., Palacios-Arreola M., Nava-Castro K., Castro J., Morales-Montor J.. The role of cytokines in breast cancer development and progression. J. Interferon Cytokine Res. 2015; 35:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mohiuddin M., Pan H., Hung Y., Huang G.. Control of growth and inflammatory response of macrophages and foam cells with nanotopography. Nanoscale Res. Lett. 2012; 7:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fajardo A., Piazza G., Tinsley H.. The role of cyclic nucleotide signaling pathways in cancer: targets for prevention and treatment. Cancers. 2014; 6:436–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frantz C., Stewart K., Weaver V.. The extracellular matrix at a glance. J. Cell Sci. 2010; 123:4195–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Warde-Farley D., Donaldson S., Comes O., Zuberi K., Badrawi R., Chao P., Franz M., Grouios C., Kazi F., Lopes C. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010; 38:214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoeben A., Landuyt B., Highley M., Wildiers H., Van Oosterom A., De Bruijn E.. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004; 56:549–580. [DOI] [PubMed] [Google Scholar]
- 43. Conway E., Collen D., Carmeliet P.. Molecular mechanisms of blood vessel growth. Cardiovasc. Res. 2001; 49:507–521. [DOI] [PubMed] [Google Scholar]
- 44. Sprague A., Khalil R.. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009; 78:539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nishida N., Yano H., Nishida T., Kamura T., Kojiro M.. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006; 2:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang Y., Zhao R., Zhe H.. The emerging role of CaMKII in cancer. Oncotarget. 2015; 6:11725–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Villanueva J., Vultur A., Lee J., Somasundaram R., Fukunaga-Kalabis M., Cipolla A., Wubbenhorst B., Xu X., Gimotty P., Kee D. et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010; 18:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Puzanov I., Flaherty K.. Targeted molecular therapy in melanoma. Semin. Cutan. Med. Surg. 2010; 29:196–201. [DOI] [PubMed] [Google Scholar]
- 49. Long G., Stroyakovskiy D., Gogas H., Levchenko E., de Braud F., Larkin J., Garbe C., Jouary T., Hauschild A., Grob J. et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014; 371:1877–1888. [DOI] [PubMed] [Google Scholar]
- 50. Pierce S. Lipid rafts and B-cell activation. Nat. Rev. Immunol. 2002; 2:96–105. [DOI] [PubMed] [Google Scholar]
- 51. Kanehisa M., Goto S.. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McKay M., Morrison D.. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007; 26:3113–3121. [DOI] [PubMed] [Google Scholar]
- 53. Kouzarides T. Chromatin modifications and their function. Cell. 2007; 128:693–705. [DOI] [PubMed] [Google Scholar]
- 54. Carbon S., Ireland A., Mungall C., Shu S., Marshall B., Lewis S. the AmiGO Hub; the Web Presence Working Group . AmiGO: online access to ontology and annotation data. Bioinformatics. 2009; 25:288–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anjum R., Blenis J.. The RSK family of kinases: emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008; 9:747–758. [DOI] [PubMed] [Google Scholar]
- 56. Du J., Tong A., Wang F., Cui Y., Li C., Zhang Y., Yan Z.. The roles of PI3K/AKT/mTOR and MAPK/ERK signaling pathways in human pheochromocytomas. Int. J. Endocrinol. 2016; 2016:5286972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee E., Kim J., Kang Y., Ahn J., Kim J., Kim B., Choi H., Jeong M., Cho S.. Interplay between PI3K/Akt and MAPK signaling pathways in DNA-damaging drug-induced apoptosis. Biochim. Biophys. Acta. 2006; 1763:958–968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A MATLAB package NetNMF is available at http://page.amss.ac.cn/shihua.zhang/.